

Abstract
Intestinal Tuft cells sense luminal contents to influence the mucosal immune response against eukaryotic infection. Paneth cells secrete antimicrobial proteins as part of the mucosal protective barrier. Defects in Tuft and Paneth cells occur commonly in various gut mucosal disorders. MicroRNA-195 (miR-195) regulates the stability and translation of target mRNAs and is involved in many aspects of cell processes and pathologies. Here, we reported the posttranscriptional mechanisms by which miR-195 regulates Tuft and Paneth cell function in the small intestinal epithelium. Mucosal tissues from intestinal epithelial tissue-specific miR-195 transgenic (miR195-Tg) mice had reduced numbers of double cortin-like kinase 1 (DCLK1)-positive (Tuft) and lysozyme-positive (Paneth) cells, compared with tissues from control mice, but there were no effects on Goblet cells and enterocytes. Intestinal organoids expressing higher miR-195 levels from miR195-Tg mice also exhibited fewer Tuft and Paneth cells. Transgenic expression of miR-195 in mice failed to alter growth of the small intestinal mucosa but increased vulnerability of the gut barrier in response to lipopolysaccharide (LPS). Studies aimed at investigating the mechanism underlying regulation of Tuft cells revealed that miR-195 directly interacted with the Dclk1 mRNA via its 3′-untranslated region and inhibited DCLK1 translation. Interestingly, the RNA-binding protein HuR competed with miR-195 for binding Dclk1 mRNA and increased DCLK1 expression. These results indicate that miR-195 suppresses the function of Tuft and Paneth cells in the small intestinal epithelium and further demonstrate that increased miR-195 disrupts Tuft cell function by inhibiting DCLK1 translation via interaction with HuR.
Keywords: Goblet cells, intestinal epithelium homeostasis, noncoding RNAs, Paneth cells, posttranscriptional regulation
INTRODUCTION
The epithelium lines the luminal surface of the intestinal mucosa and forms a physical and biochemical barrier that defends against luminal stimuli and the microbiome through several mechanisms. The intestinal protective barrier is a specialized complex consisting of multiple elements including the epithelial layer and immune system (1, 2). Tuft cells in the intestinal epithelium are chemosensory cells characterized by long and thick microvilli on their apical sides and identified by the presence of double cortin-like kinase 1 (DCLK1) (3). Intestinal DCLK1-positive Tuft cells sense luminal contents to influence the mucosal immune response after eukaryotic infection and are involved in recovery of the intestinal epithelium from damage (4, 5). Paneth cells produce high quantities of defensins and other antibiotic proteins such as lysozyme, Reg3 lectins, and phospholipase A2 when exposed to pathogenic bacteria and bacterial products (6), and they also act as a niche for intestinal stem cells to regulate the renewal of the intestinal epithelium (7). Like other differentiated intestinal epithelial cells (IECs), Tuft and Paneth cells originate from a shared progenitor located above the stem cell niche (8). During differentiation, Tuft cells migrate into both crypts and villi but are dramatically more prevalent in the villous area, whereas Paneth cells move back to the base of crypts and become interspersed between the stem cells (3, 6, 7). Defects in the development of Tuft and Paneth cells compromise the intestinal epithelial integrity and host defense, thus contributing to a wide range of gut mucosal diseases (4, 9–11).
The gene expression programs that control the intestinal epithelium homeostasis and barrier function are tightly regulated at the transcriptional level, but the posttranscriptional events, particularly altered mRNA stability and translation, play essential roles in the regulation of gut mucosal regeneration, protection, and permeability (12–15). The stability and translation of mRNAs are controlled through the interaction of specific mRNA sequences (cis elements) with specific trans-acting factors including microRNAs (miRNAs) and RNA-binding proteins (RBPs) (16–18). Associations of mRNAs with miRNAs and/or RBPs via cis elements, frequently present at the 3′-untranslated region (3′-UTR) of the mRNA, can alter the production levels of target transcripts (19). miRNAs also interact functionally with RBPs to jointly regulate shared target mRNAs, often synergistically or antagonistically (13, 20, 21). Transgenic expression and targeted deletion of given miRNAs or RBPs in mice affected intestinal mucosal development and differentiation, disrupted integrity of the intestinal epithelium, and caused mucosal pathologies (22, 23). For example, transgene-driven overexpression of miR-222 in IECs led to small intestinal mucosal atrophy, inhibited repair of damaged mucosa, and delayed recovery of gut barrier function after exposure to septic stress (24). On the other hand, conditional deletion of RBP HuR in mice inhibited Paneth cell function by impairing the membrane localization of TLR2 (23). Deregulation of miRNAs and RBPs occurs commonly in many human gut mucosal disorders such as colonic cancer, leaky gut, and inflammatory bowel disease (22, 25, 26).
miR-195 is evolutionally conserved among different species and is involved in different cellular processes (27, 28). miR-195 inhibits cell proliferation by reducing the levels of cyclin-dependent kinase 4 (CDK4), cyclin D1 (CCND1), CDK6, and WEE1 (28, 29), promotes apoptosis by lowering SIRT1 abundance (30), and affects cell migration and cancer invasion by modulating expression of ActRIIA (31). Our previous study revealed that the intestinal epithelium expresses miR-195 and that increasing the levels of cellular miR-195 represses IEC migration over the denuded area after wounding in an in vitro epithelial repair model by destabilizing the mRNA encoding STIM1, a protein essential for stored-operated Ca2+ influx in IECs after wounding (32, 33). Increased miR-195 also inactivates IGF signaling by repressing translation of the IGF2-receptor (21) but the levels of cellular miR-195 can be decreased by long noncoding RNA (lncRNA) uc.173 (34). However, most studies investigating miR-195 function in the intestine were conducted in cultured IECs, and the exact in vivo function of miR-195 in the intestinal epithelium remains to be fully elucidated. Using a gain-of-function transgenic approach, we found that intestinal epithelial tissue-specific overexpression of miR-195 in mice resulted in defects in Tuft and Paneth cells in vivo and ex vivo. Our results further indicate that miR-195 inhibited Tuft cell function by repressing the translation of DCLK1, and that this repression was antagonized by HuR.
MATERIALS AND METHODS
Chemicals and Cell Cultures
Tissue culture medium and dialyzed fetal bovine serum were from Invitrogen (Carlsbad, CA) and biochemicals were from Sigma (St. Louis, MO). Pre-miR miRNA precursor of miR-195 was purchased from Ambion (Austin, TX). Biotin-labeled miRNA-195 was custom-made by Dharmacon (Lafayette, CO). The antibodies recognizing DCLK1 (known as DCAMKL1, Cat. No. ab31704), E-Cadherin (Cat. No. 610182), HSC70 (Cat. No. ab19136), lysozyme (Cat. No. ab108508, for immunoblotting), and villin (Cat. No. ab130751) were obtained from Abcam and BD Biosciences. Antibodies recognizing HuR (Cat. No. sc-5261), GAPDH (Cat. No. sc-32233), and lysozyme (Cat. No. PA5-16668, for immunostaining) were purchased from Santa Cruz biotechnology and Invitrogen. Antibodies against BrdU (Cat. No. 5292), PCNA (Cat. No. 2586), and mucin 2 (Cat. No. NBP1-31231) were from Cell Signaling and Novus Biologicals. Secondary antibodies such as anti-mouse IgG horseradish peroxidase (HRP) (Cat. No. 7076), anti -rabbit IgG HRP (Cat. No. 7074), and anti- rat IgG HRP (Cat. No. 7077) were from Cell Signaling Technologies. All antibodies used in this study were thoroughly validated for species specificity. Relative protein levels were analyzed by using Biorad Chemidoc and XRS system equipped with Image laboratory software (v. 4.1). We also used “Quantity tool” to determine the band intensity volume; the values were normalized with internal loading control either GAPDH or HSC70 (stable housekeeper proteins). Human colorectal adenocarcinoma (Caco-2) cells (Cat. No. HTB-37) and human embryonic kidney (HEK-293) cells (Cat. No. CRL-1573) were purchased from the American Type Culture Collection (Manassas, VA) and were maintained under standard culture conditions (21, 34).
Animal Experiments
All animal experiments were performed in accordance with the National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and Use Committee of University Maryland School of Medicine and Baltimore VA hospital. Intestinal epithelial tissue-specific miR-195 transgenic (miR195-Tg) mice were generated by using A33-promoter and following the strategy as we used in the generation of miR-222 mice (24). The 748-bp fragment including the mouse miR-195 locus bearing primary miR-195 (pri-miR-195) sequence and human β-globin intron (222-bp 5′ upstream sequence and 259-bp 3′ sequence) were cloned into the pIRES-AcGFP1-Nuc vector using miR-195 cloning primer set (Table 1), and the final miR-195 expression vector, A33-miR195-GFP, was used for microinjection into fertilized eggs (24, 35). Transgenic founders on pure C57BL/6J background were established by pronuclear injection through a commercial service provided by Genome Modification Facility at Harvard University. Genotyping was performed by PCR in DNA extracted by tail clippings to identify the first generation of recombinant mice with miR195/GFP bicistronic RNA. Two founders were further characterized for the transmission to subsequently establish transgenic colonies. Both male and female, age-matched (6–8 wk old; weighing between 18 and 22 g) miR195-Tg and control littermate, and wild-type C57BL/6J mice (purchased from the Jackson Laboratory, Bar Harbor, ME) were housed in a specific pathogen-free breeding barrier and cared for by trained technicians and veterinarians. Animals were deprived of food but allowed free access to tap water for 48 h in the fasting model. To examine gut mucosal growth, BrdU was incorporated in intestinal mucosa by intraperitoneal (ip) injection of 2 mg BrdU (Sigma, St. Louis, MO) in phosphate-buffered saline. Two portions of the small intestine taken from 0.5 cm distal to the ligament of Trietz were removed, one for histological examination and the other for extraction of protein and RNA. The mucosa was scraped with a glass slide for various measurements as described previously (2, 36). Representative results from two independent founders were reported here and compared with those obtained from control littermate mice.
Table 1.
Primers used for DCLK1 luciferase reporters and miR-195 construct
| Primers | Sequences |
|---|---|
| Luciferase assay | |
| hDCLK1 3′UTR-WT | |
| Forward | 5′-GCGCCTCGAGGGTACAAGGCGCAGCCAG-3′ |
| Reverse | 5′-GCGCGTCGACGTGTTATCACTTCCGGATGC-3′ |
| hDCLK1 3′UTR-F1 | |
| Forward | 5′-GCGCCTCGAGGGTACAAGGCGCAGCCAG-3′ |
| Reverse | 5′-GCGCGTCGACTTCCAGCCCTGCAGAGCACC-3′ |
| hDCLK1 3′UTR-F2 | |
| Forward | 5′-GCGCCTCGAGGGAATTTCAAACAGAAAGAGGG-3′ |
| Reverse | 5′-GCGCGTCGACGTGTTATCACTTCCGGATGC-3′ |
| Cloning | |
| miR195 Forward | 5′-CGACGTTTAAACGCGGATCCAGATCTGAA-3′ |
| miR195 Reverse | 5′-CTGGGTAAAAGCTAGCGAATTCCGTTCC-3′ |
| Genotyping | |
| Forward | 5′-AGAAACTGGGCTTGTCGAGACAGA-3′ |
| Reverse | 5′-AAGACTCTACTTTGCTCTGTGGGG-3′ |
DCLK1, double cortin-like kinase 1; UTR, untranslated region; WT, wild type.
Intestinal Organoid Culture
Isolation and culture of primary enterocytes were conducted following the method provided by Stem Cell Technologies (Cambridge, MA) with minor modifications as described previously (14, 37). Briefly, primary crypts were released from the small intestinal mucosa in mice; then, the isolated crypts were mixed with Matrigel and cultured in Advanced Dulbecco’s modified Eagle medium/F12 medium. The growth of intestinal organoids was examined under phase-contrast microscopy.
Histology and Immunofluorescence Staining
Dissected and opened intestines were mounted onto a solid surface and fixed in formalin and paraffin. Sections of 5 μm thickness were stained with hematoxylin-eosin (H&E) for general histology. Using a grade micrometer eyepiece, the overall length of villus and crypts of each section was measured, and the villus/crypt ratio was calculated. The immunofluorescence staining procedure was carried out according to the method we have described previously (2, 14). For experiments using mucosal tissue samples from mice, more than five slides (5-μm-thickness section) in each tissue sample were prepared for immunofluorescence staining. For studies in cultured intestinal organoids, the slides were fixed in 3.7% formaldehyde in phosphate-buffered saline and rehydrated. All slides were incubated with a primary antibody recognizing DCLK1, lysozyme, mucin 2, or E-cadherin in blocking buffer overnight and then incubated with secondary antibody conjugated with Alexa Fluor-594 (Cat. No. A32754, Molecular Probes, Eugene, OR) for 2 h at room temperature. After rinsing three times, the slides were incubated with DAPI (Cat. No. D1306, Molecular Probes) at the concentration of 1 μM for 10 min to stain cell nuclei. Finally, the slides were washed, mounted, and viewed through a Zeiss confocal microscope (model LSM700). Measurements of apoptotic cell death by TUNEL staining were performed as described (24). Slides were examined in a blinded fashion by coding them, and only after examination was complete were they decoded. Images were processed using Photoshop software (Adobe, San Jose, CA).
Assays of Gut Permeability
FITC-conjugated dextran dissolved in water (Cat. No. FD4, Sigma; 4KD; 600 mg/kg) was administered to mice via gavage as described (38). Blood was collected 4 h thereafter via cardiac puncture. The serum concentration of the FITC-dextran was determined using a plate reader with an excitation wavelength at 490-nm and an emission wavelength of 530-nm. The concentration of FITC-dextran in sera was determined by comparison to the FITC-dextran standard curve.
Plasmid Construction
The fragments of Dclk3 3′-UTR and its deletion mutation were subcloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Cat. No. P1330, Promega, Madison, WI) to generate the pmirGLO-Luc-Dclk1 3’UTR reporter constructs as described (14, 33). The primer sequences for generating these constructs are provided in Table 1. Transient transfections in HEK-293 cells were performed using the Lipofectamine Reagent as recommended by the manufacturer, and the levels of firefly luciferase activity were normalized to Renilla luciferase activity.
Reverse Transcription and qPCR Analysis
Total RNA was isolated by using RNeasy mini kit (Cat. No. t4104, Qiagen, Valencia, CA), and reverse transcription and PCR amplification reactions were performed as described (36). The levels of Gapdh PCR product were examined to monitor the evenness in RNA input in RT-qPCR samples. RT-qPCR analysis was conducted using 7500-Fast Real-Time PCR Systems with specific primers, probes, and software (Applied Biosystems, Foster City, CA). For miRNA studies, the levels of miR-195 were also quantified by reverse transcription (RT)-qPCR by using Taqman MicroRNA assay; small nuclear RNA (snRNA) U6 was used as endogenous control.
Western Blot Analysis
Whole cell lysates were prepared by using the RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl) containing 1% sodium dodecyl sulfate (SDS), sonicated, and centrifuged at 4°C for 15 min. The supernatants were boiled and size-fractionated by SDS-PAGE. After the blots were incubated with primary antibody and then secondary antibodies, immunocomplexes were developed by using chemiluminescence.
Biotin Labeled miR-195 Pulldown Assays
Biotin-labeled miR-195 was transfected into HEK-293 cells and 24 h later whole cell lysates were collected, mixed with Streptavidin-Dynal beads (Cat. No. 11205 D, Invitrogen, Carlsbad, CA) and incubated at 4°C with rotation overnight (33, 39). After the beads were washed thoroughly, the bead-bound RNA was isolated and subjected to RT-qPCR analysis. Input RNA was extracted and served as control.
Assays of Newly Translated Protein and Polysome Analysis
New synthesis of nascent DCLK1 protein was detected by Click-iT protein analysis detection kit (Cat. No. C33372, Life technologies, Grand Island, NY) and performed following the company’s manual. Briefly, HEK-293 cells were incubated in methionine-free medium and then exposed to L-azidohomoalanine (AHA). After mixing cell lysates with the reaction buffer for 20 min, the biotin-alkyne/azide-modified protein complex was pulled down using paramagnetic Streptavidin-conjugated Dynabeads. The pull-down material was resolved by 10% SDS-PAGE and analyzed by Western immunoblotting analysis using antibodies against DCLK1 or GAPDH.
Polysome analysis was performed as described (40). Briefly, HEK-293 cells at ∼70% confluence were incubated in 0.1 mg/mL cycloheximide, then lifted by scraping in PEB lysis buffer. Nuclei were pelleted, and the resulting supernatant was centrifuged through a 10%–50% linear sucrose gradient to fractionate cytoplasmic components according to their molecular weights. The eluted fractions were prepared with a fraction collector (Brandel, Gaithersburg, MD), and their quality was monitored at 254 nm using a UV-6 detector (ISCO, Louisville, KY). After RNA in each fraction was extracted, the levels of each individual mRNA were quantified by RT-qPCR in each of the fractions.
Statistical Analysis
All values were expressed as the means ± SE from five animals or three separate experiments. Unpaired, two-tailed Student’s t test was used when indicated with P < 0.05 considered statistically significant (41). The statistical software used was GraphPad Instat Prism 5 (San Diego, CA).
RESULTS
MiR-195 Expression in Responding to Stress and a Model of miR-195 Transgenic Expression
To begin to investigate the involvement of miR-195 in intestinal mucosal homeostasis and pathologies, we first examined changes in miR-195 expression in the small intestinal mucosa of wild-type mice exposed to food starvation. As we and others reported previously (42, 43), fasting for 48 hours inhibited growth of the small intestinal mucosa in mice, as indicated by decreases in both the proliferating crypt population (marked by BrdU) and the lengths of villi and crypts. Fasting for 24 h and 48 h also increased mucosal miR-195 abundance in the small intestine (Fig. 1A), whereas it decreased the levels of mucosal miR-471, miR-291a-5p, miR-675, and miR-341. Second, analysis of the changes in miR-195 in cultured IECs revealed that both inhibition of proliferation and disruption of the intestinal epithelial barrier function by depleting cellular polyamines with D,L-α-difluoromethylornithine (DFMO, a specific inhibitor of polyamine biosynthesis) (44) or by silencing the lncRNA uc.173 were also associated with an increase in the levels of cellular miR-195. Consistent with previous findings (45), exposure to DFMO for 6 days almost completely depleted the cellular polyamines putrescine and spermidine. Decreasing the levels of cellular polyamines by DFMO significantly increased the levels of cellular miR-195, which was prevented by exogenous putrescine given together with DFMO (Fig. 1B, left). Administration of putrescine also restored cell proliferation and epithelial barrier function in DFMO-treated cells, as reported (46, 47). In addition, silencing uc.173 by transfection with LNA-modified anti-uc.173 oligonucleotides caused growth arrest of IECs and led to epithelial barrier dysfunction (34, 39), which was also associated with an increase in the levels of cellular miR-195 (Fig. 1B, right). Together, these results obtained from in vivo and in vitro systems suggest that miR-195 may be involved in sustaining homeostasis of the intestinal epithelium and may play a role in gut mucosal pathologies in response to stress.
Figure 1.

MiR-195 levels in the intestinal epithelium responding to critical stress and characterization of miR195-Tg mice. A: levels of tissue miR-195 in the small intestinal mucosa of mice fasted for 24 h and 48 h as measured by RT-qPCR analysis. Total RNA was isolated and used in RT-qPCR reactions, and equal loading was monitored by assessing Gapdh mRNA levels. Values are the means ± SE (n = 5 mice). *P < 0.05 compared with control mice. B: levels of cellular miR-195 in polyamine-deficient (left) and uc.173-silenced (right) cells. Caco-2 cells were treated with DFMO with or without putrescine (Put) for 6 days or 48 h after transfection with anti-uc.173 oligo. Values are the means ± SE (n = 3 replicates). *P < 0.05 compared with controls. C: the A33/miR195/GFP construct used to be injected into fertilized oocytes for generating miR195-Tg mice. D: levels of miR-195 in the mucosa of small intestine, cecum, colon, and stomach and tissues of heart, liver, and spleen. *P < 0.05 compared with control littermates (n = 4 mice). E and F: mouse and gastrointestinal gross morphology and body weights of age- and sex-matched littermate and miR195-Tg mice. Values are the means ± SE (n = 5 mice). Statistical significance was analyzed using unpaired, two-tailed Student’s t test. DCLK1, double cortin-like kinase 1; miR195-Tg, miR-195 transgenic; LncRNAs, long noncoding RNAs; miRNAs, microRNAs; RBPs, RNA-binding proteins; q, quantitative; RT, reverse transcription.
To examine the function of miR-195 in intestinal epithelium in vivo, we used a gain-of-function transgenic approach and generated miR-195 transgenic (miR195-Tg) mice. The A33 promoter was used to drive intestinal epithelial tissue-specific overexpression of genomic miR-195 precursor (Fig. 1C), as described previously (24). Age-matched miR195-Tg mice and littermate controls (male and female, 3- or 4-mo-old) were used for comparison of phenotype. MiR195-Tg mice exhibited a specific overexpression of miR195/GFP bicistronic RNA in the mucosal tissues of the small intestine, but not in the mucosa of colon, caecum, or stomach, or in tissues from heart, liver, or spleen as measured by in situ detection using anti-GFP antibody. Because primary miR-195 provides two forms of mature miRNAs, miR-195-5p and miR-195-3p, we examined changes in the levels of both miRNAs in different tissues by RT-qPCR analysis. The levels of miR-195-5p and miR-195-3p in the small intestinal mucosa of miR195-Tg mice increased by ∼3-fold and 8-fold, respectively, when compared with those observed in control littermates (Fig. 1D). On the other hand, there were no significant changes in the levels of miR-195-5p or miR-195-3p in the mucosa of cecum, colon, or stomach, or in tissues from heart, liver, or spleen in miR195-Tg mice compared with littermates. MiR195-Tg mice looked normal in general (Fig. 1E, top) and there were no significant differences in gastrointestinal gross morphology (Fig. 1E, bottom), body weight (Fig. 1F), reproductivity, or general appearance between miR195-Tg mice and littermate controls. These results indicate that miR195-Tg mouse is a suitable tissue-specific miR-195 transgenic model for studying Tuft and Paneth cells in the intestinal epithelium.
Transgenic Expression of miR-195 in Mice Causes Defects in Tuft and Paneth Cells
Intestinal epithelium-specific transgenic expression of miR-195 in mice resulted in defects in Tuft and Paneth cells in the small intestinal mucosa, as examined by detecting DCLK1 and lysozyme using immunofluorescence analysis. Staining of whole mounts of the small intestine revealed that in control mice (“Littermates”), DCLK1-positive cells (Tuft cells) were predominantly distributed at the villous regions (Fig. 2Aa), and lysozyme-positive cells (Paneth cells) were located at the base of the crypt areas (Fig. 2Ba). By contrast, the levels of DCLK1-positive Tuft cells and lysozyme-positive Paneth cells in the small intestinal mucosa decreased dramatically in miR195-Tg mice relative to control littermate mice (Fig. 2, Ab and Bb). Quantification of this reduction indicated that the numbers of intestinal Tuft and Paneth cells in miR195-Tg mice decreased by ∼85% and ∼45%, respectively, compared with control littermate mice (Fig. 2, Ac and Bc). Moreover, the levels of DCLK1 and lysozyme proteins in the small intestinal mucosa also decreased in miR195-Tg mice as measured by immunoblotting analysis (Fig. 2, Ad and Bd). On the other hand, transgenic miR-195 expression failed to alter the function of Goblet cells, as there were no differences in the number of Goblet (Alcian blue-positive) cells in the small intestinal mucosa between miR195-Tg mice and control littermates (Fig. 2C). miR-195 overexpression in mice did not affect enterocyte differentiation in the small intestinal mucosa as measured by villin immunostaining analysis (Fig. 2D). Another line of miR195-Tg mice generated from an independent founder was also examined and exhibited similar defects in Tuft and Paneth cells in the small intestinal mucosa (data not shown).
Figure 2.

Transgenic expression of miR-195 in mice reduces Tuft and Paneth cells in the small intestinal epithelium. A: immunostaining of Tuft cells (DCLK1-positive cells) and DCLK1 levels in the small intestinal mucosa: littermates (a); miR195-Tg mice (b); quantitative data of Tuft cells (c); and representative immunoblots of DCLK1 (d). Red, DCLK1; and green, E-cadherin (E-cad). Scale bars: 50 μm. Values are the means ± SE (n = 5 mice). *P < 0.05 compared with littermate mice. B: changes in Paneth cells (lysozyme-positive cells) and lysozyme levels in the intestinal mucosa described in A. Red, lysozyme; and green, E-cad. Scale bars: 50 μm. *P < 0.05 compared with littermate mice (n = 5 mice). C and D: Goblet cells and enterocytes in the intestinal mucosa described in A, as examined by alcian blue staining (blue) and villin immunostaining (red), respectively. Experiments were repeated in five littermate or miR195-Tg mice and showed similar results. Statistical significance was analyzed using unpaired, two-tailed Student’s t test. DCLK1, double cortin-like kinase 1; miR195-Tg, miR-195 transgenic; miRNAs, microRNAs.
Consistent with the in vivo findings, we further found that there were obvious abnormalities in Tuft and Paneth cells in the intestinal organoids from miR195-Tg mice. As shown, DCLK1-positive and lysozyme-positive cells were enriched in the intestinal organoids from control littermates (Fig. 3Aa) but decreased dramatically in the organoids from miR195-Tg mice (Fig. 3Ab). DCLK1-positive cells were almost undetectable in the organoids from miR195-Tg mice, and the numbers of lysozyme-positive cells decreased by ∼65%, compared with those observed in the organoids from control littermates (Fig. 3Ac). Again, increased levels of miR-195 did not appear to affect Goblet cell development and enterocyte differentiation ex vivo, as the numbers of Goblet cells and villin staining in the miR-195-elevated organoids from miR195-Tg mice were indistinguishable from those of the organoids from control littermates (Fig. 3B). These results indicate that locally increasing the levels of miR-195 in the intestinal epithelium specifically decreases the numbers of Tuft and Paneth cells in mice.
Figure 3.

Increasing miR-195 reduces the numbers of Tuft and Paneth cells in intestinal organoids. A: immunostaining of Tuft (DCLK1-positive) and Paneth (lysozyme-positive) cells in intestinal organoids: littermates (a); miR195-Tg mice (b); and quantitative data of Tuft and Paneth cells (c). Primary enterocytes were isolated from the proximal small intestine, and changes in Tuft and Paneth cells were examined by immunostaining assays on day 5 after primary cultures. Values are the means ± SE (n = 5 replicates). *P < 0.05 compared with littermate mice. Scale bar: 25 μm. B: Goblet cells (Mucin 2-positive cells) and enterocyte differentiation (marked by villin immunostaining) in intestinal organoids described in A. Scale bar: 25 μm. Experiments were repeated in five littermate or miR195-Tg mice and showed similar results. Statistical significance was analyzed using unpaired, two-tailed Student’s t test. DCLK1, double cortin-like kinase 1; miR195-Tg, miR-195 transgenic.
Against expectation, transgenic expression of miR-195 in mice did not affect the renewal of the intestinal epithelium in vivo. There were no significant differences in histological features of the small intestinal mucosa (Fig. 4A, top), the proliferating crypt cell population (BrdU-positive cells per crypt: 22 ± 4 in littermates and 21 ± 3 in miR195-Tg mice, n = 5; Fig. 4A, bottom), the lengths of villi and crypts (Fig. 4B, top), or the levels of proliferation marker PCNA (Fig. 4B, bottom) between miR195-Tg mice and control littermates. In an ex vivo model, organoids from miR195-Tg mice also exhibited no difference in growth (Fig. 4C). As shown, an organoid was initiated from tiny isolated crypts, but it grew rapidly into multiple cells and buds by 3 days in culture, without noticeable differences in the sizes or surface areas of intestinal organoids from miR195-Tg mice compared with organoids from control littermates. We also examined the colonic mucosa of miR195-Tg mice and found that there were no differences in mucosal growth in the colon between miR195-Tg mice and control littermates (data not shown). Although transgenic expression of miR-195 in mice did not alter basal level of gut permeability as measured by FITC-conjugated dextran assays (Fig. 4D), it increased vulnerability of the gut barrier in response to bacterial product lipopolysaccharide (LPS), as shown by a significant increase in gut permeability in miR195-Tg mice compared with littermates after exposure to same doses of LPS (Fig. 4E). In addition, transgenic miR-195 expression did not directly induce apoptosis in the intestinal epithelium, as there was no significant increase in cell death observed in miR195-Tg mice. Rates of apoptotic cell death were <1% (n = 5) in both littermate and miR195-Tg mice as examined by TUNEL staining.
Figure 4.

Transgenic expression of miR-195 fails to alter growth of the small intestinal mucosa but increases vulnerability of the gut barrier to lipopolysaccharide (LPS). A: photomicrographs of hematoxylin and eosin staining of the small intestine (top) and proliferating cells in the crypts (bottom) as measured by BrdU labeling assay: littermates (a); and miR195-Tg mice (b). BrdU (1 h postinjection, S phase), green. Scale bar, 50 µm. All experiments were repeated in five littermate or miR195-Tg mice and showed similar results. B: the length of crypts and villi (top) and immunoblots of PCNA and HuR (bottom) in the mucosa described in A. Values are the means ± SE (n = 6 mice). C: growth of intestinal organoids isolated from the proximal small intestine: littermate (a); and miR195-Tg mice (b). Images were taken at different times after cultures. Three separate experiments were performed and showed similar results. Scale bar: 100 μm. D: gut permeability in the mice described in A. FITC dextran was given orally, and blood samples were collected 4 h thereafter for measurement. Values are the means ± SE (n = 5 replicates). E: increased vulnerability of the gut barrier in response to LPS. Mice were intraperitoneally injected with LPS (0.1 mg/kg) in 200 µL PBS once daily for 5 days. Gut permeability was measured 24 h after treatment with last dose of LPS. Values are the means ± SE (n = 5 replicates). *P < 0.05 compared with PBS or littermates exposed to LPS. Statistical significance was analyzed using unpaired, two-tailed Student’s t test. miR195-Tg, miR-195 transgenic; LncRNAs, long noncoding RNAs; miRNAs, microRNAs; q, quantitative; RT, reverse transcription.
miR-195 Interacts with and Represses Dclk1 mRNA Translation
We sought to further investigate the mechanisms whereby miR-195 regulated the development of Tuft cells. As DCLK1 is specifically expressed in Tuft cells in the intestinal epithelium and plays an essential role in maintaining the morphological and molecular characteristics of Tuft cells (3, 5), we tested the possibility that miR-195 regulates Tuft cell function by altering DCLK1 expression levels. To do so, cultured HEK-293 cells were used in this study, because they express high levels of DCLK1 (48) and they are easily transfected. First, we examined the association of miR-195 with the Dclk1 mRNA by RNA pulldown assays using biotin-labeled miR-195, as reported previously (33). Twenty-four hours after transfection, miR-195 levels increased significantly, whereas the levels of the housekeeping noncoding RNA U6 did not (Fig. 5A). Dclk1 mRNA was enriched in the materials pulled down by transfected biotin-miR-195 but not from cells that had been transfected with a control biotinylated scramble RNA (Fig. 5Ba). The association of miR-195 with the Dclk1 mRNA was specific, as increasing the levels of biotin-miR-195 did not increase its interaction with Myc mRNA. In addition, transfection with biotin-labeled miR-195 failed to alter the steady-state levels of total Dclk1 and Myc mRNAs (Fig. 5Bb). We also examined the interaction of miR-222 with Dclk1 mRNA and found that increasing the levels of biotin-miR-222 did not alter its association with the Dclk1 mRNA.
Figure 5.

MiR-195 inhibits translation of DCLK1. A: levels of miR-195 in HEK-293 cells transfected with biotinylated miR-195 for 24 h. Values are the means ± SE (n = 3 replicates). *P < 0.05 compared with cells transfected with scramble oligomer. B: binding of biotinylated miR-195 to mRNAs encoding DCLK1 and Myc: levels of mRNAs in the materials pulled down by biotin-miR-195 (a); and levels of total input mRNAs (b). *P < 0.05 compared with scramble (n = 3 replicates). C: levels of miR-195 in cells transfected with pre-miR-195 for 48 h. *P < 0.05 compared with control (n = 3 replicates). D and E: changes in the levels of DCLK1 protein and Dclk1 mRNA in cells treated as described in C. F: newly synthesized DCLK1 protein as measured by L-azidohomoalaine (AHA) incorporation assays in cells described in C. Three separate experments were performed and showed similar results. G: distribution of Dclk1 (a) and Gapdh (b) mRNAs in each gradient fraction of polysomal profiles prepared from cells described in C. H: Levels of the Luc reporter activity 48 h after transfection with pre-miR-195. Left, schematic of different chimeric firefly luciferase reporters bearing the wild-type Dclk1 3′-UTR or its deletion mutation. *P < 0.05 compared with control (n = 3 replicates). Statistical significance was analyzed using unpaired, two-tailed Student’s t test. DCLK1, double cortin-like kinase 1; miR195-Tg, miR-195 transgenic; miRNAs, microRNAs.
Second, we investigated the functional consequence of the association of miR-195-Dclk1 mRNA by increasing the levels of cellular miR-195 through transfection of the miR-195 precursor (pre-miR-195). Cells transfected with pre-miR-195 for 48 h showed dramatic increase in the levels of miR-195 compared with the levels seen in cells transfected with control vector (Fig. 5C). Ectopically expressed miR-195 reduced the levels of DCLK1 and STIM1 (positive control) proteins without effect on HuR (Fig. 5D). On the other hand, miR-195 overexpression did not alter Dclk1 mRNA abundance (Fig. 5E). To examine whether miR-195 alters the translation of Dclk1 mRNA, we examined changes in the level of nascent DCLK1 protein synthesis after transfection with pre-miR-195 and found that newly synthesized DCLK1 protein decreased in cells overexpressing miR-195, compared with cells transfected with control vector (Fig. 5F); this effect was specific, as the levels of nascent GAPDH, a housekeeping protein, were not affected by transfection of pre-miR-195. To further define the role of miR-195 in the regulation of DCLK1 translation, we examined the relative distribution of Dclk1 mRNA in polyribosome gradients prepared from cytoplasmic lysates of HEK-293 cells. Although increasing the levels of miR-195 did not affect global polysomal profiles as reported (21), the abundance of Dclk1 mRNA associated with fractions 9–11, which contained large polyribosomes and hence were presumed to be actively translating, decreased in cells overexpressing miR-195, with a robust shift of Dclk1 mRNAs to less-translating fractions (fractions 4–6; Fig. 5G, left). In contrast, Gapdh mRNA, encoding GAPDH, distributed similarly in both groups (Fig. 5G, right).
Third, we determined whether repression of DCLK1 translation by miR-195 was mediated through specific binding sites in the Dclk1 mRNA. As three computationally predicted miR-195 binding sites are located at the Dclk1 3′-UTR, fractions of full-length wild type (WT) 3′-UTR and partial transcripts of the 3′-UTR with or without the predicted binding sites were cloned into the pmirGLO dual-luciferase miRNA target expression vector to generate pmirGLO-Luc reporter constructs (Fig. 5H, schematic). Importantly, increasing the levels of miR-195 by transfection with pre-miR-195 selectively decreased the levels of Luc-3′UTR-WT luciferase reporter activity and Luc-3′UTR-F1 bearing with miR-195 binding sites, but failed to inhibit the activities of Luc-3′UTR-F2 containing no miR-195 sites (Fig. 5H, right). Taken together, these results indicate that miR-195 interacts with Dclk1 mRNA via the 3′-UTR and inhibits DCLK1 translation.
Competitive Binding of miR-195 and HuR to Dclk1 mRNA Regulates DCLK1 Translation
We sought to look deeper into the mechanism whereby miR-195 suppresses DCLK1 translation. In light of earlier studies showing that miR-195 and the RBP HuR competitively regulated expression of shared target transcript (Stim1 mRNA) (33), we postulated that miR-195 might also compete with HuR to regulate DCLK1 translation. Ribonucleoprotein immunoprecipitation (RIP) analysis using an anti-HuR antibody followed by measurement of Dclk1 mRNA levels by RT-qPCR analysis in the IP material revealed that Dclk1 mRNA associated with HuR, as Dclk1 mRNA was highly enriched in HuR IP relative to IgG IP samples (Fig. 6A). HuR binding to the Occludin mRNA served as a positive control in this study (38). To determine the impact of HuR-Dclk1 mRNA interaction on DCLK1 expression, we found that HuR silencing by transfection with HuR-directed siRNA (siHuR) decreased the levels of DCLK1 and p53 (positive control) proteins (Fig. 6B), although it did not alter total miR-195 abundance (data not shown).
Figure 6.
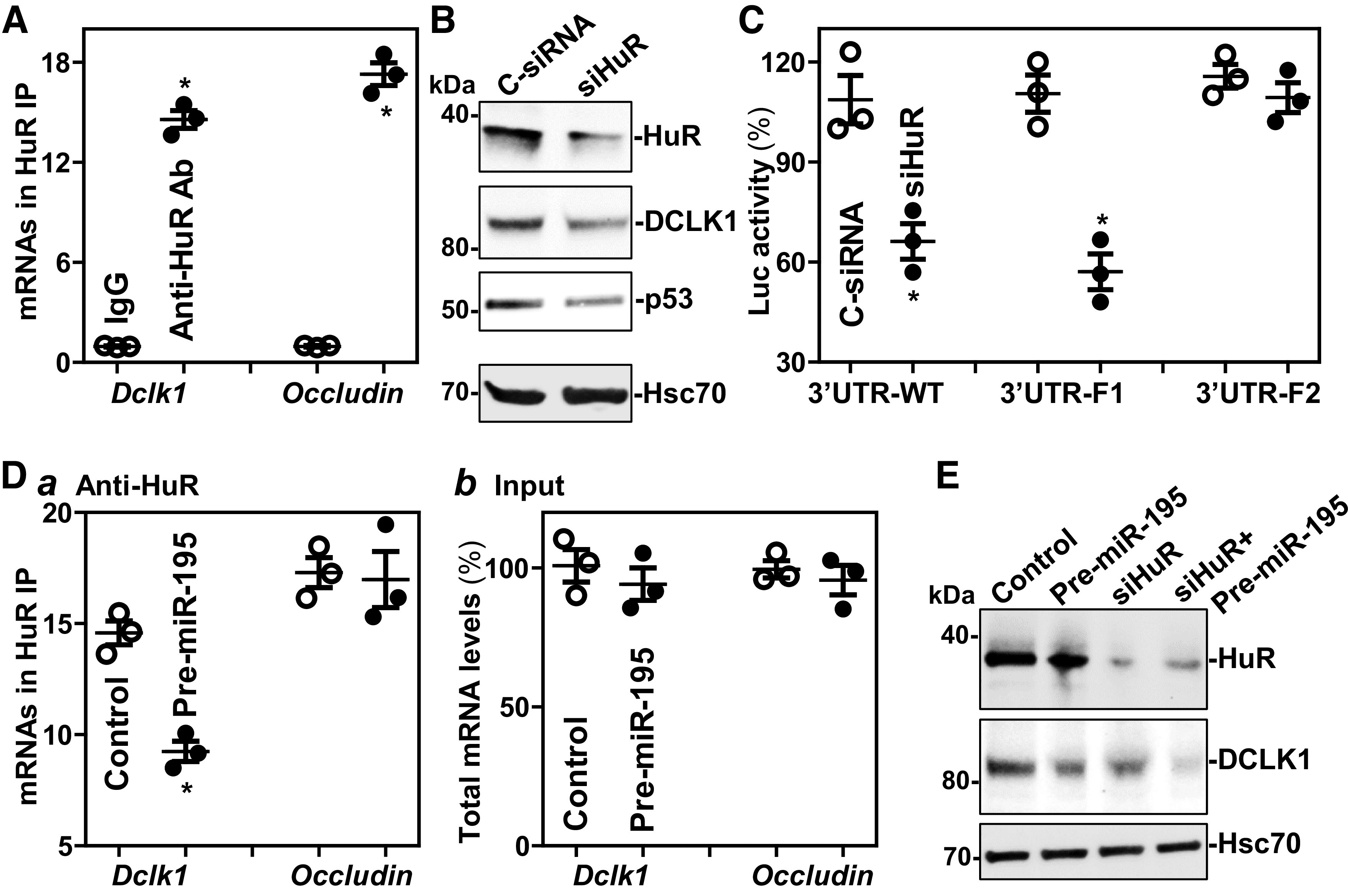
miR-195 represses DCLK1 expression by interacting with HuR. A: association of endogenous HuR with endogenous Dclk1 mRNA in cultured HEK-293 cells. After IP of RNA-protein complexes from cell lysates using either anti-HuR antibody (Ab) or control IgG, RNA was isolated and used for RT-qPCR analysis. Values are the means ± SE (n = 3 replicates). *P < 0.05 compared with IgG IP. B: immunoblots of HuR and DCLK1 in cells transfected with siHuR or C-siRNA for 48 h. Three separate experments were performed and showed similar results. C: changes in activities of luciferase reporters containing WT or mutated Dclk1 3’UTR in cells treated as described in B. *P < 0.05 compared with C-siRNA (n = 3 replicates). D: changes in the binding of HuR to Dclk1 mRNA in cells transfected with pre-miR-195 for 48 h: levels of mRNAs in the materials pulled down by anti-HuR Ab (a); and levels of total input mRNAs (b). *P < 0.05 compared with control (n = 3 replicates). E: immunoblots of DCLK1 in cells transfected with pre-miR-195 or siHuR alone or cotransfected with pre-miR-195 and siHuR for 48 h. Statistical significance was analyzed using unpaired, two-tailed Student’s t test. DCLK1, double cortin-like kinase 1; miR195-Tg, miR-195 transgenic; LncRNAs, long noncoding RNAs; miRNAs, microRNAs; RBPs, RNA-binding proteins; q, quantitative; RT, reverse transcription; WT, wild type.
Studies using Dclk1 3′UTR-Luc-reporter constructs showed that HuR regulated DCLK1 expression also by interacting with the proximal region of the Dclk1 3′-UTR, as HuR silencing decreased the levels of Luc-3′UTR-WT and Luc-3′UTR-F1, but not the activity of Luc-3′UTR-F2 (Fig. 6C). As both miR-195 and HuR interacted with Luc-3′UTR-F1 (Fig. 5H and Fig. 6C), we further examined the effect of ectopically expressing miR-195 on HuR binding to the Dclk1 mRNA. As shown, increased miR-195 by transfection with pre-miR-195 decreased the levels of HuR-Dclk1 mRNA complex, but it failed to alter HuR interaction with Occludin mRNA (Fig. 6D). Moreover, HuR silencing and miR-195 overexpression synergistically repressed DCLK1 expression (Fig. 6E). These results strongly suggest that miR-195 and HuR compete for association with the Dclk1 mRNA and that induced miR-195 inhibits DCLK1 translation partially by displacing HuR.
DISCUSSION
Intestinal Tuft and Paneth cells play an important role in sustaining homeostasis of the gut epithelium by enhancing host defense against invading bacteria and other pathogens (3, 6, 7), but the exact mechanisms that regulate the function of Tuft and Paneth cells are poorly understood. In this study, using a gain-of-function transgenic approach in vivo, we discovered that miR-195 regulates the function of Tuft and Paneth cells. Transgene-driven overexpression of miR-195 in mice caused defects in Tuft and Paneth cells in the small intestinal mucosa, although there were no effects on Goblet cells or enterocytes. In an ex vivo model, miR-195-overexpressing enteroids from miR195-Tg mice also exhibited dysfunction of Tuft and Paneth cells. Interestingly, miR-195 regulated Tuft cell function by posttranscriptionally controlling DCLK1 expression levels. miR-195 directly interacted with the Dclk1 mRNA via the 3′-UTR and inhibited its translation without effect on Dclk1 mRNA levels. These findings represent a conceptual advance linking miR-195 to the regulation of Tuft and Paneth cells and further implicate miR-195 in the homeostatic control of host defense and mucosal inflammation in the intestinal tract.
The results reported here are the first demonstration that miR-195 represses the development of Tuft and Paneth cells in the small intestinal epithelium. As shown, miR195-Tg mice exhibited abnormalities in intestinal Tuft and Paneth cells as evidenced by a significant decrease in the number of DCLK1-positive cells and lysozyme-positive cells in vivo and ex vivo (Figs. 2 and 3). The inhibitory effect of miR-195 overexpression on Tuft and Paneth cell function was not surprising, because miR-195 was shown to modulate expression of several migration-associated (31, 33) and proliferation-relevant proteins (28, 29, 49) that are intimately involved in the maturation and differentiation of the gut epithelium. This notion is further supported by our studies in cultured cells showing that increasing miR-195 levels repressed the translation of the Tuft cell resident protein DCLK1. Although the mechanism responsible for Paneth cell dysfunction in miR195-Tg mice was not identified, we recently found that HuR is essential for normal Paneth cell function in the intestinal epithelium (14) and that miR-195 and HuR have antagonizing impacts on the stability and translation of target mRNAs (33). However, the findings in cultured cells do not fully agree with the results in mice: in cultured cells, miR-195 represses cell proliferation (21, 34), whereas in miR-195-overexpressing mice and in ex vivo organoids there was no difference in the renewal of small intestinal epithelium or in the rates of mucosal growth (Fig. 4). The exact reasons for the differences seen in vivo, ex vivo, and in culture remain unclear, but tissue environment, magnitude of miR-195 abundance, and cellular context likely contribute to the distinct responses, as seen in other discrepant models (50).
Another important finding of this study is that both miR-195 and HuR interacted with the Dclk1 3′-UTR and regulated DCLK1 translation competitively in opposite directions. It has been reported that miR-195 regulates the DCLK1 expression in pancreatic cancer cells (51), but our current studies further show that HuR is a novel regulator of DCLK1 expression and that miR-195 inhibits DCLK1 translation by preventing HuR binding to the Dclk1 mRNA. Through the use of various ectopic luciferase reporters bearing partial transcripts spanning the Dclk1 3′-UTR, we found that the proximal region of the Dclk1 3'-UTR was the predominant site through which both miR-195 and HuR regulate DCLK1 translation (Fig. 6). Both miR-195 and HuR have been found to associate extensively with the 3′-UTRs of their target transcripts (21, 27, 33, 40), although in some instances they regulate target mRNAs by interacting with the coding region (14). Several other studies have also revealed that HuR antagonizes the regulatory actions of some miRNAs. For example, HuR prevents miR-122-mediated repression of CAT-1 expression (52), antagonizes miR-548c-3p to regulate the expression of TOP2A (53), blocks miR-494 to regulate the expression of nucleolin (54), and co-operates with let-7 in repressing c-Myc expression (55). Although the exact process by which HuR competes with miR-195 for association with Dclk1 mRNA remains unclear at present, our results show that binding sites for miR-195 and HuR are present at 3′-UTR-F1, suggesting that miR-195 and HuR might compete via their physical interaction with Dclk1 mRNA. However, HuR and the coregulatory miRNAs can also bind at distances up to several hundreds or thousands of bases apart in some targets (52–54). In this regard, we have reported that the miR-195 site and two HuR sites are located at 153 and 1027 bases apart on the stim1 3′-UTR, respectively, but miR-195 and HuR compete to modulate Stim1 mRNA stability antagonistically (33). Another possibility is that HuR may affect the processing of pre-miR-195, but our preliminary studies revealed that ectopically expressed HuR did not alter the levels of mature miR-195 in IECs (data not shown).
The exact mechanism involved in competitive regulation of DCLK1 translation by miR-195 and HuR remains unknown. An increasing body of evidence suggests that miRNA-mRNA or RBP-mRNA interactions can alter the recruitment of target mRNAs to processing bodies (P-bodies) (56, 57), where mRNAs are sorted for translational repression and/or degradation (58). Several miRNAs including miR-195 and miR-222 are shown to promote translocation of target mRNAs to P-bodies, thus decreasing the stability and translation of target transcripts, but HuR blocks mRNA translocation to P-bodies (33, 57–59). Our previous study revealed that ectopic miR-195 overexpression increases Stim1 mRNA levels in P-bodies, whereas increasing the levels of HuR prevents the colocalization of Stim1 mRNA in P-bodies in cells overexpressing miR-195, suggesting that HuR competes with miR-195 for the binding to Stim1 mRNA and thereby prevents the translocation of Stim1 to P-bodies by miR-195 (33). By contrast, the RBP CUGBP1 and miR-222 instead inhibit translation of the Cdk4 mRNA by cooperatively promoting the translocation of Cdk4 mRNA to P-bodies (15). Efforts are ongoing in our laboratory to investigate whether miR-195-Dclk1 mRNA association increases the subcellular localization of Dclk1 mRNA to P-bodies or if HuR inhibits the recruitment of Dclk1 mRNA to P-bodies by replacing miR-195.
In summary, we have established an in vivo function for miR-195 as a posttranscriptional regulator of intestinal Tuft and Paneth cells. The actions of miR-195 on Tuft cells are mediated at least partially by repressing DCLK1 translation, a process that is competed by HuR. As miR-195 dysregulation and defects in Tuft and Paneth cells occur commonly in patients with inflammation and infection of the gut mucosa (9, 14, 15, 60), our results identify new potential therapeutic targets for interventions to enhance the intestinal protective barrier.
GRANTS
This work was supported by Merit Review Awards (to J.-Y.W. and J.N.R.) from US Department of Veterans Affairs; National Institutes of Health (NIH) Grants DK57819, DK61972, and DK68491 (to J.Y.W.); and funding from the National Institute on Aging-Intramural Research Program, NIH [Z01-AG000393 (to M.G.)].
DISCLOSURES
J.-Y. Wang is a Senior Research Career Scientist at the Biomedical Laboratory Research and Development Service (US Department of Veterans Affairs). None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
M.S.K., H.K.C., and J.-Y.W. conceived and designed research; M.S.K., H.K.C., L.X., T.-X.Y., S.R.W., J.-J.P., and J.N.R. performed experiments; M.S.K., H.K.C., L.X., T.-X.Y., J.-J.P., J.N.R., M.G., and J.-Y.W. analyzed data; H.K.C., J.N.R., M.G., and J.-Y.W. interpreted results of experiments; J.-Y.W. prepared figures; M.S.K. and J.-Y.W. drafted manuscript; M.G. and J.-Y.W. edited and revised manuscript; M.S.K., H.K.C., L.X., T.-X.Y., S.R.W., J.-J.P., J.N.R., M.G., and J.-Y.W. approved final version of manuscript.
REFERENCES
- 1.Camilleri M. Leaky gut: mechanisms, measurement and clinical implications in humans. Gut 68: 1516–1526, 2019. doi: 10.1136/gutjnl-2019-318427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu T-X, Chung HK, Xiao L, Piao J-J, Lan S, Jaladanki SK, Turner DJ, Raufman J-P, Gorospe M, Wang J-Y. Long noncoding RNA H19 impairs the intestinal barrier by suppressing autophagy and lowering Paneth and goblet cell function. Cell Mol Gastroenterol Hepatol 9: 611–625, 2020. doi: 10.1016/j.jcmgh.2019.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee A, McKinley ET, von Moltke J, Coffey RJ, Lau KS. Interpreting heterogeneity in intestinal tuft cell structure and function. J Clin Invest 128: 1711–1719, 2018. doi: 10.1172/jci120330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerjee A, Herring CA, Chen B, Kim H, Simmons AJ, Southard-Smith AN, Allaman MM, White JR, Macedonia MC, Mckinley ET. Succinate produced by intestinal microbes promotes specification of Tuft cells to suppress ileal inflammation. Gastroenterology 159: 2101–2115.e5, 2020. doi: 10.1053/j.gastro.2020.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.May R, Qu D, Weygant N, Chandrakesan P, Ali N, Lightfoot SA, Li L, Sureban SM, Houchen CW. Brief report: Dclk1 deletion in Tuft cells results in impaired epithelial repair after radiation injury. Stem Cells 32: 822–827, 2014. doi: 10.1002/stem.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lueschow SR, McElroy SJ. The Paneth cell: the curator and defender of the immature small intestine. Front Immunol 11: 587, 2020. doi: 10.3389/fimmu.2020.00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418, 2011. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Es JH, Sato T, van de Wetering M, Lyubimova A, Yee Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van den Born M, Korving J, Martens ACM, Barker N, van Oudenaarden A, Clevers H. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 14: 1099–1104, 2012. doi: 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson DN, Panopoulos M, Neumann WL, Turner K, Cantarel BL, Thompson-Snipes L, Dassopoulos T, Feagins LA, Souza RF, Mills JC, Blumberg RS, Venuprasad K, Thompson WE, Theiss AL. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 69: 1928–1938, 2020. doi: 10.1136/gutjnl-2019-319523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones JC, Brindley CD, Elder NH, Myers MG Jr, Rajala MW, Dekaney CM, McNamee EN, Frey MR, Shroyer NF, Dempsey PJ. Cellular plasticity of Defa4Cre-expressing Paneth cells in response to notch activation and intestinal injury. Cell Mol Gastroenterol Hepatol 7: 533–554, 2019. doi: 10.1016/j.jcmgh.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Es JH, Wiebrands K, López-Iglesias C, van de Wetering M, Zeinstra L, van den Born M, Korving J, Sasaki N, Peters PJ, van Oudenaarden A, Clevers H. Enteroendocrine and tuft cells support Lgr5 stem cells on Paneth cell depletion. Proc Natl Acad Sci USA 116: 26599–26605, 2019. doi: 10.1073/pnas.1801888117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giammanco A, Blanc V, Montenegro G, Klos C, Xie Y, Kennedy S, Luo J, Chang S-H, Hla T, Nalbantoglu ILKe, Dharmarajan S, Davidson NO. Intestinal epithelial HuR modulates distinct pathways of proliferation and apoptosis and attenuates small intestinal and colonic tumor development. Cancer Res 74: 5322–5335, 2014. doi: 10.1158/0008-5472.can-14-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J-Y, Xiao L, Wang J-Y. Posttranscriptional regulation of intestinal epithelial integrity by noncoding RNAs. Wiley Interdiscip Rev RNA 8: 10.1002/wrna.1399, 2017. doi: 10.1002/wrna.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao L, Gorospe M, Wang J-Y. Long noncoding RNAs in intestinal epithelium homeostasis. Am J Physiol Cell Physiol 317: C93–C100, 2019. doi: 10.1152/ajpcell.00092.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou L, Xiong X, Wang K, Yin Y. MicroRNAs in the intestine: role in renewal, homeostasis, and inflammation. Curr Mol Med 18: 190–198, 2018. doi: 10.2174/1566524018666180907163638. [DOI] [PubMed] [Google Scholar]
- 16.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233, 2009. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, Rajewsky N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell 43: 340–352, 2011. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Turner M, Diaz-Munoz MD. RNA-binding proteins control gene expression and cell fate in the immune system. Nat Immunol 19: 120–129, 2018. doi: 10.1038/s41590-017-0028-4. [DOI] [PubMed] [Google Scholar]
- 19.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8: 113–126, 2007. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 20.Li XX, Xiao L, Chung HK, Ma XX, Liu X, Song JL, Jin CZ, Rao JN, Gorospe M, Wang JY. Interaction between HuR and circPABPN1 modulates autophagy in the intestinal epithelium by altering ATG16L1 translation. Mol Cell Biol 40: e00492–19, 2020. doi: 10.1128/mcb.00492-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Zhang Y, Xiao L, Yu T-X, Li J-Z, Rao JN, Turner DJ, Gorospe M, Wang J-Y. Cooperative repression of insulin-like growth factor type 2 receptor translation by microRNA 195 and RNA-binding protein CUGBP1. Mol Cell Biol 37: e00225–17, 2017. doi: 10.1128/mcb.00225-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li XX, Rao JN, Wang J-Y. Posttranscriptional regulation of gut epithelium homeostasis by RNA-binding proteins and long noncoding RNAs. In: Encyclopedia of Gastroenterology (2nd ed.). Cambridge, MA: Academic Press, 2020, p. 247–256. doi: 10.1016/B978-0-12-801238-3.11106-7. [DOI] [Google Scholar]
- 23.Xiao L, Li X-X, Chung HK, Kalakonda S, Cai J-Z, Cao S, Chen N, Liu Y, Rao JN, Wang H-Y, Gorospe M, Wang J-Y. RNA-binding protein HuR regulates Paneth cell function by altering membrane localization of TLR2 via post-transcriptional control of CNPY3. Gastroenterology 157: 731–743, 2019. doi: 10.1053/j.gastro.2019.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung HK, Chen Y, Rao JN, Liu L, Xiao L, Turner DJ, Yang P, Gorospe M, Wang J-Y. Transgenic expression of miR-222 disrupts intestinal epithelial regeneration by targeting multiple genes including Frizzled-7. Mol Med 21: 676–687, 2015. doi: 10.2119/molmed.2015.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M Jr, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell 43: 327–339, 2011. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rawat M, Nighot M, Al-Sadi R, Gupta Y, Viszwapriya D, Yochum G, Koltun W, Ma TY. IL1B increases intestinal tight junction permeability by up-regulation of miR200C-3p, which degrades occludin mRNA. Gastroenterology 159: 1375–1389, 2020. doi: 10.1053/j.gastro.2020.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhattacharya A, Schmitz U, Wolkenhauer O, Schönherr M, Raatz Y, Kunz M. Regulation of cell cycle checkpoint kinase WEE1 by miR-195 in malignant melanoma. Oncogene 32: 3175–3183, 2013. doi: 10.1038/onc.2012.324. [DOI] [PubMed] [Google Scholar]
- 28.Xu T, Zhu Y, Xiong Y, Ge Y-Y, Yun J-P, Zhuang S-M. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology 50: 113–121, 2009. doi: 10.1002/hep.22919. [DOI] [PubMed] [Google Scholar]
- 29.Lin Y, Wu J, Chen H, Mao Y, Liu Y, Mao Q, Yang K, Zheng X, Xie L. Cyclin-dependent kinase 4 is a novel target in micoRNA-195-mediated cell cycle arrest in bladder cancer cells. FEBS Lett 586: 442–447, 2012. doi: 10.1016/j.febslet.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 30.Zhu H, Yang Y, Wang Y, Li J, Schiller PW, Peng T. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc Res 92: 75–84, 2011. doi: 10.1093/cvr/cvr145. [DOI] [PubMed] [Google Scholar]
- 31.Bai Y, Yang W, Yang H-X, Liao Q, Ye G, Fu G, Ji L, Xu P, Wang H, Li Y-X, Peng C, Wang Y-L. Downregulated miR-195 detected in preeclamptic placenta affects trophoblast cell invasion via modulating ActRIIA expression. PLoS One 7: e38875, 2012. doi: 10.1371/journal.pone.0038875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao JN, Rathor N, Zou T, Liu L, Xiao L, Yu T-X, Cui Y-H, Wang J-Y. STIM1 translocation to the plasma membrane enhances intestinal epithelial restitution by inducing TRPC1-mediated Ca2+ signaling after wounding. Am J Physiol Cell Physiol 299: C579–C588, 2010. doi: 10.1152/ajpcell.00066.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhuang R, Rao JN, Zou T, Liu L, Xiao L, Cao S, Hansraj NZ, Gorospe M, Wang J-Y. miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration. Nucleic Acids Res 41: 7905–7919, 2013. doi: 10.1093/nar/gkt565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao L, Wu J, Wang JY, Chung HK, Kalakonda S, Rao JN, Gorospe M, Wang JY. Long noncoding RNA uc.173 promotes renewal of the intestinal mucosa by inducing degradation of microRNA 195. Gastroenterology 154: 599–611, 2018. doi: 10.1053/j.gastro.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flentjar N, Chu P-Y, Ng AY-N, Johnstone CN, Heath JK, Ernst M, Hertzog PJ, Pritchard MA. TGF-βRII rescues development of small intestinal epithelial cells in Elf3-deficient mice. Gastroenterology 132: 1410–1419, 2007. doi: 10.1053/j.gastro.2007.02.054. [DOI] [PubMed] [Google Scholar]
- 36.Xiao L, Rao JN, Cao S, Liu L, Chung HK, Zhang Y, Zhang J, Liu Y, Gorospe M, Wang J-Y. Long noncoding RNA SPRY4-IT1 regulates intestinal epithelial barrier function by modulating the expression levels of tight junction proteins. Mol Biol Cell 27: 617–626, 2016. doi: 10.1091/mbc.e15-10-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, Ivanov JA, Fu Y-Y, Takashima S, Hua G, Martin ML, O’Rourke KP, Lo Y-H, Mokry M, Romera-Hernandez M, Cupedo T, Dow LE, Nieuwenhuis EE, Shroyer NF, Liu C, Kolesnick R, van den Brink MRM, Hanash AM. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528: 560–564, 2015. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu T-X, Wang P-Y, Rao JN, Zou T, Liu L, Xiao L, Gorospe M, Wang J-Y. Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function. Nucleic Acids Res 39: 8472–8487, 2011. doi: 10.1093/nar/gkr567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J-Y, Xiao L, Zhang YZ, Chung H, Rao JN, Gorospe M, Wang JY. Regulation of intestinal epithelial barrier function by long noncoding RNA uc.173 through interaction with microRNA 29b. Mol Cell Biol 38: e00010–18, 2018. doi: 10.1128/mcb.00010-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L, Christodoulou-Vafeiadou E, Rao JN, Zou T, Xiao L, Chung HK, Yang H, Gorospe M, Kontoyiannis D, Wang J-Y. RNA-binding protein HuR promotes growth of small intestinal mucosa by activating the Wnt signaling pathway. Mol Biol Cell 25: 3308–3318, 2014. doi: 10.1091/mbc.e14-03-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harter HL. Critical values for Duncan’s new multiple range test. Biometrics 16: 671–685, 1960. doi: 10.2307/2527770. [DOI] [Google Scholar]
- 42.Freeman JJ, Feng Y, Demehri FR, Dempsey PJ, Teitelbaum DH. TPN-associated intestinal epithelial cell atrophy is modulated by TLR4/EGF signaling pathways. FASEB J 29: 2943–2958, 2015. doi: 10.1096/fj.14-269480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao L, Rao JN, Zou T, Liu L, Cao S, Martindale JL, Su W, Chung HK, Gorospe M, Wang J-Y. miR-29b represses intestinal mucosal growth by inhibiting translation of cyclin-dependent kinase 2. Mol Biol Cell 24: 3038–3046, 2013. doi: 10.1091/mbc.e13-05-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao JN, Xiao L, Wang J-Y. Polyamines in gut epithelial renewal and barrier function. Physiology (Bethesda) 35: 328–337, 2020. doi: 10.1152/physiol.00011.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L, Li L, Rao JN, Zou T, Zhang HM, Boneva D, Bernard MS, Wang JY. Polyamine-modulated expression of c-myc plays a critical role in stimulation of normal intestinal epithelial cell proliferation. Am J Physiol Cell Physiol 288: C89–C99, 2005. doi: 10.1152/ajpcell.00326.2004. [DOI] [PubMed] [Google Scholar]
- 46.Liu L, Guo X, Rao JN, Zou T, Xiao L, Yu T, Timmons JA, Turner DJ, Wang J-Y. Polyamines regulate E-cadherin transcription through c-Myc modulating intestinal epithelial barrier function. Am J Physiol Cell Physiol 296: C801–C810, 2009. doi: 10.1152/ajpcell.00620.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JY, McCormack SA, Viar MJ, Wang H, Tzen CY, Scott RE, Johnson LR. Decreased expression of protooncogenes c-fos, c-myc, and c-jun following polyamine depletion in IEC-6 cells. Am J Physiol Gastrointest Liver Physiol 265: G331–G338, 1993. doi: 10.1152/ajpgi.1993.265.2.g331. [DOI] [PubMed] [Google Scholar]
- 48.Sarkar S, Popov VL, O’Connell MR, Stevenson HL, Lee BS, Obeid RA, Luthra GK, Singh P. A novel antibody against cancer stem cell biomarker, DCLK1-S, is potentially useful for assessing colon cancer risk after screening colonoscopy. Lab Invest 97: 1245–1261, 2017. doi: 10.1038/labinvest.2017.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang X, Ji R, Liao X, Castillero E, Kennel PJ, Brunjes DL, Franz M, Möbius-Winkler S, Drosatos K, George I, Chen EI, Colombo PC, Schulze PC. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation 137: 2052–2067, 2018. doi: 10.1161/circulationaha.117.030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferrad M, Ghazzaui N, Issaoui H, Cook-Moreau J, Denizot Y. Mouse models of c-myc deregulation driven by IgH locus enhancers as models of B-cell lymphomagenesis. Front Immunol 11: 1564, 2020. doi: 10.3389/fimmu.2020.01564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou B, Sun C, Hu X, Zhan H, Zou H, Feng Y, Qiu F, Zhang S, Wu L, Zhang B. MicroRNA-195 suppresses the progression of pancreatic cancer by targeting DCLK1. Cell Physiol Biochem 44: 1867–1881, 2017. doi: 10.1159/000485876. [DOI] [PubMed] [Google Scholar]
- 52.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125: 1111–1124, 2006. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 53.Srikantan S, Abdelmohsen K, Lee EK, Tominaga K, Subaran SS, Kuwano Y, Kulshrestha R, Panchakshari R, Kim HH, Yang X, Martindale JL, Marasa BS, Kim MM, Wersto RP, Indig FE, Chowdhury D, Gorospe M. Translational control of TOP2A influences doxorubicin efficacy. Mol Cell Biol 31: 3790–3801, 2011. doi: 10.1128/mcb.05639-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tominaga K, Srikantan S, Lee EK, Subaran SS, Martindale JL, Abdelmohsen K, Gorospe M. Competitive regulation of nucleolin expression by HuR and miR-494. Mol Cell Biol 31: 4219–4231, 2011. doi: 10.1128/mcb.05955-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23: 1743–1748, 2009. doi: 10.1101/gad.1812509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riggs CL, Kedersha N, Ivanov P, Anderson P. Mammalian stress granules and P bodies at a glance. J Cell Sci 133: jcs242487, 2020. doi: 10.1242/jcs.242487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Youn J-Y, Dyakov BJA, Zhang J, Knight JDR, Vernon RM, Forman-Kay JD, Gingras A-C. Properties of stress granule and P-body proteomes. Mol Cell 76: 286–294, 2019. doi: 10.1016/j.molcel.2019.09.014. [DOI] [PubMed] [Google Scholar]
- 58.Ivanov P, Kedersha N, Anderson P. Stress granules and processing bodies in translational control. Cold Spring Harb Perspect Biol 11: a032813, 2019. doi: 10.1101/cshperspect.a032813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao L, Cui Y-H, Rao JN, Zou T, Liu L, Smith A, Turner DJ, Gorospe M, Wang J-Y. Regulation of cyclin-dependent kinase 4 translation through CUG-binding protein 1 and microRNA-222 by polyamines. Mol Biol Cell 22: 3055–3069, 2011. doi: 10.1091/mbc.e11-01-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosen MJ. Could a small population of epithelial cells get “Tuft” with Crohn’s disease? Gastroenterology 159: 2025–2027, 2020. doi: 10.1053/j.gastro.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]