

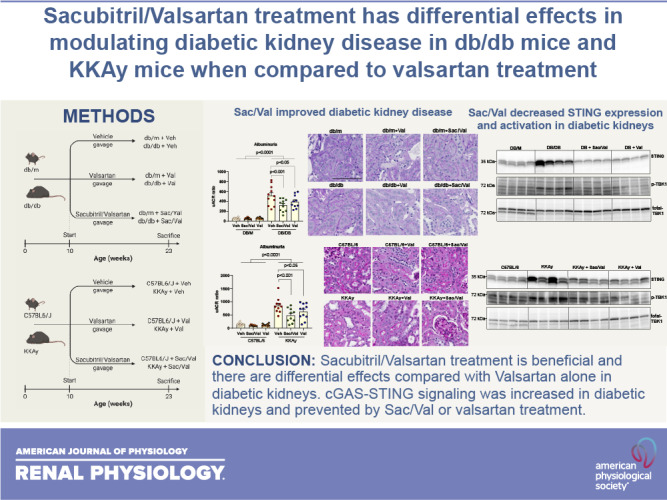
Keywords: cGMP-AMP synthase, diabetic kidney disease, sacubitril, stimulator of interferon genes, valsartan
Abstract
Although renin-angiotensin blockade has shown beneficial outcomes in patients with diabetes, renal injury progresses in most of these patients. Therefore, there remains a need for new therapeutic targets in diabetic kidney disease. Enhancement of vasoactive peptides, such as natriuretic peptides, via neprilysin inhibition, has been a new approach. A first-in-class drug, sacubitril/valsartan (Sac/Val), a combination of the angiotensin II receptor blocker Val and neprilysin inhibitor prodrug Sac, has been shown to be more effective than renin-angiotensin blockade alone in the treatment of heart failure with reduced ejection fraction. In this study, we tested the effects of Sac/Val in diabetic kidney disease. We administered Sac/Val or Val to two type 2 diabetes mouse models, db/db mice or KKAy mice. After 3 mo of treatment, Sac/Val attenuated the progression of proteinuria, glomerulosclerosis, and podocyte loss in both models of diabetic mice. Val shared a similar improvement but to a lesser degree in some parameters compared with Sac/Val. Sac/Val but not Val decreased the blood glucose level in KKAy mice. Sac/Val exerted renal protection through coordinated effects on antioxidative stress and anti-inflammation. In both diabetic models, we revealed a new mechanism to cause inflammation, self-DNA-activated cGMP-AMP synthase-stimulator of interferon genes (cGAS-STING) signaling, which was activated in diabetic kidneys and prevented by Sac/Val or Val treatment. The present data suggest that Sac/Val has sufficient therapeutical potential to counter the pathophysiological effects of diabetic kidney disease, and its effectiveness could be better than Val alone.
NEW & NOTEWORTHY The first-in-class drug sacubitril/valsartan, a combination of the angiotensin II receptor blocker valsartan and neprilysin inhibitor sacubitril, was tested for its effects in diabetic kidney disease using db/db mice and KKAy mice. We found that Sac/Val has sufficient therapeutical potential to counter the pathophysiological effects of diabetic kidney disease. We further revealed a new mechanism to cause inflammation, self-DNA-activated cGAS-STING signaling, which was activated in diabetic kidneys and prevented by sacubitril/valsartan or valsartan treatment.
INTRODUCTION
Diabetes mellitus is the leading cause of cardiovascular and renal disease in the United States (1–3). This is of increasing concern, because as many as one in four Americans are expected to become diabetic by the year 2050 (4, 5).
Despite all of the beneficial interventions implemented in patients with diabetes, including tight glucose control, stringent blood pressure control, angiotensin-converting enzyme inhibition (ACEI), angiotensin II receptor blocker (ARB), or mineralocorticoid receptor antagonism, renal injury progresses in most of these patients (6, 7). Therefore, there remains a need for new therapeutic targets in diabetic kidney disease. Recently, glucagon-like peptide-1 receptor agonists and sodium-glucose cotransporter 2 (SGLT2) inhibitors are important emerging therapeutic tools for patients with diabetic kidney disease (8, 9).
One additional novel therapeutic approach is enhancement of vasodilatory peptides, such as natriuretic peptides (NPs), as a complement to the approach using vasoconstriction blocking with ACEI/ARB. The NP system is an important endocrine, autocrine, and paracrine system consisting of a family of peptides including atrial NP (10), B-type NP (11), and C-type NP. These hormones bind to their receptors to activate guanylate cyclase, increasing levels of cGMP and its effector protein kinase G. This induces natriuresis, diuresis, vasodilation, and the sympathetic nervous system to regulate blood pressure and fluid homeostasis (12–14). In addition, there is evidence suggesting that NPs can influence glucose and fat metabolism by increasing adiponectin levels (15) and can protect against the onset of diabetes (16). Therefore, modulation of NPs can address many of the underlying pathological processes involved in cardiorenal diseases such as hypertension, heart failure, and chronic kidney disease (CKD).
Direct administration of NPs is difficult because oral delivery is ineffective and parenteral delivery is problematic (17, 18). Blockade of NP breakdown has, therefore, been investigated. The level of endogenous NPs can be enhanced via inhibition of neprilysin, a neutral endopeptidase that catalyzes the degradation of NPs (19). Neprilysin inhibition can activate the renin-angiotensin-aldosterone system (RAAS) (20). To be clinically beneficial, neprilysin inhibition requires concomitant inhibition of the RAAS. Since ARBs have a lesser effect on bradykinin (21) and lower risk of angioedema compared with ACEI (22), the first-in-class drug sacubitril/valsartan (Sac/Val; Entresto, Novartis) combines the ARB Val and neprilysin inhibitor prodrug Sac in a 1:1 molar ratio (23). Sac/Val has been shown to be more effective in the treatment of heart failure with reduced ejection fraction than standard monotherapy with ACEIs (24). In a rat model of CKD, Sac/Val attenuated CKD-related cardiomyopathy, cardiac hypertrophy, and fibrosis (25) and improved renal function in another report (26). Sac/Val also alleviated kidney injury in a salt-sensitive hypertension model (27). However, the effect of Sac/Val in diabetic kidney disease has not been fully investigated and compared with monotherapy with ACEI/ARB.
In the present study, we administered Sac/Val in a type 2 diabetes animal model including db/db mice and KKAy mice to evaluate its effects in diabetic nephropathy. In addition, we included a group of diabetic animals treated with Val alone to further delineate any potential benefit of Sac/Val beyond that conferred by RAAS blockade alone.
MATERIALS AND METHODS
Animal Study Design
To evaluate the effects of Sac/Val (neprilysin inhibitor Sac and ARB Val), we studied two different established mouse models of diabetic kidney disease, db/db mice homozygous for a spontaneous mutation of the leptin receptor (Leprdb) (28) and KKAy mice with heterozygous mutation of the agouti gene (KK-Ay) (29).
In the first set of animal experiments, 10-wk-old male diabetic (db/db, Cat. No. 000642) and nondiabetic (db/m, Cat. No. 000662) control mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in an animal care facility with a 12:12-h light-dark cycle. All mice were fed standard rodent chow diet (Cat. No. 5053, PicoLab Rodent Diet) along with free access to drinking water. Both Val and Sac/Val compounds were received from Novartis (Basel, Switzerland). All db/m and db/db mice were randomly classified into six groups (n = 10 or 11 mice/group). Both db/m and db/db mice were administered 1) vehicle (PBS), 2) Val (30 mg/kg), or 3) Sac/Val (60 mg/kg) via oral gavage for 90 days.
At the end of the treatment period when the mice were 20 wk old, we measured blood pressure (BP-2000, Visitech Systems) via the tail-cuff method. One week before the end of the study, mice were placed in metabolic cages for a 24-h urine collection for the assessment of albuminuria, kidney injury molecule-1 (KIM-1), neutrophil gelatinase-associated lipocalin (NGAL), and thiobarbituric acid-reactive substances (TBARS). All mice were euthanized at 23 wk of age, and heparinized plasma and kidneys were harvested for further processing, histology, and biochemical analysis.
In the second set of animal experiments, we used 10-wk-old male KKAy (CLEA) mice and age-matched C57BL6/J mice (Jackson Laboratory), which were used as control mice. Housing and feeding conditions were maintained similar to the first set of experimental mice. KKAy and C57BL6/J mice were administered 1) vehicle (PBS), 2) Val (30 mg/kg body wt), or 3) Sac/Val (60 mg/kg body wt) via oral gavage for 90 days. All mice were euthanized at 23 wk of age, and tissue collection and biochemical assays were conducted similar to the first set of experimental mice.
All animal experiments were conducted according to the guidelines for the Institutional Animal Care and Use Committee of Georgetown University (Washington, DC). Entire methods were carried out in accordance with relevant guidelines and regulations.
Measurement of Plasma and Urine Biochemistry
Blood glucose levels were assessed with a glucometer (Elite XL, Bayer, Tarrytown, NY). Plasma blood urea nitrogen (BUN) was measured with a colorimetric QuantiChrom assay kit (Bioassay Systems, Hayward, CA). Plasma insulin (Cat. No. 90080) and leptin (Cat. No. 90030) levels were determined in accordance with the mouse ELISA kit (Crystal Chem, Elk Grove Village, IL). Plasma concentrations of total cholesterol and triglyceride levels were determined with the calorimetric assay kit provided by Pointe Scientific (Canton, MI). Urinary albumin and creatinine concentrations were quantified according to assay procedures provided by the commercially available Albuwell ELISA kit (Exocell, Ethos Biosciences, Philadelphia, PA) and Bioassay Systems, respectively. Urine TBARS was quantitatively measured by a QuantiChrom TBARS assay kit (Bioassay Systems). Urinary KIM-1 (Cat. No. ab213477) and NGAL (Cat. No. ab199083) concentrations were assessed quantitatively with a mouse ELISA kit (Abcam, Cambridge, MA).
RNA Extraction and Quantitative Real-Time PCR
Total RNA was prepared using the RNeasy Mini kit (Qiagen, Gaithersburg, MD) according to the manufacturer’s instructions. Isolated total RNA was reverse transcribed into cDNA using the high-capacity cDNA reverse transcription kit (Cat. No. 4374967), and quantitative PCRs were performed as previously described (30–32).
Western Blot Analysis
Cortical homogenate protein content was measured by the bicinchoninic acid (BCA) assay (ThermoFisher Scientific, Waltham, MA). Equal amounts of total protein were separated by SDS-PAGE gels and transferred onto PVDF membranes. Antibodies against 4-hydroxynonenal (4-HNE; 1:1,000, Alpha Diagnostic, San Antonio, TX), monocyte chemoattractant protein-1 (MCP-1; 1:1,000, Proteintech, Rosemont, IL), stimulator of interferon genes (STING; 1:1,000, Cell Signaling, Danver, MA), phospho-p65 and total p65 (1:1,000, Cell Signaling), phospho-TANK-binding kinase 1 (TBK1)/total TBK1 (1:1,000, Cell Signaling), phospho-interferon regulatory factor 3 (IRF3)/total IRF3 (1:1,000, Cell Signaling), phospho-Stat3 and total Stat3 (1:1,000, Cell Signaling), and absent in melanoma 2 (AIM2; 1:1,000, Cell Signaling) were used for Western blot analysis. After horseradish peroxidase-conjugated secondary antibodies, the immune complexes were detected by chemiluminescence captured on a C300 analyzer (Azure Biosystems, Dublin, CA). Blots were quantified against total protein using ImageJ 1.44 software.
Mitochondrial Enzymatic Complex Activity Assay
Kidney tissue (100 mg) was homogenized in mitochondrial assay buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, and 1 mM EGTA) with 0.2% fatty acid-free BSA. The mitochondrial fraction was isolated according to a previously described protocol (31). Isolated mitochondria were subjected to an enzymatic complex I assay with a kit (NADH dehydrogenase, Cat. No. ab109721) purchased from Abcam.
Analysis of Mitochondrial DNA Damage
Total DNA from kidney tissue was isolated by using Genomic Tip (Qiagen, Valencia, CA), and its concentration was measured by PicoGreen dye (Invitrogen, Carlsbad, CA). DNA (15 ng) was used to amplify a 10-kb-long mitochondria DNA target followed by real-time PCR-based quantification to determine mitochondrial DNA lesions, as previously described (33).
Histological Analysis and Morphometry
Sections (4 μm thick) cut from 10% formalin-fixed, paraffin-embedded kidney samples were used for periodic acid-Schiff (PAS) staining. Quantifications were performed in a masked manner. Using coronal sections of the kidney, 30 consecutive glomeruli per mouse, with four mice per group, were examined for evaluation of glomerular mesangial expansion. The index of mesangial expansion was defined as the ratio of mesangial area to glomerular tuft area. The mesangial area was determined by assessment of PAS-positive and nucleus-free areas in the mesangium using the ScanScope image analyzer (Aperio Technologies, Vista, CA).
Immunofluorescence Microscopy
Frozen sections were used for immunostaining for synaptopodin (Sigma, St. Louis, MO) and fibronectin (Sigma) and imaged with a laser scanning confocal microscope (SP8, Leica Biosystems, Buffalo Grove, IL). The expression level was quantified as the sum of pixel values per glomerular area using ImageJ 1.44 software.
Statistical Analysis
All results are calculated and presented as means ± SE. n means biological replicates, i.e., animals, in this study. Two-way ANOVA followed by Student–Newman–Keuls post hoc analysis were used to analyze the variance among multiple groups and between two groups if treatment for both nondiabetic and diabetic strains was involved. Otherwise, one-way ANOVA with Student–Newman–Keuls post hoc analysis was used for multiple-group comparisons. Statistically significant differences were indicated as at least P < 0.05. The GraphPad Prism 8.1.2 software package was used for statistical analysis (San Diego, CA).
RESULTS
Sac/Val Treatment Improved Metabolic Parameters, Blood Pressure, and Albuminuria in db/db Mice
We treated 10-wk-old male db/db mice with Sac/Val for 3 mo by gavage. We also included Val for comparison. At the end of the study, compared with db/db control mice, we found that Sac/Val decreased kidney weight (normalized to body weight) but did not change body weight, food intake, or blood glucose in db/db mice, similar to Val (Table 1). Although db/db mice did not show elevated systolic blood pressure compared with nondiabetic db/m controls, Sac/Val or Val reduced blood pressure significantly in db/db mice but not in db/m mice (Table 1) compared with untreated db/db or db/m control mice, respectively. Serum lipids were higher in db/db mice than in db/m mice, and the treatment did not change the levels in either strain. We also found that Sac/Val-treated but not Val-treated db/db mice had a lower mean serum BUN level compared with db/db control mice (Table 1).
Table 1.
Effect of Val and Sac/Val on physical and biochemical characteristics in nondiabetic (db/m) and diabetic (db/db) mice
| db/m | db/m + Sac/Val | db/m + Val | db/db | db/db + Sac/Val | db/db + Val | |
|---|---|---|---|---|---|---|
| Body weight, g | 33.3 ± 3 | 30.9 ± 2.9 | 30.7 ± 2.9 | 45.8 ± 3a | 47.3 ± 2.9 | 39.3 ± 3b |
| Food intake, g/day | 3.7 ± 0.3 | 3.8 ± 0.3 | 3.9 ± 0.3 | 6.3 ± 0.3a | 6.02 ± 0.3 | 6.1 ± 0.2 |
| Kidney weight/body weight, mg/g | 0.01 ± 0.002 | 0.01 ± 0.02 | 0.01 ± 0.02 | 0.016 ± 0.02C | 0.009 ± 0.02d | 0.01 ± 0.02d |
| Systolic blood pressure, mmHg | 113 ± 4.2 | 109.6 ± 4.1 | 109.3 ± 4.1 | 116.2 ± 4.2 | 103.8 ± 4.2d | 103.2 ± 4.3e |
| Glucose, mg/dL | 159.2 ± 8.2 | 150.5 ± 8.2 | 159.7 ± 8.2 | 495.3 ± 8.2a | 490.9 ± 8.4 | 497.1 ± 8.4 |
| Blood urea nitrogen, mg/dL | 43.6 ± 4.2 | 43.3 ± 4.2 | 42.3 ± 4.2 | 62.7 ± 4.2a | 51.6 ± 4.2e,f | 60.3 ± 4.2 |
| Triglycerides, mg/dL | 59.6 ± 19.8 | 52.8 ± 19.3 | 59.7 ± 19.3 | 181.8 ± 19.8a | 174.5 ± 20 | 143.3 ± 20 |
| Cholesterol, mg/dL | 103.8 ± 9.0 | 106.4 ± 8.7 | 103.2 ± 8.7 | 154.2 ± 9.0a | 132.9 ± 9.2 | 149.6 ± 9.4 |
All data analyses are presented as means ± SE for n = 9 or 10 mice in each group. Sac, sacubitril; Val, valsartan. aP < 0.0001 v. db/m; bP < 0.05 vs. db/db + Sac/Val; cP < 0.05 vs. db/m; dP < 0.01 vs. db/db; eP < 0.05 vs. db/db; fP < 0.05 vs. db/db + Val.
We collected 24-h urine to check the effect of the treatment on urine parameters. As a hallmark of diabetic nephropathy, albuminuria was dramatically higher in db/db mice for 10-fold more than that in db/m controls. Both treatments significantly improved albuminuria in db/db mice (Fig. 1A). We also checked the kidney injury markers Kim-1 and NGAL in urine samples. The Val-treated group showed a lower Kim-1 level, whereas the Sac/Val-treated group exhibited lower levels in both Kim-1 and NGAL in db/db urine (Fig. 1A). The decrease in albuminuria is likely related to improved podocyte function as podocyte density shown with podocyte marker synaptopodin immunofluorescence was significantly improved in both Sac/Val-treated and Val-treated db/db groups (Fig. 1B).
Figure 1.
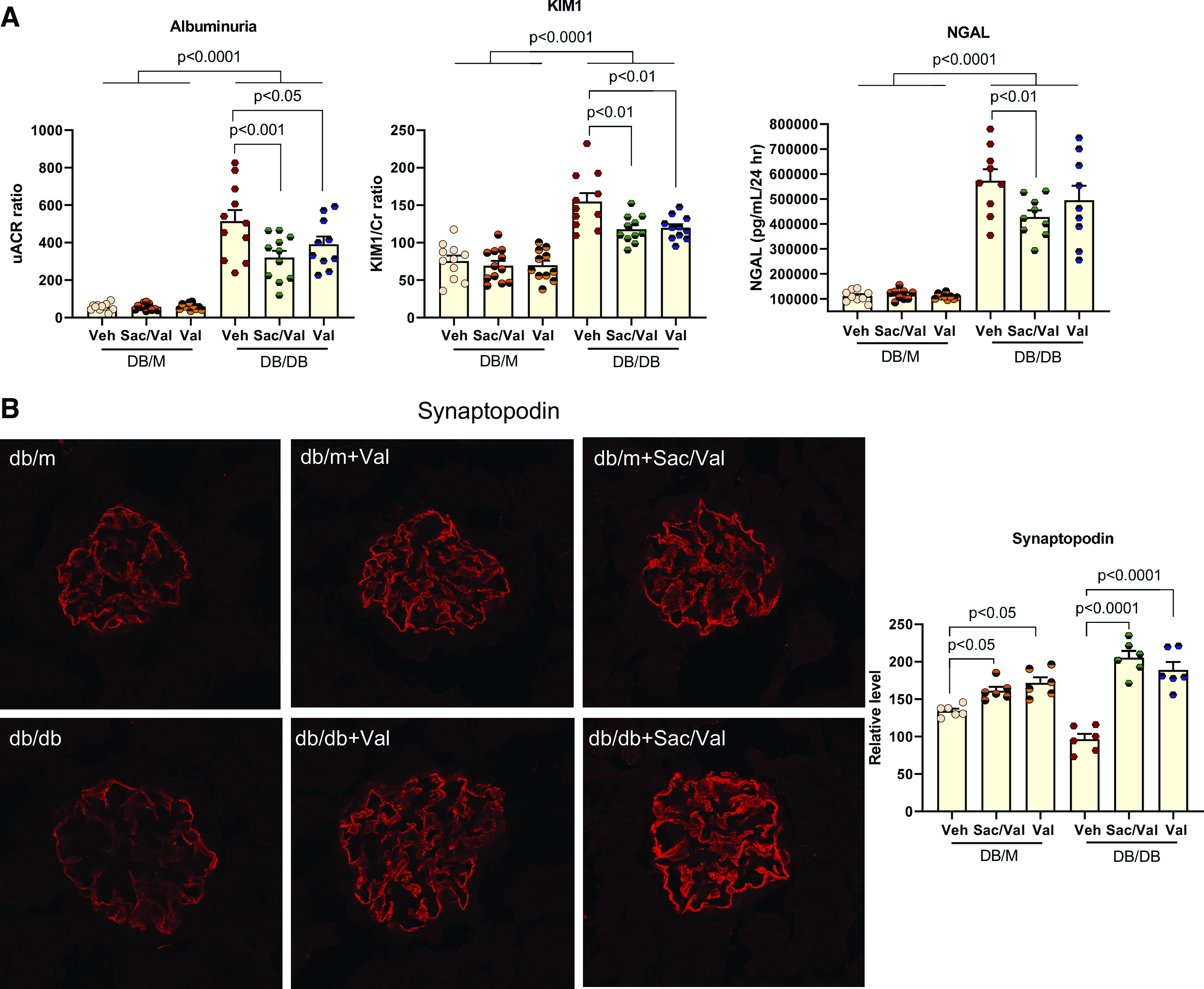
Sacubitril/valsartan (Sac/Val) treatment improved urine parameters and podocyte density in db/db mice. A: urinary albumin, kidney injury molecule-1 (KIM-1), and neutrophil gelatinase-associated lipocalin (NGAL) were measured in 24-h urine. Urinary albumin, KIM-1, and NGAL excretion were markedly increased in db/db mice compared with nondiabetic db/m controls. Administration with Sac/Val and Val significantly decreased urinary albumin, KIM-1, and NGAL levels; n = 10 or 11 per group. B: immunofluorescence staining of kidney sections for the podocyte marker synaptopodin showed decreased podocyte density represented by the ratio of the synaptopodin-positive area vs. glomerular tuft area in db/db kidneys, which was prevented by Sac/Val or Val treatment; n = 6 per group. Data are presented as means ± SE. uACR, urine albumin-to-creatinine (Cr) ratio; Veh, vehicle.
Sac/Val Treatment Improved Glomerulosclerosis in db/db Mice
Kidneys in db/db mice have increased glomerular area and mesangial expansion determined with PAS staining (Fig. 2A). Neither treatment altered glomerular area for db/db mice. In contrast, Sac/Val-treated and Val-treated db/db mice had less mesangial expansion compared with untreated db/db mice (Fig. 2A).
Figure 2.
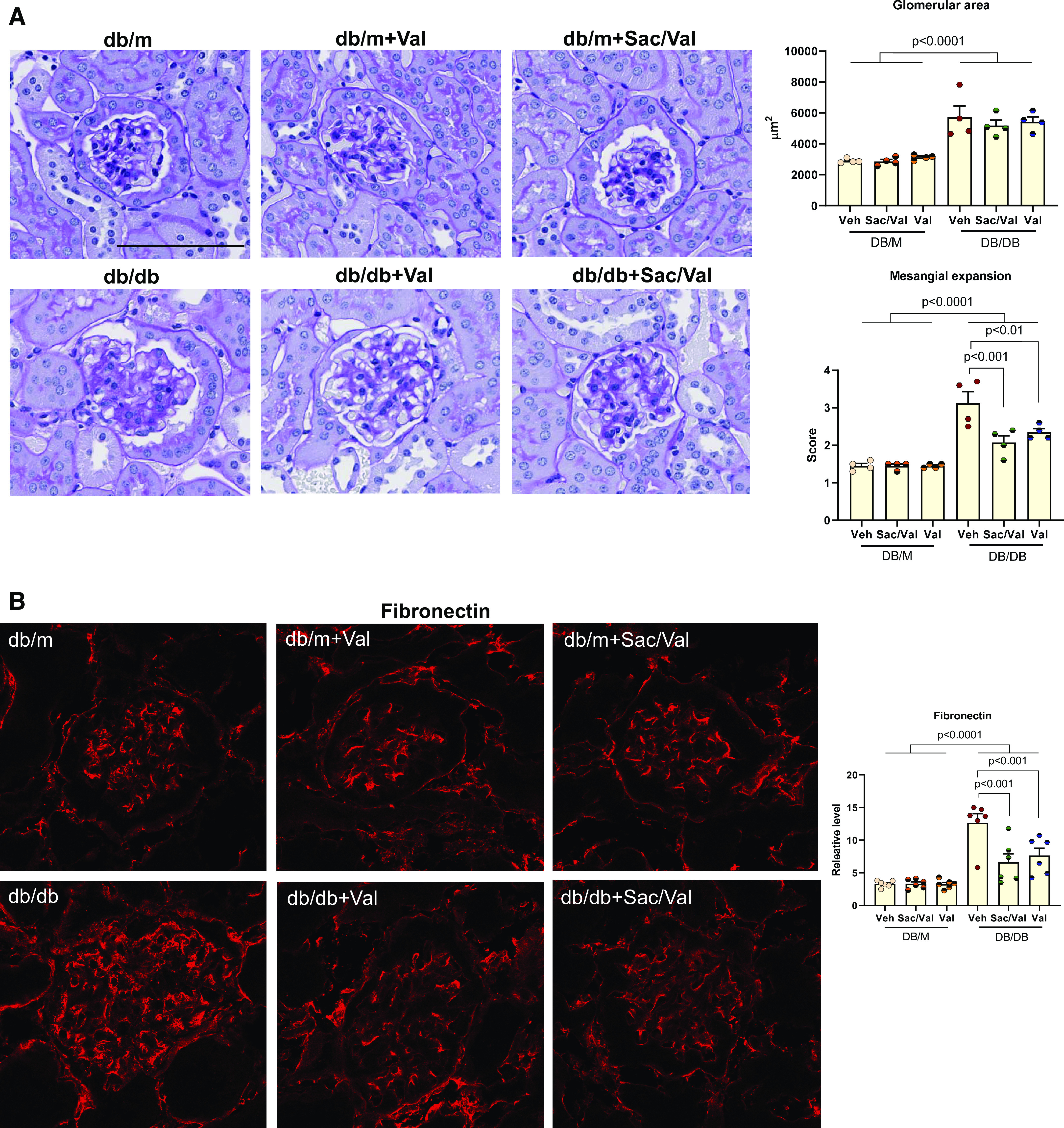
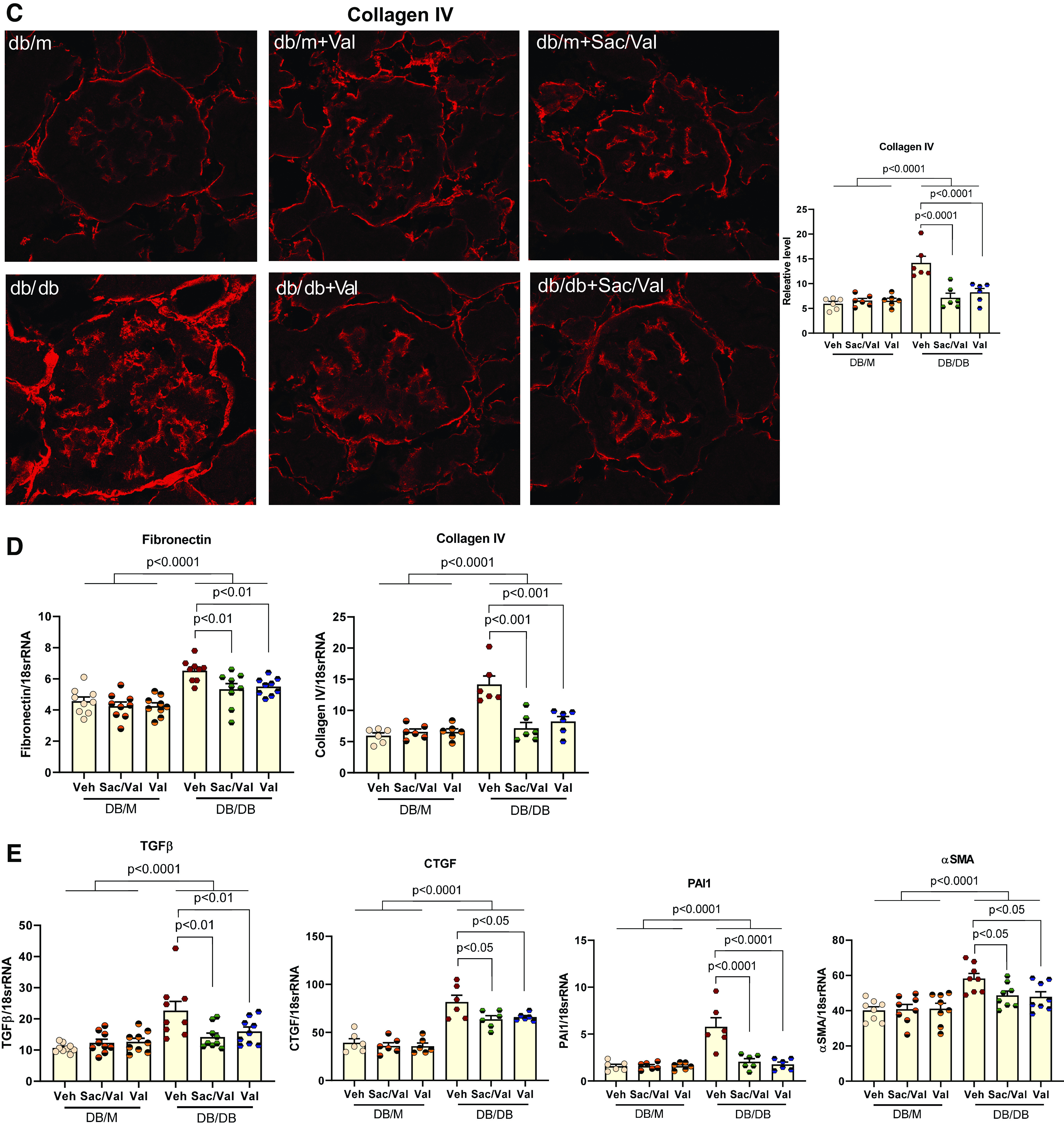
Sacubitril/valsartan (Sac/Val) treatment improved glomerulosclerosis in db/db mice. A: representative images of periodic acid-Schiff staining of kidney sections. The mesangial expansion score was defined by the percentage of the mesangial area in the glomerular tuft area. The mesangial area was determined by assessment of periodic acid-Schiff-positive and nucleus-free areas in the mesangium; n = 4 per group. Scale bar = 100 µm. B and C: immunofluorescence staining of kidney sections for fibronectin (B) and collagen type IV (C). Data were quantified using the ratio of total pixel intensity vs. glomerular area; n = 6 per group. D: quantitative PCR analysis of mRNA levels of fibronectin and collagen type IV in the kidneys from each group; n = 6−9 per group. E: quantitative PCR analysis of mRNA levels of genes involved in profibrotic factors and signaling pathways, transforming growth factor-β (TGF-β), connective tissue growth factor (CTGF), plasminogen activator inhibitor-1 (PAI1), and α-smooth muscle actin (α-SMA); n = 6−9 per group. Data are presented as means ± SE. Veh, vehicle.
We further examined by immunofluorescence the expression of fibronectin and collagen type IV, both of which represent the major components in the extracellular matrix released by mesangial cells in the glomeruli and contribute to glomerulosclerosis. As shown in Fig. 2, B and C, both Sac/Val and Val treatment resulted in lower expression of fibronectin and collagen type IV in glomeruli. These findings were consistent with parallel decreases in their mRNA levels (Fig. 2D). Activation of profibrotic factors and signaling pathways will lead to increased expression of extracellular matrix proteins. In this regard, we found that transforming growth factor-β (TGF-β), connective tissue growth factor (CTGF), plasminogen activator inhibitor-1 (PAI1), and α-smooth muscle actin (α-SMA) were all increased in db/db kidneys, and the treatments significantly reduced their expression (Fig. 2E).
Sac/Val Treatment Improved Oxidative Stress in db/db Mice
In urine from db/db mice, there was a higher level of the oxidative stress marker TBARS (Fig. 3A) compared with that from db/m mice. Both Sac/Val and Val treatment reduced urinary TBARS significantly compared with that reported in db/db control mice. Levels of another oxidative stress marker, 4-HNE, were measured with Western blot analysis, and both treatments attenuated the increase of 4-HNE in db/db kidneys (Fig. 3B). Furthermore, Sac/Val also reduced the NADPH oxidase 4 (Nox4) mRNA level in db/db kidneys compared with in db/db control mice kidneys (Fig. 3C). Val treatment did not change the Nox4 level.
Figure 3.
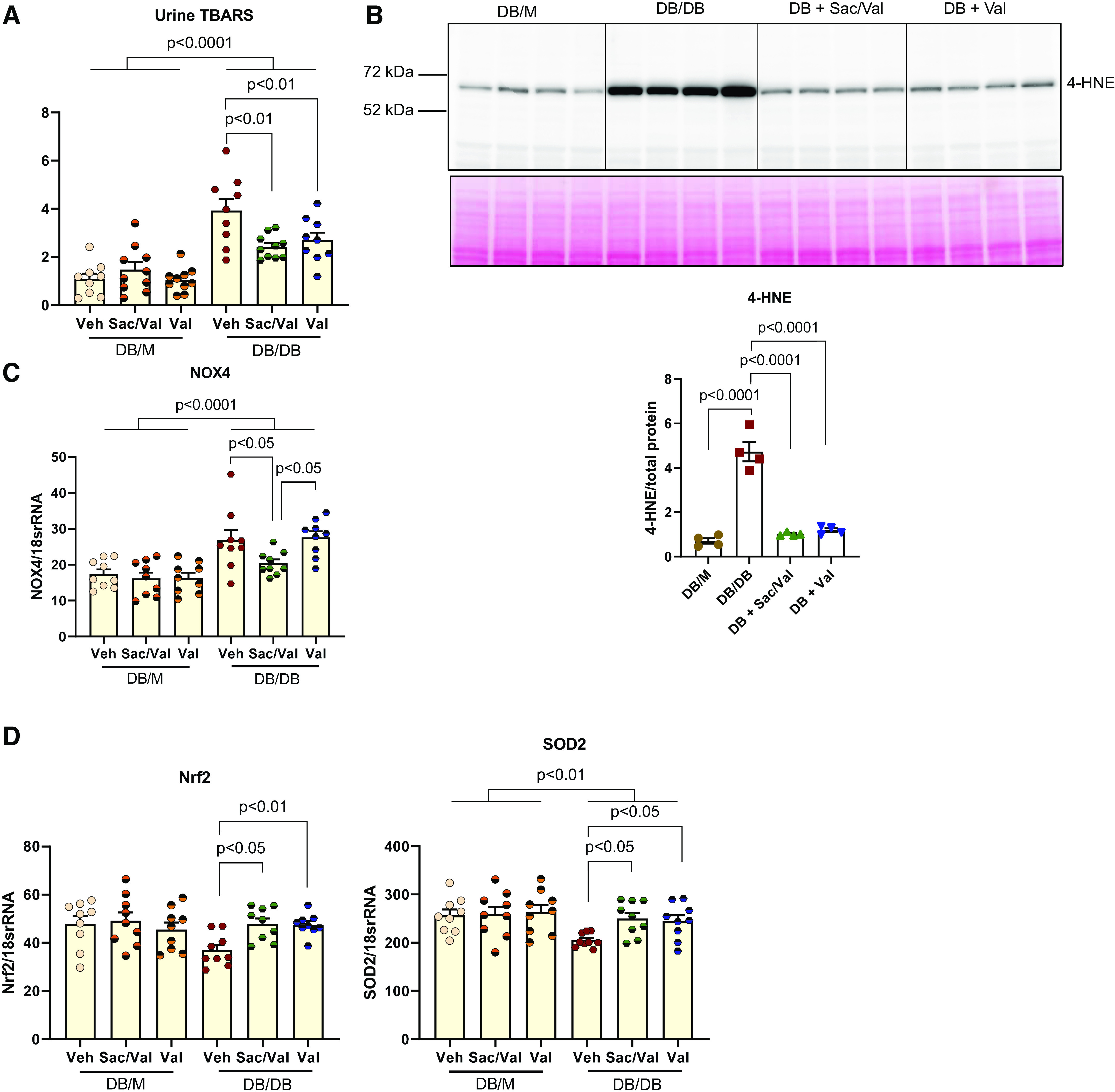
Treatment of sacubitril/valsartan (Sac/Val) in diabetic mice ameliorates oxidative damage in the kidney. A: urinary thiobarbituric acid-reactive substances (TBARS) were excreted significantly higher in db/db mice and markedly reduced upon Sac/Val and Val administration; n = 9 or 10 per group. B: representative Western blot showed increased oxidative stress marker 4-hydroxynonenal (4-HNE) protein expression in db/db kidneys; this was abrogated with Sac/Val or Val. Total protein was used as a loading control; n = 4 per group. C: quantitative PCR analysis of NADPH oxidase 4 (NOX4) mRNA. D: quantitative PCR analysis of antioxidative marker nuclear factor erythroid 2-related factor 2 (Nrf2) and superoxide dismutase 2 (SOD2) mRNA levels; n = 9 or 10 per group. Data are presented as means ± SE. Veh, vehicle.
In addition, we examined antioxidative pathways. Both Sac/Val and Val treatment groups displayed increases in gene expression of the master antioxidative regulator nuclear factor erythroid 2-related factor 2 (Nrf2) and mitochondrial antioxidative enzyme superoxide dismutase 2 (SOD2) in db/db kidneys (Fig. 3D).
Sac/Val Treatment Improved Inflammation in db/db Mice
Sac/Val-treated and Val-treated mice had lower renal expression of inflammatory cytokine MCP-1 and Toll-like receptor (TLR)2 compared with untreated diabetic mice (Fig. 4, A and B). This was consistent with the inhibition by Sac/Val and Val treatment of NF-κB activity as determined by level of phospho-p65 (Fig. 4C). Recently, the cGMP-AMP synthase (cGAS)-STING pathway has been discovered as a link to the trigger of inflammation. We found that STING expression was increased in db/db kidneys and normalized by both Sac/Val and Val treatment (Fig. 4D). To examine whether STING was indeed activated in diabetic kidneys, we checked the STING direct target TBK1/IRF3 and found that their active forms, phospho-TBK1 or phospho-IRF3, were increased in db/db kidneys (Fig. 4, E and F), indicating STING activation. Expression of phospho-TBK1 or phospho-IRF3 was decreased in both treatment groups (Fig. 4, E and F). In addition, phospho-Stat3 as another downstream effector of STING activation was downregulated by both Sac/Val and Val treatments (Fig. 4G). STING activation is known to be related to self-DNA released into the cytosol. To find out whether other self-DNA-sensing mechanisms are involved, we also looked at the expression of TLR9 and AIM2. TLR9 expression was not changed in db/db kidneys, although the treatment did reduce its expression (Fig. 4H). There was a modest increase in the AIM2 protein level in db/db kidneys, and Val but not Sac/Val treatment had an effect in decreasing its expression (Fig. 4H).
Figure 4.

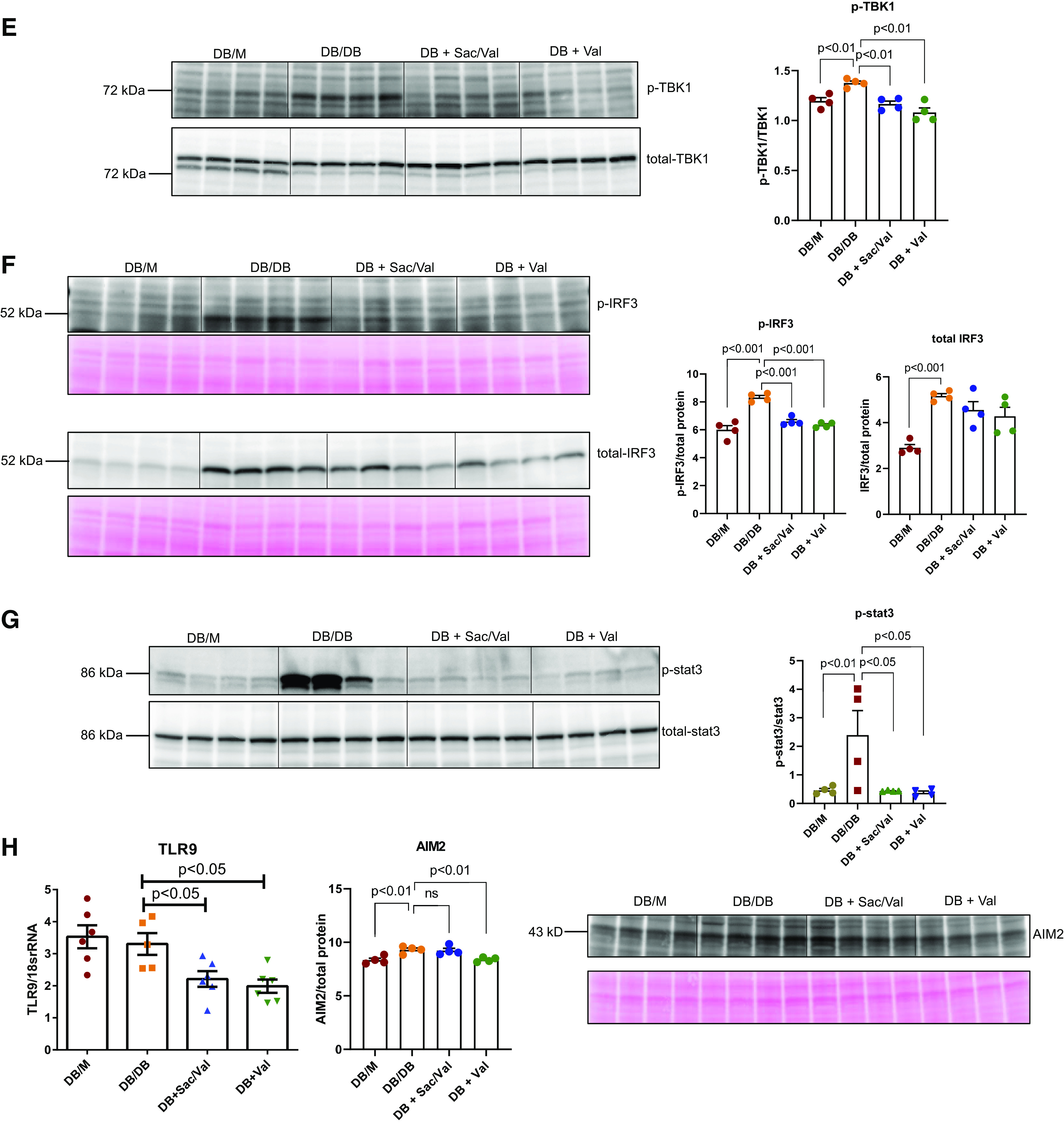
Administration of sacubitril/valsartan (Sac/Val) improved inflammation in db/db mice. A: monocyte chemoattractant protein-1 (MCP-1) mRNA levels and protein abundance were increased in db/db mice and significantly abolished with Sac/Val or Val treatment; n = 4−10 per group. B: quantitative PCR analysis of Toll-like receptor (TLR)2 mRNA; n = 6 per group. C − E: representative Western blot images of phosphorylated p65 as a marker for NF-κB activity (C), stimulator of interferon genes (STING; D), phosphorylated TANK-binding kinase 1 (TBK1; E), phosphorylated interferon regulatory factor 3 (IRF3; F), and phosphorylated Stat3 protein (G) normalized to total p65, TBK1, or Stat3 in C, E, and G or total protein in D and F, respectively; n = 4 per group. H: quantitative PCR analysis of TLR9 mRNA and representative Western blot images of absent in melanoma 2 (AIM2); n = 4−6 per group. Data are presented as means ± SE. Veh, vehicle.
Sac/Val Treatment Improved Mitochondria Function in db/db Mice
Damaged mitochondrial DNA could be a source of self-DNA leakage to activate a DNA sensor such as cGAS-STING (34). We isolated DNA from the kidneys to check their mitochondrial DNA lesions. Diabetic kidneys showed significantly increased mitochondrial DNA lesions, which was reversed by Sac/Val or Val treatment (Fig. 5A). In line with the mitochondrial DNA damage, we found decreased complex I activity in db/db kidneys, which was restored by Sac/Val or Val treatment (Fig. 5B). This improvement was also associated with increased transcription factor A, mitochondrial (Tfam), or medium-chain acyl-CoA dehydrogenase (MCAD) expression (Fig. 5C) with Sac/Val treatment, suggesting its role in maintaining mitochondrial biogenesis and fatty acid oxidation in db/db kidneys.
Figure 5.

Treatment with sacubitril/valsartan (Sac/Val) improved mitochondrial function in db/db mice. A: mitochondrial DNA (mtDNA) damage was increased in db/db kidneys and reversed in Sac/Val-treated or Val-treated groups; n = 6 per group. B: mitochondrial complex I enzyme activity was significantly diminished in db/db kidneys, which was restored in Sac/Val-treated or Val-treated groups; n = 6−8 per group. C: quantitative PCR analysis of mitochondrial biogenesis regulator transcription factor A, mitochondrial (Tfam), and fatty acid oxidation enzyme medium-chain acyl-CoA dehydrogenase (MCAD) mRNA levels; n = 6 per group. Data are presented as means ± SE. Veh, vehicle.
Sac/Val Treatment Improved Diabetic Nephropathy in KKAy Mice
Body weight and food intake were similar for all three groups of KKAy mice after 3 mo of treatment. Both Sac/Val-treated and Val-treated KKAy mice had lower mean systolic blood pressures than untreated mice. Sac/Val-treated mice had lower kidney weight-to-body weight ratios and BUN levels. Furthermore, in contrast to db/db mice, Sac/Val-treated KKAy mice had lower levels of blood glucose and serum lipids than untreated KKAy mice. This latter effect appeared to be due to Sac, since Val-treated mice did not have serum BUN, blood glucose, and serum lipid levels (Table 2) or serum insulin and leptin levels (Fig. 6A) different from untreated KKAy mice.
Table 2.
Effect of Val and Sac/Val on physical and biochemical characteristics in nondiabetic (C57BL/6) and diabetic (KKAy) mice
| C57BL/6 | C57BL/6 + Sac/Val | C57BL/6 + Val | KKAy | KKAy + Sac/Val | KKAy + Val | |
|---|---|---|---|---|---|---|
| Body weight, g | 27.3 ± 0.9 | 30.1 ± 0.9 | 30.9 ± 0.9 | 43.5 ± 0.9a | 42.1 ± 0.9 | 43.3 ± 0.9 |
| Food intake, g | 3.75 ± 0.29 | 3.6 ± 0.29 | 3.56 ± 0.29 | 6.3 ± 0.29a | 6.2 ± 0.29 | 5.9 ± 0.29 |
| Kidney weight/body weight, mg/g | 0.01 ± 0.007 | 0.01 ± 0.007 | 0.01 ± 0.007 | 0.016 ± 0.007b | 0.013 ± 0.007c | 0.014 ± 0.007 |
| Systolic blood pressure, mmHg | 112.4 ± 4.3 | 110.8 ± 4.3 | 113.4 ± 4.3 | 109.3 ± 4.3 | 92.6 ± 4.3d | 94.4 ± 4.3d |
| Glucose, mg/dL | 183.8 ± 27.8 | 161.3 ± 27.8 | 174.2 ± 27.8 | 325.5 ± 27.8a | 185.7 ± 28.5e,f | 278.8 ± 28.5 |
| Blood urea nitrogen, mg/dL | 44.9 ± 7.8 | 45.8 ± 7.8 | 46.1 ± 7.8 | 87.1 ± 7.8a | 66.8 ± 7.8g,f | 91.02 ± 7.8 |
| Triglycerides, mg/dL | 48.4 ± 15.9 | 42.6 ± 15.9 | 49.7 ± 15.9 | 244.1 ± 15.9a | 142.8 ± 15.9d,f | 192 ± 15.9c |
| Cholesterol, mg/dL | 114.9 ± 8.9 | 114.5 ± 8.9 | 114.1 ± 8.9 | 172 ± 8.9a | 136.2 ± 8.9d | 154.1 ± 8.9 |
All data analyses are presented as means ± SE for n = 9 or 10 mice in each group. Sac, sacubitril; Val, valsartan. aP < 0.0001 vs. C57BL6/J; bP < 0.001 vs. C57BL6/J; cP < 0.01 vs. KKAy; dP < 0.001 vs. KKAy; eP < 0.0001 vs. KKAy; fP < 0.01 vs. KKAy + Val; gP < 0.05 vs. KKAy.
Figure 6.
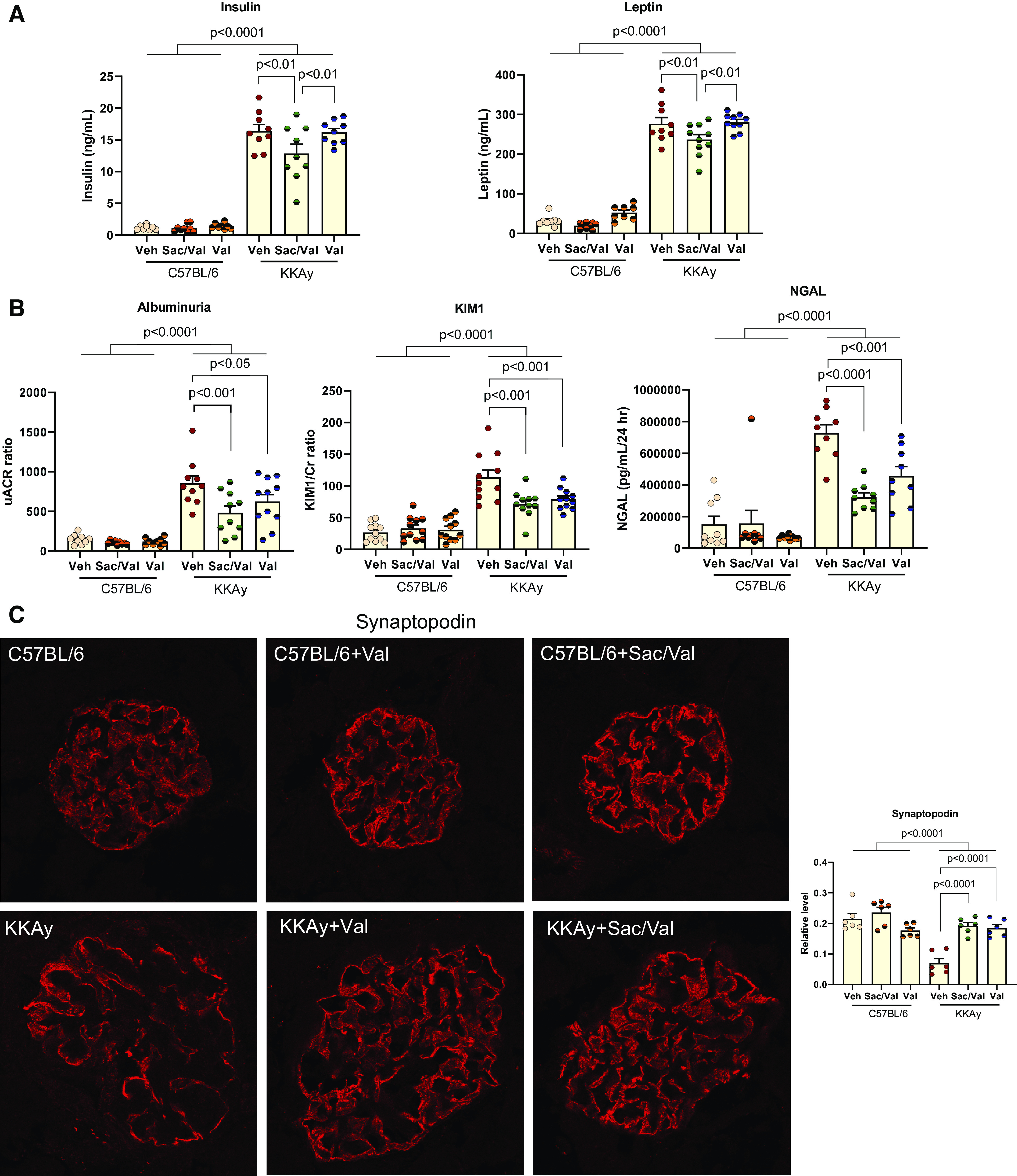
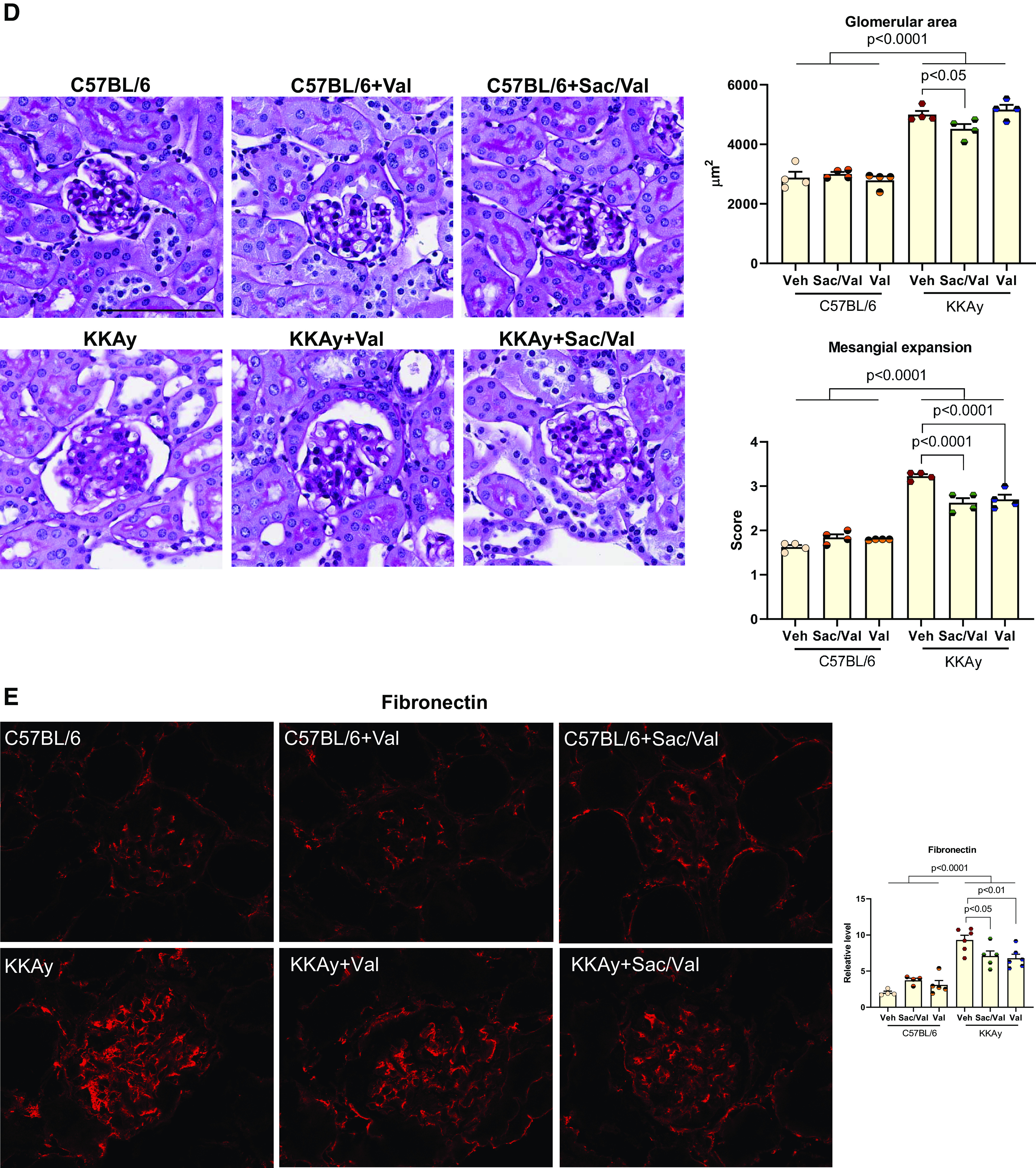

Administration of sacubitril/valsartan (Sac/Val) improved diabetic nephropathy in KKAy mice. A: serum insulin and leptin hormone levels were higher in KKAy mice and markedly reduced in KKAy mice administered with Sac/Val but not Val; n = 10 or 11 per group. B: excretion of urinary albumin, kidney injury molecule-1 (KIM-1), and neutrophil gelatinase-associated lipocalin (NGAL) levels were measured with 24-h urine from KKAy mice; n = 10 or 11 per group. C: immunofluorescence staining of kidney sections for the podocyte marker synaptopodin; n = 6 per group. D: representative images of periodic acid-Schiff staining of kidney sections. The mesangial expansion score was defined by the percentage of the mesangial area in the glomerular tuft area. The mesangial area was determined by assessment of periodic acid-Schiff-positive and nucleus-free areas in the mesangium; n = 4 per group. Scale bar = 100 µm. E and F: immunofluorescence staining of kidney sections for fibronectin (E) and collagen type IV (F). Data were quantified using the ratio of total pixel intensity vs. glomerular area; n = 6 per group. G: quantitative PCR analysis of mRNA of transforming growth factor-β (TGF-β), plasminogen activator inhibitor-1 (PAI1), fibronectin, collagen type IV-α3 (Col4a3), and α-smooth muscle actin (α-SMA); n = 6−9 per group. Data are presented as means ± SE. Veh, vehicle.
Both Sac/Val-treated and Val-treated groups had lower urinary albumin, KIM-1, and NGAL levels compared with untreated mice (Fig. 6B). Similar to the db/db experiments, both Sac/Val and Val treatments restored podocyte density in KKAy mice (Fig. 6C).
KKAy kidneys had a higher glomerular area, which was decreased with Sac/Val but not Val treatment (Fig. 6D). However, both Sac/Val and Val had lower mesangial expansion as shown by PAS staining (Fig. 6D), in line with the same changes in fibronectin and collagen type IV staining (Fig. 6, E and F), as well as mRNA expression levels of TGF-β, PAI1, fibronectin, collagen type IV, and α-SMA (Fig. 6G).
Both Sac/Val and Val had a lower level of oxidative stress markers 4-HNE and Nox4 (Fig. 7). We further found that both treatments increased Nrf2 and SOD2 expression to restore the antioxidative response (Fig. 7).
Figure 7.

Treatment with sacubitril/valsartan (Sac/Val) decreased oxidative stress in the kidneys of KKAy mice. A: immunoblot images from the total kidney indicated that 4-hydroxynonenal (4-HNE) protein expression was higher in KKAy mice and that treatment with Sac/Val or Val suppressed their abundance; n = 4 per group. B: quantitative PCR analysis of NADPH oxidase 4 (Nox4), nuclear factor erythroid 2-related factor 2 (Nrf2), and superoxide dismutase 2 (SOD2) mRNA levels; n = 8 per group. Data are presented as means ± SE. Veh, vehicle.
Sac/Val seemed to have more anti-inflammatory effects than Val in KKAy kidneys. Sac/Val-treated mice had lower mRNA levels of MCP-1, TLR2, cGAS, and STING. In contrast, only MCP-1 and STING mRNA levels were lower in the Val-treated group (Fig. 8A). Both Sac/Val and Val treatments resulted in similar decreases in STING and phospho-Stat3 protein abundance (Fig. 8B). STING activation, as represented by phospho-TBK1 or phospho-IRF3 levels, was also decreased in Sac/Val-treated and Val-treated db/db kidneys (Fig. 8C). However, other DNA sensors, such as TLR9 and AIM2, seemed to be not activated in KKAy kidneys (Fig. 8D).
Figure 8.

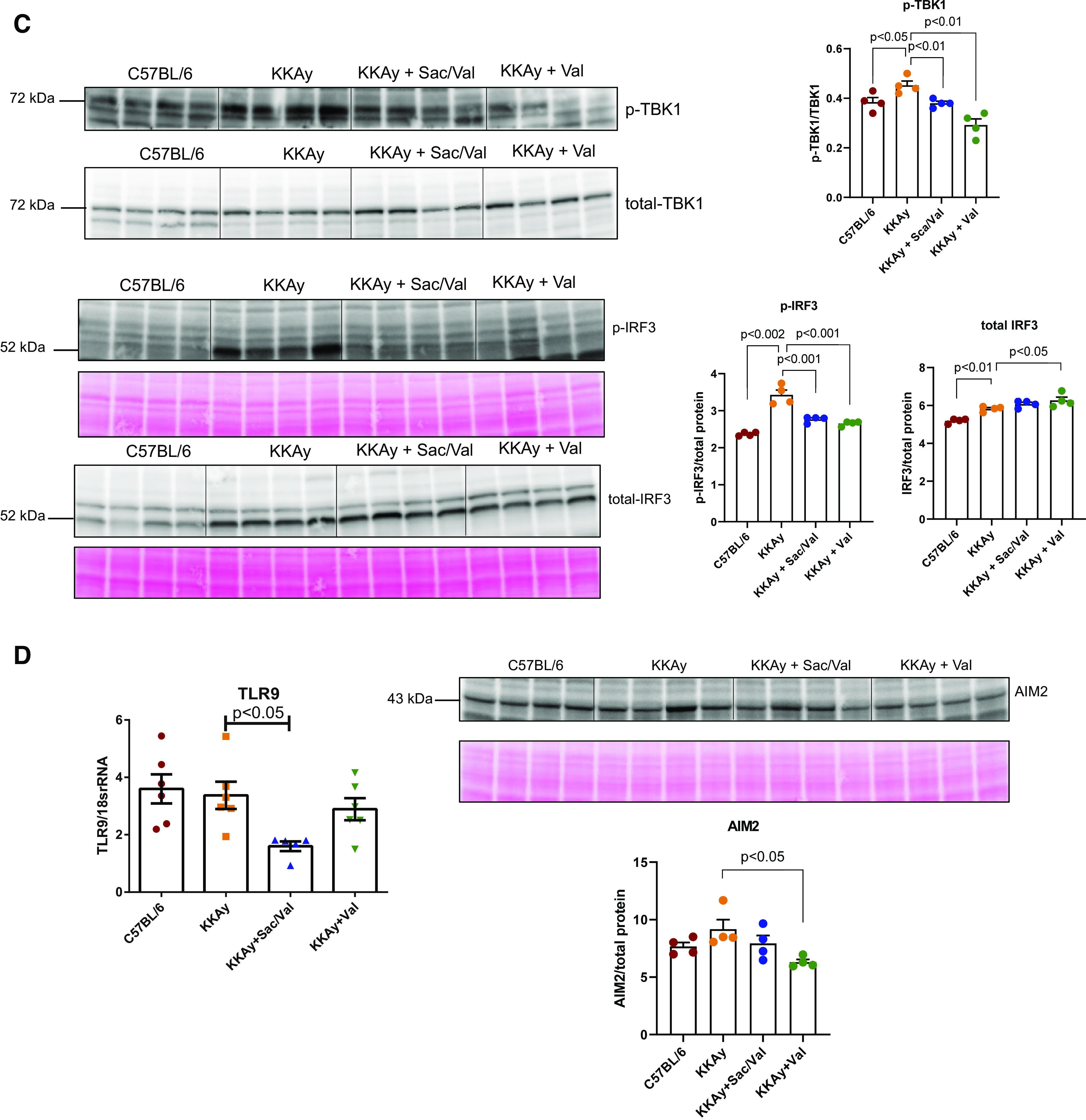
Sacubitril/valsartan (Sac/Val) decreased inflammation in the kidneys of KKAy mice. A: quantitative PCR analysis of monocyte chemoattractant protein-1 (MCP-1), Toll-like receptor (TLR)2, cGMP-AMP synthase (cGAS), and stimulator of interferon genes (STING) transcript levels; n = 6−8 per group. B: representative Western blot images of STING normalized to total protein and phosphorylated Stat3 protein normalized to total Stat3; n = 4 per group. C: representative Western blot images of phosphorylated TANK-binding kinase 1 (TBK1) normalized to total TBK1 and phosphorylated IRF3 normalized to total interferon regulatory factor 3 (IRF3); n = 4 per group. D: quantitative PCR analysis of TLR9 mRNA and representative Western blot images of absent in melanoma 2 (AIM2); n = 4−6 per group. Data are presented as means ± SE. Veh, vehicle.
Unlike db/db kidneys, KKAy kidneys did not exhibit changes in mitochondrial complex I activity, Tfam, or MCAD mRNA levels. Furthermore, Sac/Val or Val had no effects on complex I activity, Tfam, or MCAD mRNA levels.
DISCUSSION
Although there are plenty of data to support the beneficial role of Sac/Val in treating heart failure and CKD in animal models and patients (25, 26, 35, 36), no full investigation has been done in diabetic kidney disease, especially that induced by type 2 diabetes. The present study reveals the renoprotective effects of Sac/Val in type 2 diabetic mice. Importantly, Sac/Val attenuated the progression of proteinuria, mesangial expansion, and podocyte loss in diabetic mice.
We used two models of diabetes in this study. db/db mice carry a leptin receptor mutation and are spontaneous/congenital diabetic because of leptin signaling abnormalities (28). However, their monogenic inheritance pattern is in contrast to the polygenic etiology of human type 2 diabetes. To circumvent this limitation, we introduced another mouse model of type 2 diabetes, KKAy mice, which develop spontaneous diabetes of polygenic origin (29). The current study showed that Sac/Val or Val-alone treatment improved proteinuria, podocyte loss, and glomerulosclerosis in both models, but it was associated with differential metabolic changes. Blood glucose was almost normalized with Sac/Val treatment in KKAy mice, but Val alone did not change the glucose level. As a result, serum insulin and leptin levels were significantly decreased with Sac/Val treatment. This is consistent with a previous report showing regulation of insulin secretion and pancreatic β-cells by NPs (37). Sac/Val also reduced serum triglycerides and cholesterol levels in KKAy mice. Val alone lowered only serum triglyceride levels, to a lesser degree than Sac/Val. In db/db mice, the glucose-lowering or lipid-lowering effect was not seen with either Sac/Val or Val.
We further explored the potential mechanism by which Sac/Val exerts its protection in the kidney. We found that Sac/Val can reduce oxidative stress and inflammation in both db/db mice and KKAy mice. For some parameters, Val alone did not result in similar amelioration; instead, it showed a significant difference from Sac/Val, such as NOX4 mRNA expression in db/db mice and MCP-1/TLR2/cGAS mRNA expression in KKAy mice. Other reports have also noticed these renal protective mechanisms by Sac/Val in CKD rats (26) and type 1 diabetic rats (38, 39). However, we have novel findings in this study that the inflammation in the kidneys of db/db mice and KKAy mice might be caused by the activation of nucleic acid sensors. We checked the current known DNA sensors cGAS-STING, TLR9, and AIM2. cGAS-STING has shown the most remarkable change in both db/db and KKAy kidneys. DNA damage or mitochondrial dysfunction can release self-DNA into the cytosol to activate cGAS, leading to the generation of a second messenger, cGAMP, which subsequently induces a type I interferon response or NF-κB activation via STING (34, 40–47). NF-κB is a crucial transcription factor responsible for the expression of proinflammatory cytokines in diabetic kidneys (48–50). The cGAS-STING pathway thus provides a novel mechanism for inflammation in diabetic nephropathy. In previous studies, there was evidence of mitochondrial damage, increased cGAS-STING signaling, and inflammation associated with fibrosis in patients with CKD or CKD animal models, and pharmacological inhibition of STING ameliorated kidney fibrosis in mouse models (51, 52). In the present study, we further found that both Sac/Val and Val prevented the STING increase in db/db kidneys or KKAy kidneys. In db/db mice, we found increased mitochondrial DNA damage and decreased complex I activity, suggesting mitochondrial dysfunction, which may explain the activation of cGAS-STING. Both Sac/Val and Val treatment can restore the mitochondrial DNA damage and complex I activity, but Sac/Val seems to improve other mitochondria functions, such as mitochondria biogenesis and fatty acid oxidation, in db/db kidneys. As a support to our findings, it has been observed that the RAAS can induce mitochondria dysfunction and oxidative stress (53), and NPs have been reported to upregulate mitochondrial respiration and fatty acid oxidation (54). In KKAy mice, we did not observe mitochondria dysfunction in this study. However, we cannot rule out damage in glomerular cells that is usually masked by using the kidney cortex instead of isolated glomeruli.
In summary, we have shown that Sac/Val treatment is beneficial and that there are differential effects of Sac/Val compared with Val alone in diabetic kidneys. Pathologically, db/db mice and KKAy mice represent early stages of human diabetic nephropathy (29, 55), and they do not have hypertension. With the advanced disease model showing a full-blown phenotype of human diabetic nephropathy, such as endothelial nitric oxide synthase knockout mice in the db/db background (56), we expect there would be more synergistic effects of dual blockade of the RAAS and neprilysin to be achieved.
GRANTS
This work was funded by Novartis through an Investigator Initiated Trial (LCZ696BUSNC07T) (to M.L.), by the National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases Grant 5R01DK116567 (to M.L.) and National Center for Advancing Translational Sciences Grant TL1TR001431 (to B.A.J.), and by the American Heart Association Postdoctoral Fellowship 19POST34381041 (to K.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
X.X.W. and M.L. conceived and designed research; K.M., B.A.J., and X.X.W. performed experiments; K.M. and X.X.W. analyzed data; K.M., X.X.W., and M.L. interpreted results of experiments; K.M. and X.X.W. prepared figures; K.M. and X.X.W. drafted manuscript; K.M., X.X.W., and M.L. edited and revised manuscript; K.M., X.X.W., and M.L. approved final version of manuscript.
REFERENCES
- 1.Breyer MD, Coffman TM, Flessner MF, Fried LF, Harris RC, Ketchum CJ, Kretzler M, Nelson RG, Sedor JR, Susztak K; Kidney Research National Dialogue (KRND). Diabetic nephropathy: a national dialogue. Clin J Am Soc Nephrol 8: 1603–1605, 2013. doi: 10.2215/CJN.03640413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest 124: 2333–2340, 2014. doi: 10.1172/JCI72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, Rossing P, Groop PH, Cooper ME. Diabetic kidney disease. Nat Rev Dis Primers 1: 15018, 2015. doi: 10.1038/nrdp.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 8: 29, 2010. doi: 10.1186/1478-7954-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D, Williams DE, Geiss L. Changes in diabetes-related complications in the United States, 1990-2010. N Engl J Med 370: 1514–1523, 2014. doi: 10.1056/NEJMoa1310799. [DOI] [PubMed] [Google Scholar]
- 6.de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 305: 2532–2539, 2011. doi: 10.1001/jama.2011.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosolowsky ET, Skupien J, Smiles AM, Niewczas M, Roshan B, Stanton R, Eckfeldt JH, Warram JH, Krolewski AS. Risk for ESRD in type 1 diabetes remains high despite renoprotection. J Am Soc Nephrol 22: 545–553, 2011. doi: 10.1681/ASN.2010040354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dieter BP, Alicic RZ, Tuttle KR. GLP-1 receptor agonists in diabetic kidney disease: from the patient-side to the bench-side. Am J Physiol Renal Physiol 315: F1519–F1525, 2018. doi: 10.1152/ajprenal.00211.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satirapoj B. Sodium-glucose cotransporter 2 inhibitors with renoprotective effects. Kidney Dis (Basel) 3: 24–32, 2017. doi: 10.1159/000471765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci 28: 89–94, 1981. doi: 10.1016/0024-3205(81)90370-2. [DOI] [PubMed] [Google Scholar]
- 11.Sudoh T, Kangawa K, Minamino N, Matsuo H. A new natriuretic peptide in porcine brain. Nature 332: 78–81, 1988. doi: 10.1038/332078a0. [DOI] [PubMed] [Google Scholar]
- 12.Boerrigter G, Burnett JC Jr.. Recent advances in natriuretic peptides in congestive heart failure. Expert Opin Investig Drugs 13: 643–652, 2004. doi: 10.1517/13543784.13.6.643. [DOI] [PubMed] [Google Scholar]
- 13.Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev 27: 47–72, 2006. doi: 10.1210/er.2005-0014. [DOI] [PubMed] [Google Scholar]
- 14.Zois NE, Bartels ED, Hunter I, Kousholt BS, Olsen LH, Goetze JP. Natriuretic peptides in cardiometabolic regulation and disease. Nat Rev Cardiol 11: 403–412, 2014. doi: 10.1038/nrcardio.2014.64. [DOI] [PubMed] [Google Scholar]
- 15.Birkenfeld AL, Boschmann M, Engeli S, Moro C, Arafat AM, Luft FC, Jordan J. Atrial natriuretic peptide and adiponectin interactions in man. PLoS One 7: e43238, 2012. doi: 10.1371/journal.pone.0043238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jujic A, Nilsson PM, Engstrom G, Hedblad B, Melander O, Magnusson M. Atrial natriuretic peptide and type 2 diabetes development–biomarker and genotype association study. PLoS One 9: e89201, 2014. doi: 10.1371/journal.pone.0089201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med 365: 32–43, 2011. [Erratum in N Engl J Med 365: 773, 2011]. doi: 10.1056/NEJMoa1100171. [DOI] [PubMed] [Google Scholar]
- 18.Wang DJ, Dowling TC, Meadows D, Ayala T, Marshall J, Minshall S, Greenberg N, Thattassery E, Fisher ML, Rao K, Gottlieb SS. Nesiritide does not improve renal function in patients with chronic heart failure and worsening serum creatinine. Circulation 110: 1620–1625, 2004. doi: 10.1161/01.CIR.0000141829.04031.25. [DOI] [PubMed] [Google Scholar]
- 19.Ferro CJ, Spratt JC, Haynes WG, Webb DJ. Inhibition of neutral endopeptidase causes vasoconstriction of human resistance vessels in vivo. Circulation 97: 2323–2330, 1998. doi: 10.1161/01.cir.97.23.2323. [DOI] [PubMed] [Google Scholar]
- 20.Margulies KB, Perrella MA, McKinley LJ, Burnett JC Jr.. Angiotensin inhibition potentiates the renal responses to neutral endopeptidase inhibition in dogs with congestive heart failure. J Clin Invest 88: 1636–1642, 1991. doi: 10.1172/JCI115477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell DJ, Krum H, Esler MD. Losartan increases bradykinin levels in hypertensive humans. Circulation 111: 315–320, 2005. doi: 10.1161/01.CIR.0000153269.07762.3B. [DOI] [PubMed] [Google Scholar]
- 22.Makani H, Messerli FH, Romero J, Wever-Pinzon O, Korniyenko A, Berrios RS; Sripal Bangalore. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 110: 383–391, 2012. doi: 10.1016/j.amjcard.2012.03.034. [DOI] [PubMed] [Google Scholar]
- 23.Gu J, Noe A, Chandra P, Al-Fayoumi S, Ligueros-Saylan M, Sarangapani R, Maahs S, Ksander G, Rigel DF, Jeng AY, Lin TH, Zheng W, Dole WP. Pharmacokinetics and pharmacodynamics of LCZ696, a novel dual-acting angiotensin receptor-neprilysin inhibitor (ARNi). J Clin Pharmacol 50: 401–414, 2010. doi: 10.1177/0091270009343932. [DOI] [PubMed] [Google Scholar]
- 24.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 371: 993–1004, 2014. doi: 10.1056/NEJMoa1409077. [DOI] [PubMed] [Google Scholar]
- 25.Suematsu Y, Jing W, Nunes A, Kashyap ML, Khazaeli M, Vaziri ND, Moradi H. LCZ696 (sacubitril/valsartan), an angiotensin-receptor neprilysin inhibitor, attenuates cardiac hypertrophy, fibrosis, and vasculopathy in a rat model of chronic kidney disease. J Card Fail 24: 266–275, 2018. doi: 10.1016/j.cardfail.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 26.Jing W, Vaziri ND, Nunes A, Suematsu Y, Farzaneh T, Khazaeli M, Moradi H. LCZ696 (sacubitril/valsartan) ameliorates oxidative stress, inflammation, fibrosis and improves renal function beyond angiotensin receptor blockade in CKD. Am J Transl Res 9: 5473–5484, 2017. [PMC free article] [PubMed] [Google Scholar]
- 27.Polina I, Domondon M, Fox R, Sudarikova AV, Troncoso M, Vasileva VY, Kashyrina Y, Gooz MB, Schibalski RS, DeLeon-Pennell KY, Fitzgibbon WR, Ilatovskaya DV. Differential effects of low-dose sacubitril and/or valsartan on renal disease in salt-sensitive hypertension. Am J Physiol Renal Physiol 319: F63–F75, 2020. doi: 10.1152/ajprenal.00125.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev 10: 131–145, 2014. doi: 10.2174/1573399810666140508121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomino Y. Lessons from the KK-Ay mouse, a spontaneous animal model for the treatment of human type 2 diabetic nephropathy. Nephrourol Mon 4: 524–529, 2012. doi: 10.5812/numonthly.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang XX, Edelstein MH, Gafter U, Qiu L, Luo Y, Dobrinskikh E, Lucia S, Adorini L, D'Agati VD, Levi J, Rosenberg A, Kopp JB, Gius DR, Saleem MA, Levi M. G protein-coupled bile acid receptor TGR5 activation inhibits kidney disease in obesity and diabetes. J Am Soc Nephrol 27: 1362–1378, 2016. doi: 10.1681/ASN.2014121271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang XX, Luo Y, Wang D, Adorini L, Pruzanski M, Dobrinskikh E, Levi M. A dual agonist of farnesoid X receptor (FXR) and the G protein-coupled receptor TGR5, INT-767, reverses age-related kidney disease in mice. J Biol Chem 292: 12018–12024, 2017. doi: 10.1074/jbc.C117.794982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang XX, Wang D, Luo Y, Myakala K, Dobrinskikh E, Rosenberg AZ, Levi J, Kopp JB, Field A, Hill A, Lucia S, Qiu L, Jiang T, Peng Y, Orlicky D, Garcia G, Herman-Edelstein M, D'Agati V, Henriksen K, Adorini L, Pruzanski M, Xie C, Krausz KW, Gonzalez FJ, Ranjit S, Dvornikov A, Gratton E, Levi M. FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity. J Am Soc Nephrol 29: 118–137, 2018. doi: 10.1681/ASN.2017020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furda A, Santos JH, Meyer JN, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol 1105: 419–437, 2014. doi: 10.1007/978-1-62703-739-6_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215: 1287–1299, 2018. doi: 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang H, Zhang J, Zhang X, Qin G, Wang K, Deng Z, Fang Y, Chen G. Effects of sacubitril/valsartan in patients with heart failure and chronic kidney disease: a meta-analysis. Eur J Pharmacol 884: 173444, 2020. doi: 10.1016/j.ejphar.2020.173444. [DOI] [PubMed] [Google Scholar]
- 36.Menendez JT. The mechanism of action of LCZ696. Card Fail Rev 2: 40–46, 2016. doi: 10.15420/cfr.2016:1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Undank S, Kaiser J, Sikimic J, Dufer M, Krippeit-Drews P, Drews G. Atrial natriuretic peptide affects stimulus-secretion coupling of pancreatic beta-cells. Diabetes 66: 2840–2848, 2017. doi: 10.2337/db17-0392. [DOI] [PubMed] [Google Scholar]
- 38.Mohany M, Alanazi AZ, Alqahtani F, Belali OM, Ahmed MM, Al-Rejaie SS. LCZ696 mitigates diabetic-induced nephropathy through inhibiting oxidative stress, NF-kappaB mediated inflammation and glomerulosclerosis in rats. PeerJ 8: e9196, 2020. doi: 10.7717/peerj.9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uijl E, Hart T, Roksnoer DC, Groningen LCW, van Veghel MCC, Garrelds R, de Vries IM, van der Vlag R, Zietse J, Nijenhuis R, Joles T, Hoorn JA, Danser EJ, Angiotensin AHJ. neprilysin inhibition confers renoprotection in rats with diabetes and hypertension by limiting podocyte injury. J Hypertens 38: 755–764, 2020. doi: 10.1097/HJH.0000000000002326. [DOI] [PubMed] [Google Scholar]
- 40.Ablasser A, Chen ZJ. cGAS in action: expanding roles in immunity and inflammation. Science 363: eaat8657, 2019. doi: 10.1126/science.aat8657. [DOI] [PubMed] [Google Scholar]
- 41.Bai J, Liu F. The cGAS-cGAMP-STING pathway: a molecular link between immunity and metabolism. Diabetes 68: 1099–1108, 2019. doi: 10.2337/dbi18-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461: 788–792, 2009. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 20: 657–674, 2019. doi: 10.1038/s41576-019-0151-1. [DOI] [PubMed] [Google Scholar]
- 44.Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep 21: e49799, 2020. doi: 10.15252/embr.201949799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791, 2013. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17: 363–375, 2017. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zierhut C, Funabiki H. Regulation and consequences of cGAS activation by self-DNA. Trends Cell Biol 30: 594–605, 2020. doi: 10.1016/j.tcb.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 50: 2792–2808, 2001. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 49.Coto E, Diaz-Corte C, Tranche S, Gomez J, Alonso B, Iglesias S, Reguero JR, Lopez-Larrea C, Coto-Segura P. Gene variants in the NF-KB pathway (NFKB1, NFKBIA, NFKBIZ) and their association with type 2 diabetes and impaired renal function. Hum Immunol 79: 494–498, 2018. doi: 10.1016/j.humimm.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 50.Schmid H, Boucherot A, Yasuda Y, Henger A, Brunner B, Eichinger F, Nitsche A, Kiss E, Bleich M, Grone HJ, Nelson PJ, Schlondorff D, Cohen CD, Kretzler M; European Renal cDNA Bank (ERCB) Consortium. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes 55: 2993–3003, 2006. doi: 10.2337/db06-0477. [DOI] [PubMed] [Google Scholar]
- 51.Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, Kaufman BA, Park J, Pei L, Baur J, Palmer M, Susztak K. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab 30: 784–799.e785, 2019. doi: 10.1016/j.cmet.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, Fujii R, Ishidate F, Tanaka T, Tanaka Y, Hirokawa N, Nangaku M, Inagi R. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep 29: 1261–1273.e1266, 2019. doi: 10.1016/j.celrep.2019.09.050. [DOI] [PubMed] [Google Scholar]
- 53.Ramalingam L, Menikdiwela K, LeMieux M, Dufour JM, Kaur G, Kalupahana N, Moustaid-Moussa N. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim Biophys Acta Mol Basis Dis 1863: 1106–1114, 2017. doi: 10.1016/j.bbadis.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 54.Domondon M, Nikiforova AB, DeLeon-Pennell KY, Ilatovskaya DV. Regulation of mitochondria function by natriuretic peptides. Am J Physiol Renal Physiol 317: F1164–F1168, 2019. doi: 10.1152/ajprenal.00384.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vallon V, Komers R. Pathophysiology of the diabetic kidney. Compr Physiol 1: 1175–1232, 2011. doi: 10.1002/cphy.c100049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brosius FC, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T; Animal Models of Diabetic Complications Consortium. Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]