

Abstract
Inward rectifying potassium (Kir) channels play important roles in both excitable and nonexcitable cells of various organ systems and could represent valuable new drug targets for cardiovascular, metabolic, immune, and neurological diseases. In nonexcitable epithelial cells of the kidney tubule, for example, Kir1.1 (KCNJ1) and Kir4.1 (KCNJ10) are linked to sodium reabsorption in the thick ascending limb of Henle’s loop and distal convoluted tubule, respectively, and have been explored as novel-mechanism diuretic targets for managing hypertension and edema. G protein-coupled Kir channels (Kir3) channels expressed in the central nervous system are critical effectors of numerous signal transduction pathways underlying analgesia, addiction, and respiratory-depressive effects of opioids. The historical dearth of pharmacological tool compounds for exploring the therapeutic potential of Kir channels has led to a molecular target-based approach using high-throughput screen (HTS) of small-molecule libraries and medicinal chemistry to develop “next-generation” Kir channel modulators that are both potent and specific for their targets. In this article, we review recent efforts focused specifically on discovery and improvement of target-selective molecular probes. The reader is introduced to fluorescence-based thallium flux assays that have enabled much of this work and then provided with an overview of progress made toward developing modulators of Kir1.1 (VU590, VU591), Kir2.x (ML133), Kir3.X (ML297, GAT1508, GiGA1, VU059331), Kir4.1 (VU0134992), and Kir7.1 (ML418). We discuss what is known about the small molecules’ molecular mechanisms of action, in vitro and in vivo pharmacology, and then close with our view of what critical work remains to be done.
Keywords: drug discovery, high-throughput screening, medicinal chemistry, small molecules
OVERVIEW OF Kir CHANNEL STRUCTURE-FUNCTION RELATIONSHIPS
Most Kir channels exist as tetrameric complexes comprised of either identical (homomeric) or similar (heteromeric) subunits arranged around a centrally located, water-filled pore that serves to conduct potassium ions down their electrochemical gradient. The electrical currents generated by Kir channels are used by cells to carry out important functions in various tissues and organs, making them attractive drug targets for treating certain diseases. The so-called KATP channels of the Kir6 subfamily form hetero-octameric complexes with sulfonylurea receptors (i.e., SUR1, SUR2A, SUR2B) that imbue sensitivity of the channels to intracellular nucleotides and magnesium. Given their importance as drug targets for treating type 2 diabetes, the physiology, pathophysiology, and pharmacology of KATP channels have been reviewed extensively elsewhere (1–4) and will not be discussed further here.
Kir channel structures are relatively simple compared to members of the voltage-gated potassium channel (Kv) superfamily (5, 6), and this has important implications for the pharmacology of the Kir channel family (discussed below). Each subunit contains only ∼1,000 amino acids, possesses only two transmembrane domains (as opposed to six for Kv channels), and lacks a charged S4 domain that confers voltage-dependent gating to Kv channels (Fig. 1A). The biophysical and namesake property termed “inward rectification” (or “anomalous rectification”) refers to the propensity of Kir channels under voltage-clamp conditions to pass ionic current in the inward direction more readily than in the outward direction (Fig. 1B) (8). The mechanism of limiting outward potassium current flow involves the voltage-dependent blockade of the channel pore by intracellular polyamines, such as spermine and putrescine, and the divalent cation, magnesium (9–17). These cationic molecules interact electrostatically with negatively charged residues lining the membrane-spanning pore. Mutating a single aspartate residue (i.e., D172) in the classical strong inward rectifier, Kir2.1, to an uncharged asparagine residue, changes Kir2.1 into a relatively weak rectifier (13), whereas mutating the equivalent asparagine (i.e., N171) residue into an aspartate residue, turns the weak rectifier, Kir1.1, into a strong rectifier (15). Although there are other residues lining the channel pore that contribute to the rectification properties of a channel, residues at this position seem to play a dominant role in channel rectification and are therefore often called the “rectification controller.” These residues play a key role in determining the small-molecule pharmacology of Kir1.1, Kir2.1, Kir3.1, Kir4.1, and Kir7.1 (18–23).
Figure 1.

Overview of inward rectifier potassium channel structure and function. A: side- (top) and top-down (bottom) views of a Kir2.2 crystal structure model (7). Each of the four subunits is shown with a different color. The top-down view highlights the central location of ion-conduction pore. B: current-to-voltage relationship of Kir2.1 (strong rectifier) and Kir1.1 (weak rectifier) current recorded between -120 mV and +120 mV in the whole cell configuration of the patch-clamp technique. The cartoon figures show the direction of potassium ion flow at voltages more negative (bottom left) or more positive (top) than the Nernst potential for potassium, before (top left) and after (top right) intracellular block by magnesium (blue sphere) present in the pipette solution. The voltage-dependent block of Kir2.1 and positive voltages is responsible for the observed inward rectification. The weaker pore block by magnesium is responsible for weaker rectification observed in Kir1.1.
Unlike most of the other members of the Kir channel family, Kir3 channels (Kir3.1–Kir3.4) are regulated directly through interactions with G proteins of Gi/o-coupled G protein-coupled receptors (GPCRs). It is through this regulation the Kir3 channels derive their more common name, G protein gated, inward rectifying potassium (K+) channels (GIRKs). Activation of the Gi/o-coupled GPCR results in a conformational change in the interaction between GIRK α-subunits and the β/ɣ-subunits of the G protein complex, resulting in increased channel activity. A physiologically important example of this mechanism is the M2 muscarinic acetylcholine receptor-dependent modulation of heart rate. This theme is repeated throughout many types of excitable cells including neurons and hormone-secreting endocrine cells. The details of GPCR regulation of GIRK channels and the physiological and pharmacological consequences of such have been extensively reviewed elsewhere.(5, 24, 25) and will not be dealt with in detail in this review.
CLASSICAL PHARMACOLOGICAL TOOLS ARE WEAK AND NONSPECIFIC
Since Dr. Susumu Hagiwara first demonstrated that anomalously rectifying potassium currents in starfish eggs are inhibited by micromolar concentrations of barium (26), this divalent cation has become a favorite go-to blocker for studying Kir channels in diverse model systems. However, barium is poorly selectivity within the Kir channel family and has pleiotropic effects on numerous ion transport mechanisms (27–29). In this review, we focus on synthetic small molecules that were discovered or improved upon from efforts focused on the development of Kir-specific probes.
THE EVOLUTION OF HIGH-THROUGHPUT SCREENING PLATFORMS FOR POTASSIUM CHANNELS
Early efforts to develop pharmacological modulators of various types of ion channels was hampered by the lack of technologies that enable screening large libraries consisting of thousands or even millions of compounds. Indeed, manual voltage-clamp electrophysiology, still to this day considered the gold-standard method for measuring ion channel activity, is slow, labor intensive, and allows only one compound to be evaluated at a time. Radioligand displacement assays can be useful for high-throughput screening of drug-like chemical libraries, but the technique is limited in that it requires a radioligand and does not allow the measurement of a compound’s effect on channel activity. Some of the first efforts to develop activity-based high-throughput screening assays for potassium channels centered around measurements of radioactive rubidium flux through potassium channels (30). Although this technology, provided a medium-throughput method for screening thousands of compounds-per-day in 96-well plates, it was still too slow to keep pace with the rapidly growing chemical libraries in the late 1990s. Furthermore, the assay used a relatively strongly emitting radioisotope, requiring dedicated laboratory equipment, and they exhibited poor temporal resolution. In the late 1990s, a nonradioactive method of rubidium flux was developed using atomic absorption spectroscopy (31), but it was still limited in both throughput and temporal resolution.
The development of ion- or plasma membrane potential-sensitive, fluorescent sensors and the fluorometric imaging plate reader (FLIPR) (32), which is capable of measuring fluorescence from entire multiwell plates simultaneously, led to a revolution in target-based drug discovery for ion channels. Calcium-sensitive, fluorescent sensors like Fluo-3 and Fluo-4 transformed screening for modulators of GPCRs (33). Fluorescent, intracellular potassium sensors were developed (34) but were not widely used due to problems related to their low sensitivity and spectral properties. Plasma membrane potential-sensitive, fluorescent sensors were more widely used for measuring potassium channel activity (35); however, they also had considerable limitations. For example, the fact that these dyes detect changes in membrane potential resulting from ion flux and do not measure ion flux directly has disadvantages, which affect their utility. Thus, these assays are more susceptible to false negative and false positive effects. Although rubidium flux and plasma membrane potential sensor-based assays were both successfully used for medium- and high-throughput screening for potassium channel modulators, for many years there remained a gap between HTS-capable potassium channel assays and measurement of actual ion flux through potassium channels.
Approximately 15 years ago, the thallium flux technique was introduced (36) to enable a low-cost, fluorescence-based, nonradioactive, FLIPR-compatible assay capable of being scaled to use with the largest compound collections. Unlike plasma membrane potential sensitive sensor-based assays, the thallium-flux assay took advantage of the fact that thallium(I) readily passes through the pores of potassium channels and that fluorescent, thallium-sensitive sensors can be used to detect and quantify its influx into cells in a way analogous to calcium-sensitive fluorescent sensors. We will discuss efforts by us and other investigator to develop potent and specific “next-generation” Kir channel modulators using thallium flux-based HTS of compound libraries and medicinal chemistry for probe optimization.
NEXT-GENERATION Kir CHANNEL MODULATORS
VU590: The First Small-Molecule Inhibitor of Kir1.1 and Kir7.1
The first member of the Kir channel family to be cloned and functionally characterized was Kir1.1, which is encoded by the gene KCNJ1 on chromosome 11 (human) (37). Kir1.1 is commonly referred to as the renal outer medullary K (ROMK) channel due to its prominent and exclusive expression in renal tubules (38–40). The homomeric channel is expressed in the luminal membrane of the thick ascending limb (TAL) of Henle’s loop, the connecting tubule, and the cortical collecting duct (Fig. 2). In the TAL, Kir1.1 recycles potassium into the tubule lumen to energize the Na-K-2Cl co-transporter, NKCC2, which is critically involved in NaCl reabsorption and is the molecular target for the loop diuretics (41).
Figure 2.
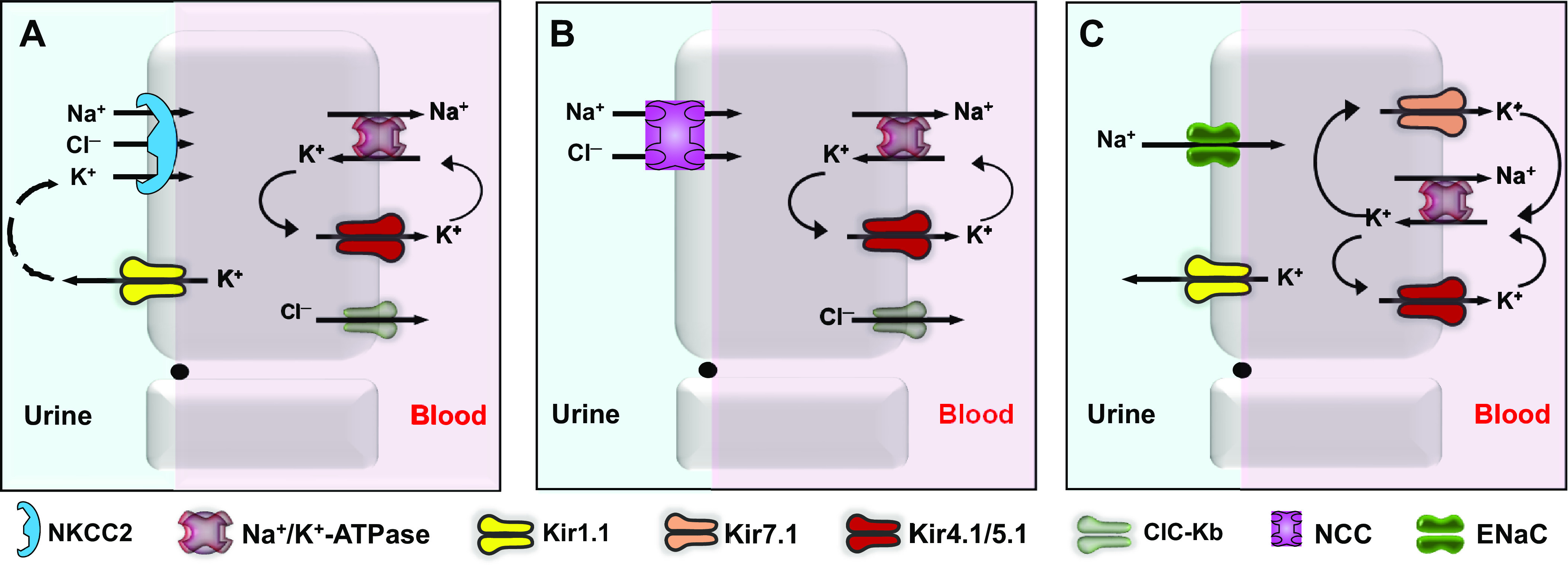
Molecular physiology of ion transport in the nephron. A: sodium chloride reabsorption in the thick ascending limb of Henle is mediated by the luminal sodium-potassium-chloride co-transporter, NKCC2. Kir1.1 (ROMK) recycles potassium into the tubule lumen to energize NKCC2. Sodium and chloride exit the basolateral membrane via the sodium pump and ClC-Kb chloride channels, respectively. B: the sodium-chloride co-transporter (NCC) mediates sodium chloride reabsorption in the distal convoluted tubule. Basolateral Kir4.1/5.1. channels hyperpolarize the membrane potential and promote chloride exit through ClC-Kb, which in turn stimulate WNK-SPAK kinase-dependent phosphorylation and activity of NCC. C: in the cortical collecting duct, luminal Kir1.1 channels hyperpolarize the apical membrane potential and create a favorable electrochemical driving force for sodium reabsorption through epithelial sodium channels (ENaC). Basolateral Kir4.1/5.1 and Kir7.1 channels also hyperpolarize the membrane potential and recycle potassium across the membrane to maintain the activity of the sodium pump. NKCC2, sodium-potassium-2-chloride.
Kir1.1 mediates potassium secretion in the connecting tubule and cortical and medullary collecting duct (Fig. 2). In the connecting tubule and collecting duct, Kir1.1 is electrically coupled to a sodium reabsorption pathway mediated by the epithelial sodium channel (ENaC), in which sodium reabsorption depolarizes the luminal membrane potential and thereby increases the electrochemical driving force for Kir1.1-dependent potassium secretion (42).
Kir1.1’s dual role in regulating sodium reabsorption in the thick ascending limb and potassium secretion in the distal nephron led us and others in the pharmaceutical industry to postulate that Kir1.1 might represent a novel diuretic target whose pharmacological blockade could induce natriuresis and diuresis without evoking potassium wasting. This notion was supported by human genetic data from the Framingham cohort study, showing that heterozygous carriers of loss-of-function mutations in KCNJ1 have lower blood pressure and are protected from age-dependent increases in blood pressure (43). However, because the molecular pharmacology of Kir1.1 when we started work in this area consisted of the peptide toxin, Tertiapin-Q (44), the tools for critically evaluating the safety and efficacy of inhibiting Kir1.1 were unavailable. We therefore set out to develop these tools using a molecular target based HTS approach.
We screened ∼225,000 compounds from the NIH Molecular Libraries Screening Center Network (MLSCN) library for small-molecule modulators of Kir1.1 using a fluorescence-based thallium flux assay. The first inhibitor we published, termed VU590 (Fig. 3), was a hit from the primary screen that inhibits Kir1.1 with an IC50 of 240 nM (45). Follow-up experiments revealed that VU590 had no effect on Kir2.1 and Kir4.1, but inhibited Kir7.1 with an IC50 of 8 µM. Both Kir1.1 and Kir7.1 exhibit voltage- and potassium-dependent block in such a way that suggested VU590 is a pore blocker that accesses its binding site through the cytoplasmic pore of the channel. We therefore performed scanning mutagenesis of the membrane-spanning pore, patch-clamp analysis of putative binding mutants, and molecular modeling to identify the functional binding site for VU590 in Kir1.1 and Kir7.1 (19). The only pore-lining residue evaluated that participates in VU590 block of Kir1.1 was asparagine 171 (N171), the so-called “rectification controller” discussed above. Mutation of N171 to either aspartate (N171D) or glutamate (N171E) led to a greater than 70-fold reduction in VU590 sensitivity. Interestingly, mutation of N171 to the structurally related (i.e., one carbon-length difference) amino acid glutamine (N171Q) led to a twofold loss in VU590 sensitivity. This latter observation is consistent with a model in which the amino acid structure and not simply charge at position 171 is an important determinant VU590 block. Surprisingly, we also found that this site could not only accommodate a bulky tyrosine residue at this position, but that this substitution (N171Y) increased VU590 block. Altogether, these data argue that N171 represents a physical binding site for VU590 in the Kir1.1 channel pore (Fig. 4) (19).
Figure 3.

Summary of next generation Kir channel modulators. Kir, inward rectifier potassium. (Reference sources listed in rightmost column in numerical order: 20, 23, 45, 46, 47, 88, 93, 94, 112, 116, 143.)
Figure 4.

VU590 functional binding sites in Kir1.1 and Kir7.1. Site-directed mutagenesis, voltage-clamp electrophysiology, and molecular modeling were used to identify Kir1.1-N171 (A) and Kir7.1-E149 and A150 (B) as essential for optimal block of the channels. These studies also revealed that Kir7.1-T153 creates an energetic barrier to small-molecule inhibitors accessing their deeper binding site near E149 and A150. Kir, inward rectifier potassium. [Reproduced from Kharade et al. (19) with permission.]
We were curious whether VU590 blocked Kir7.1 through a similar molecular mechanism and reasoned that the glutamate residue (Kir7.1-E149) in the equivalent position of Kir1.1-N171 accounted for the lower sensitivity to VU590. We hypothesized that mutation of E149 to an uncharged residue such as asparagine or glutamine would enhance block by VU590. Unfortunately, mutation of E149 to asparagine (Kir7.1-E149N) did not produce functional channels; however, the E149Q mutation led to a dramatic loss, not gain, of VU590 sensitivity. Shortening the side chain length by one carbon while maintaining the negative charge with an aspartate substitution (E149D) led to a significant increase in VU590 sensitivity, suggesting as we observed in Kir1.1 that this site of the channel is a physical binding site for VU590. Consistent with this model, mutating the neighboring alanine residue to a serine (A150S) also led to an abrogation of VU590 sensitivity.
Extending our mutagenesis analysis toward the cytoplasmic end of the channel pore revealed that mutation of threonine 153 to cysteine (T153C) led to a significant increase in block by VU590. Because T153 is predicted from homology modeling to reside only 6 Å and 8 Å away from A150 and E149, respectively, and the molecular dimensions of a low-energy conformer of VU590 (5 Å × 6 Å × 8 Å) approximate these distances, we postulated that there might be a functional interaction between VU590 and the three residues. Indeed, we found that the T153C mutation restored full VU590 potency to Kir7.1 channels also carrying E149Q or A150S mutations, suggesting that in the presence of a compromised binding site, T153 creates an energetic barrier that exacerbates low-affinity block (19).
If this model is correct, we postulated that the T153 barrier might impact other small molecules that bind in the same location but with different affinities. To test this hypothesis, we turned to two structurally related Kir7.1 inhibitors termed VU714 (IC50 = 1.5 µM) (Fig. 3) and ML418 (IC50 = 310 nM) (Fig. 3), the latter of which requires both E149 and A150 for fully efficacious block. Consistent with the model, the T153C mutation had no effect on the higher-affinity blocker ML418 but led to a significant increase in block by VU714. Taken together, these studies revealed a previously unrecognized role of T153 located at the base of the Kir7.1 channel pore in determining the pharmacological properties of intracellular pore blockers. In future high-throughput library screens, employing the Kir7.1-T153C mutant as a surrogate for the wild-type channel might help identify lower-potency pore blockers without affecting the ability to identify higher-affinity blockers (19).
VU591: The First Selective Inhibitor of Kir1.1
Because VU590 exhibited off-target activity toward Kir7.1 and therefore could not be used to interrogate the therapeutic potential of Kir1.1 as a diuretic target, we set out to develop a more selective inhibitors of Kir1.1 that might be used in vivo. One hit from the primary screen was bis-nitro-benzimidazole (BNBI), which exhibited some structural similarities with VU590. Both compounds contain nitro groups flanking an intervening linker region containing oxygen and/or nitrogen atoms, which gave us confidence that BNBI was a bona fide inhibitor of Kir1.1. Unfortunately, BNBI inhibits Kir1.1 with only weak potency (IC50 = 8 µM) compared to VU590. Importantly, however, BNBI had no effects on Kir7.1 at doses up to 100 µM. This indicated that although BNBI is a weak inhibitor of Kir1.1, it might be selective and serve as a good synthetic starting point for the development of more potent and specific analogs with medicinal chemistry. One key difference between BNBI and VU590 was the absence of an ether oxygen in the BNBI linker that might provide structural flexibility and/or polarity present in VU590 needed for high-affinity block. Strikingly, the addition of an ether oxygen in BNBI led to a 30-fold improvement in potency toward Kir1.1 (IC50 = 240 nM) without affecting selectivity over Kir7.1. Evaluating the ancillary pharmacology of this BNBI analog in patch-clamp experiments or radioligand biding assays revealed that it is selective for Kir1.1 over more than 65 potential ion channels, transporters, and receptors. We called this inhibitor VU591 (46) (Fig. 3).
Like VU590, VU591 exhibited voltage- and potassium-dependent block of Kir1.1, suggesting it too is a pore blocker. An unbiased computational approach was used to identify potential binding sites for subsequent interrogation with site-directed mutagenesis and patch-clamp analysis (21). An ensemble of 10 models with distinct conformations was constructed using crystal structures that were available at the time. 99,000 docking simulations of 200 low-energy conformations of VU591 were performed starting from three different locations in the membrane pore. Cluster analysis of the best poses revealed two potential binding sites in the pore: A “lower” site situated near the pore-cytoplasmic domain interface, and an “upper” site located in the center of the membrane (Fig. 5A). Scanning mutagenesis revealed that no pore-facing residues in the lower site are required for potent block of Kir1.1 by VU591. However, mutation of valine 168 (V168) or N171 led to a dramatic loss of VU591 activity. Shortening the side chain length at position 168 to an alanine (V168A) led to a fivefold reduction in potency, whereas lengthening it with leucine (V168L) led to a greater than 168-fold loss of activity. Mutation of N171 to N171D or N171E led to a complete abrogation of VU591 activity, whereas the more conservative mutation, N17Q, led to only a fivefold loss of VU591 sensitivity. The double mutation, V168A/N171Q, led to an additive loss of channel sensitivity, suggesting both residues mediate VU591 block of the channel pore. Closer inspection of a low-energy binding pose of VU591 in the pore (Fig. 5B) raised two intriguing mechanistic possibilities, which are not mutually exclusive. First, the nitro groups flanking the VU591 linker, which were essential for high-affinity block by VU590, are positioned within 5 Å of both residues and could therefore mediate interaction with the channel pore. The second detail is that VU591 must assume a folded “horseshoe” shape to be accommodated at that binding site. It is possible that the ether oxygen that improved potency over BNBI might allow for conformational flexibility needed to bind with high affinity to V168 and N171 (Fig. 5B).
Figure 5.

Identification of the VU591 functional binding site in Kir1.1. A: homology modeling and in silico docking were used to identify energetically favorable VU591 docking sites in putative “upper” and “lower” pore locations that were subsequently tested with mutagenesis and patch-clamp electrophysiology. No mutations in the lower site alter VU591 block; however, mutation of V168 and N171 led to a loss of VU591 potency. B: close-up view of VU591 docked into the upper site interacting with V168 and N171. Kir, inward rectifier potassium. [Reproduced from Swale et al. (21) with permission from Elsevier.]
Merck Kir1.1 Inhibitor Compound A: Therapeutic Proof-of-Concept and Renal Mechanisms of Action
In 2012, the ion channel group at Merck began publishing their work to develop of structurally diverse Kir1.1 inhibitors for use as novel mechanism diuretics (47). Interestingly, the first potent Kir1.1 inhibitor disclosed was a symmetrical small molecule with a nitrogen-containing linker flanked by two nitro groups, two key properties that are shared by VU591. We subsequently confirmed that, like VU590 and VU591, an optimized analog of this Merck compound, termed Compound A (Fig. 3), requires N171 for high-affinity Kir1.1 block (unpublished observations). Importantly, the Merck group demonstrated for the first time that pharmacological inhibition of Kir1.1 induces natriuresis and diuresis without causing potassium wasting, lowers blood pressure, and protects the kidney from hypertension-induced kidney injury (48, 49).
In light of this seminal data, we attributed the failure of VU591 to induce renal excretion to its poor metabolic stability and high serum protein binding. We therefore decided to use Compound A to better understand the renal mechanisms by which Kir1.1 inhibition leads to a potassium-sparing diuresis. We administered Compound A either alone or in combination with diuretics that act on discrete segments of the nephron and evaluated the effects on urine volume and electrolyte composition (50). The NKCC2 inhibitor, bumetanide, was used as an inhibitor of sodium reabsorption in the thick ascending limb, whereas hydrochlorothiazide (HCTZ), an inhibitor of the sodium-chloride co-transporter (NCC), was used to inhibit sodium reabsorption in the distal convoluted tubule. The ENaC inhibitors, amiloride or benzamil, were used to inhibition sodium reabsorption in the collecting duct. We reasoned that an additive or synergistic effect of Compound A with a diuretic would be evidence of the two compounds inhibiting sodium reabsorption in different nephron segments, whereas a lack of synergy would suggest that both compounds act on the same segment. Briefly, we found a synergistic effect between Compound A and HCTZ, which was expected since Kir1.1 is not co-expressed with NCC in the distal convoluted tubule. However, we did not observe synergy between Compound A and bumetanide or amiloride/benzamil, suggesting that Compound A inhibits sodium reabsorption in the thick ascending limb and collecting duct. Interestingly, Compound A was able to block the kaliuresis induced by bumetanide, confirming that at least one source of potassium wasting with loop diuretic administration is unopposed Kir1.1-mediated potassium secretion in the thick ascending limb. Compound A also inhibited potassium wasting induced by HCTZ and the residual potassium secretion in the presence of amiloride. Thus, the combined inhibition of sodium reabsorption in the thick ascending limb and inhibition of potassium secretion in the TAL and collecting duct account for the potassium-sparing diuretic effects of Compound A (50).
ML133: A Selective Inhibitor of Kir2.x Channels
The second member of the inward rectifying channel family to be cloned, Kir2.1 (51), is encoded by the KCNJ2 gene. The structure, function, and regulation of the channel are reviewed in Refs. 5 and 52. The Kir2.1 channel is expressed in numerous tissues including macrophages (51), heart (where it underlies IK1) (53), vasculature (54), microglia (55), and neurons (56, 57) and neutrophils (58). In these tissues, it plays a wide variety of roles including setting resting membrane potential (59), microglial patterning and development, signaling and migration (60), and smooth muscle proliferation and migration (61). Kir2.1 is known to co-assemble with a number of other members of the Kir2 channel family including Kir2.2, Kir2.3, and Kir2.4 (62, 63). Mutations in Kir2.1 channels have been linked to a variety of disorders including periodic paralysis, Anderson–Tawil, and short-QT syndromes (52, 64, 65). Kir2.1 has also been observed to be upregulated in small cell lung cancer where it may play a role in the development of drug resistance (66).
Although a number of compounds have been reported to be inhibitors (67–73) and in one case an activator of Kir2.x channels (74), they had all been previously described as modulators of other targets of a variety from target classes. However, ML133 (Fig. 3) was developed from a compound discovered through a Kir2.1-focused HTS (23) using a fluorescence-based thallium flux assay. The screen was funded through the Molecular Libraries Screening Center Network (MLSCN).
Initial characterization of ML113 revealed a potency in the low micromolar range. ML133 contains a basic nitrogen (pKa ∼ 8.8), which led Wang et al. (23) to explore whether pH has any effect on ML133’s ability to inhibit Kir2.1 channels. They observed that ML133 potency was strongly affected by pH, whereas Kir2.1 activity was not sensitive to changes in pH over the range tested. At pH 8.5, ML133 had a measure potency of 290 nM, whereas its potency was 1.8 µM and 10 µM at pH 7.4 and pH 6.5, respectively. These observations together with additional data led the investigators to conclude that it is the neutral form of ML133 that penetrates the membrane and that increasing the extracellular pH results in concentration of ML133 inside the cell.
ML133 was reported to possess a clean ancillary pharmacology profile based primarily on data from a radioligand-binding panel but also specifically including a lack of activity and N- and L-type calcium channels as well as Kv11.1 (hERG). Among other members of the Kir family that were evaluated, ML133 was at least 10-fold more potent at inhibiting Kir2.1 (1.8 µM) compared to Kir1.1 (>300 µM), Kir4.1 (76 µM), and Kir7.1 (33 µM). However, ML113 does inhibit other members of the Kir2 family, Kir2.2, Kir2.3, and Kir2.6, with potencies similar for inhibition of Kir2.1, in the low uM range.
In order to investigate structural determinants of ML133 function and specificity, Wang et al. (23) used Kir1.1 and Kir2.1 chimeric channels to identify D172 and I176 as critical for Kir2.1 inhibition. It is intriguing to note that this critical aspartate residue is in the same position as the asparagine residue found to be critical for VU590 function, namely the “rectification controller.” This finding suggests that this region of Kir channels may be a privileged position for inhibitor function. Although VU590’s effect on ROMK activity required an asparagine at this position, ML133 showed preference for aspartate.
Unfortunately, ML133 was found to have very high plasma protein binding and high intrinsic clearance. Even though these properties limit its utility to that of an in vitro probe, ML133 has seen widespread usage to investigate the broad diversity of roles Kir2 family members play: microglia (60, 75, 76), regulation of neuronal process (57, 77), modulation of autophagy in endothelial progenitor cells (78), sour taste transduction (79), effects on protective effects or ischemic preconditioning in cardiac ventricle (80), and to help validate the expression of Kir2.1 channels in neutrophils (58) and colon (81). Further refinement of ML133 analogs might result in the development of compounds with properties suitable for in vivo administration further expanding our ability to probe the function of Kir2.1 and other members of the Kir2 family and that these studies might reveal potential therapeutic utility of Kir2 inhibition.
ML297, VU0810464, GAT1508, and GiGA1: Selective GIRK1-Containing GIRK Activators
Continuing the theme from Kir1.1 and Kir2.1, until recently the pharmacology of GIRK channels was very poorly developed (82). The molecules published were limited to mostly poorly selective inhibitors with NTC 801 (83), being a notable exception. NTC 801 is a discontinued clinical candidate for the treatment of atrial fibrillation. It shows good specificity for inhibiting GIRK channels with low nanomolar potency. It was demonstrated to be safe in humans but in a limited clinical trial, it did not demonstrate an ability to treat atrial fibrillation.
The GIRK activator landscape was even sparser than the inhibitor landscape. As described above, the activity of GIRK channels is regulated in a positive fashion by Gi/o-coupled GPCRs. GIRK channels are also known to be positively modulated by intracellular Na+. However, the only reported exogenous GIRK activators were ethanol (84) the natural product, naringin, which activates GIRK with very low potency (85), and some compounds of undisclosed structure (86).
From an HTS that used GIRK channel activity as a readout for GPCR activity (87), a number of compounds were discovered that activated GIRK channels in the absence of an active Gi/o-couple GPCR. The discovery of these molecules led to the development of ML297 (88) (Fig. 3) and subsequently to a large number of related compounds including closely structurally related GIRK channel inhibitors (89). ML297 exhibited a potency of ∼300 nM and ∼ 5-fold preference for activating heteromeric channels containing the GIRK1 and GIRK2 subunits compared to channels comprised of GIRK1 and GIRK4 or GIRK1 and GIRK3. Surprisingly, ML297 was only able to activate heteromeric GIRK channels, which contain Kir3.1 (GIRK 1). Since GIRK 1 does not form channels on its own, ML297 is able to activate GIRK channels comprised of Kir3.1+ either Kir3.2, Kir3.3, or Kir3.4. A mutagenesis study revealed that two amino acids in GIRK 1 are responsible for ML297’s ability to activate GIRK1 containing channels (90) (Fig. 6). Wydeven et al. (90) demonstrated that transfer of both F137 and D173 residues from GIRK1 to their homologous positions in GIRK2 results in ML297 being able to activate heteromeric GIRK2/GIRK2-double mutant channels. The F137 residue has been previously identified as important for channel gating (91) and critical for preventing GIRK1 from forming functional homomeric channels (92). Among all of the inward rectifier channels, GIRK1 is unique in that it is the only one which contains a phenylalanine at this position. All other Kir channels contain a serine at this position, except for Kir2.4 that contains an alanine. The other key residue is an aspartate, D173. In GIRK2, the analogous amino acid residue is an asparagine. In repetition with a theme previously established with Kir1.1 and Kir2 modulators, this “rectification controller” position appears to also be a “pharmacology controller” position for a number of Kir modulators. Although the effect of ML297 on GIRK1-containing channels has not been investigated in as much mechanistic detail as the effect of flecainide on Kir2.1 (74), like flecainide, ML297 increases the open probability and weakens the inward rectification of the channel.
Figure 6.
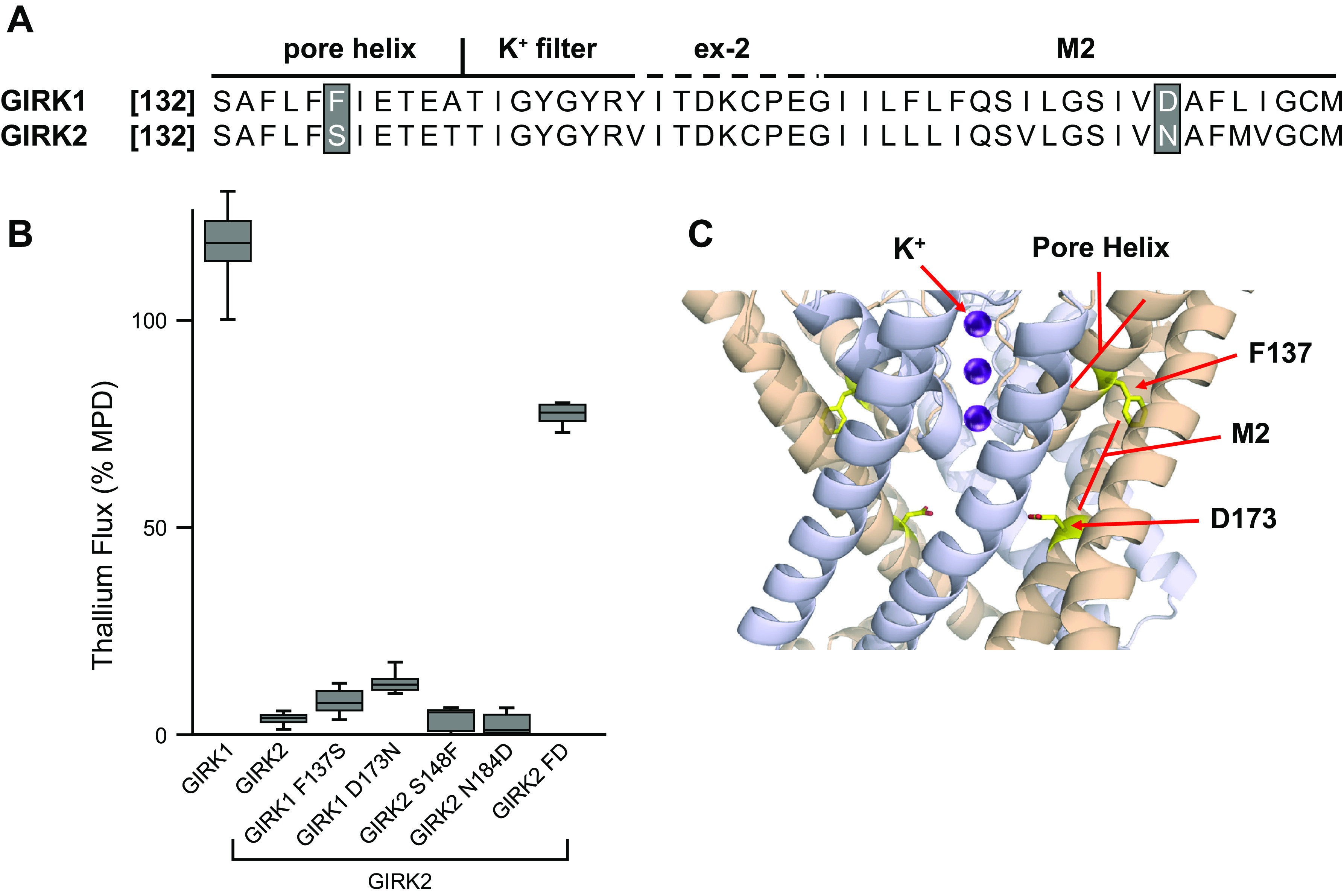
Identifications of amino acids critical for activation of GIRK channels by ML297. Shown in A are the aligned amino acid sequences of GIRK1 and GIRK2 highlighting the two positions in the sequence that were determined as critical for ML297 efficacy. Shown in B are the results of thallium flux assays from HEK-293 cells co-expressing wild-type GIRK2 along with either wild-type GIRK1, wild-type GIRK2, mutant GIRK1, or mutant GIRK2 channels. Mutation of either phenylalanine F137 or aspirate D173 in GIRK1 resulted in loss of ML297 efficacy. ML297 efficacy was only observed with co-expression of either wild-type GIRK2 along with wild-type GIRK1 or wild-type GIRK2 along with a GIRK2 mutant containing both residues, F148 and D184 (correspond to residues F137 and F173 found in GIRK1). Shown in C are the positions of GIRK1 F137 and D173 in a computer model of heteromeric GIRK1/GIRK2 based on the crystal structure of homomeric GIRK2. GIRK, G protein-coupled inward rectifier potassium. [Elements of this figure were derived from information previously published by Wydeven et al. (90).]
Although F137 and D173 are required for ML297’s ability to activate GIRK1-containing GIRK channels, it remains to be determined conclusively whether these amino acids interact directly with ML297. Computer modeling, mutagenesis, and medicinal chemistry studies have begun to shed more light on the role of these and other amino acid residues in activation of GIRK by ML297 and the closely structurally related compound, GAT1508 (93). These studies implicated additional residues as important for the preference for some compounds, like GAT1508 GIRK1/GIRK2 heteromers compared to GIRK1/GIRK4 heteromers.
Recently, Zhao et al. (94) reported the discovery of a new GIRK activator, GiGA1 (Fig. 3) discovered using computer modeling and virtual high-throughput screening. They found that, unlike other previously described compounds that require F137 and D173 in the GIRK1 subunit, GiGA1 was not sensitive to mutations of these residues. These data, the fact that the GiGA1 was discovered using a model of GIRK2 alcohol binding pocket, and that residues known to be important for the GIRK activating properties of alcohols also affect the ability of GiGA1 to activate GIRK channels, argue for ML297 and related compounds being functionally distinct from GiGA1.
Since its discovery, ML297 and other more recently discovered GIRK activators have been used by a number of groups to probe the function of GIRK channels in isolated cells, tissues, and in vivo. These studies include demonstration that pharmacologically increasing the activity of GIRK channels can produce efficacy rodent models of numerous disorders: anti-seizure (88, 94,95), anxiolytic (90, 96), anti-nociceptive (97, 98), facilitation of conditioned fear extinction (93), promotion of non-REM sleep (99), and rescue of amyloid-β-evoked deficits in hippocampal function (100, 101). These tools have also been used extensively to probe GIRK’s role in wide variety of systems (102–111). Continued work on these molecules has been focused on improving their drug-like properties and selectivity for the major neuronal GIRK heteromer, GIRK1/2 and away from the primary cardiac form, GIRK1/4: VU0810464 and GAT1508 (93, 96, 112) (Fig. 3).
Although largely overlooked, early medicinal chemistry studies on ML297 identified very closely structurally related compounds, which were GIRK inhibitors, not GIRK activators. Wen et al. (89) reported that the substitution of the methyl at the 3-pyridyl position in ML297 for isopropyl results in inhibition of GIRK channels, whereas the potency remains similar. In this study, the investigators observed that other substitutions at the 3-pyridyl position, but not other positions on the molecule resulted in GIRK inhibition. As is the case with the closely related GIRK channel activators, the efficacy of these ML297-like inhibitors requires the GIRK1 F137 and D173 residues, and thus, these compounds do not inhibit non-GIRK1-containing GIRK channels.
VU0529331 and Ivermectin Activators of Non-GIRK1-Containing GIRK Channels
As described above, ML297 and related compounds are unable to activate GIRK channels that do not contain the GIRK1 subunit. However, there are GIRK channels in the body which do not contain GIRK1 subunits. Although these non-GIRK-1 containing channels exhibit much more restricted expression than the GIRK1-containing GIRK channels, non-GIRK1-containing channels are expressed in some intriguing locations including the ventral tegmental area and the substantia nigra, reviewed in Ref. (24). Recently, compounds capable of activating non-GIRK1-containing GIRKs have been discovered. Using a compound library screening approaches, two groups of investigators discovered that the insecticide ivermectin is able to activate GIRK channels (113, 114). Unfortunately, the utility of this compound is limited because it is known to modulate the activity of a wide variety of ion channels include voltage-gated sodium channels, GABAa and glycine receptors (115). More recently, VU0529331 (Fig. 3) was identified using a thallium flux-based HTS focused specifically on discovering modulators capable of activating non-GIRK1-containing GIRKs (116). Although VU0529331 shows considerably better specificity for activating GIRK channels when compared to ivermectin, relatively low potency (∼ 5 µM) has limited its utility to isolated cell systems. Unfortunately, ivermectin and VU0529331 also activate GIRK1-containing GIRK channels, further limiting their utility as tools to selectively probe non-GIRK1-containing GIRK channels. Unsurprisingly, considering that VU0529331 and Ivermectin can activate GIRK1 and non-GIRK1-containing GIRKs, neither of their ability to activate GIRK channels depends on the presence of the F137 and D173 amino acid residues found in GIRK1 channels. In the future, improvements in VU0529331 may lead to compounds that are useful for probing the functions of non-GiRK1-containing GIRK channels in brain slices or potentially when precisely injected into specific brain regions, but until compounds capable which are selective only for non-GIRK1-containing GIRK channels are developed, the specific roles and therapeutic potential of pharmacological modulation of these channels remains out of reach. As a final note, Zhao et al. (94) report the identification of hits from a virtual screen that have some capacity to activate GIRK2 homomeric channels, but their structures were not revealed and their capacity to activated non-GIRK1-containing GIRKs was not described in detail.
VU0134992: The First Homomeric Kir4.1-Preferring Inhibitor
Kir4.1 is expressed in the kidney tubule epithelium as well as astrocytes and glial cells of the nervous system (91, 117–122). Kir4.1 can assemble into a homotetramer or heterotetramer with Kir5.1 (KCNJ16), with each subtype exhibiting unique functional, regulatory, and pharmacological properties (123–125). One of the best-known functions of homomeric Kir4.1 is its role in “spatial buffering” in the brain and eye, where the channel promotes the uptake of excess extracellular potassium into astroglial cells to maintain normal electrical activity of neurons (119, 126). Changes in the expression levels of Kir4.1 in astrocytes have been associated with treatment-resistant depression (127, 128) and epilepsy (129–131), suggesting Kir4.1 might underlie the etiology of these diseases. Consistent with its known roles in these organ systems, heritable loss-of-function mutations in KCNJ10 lead to SeSAME or EAST syndrome, a mixed neurological and kidney disease characterized by epilepsy, ataxia, sensorineural deafness, and renal salt wasting (132, 133).
The major subtype in the renal tubule is the heteromeric Kir4.1/5.1 form of channel, which is expressed on the basolateral membrane of the thick ascending limb of Henle, distal convoluted tubule, collecting duct (117, 134, 135). A great deal of progress has been made recently in understanding the physiology of Kir4.1/5.1 in the distal convoluted tubule, a tubule segment that plays a critical role in sensing plasma potassium and regulating sodium chloride reabsorption mediated by the thiazide-inhibitable sodium-chloride co-transporter NCC (Fig. 2). Genetic and crude pharmacology data support a model in which Kir4.1/5.1 channels hyperpolarize the basolateral membrane potential, creating a favorable electrochemical gradient that promotes chloride exit via basolateral ClC-Kb channels. Knockout or barium inhibition of Kir4.1/5.1 depolarizes the membrane potential, inhibits chloride exit, and increases intracellular chloride concentrations, which, in turn, inhibits a WNK-SPAK/OSR1 kinase pathway that regulates the phosphorylation state and activity of NCC (136–138). In the collecting duct, there appears to be a relatively minor population of homomeric Kir4.1 channels whose functions have not been clearly elucidated (139) (Fig. 2).
There are a number of neurological drugs (e.g., selective serotonin reuptake inhibitors and tricyclic antidepressants) that inhibit homomeric Kir4.1 channels; however, they block Kir4.1 at concentrations in the tens of micromolar (140–142). As a first step toward developing more potent and specific small-molecule tools for probing the physiology and therapeutic value of Kir4.1, we screened 76,574 compounds from the VICB library for modulators of homomeric Kir4.1 channels using a thallium flux assay (143). Putative inhibitors and potentiators of Kir4.1 were identified in the screen. The most potent confirmed inhibitor was 2-(2-bromo-4-isopropylphenoxy)-N-(2,2,6,6-tetramethylpiperidin-4-yl)acetamide, which we termed VU0134992 (Fig. 3). In whole cell patch-clamp experiments, VU0134992 inhibits Kir4.1 with an IC50 of ∼1 µM and affords 9-fold to 22-fold selectivity over Kir4.1/5.1 at -120 mV and +120 mV, respectively. The voltage dependence of block reflects the location of the VU0134992 functional binding site in the ion-conduction pathway, which we localized to E158 and I159 using site-directed mutagenesis and electrophysiology. It is interesting to note that E158 is the “rectification controller” in Kir4.1 and is analogous to Kir1.1-N171 required for block by VU590 and VU591.
Despite its modest potency, VU0134992 had a favorable pharmacokinetic profile that suggested it might be useful for determining if inhibition of Kir4.1 induces diuresis in a rat model. Rats were administered escalating doses of VU0134992, volume loaded with saline, and then placed in metabolic cages for urine collection over a 4-h period. No effect on urine production was observed at a dose of 10 mg/kg; however, significant increases in urine volume, sodium excretion, and potassium excretion were observed at 50 mg/kg and 100 mg/kg (143). This electrolyte profile is consistent with what is known about the physiology of Kir4.1 in the distal nephron. Inhibition of Kir4.1 and possibly some Kir4.1/5.1 channels should 1) inhibit NCC-mediated sodium chloride reabsorption in the distal convoluted tubule, 2) promote flow-stimulated potassium excretion in the collecting duct, and 3) possibly limit ENaC-mediated sodium reabsorption in the collecting duct. Dissecting the relative contributions of Kir4.1 and Kir4.1/5.1 channels in the diuretic response will require the development of Kir4.1/5.1 as well.
ML418: The First Submicromolar Inhibitor of Kir7.1
Kir7.1 (KCNJ13) is one of the most recently cloned members of the Kir channel family and perhaps the most poorly understood. The channel is broadly expressed as a homotetramer in epithelial cells of the eye, gut, kidney, choroid plexus, and respiratory tract, as well as certain brain neurons where it participates in melanocortin signaling (144–153). Kir7.1’s low sensitivity to barium and unusually small unitary conductance (i.e., 50 femptoSiemens) (152) have made dissecting the physiological functions of the channel in native cells difficult. Adding to this problem, genetic ablation of KCNJ13 leads to lethality 24 h after birth due at least in part to defective tracheal and lung development (154). This scenario highlights the need for potent and specific pharmacological tools for modulating Kir7.1 channel activity in genetically wild type animals.
As noted earlier, VU590 was the first moderately potent (IC50 = 8 µM) and moderately selective (also inhibits Kir1.1 with IC50 = 240 nM) inhibitor of Kir7.1 to be published. Despite these shortcomings, VU590 has been used successfully by several groups to assess the function and physiology of Kir7.1 in various tissues (145, 154–157). In an effort to develop more precise Kir7.1 inhibitors, we performed a high-throughput screen of 5,230 compounds from the VICB library using a mutant of Kir7.1 in which methionine 127 was mutated to an arginine (Kir7.1-M125R) to improve thallium flux through the channel (20). The most potent and selective inhibitor discovered in the screen was a compound termed VU714 (Fig. 3), which inhibited Kir7.1 with an IC50 of 1.5 µM but exhibited off-target activity toward Kir1.1, Kir4.1, and Kir6.2/SUR1. Scanning mutagenesis of pore-lining residues identified E149 and A158 as being essential for potent block of Kir7.1. Exploring the structure-activity relationships with the synthesis of ∼30 VU714 analogs led to the development of a compound termed ML418 (Fig. 3) exhibiting ∼5-fold improved potency (i.e., IC50 = 310 nM) and improved selectivity (i.e., no activity toward Kir1.1, Kir2.1, Kir4.1, but still inhibits Kir6.2/SUR1). The pharmacokinetic properties and CNS penetrance of ML418 should support its use for in vivo studies in the brain and periphery (20).
SUMMARY AND PERSPECTIVES
We have described the current state-of-the-art of Kir channel pharmacology developed using a molecular target-based approach that leverages heterologous channel expression, high-throughput screening of small-molecule libraries, and compound optimization with medicinal chemistry. These efforts have led to first-in-class small-molecule inhibitors of Kir1.1, Kir2.x, Kir4.1, and Kir7.1, as well as activators of Kir3 GIRK channels. Some of these compounds have been used to show proof-of-concept of therapeutic potential of Kir channel targets for managing hypertension (Kir1.1, Kir4.1), epilepsy (Kir3), and pain (Kir3). Yet, there is still a lot of work to be done to more fully advance the molecular pharmacology of the Kir channel family. For example, although ML133 is selective for members of the Kir2.X family, there are currently no inhibitors that can distinguish between homomeric or heteromeric subfamily members, nor have any selective potentiators of Kir2.X been identified. Although VU0134992 shows that selective inhibitors of Kir4.1 can be developed, its moderate potency (IC50 = 1 µM), moderate selectivity over Kir4.1/5.1 channels, and poor CNS penetrance create limitations for fully exploring the therapeutic potential of Kir4.1 in hypertension and CNS disorders. It will be critically important to develop selective inhibitors and activators of heteromeric Kir4.1/5.1 to begin addressing the druggability of this Kir channel subtype in the renal tubule and brain. Finally, although in vivo efficacy of GIRK activators has been attained, much work remains to be done to develop compounds with properties suitable for clinical study as well as a more thorough evaluation of potential side effect liabilities associated with increased systemic GIRK activation. The progress described herein clearly illustrates that modern drug discovery approaches can be harnessed in an academic setting to build a useful toolkit of next-generation Kir channel probes.
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01DK120821 (to J.S.D) and funds from Vanderbilt University Department of Pharmacology and Institute of Chemical Biology (to C.D.W.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.D.W. and J.S.D. prepared figures; drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
REFERENCES
- 1.Kharade SV, Nichols C, Denton JS. The shifting landscape of KATP channelopathies and the need for ‘sharper’ therapeutics. Future Med Chem 8: 789–802, 2016. doi: 10.4155/fmc-2016-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res 112: 1059–1072, 2013. doi: 10.1161/CIRCRESAHA.112.300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nichols CG, Kj Enkvetchakul D, Flagg TP. KATP channels: from structure to disease. Biological Membranes 23: 101–110, 2006. https://www.researchgate.net/publication/283146317_KATP_channels_From_structure_to_disease. [Google Scholar]
- 4.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 440: 470–476, 2006. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 5.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 6.Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol 59: 171–191, 1997. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 7.Tao X, Avalos JL, Chen J, MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 326: 1668–1674, 2009. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagiwara S, Takahashi K. The anomalous rectification and cation selectivity of the membrane of a starfish egg cell. J Membr Biol 18: 61–80, 1974. doi: 10.1007/BF01870103. [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Jan YN, Jan LY. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron 14: 1047–1054, 1995. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]
- 10.Lopatin AN, Makhina EN, Nichols CG. The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. J Gen Physiol 106: 923–955, 1995. doi: 10.1085/jgp.106.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fakler B, Brandle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell 80: 149–154, 1995. doi: 10.1016/0092-8674(95)90459-x. [DOI] [PubMed] [Google Scholar]
- 12.Wible BA, Taglialatela M, Ficker E, Brown AM. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature 371: 246–249, 1994. doi: 10.1038/371246a0. [DOI] [PubMed] [Google Scholar]
- 13.Stanfield PR, Davies NW, Shelton PA, Sutcliffe MJ, Khan IA, Brammar WJ, Conley EC. A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+ of the inward rectifier, IRK1. J Physiol 478: 1–6, 1994. doi: 10.1113/jphysiol.1994.sp020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nichols CG, Ho K, Hebert S. Mg(2+)-dependent inward rectification of ROMK1 potassium channels expressed in Xenopus oocytes. J Physiol 476: 399–409, 1994. doi: 10.1113/jphysiol.1994.sp020141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Z, MacKinnon R. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature 371: 243–246, 1994. doi: 10.1038/371243a0. [DOI] [PubMed] [Google Scholar]
- 16.Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372: 366–369, 1994. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- 17.Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science 266: 1068–1072, 1994. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- 18.Kharade SV, Sanchez-Andres JV, Fulton MG, Shelton EL, Blobaum AL, Engers DW, Hofmann CS, Dadi PK, Lantier L, Jacobson DA, Lindsley CW, Denton JS. Structure-activity relationships, pharmacokinetics, and pharmacodynamics of the Kir6.2/SUR1-specific channel opener VU0071063. J Pharmacol Exp Ther 370: 350–359, 2019. doi: 10.1124/jpet.119.257204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kharade S, Sheehan J, Figueroa E, Meiler J, Denton J. Pore polarity and charge determine differential block of Kir1.1 and Kir7.1 potassium channels by the small-molecule inhibitor VU590. Mol Pharmacol 92: 338–346, 2017. doi: 10.1124/mol.117.108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swale DR, Kurata H, Kharade SV, Sheehan J, Raphemot R, Voigtritter KR, Figueroa EE, Meiler J, Blobaum AL, Lindsley CW, Hopkins CR, Denton JS. ML418: the first selective, sub-micromolar pore blocker of Kir7.1 potassium channels. ACS Chem Neurosci 7: 1013–1023, 2016. doi: 10.1021/acschemneuro.6b00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swale DR, Sheehan JH, Banerjee S, Husni AS, Nguyen TT, Meiler J, Denton JS. Computational and functional analyses of a small-molecule binding site in ROMK. Biophys J 108: 1094–1103, 2015. doi: 10.1016/j.bpj.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swale DR, Kharade SV, Denton JS. Cardiac and renal inward rectifier potassium channel pharmacology: emerging tools for integrative physiology and therapeutics. Curr Opin Pharmacol 15: 7–15, 2014. doi: 10.1016/j.coph.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang HR, Wu M, Yu H, Long S, Stevens A, Engers DW, Sackin H, Daniels JS, Dawson ES, Hopkins CR, Lindsley CW, Li M, McManus OB. Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol 6: 845–856, 2011. doi: 10.1021/cb200146a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11: 301–315, 2010. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lujan R, Marron Fernandez de Velasco E, Aguado C, Wickman K. New insights into the therapeutic potential of Girk channels. Trends Neurosci 37: 20–29, 2014. doi: 10.1016/j.tins.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagiwara S, Miyazaki S, Moody W, Patlak J. Blocking effects of barium and hydrogen ions on the potassium current during anomalous rectification in the starfish egg. J Physiol 279: 167–185, 1978. doi: 10.1113/jphysiol.1978.sp012338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neyton J, Miller C. Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+ -activated K+ channel. J Gen Physiol 92: 569–586, 1988. doi: 10.1085/jgp.92.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neyton J, Miller C. Potassium blocks barium permeation through a calcium-activated potassium channel. J Gen Physiol 92: 549–567, 1988. doi: 10.1085/jgp.92.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma XY, Yu JM, Zhang SZ, Liu XY, Wu BH, Wei XL, Yan JQ, Sun HL, Yan HT, Zheng JQ. External Ba2+ block of the two-pore domain potassium channel TREK-1 defines conformational transition in its selectivity filter. J Biol Chem 286: 39813–39822, 2011. doi: 10.1074/jbc.M111.264788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weir SW, Weston AH. The effects of BRL 34915 and nicorandil on electrical and mechanical activity and on 86Rb efflux in rat blood vessels. Br J Pharmacol 88: 121–128, 1986. doi: 10.1111/j.1476-5381.1986.tb09478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terstappen GC. Functional analysis of native and recombinant ion channels using a high-capacity nonradioactive rubidium efflux assay. Anal Biochem 272: 149–155, 1999. doi: 10.1006/abio.1999.4179. [DOI] [PubMed] [Google Scholar]
- 32.Schroeder KS, Neagle BD. FLIPR: a new instrument for accurate, high throughput optical screening. J Biomol Screen 1: 75–80, 1996. doi: 10.1177/108705719600100205. [DOI] [Google Scholar]
- 33.Minta A, Kao JP, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem 264: 8171–8178, 1989. [PubMed] [Google Scholar]
- 34.Minta A, Tsien RY. Fluorescent indicators for cytosolic sodium. J Biol Chem 264: 19449–19457, 1989. [PubMed] [Google Scholar]
- 35.Holevinsky KO, Fan Z, Frame M, Makielski JC, Groppi V, Nelson DJ. ATP-sensitive K+ channel opener acts as a potent Cl− channel inhibitor in vascular smooth muscle cells. J Membr Biol 137: 59–70, 1994. doi: 10.1007/BF00234998. [DOI] [PubMed] [Google Scholar]
- 36.Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J Biomol Screen 9: 671–677, 2004. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 37.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362: 31–38, 1993. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 38.Lee WS, Hebert SC. ROMK inwardly rectifying ATP-sensitive K+ channel. I. Expression in rat distal nephron segments. Am J Physiol Renal Physiol 268: F1124–F1131, 1995. doi: 10.1152/ajprenal.1995.268.6.F1124. [DOI] [PubMed] [Google Scholar]
- 39.Boim MA, Ho K, Shuck ME, Bienkowski MJ, Block JH, Slightom JL, Yang Y, Brenner BM, Hebert SC. ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms. Am J Physiol Renal Physiol 268: F1132–F1140, 1995. doi: 10.1152/ajprenal.1995.268.6.F1132. [DOI] [PubMed] [Google Scholar]
- 40.Hebert SC, Ho K. Structure and functional properties of an inwardly rectifying ATP-regulated K+ channel from rat kidney. Ren Physiol Biochem 17: 143–147, 1994. doi: 10.1159/000173804. [DOI] [PubMed] [Google Scholar]
- 41.Greger R. Ion transport mechanisms in thick ascending limb of Henle's loop of mammalian nephron. Physiol Rev 65: 760–797, 1985. doi: 10.1152/physrev.1985.65.3.760. [DOI] [PubMed] [Google Scholar]
- 42.Koeppen BM, Biagi BA, Giebisch GH. Intracellular microelectrode characterization of the rabbit cortical collecting duct. Am J Physiol Renal Physiol 244: F35–F47, 1983. doi: 10.1152/ajprenal.1983.244.1.F35. [DOI] [PubMed] [Google Scholar]
- 43.Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 40: 592–599, 2008. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin W, Lu Z. A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry 37: 13291–13299, 1998. doi: 10.1021/bi981178p. [DOI] [PubMed] [Google Scholar]
- 45.Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol 76: 1094–1103, 2009. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhave G, Chauder BA, Liu W, Dawson ES, Kadakia R, Nguyen TT, Lewis LM, Meiler J, Weaver CD, Satlin LM, Lindsley CW, Denton JS. Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol Pharmacol 79: 42–50, 2011. doi: 10.1124/mol.110.066928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang H, Walsh SP, Yan Y, de Jesus RK, Shahripour A, Teumelsan N, Zhu Y, Ha S, Owens KA, Thomas-Fowlkes BS, Felix JP, Liu J, Kohler M, Priest BT, Bailey T, Brochu R, Alonso-Galicia M, Kaczorowski GJ, Roy S, Yang L, Mills SG, Garcia ML, Pasternak A. Discovery of selective small molecule ROMK inhibitors as potential new mechanism diuretics. ACS Med Chem Lett 3: 367–372, 2012. doi: 10.1021/ml3000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia ML, Priest BT, Alonso-Galicia M, Zhou X, Felix JP, Brochu RM, Bailey T, Thomas-Fowlkes B, Liu J, Swensen A, Pai LY, Xiao J, Hernandez M, Hoagland K, Owens K, Tang H, de Jesus RK, Roy S, Kaczorowski GJ, Pasternak A. Pharmacologic inhibition of the renal outer medullary potassium channel causes diuresis and natriuresis in the absence of kaliuresis. J Pharmacol Exp Ther 348: 153–164, 2014. doi: 10.1124/jpet.113.208603. [DOI] [PubMed] [Google Scholar]
- 49.Tang H, de Jesus RK, Walsh SP, Zhu Y, Yan Y, Priest BT, Swensen AM, Alonso-Galicia M, Felix JP, Brochu RM, Bailey T, Thomas-Fowlkes B, Zhou X, Pai LY, Hampton C, Hernandez M, Owens K, Roy S, Kaczorowski GJ, Yang L, Garcia ML, Pasternak A. Discovery of a novel sub-class of ROMK channel inhibitors typified by 5-(2-(4-(2-(4-(1H-Tetrazol-1-yl)phenyl)acetyl)piperazin-1-yl)ethyl)isobenzofuran- 1(3H)-one. Bioorg Med Chem Lett 23: 5829–5832, 2013. doi: 10.1016/j.bmcl.2013.08.104. [DOI] [PubMed] [Google Scholar]
- 50.Kharade SV, Flores D, Lindsley CW, Satlin LM, Denton JS. ROMK inhibitor actions in the nephron probed with diuretics. Am J Physiol Renal Physiol 310: F732–F737, 2015. doi: 10.1152/ajprenal.00423.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature 362: 127–133, 1993. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- 52.Ambrosini E, Sicca F, Brignone MS, D'Adamo MC, Napolitano C, Servettini I, Moro F, Ruan Y, Guglielmi L, Pieroni S, Servillo G, Lanciotti A, Valvo G, Catacuzzeno L, Franciolini F, Molinari P, Marchese M, Grottesi A, Guerrini R, Santorelli FM, Priori S, Pessia M. Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype. Hum Mol Genet 23: 4875–4886, 2014. doi: 10.1093/hmg/ddu201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miake J, Marban E, Nuss HB. Functional role of inward rectifier current in heart probed by Kir2.1 overexpression and dominant-negative suppression. J Clin Invest 111: 1529–1536, 2003. doi: 10.1172/JCI17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bradley KK, Jaggar JH, Bonev AD, Heppner TJ, Flynn ER, Nelson MT, Horowitz B. Kir2.1 encodes the inward rectifier potassium channel in rat arterial smooth muscle cells. J Physiol 515: 639–651, 1999. doi: 10.1111/j.1469-7793.1999.639ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen HM, Blomster LV, Christophersen P, Wulff H. Potassium channel expression and function in microglia: Plasticity and possible species variations. Channels (Austin) 11: 305–315, 2017. doi: 10.1080/19336950.2017.1300738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murata Y, Yasaka T, Takano M, Ishihara K. Neuronal and glial expression of inward rectifier potassium channel subunits Kir2.x in rat dorsal root ganglion and spinal cord. Neurosci Lett 617: 59–65, 2016. doi: 10.1016/j.neulet.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 57.Ford NC, Baccei ML. Inward-rectifying K+ (Kir2) leak conductance dampens the excitability of lamina I projection neurons in the neonatal rat. Neuroscience 339: 502–510, 2016. doi: 10.1016/j.neuroscience.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masia R, Krause DS, Yellen G. The inward rectifier potassium channel Kir2.1 is expressed in mouse neutrophils from bone marrow and liver. Am J Physiol Cell Physiol 308: C264–C276, 2015. doi: 10.1152/ajpcell.00176.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen K, Zuo D, Liu Z, Chen H. Kir2.1 channels set two levels of resting membrane potential with inward rectification. Pflugers Arch 470: 599–611, 2018. doi: 10.1007/s00424-017-2099-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lam D, Schlichter LC. Expression and contributions of the Kir2.1 inward-rectifier K+ channel to proliferation, migration and chemotaxis of microglia in unstimulated and anti-inflammatory states. Front Cell Neurosci 9: 185, 2015. doi: 10.3389/fncel.2015.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiao Y, Tang C, Wang Q, Wang D, Yan G, Zhu B. Kir2.1 regulates rat smooth muscle cell proliferation, migration, and post-injury carotid neointimal formation. Biochem Biophys Res Commun 477: 774–780, 2016. doi: 10.1016/j.bbrc.2016.06.134. [DOI] [PubMed] [Google Scholar]
- 62.Preisig-Muller R, Schlichthorl G, Goerge T, Heinen S, Bruggemann A, Rajan S, Derst C, Veh RW, Daut J. Heteromerization of Kir2.x potassium channels contributes to the phenotype of Andersen’s syndrome. Proc Natl Acad Sci USA 99: 7774–7779, 2002. doi: 10.1073/pnas.102609499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schram G, Melnyk P, Pourrier M, Wang Z, Nattel S. Kir2.4 and Kir2.1 K+ channel subunits co-assemble: a potential new contributor to inward rectifier current heterogeneity. J Physiol 544: 337–349, 2002. doi: 10.1113/jphysiol.2002.026047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105: 511–519, 2001. doi: 10.1016/S0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 65.Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 96: 800–807, 2005. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 66.Liu H, Huang J, Peng J, Wu X, Zhang Y, Zhu W, Guo L. Upregulation of the inwardly rectifying potassium channel Kir2.1 (KCNJ2) modulates multidrug resistance of small-cell lung cancer under the regulation of miR-7 and the Ras/MAPK pathway. Mol Cancer 14: 59, 2015. doi: 10.1186/s12943-015-0298-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lopez-Izquierdo A, Ponce-Balbuena D, Moreno-Galindo EG, Arechiga-Figueroa IA, Rodriguez-Martinez M, Ferrer T, Rodriguez-Menchaca AA, Sanchez-Chapula JA. The antimalarial drug mefloquine inhibits cardiac inward rectifier K+ channels: evidence for interference in PIP2-channel interaction. J Cardiovasc Pharmacol 57: 407–415, 2011. doi: 10.1097/FJC.0b013e31820b7c03. [DOI] [PubMed] [Google Scholar]
- 68.Rodriguez-Menchaca AA, Navarro-Polanco RA, Ferrer-Villada T, Rupp J, Sachse FB, Tristani-Firouzi M, Sanchez-Chapula JA. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc Natl Acad Sci USA 105: 1364–1368, 2008. doi: 10.1073/pnas.0708153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ponce-Balbuena D, Lopez-Izquierdo A, Ferrer T, Rodriguez-Menchaca AA, Arechiga-Figueroa IA, Sanchez-Chapula JA. Tamoxifen inhibits inward rectifier K+ 2.x family of inward rectifier channels by interfering with phosphatidylinositol 4,5-bisphosphate-channel interactions. J Pharmacol Exp Ther 331: 563–573, 2009. doi: 10.1124/jpet.109.156075. [DOI] [PubMed] [Google Scholar]
- 70.Scherer D, Schworm B, Seyler C, Xynogalos P, Scholz EP, Thomas D, Katus HA, Zitron E. Inhibition of inwardly rectifying Kir2.x channels by the novel anti-cancer agent gambogic acid depends on both pore block and PIP2 interference. Naunyn-Schmiedeberg's Arch Pharmacol 390: 701–710, 2017. doi: 10.1007/s00210-017-1372-5. [DOI] [PubMed] [Google Scholar]
- 71.de Boer TP, Nalos L, Stary A, Kok B, Houtman MJ, Antoons G, van Veen TA, Beekman JD, de Groot BL, Opthof T, Rook MB, Vos MA, van der Heyden MA. The anti-protozoal drug pentamidine blocks KIR2.x-mediated inward rectifier current by entering the cytoplasmic pore region of the channel. Br J Pharmacol 159: 1532–1541, 2010. doi: 10.1111/j.1476-5381.2010.00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ren S, Pang C, Huang Y, Xing C, Zhan Y, An H. Hydrocinnamic acid inhibits the currents of WT and SQT3 syndrome-related mutants of Kir2.1 channel. J Membr Biol 250: 425–432, 2017. doi: 10.1007/s00232-017-9964-z. [DOI] [PubMed] [Google Scholar]
- 73.Takanari H, Nalos L, Stary-Weinzinger A, de Git KC, Varkevisser R, Linder T, Houtman MJ, Peschar M, de Boer TP, Tidwell RR, Rook MB, Vos MA, van der Heyden MA. Efficient and specific cardiac IK1 inhibition by a new pentamidine analogue. Cardiovasc Res 99: 203–214, 2013. doi: 10.1093/cvr/cvt103. [DOI] [PubMed] [Google Scholar]
- 74.Caballero R, Dolz-Gaiton P, Gomez R, Amoros I, Barana A, Gonzalez de la Fuente M, Osuna L, Duarte J, Lopez-Izquierdo A, Moraleda I, Galvez E, Sanchez-Chapula JA, Tamargo J, Delpon E. Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc Natl Acad Sci USA 107: 15631–15636, 2010. doi: 10.1073/pnas.1004021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gattlen C, Deftu AF, Tonello R, Ling Y, Berta T, Ristoiu V, Suter MR. The inhibition of Kir2.1 potassium channels depolarizes spinal microglial cells, reduces their proliferation, and attenuates neuropathic pain. Glia 68: 2119–2135, 2020. doi: 10.1002/glia.23831. [DOI] [PubMed] [Google Scholar]
- 76.Spencer NG, Schilling T, Miralles F, Eder C. Mechanisms underlying interferon-gamma-induced priming of microglial reactive oxygen species production. PLoS One 11: e0162497, 2016. doi: 10.1371/journal.pone.0162497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shi Y, Chen Y, Wang Y. Kir2.1 channel regulation of glycinergic transmission selectively contributes to dynamic mechanical allodynia in a mouse model of spared nerve injury. Neurosci Bull 35: 301–314, 2019. doi: 10.1007/s12264-018-0285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, Cui X, Li X, Yan H, Li H, Guan X, Wang Y, Liu S, Qin X, Cheng M. Inhibition of Kir2.1 channel-induced depolarization promotes cell biological activity and differentiation by modulating autophagy in late endothelial progenitor cells. J Mol Cell Cardiol 127: 57–66, 2019. doi: 10.1016/j.yjmcc.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 79.Ye W, Chang RB, Bushman JD, Tu YH, Mulhall EM, Wilson CE, Cooper AJ, Chick WS, Hill-Eubanks DC, Nelson MT, Kinnamon SC, Liman ER. The K+ channel KIR2.1 functions in tandem with proton influx to mediate sour taste transduction. Proc Natl Acad Sci USA 113: E229–E238, 2016. doi: 10.1073/pnas.1514282112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Diaz RJ, Zobel C, Cho HC, Batthish M, Hinek A, Backx PH, Wilson GJ. Selective inhibition of inward rectifier K+ channels (Kir2.1 or Kir2.2) abolishes protection by ischemic preconditioning in rabbit ventricular cardiomyocytes. Circ Res 95: 325–332, 2004. doi: 10.1161/01.RES.0000137727.34938.35. [DOI] [PubMed] [Google Scholar]
- 81.Huang X, Lee SH, Lu H, Sanders KM, Koh SD. Molecular and functional characterization of inwardly rectifying K+ currents in murine proximal colon. J Physiol 596: 379–391, 2018. doi: 10.1113/JP275234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhave G, Lonergan D, Chauder BA, Denton JS. Small-molecule modulators of inward rectifier K+ channels: recent advances and future possibilities. Future Med Chem 2: 757–774, 2010. doi: 10.4155/fmc.10.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Machida T, Hashimoto N, Kuwahara I, Ogino Y, Matsuura J, Yamamoto W, Itano Y, Zamma A, Matsumoto R, Kamon J, Kobayashi T, Ishiwata N, Yamashita T, Ogura T, Nakaya H. Effects of a highly selective acetylcholine-activated K+ channel blocker on experimental atrial fibrillation. Circ Arrhythm Electrophysiol 4: 94–102, 2011. doi: 10.1161/CIRCEP.110.951608. [DOI] [PubMed] [Google Scholar]
- 84.Kobayashi T, Ikeda K, Kojima H, Niki H, Yano R, Yoshioka T, Kumanishi T. Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat Neurosci 2: 1091–1097, 1999. doi: 10.1038/16019. [DOI] [PubMed] [Google Scholar]
- 85.Yow TT, Pera E, Absalom N, Heblinski M, Johnston GA, Hanrahan JR, Chebib M. Naringin directly activates inwardly rectifying potassium channels at an overlapping binding site to tertiapin-Q. Br J Pharmacol 163: 1017–1033, 2011. doi: 10.1111/j.1476-5381.2011.01315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nishizawa D, Gajya N, Ikeda K. Identification of selective agonists and antagonists to g protein-activated inwardly rectifying potassium channels: candidate medicines for drug dependence and pain. Curr Neuropharmacol 9: 113–117, 2011. doi: 10.2174/157015911795017227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jalan-Sakrikar N, Roper-Field J, Klar R, Mattman M, Walker AG, Zamorano R, Xiang Z, Byers CF, Blobaum AL, Engers D, Weaver CD, Days E, Utley TJ, Melancon B, Daniels JS, Wood MR, Lindsley CW, Conn PJ, Hopkins CR, Niswender CM. The discovery and characterization of a centrally penetrant (ML396) and a peripherally restricted (ML397) pan-Group III mGlu positive allosteric modulators. In: Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Centre for Biotechnology Information, 2010. [PubMed] [Google Scholar]
- 88.Kaufmann K, Romaine I, Days E, Pascual C, Malik A, Yang L, Zou B, Du Y, Sliwoski G, Morrison RD, Denton J, Niswender CM, Daniels JS, Sulikowski GA, Xie XS, Lindsley CW, Weaver CD. ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem Neurosci 4: 1278–1286, 2013. doi: 10.1021/cn400062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wen W, Wu W, Romaine IM, Kaufmann K, Du Y, Sulikowski GA, Weaver CD, Lindsley CW. Discovery of ‘molecular switches’ within a GIRK activator scaffold that afford selective GIRK inhibitors. Bioorg Med Chem Lett 23: 4562–4566, 2013. doi: 10.1016/j.bmcl.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wydeven N, Marron Fernandez de Velasco E, Du Y, Benneyworth MA, Hearing MC, Fischer RA, Thomas MJ, Weaver CD, Wickman K. Mechanisms underlying the activation of G-protein-gated inwardly rectifying K+ (GIRK) channels by the novel anxiolytic drug, ML297. Proc Natl Acad Sci USA 111: 10755–10760, 2014. doi: 10.1073/pnas.1405190111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci 20: 5733–5740, 2000. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chan KW, Sui JL, Vivaudou M, Logothetis DE. Control of channel activity through a unique amino acid residue of a G protein-gated inwardly rectifying K+ channel subunit. Proc Natl Acad Sci USA 93: 14193–14198, 1996. doi: 10.1073/pnas.93.24.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu Y, Cantwell L, Molosh AI, Plant LD, Gazgalis D, Fitz SD, Dustrude ET, Yang Y, Kawano T, Garai S, Noujaim SF, Shekhar A, Logothetis DE, Thakur GA. The small molecule GAT1508 activates brain-specific GIRK1/2 channel heteromers and facilitates conditioned fear extinction in rodents. J Biol Chem 295: 3614–3634, 2020. doi: 10.1074/jbc.RA119.011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao Y, Ung PM-U, Zahoránszky-Kőhalmi G, Zakharov AV, Martinez NJ, Simeonov A, Glaaser IW, Rai G, Schlessinger A, Marugan JJ, Slesinger PA. Identification of a G-protein-independent activator of GIRK channels. Cell Rep 31: 107770, 2020. doi: 10.1016/j.celrep.2020.107770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang Y, Zhang Y, Kong S, Zang K, Jiang S, Wan L, Chen L, Wang G, Jiang M, Wang X, Hu J, Wang Y. GIRK1-mediated inwardly rectifying potassium current suppresses the epileptiform burst activities and the potential antiepileptic effect of ML297. Biomed Pharmacother 101: 362–370, 2018. doi: 10.1016/j.biopha.2018.02.114. [DOI] [PubMed] [Google Scholar]
- 96.Vo BN, Abney KK, Anderson A, Marron Fernandez de Velasco E, Benneyworth MA, Daniels JS, Morrison RD, Hopkins CR, Weaver CD, Wickman K. VU0810464, a non-urea G protein-gated inwardly rectifying K+ (Kir 3/GIRK) channel activator, exhibits enhanced selectivity for neuronal Kir 3 channels and reduces stress-induced hyperthermia in mice. Br J Pharmacol 176: 2238–2249, 2019. doi: 10.1111/bph.14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abney KK, Bubser M, Du Y, Kozek KA, Bridges TM, Linsdley CW, Daniels JS, Morrison RD, Wickman K, Hopkins CR, Jones CK, Weaver CD. Analgesic effects of the GIRK activator, VU0466551, alone and in combination with morphine in acute and persistent pain models. ACS Chem Neurosci 10: 1294–1299, 2019. [Erratum in ACS Chem Neurosci 10: 2621, 2019]. doi: 10.1021/acschemneuro.8b00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kimura M, Shiokawa H, Karashima Y, Sumie M, Hoka S, Yamaura K. Antinociceptive effect of selective G protein-gated inwardly rectifying K+ channel agonist ML297 in the rat spinal cord. PLoS One 15: e0239094, 2020. doi: 10.1371/journal.pone.0239094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zou B, Cao WS, Guan Z, Xiao K, Pascual C, Xie J, Zhang J, Xie J, Kayser F, Lindsley CW, Weaver CD, Fang J, Xie XS. Direct activation of G-protein-gated inward rectifying K+ channels promotes nonrapid eye movement sleep. Sleep 42: zsy244, 2019. doi: 10.1093/sleep/zsy244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sanchez-Rodriguez I, Djebari S, Temprano-Carazo S, Vega-Avelaira D, Jimenez-Herrera R, Iborra-Lazaro G, Yajeya J, Jimenez-Diaz L, Navarro-Lopez JD. Hippocampal long-term synaptic depression and memory deficits induced in early amyloidopathy are prevented by enhancing G-protein-gated inwardly rectifying potassium channel activity. J Neurochem 153: 362–376, 2020. doi: 10.1111/jnc.14946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sanchez-Rodriguez I, Temprano-Carazo S, Najera A, Djebari S, Yajeya J, Gruart A, Delgado-Garcia JM, Jimenez-Diaz L, Navarro-Lopez JD. Activation of G-protein-gated inwardly rectifying potassium (Kir3/GirK) channels rescues hippocampal functions in a mouse model of early amyloid-β pathology. Sci Rep 7: 14658, 2017. doi: 10.1038/s41598-017-15306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nimitvilai S, Lopez MF, Mulholland PJ, Woodward JJ. Ethanol dependence abolishes monoamine and GIRK (Kir3) channel inhibition of orbitofrontal cortex excitability. Neuropsychopharmacology 42: 1800–1812, 2017. doi: 10.1038/npp.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nimitvilai S, Lopez MF, Woodward JJ. Effects of monoamines on the intrinsic excitability of lateral orbitofrontal cortex neurons in alcohol-dependent and non-dependent female mice. Neuropharmacology 137: 1–12, 2018. doi: 10.1016/j.neuropharm.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brewer CL, Baccei ML. Enhanced postsynaptic GABAB receptor signaling in adult spinal projection neurons after neonatal injury. Neuroscience 384: 329–339, 2018. doi: 10.1016/j.neuroscience.2018.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lippiello P, Hoxha E, Tempia F, Miniaci MC. GIRK1-mediated inwardly rectifying potassium current is a candidate mechanism behind Purkinje cell excitability. Cerebellum 19: 751–761, 2020. doi: 10.1007/s12311-020-01158-y. [DOI] [PubMed] [Google Scholar]
- 106.Psichas A, Glass LL, Sharp SJ, Reimann F, Gribble FM. Galanin inhibits GLP-1 and GIP secretion via the GAL1 receptor in enteroendocrine L and K cells. Br J Pharmacol 173: 888–898, 2016. doi: 10.1111/bph.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marron Fernandez de Velasco E, Zhang L, N. Vo B, Tipps M, Farris S, Xia Z, Anderson A, Carlblom N, Weaver CD, Dudek SM, Wickman K. GIRK2 splice variants and neuronal G protein-gated K+ channels: implications for channel function and behavior. Sci Rep 7: 1639, 2017. doi: 10.1038/s41598-017-01820-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gonzalez JC, Epps SA, Markwardt SJ, Wadiche JI, Overstreet-Wadiche L. Constitutive and synaptic activation of GIRK channels differentiates mature and newborn dentate granule cells. J Neurosci 38: 6513–6526, 2018. doi: 10.1523/JNEUROSCI.0674-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Constantin S, Wray S. Nociceptin/orphanin-FQ inhibits gonadotropin-releasing hormone neurons via G-protein-gated inwardly rectifying potassium channels. eNeuro 5: ENEURO.0161-18.2018, 2018. doi: 10.1523/ENEURO.0161-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wei AD, Ramirez JM. Presynaptic mechanisms and KCNQ potassium channels modulate opioid depression of respiratory drive. Front Physiol 10: 1407, 2019. doi: 10.3389/fphys.2019.01407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kotecki L, Hearing M, McCall NM, Marron Fernandez de Velasco E, Pravetoni M, Arora D, Victoria NC, Munoz MB, Xia Z, Slesinger PA, Weaver CD, Wickman K. GIRK channels modulate opioid-induced motor activity in a cell type- and subunit-dependent manner. J Neurosci 35: 7131–7142, 2015. doi: 10.1523/JNEUROSCI.5051-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wieting JM, Vadukoot AK, Sharma S, Abney KK, Bridges TM, Daniels JS, Morrison RD, Wickman K, Weaver CD, Hopkins CR. Discovery and characterization of 1H-pyrazol-5-yl-2-phenylacetamides as novel, non-urea-containing GIRK1/2 potassium channel activators. ACS Chem Neurosci 8: 1873–1879, 2017. doi: 10.1021/acschemneuro.7b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen IS, Tateyama M, Fukata Y, Uesugi M, Kubo Y. Ivermectin activates GIRK channels in a PIP2-dependent, Gβγ-independent manner and an amino acid residue at the slide helix governs the activation. J Physiol 595: 5895–5912, 2017. doi: 10.1113/JP274871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Su Z, Brown EC, Wang W, MacKinnon R. Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc Natl Acad Sci USA 113: 5748–5753, 2016. doi: 10.1073/pnas.1602815113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen IS, Kubo Y. Ivermectin and its target molecules: shared and unique modulation mechanisms of ion channels and receptors by ivermectin. J Physiol 596: 1833–1845, 2018. doi: 10.1113/JP275236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kozek KA, Du Y, Sharma S, Prael FJ, Spitznagel BD, Kharade SV, Denton JS, Hopkins CR, Weaver CD. Discovery and characterization of VU0529331, a synthetic small-molecule activator of homomeric g protein-gated, inwardly rectifying, potassium (GIRK) channels. ACS Chem Neurosci 10: 358–370, 2019. doi: 10.1021/acschemneuro.8b00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Su XT, Wang WH. The expression, regulation, and function of Kir4.1 (Kcnj10) in the mammalian kidney. Am J Physiol Renal Physiol 311: F12–F15, 2016. doi: 10.1152/ajprenal.00112.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am J Physiol Renal Physiol 308: F1288–F1296, 2015. doi: 10.1152/ajprenal.00687.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27: 11354–11365, 2007. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Olsen ML, Higashimori H, Campbell SL, Hablitz JJ, Sontheimer H. Functional expression of Kir4.1 channels in spinal cord astrocytes. Glia 53: 516–528, 2006. doi: 10.1002/glia.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci 21: 5429–5438, 2001. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonosaki A, Tomoike H, Kurachi Y. Immunolocalization of an inwardly rectifying K+ channel, KAB-2 (Kir4.1), in the basolateral membrane of renal distal tubular epithelia. FEBS Lett 388: 11–15, 1996. doi: 10.1016/0014-5793(96)00502-9. [DOI] [PubMed] [Google Scholar]
- 123.Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, Sohet F, Eladari D, Houillier P, Lourdel S, Teulon J, Tucker SJ. Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc Natl Acad Sci USA 108: 10361–10366, 2011. doi: 10.1073/pnas.1101400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pessia M, Imbrici P, D'Adamo MC, Salvatore L, Tucker SJ. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol 532: 359–367, 2001. doi: 10.1111/j.1469-7793.2001.0359f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J 15: 2980–2987, 1996. [PMC free article] [PubMed] [Google Scholar]
- 126.Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem 107: 589–601, 2008. doi: 10.1111/j.1471-4159.2008.05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cui Y, Yang Y, Ni Z, Dong Y, Cai G, Foncelle A, Ma S, Sang K, Tang S, Li Y, Shen Y, Berry H, Wu S, Hu H. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 554: 323–327, 2018. doi: 10.1038/nature25752. [DOI] [PubMed] [Google Scholar]
- 128.Xiong Z, Zhang K, Ren Q, Chang L, Chen J, Hashimoto K. Increased expression of inwardly rectifying Kir4.1 channel in the parietal cortex from patients with major depressive disorder. J Affect Disord 245: 265–269, 2019. doi: 10.1016/j.jad.2018.11.016. [DOI] [PubMed] [Google Scholar]
- 129.Villa C, Combi R. Potassium channels and human epileptic phenotypes: an updated overview. Front Cell Neurosci 10: 81, 2016. doi: 10.3389/fncel.2016.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci 29: 10588–10599, 2009. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, Schramm J, Steinhauser C. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci 12: 2087–2096, 2000. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- 132.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van't Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360: 1960–1970, 2009. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 106: 5842–5847, 2009. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Welling PA. Roles and regulation of renal K channels. Annu Rev Physiol 78: 415–435, 2016. doi: 10.1146/annurev-physiol-021115-105423. [DOI] [PubMed] [Google Scholar]
- 135.Palygin O, Pochynyuk O, Staruschenko A. Distal tubule basolateral potassium channels: cellular and molecular mechanisms of regulation. Curr Opin Nephrol Hypertens 27: 373–378, 2018. doi: 10.1097/MNH.0000000000000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wu P, Gao ZX, Su XT, Wang MX, Wang WH, Lin DH. Kir4.1/Kir5.1 activity is essential for dietary sodium intake-induced modulation of Na-Cl cotransporter. J Am Soc Nephrol 30: 216–227, 2019. doi: 10.1681/ASN.2018080799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wu P, Gao ZX, Zhang DD, Su XT, Wang WH, Lin DH. Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J Am Soc Nephrol 30: 1425–1438, 2019. doi: 10.1681/ASN.2019010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Penton D, Czogalla J, Wengi A, Himmerkus N, Loffing-Cueni D, Carrel M, Rajaram RD, Staub O, Bleich M, Schweda F, Loffing J. Extracellular K+ rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl− -dependent and independent mechanisms. J Physiol 594: 6319–6331, 2016. doi: 10.1113/JP272504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. Am J Physiol Renal Physiol 305: F1277–F1287, 2013. doi: 10.1152/ajprenal.00363.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol 75: 1287–1295, 2009. [Erratum in Mol Pharmacol 76: 451, 2009]. doi: 10.1124/mol.108.052936. [DOI] [PubMed] [Google Scholar]
- 141.Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, Kurachi Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J Pharmacol Exp Ther 320: 573–580, 2007. doi: 10.1124/jpet.106.112094. [DOI] [PubMed] [Google Scholar]
- 142.Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178: 44–51, 2007. doi: 10.1016/j.brainres.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 143.Kharade SV, Kurata H, Bender AM, Blobaum AL, Figueroa EE, Duran A, Kramer M, Days E, Vinson P, Flores D, Satlin LM, Meiler J, Weaver CD, Lindsley CW, Hopkins CR, Denton JS. Discovery, characterization, and effects on renal fluid and electrolyte excretion of the Kir4.1 potassium channel pore blocker, VU0134992. Mol Pharmacol 94: 926–937, 2018. doi: 10.1124/mol.118.112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pattnaik BR, Shahi PK, Marino MJ, Liu X, York N, Brar S, Chiang J, Pillers AM, Traboulsi EI. A novel KCNJ13 nonsense mutation and loss of Kir7.1 channel function causes Leber congenital amaurosis (LCA16). Hum Mutat 36: 720–727, 2015. doi: 10.1002/humu.22807. [DOI] [PubMed] [Google Scholar]
- 145.Ghamari-Langroudi M, Digby GJ, Sebag JA, Millhauser GL, Palomino R, Matthews R, Gillyard T, Panaro BL, Tough IR, Cox HM, Denton JS, Cone RD. G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature 520: 94–98, 2015. doi: 10.1038/nature14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pattnaik BR, Tokarz S, Asuma MP, Schroeder T, Sharma A, Mitchell JC, Edwards AO, Pillers DA. Snowflake vitreoretinal degeneration (SVD) mutation R162W provides new insights into Kir7.1 ion channel structure and function. PLoS One 8: e71744, 2013. doi: 10.1371/journal.pone.0071744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang D, Zhang X, Hughes BA. Expression of inwardly rectifying potassium channel subunits in native human retinal pigment epithelium. Exp Eye Res 87: 176–183, 2008. doi: 10.1016/j.exer.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hejtmancik JF, Jiao X, Li A, Sergeev YV, Ding X, Sharma AK, Chan CC, Medina I, Edwards AO. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am J Hum Genet 82: 174–180, 2008. doi: 10.1016/j.ajhg.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Derst C, Hirsch JR, Preisig-Muller R, Wischmeyer E, Karschin A, Doring F, Thomzig A, Veh RW, Schlatter E, Kummer W, Daut J. Cellular localization of the potassium channel Kir7.1 in guinea pig and human kidney. Kidney Int 59: 2197–2205, 2001. doi: 10.1046/j.1523-1755.2001.00735.x. [DOI] [PubMed] [Google Scholar]
- 150.Ookata K, Tojo A, Suzuki Y, Nakamura N, Kimura K, Wilcox CS, Hirose S. Localization of inward rectifier potassium channel Kir7.1 in the basolateral membrane of distal nephron and collecting duct. J Am Soc Nephrol 11: 1987–1994, 2000. [DOI] [PubMed] [Google Scholar]
- 151.Nakamura N, Suzuki Y, Sakuta H, Ookata K, Kawahara K, Hirose S. Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+,K+-ATPase. Biochem J 342: 329–336, 1999. [PMC free article] [PubMed] [Google Scholar]
- 152.Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. A novel inward rectifier K+ channel with unique pore properties. Neuron 20: 995–1005, 1998. doi: 10.1016/s0896-6273(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 153.Doring F, Derst C, Wischmeyer E, Karschin C, Schneggenburger R, Daut J, Karschin A. The epithelial inward rectifier channel Kir7.1 displays unusual K+ permeation properties. J Neurosci 18: 8625–8636, 1998. doi: 10.1523/JNEUROSCI.18-21-08625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yin W, Kim HT, Wang S, Gunawan F, Wang L, Kishimoto K, Zhong H, Roman D, Preussner J, Guenther S, Graef V, Buettner C, Grohmann B, Looso M, Morimoto M, Mardon G, Offermanns S, Stainier DYR. The potassium channel KCNJ13 is essential for smooth muscle cytoskeletal organization during mouse tracheal tubulogenesis. Nat Commun 9: 2815, 2018. doi: 10.1038/s41467-018-05043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McCloskey C, Rada C, Bailey E, McCavera S, van den Berg HA, Atia J, Rand DA, Shmygol A, Chan YW, Quenby S, Brosens JJ, Vatish M, Zhang J, Denton JS, Taggart MJ, Kettleborough C, Tickle D, Jerman J, Wright P, Dale T, Kanumilli S, Trezise DJ, Thornton S, Brown P, Catalano R, Lin N, England SK, Blanks AM. The inwardly rectifying K+ channel KIR7.1 controls uterine excitability throughout pregnancy. EMBO Mol Med 6: 1161–1174, 2014. doi: 10.15252/emmm.201403944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shahi PK, Liu X, Aul B, Moyer A, Pattnaik A, Denton J, Pillers DM, Pattnaik BR. Abnormal electroretinogram after Kir7.1 channel suppression suggests role in retinal electrophysiology. Sci Rep 7: 10651, 2017. doi: 10.1038/s41598-017-11034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Papanikolaou M, Butt AM, Lewis A. A critical role for the inward rectifying potassium channel Kir7.1 in oligodendrocytes of the mouse optic nerve. Brain Struct Funct 225: 925–934, 2020. doi: 10.1007/s00429-020-02043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]