

Abstract
Background
Multiple myeloma (MM) is a hematologic malignancy characterized by the accumulation of aberrant plasma cells within the bone marrow. The high frequent mutation of family with sequence similarity 46, member C (FAM46C) is closely related with the occurrence and progression of MM. Recently, FAM46C has been identified as a non‐canonical poly(A) polymerase (PAP) that functions as a tumor suppressor in MM. This study aimed to elucidate the structural features of this novel non‐canonical PAP and how MM‐related mutations affect the structural and biochemical properties of FAM46C, eventually advancing our understandings towards FAM46C mutation‐related MM occurrence.
Methods
We purified and crystallized a mammalian FAM46C construct, and solved its structure. Next, we characterized the property of FAM46C as a PAP through a combination of structural analysis, site‐directed mutagenesis and biochemical assays, and by comparison with its homolog FAM46B. Finally, we structurally analyzed MM‐related FAM46C mutations and tested the enzymatic activity of corresponding mutants.
Results
We determined the crystal structure of a mammalian FAM46C protein at 2.35 Å, and confirmed that FAM46C preferentially consumed adenosine triphosphate (ATP) and extended A‐rich RNA substrates. FAM46C showed a weaker PAP activity than its homolog FAM46B, and this difference was largely dependent on the residue variance at particular sites. Of them, residues at positions 77, 290, and 298 of mouse FAM46C were most important for the divergence in enzymatic activity. Among the MM‐associated FAM46C mutants, those residing at the catalytic site (D90G and D90H) or putative RNA‐binding site (I155L, S156F, D182Y, F184L, Y247V, and M270V) showed abolished or compromised PAP activity of FAM46C, while N72A and S248A did not severely affect the PAP activity. FAM46C mutants D90G, D90H, I155L, S156F, F184L, Y247V, and M270V had significantly lower inhibitory effect on apoptosis of RPMI‐8226 cells as compared to wild‐type FAM46C.
Conclusions
FAM46C is a prokaryotic‐like PAP with preference for A‐rich RNA substrates, and showed distinct enzymatic efficiency with its homolog FAM46B. The MM‐related missense mutations of FAM46C lead to various structural and biochemical outcomes to the protein.
Keywords: apoptosis, crystal structure, FAM46C, miRNA, multiple myeloma, poly(A) polymerase, PTEN
Mammalian FAM46C is a unique non‐canonical poly(A) polymerase (PAP) with a bacterial‐like protein fold.
FAM46C differs from its homolog FAM46B in PAP activity due to the divergence of amino acid residues at several critical sites.
The structural‐functional characterization of MM‐related FAM46C mutations that may be used for prognosis in the future.

1. BACKGROUND
Multiple myeloma (MM), also known as Kahler's disease, is a hematological malignancy of mature B‐lymphoid cells [1]. MM is usually beginning with the early premalignant phase called monoclonal gammopathy of undetermined significance followed by an asymptomatic condition termed smoldering MM, then advances to an active MM state and finally proceeds to extramedullary or plasma cell leukemia [2, 3]. The common clinical manifestations of symptomatic MM are anemia, hypercalcemia, osteopenia, and end‐organ damage including renal failure and cardiac dysfunction [1, 4]. Despite the pathogenesis of MM is still incompletely understood, cytogenetic aberrations are recognized as crucial causal driver to MM. The frequent mutations of KRAS, NRAS, BRAF, and TP53 as well as the dysregulation of MYC are identified as important pathogenic factors through genome sequencing, fluorescence in situ hybridization (FISH), and karyotyping analyses of MM patients or tumors and cell lines [5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16].
The family with sequence similarity 46, member C (FAM46C, also termed terminal nucleotidyltransferase 5C [TENT5C]) gene, located at 1p12, is closely associated to the occurrence of MM [15, 17]. FAM46C is found to be frequently mutated (∼3.4%−35%) or deleted (∼19%) in clinical investigations of MM patients [5, 10, 13, 15, 17, 18, 19]. These MM‐related genetic defects in FAM46C are comprised of frame‐shift, missense, and nonsense mutations across the coding sequence, especially in the N‐terminal region [20]. The copy number changes of FAM46C, including hemizygous or homozygous deletion and hyperdiploidy, are closely correlated to the relapse and adverse survival of MM [5, 17, 21]. Moreover, the translocation at 8q24, for which the FAM46C gene is involved, has been identified to yield a super‐enhancer of the proto‐oncogene MYC in the aggressive MM phenotype [7, 22‐24]. In human myeloma cell lines, mutation of FAM46C has been found to promote the resistance to panobinostat, a histone deacetylase inhibitor used in MM treatment [16]. Myeloma cell lines with depletion of FAM46C develop resistance to dexamethasone and lenaidomide [21]. In addition, FAM46C has been reported to be indispensable for erythroid differentiation, and functional deficiency of FAM46C results in anemia in mammalian tissue culture [25].
Recently, FAM46C has been characterized as a non‐canonical poly(A) polymerase (PAP) that functions as a tumor suppressor against B‐lymphocyte lineage originated MM [26]. In an human myeloma cell line model, knockout of FAM46C up‐regulates oncogenic long noncoding RNA (lncRNA) MALAT1 and promotes cell migration in a phosphatidylinositol 3‐kinase (PI3K)‐dependent manner [27]. During plasma cell differentiation, FAM46C is up‐regulated and directly controls antibody production [27, 28]. FAM46C‐knockout (KO) mice have lower immunoglobulin (Ig) level and diminished T‐cell‐independent immune response as compared with wild‐type [28]. The polyadenylation activity of FAM46C is thought to be essential in this process by influencing the poly(A) tail length, hence the steady‐state levels of the mRNAs of Ig [27, 28]. By forming complex with endoplasmic reticulum (ER)‐associated fibronectin type‐III domain‐containing protein A (FNDC3A) and FNDC3B, FAM46C remodels the trafficking and secretion of myeloma cells, which induces ER stress and impairs autophagy, eventually causing apoptosis [29, 30]. Moreover, FAM46C is reported to physically interact with polo‐like kinase 4 (PLK4) and inhibits its kinase activity, thereby suppressing colorectal cancer in a PAP activity‐independent manner [31]. The tumor suppressor function of FAM46C is also implicated in prostatic cancer [32], gastric cancer [33], hepatocellular carcinoma [34], and oral squamous cell carcinoma [35]. Given the close association with MM and other types of cancer, the structural and biochemical features of FAM46C that underlies its cellular function and disease‐related mutations remain largely unknown.
FAM46C is a member of the metazoan‐specific FAM46 family and shares high sequence identity (>50%) with other three paralogs FAM46A, FAM46B, and FAM46D [36]. The FAM46 proteins belong to the nucleotidyltransferase (NTase) superfamily responsible for the addition of nucleotides to a variety of substrates, including bases/nucleic acids, amino acids/peptides, and other biomolecules [37]. NTases all contain a conserved classical fold, characterized by an αβαβαβα topology [37]. We have previously reported the structural and biochemical properties of FAM46B, which resembles prokaryotic PAPs and shows potent in vitro polyadenylation activity on oligo‐A substrates [38]. Compared to FAM46B, FAM46C is a considerably weaker PAP [38]. This research aimed to understand the structural difference that governs the enzymatic divergence of the FAM46 proteins and its implication in the occurrence and progression of MM.
2. MATERIALS AND METHODS
2.1. Protein expression and purification
The cDNAs of Homo sapiens FAM46C (hsFAM46C) and Mus musculus FAM46C (mmFAM46C) were purchased from Youbio Co. (Changsha, Hunan, China). All constructs were individually cloned into a modified pET28 vector with a PreScission protease (PSP) cleavage site (acquired from Prof. Oliver Daumke's lab at Max‐Delbrück‐Center for Molecular Medicine, Berlin, Germany). The primers used here, and others in this study, are listed in Supplementary Table S1. The mutants were generated by site‐directed mutagenesis on the recombinant wild‐type mmFAM46C plasmid. Escherichia coli (E. coli) Rosetta DE3 (AlpaLife, Shenzhen, Guangdong, China) transformed with recombinant plasmids were cultured at 37°C in Terrific Broth (TB) medium to an optic density at 600 nm (OD600) of 0.6, and then induced with 0.1 mmol/L isopropyl‐1‐thio‐β‐D‐thiogalactopyranoside (IPTG, Sigma‐Aldrich, St Louis, MO, USA). After an overnight growth at 18°C, the bacteria were harvested and resuspended in 50 mmol/L HEPES pH 7.5, 500 mmol/L NaCl, 30 mmol/L imidazole, 5 mmol/L MgCl2, 5 mmol/L KCl, 1 μmol/L DNase I, 1 mmol/L phenylmethanesulfonylfluoride (PMSF), and 2 mmol/L β‐mercaptoethanol (β‐ME). The following protein purification steps were all carried out at 4°C. After homogenized by high pressure cell disruption followed by centrifuging at 40,000 × g for 60 min, the supernatant was filtered and applied onto a Ni‐NTA column (GE Healthcare, Pittsburgh, PA, USA) equilibrated with 20 mmol/L HEPES pH 7.5, 500 mmol/L NaCl, 30 mmol/L imidazole, 2 mmol/L MgCl2, 2 mmol/L KCl, and 2 mmol/L β‐ME. The column was extensively washed with the equilibrated buffer and shortly with 20 mmol/L HEPES pH 7.5, 500 mmol/L NaCl, 70 mmol/L imidazole, 2 mmol/L MgCl2, 2 mmol/L KCl, and 2 mmol/L β‐ME. Target proteins were eluted with 20 mmol/L HEPES pH 7.5, 500 mmol/L NaCl, 300 mmol/L imidazole, 2 mmol/L MgCl2, 2 mmol/L KCl, and 2 mmol/L β‐ME. Subsequently, proteins were incubated with glutathione S‐transferase (GST)‐fused PSP to remove the His6‐tag and dialyzed overnight against the dialysis buffer containing 20 mmol/L HEPES pH 7.5, 300 mmol/L NaCl, 2 mmol/L MgCl2, 2 mmol/L KCl, and 2 mmol/L β‐ME. After dialysis, PSP was removed using a GST column (GE Healthcare). The proteins without His6‐tag were further purified using a second Ni‐NTA column (GE Healthcare) as described earlier [38]. The proteins were then concentrated and loaded into a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) equilibrated with the gel filtration buffer containing 20 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, 2 mmol/L MgCl2, 2 mmol/L KCl, and 1 mmol/L dithiothreitol (DTT). Finally, the peak fractions corresponding to monomeric FAM46C constructs were collected, concentrated, and stored at ‐80°C if not used immediately.
2.2. Crystallization and structure determination
Crystallization was performed by vapor‐diffusion method at 18°C by mixing 16 mg/mL mmFAM46C1‐349 with an equal volume of the reservoir solution. Crystals were obtained from the reservoir solution containing 0.2 M sodium citrate tribasic pH 6.5 and 16% (w/v) PEG3350 at 18°C. During flash‐cooling of the crystals in liquid N2, a cryo‐solution containing 0.2 M sodium citrate tribasic pH 6.5, 16% (w/v) PEG3350, and 25% (v/v) glycerol was used. The X‐ray diffraction data sets for native and SeMet‐substituted crystals were collected at beamline BL17U1 of the Shanghai Synchrotron Radiation Facility [39]. Data sets were processed using the XDS program suite [40]. Initial phases were obtained by the single‐anomalous dispersion method using Phaser [41]. An initial model was manually built with COOT [42] and refined with Refmac5 [43] and Phenix [44]. Structural validation was carried out using the MolProbity server (http://molprobity.biochem.duke.edu/). Structural illustrations were prepared using the PyMOL Molecular Graphics Systems (version 2.3.0, Schrödinger LLC, New York, NY, USA; http://www.pymol.org/). The Ramachandran statistics of the final structural model for mmFAM46C1‐349 are as follows: 97.25% in favoured region, 2.75% allowed, and 0% outlier. X‐ray data collection and refinement statistics can be found in Table 1.
TABLE 1.
Crystallographic data collection and refinement
| mmFAM46CΔ # | ||
|---|---|---|
| Data collection | ||
| Data set | Native | SeMet derivative |
| Space group | P212121 | P212121 |
| Unit cell | ||
| a, b, c (Å) | 51.8, 59.2, 173.4 | 52.5, 60.2, 172.9 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.97898 | 0.97898 |
| Resolution (Å) | 41.36−2.35 (2.49−2.35)* | 86.59−2.81 (2.97−2.81) |
| No. reflections | 42201 (6828) | 25369 (4162) |
| R sym | 0.051 (0.504) | 0.078 (0.347) |
| I/σ(I) | 13.33 (2.28) | 11.54 (3.19) |
| Completeness (%) | 98.3 (99.0) | 97.9 (99.1) |
| Redundancy | 3.15 (3.19) | 3.73 (3.85) |
| Refinement | ||
| No. reflections | 22649 (2210) | |
| Resolution (Å) | 25.97−2.35 (2.43−2.35) | |
| R work/R free | 0.218/0.246 | |
| No. atoms | ||
| Protein | 2685 | |
| Ligand/ion | 7 | |
| Water | 24 | |
| B‐factors | ||
| Protein | 75.2 | |
| Ligand/ion | 113.7 | |
| Water | 73.5 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.001 | |
| Bond angles (°) | 0.378 | |
| Ramachandran | ||
| Favored (%) | 97.25 | |
| Outliers (%) | 0 | |
mm denotes Mus Musculus.
Numbers in parentheses represent values from the highest resolution shell.
2.3. NMP incorporation assay
For NMP incorporation, 5 μmol/L protein was incubated with 5 μmol/L 5’‐biotinylated 15‐mer RNA primer (Bioneer, Daejeon, Korea) and 500 μmol/L NTP in a buffer containing 20 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, 2 mmol/L MgCl2, 0.2 mmol/L MnCl2, 2 mmol/L KCl, and 1 mmol/L DTT for 60 min at 37°C. In the assay for checking the PAP activity of FAM46B‐related FAM46C mutants, MnCl2 was not included in the reaction buffer. The mixtures were subjected to a 20% urea‐polyacrylamide gel electrophoresis (PAGE) and the biotinylated RNA substrates and products were visualized using Chemiluminescent Nucleic Acid Detection Module kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer's instructions.
2.4. Surface conservation plot
Protein sequences were downloaded from Uniprot (https://www.uniprot.org/) and NCBI protein database (https://www.ncbi.nlm.nih.gov/protein). All sequences were aligned using ClustalW [45]. The sequences include Homo sapiens FAM46B (hsFAM6B) (UniProt accession: Q96A09) and FAM46C (Q5VWP2), Mus musculus FAM46B (Q8C152) and FAM46C (Q5SSF7), Rattus norvegicus FAM46B (B0BNK8) and FAM46C (Q5XIV0), Bos taurus FAM46B (Q29RH2), Xenopus tropicalis FAM46B (RefSeq: NP_001017206.1), Gallus gallus FAM46C (Q5ZL95) and Danio rerio FAM46C (Q7ZUP1). The alignment result and structure of mmFAM46C were uploaded to the online ConSurf Server (http://conseq.tau.ac.il) [46] to compute conservation scores for the residues. Surface plot of FAM46C with conservation score‐based coloring was generated using PyMOL.
2.5. Structural modeling
Structure coordinates of hsFAM46B were generated by homology modeling using the Xenopus tropicalis FAM46B (xtFAM46B) (Protein Data Bank [PDB] ID: 6JYJ) as the template via SWISS‐MODEL server (http://swissmodel.expasy.org/) [47]. The three‐dimensional structural models of hsFAM46C, each with an MM‐associated mutation, were individually generated using the recently reported hsFAM46C (PDB ID: 6W36) as the template via the SWISS‐MODEL server.
2.6. ATP consumption assay
For ATP consumption, 16 μmol/L xtFAM46B was incubated with 80 μmol/L A15 primer and 80 μmol/L ATP in a buffer containing 20 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, 2 mmol/L MgCl2, and 2 mmol/L KCl at 37°C for 20 min. The mixture was then analyzed by an HPLC system (Agilent, Santa Clara, CA, USA) equipped with a reverse phase C18 ODS‐2 Hypersil analytical column preceded by a C18 guard column (Thermo Scientific), with 100 mmol/L potassium phosphate pH 6.5, 10 mmol/L tetrabutyl ammonium bromide, and 7.5% acetonitrile as running buffer. ATP was detected by absorption at 254 nm and quantified by integration of the corresponding peaks.
2.7. Cell culture
Human MM cell line RPMI‐8226 was purchased from Cell Bank of Shanghai Institute of Cell Biology (Chinese Academy of Medical Science, Shanghai, China). Unless specified, cells were cultivated in special culture medium (CM‐0564, Procell, Wuhan, Hubei, China) and maintained at 37°C in a humidified 5% CO2 incubator. The culture media was half replenished every 48 hours. The cell line is free of mycoplasma.
2.8. Knockout of FAM46C gene
FAM46C gene was knocked out from RPMI‐8226 cells using CRISPR‐Cas9 system. The sgRNA was cloned into a pSpCas9(BB)‐2A‐mCherry(px458) vector from Prof. Jichang Wang's lab at Zhongshan School of Medicine, Sun Yat‐sen University (Guangzhou, Guangdong, China). The recombinant plasmid was amplified in E. coli DH5α (AlpaLife, Shenzhen, Guangdong, China) and transfected into RPMI‐8226 cells with LipofectamineTM 2000 (Invitrogen, Carlsbad, CA, USA) according to manufacturer's instructions. The cells were transferred into 12‐well plates and cultured for 48 hours. RPMI‐8226 cells carrying recombinant pSpCas9(BB)‐2A‐mCherry(px458)‐gRNA FAM46C plasmid were sorted using a fluorescent activity cell sorters (FACS) MoFlo flow cytometer (Beckman, LA, CA, USA) at a wavelength of 561 nm and allocated to 96‐well plates. After 2‐month culturing of these single clones, knockout of FAM46C was examined by Sanger sequencing and Western blotting.
2.9. Western blotting
Cells were lysed on ice in RIPA (Beyotime, Shanghai, China) containing protease inhibitor cocktail (Roche, Basel, Switzerland) and PMSF (Sigma‐Aldrich) for 30 min. Total cell extracts were separated by 15% sodium dodecyl sulfate (SDS)‐PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Burlington, MA, USA). The membranes were blocked with 5% skim milk and probed with the primary antibodies, namely rabbit anti‐FAM46C polyclonal antibody (1:1000, 25038‐1‐AP, Rosemont, IL, USA) and rabbit anti‐GAPDH polyclonal antibody (1:5000, SAB2100894, Sigma‐Aldrich). The blots were then incubated with species‐specific HRP‐conjugated secondary antibodies and visualized by enhanced chemiluminescence (ECL, Tanon, Shanghai, China).
2.10. Tumor biology assays
The cDNA of wild‐type FAM46C was cloned into a pcDNA3.1 vector, and mutants were generated by site‐directed mutagenesis. The recombinant plasmids were individually transfected into FAM46C‐knockout RPMI‐8226 cells. For proliferation assay, 3 × 103 RPMI‐8226 cells per well were planted in 96‐well plates in RPMI‐1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco). At 24, 48, 72, 96, and 120 hours after transfection, cells were collected and treated using Cell Counting Kit‐8 (DOJINDO, Shanghai, China) according to manufacturer's instructions. After 2.5 hours culturing, the cells were applied to a microplate reader (BioTek, Winooski, VT, USA) for examining their viability at a wavelength of 450 nm. For apoptosis assay, cells were washed with cold phosphate‐buffered saline and stained using Annexin V‐APC/PI apoptosis kit (MultiSciences, Hangzhou, Zhejiang, China) according to manufacturer's instructions before analyzed by flow cytometry (cytoFLEX, Beckman). For migration assay, 3.5 × 105 cells per well were planted to upper chamber of a transwell system (8.0 μm pore size, REF 353097, Cell Culture Insert, Thermo Fisher Scientific) containing 250 μL RPMI‐1640 medium. The lower chamber contained 750 μL RPMI‐1640 medium supplemented with 20% FBS. After 24‐hour incubation, the numbers of cells in the lower chamber were determined by flow cytometry.
2.11. Poly(A) tail length determination
Total RNA was extracted using RNA Quick Purification Kit (ES Science, Shanghai, China). The poly(A) tail length of PTEN mRNA was analyzed using the USB® Poly(A) Tail‐length assay kit from Affymetrix (Thermo Fisher Scientific).
2.12. RNA half‐life analysis
FAM46C‐knockout RPMI‐8226 cells transduced with wild‐type FAM46C or mutants were planted into 6‐well plates (0.5 × 106 cells per well). For RNA half‐life determination, cells were treated with 5 μg/mL actinomycin D and collected at indicated time points (1, 2, 3, 6, 12, 24 hours). The total RNA was extracted by miRNeasy Kit (Qiagen, Germantown, MD, USA) and quantified by RT‐PCR using a CFX96 Real‐Time PCR System (Bio‐Rad, Hercules, CA, USA). The mRNA decay and half‐life were calculated as described earlier [48].
2.13. Statistical analysis
Data are presented as mean ±standard deviation (SD) of experimental triplicates, unless specified. Student's t‐test was applied to compare differences among the experimental groups using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA).
3. RESULTS
3.1. Overall structure of FAM46C
Full‐length hsFAM46C and mmFAM46C were recombinantly expressed and purified from E. coli, but these proteins did not yield crystals. We then carried out construct optimization, and obtained crystals of a truncated version of mmFAM46C with deletion of 42 residues from the C‐terminus (Figure 1A). This construct, termed mmFAM46CΔ, showed concentration‐dependent monomer‐dimer equilibrium in the size‐exclusion chromatography (Supplementary Figure S1). We determined a 2.35 Å structure nucleotide‐free mmFAM46CΔ with a R free of 0.246 (Table 1). One mmFAM46CΔ molecule was present in the asymmetric unit of the crystal structure. In the refined structural model, residues 1−9, 123−125 and 344−349 are missing possibly due to disordered configuration. mmFAM46CΔ is composed of an N‐terminal catalytic domain (NCD, residues 10−229) and a C‐terminal helical domain (HD, residues 230−343) (Figure 1A and B). The two domains form a prominent central cleft with a surface area of 1744.6 Å2 and a volume of 1786.1 Å3, which embraces the putative catalytic site (Figure 1C). A conserved salt bridge between Arg175 and Glu238, as well as the hydrogen bond formed by Lys89 and Ser297, are present flanking the central cleft (Figure 1D and E). An extensive hydrophobic network formed by residues from both NCD and HD buttresses the bottom of the central cleft (Figure 1F).
FIGURE 1.
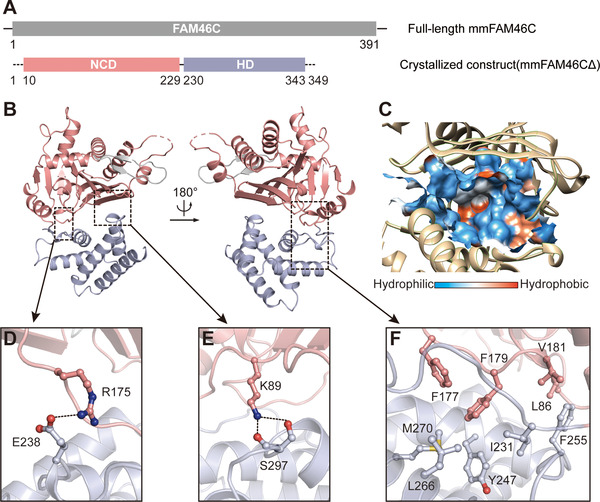
Overall structure of FAM46C. (A) Schematic representation of the domain structure of Mus musculus family with sequence similarity 46, member C (mmFAM46C). The N‐terminal catalytic domain (NCD) and C‐terminal helical domain (HD) are colored in pink and light blue, respectively. The numbers indicate the full‐length and the crystallized constructs of mmFAM46C. (B) The cartoon model of the crystal structure of mmFAM46CΔ. Domains are colored as in panel A. (C) The surface hydrophobicity plot of the central cavity. (D−F) Interactions between NCD and HD. Side chains of involved residues are shown as ball‐and‐stick models in the same color as the domains they belong to. Hydrogen bonds and salt bridge are indicated by the dashed lines.
The NCD consists of an eight‐stranded β‐sheet flanked by seven α‐helices and two FAM46 family‐specific short β‐strands [38] (Figure 2A and B). The classical NTase fold residing in NCD is formed by the secondary structural elements α2, β2, α3, β3, α4, β6, and α6, containing the NTase consensus catalytic motifs Gly73, Ser74, Asp90, Asp92, and Glu166 (Figure 2C) responsible for the coordination and hydrolysis of the nucleotide. The HD is featured by a pile of five α‐helices (Figure 2B and D). These α‐helices are compactly assembled via extensive hydrophobic interactions (Supplementary Figure S2). As FAM46B, FAM46C displays close topological relationship with bacterial PAP and CCA‐adding enzymes (Supplementary Figure S3).
FIGURE 2.
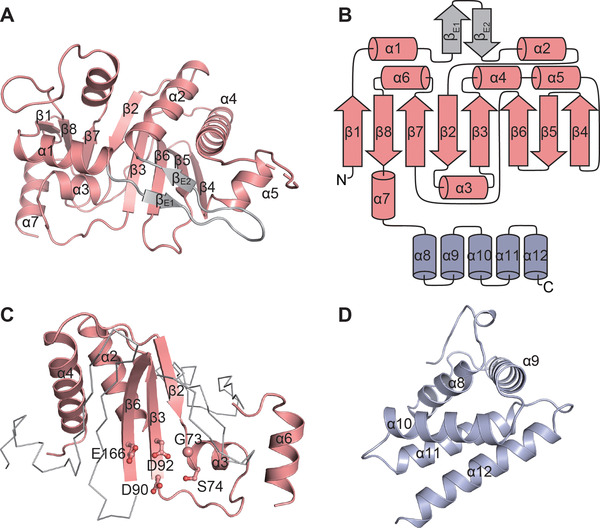
The domains of FAM46C. (A) Overall structure of NCD with secondary elements assigned. (B) The topology diagram of the mmFAM46CΔ. (C) The nucleotidyltransferase (NTase) fold of mmFAM46C. The consensus NTase motif is highlighted as cartoon representation in pink. The conserved key residues are shown as ball‐and‐stick models. (D) Overall structure of HD with secondary elements assigned
3.2. Structural comparison between FAM46C and FAM46B
FAM46C and FAM46B are highly similar in architecture. Structural superposition between mmFAM46C and xtFAM46B yielded a root‐mean‐square deviation (RMSD) of 1.4 Å for aligned Cα atoms (Figure 3A). The structural features mentioned above, including the inter‐ and intra‐domain interactions, are also present in FAM46B [38]. A major difference between the two proteins is found at the linker region between NTD and CD, where residues Gln176 and Phe177 of mmFAM46C show a ∼4 Å reposition relative to their counterparts Gln177 and Phe178 in xtFAM46B (Figure 3B). This conformational divergence leads to the differences in the outline of the central clefts between the two proteins (Figure 3C).
FIGURE 3.
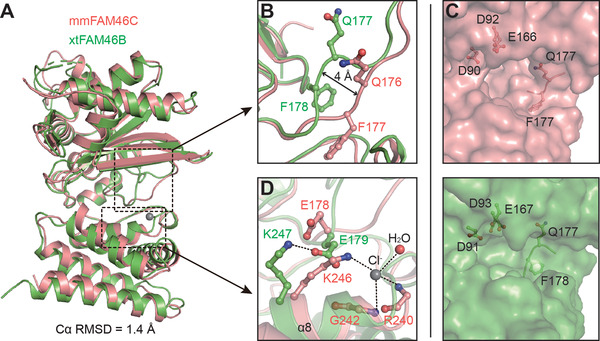
Structure comparison of mmFAM46C and xtFAM46B. (A) mmFAM46C (pink) and Xenopus tropicalis (xt) FAM46B (PDB ID: 6JYJ, green) are superimposed. (B) Difference between mmFAM46C and xtFAM46B at the loop linking NCD and HD. Involved residues are shown as ball‐and‐stick models. (C) Comparison of the central cleft outlines of mmFAM46C and xtFAM46B. (D) mmFAM46C does not possess the inter‐domain salt bridge as in xtFAM46B. The putative chloride ion in the mmFAM46C structure is shown as a gray sphere
In xtFAM46B, Glu179 on the NTD‐HD linker region forms a salt bridge with Lys247, which is a conserved association also found in E. coli PAP I (ecPAP) and Thermotoga maritima CCA‐adding enzyme (tmCCA) [38, 49, 50]. In mmFAM46C, however, this salt bridge is not present between corresponding residues Glu178 and Lys246 in mmFAM46C. The side chains of the catalytic residue Glu178 is differently orientated with regard to xtFAM46B‐Glu179, and Lys246 is directed to a putative chloride ion at the beginning of α8 (Figure 3D).
3.3. The PAP activity of FAM46C
We checked the PAP activity of FAM46C constructs by a nucleotide incorporation assay using 5’ biotinylated A15 oligo. As some non‐classical PAPs are able to add other nucleotides to the RNA substrates, ATP, UTP, CTP, and GTP were tested in parallel in the NMP incorporation assay. For all three constructs, namely mmFAM46C, mmFAM46CΔ, and hsFAM46C, A15 was elongated only in the presence of ATP, but not of other nucleotides (Figure 4A). mmFAM46C and mmFAM46CΔ showed similar polyadenylation efficiency, suggesting that the 42 residues at the C terminus deleted from mmFAM46C are not essential for the PAP activity. In addition, hsFAM46C showed a slightly lower PAP activity than mmFAN46C. From here on, mmFAM46C was used for all the biochemical assays, and thus was referred as FAM46C in the rest of the paper, unless specified.
FIGURE 4.
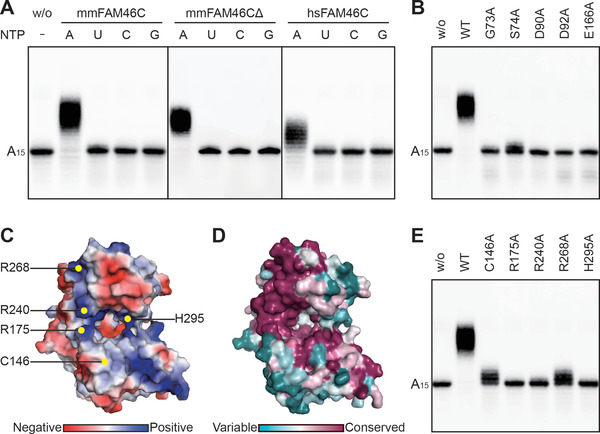
The catalytic features of FAM46C as a PAP. (A) NMP incorporation for various FAM46C constructs in the presence of different nucleotides. (B) AMP incorporation for mmFAM46C mutants on conserved key residues. WT indicates the wild‐type mmFAM46C. (C) Electrostatic potential of mmFAM46CΔ. Putative RNA‐binding sites are indicated. (D) Surface conservation plot of FAM46C within the FAM46 family. (E) AMP incorporation for FAM46C mutants at putative RNA‐binding sites
Next, we verified the residues that are important for the PAP activity of FAM46C. Mutating any of the aforementioned consensus catalytic residues to alanine abolished PAP activity of FAM46C (Figures 2C and 4B). By analyzing the electrostatic potential of FAM46C, we found a positively charged area surrounding the central cleft (Figure 4C), which may be critical for substrate RNA binding. According to the surface conservation plot, this area is relatively conserved in the FAM46 family (Figure 4D). By comparison with corresponding regions in prokaryotic PAP which are suggested to mediate RNA binding, we mutated four positively charged residues (Arg175, Arg240, Arg268, His295) and a cysteine (Cys146) of FAM6C individually to alanine, and tested them in the AMP incorporation assay. These mutants unexceptionally showed compromised PAP activity: FAM46C(R175A), FAM46C(R240A), and FAM46C(H295A) were inactive, whereas FAM46C(C146A) and FAM46C(R268A) only slightly extended the biotinylated A15 primer (Figure 4E). These results indicate that these residues may be involved in substrate RNA binding.
3.4. The substrate preference of FAM46C
Many non‐canonical PAPs can extend the 3’ end of various substrate RNAs with different nucleotides, which underlies their biological functions. For example, mammalian PAP germline development defective‐2 (GLD‐2) was recently shown to be able to adenylate and uridylate RNA oligos with diverse sequences, and this feature is consistent with its cellular role, a RNA‐processing enzyme for both mRNAs and microRNAs (miRNAs) [51]. Another two groups of non‐canonical PAPs, yeast Cid1 and mammalian terminal uridylyltransferase 4/7 (TUT4/7), specifically add uridine to various RNA substrates including mRNAs, miRNAs, or U6 small nuclear RNA [52, 53, 54, 55]. Thus, to understand the function of FAM46C in a physiological and disease‐related context, it would be necessary to investigate its substrate preference. Since FAM46C showed a strict preference for ATP over other NTPs, we tested preference of FAM46C for RNA substrates by AMP incorporation assay. Eight 15‐mer biotinylated RNA primers with different compositions of bases were used in the assay. In terms of the product length, the A15 group was the longest, followed by the U10A5 group and A14U group (Figure 5A). Weak AMP incorporation was also observed for U14A and R14A groups (Figure 5A). These results collectively demonstrated that [1] RNA oligos with an A as the last base at the 3’ end can be extended by FAM46C and [2] RNA oligos ending with U can also be extended by FAM46C, provided that the sequence is A‐rich. In addition, the distance of the A‐rich sequence to the 3’ end is an important determinant for the efficiency of polyadenylation (based on the comparison between the A14U and A10U5 groups). Next, we checked whether FAM46C works on certain miRNA substrates. Let‐7b‐5a, an miRNA with proliferation suppression function in MM [56], was applied in the NMP incorporation assay. The result indicated that FAM46C did not extend let‐7b‐5a in the presence of various nucleotides (Figure 5B). Thus, FAM46C seems to have a substrate specificity on RNA molecules with poly(A) tails.
FIGURE 5.
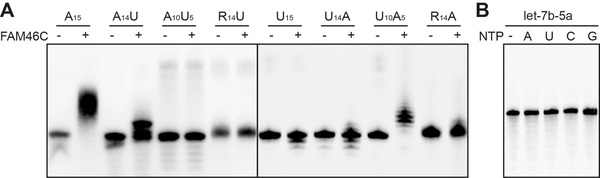
Substrate preference of FAM46C. (A) AMP incorporation assay shows the preference of FAM46C on various 15‐mer biotinylated RNA substrates. (B) FAM46C shows no NTase activity towards let‐7b‐5a
3.5. Structural basis for the enzymatic difference between FAM46C and FAM46B
As evidenced by our previous study, in either Mg2+‐ or Mn2+‐present conditions, FAM46C showed substantially weaker PAP activity as compared to FAM46B [38]. While the two FAM46 proteins share high sequence and structural similarity (Figures 3A and 6), we inferred that the PAP activity difference between the two FAM46 proteins may be relevant to, besides the aforementioned conformational divergence (Figure 3B and C), the variance of individual residues at certain sites involved in the catalytic process. To reveal these sites, we mapped the surface residues inside and around the central cavity that are not conserved between FAM46B and FAM46C, and screened these sites by individually replacing the corresponding residues of FAM46C with their counterparts in FAM46B (Figures 6 and 7A). A total of nine FAM46C mutants were tested in the AMP incorporation assay. Compared with wild‐type FAM46C, mutants FAM46C(G77S), FAM46C(T290R), and FAM46C(D298G) showed increased PAP activities that are comparable to FAM46B (Figure 7B). In particular, the N72H mutation significantly boosted the PAP activity of FAM46C, far exceeding the enzymatic potency of FAM46B (Figure 7B). Consistent with this, the corresponding site on xtFAM46B is an aspartate as for FAM46C, and xtFAM46B also shows extremely weak PAP activity (Figure 6 and Supplementary Figure S4). Overall, these non‐conserved sites may largely dictate the PAP activity difference between FAM46C and FAM46B.
FIGURE 6.

Sequence alignment of FAM46B and FAM46C from various species. The sequences of Homo sapiens (hs) FAM46B (UniProt accession: Q96A09) and FAM46C (Q5VWP2), mmFAM46B (Q8C152) and FAM46C (Q5SSF7), Rattus norvegicus (rn) FAM46B (B0BNK8) and FAM46C (Q5XIV0), Bos taurus (bt) FAM46B (Q29RH2), xtFAM46B (RefSeq: NP_001017206.1), Gallus gallus (gg) FAM46C (Q5ZL95) and Danio rerio (dr) FAM46C (Q7ZUP1) are aligned. α‐helices are shown as cylinders and β‐strands as cylinders and arrows above the sequences. The secondary structure signs are colored as in Figure 2B. Regions not resolved in the crystal structure are indicated by dashed lines
FIGURE 7.
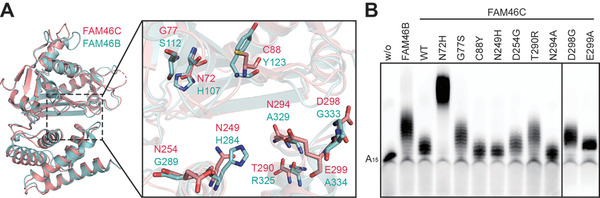
Structural basis for the enzymatic difference between FAM46C and FAM46B. (A) The superimposed crystal structures of mmFAM46C (pink) and modeled hsFAM46B (cyan). The divergent residues at putative RNA‐binding sites are shown as ball‐and‐stick models. (B) The PAP activities of FAM46C‐FAM46B switching mutants regarding the putative RNA‐binding sites. Mutants were based on the mmFAM46C construct, and the target amino acids of the mutated sites are their counterparts in hsFAM46B
3.6. Structural and biochemical investigations of MM‐related FAM46C mutations
Taking advantage of the crystal structure of FAM46C, we plotted 10 MM‐related somatic single‐point mutations in a three‐dimensional landscape. Information of these disease‐related mutations was gleaned from published results and the public database COSMIC (https://cancer.sanger.ac.uk/cosmic), namely N72A, D90G, D90H, I155L, S156F, D182Y, F184L, Y247V, S248A, and M270V (Figure 8A). Among these mutants, D90G and D90H impair the catalytic motif, and thus affect the binding of the nucleotide (Figure 8B). Mutants I155L, F184L, Y247V, and M270V do not alter the polarity of the original side chain, and therefore the interactions of these residues with other surrounding hydrophobic residues are likely to be retained. However, for these four mutants, changes in side chain size or shape might somewhat disturb the overall folding of the FAM46C protein (Figure 8C−F). Ser156 is a buried residue encompassed by a polar environment (Figure 8G). Its mutation to phenylalanine is chemically disfavored with regard to both polarity and size, and may harm the stability of FAM46C. The D182Y mutation may cause a similar problem (Figure 8H). Asn72 is a surface‐exposed residue with no interaction with other residues (Figure 8B), and Ser248 is surrounded by several hydrophobic residues (Figure 8I). Mutation of these two residues to a smaller weak‐polar alanine should not impair the stability of the protein. The positions and chemical properties of these mutants are summarized in Table 2.
FIGURE 8.
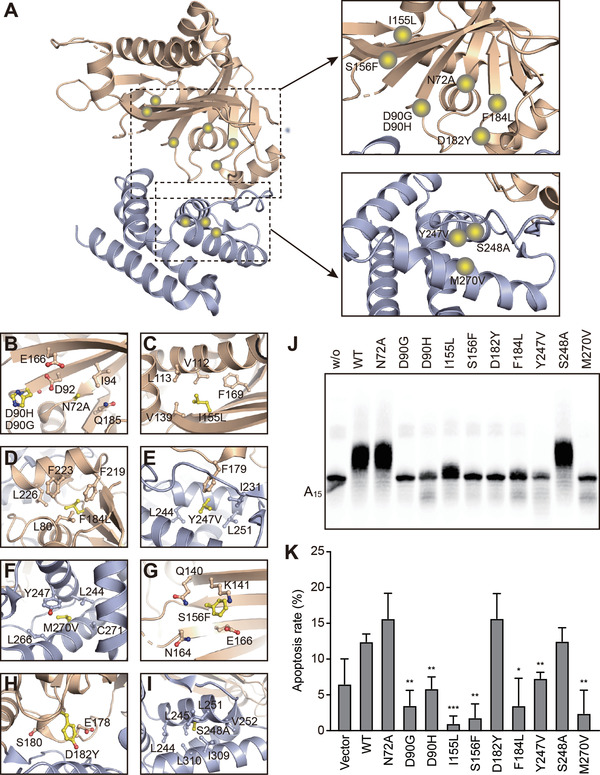
Analysis of selected MM‐associated FAM46C mutations. (A) Overview of the distribution of MM‐associated mutations on hsFAM46C. The NCD and HD are shown as wheat and light blue, respectively. Mutations are indicated by yellow spheres. (B−I) Local environment of the MM‐associated hsFAM46C mutation sites. The mutated residues are shown in yellow, and surrounding residues are colored as the domains they belong to. (J) AMP incorporation assay for MM‐associated FAM46C mutants. Mutants were based on the mmFAM46C construct. (K) Apoptosis rates of FAM46C‐knockout RPMI‐8226 cells transduced with wild‐type FAM46C or mutants. Error bar indicate standard deviation of experimental triplicates. *P < 0.05, **P < 0.01, ***P < 0.001
TABLE 2.
Summary and structural analysis of the MM‐related FAM46C mutations
| Mutant | Position | Polarity before mutation | Polarity after mutation |
|---|---|---|---|
| N72A | Exposed | Polar uncharged | Hydrophobic |
| D90G | Exposed | Negatively charged | Special |
| D90H | Exposed | Negatively charged | Positively charged |
| I155L | Buried | Hydrophobic | Hydrophobic |
| S156F | Buried | Polar uncharged | Hydrophobic |
| D182Y | Exposed | Negatively charged | Hydrophobic |
| F184L | Buried | Hydrophobic | Hydrophobic |
| Y247V | Buried | Hydrophobic | Hydrophobic |
| S248A | Buried | Polar uncharged | Hydrophobic |
| M270V | Buried | Hydrophobic | Hydrophobic |
We tested these mutants in the AMP incorporation assay. As expected, mutants predicted to cause severe problems, namely D90G, D90H, S156F, and D182Y, showed negligible PAP activity, whereas mutants N72A and S248A supposed to be tolerable by FAM46C were equally active as wild‐type (Figure 8J). In addition, the three hydrophobic‐to‐hydrophobic mutants (F184L, Y247V, and M270V) showed diminished PAP activity, suggesting that these mutations did perturb the stability of the FAM46C protein. Next, we checked the tumor suppression activity of these mutants by individually overexpressing them in FAM46C‐knockout RPMI‐8226 cells (Supplementary Figure S5). Except N72A, D182Y, and S248A, other mutants showed reduced activity in inducing cell apoptosis as compared to wild‐type (Figure 8K, Supplementary Figure S6A). Besides D182Y, this result was in agreement with the PAP activity changes of FAM46C caused by these mutations. On the other hand, none of the mutants affected the proliferation or migration of tumor cells (Supplementary Figure S6B‐D). Overall, the different behaviors of these MM‐related mutants in biochemical properties imply a complicated mechanism of FAM46C‐dictated MM onset and progress.
3.7. Effect of FAM46C mutations on PTEN mRNA stability
It was reported recently that loss of FAM46C reduced the activity of phosphatase and tensin homolog (PTEN), thereby activating the PI3K‐protein kinase B (Akt) signaling pathway and promoting the malignancy of MM cells [57]. A similar mechanism of FAM46C in regulating the PTEN/Akt axis was also observed in prostate cancer [32]. To investigate whether FAM46C has a direct impact on the extension of PTEN mRNA, we measured its poly(A) tail length in the presence and absence of FAM46C. For FAM46C‐knockout RPMI‐8226 cells, re‐introduction of FAM46C did not alter the tail length of PTEN mRNA. Likewise, N72A and I155L, the FAM46C mutants either competent or deficient in PAP/tumor suppression activity, also showed no effect (Supplementary Figure S7A). Furthermore, the stability of PTEN mRNA was not significantly affected by wild‐type FAM46C or mutants in FAM46C‐knockout RPMI‐8226 cells (Supplementary Figure S7B and C). These data suggest that the regulation of FAM46C on PTEN is not a consequence of FAM46C‐dependent PTEN mRNA polyadenylation. This is in accordance with the earlier report that FAM46C controlled protein level of PTEN by inhibiting PTEN ubiquitination [32].
4. DISCUSSION
In this study, we have revealed the crystal structure of the metazoan‐specific non‐canonical PAP FAM46C. Like its homolog FAM46B, FAM46C is featured by the topological resemblance with prokaryotic PAPs and CCAs, and a relatively large NTD compared to other eukaryotic PAPs. Given the high structural similarity, substantial difference in PAP activity is observed between FAM46C and FAM46B [38]. The two FAM46 proteins display differences in the local conformation and residues at specific sites that may account for their divergent enzymatic properties. In terms of conformational difference, FAM46B as well as prokaryotic PAPs and CCAs possess an inter‐domain salt bridge that involves one of the consensus catalytic residues, but mmFAM46C lacks it (Figure 3B). However, this salt bridge remains intact in the recently reported hsFAM46C structure (Supplementary Figure S8) [58]. While human and mouse FAM46C showed similar PAP activity [38], it seems that such local conformational variation in the ligand‐free state does not account for PAP efficiency. On the other hand, switching certain residues of FAM46C, namely Gly77, Thr290, and Asn298, to their counterparts in FAM46B promoted its PAP efficiency (Figure 7B), suggesting that the residue variation at these sites are responsible for the in vitro enzymatic activity discrepancy between the two proteins. Interestingly, these sites are not strictly conserved in FAM46B or FAM46C among the species (Figure 6). This implies that FAM46 proteins in different species may have distinct PAP efficiency to meet various biological needs during evolution.
Many eukaryotic non‐canonical PAPs are able to catalyze the 3’ end extension of various RNA substrates. For example, the well‐studied GLD‐2 can polyadenylate mRNAs and adenylate/uridylate miRNAs in Huh7 cells [59, 60, 61, 62, 63]. Although the partners are known to be important in regulating the activity and substrate specificity of PAPs [64], the feature of substrate promiscuity for GLD‐2 seems to be intrinsic according to the in vitro elongation experiment [51]. TUT7, whose catalytic core is structurally similar to GLD‐2, can also work on miRNA with various sequences [54, 55, 65, 66]. With substantial structural difference from GLD‐2, FAM46C and FAM46B seem to preferentially polyadenylate on RNA substrates with poly(A) tails (Figure 4A) [51]. This relatively strict in vitro substrate preference indicates that FAM46 proteins mainly work on mRNA processing. In addition, polyadenylate‐binding protein‐1 (PABP‐1) and BRCA2 and CDKN1A interacting protein (BCCIP) were reported to be a functional partner for FAM46C in SKMM1 and H929 cells [26]. Whether they promote the PAP activity or determine the substrate preference of FAM46C is still unknown. It is important to biochemically investigate the regulatory role of the reported partners on the enzymatic properties of FAM46C. A recent study suggested that FAM46C was specifically expressed during the early development of human and mouse embryos [38]. In humans, FAM46C shows a high relative expression level in the zygote, which maintains steady until the morula stage [67, 68]. This spatiotemporal expression profile, together with its intrinsic PAP activity and subcellular localization, implicates a role of FAM46C in stabilizing cytoplasmic mRNA through extending their poly(A) tails during mammalian early development.
Sequencing data have linked missense mutations of FAM46C with the occurrence and progression of MM [5, 8, 15, 17]. In terms of in vitro PAP activity, the FAM46C constructs each bearing a selected MM‐related mutation displayed quite different phenotypes (Figure 8J). For those mutations that deprive FAM46C of the PAP activity, it is conceivable that the decreased level of polyadenylation on target mRNAs of important tumor suppressors, e.g., Ig antibodies [27], may be a direct consequence related to the occurrence of MM. For mutants FAM46C(N72A) and FAM46C(S248A) that maintain the wild‐type PAP activity, the loss of function in the cellular level might result from the disruption of the interaction between FAM46C and its partners. For the unusual phenotype of mutant FAM46C(D182Y), which lacks PAP in vitro activity but remains active in killing tumor cells, a possible explanation is that the PAP activity is somehow regained from certain FAM46C partners in the cell.
5. CONCLUSIONS
Overall, the mechanism underlying the relationship between different FAM46C missense mutations and MM occurrence/progression is diverse and requires further investigation, which is needed for the development of FAM46C‐based precision clinical approaches against MM.
AVAILABILITY OF DATA AND MATERIALS
The X‐ray crystallographic coordinates and structure factor files for the mmFAM46CΔ structure have been deposited in the Protein Data Bank (PDB) under the accession number 7CQZ. The key raw data have been deposited into the Research Data Deposit (www.researchdata.org.cn), with the approval number of RDDB2021001100.
COMPETING INTERESTS
All authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
S.G. conceived project. J.L.H. crystallized mmFAM46C. J.L.H. and Y.C. collected the X‐ray diffraction data. H.Z., S.L. and S.G. solved the structure. H.Z., J.L.H., Y.T.W., X.Y.M., B.Y. and H.H. performed the biochemical experiments. S.H.Z. did cell biology assays. H.Z., S.H.Z., J.L.H. and S.G. wrote the paper.
Supporting information
Supporting information.
Supplementary Figure S1. Properties and crystals of FAM46C. (A) The size‐exclusion chromatographs of different FAM46C constructs. mmFAM46CΔ that has yielded crystals displays a mixed monomer‐dimer states in solution. (B) SDS‐PAGE analysis of the FAM46C collected and concentrated after the gel filtration. (C) The crystals of mmFAM46CΔ observed under a Leica M80 stereo light microscope.
Supplementary Figure S2. Hydrophobic interactions that stabilize HD. Residues involved in the hydrophobic network interactions are shown as ball‐and‐stick models. The secondary structural elements of HD are labeled.
Supplementary Figure S3. Structural comparison between FAM46C and ecPAP/tmCCA. The structure of mmFAM46CΔ (pink) is individually superimposed with those of ecPAP (PDB ID: 3AQM, marine, left) and tmCCA (PDB ID: 3H39, teal, right). The RMSD values of aligned Cα atoms are indicated.
Supplementary Figure S4. The ATP consumption of xtFAM46B. The ATP consumption of xtFAM46B measured by an HPLC‐based assay is shown. The area of the ATP peak did not significantly change before and after incubation with xtFAM46B, suggesting a weak PAP activity of xtFAM46B.
Supplementary Figure S5. Generation and verification of FAM46C‐knockout cells. (A) Design of sgRNA for knockout of FAM46C. The target position and sequence of the sgRNA are shown. (B) Sanger sequencing of CRISPR‐Cas9 mediated FAM46C‐knockout RPMI‐8226 cells shows successful genome editing. KO1, KO2, and KO3 denote three single clones that were used as parallel experimental groups in the cell biology assays. (C) Western blotting confirms the absence of FAM46C protein from the three clones of FAM46C‐knockout RPMI‐8226 cells.
Supplementary Figure S6. Tumor suppression activity of wild‐type FAM46C and mutants. (A) Representative flow cytometry graphs of apoptosis assay. (B) Proliferation assay for FAM46C‐knockout RPMI‐8226 cells transduced with wild‐type FAM46C or mutants. Error bars indicate standard error of mean of experimental triplicates. n.s., not significant. (C) Migration assay for wild‐type FAM46C and mutants. FAM46C‐knockout RPMI‐8226 cells were transduced with wild‐type FAM46C or mutants. Error bars indicate standard deviation of experimental triplicates. (D) Representative flow cytometry graphs of migration assay. SSC, side scatters; FSC, forward scatter.
Supplementary Figure S7. FAM46C does not affect PTEN mRNA stability. (A) PCR result shows the poly(A) tail lengths of PTEN mRNA in RPMI‐8226 cells transduced with wild‐type FAM46C or mutants. (B) Stability of PTEN mRNA affected by wild‐type FAM46C or mutants. (C) Calculation of RNA half‐life values shown in panel B. Error bars indicate standard error of mean of experimental triplicates.
Supplementary Figure S8. Structural comparison between hsFAM46C and mmFAM46C. Structures of hsFAM46C (PDB ID, 6W36, wheat) and mmFAM46C (pink) are superimposed. The conserved inter‐domain salt bridge formed by Glu178 and Lys246 are specified. The putative chloride ion in the mmFAM46C structure is shown as a gray sphere.
Supplementary Table S1. The primers used in the study
ACKNOWLEDGEMENTS
We thank the staff of the Shanghai Synchrotron Radiation Facility for the help with the collection of crystal diffraction data. This study was supported by the National Key R&D Program of China [2018YFA0508300], National Natural Science Foundation of China [81772977, 31722016, 31470729], Natural Science Foundation of Guangdong Province [2019TX05Y598, 2014TQ01R584, 2014A030312015], and Innovative Team Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory [2018GZR110103002].
Zhang H, Zhang S‐H, Hu J‐L, Wu Y‐T, Ma X‐Y, Chen Y, et al. Structural and functional characterization of multiple myeloma associated cytoplasmic poly(A) polymerase FAM46C. Cancer Commun. 2021;41:615–630. 10.1002/cac2.12163
REFERENCES
- 1. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet North Am Ed. 2015;385(9983):2197‐208. [DOI] [PubMed] [Google Scholar]
- 2. Braggio E, Kortüm KM, Stewart AK. SnapShot: Multiple Myeloma. Cancer Cell. 2015;28(5):678‐e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weaver CJ, Tariman JD. Multiple Myeloma Genomics: A Systematic Review. Semin Oncol Nurs. 2017;33(3):237‐53. [DOI] [PubMed] [Google Scholar]
- 4. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos M‐V, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538‐e48. [DOI] [PubMed] [Google Scholar]
- 5. Boyd KD, Ross FM, Walker BA, Wardell CP, Tapper WJ, Chiecchio L, et al. Mapping of Chromosome 1p Deletions in Myeloma Identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as Being Genes in Regions Associated with Adverse Survival. Clin Cancer Res. 2011;17(24):7776‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Affer M, Chesi M, Chen WG, Keats JJ, Demchenko YN, Roschke AV, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super‐enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014;28(8):1725‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chesi M, Bergsagel PL. Advances in the pathogenesis and diagnosis of multiple myeloma. Int J Lab Hematol. 2015;37(S1):108‐14. [DOI] [PubMed] [Google Scholar]
- 9. Kortüm KM, Langer C, Monge J, Bruins L, Egan JB, Zhu YX, et al. Targeted sequencing using a 47 gene multiple myeloma mutation panel (M3P) in ‐17p high risk disease. Br J Haematol. 2015;168(4):507‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kortüm KM, Langer C, Monge J, Bruins L, Zhu YX, Shi CX, et al. Longitudinal analysis of 25 sequential sample‐pairs using a custom multiple myeloma mutation sequencing panel (M3P). Ann Hematol. 2015;94(7):1205‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li F, Hu L, Xu Y, Li Z, Yi S, Gu Z, et al. Identification of characteristic and prognostic values of chromosome 1p abnormality by multi‐gene fluorescence in situ hybridization in multiple myeloma. Leukemia. 2015;30(5):1197‐201. [DOI] [PubMed] [Google Scholar]
- 12. Fakhri B, Vij R. Clonal Evolution in Multiple Myeloma. Clin Lymphoma Myeloma Leuk. 2016;16(S1):S130‐4. [DOI] [PubMed] [Google Scholar]
- 13. White BS, Lanc I, O'Neal J, Gupta H, Fulton RS, Schmidt H, et al. A multiple myeloma‐specific capture sequencing platform discovers novel translocations and frequent, risk‐associated point mutations in IGLL5. Blood Cancer J. 2018;8(3):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Y, Chen W, Wang J. Progress in the identification of gene mutations involved in multiple myeloma. OncoTargets and therapy. 2019;12:4075‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Y, Chen W, Wang J. Mutations In Thirty Hotspot Genes In Newly Diagnosed Chinese Multiple Myeloma Patients. OncoTargets and therapy. 2019;12:9999‐10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vikova V, Jourdan M, Robert N, Requirand G, Boireau S, Bruyer A, et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics. 2019;9(2):540‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol. 2015;33(33):3911‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ryland GL, Jones K, Chin M, Markham J, Aydogan E, Kankanige Y, et al. Novel genomic findings in multiple myeloma identified through routine diagnostic sequencing. J Clin Pathol. 2018;71(10):895‐9. [DOI] [PubMed] [Google Scholar]
- 19. Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD, et al. A compendium of myeloma‐associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010;116(15):e56‐e65. [DOI] [PubMed] [Google Scholar]
- 20. Barbieri M, Manzoni M, Fabris S, Ciceri G, Todoerti K, Simeon V, et al. Compendium of FAM46C gene mutations in plasma cell dyscrasias. Br J Haematol. 2016;174(4):642‐5. [DOI] [PubMed] [Google Scholar]
- 21. Zhu YX, Shi C‐X, Bruins LA, Jedlowski P, Wang X, Kortüm KM, et al. Loss of FAM46C Promotes Cell Survival in Myeloma. Cancer Res. 2017;77(16):4317‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walker BA, Wardell CP, Brioli A, Boyle E, Kaiser MF, Begum DB, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014;4(3):e191‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kastritis E, Dimopoulos MA. Appraising myc involvement in high risk myeloma. Leuk Lymphoma. 2015;56(3):551‐2. [DOI] [PubMed] [Google Scholar]
- 24. Mikulasova A, Ashby C, Tytarenko RG, Qu P, Rosenthal A, Dent JA, et al. Microhomology‐mediated end joining drives complex rearrangements and overexpression of MYC and PVT1 in multiple myeloma. Haematologica. 2020;105(4):1055‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. M. T. The molecular cloning and characterization of Fam46c RNA stability factor. 2010.
- 26. Mroczek S, Chlebowska J, Kuliński TM, Gewartowska O, Gruchota J, Cysewski D, et al. The non‐canonical poly(A) polymerase FAM46C acts as an onco‐suppressor in multiple myeloma. Nat Commun. 2017;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herrero AB, Quwaider D, Corchete LA, Mateos MV, García‐Sanz R, Gutiérrez NC. FAM46C controls antibody production by the polyadenylation of immunoglobulin mRNAs and inhibits cell migration in multiple myeloma. J Cell Mol Med. 2020;24(7):4171‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bilska A, Kusio‐Kobiałka M, Krawczyk PS, Gewartowska O, Tarkowski B, Kobyłecki K, et al. Immunoglobulin expression and the humoral immune response is regulated by the non‐canonical poly(A) polymerase TENT5C. Nat Commun. 2020;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manfrini N, Mancino M, Miluzio A, Oliveto S, Balestra M, Calamita P, et al. FAM46C and FNDC3A Are Multiple Myeloma Tumor Suppressors That Act in Concert to Impair Clearing of Protein Aggregates and Autophagy. Cancer Res. 2020;80(21):4693‐706. [DOI] [PubMed] [Google Scholar]
- 30. Fucci C, Resnati M, Riva E, Perini T, Ruggieri E, Orfanelli U, et al. The Interaction of the Tumor Suppressor FAM46C with p62 and FNDC3 Proteins Integrates Protein and Secretory Homeostasis. Cell Rep. 2020;32(12):108162. [DOI] [PubMed] [Google Scholar]
- 31. Kazazian K, Haffani Y, Ng D, Lee CMM, Johnston W, Kim M, et al. FAM46C/TENT5C functions as a tumor suppressor through inhibition of Plk4 activity. Communications biology. 2020;3(1):448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma L, He H, Jiang K, Jiang P, He H, Feng S, et al. FAM46C inhibits cell proliferation and cell cycle progression and promotes apoptosis through PTEN/AKT signaling pathway and is associated with chemosensitivity in prostate cancer. Aging. 2020;12(7):6352‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi J, Zhu Q, Wu J, Zhu P. FAM46C suppresses gastric cancer by inhibition of Wnt/beta‐catenin. Frontiers in bioscience (Landmark edition). 2020;25:549‐63. [DOI] [PubMed] [Google Scholar]
- 34. Zhang QY, Yue XQ, Jiang YP, Han T, Xin HL. FAM46C is critical for the anti‐proliferation and pro‐apoptotic effects of norcantharidin in hepatocellular carcinoma cells. Sci Rep. 2017;7(1):396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhuang X, Lu M. The potential functions of FAM46C in oral squamous cell carcinoma. OncoTargets and therapy. 2018;11:8915‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuchta K, Muszewska A, Knizewski L, Steczkiewicz K, Wyrwicz LS, Pawlowski K, et al. FAM46 proteins are novel eukaryotic non‐canonical poly(A) polymerases. Nucleic Acids Res. 2016;44(8):3534‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuchta K, Knizewski L, Wyrwicz LS, Rychlewski L, Ginalski K. Comprehensive classification of nucleotidyltransferase fold proteins: identification of novel families and their representatives in human. Nucleic Acids Res. 2009;37(22):7701‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu JL, Liang H, Zhang H, Yang MZ, Sun W, Zhang P, et al. FAM46B is a prokaryotic‐like cytoplasmic poly(A) polymerase essential in human embryonic stem cells. Nucleic Acids Res. 2020;48(5):2733‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Q‐S, Zhang K‐H, Cui Y, Wang Z‐J, Pan Q‐Y, Liu K, et al. Upgrade of macromolecular crystallography beamline BL17U1 at SSRF. Nucl Sci Tech. 2018;29(5):68. [Google Scholar]
- 40. Kabsch W. Xds. Acta Crystallographica Section D Biological Crystallography. 2010;66(2):125‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(Pt 4):658‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P, Cowtan K. Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126‐32. [DOI] [PubMed] [Google Scholar]
- 43. Murshudov GN, Vagin Aa Fau ‐ Dodson EJ, Dodson EJ. Refinement of macromolecular structures by the maximum‐likelihood method. (0907‐4449 (Print)). [DOI] [PubMed]
- 44. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallographica Section D Biological Crystallography. 2010;66(2):213‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;00(1):2.3.1‐22. [DOI] [PubMed] [Google Scholar]
- 46. Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296‐W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Publisher Correction: Recognition of RNA N(6)‐methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2020;22(10):1288. [DOI] [PubMed] [Google Scholar]
- 49. Toh Y, Takeshita D, Nagaike T, Numata T, Tomita K. Mechanism for the alteration of the substrate specificities of template‐independent RNA polymerases. Structure. 2011;19(2):232‐43. [DOI] [PubMed] [Google Scholar]
- 50. Toh Y, Takeshita D, Numata T, Fukai S, Nureki O, Tomita K. Mechanism for the definition of elongation and termination by the class II CCA‐adding enzyme. EMBO J. 2009;28(21):3353‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ma XY, Zhang H, Feng JX, Hu JL, Yu B, Luo L, et al. Structures of mammalian GLD‐2 proteins reveal molecular basis of their functional diversity in mRNA and microRNA processing. Nucleic Acids Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rissland OS, Mikulasova A, Norbury CJ. Efficient RNA polyuridylation by noncanonical poly(A) polymerases. Mol Cell Biol. 2007;27(10):3612‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Faehnle CR, Walleshauser J, Joshua‐Tor L. Multi‐domain utilization by TUT4 and TUT7 in control of let‐7 biogenesis. Nat Struct Mol Biol. 2017;24(8):658‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lim J, Ha M, Chang H, Kwon SC, Simanshu Dhirendra K, Patel Dinshaw J, et al. Uridylation by TUT4 and TUT7 Marks mRNA for Degradation. Cell. 2014;159(6):1365‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim B, Ha M, Loeff L, Chang H, Simanshu DK, Li S, et al. TUT7 controls the fate of precursor microRNAs by using three different uridylation mechanisms. EMBO J. 2015;34(13):1801‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xu H, Liu C, Zhang Y, Guo X, Liu Z, Luo Z, et al. Let‐7b‐5p regulates proliferation and apoptosis in multiple myeloma by targeting IGF1R. Acta Biochim Biophys Sin. 2014;46(11):965‐72. [DOI] [PubMed] [Google Scholar]
- 57. Kanasugi J, Hanamura I, Ota A, Karnan S, Lam VQ, Mizuno S, et al. Biallelic loss of FAM46C triggers tumor growth with concomitant activation of Akt signaling in multiple myeloma cells. Cancer Sci. 2020;111(5):1663‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen H, Lu D, Shang G, Gao G, Zhang X. Structural and functional analyses of the FAM46C/Plk4 complex. Structure. 2020;28(8):910‐21.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kwak JE, Wang L, Ballantyne S, Kimble J, Wickens M. Mammalian GLD‐2 homologs are poly(A) polymerases. Proc Natl Acad Sci. 2004;101(13):4407‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Katoh T, Sakaguchi Y, Miyauchi K, Suzuki T, Kashiwabara S, Baba T, et al. Selective stabilization of mammalian microRNAs by 3' adenylation mediated by the cytoplasmic poly(A) polymerase GLD‐2. Genes Dev. 2009;23(4):433‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. D'Ambrogio A, Gu W, Udagawa T, Mello CC, Richter JD. Specific miRNA stabilization by Gld2‐catalyzed monoadenylation. Cell Rep. 2012;2(6):1537‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chung CZ, Jo DH, Heinemann IU. Nucleotide specificity of the human terminal nucleotidyltransferase Gld2 (TUT2). RNA. 2016;22(8):1239‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hojo H, Yashiro Y, Noda Y, Ogami K, Yamagishi R, Okada S, et al. The RNA‐binding protein QKI‐7 recruits the poly(A) polymerase GLD‐2 for 3' adenylation and selective stabilization of microRNA‐122. J Biol Chem. 2020;295(2):390‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schmidt MJ, Norbury CJ. Polyadenylation and beyond: emerging roles for noncanonical poly(A) polymerases. Wiley Interdiscip Rev RNA. 2010;1(1):142‐51. [DOI] [PubMed] [Google Scholar]
- 65. Thornton JE, Chang HM, Piskounova E, Gregory RI. Lin28‐mediated control of let‐7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA. 2012;18(10):1875‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Trippe R, Guschina E, Hossbach M, Urlaub H, Luhrmann R, Benecke BJ. Identification, cloning, and functional analysis of the human U6 snRNA‐specific terminal uridylyl transferase. RNA. 2006;12(8):1494‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yan L, Yang M, Guo H, Yang L, Wu J, Li R, et al. Single‐cell RNA‐Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20(9):1131‐9. [DOI] [PubMed] [Google Scholar]
- 68. Dang Y, Yan L, Hu B, Fan X, Ren Y, Li R, et al. Tracing the expression of circular RNAs in human pre‐implantation embryos. Genome Biol. 2016;17(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supplementary Figure S1. Properties and crystals of FAM46C. (A) The size‐exclusion chromatographs of different FAM46C constructs. mmFAM46CΔ that has yielded crystals displays a mixed monomer‐dimer states in solution. (B) SDS‐PAGE analysis of the FAM46C collected and concentrated after the gel filtration. (C) The crystals of mmFAM46CΔ observed under a Leica M80 stereo light microscope.
Supplementary Figure S2. Hydrophobic interactions that stabilize HD. Residues involved in the hydrophobic network interactions are shown as ball‐and‐stick models. The secondary structural elements of HD are labeled.
Supplementary Figure S3. Structural comparison between FAM46C and ecPAP/tmCCA. The structure of mmFAM46CΔ (pink) is individually superimposed with those of ecPAP (PDB ID: 3AQM, marine, left) and tmCCA (PDB ID: 3H39, teal, right). The RMSD values of aligned Cα atoms are indicated.
Supplementary Figure S4. The ATP consumption of xtFAM46B. The ATP consumption of xtFAM46B measured by an HPLC‐based assay is shown. The area of the ATP peak did not significantly change before and after incubation with xtFAM46B, suggesting a weak PAP activity of xtFAM46B.
Supplementary Figure S5. Generation and verification of FAM46C‐knockout cells. (A) Design of sgRNA for knockout of FAM46C. The target position and sequence of the sgRNA are shown. (B) Sanger sequencing of CRISPR‐Cas9 mediated FAM46C‐knockout RPMI‐8226 cells shows successful genome editing. KO1, KO2, and KO3 denote three single clones that were used as parallel experimental groups in the cell biology assays. (C) Western blotting confirms the absence of FAM46C protein from the three clones of FAM46C‐knockout RPMI‐8226 cells.
Supplementary Figure S6. Tumor suppression activity of wild‐type FAM46C and mutants. (A) Representative flow cytometry graphs of apoptosis assay. (B) Proliferation assay for FAM46C‐knockout RPMI‐8226 cells transduced with wild‐type FAM46C or mutants. Error bars indicate standard error of mean of experimental triplicates. n.s., not significant. (C) Migration assay for wild‐type FAM46C and mutants. FAM46C‐knockout RPMI‐8226 cells were transduced with wild‐type FAM46C or mutants. Error bars indicate standard deviation of experimental triplicates. (D) Representative flow cytometry graphs of migration assay. SSC, side scatters; FSC, forward scatter.
Supplementary Figure S7. FAM46C does not affect PTEN mRNA stability. (A) PCR result shows the poly(A) tail lengths of PTEN mRNA in RPMI‐8226 cells transduced with wild‐type FAM46C or mutants. (B) Stability of PTEN mRNA affected by wild‐type FAM46C or mutants. (C) Calculation of RNA half‐life values shown in panel B. Error bars indicate standard error of mean of experimental triplicates.
Supplementary Figure S8. Structural comparison between hsFAM46C and mmFAM46C. Structures of hsFAM46C (PDB ID, 6W36, wheat) and mmFAM46C (pink) are superimposed. The conserved inter‐domain salt bridge formed by Glu178 and Lys246 are specified. The putative chloride ion in the mmFAM46C structure is shown as a gray sphere.
Supplementary Table S1. The primers used in the study
Data Availability Statement
The X‐ray crystallographic coordinates and structure factor files for the mmFAM46CΔ structure have been deposited in the Protein Data Bank (PDB) under the accession number 7CQZ. The key raw data have been deposited into the Research Data Deposit (www.researchdata.org.cn), with the approval number of RDDB2021001100.