

Keywords: aldosterone-sensitive distal nephron, epithelial Na+ channel, Kir4.1/Kir5.1 heterotetramer, renal K+ excretion, thiazide-sensitive Na+-Cl− cotransporter
Abstract
High sodium (HS) intake inhibited epithelial Na+ channel (ENaC) in the aldosterone-sensitive distal nephron and Na+-Cl− cotransporter (NCC) by suppressing basolateral Kir4.1/Kir5.1 in the distal convoluted tubule (DCT), thereby increasing renal Na+ excretion but not affecting K+ excretion. The aim of the present study was to explore whether deletion of Kir5.1 compromises the inhibitory effect of HS on NCC expression/activity and renal K+ excretion. Patch-clamp experiments demonstrated that HS failed to inhibit DCT basolateral K+ channels and did not depolarize K+ current reversal potential of the DCT in Kir5.1 knockout (KO) mice. Moreover, deletion of Kir5.1 not only increased the expression of Kir4.1, phospho-NCC, and total NCC but also abolished the inhibitory effect of HS on the expression of Kir4.1, phospho-NCC, and total NCC and thiazide-induced natriuresis. Also, low sodium-induced stimulation of NCC expression/activity and basolateral K+ channels in the DCT were absent in Kir5.1 KO mice. Deletion of Kir5.1 decreased ENaC currents in the late DCT, and HS further inhibited ENaC activity in Kir5.1 KO mice. Finally, measurement of the basal renal K+ excretion rate with the modified renal clearance method demonstrated that long-term HS inhibited the renal K+ excretion rate and steadily increased plasma K+ levels in Kir5.1 KO mice but not in wild-type mice. We conclude that Kir5.1 plays an important role in mediating the effect of HS intake on basolateral K+ channels in the DCT and NCC activity/expression. Kir5.1 is involved in maintaining renal ability of K+ excretion during HS intake.
NEW & NOTEWORTHY Kir5.1 plays an important role in mediating the effect of high sodium intake on basolateral K+ channels in the distal convoluted tubule and Na+-Cl− cotransporter activity/expression.
INTRODUCTION
We have previously demonstrated that dietary sodium intake regulated basolateral Kir4.1/Kir5.1 channel activity in the distal convoluted tubule (DCT) such that low sodium (LS) intake stimulated, whereas high sodium (HS) intake inhibited, Kir4.1/Kir5.1 in the DCT (1). HS-induced inhibition of basolateral Kir4.1/Kir5.1 in the DCT was essential for HS-induced inhibition of the Na+-Cl− cotransporter (NCC). The Kir4.1/Kir5.1 heterotetramer is a major type of K+ channel expressed in the basolateral membrane of the DCT and collecting duct (2–6). Although Kir4.1 is a key component for providing K+ conductance of the basolateral Kir4.1/Kir5.1 heterotetramer (7–9), Kir5.1 is an important regulatory subunit of the basolateral K+ channel of the DCT (2, 10–14). For instance, Kir5.1 regulates Kir4.1 expression/activity by facilitating neural precursor cell expressed, developmentally downregulated protein 4-2 (Nedd4-2)-dependent ubiquitination of Kir4.1 (14). We have previously demonstrated that deletion of Kir5.1 abolished the effect of high dietary K+ (HK) intake on the basolateral K+ conductance of the DCT and NCC activity (15), suggesting that Kir5.1 plays a role in mediating HK-induced inhibition of basolateral K+ channels and NCC. Since HS also inhibits basolateral K+ channels of the DCT and NCC, the first aim of the present study was to explore whether Kir5.1 deletion compromises the effect of HS on basolateral K+ conductance and NCC activity.
NCC is the target of thiazide diuretic and plays an important role not only in regulating renal Na+ reabsorption but also in regulating renal K+ excretion by controlling Na+ and fluid volume delivery to the late distal nephron (16–21). For instance, despite high epithelial Na+ channel (ENaC) activity in the collecting duct induced by LS intake (22–24), ENaC-dependent K+ excretion is not increased due to LS-induced stimulation of the Na+-driven Cl−/HCO3− exchanger in the collecting duct and NCC activity in the DCT (25–27). High NCC activity induced by LS should enhance Na+ reabsorption in the DCT, thereby decreasing Na+ delivery to the late distal nephron and preventing excessive ENaC-dependent K+ excretion. In contrast, HS intake suppresses NCC activity and increases Na+ delivery to the late distal nephron, thereby facilitating renal Na+ excretion. Since HS intake is expected to inhibit ENaC activity in the aldosterone-sensitive distal nephron (ASDN) (23, 28), high Na+ and fluid delivery to the late ASDN should offset the negative effect of low ENaC activity on renal K+ excretion, thereby maintaining K+ homeostasis. Indeed, a previous in vivo study has demonstrated that dietary Na+ intake did not affect the renal K+ secretion rate and plasma K+ levels in animals with an intact renin-angiotensin-aldosterone system (29). Thus, if Kir5.1 is involved in mediating the effect of HS intake on NCC activity, deletion of Kir5.1 should also affect renal K+ excretion and K+ homeostasis during increasing dietary Na+ intake. Thus, the second aim of the present study was to examine whether deletion of Kir5.1 in the kidney impairs renal K+ excretion in animals on HS.
METHODS
Animals
We used Kcnj16+/+ or Kir5.1 wild-type (WT) mice (which were obtained from the original Kcnj16+/− mice) and Kcnj16−/− [Kir5.1 knockout (KO)] mice on the C57bl/6j background. We purchased male and female Kcnj16+/− mice from the Mary Lyon Center (Oxfordshire, UK) for breeding in the animal facility of New York Medical College (NYMC). The genotyping primers were as follows: forward F1 (WT), 5′- CTGCTTGCAGTTTGAAGGAAG-3′; forward F2 (Kir5.1 KO), 5′- AGGGGGAGGATTGGGAAGACAATAGCA-3′, and reverse, 5′- CATTCATCTTGTGGGGACAGGACGGTCT-3′, yielding 225-bp (WT) and 325-bp (KO) products.
Deletion of Kir5.1 protein was confirmed by Western blot analysis (Fig. 1A). Quantitative RT-PCR experiments also showed that mRNA expression of Kcnj16 to Gapdh in Kcnj16−/− mice was almost absent (0.08% ± 0.02% of WT), as confirmed by an agarose gel (Fig. 1B). Animals were housed in the NYMC animal facility with lights on at 7:00 AM and off at 7:00 PM. Mice had unlimited access to water and rodent chow. To study the effect of dietary Na+ intake on renal K+ excretion, mice were fed with the control diet (0.8% K+ and 0.4% Na+) (provided by the NYMC animal facility), HS diet (4.0% Na+ and 0.8% K+ diet, Catalog No. TD.92034), or LS diet (<0.01%−0.02% Na+ and 0.8% K+, Catalog No. TD.90228). HS and LS diets were purchased from Envigo-Teklad Diets (Madison, WI). All procedures were reviewed and approved by the Institutional Animal Care and Use Committee.
Figure 1.

Expression of Kir5.1 and Kcnj16 mRNA in Kcnj16+/+ and Kcnj16−/− mice. A: Western blots showing the expression of Kir5.1 (indicated by the arrow) and GAPDH in Kcnj16+/+ and Kcnj16−/− mice (n = 6 mice), respectively. B: agarose gel showing the expression of Gapdh and Kcnj16 mRNA in Kcnj16+/+ and Kcnj16−/− mice (n = 4 mice), respectively. C: bar graph summarizing the normalized expression of Kinj16 to Gapdh mRNA in Kcnj16+/+ and Kcnj16−/− mice. IB, immunoblot.
Quantitative RT-PCR to Detect Kcnj16 and Gapdh in Renal Tubules
Total RNA was extracted from 50 µg of the kidney cortex with the RNeasy Mini Kit (QIAGEN), and cDNA was then synthesized from 1 µg of total RNA with the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). We used 0.2 µg of cDNA as the template for detecting Kcnj16 expression with Maxima SYBR Green/ROX qPCR Master Mix (2×, Thermo Fisher Scientific). The quantitative PCR primer sequences for Kcnj16 were forward 5′- TGCTTGTTGGCTCACTGGAA-3′ and reverse 5′- GGGCATTCAGAGGAGTCCG-3′ (yielding a 135-bp product). The quantitative PCR primer sequences for GAPDH were forward 5′- TCACCACCATGGAGAAGGC-3′ and reverse 5′- GCTAAGCAGTTGGTGGTGCA-3′ (yielding a 168-bp product). We used the 2−ΔΔCT method (where CT is threshold cycle) to conduct quantitative analysis of Kcnj16 to Gapdh mRNA expression, and the specificity of PCR products was confirmed by agarose gel and dissociation curves.
Isolation of the DCT for Patch-Clamp Experiments
We used 8- to 10-wk-old male and female Kir5.1 KO and Kcnj16+/+ (control or WT) mice for dissecting the renal DCT, harvesting renal tissues, and collecting plasma samples. For collecting plasma samples through cardiac puncture, mice were anesthetized with isoflurane inhalation. Mice were then euthanized by CO2 inhalation plus cervical dislocation to harvest the kidneys. After opening the abdomen to expose the left kidney, we perfused the left kidney with 2-mL L-15 medium (Life Technology) containing collagenase type 2 (250 U/mL). The collagenase-perfused kidney was then removed for further dissection. The renal cortex was separated and cut into small pieces for additional incubation in collagenase-containing L-15 media for 30–50 min at 37°C. The tissue was then washed three times with fresh L-15 medium and transferred to an ice-cold chamber for dissection. The isolated tubules were placed on a small coverglass coated with poly-l-lysine, and the coverglass was placed on a chamber mounted on an inverted microscope.
Electrophysiology
All patch-clamp experiments for the measurement of basolateral K+ channel activity were performed in both male and female mice. As we did not find a significant difference, data were pooled.
Single-channel recording.
Single-channel patch-clamp experiments were performed in the basolateral membrane of both the early DCT (DCT1) and late DCT (DCT2). Single K+ channel currents were recorded with an Axon200B amplifier (Axon), low-pass filtered at 1 kHz, and digitized by an Axon interface (Digidata 1332) with a sampling rate of 4 kHz. The pipette solution for the single-channel recording contained (in mM) 140 KCl, 2 MgCl2, 1 EGTA, and 10 HEPES (titrated with KOH to pH 7.4), and the bath solution contained (in mM) 135 NaCl, 5 KCl, 2 MgCl2, 1.8 CaCl2, 5 glucose, and 10 HEPES (titrated with NaOH to pH 7.4). For the calculation of channel numbers, we selected a channel recording that was at least 10-min long and had less than three current levels to determine the close line. We determined the channel open probability (Po) from the channel number (N) and NPo (the product of N and Po), which was calculated from data samples of 60-s duration at the steady state. NPo was determined using the following equation:
where ti is the fractional open time spent at each of the observed current levels. Channel conductance was determined by measuring current amplitudes over several voltages.
Whole cell recording.
We measured whole cell K+ currents and K+ current reversal potentials with an Axon 200A amplifier only in the DCT1. The reasons for conducting experiments only in the DCT1 were 1) basolateral K+ currents in the DCT1 and DCT2 were similar in both WT and Kir5.1 KO mice on different Na+ diets for 7 days and 2) Kir4.1/Kir5.1 is only type of K+ channel expressed in the DCT1 (no renal outer medullary K+ channels in the apical membrane). Accordingly, Ba2+-sensitive K+ currents are equal to Kir4.1/Kir5.1 (in WT mice) or Kir4.1-mediated K+ currents (in Kir5.1 KO mice). To measure K+ current reversal potential, the tip of the pipette was filled with pipette solution containing (in mM) 140 KCl, 2 MgCl2, 1 EGTA, and 10 HEPES (pH 7.4). The pipette was then back filled with pipette solution containing amphotericin B (20 μg/0.1 mL). The bath solution was the same as the solution we used to perform single-channel recordings. For the measurement of whole cell Ba2+-sensitive K+ currents, the bath solution contained (in mM) 140 KCl, 2 MgCl2, 1.8 CaCl2, and 10 HEPES (pH 7.4). After a high resistance seal had been formed, membrane capacitance was monitored until the whole cell patch configuration was formed. DCT membrane capacitance was compensated using the offsetting function of the amplifier, and values to offset membrane capacitance were used as an index of cell surface size. Because variations of mean DCT cell capacitance (around 20 pF) among cells were <1 pF (suggesting that errors of K+ currents/density measurements induced by different cell sizes were <5%) (24), we present the results as currents (pA) per cell rather than pA/pF. We measured whole cell currents twice (before and after 0.1 mM Ba2+), and Ba2+-sensitive K+ currents were obtained by subtracting Ba2+-insensitive currents from total currents. Currents were low-pass filtered at 1 kHz and digitized by an Axon interface with a sampling rate of 4 kHz (Digidata 1440A). Data were analyzed using pClamp software system 9.0 (Axon).
We studied ENaC with perforated whole cell recordings in the split-open DCT tubule [the last 100 µm of the DCT before the start of the connecting tubule (CNT)] using a gap-free protocol because we can continuously monitor amiloride-sensitive Na+ currents (without the interference of Cl− currents). Since the diameter of the DCT2 is normally larger than the CNT (24), this anatomic characterization has been used to determine the end of the DCT or the start of the CNT. However, it was not always obvious to identify the beginning of the CNT in some cases. Thus, it is very likely that some experiments were actually performed in the early CNT. Thus, we have referred that the study was performed in the DCT2/CNT. ENaC currents were determined by adding amiloride (10 µΜ) in the bath solution. The pipette solution contained (in mM) 125 K-gluconate, 15 KCl, 2 MgATP, 1 EGTA, and 10 HEPES (pH 7.4), whereas the bath solution contained 130 Na-gluconate, 10 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, and 5 HEPES (pH 7.4).
Immunoblot Analysis
Protein extract harvested from the renal cortex was homogenized in buffer containing 250 mM sucrose, 50 mM Tris·HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, and 1 mM DTT supplemented with phosphatase and protease inhibitor cocktails (Sigma). Protein (40–60 µg) was separated on a 4%–12% (wt/vol) Tris-glycine gel (Thermo Fisher Scientific) and transferred to a nitrocellulose membrane. Membranes were incubated for 1 h with LI-COR blocking buffer (PBS) and then incubated overnight at 4°C with primary antibodies (Table 1). An Odyssey infrared imaging system (LI-COR) was used to capture the images at wavelengths of 680 or 800 nm.
Table 1.
Antibodies used for Western blots
| Antibody | Species | Dilution for Western Blot | Source |
|---|---|---|---|
| pT53-NCC | Rabbit | 1:3,000 | PhosphoSolutions (30) |
| NCC | Rabbit | 1:2,000 | Millipore (30) |
| ENaCα | Rabbit | 1:1,000 | StressMarq (24) |
| ENaCβ | Rabbit | 1:1,000 | StressMarq (24) |
| ENaCγ | Rabbit | 1:1,000 | StressMarq (24) |
| Kir4.1 (APC-035) | Rabbit | 1:200 | Alomone Labs (7) |
| GAPDH | Rabbit | 1:1,000 | Cell Signaling (15) |
ENaC, epithelial Na+ channel; NCC, Na+-Cl− cotransporter; pT53-NCC, phospho-Thr53 NCC.
Procedures for Modified Renal Clearance
Animals were anesthetized with Inactin at 100 mg/kg by peritoneal injection. Mice were placed on a heated small blanket to maintain body temperature at 37°C. The trachea was cannulated to clear any mucus that may be produced during the experiment. A carotid artery was catheterized with PE-10 tubing for blood collection; a jugular vein was also cannulated for intravenous infusion. The bladder was exposed and catheterized via a suprapubic incision with a 10-cm piece of PE-10 tubing for urine collections. After completion of surgery, isotonic saline was given intravenously for 4 h (0.25–0.3 mL/1 h and total 1.0–1.2 mL of 0.9% saline) to replace surgical fluid losses and to maintain hemodynamics. Urine collections started 1 h after the infusion of 0.3 mL of saline, and six total collections (every 30 min) were performed [two collections before and four collections after hydrochlorothiazide (HCTZ) at 30 mg/kg body wt]. For measuring the basal level of urinary K+ excretion, urine samples were collected every 30 min for six total collections and samples were pooled. Plasma and urine Na+ and K+ concentrations were measured using a dual-channel flame photometer with an internal lithium standard (Cole-Parmer Instrument, Vernon Hills, IL).
Materials
HCTZ and amiloride hydrochloride hydrate were purchased from Sigma-Aldrich (St. Louis, MO) and resolved in saline or NaCl bath solution, respectively. The information regarding antibodies used for Western blots is included in Table 1. Plasma aldosterone was measured by ELISA (Catalog No. ADI-900-173, Enzo Life Sciences) using plasma diluted 1:25 or 1:200.
Statistical Analysis
We used SigmaPlot software to conduct the statistical analysis. For analyzing the values between two groups, we used a t test; for comparisons of the values within the same group, we used a paired t test. We used one-way or two-way ANOVA for analyzing results of more than two groups (significant difference was determined after a Holm–Sidak test as post hoc analysis). P values of <0.05 were considered statistically significant. Data are presented as means ± SE.
RESULTS
We first used single-channel recording to examine the effect of dietary Na+ intake on the basolateral 40-pS K+ channel, the only K+ channel type (Kir4.1/Kir5.1) in the DCT of WT mice, and the basolateral 20-pS K+ channel, the only K+ channel type (Kir4.1 homotetramer) in the DCT of Kir5.1 KO mice (8, 31). Figure 2, A and B, shows recordings made in a cell-attached patch showing the effect of HS, normal sodium (NS), and LS on 40-pS K+ channel activity in WT mice and on the 20-pS K+ channel in Kir5.1 KO mice, respectively. Results from eight patches (DCT tubules in 5 mice) are shown in Fig. 2C, demonstrating that mean NPo (the product of N and Po) in mice on LS (1.96 ± 0.08) was significantly higher than those on NS (1.43 ± 0.07) and HS (1.07 ± 0.08). In contrast, the effect of dietary Na+ intake on the basolateral 20-pS K+ channel was absent in the DCT of Kir5.1 KO mice (HS: 1.90 ± 0.11, NS: 1.95 ± 0.11, and LS: 2.05 ± 0.12). The notion that deletion of Kir5.1 abolishes HS-induced inhibition of Kir4.1 was also confirmed by Western blot analysis showing that HS for 7 days decreased Kir4.1 monomer expression by 25% ± 2% (Fig. 2D). Kir5.1 deletion not only increased the expression of Kir4.1 monomer (155% of WT) but also abolished the effect of HS on Kir4.1 expression (Fig. 2D). However, because the tissues from the renal cortex were used for Western blots, deletion of the Kir5.1-induced increase in Kir4.1 monomer should also be the result of increasing Kir4.1 expression not only in the DCT but also in part of the thick ascending limb and in the cortical collecting duct (32, 33).
Figure 2.

Deletion of Kir5.1 abolishes the effect of dietary Na+ intake on basolateral K+ channel activity in the distal convoluted tubule (DCT). Representative single-channel recordings showing basolateral K+ channel activity in the DCT of wild-type (WT; A) and Kir5.1 knockout (KO) mice (B) on low sodium (LS), normal sodium (NS), and high sodium (HS) for 7 days. Experiments were performed in cell-attached patches with bath solution containing 135 mM NaCl and 5 mM KCl and pipette solution containing 140 mM KCl. The channel closed level is indicated by “C,” and the holding potential was 0 mV. C: two scatterplots showing mean values (left side of each column) and each data point of basolateral K+ channel activity (NPo) in Kir5.1 WT and Kir5.1 KO mice on different Na+ diets. Data from male and female mice are indicated by triangles and circles, respectively. *Significance as determined by one-way ANOVA. D, left: Western blots showing the expression of Kir4.1 monomer (indicated by an arrow) and GAPDH in Kcnj16+/+ mice (control or WT) and Kcnj16−/− mice on NS or HS (7 days), respectively. Results from three male mice on NS or HS for 7 days are summarized in the bar graph (right). *Significant difference as determined by two-way ANOVA.
We then measured Ba2+-sensitive K+ currents of the DCT1 in WT and Kir5.1 KO mice on NS and HS for 7 days using perforated whole cell recordings. Figure 3A shows two sets of traces demonstrating whole cell K+ currents measured with a step protocol (−100 to 60 mV) in the DCT of WT and Kir5.1 KO mice (male and female) on LS, NS, and HS diets for 7 days, respectively. Figure 3B shows a scatterplot summarizing the results of experiments in which K+ currents were measured at −60 mV. Although the deletion of Kir5.1 increased basolateral K+ currents in the DCT (WT: 1,243 ± 40 pA and Kir5.1 KO: 1,990 ± 120 pA), the effect of dietary Na+ intake on the basolateral K+ channel was completely absent in Kir5.1 KO mice (HS: 2,100 ± 95 pA and LS: 2,090 ± 70 pA). In contrast, we confirmed the previous finding that HS decreased (635 ± 25 pA), whereas LS increased (2,060 ± 103 pA), K+ currents in WT mice (1).
Figure 3.

Deletion of Kir5.1 abolishes the effect of dietary Na+ intake on basolateral K+ conductance. A: set of recordings showing Ba2+-sensitive K+ currents measured with the step protocol from −100 to 60 mV in the early distal convoluted tubule (DCT1) of wild-type (WT) and Kir5.1 knockout (KO) mice on low-sodium (LS), normal-sodium (NS), and high-sodium (HS) diets for 7 days, respectively. B: scatter graph summarizing values measured at −60 mV in the DCT1 of WT and Kir5.1 KO mice on different Na+ diets with whole cell recordings. Mean values and SEs are shown on the left of each column. *Significant difference as determined by two-way ANOVA. Data from male and female mice are indicated by triangles and circles, respectively.
The view that Kir5.1 plays a role in mediating dietary Na+ intake on basolateral K+ channels in the DCT was also suggested by measurement of K+ current reversal potentials (an index of membrane potential) in the DCT1 of WT and Kir5.1 KO mice on different Na+ diets. Figure 4, A and B, shows two sets of typical traces of K+ current reversal potentials in the DCT from WT and Kir5.1 KO mice (male and female) on NS, HS (7 days), and LS (7 days), respectively. Figure 4C shows a scatterplot demonstrating the value of each measurement and mean value of eight experiments (tubules). K+ current reversal potentials of the DCT of WT mice on NS was −62.3 ± 1 mV, and it was −71.3 ± 1 mV in the DCT of Kir5.1 KO mice on NS, suggesting that deletion of Kir5.1 hyperpolarized the membrane of the DCT. Moreover, dietary Na+ intake failed to significantly alter K+ current reversal potential of the DCT in Kir5.1 KO mice (HS: −70.8 ± 0.9 mV and LS: −71.0 ± 0.8 mV). In contrast, we confirmed the previous finding that HS decreased (−54 ± 1 mV), whereas LS increased (−72.3 ± 1.2 pA), the negativity of K+ current reversal potential in WT mice (1). These results strongly indicate that Kir5.1 is required for the effect of dietary Na+ intake on basolateral K+ conductance of the DCT and membrane potentials.
Figure 4.

Deletion of Kir5.1 eliminates the effect of dietary Na+ intake on the distal convoluted tubule (DCT) membrane potential. Perforated whole cell recordings showing K+ current (IK) reversal potential in the DCT of wild-type (WT; A) and Kir5.1 knockout (KO) mice (B) on normalsodium (NS), high-sodium (HS), and low-sodium (LS) diets for 7 days, respectively. (Traces in A and B were selected because the IK reversal potentials in these three traces were most close to the mean value.) C: scatterplot summarizing the results of experiments in which IK reversal potentials were measured in WT and Kir5.1 KO mice on different Na+ diets for 7 days. Mean values and SEs are shown on the left of each column. Each data point was obtained from one DCT, and at least four mice were used for each group. Data from male and female mice are indicated by blue triangles and red circles, respectively. *Significant difference as determined by two-way ANOVA.
We next examined the effect of dietary Na+ intake on the expression of phospho-NCC (pNCC) and total NCC in Kir5.1 KO mice. We confirmed that deletion of Kir5.1 increased the expression of pNCC (154% ± 4%) and total NCC (140% ± 3%) compared with WT mice on NS (Fig. 5, A and D). Moreover, neither HS nor LS altered the expression of pNCC and total NCC in Kir5.1 KO mice (Fig. 5, B and E). In contrast, HS (pNCC: 51% ± 3% and total NCC: 66% ± 3%, n = 6) decreased and LS (pNCC: 164% ± 7% and total NCC: 134% ± 3%, n = 6) increased the expression of pNCC and total NCC compared with NS in WT control mice (Fig. 5, C and E).
Figure 5.

Deletion of Kir5.1 abolishes the effect of dietary Na+ intake on total Na+-Cl− cotransporter (tNCC) and phospho-NCC (pNCC). A: immunoblots (IB) showing the expression of pNCC and tNCC in wild-type (WT) control and in Kir5.1 knockout (KO) mice (male) on normal sodium (NS). B: Western blots showing the expression of pNCC and tNCC in Kir5.1 KO mice (male) on high sodium (HS), NS, and low-sodium (LS) diets for 7 days. C: Western blots showing the expression of pNCC and tNCC in WT mice (male) on HS, NS, and LS diets for 7 days. D: bar graph summarizing normalized band intensity of pNCC and tNCC in Kir5.1 WT and Kir5.1 KO mice on NS (6 male mice). Significance was determined by an unpaired t test. E: two bar graphs summarizing normalized band intensity of pNCC and tNCC in Kir5.1 KO mice (6 male) or Kir5.1 WT mice (6 male) on LS (7 days), NS, and HS (7 days), respectively. Significance was determined by one-way ANOVA.
The notion that high Kir4.1 activity/expression in Kir5.1 KO mice may compromise the effect of dietary Na+ intake on NCC activity was also suggested with renal clearance experiments in which we examined HCTZ-induced (30 mg/kg body wt) Na+ excretion as a relative index of NCC activity. Figure 6A shows results from each individual experiment and mean values, and Fig. 6B shows a scatterplot demonstrating the Δ value of HCTZ-induced net Na+ excretion (before and after HCTZ). It was apparent that HCTZ-induced natriuresis in Kir5.1 KO mice on NS (Δ value: 3.42 ± 0.18 from 0.91 ± 0.07 to 4.33 ± 0.20 μeq/min/100 g body wt, n = 8) was significantly larger than in WT mice (1.69 ± 0.16 from 1.14 ± 0.08 to 2.83 ± 0.08 μeq/min/100 g body wt, n = 6). Moreover, LS did not increase Na+ excretion (3.21 ± 0.10 from 0.31 ± 0.04 to 3.62 ± 0.13 μeq/min/100 g body wt, n = 8) nor did HS decrease Na+ excretion (3.27 ± 0.18 from 1.74 ± 0.14 to 5.01 ± 0.19 μeq/min/100 g body wt, n = 8) in Kir5.1 KO mice. In contrast, we confirmed the previous finding that HS decreased (1.12 ± 0.07 from 1.95 ± 0.14 to 3.07 ± 0.13 μeq/min/100 g body wt, n = 4), whereas LS increased (3.23 ± 0.14 from 0.29 ± 0.04 to 3.52 ± 0.2 μeq/min/100 g body wt, n = 5), Na+ excretion in WT mice (1).
Figure 6.

Deletion of Kir5.1 abolishes the effect of dietary Na+ intake on Na+-Cl− cotransporter (NCC) function. A: scatterline graph showing the results of each experiment in which the effect of single-dose hydrochlorothiazide (HCTZ; 30 mg/kg body wt) on urinary Na+ excretion (ENa) within 120 min was measured with the renal clearance method in wild-type (WT) and Kir5.1 knockout (KO) mice (male) on low-sodium (LS), normal-sodium (NS), and high-sodium (HS) diets for 7 days, respectively. *Significant difference as determined by a paired t test. B: scatterplot showing HCTZ-induced net renal Na+ excretion from the above experiments. Mean values and SEs are shown on the left of each column. *Significant difference as determined by two-way ANOVA.
We next used the whole cell recording technique to examine the effect of LS and HS on amiloride-sensitive Na+ currents in the DCT2/CNT of WT and Kir5.1 KO mice (male). Figure 7A shows a set of gap-free recordings showing amiloride-sensitive (10 µM) Na+ currents at −60 mV in the DCT2/CNT measured with the perforated whole cell patch in WT and Kir5.1 KO mice on LS, NS, and HS, respectively. Figure 7B shows a scatterplot summarizing the results of experiments in which amiloride-sensitive Na+ currents were measured at −60 mV with perforated whole cell recordings in the DCT2/CNT of WT and Kir5.1 KO mice on NS, LS (7 days), or HS diets for 7 or 14 days, respectively. HS intake decreased amiloride-sensitive Na+ currents from 213 ± 10 pA (n = 8 tubules from 5 mice) to 168 ± 7 pA (n = 6 tubules from 4 mice, 7 days) and 104 ± 7 pA (n = 6 tubules from 4 mice, 14 days) in the DCT2/CNT of WT mice. Amiloride-sensitive Na+ currents were significantly smaller (93 ± 6 pA, n = 7 tubules from 4 mice) in Kir5.1 KO mice on NS than in WT mice, presumably as a compensatory inhibition of ENaC due to high NCC activity. Moreover, HS intake had a trend to further decrease amiloride-sensitive Na+ currents to 81 ± 7 pA (n = 6 tubules from 4 mice, 7 days) in the DCT2/CNT (not significant). However, HS for 14 days significantly decreased ENaC to 72 ± 5 pA (n = 7 tubules from 4 mice) in the DCT2/CNT of Kir5.1 KO mice. Also, LS did not significantly increase ENaC in the DCT2/CNT of both WT (203 ± 11 pA, n = 6 tubules of 4 mice) and Kir5.1 KO mice (94 ± 8 pA, n = 6 tubules of 4 mice) compared with the corresponding control mice on NS. (Amiloride-induced inhibition of ENaC was fully reversible after washout, although data are not shown here.) The notion that ENaC activity was inhibited in Kir5.1 KO mice was also indicated by Western blot analysis showing that ENaCβ expression was decreased in Kir5.1 KO mice on NS to 80% ± 3% of WT control mice on NS (n = 6). Moreover, HS for 14 days decreased the expression of ENaCβ by 40% ± 3% in WT mice and by 50% ± 3% in Kir5.1 KO mice (Fig. 8). However, the expression of ENaCα and ENaCγ was similar between WT and Kir5.1 KO mice. Moreover, since ENaC expression was examined in the whole renal cortex, the changes in ENaCβ expression should be a reflex of multiple tubule segments from the DCT2, CNT, and cortical collecting duct. The decrease in ENaC activity/expression in Kir5.1 KO mice was possibly due to the inhibition of renin-angiotensin II-aldosterone pathway because plasma aldosterone levels were lower in Kir5.1 KO mice than in WT mice. Figure 9A shows a table demonstrating that the plasma aldosterone level in Kir5.1 KO mice (118 ± 15 pg/mL, n = 4) was significantly lower than in WT mice (293 ± 52 pg/mL, n = 4). Moreover, HS for 14 days further significantly decreases aldosterone concentrations (WT: 134 ± 10 pg/mL and Kir5.1 KO: 70 ± 10 pg/mL).
Figure 7.

High sodium (HS)-induced decrease in epithelial Na+ channel (ENaC) activity in the distal convoluted tubule (DCT) is larger in Kir5.1 knockout (KO) mice than in wild-type (WT) mice. A: set of gap-free recordings showing amiloride-sensitive Na+ currents in the late DCT (DCT2)/connecting tubule (CNT) measured at −60 mV with the perforated whole cell patch in WT and Kir5.1 KO mice (male only) on low sodium (LS; 7 days), normal sodium (NS), and high sodium (HS; 7 days), respectively. B: scatterplot summarizing the results of experiments in which amiloride-sensitive (10 µM) Na+ currents were measured at −60 mV with perforated whole cell recordings in the DCT2/CNT of WT and Kir5.1 KO mice on NS, LS (7 days), or HS diets for 7 or 14 days, respectively. Mean values and SEs are shown on the left of each column. *P < 0.05 and **P < 0.01 as determined by two-way ANOVA.
Figure 8.

Expression of the epithelial Na+ channel (ENaC) in Kir5.1 wild-type (WT) and Kir5.1 knockout (KO) mice on normal sodium (NS) or high sodium (HS). Left: Western blots showing the expression of ENaCα, ENaCβ, and ENaCγ in Kir5.1 WT and Kir5.1 KO mice on NS or HS (14 days), respectively. Right: three bar graphs summarizing the results from six male mice showing the normalized band density of each ENaC subunit. *Significance as determined by two-way ANOVA.
Figure 9.

High sodium (HS) decreases urinary K+ excretion in Kir5.1 knockout (KO) mice. A: aldosterone concentrations in wild-type (WT) and Kir5.1 knockout (KO) mice on normal sodium (NS) and HS (14 days). #Value was significantly different from WT mice on NS; pValue was significantly different from the other groups. B: scatterplot showing the results of each experiment in which the basal level of urinary K+ excretion was measured with the renal clearance method in WT and Kir5.1 KO mice (male) on low sodium (LS; 7 days), NS, and HS (7 and 14 days). C: scatterplot showing each data point and mean value of plasma K+ concentrations in WT mice (top) and Kir5.1 KO mice (bottom) on LS (7 days), NS, and HS diets for 7 or 14 days, respectively. The mean value is shown on the left side of each column. *Significant difference. Data from male and female mice are indicated by triangles and circles, respectively.
Because HS-induced inhibition of ENaC activity/expression was larger in Kir5.1 KO mice than in WT mice, decreased ENaC activity should reduce the driving force of K+ excretion in the late ASDN. To test whether renal K+ excretion was compromised in Kir5.1 KO mice on HS, we used the renal clearance method to examine the basal level of renal K+ secretion in WT and Kir5.1 KO mice on different Na+ diets. Figure 9B shows a scatterplot demonstrating the renal K+ excretion rate in WT and Kir5.1 KO mice on LS (7 days), NS, and HS diets for 7 or 14 days. Dietary Na+ intake had no significant effect on the renal K+ excretion rate in WT mice (NS: 0.36 ± 0.04 μeq/min/100 g body wt, n = 8; 7-day HS: 0.34 ± 0.04 μeq/min/100 g body wt, n = 5; 14-day HS: 0.36 ± 0.03 μeq/min/100 g body wt, n = 5; and 7-day LS: 0.44 ± 0.08 μeq/min/100 g body wt, n = 5). Although the basal level of renal K+ excretion tended to be slightly higher (0.47 ± 0.04 μeq/min/100 g body wt, n = 9) in Kir5.1 KO mice than in WT mice on NS, the difference was not significant. However, HS intake for 7 days (0.16 ± 0.02 μeq/min/100 g body wt, n = 7) or 14 days (0.15 ± 0.02 μeq/min/100 g body wt, n = 7) significantly decreased the basal level of the urinary K+ excretion rate in Kir5.1 KO mice. In contrast, LS intake did not significantly alter the basal level of the renal K+ excretion rate (0.48 ± 0.05 μeq/min/100 g body wt, n = 6) in Kir5.1 KO mice. Consequently, plasma K+ in Kir5.1 KO mice on HS increased from 3.45 ± 0.09 mM (NS) to 3.65 ± 0.15 (HS for 7 days, n = 6, not significant) and to 4.06 ± 0.07 mM (HS for 14 days, n = 7, significant) (Fig. 9C). In contrast, plasma K+ concentrations in WT mice were not significantly different (NS: 3.90 ± 0.11 mM, HS for 7 days: 3.86 ± 0.06 mM, and HS for 14 days: 3.93 ± 0.02 mM, n = 6). Thus, long-term HS intake had no effect on plasma K+ concentrations in control mice but raised plasma K+ in Kir5.1 KO mice to the similar level of Kir5.1 WT mice. However, dietary Na+ intake did not significantly affect plasma Na+ concentration in both WT and Kir5.1 KO mice (Table 2).
Table 2.
Mean values of plasma K+ and Na+ in WT and Kir5.1 KO mice on LS (7 days), NS, and HS diets for 7 or 14 days, respectively
| WT |
Kir5.1 KO |
|||||||
|---|---|---|---|---|---|---|---|---|
| LS (7 days | NS | HS (7 days) | HS (14 days) | LS (7 days) | NS | HS (7 days) | HS (14 days) | |
| Plasma K+, mM | 3.87 ± 0.08 (n = 6) | 3.90 ± 0.11 (n = 6) | 3.86 ± 0.06 (n = 7) | 3.93 ± 0.03 (n = 6) | 3.45 ± 0.09 (n = 6)* | 3.46 ± 0.17 (n = 7)* | 3.65 ± 0.15 (n = 6) | 4.06 ± 0.07 (n = 7)† |
| Plasma Na+, mM | 143.8 ± 1.0 (n = 6) | 141.5 ± 1.0 (n = 6) | 143 ± 1.3 (n = 7) | 142 ± 1.0 (n = 6) | 139.0 ± 1.2 (n = 6)† | 142.1 ± 1.0 (n = 7) | 141.0 ± 0.7 (n = 6) | 143.3 ± 1.0 (n = 7) |
Shown are values for wild-type (WT) and Kir5.1 knockout (KO) mice on low-sodium (LS), normal-sodium (NS), and high-sodium (HS) diets for the indicated times.
Significant difference of the value compared with the corresponding WT control;
significant difference of the value compared with Kir5.1 KO mice on NS.
DISCUSSION
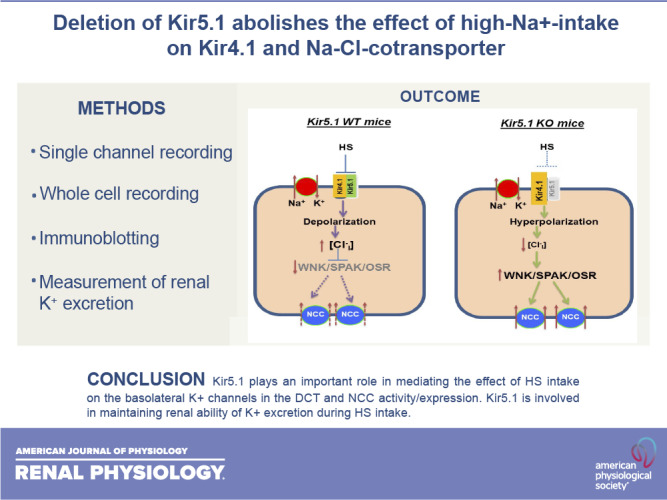
A previous study has demonstrated that dietary Na+ intake regulates basolateral K+ channel activity in the DCT such that HS inhibits, whereas LS stimulates, basolateral Kir4.1/Kir5.1 in the DCT (1). We believe that the renin-angiotensin system should be responsible for the effects of dietary salt intake on Kir4.1/Kir5.1 activity. We speculate that LS intake should activate the renin-angiotensin II signaling pathway and stimulate angiotensin II type 1a receptors in the DCT, thereby stimulating Kir4.1/Kir5.1 activity. Conversely, HS should inhibit the renin-angiotensin II signaling pathway, thereby inhibiting basolateral K+ channel activity in the DCT. One of the main findings of the present study is that the effect of dietary Na+ intake on basolateral K+ conductance in the DCT was absent in Kir5.1 KO mice despite the fact that deletion of Kir5.1 increased Kir4.1 channel activity/expression in the DCT. The basolateral 40-pS K+ channel in the DCT is composed of Kir4.1 and Kir5.1 (8, 34). However, it is well established that Kir5.1 is not able to provide K+ conductance, whereas Kir4.1 protein provides K+ conductance for Kir4.1/Kir5.1 heterotetramer in the kidney (7, 8, 31, 34). This view is indicated by the finding that deletion of Kir4.1 eliminated basolateral K+ conductance in the DCT, although Kir5.1 expression can be detected in the intracellular space of the DCT. A previous study has shown that HS intake inhibits basolateral K+ channels in the DCT. However, the finding that HS failed to depolarize the DCT membrane in Kir5.1 KO mice suggests that Kir5.1 is required for the effect of HK intake on basolateral K+ channels in the DCT. This notion was supported by three lines of evidence: 1) the HS-induced decrease in Kir4.1 expression was absent in Kir5.1 KO mice, 2) neither HS decreased nor LS increased basolateral K+ conductance of the DCT, and 3) the HS-induced decrease (depolarization) or LS-induced increase in the negativity (hyperpolarization) of the DCT membrane was also absent in Kir5.1 KO mice. Because the basolateral K+ channel of the DCT is a Kir4.1 homotetramer in Kir5.1 KO mice, whereas it is a Kir4.1/Kir5.1 heterotetramer in WT mice, this suggests that Kir5.1 is required for HS-induced inhibition of Kir4.1 activity/expression. Since Kir5.1 has been shown to be associated with Nedd4-2 in an in vitro cell model (14), we speculate that Kir5.1 may regulate basolateral K+ conductance in the DCT by participating in Nedd4-2-mediated inhibition of Kir4.1 during increased dietary Na+ intake. The second possibility is that lack of Kir5.1 may enhance Kir4.1 insertion to the plasma membrane. Relevant to this notion is that Palygin et al. (8) have suggested that Kir5.1 is required for Kir4.1/Kir5.1 trafficking.
In addition to regulation of Kir4.1/Kir5.1 in the DCT, dietary Na+ intake also regulates thiazide-sensitive NCC activity (25, 27, 30, 35). Moreover, our previous study has demonstrated that Kir4.1/Kir5.1 plays a key role in mediating dietary Na+ intake on NCC activity (1). We have shown that LS-induced stimulation of NCC was associated with increased Kir4.1/Kir5.1 activity in the DCT, whereas HS-induced inhibition of NCC was associated with decreased Kir4.1/Kir5.1 activity in the DCT. The notion that Kir4.1 plays a role in mediating the effect of dietary Na+ intake on NCC was strongly suggested by the finding that the effect of dietary Na+ intake on NCC activity was largely abolished in kidney-specific (Ks-)Kir4.1 KO mice. Now, we have demonstrated that not only Kir4.1 but also Kir5.1 plays an important role in mediating the effect of dietary Na+ intake on NCC because the effect of dietary Na+ intake on NCC activity/expression was absent in Kir5.1 KO mice. Thus, the lack of a dietary Na+ effect on NCC in Kir5.1 KO mice should be related to the absence of dietary Na+ intake-induced changes in basolateral K+ channel conductance of the DCT. Figure 10 shows a cell scheme illustrating the mechanism by which deletion of Kir5.1 impairs the effect of HS on NCC expression/activity. Deletion of Kir5.1 increases Kir4.1 expression/activity and leads to hyperpolarization of the DCT membrane, which is expected to decrease the intracellular Cl− concentration (36). A decrease in the intracellular Cl− concentration should activate with no lysine kinase (WNK)4/WNK1 (37–39). A large body of evidence has convincingly demonstrated that WNK, STE20-proline-alanine-rich kinase, and oxidative stress-responsive kinase play key roles in stimulating NCC expression and activity (20, 25, 40–44). Thus, compromised HS-induced inhibition of basolateral K+ conductance increases NCC function in Kir5.1 KO mice.
Figure 10.

Cell scheme illustrating the role of Kir5.1 in mediating the effect of high sodium (HS) on basolateral K+ conductance, membrane potential, and Na+-Cl− cotransporter (NCC) in the distal convoluted tubule of wild-type (WT) and Kir5.1 knockout (KO) mice. Solid and dotted lines represent stimulatory and inhibitory effects, respectively. The gray color of font means deletion or inhibition, and larger or smaller font size represents stimulation or inhibition of the activity. WNK/SPAK/OSR, with no lysine kinase/STE20-proline-alanine-rich kinase/oxidative stress-responsive kinase.
Although deletion of Kir5.1 increased NCC activity/expression, we and others have demonstrated that the plasma K+ concentration in Kir5.1 KO mice is only slightly lower than in control WT mice (15, 31). The relative moderate hypokalemia in Kir5.1 KO mice was most likely induced by compromised K+ transport in the proximal tubule. It has been previously reported that Kir5.1 is expressed in the proximal tubule and possibly interacts with Kir4.2 to form basolateral K+ channels in the proximal tubule (11, 13). Deletion of Kir4.2 has been reported to impair membrane transport in the proximal tubule (45, 46). Thus, it is conceivable that deletion of Kir5.1 in the proximal tubule should impair electrolyte reabsorption in the proximal tubule, thereby increasing K+ delivery to the ASDN. Indeed, a previous study has demonstrated that Kir5.1 KO mice had a slightly lower body weight, higher water intake and urine volume, and lower urine osmolality than WT mice (31). Compromised membrane transport in the proximal tubule may explain that the blood pressure in Kir5.1 KO mice or in Kir5.1-deficient Dahl salt-sensitive rats was normotensive under control conditions, although NCC activity is higher in Kir5.1-deficient animals than in WT mice (8, 31). An increase in NCC activity in the DCT of Kir5.1 KO mice should partially offset the defective effect of Kir5.1 on electrolyte transport in the proximal tubule. Indeed, although urinary K+, Ca2+, and Mg2+ excretions were higher in Kir5.1 KO mice than in Kir5.1 WT mice under control conditions, Kir5.1 KO mice have normal plasma Mg2+ and Ca2+ concentrations (31). However, Kir5.1 KO mice have metabolic acidosis and lower plasma K+ concentrations (still in normal plasma K+ ranges) than WT control mice. In contrast, deletion of renal Kir4.1 causes a significant electrolytes imbalance induced by inhibition of NCC (7).
In addition to stimulating NCC activity/function, ENaC activity in the DCT2/CNT and ENaC β-subunit expression are decreased in Kir5.1 KO mice on NS or HS due to a decreased aldosterone level. This should inhibit ENaC-dependent K+ excretion of the ASDN in Kir5.1 KO mice (47). The notion that the renal K+ excretion rate was decreased in Kir5.1 KO mice on HS was supported by the finding that long-term HS raised the plasma K+ level in Kir5.1 KO mice but not in Kir5.1 WT mice. Thus, deletion of Kir5.1 inducing a decrease in ENaC activity in the DCT2/CNT and ASDN should be one of the many factors impairing renal K+ excretion during HS. Further study is required to explore the mechanism by which long-term HS inhibits renal K+ excretion in Kir5.1 KO mice.
In contrast to our observation that HS intake decreased renal K+ excretion and increased the plasma K+ level in Kir5.1 KO mice, Palygin et al. (8) demonstrated that HS increased renal K+ excretion in Kir5.1-deficient Dahl salt-sensitive rats. However, ENaC activity in Kir5.1-deficient Dahl salt-sensitive rats was high not only under NS but also during HS. Thus, HS significantly increased mortality of Kir5.1-deficient Dahl salt-sensitive rats due to severe hypokalemia. Since either increased dietary K+ supplement or inhibition of ENaC could rescue the animals, this suggests that high ENaC activity was responsible for renal K+ wasting in Kir5.1-deficient Dahl salt-sensitive rats. In summary, our data suggest that not only Kir4.1 but also Kir5.1 plays an important role in mediating the effect of HS on NCC and in maintaining normal renal K+ excretion during HS. We conclude that Kir5.1 plays a role in mediating the effect of HS intake on basolateral K+ channels in the DCT and NCC.
GRANTS
The work was supported by National Institutes of Health Grants DK115366 (to D.-H.L.), K08DK110332 (to E. C. Ray), and DK54983 (to W.-H.W.). X.-P.D. is supported by China National Natural Science Foundation (CNSF) Grant 81900648 and by Natural Science Foundation of Jiangsu High Education Institute Grant 19KJB310019. P.W. and Z.-X.G. are supported by CNSF Grants 31971065 and 81900651.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
W.-H.W. and D.-H.L. conceived and designed research; X.-P.D., P.W., D.-D.Z., Z.-X.G., Y.X., E.C.R., W.-H.W., and D.-H.L. performed experiments; X.-P.D., P.W., D.-D.Z., Z.-X.G., Y.X., E.C.R., W.-H.W., and D.-H.L. analyzed data; X.-P.D., P.W., Z.-X.G., Y.X., E.C.R., W.-H.W., and D.-H.L. interpreted results of experiments; X.-P.D., Y.X., W.-H.W., and D.-H.L. prepared figures; W.-H.W., and D.-H.L. drafted manuscript; D.-H.L. edited and revised manuscript; X.-P.D., P.W., D.-D.Z., Z.-X.G., Y.X., W.-H.W., and D.-H.L. approved final version of manuscript.
REFERENCES
- 1.Wu P, Gao Z-X, Su X-T, Wang M-X, Wang W-H, Lin D-H. Kir4.1/Kir5.1 activity is essential for dietary sodium intake-induced modulation of Na-Cl cotransporter. J Am Soc Nephrol 30: 216–227, 2019. doi: 10.1681/ASN.2018080799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol 294: F1398–F1407, 2008. doi: 10.1152/ajprenal.00288.2007. [DOI] [PubMed] [Google Scholar]
- 3.Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A, Teulon J. An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J Physiol 538: 391–404, 2002. doi: 10.1113/jphysiol.2001.012961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su XT, Ellison DH, Wang WH. Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K+ excretion. Am J Physiol Renal Physiol 316: F582–F586, 2019. doi: 10.1152/ajprenal.00412.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaika O, Palygin O, Tomilin V, Mamenko M, Staruschenko A, Pochynyuk O. Insulin and IGF-1 activate Kir4.1/5.1 channels in cortical collecting duct principal cells to control basolateral membrane voltage. Am J Physiol Renal Physiol 310: F311–F321, 2015. doi: 10.1152/ajprenal.00436.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. Am J Physiol Renal Physiol 305: F1277–F1287, 2013. doi: 10.1152/ajprenal.00363.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang C-L, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 28: 1814–1825, 2017. doi: 10.1681/ASN.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Pochynyuk OM, Jacob HJ, Geurts AM, Hodges MR, Staruschenko A. Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight 2: e92331, 2017. doi: 10.1172/jci.insight.92331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang MX, Cuevas-Gallardo C, Su XT, Wu P, Gao Z-X, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium (K+) intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the K+ channel Kir4.1. Kidney Int 93: 893–902, 2018. doi: 10.1016/j.kint.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casamassima M, D'Adamo MC, Pessia M, Tucker SJ. Identification of a heteromeric interaction that influences the rectification, gating, and pH sensitivity of Kir4.1/Kir5.1 potassium channels. J Biol Chem 278: 43533–43540, 2003. doi: 10.1074/jbc.M306596200. [DOI] [PubMed] [Google Scholar]
- 11.Pessia M, Imbrici P, D'Adamo MC, Salvatore L, Tucker SJ. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol 532: 359–367, 2001. doi: 10.1111/j.1469-7793.2001.0359f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J Physiol 525: 587–592, 2000. doi: 10.1111/j.1469-7793.2000.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tucker SJ, Imbrici P, Salvatore L, D'Adamo MC, Pessia M. pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem 275: 16404–16407, 2000. doi: 10.1074/jbc.C000127200. [DOI] [PubMed] [Google Scholar]
- 14.Wang M-X, Su X-T, Wu P, Gao Z-X, Wang W-H, Staub O, Lin D-H. Kir5.1 regulates Nedd4-2-mediated ubiquitination of Kir4.1 in distal nephron. Am J Physiol Renal Physiol 315: F986–F996, 2018. doi: 10.1152/ajprenal.00059.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu P, Gao ZX, Zhang DD, Su XT, Wang WH, Lin DH. Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J Am Soc Nephrol 30: 1425–1438, 2019. doi: 10.1681/ASN.2019010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellison DH, Terker AS, Gamba G. Potassium and its discontents: new insight, new treatments. J Am Soc Nephrol 27: 981–989, 2016. doi: 10.1681/asn.2015070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 18.Ko B, Mistry AC, Hanson L, Mallick R, Cooke LL, Hack BK, Cunningham P, Hoover RS. A new model of the distal convoluted tubule. Am J Physiol Renal Physiol 303: F700–F710, 2012. doi: 10.1152/ajprenal.00139.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko B, Mistry AC, Hanson LN, Mallick R, Hoover RS. Mechanisms of angiotensin II stimulation of NCC are time-dependent in mDCT15 cells. Am J Physiol Renal Physiol 308: F720–F727, 2015. doi: 10.1152/ajprenal.00465.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 21.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 22.Frindt G, Ergonul Z, Palmer LG. Na channel expression and activity in the medullary collecting duct of rat kidney. Am J Physiol Renal Physiol 292: F1190–F1196, 2007. doi: 10.1152/ajprenal.00399.2006. [DOI] [PubMed] [Google Scholar]
- 23.Nesterov V, Dahlmann A, Krueger BK, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol 303: F1289–F1299, 2012. doi: 10.1152/ajprenal.00247.2012. [DOI] [PubMed] [Google Scholar]
- 24.Wu P, Gao Z, Zhang D, Duan X, Terker AS, Lin D, Ellison DH, Wang W. Effect of angiotensin II on ENaC in the distal convoluted tubule and in the cortical collecting duct of mineralocorticoid receptor deficient mice. J Am Heart Assoc 9: e014996, 2020. doi: 10.1161/JAHA.119.014996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S. Dietary salt regulates the phosphorylation of OSR1//SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409, 2008. doi: 10.1038/ki.2008.451. [DOI] [PubMed] [Google Scholar]
- 26.Leviel F, Huebner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hatim H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel AB, Frindt G, Palmer LG. Feedback inhibition of ENaC during acute sodium loading in vivo. Am J Physiol Renal Physiol 304: F222–F232, 2012. doi: 10.1152/ajprenal.00596.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young DB, Jackson TE, Tipayamontri U, Scott RC. Effects of sodium intake on steady-state potassium excretion. Am J Physiol Renal Physiol 246: F772–F778, 1984. doi: 10.1152/ajprenal.1984.246.6.F772. [DOI] [PubMed] [Google Scholar]
- 30.Yang LE, Sandberg MB, Can AD, Pihakaski-Maunsbach K, McDonough AA. Effects of dietary salt on renal Na+ transporter subcellular distribution, abundance, and phosphorylation status. Am J Physiol Renal Physiol 295: F1003–F1016, 2008. doi: 10.1152/ajprenal.90235.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, Sohet F, Eladari D, Houillier P, Lourdel S, Teulon J, Tucker SJ. Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc Natl Acad Sci USA 108: 10361–10366, 2011. doi: 10.1073/pnas.1101400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomilin VN, Zaika O, Subramanya AR, Pochynyuk O. Dietary K+ and Cl− independently regulate basolateral conductance in principal and intercalated cells of the collecting duct. Pflugers Arch 470: 339–353, 2018. doi: 10.1007/s00424-017-2084-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am J Physiol Renal Physiol 308: F1288–F1296, 2015. doi: 10.1152/ajprenal.00687.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang C, Wang L, Zhang J, Su X-T, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 111: 11864–11869, 2014. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frindt G, Yang L, Uchida S, Weinstein AM, Palmer LG. Responses of distal nephron Na+ transporters to acute volume depletion and hyperkalemia. Am J Physiol Renal Physiol 313: F62–F73, 2017. doi: 10.1152/ajprenal.00668.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su XT, Klett NJ, Sharma A, Allen CN, Wang WH, Yang CL, Ellison DH. Distal convoluted tubule Cl− concentration is modulated via K+ channels and transporters. Am J Physiol Renal Physiol 319: F534–F540, 2020. doi: 10.1152/ajprenal.00284.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, Gonzalez-Rodriguez X, Vazquez N, Rodriguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, Garcia-Valdes J, Hadchouel J, Gamba G. The effect of WNK4 on the NaCl-cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26: 1781–1786, 2014. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piala AT, Moon TM, Akella R, He HX, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphosphorylation. Sci Signal 7: ra41, 2014. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terker A-S, Zhang C, McCormick J-A, Lazelle R-A, Zhang C, Meermeier N-P, Siler D-A, Park H-J, Fu Y, Cohen D-M, Weinstein A-M, Wang WH, Yang CL, Ellison D-H. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na:Cl cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2555–2563, 2017. doi: 10.1681/asn.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Hum Mol Genet 20: 855–866, 2011. doi: 10.1093/hmg/ddq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang C-L, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ponce-Coria J, Markadieu N, Austin TM, Flammang L, Rios K, Welling PA, Delpire E. A novel Ste20-related proline/alanine-rich kinase (SPAK)-independent pathway involving calcium-binding protein 39 (Cab39) and serine threonine kinase with no lysine member 4 (WNK4) in the activation of Na-K-Cl cotransporters. J Biol Chem 289: 17680–17688, 2014. doi: 10.1074/jbc.M113.540518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bignon Y, Pinelli L, Frachon N, Lahuna O, Figueres L, Houillier P, Lourdel S, Teulon J, Paulais M. Defective bicarbonate reabsorption in Kir4.2 potassium channel deficient mice impairs acid-base balance and ammonia excretion. Kidney Int 97: 304–315, 2020. doi: 10.1016/j.kint.2019.09.028. [DOI] [PubMed] [Google Scholar]
- 46.Imenez Silva PH, Wagner CA. Potassium channels in control of renal function. Kidney Int 97: 253–255, 2020. doi: 10.1016/j.kint.2019.10.029. [DOI] [PubMed] [Google Scholar]
- 47.Frindt G, Palmer LG. K+ secretion in the rat kidney: Na+ channel-dependent and -independent mechanisms. Am J Physiol Renal Physiol 297: F389–F396, 2009. doi: 10.1152/ajprenal.90528.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]