

Abstract
The lysosomal degradation of endoplasmic reticulum (ER) fragments by autophagy, called ER-phagy or reticulophagy, occurs under normal as well as stress conditions. The recent discovery of multiple ER-phagy receptors has stimulated studies on the roles of ER-phagy. Here, we discuss how the ER-phagy receptors and the cellular components that work with these receptors mediate two important functions: ER homeostasis and ER quality control. We highlight that ER-phagy plays an important role in alleviating ER expansion induced by ER stress and acts as an alternate disposal pathway for misfolded proteins. We suggest that the latter function explains the emerging connection between ER-phagy and disease. Additional ER-phagy-associated functions and important unanswered questions are also discussed.
Keywords: endoplasmic reticulum, reticulophagy, autophagy receptor, macro-ER-phagy, micro-ER-phagy, proteostasis, human diseases
Autophagic turnover of ER fragments is mediated by ER-phagy receptors.
The endoplasmic reticulum (ER), which forms a dynamic contiguous network of interconnected flat sheets and curved tubules, serves as a protein biogenesis hub, accommodating approximately one-third of the proteome of eukaryotes (1). However, errors in protein folding, post-translational modifications, and protein complex assembly can occur and impair ER homeostasis, or “ER proteostasis” (2). During these conditions, the unfolded protein response (UPR; see Glossary) is induced and aberrant proteins are retrotranslocated into the cytoplasm via the ER Associated Degradation (ERAD) machinery and disposed of by the proteasome (2). Nevertheless, not every damaged and potentially toxic protein is degraded by ERAD, and not every misfolded protein induces the UPR (3, 4). Some proteins are targeted for degradation via alternate ER disposal pathways, such as ER-phagy, which degrade specific parts of the ER by autophagy (5–7). While ER-phagy is one component of a starvation response, recent studies have linked ER-phagy to ER quality control and other functions (5–7). The growing links between ER-phagy and homeostasis have prompted us to propose that ER-phagy pathways play a critical role in human disease. In support of this proposal, mutations in genes encoding ER-phagy components have been linked to diabetes and certain neurological disorders (6, 8).
ER-phagy employs autophagy receptors that link ER domains to the autophagy machinery (5–7). ER-phagy can be non-autophagosome-mediated (micro-ER-phagy) or autophagosome mediated (macro-ER-phagy) (5–7). Micro-ER-phagy is characterized as the piecemeal engulfment of ER fragments directly by endosomes and/or the lysosome (Figure 1). A role for the membrane remodeling complex, ESCRT-III, was recently described in this process (9). In contrast, macro-ER-phagy involves the sequestration of ER fragments into double-membrane vesicles called autophagosomes, which arise from a precursor cisterna known as the phagophore. Autophagosomes then deliver their contents to lysosomes/vacuoles for degradation (Figure 1).
Figure 1. ER-phagy uses autophagy receptors to package ER into autophagosomes.
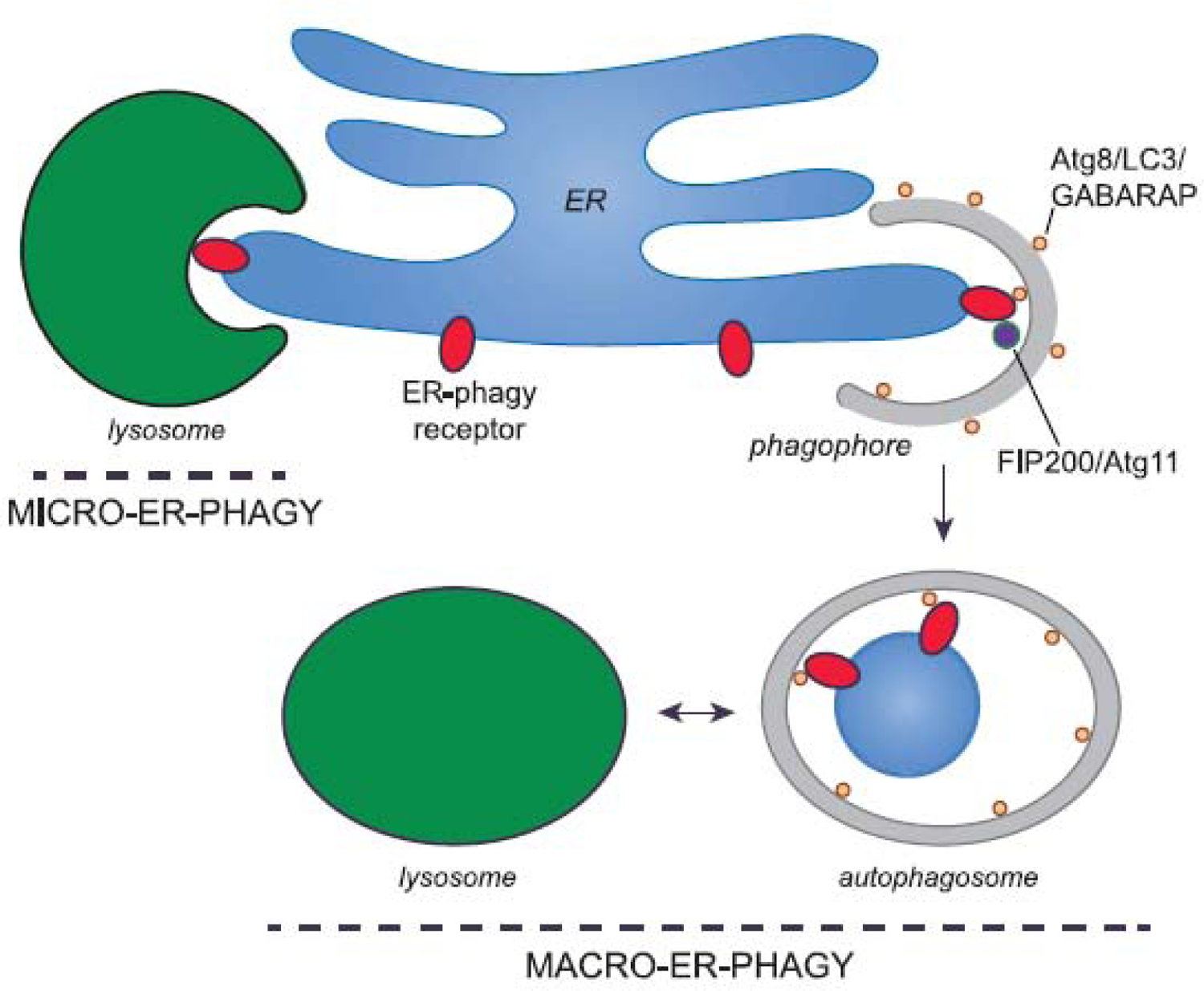
During macro-ER-phagy, ER-phagy receptors bind Atg8 in yeast, or LC3 and GABARAP proteins in mammals (tan spheres), to package ER fragments into a phagophore (grey). Some of the ER-phagy receptors also bind the autophagy-inducing ULK complex via its subunit FIP200 (or Atg11 in yeast) to coordinate the autophagy machinery with cargo sequestration. The phagophore expands and then seals to form an autophagosome that delivers ER fragments to the vacuole (yeast) or lysosome (mammals; green) for degradation. During micro-ERphagy, ER fragments are directly engulfed by lysosomes/vacuoles. While in some cases this process involves ER-phagy receptors and LC3 or GABARAP proteins (e.g. during recovER-phagy (9)), micro-ER-phagy in general does not appear to require these factors (e.g. during micro-ER-phagy induced by tunicamycin treatment in yeast (41)).
In this Opinion article, we largely focus on macro-ER-phagy and how it functions in the context of ER homeostasis and quality control. We discuss how some ER-phagy receptors act on several different pathways, and suggest that some of these pathways connect ER-phagy machinery to homeostasis and human disease. We propose that ER-phagy degrades ERAD-resistant forms of ERAD substrates. This may require a multiplicity of receptors to recognize a spectrum of misfolded proteins. Examples of ERAD-resistant misfolded proteins that are degraded by ER-phagy include mutant forms of procollagen and proinsulin discussed below (10, 11). Recent studies have also revealed that an ER-phagy receptor may act on more than one pathway (5–7). We also propose that the interaction of an ER-phagy receptor with a specific binding partner may define the pathway in which the receptor functions. The contribution of ER-phagy to ER function, and other open questions in the field, are also discussed.
ER-phagy receptors
The identification of ER-phagy receptors has accelerated progress in uncovering the many roles of ER-phagy. ER-phagy receptors are either resident ER membrane proteins or, less commonly, cytosolic proteins. ER-phagy receptors connect domains of ER sheets or tubules to the autophagosome biogenesis machinery by binding members of the ubiquitin-like Atg8 protein family, which consists of the LC3 and GABARAP proteins in mammals (Figure 1) (5–7). ER-phagy receptors bind to Atg8 proteins via an LC3-interacting region (LIR) motif, also known as an Atg8-interacting motif (AIM) in yeast (5–7). Some ER-phagy receptors also bind FIP200 (Atg11 in yeast), a component of the autophagy-inducing ULK complex (12–15). In this section, we will introduce the known ER-phagy receptors and then discuss their contribution to ER homeostasis and ER quality control. We suggest that compromised ER homeostasis in individuals with mutations in genes encoding receptors or other components of the ER-phagy machinery contributes to disease.
To date, there are two known resident ER membrane receptors in Saccharomyces cerevisiae (Atg39 and Atg40) and six in mammalian cells (FAM134B, RTN3L SEC62, CCPG1, ATL3, and TEX264) (Figures 2–4) (15–22). Each ER-phagy receptor principally resides in a unique domain of the ER (Figure 2) and/or tissue, and responds to a variety of cell stresses that include starvation, misfolded protein accumulation, and imbalances in lumenal calcium (5–7). For example, SEC62 mediates ER-phagy in flat ER sheets, whereas FAM134B functions on the curved edges of the sheets (Figure 2). Additionally, RTN3L and ATL3 carry out ER-phagy on ER tubules (Figure 2). RTN3L localizes to tubules, while ATL3 is found at tubule junctions (17–19, 22). Some receptors, like FAM134B, have isoforms (FAM134A and FAM134C) that also bind LC3 (17). Moreover, RTN3L and ATL3 may possess redundant functions as RTN3L overexpression was reported to suppress the ER-phagy defect in ATL3-depleted cells (22). ATL1, ATL2, and ATL3 were all reported to substitute for each other during ER-phagy (23). The ATL proteins have been proposed to remodel the ER to facilitate fragmentation during autophagosomal engulfment (23), but this role appears to be unrelated to the adaptor function of ATL3 (22).
Figure 2. ER-phagy receptors reside in different ER subdomains.
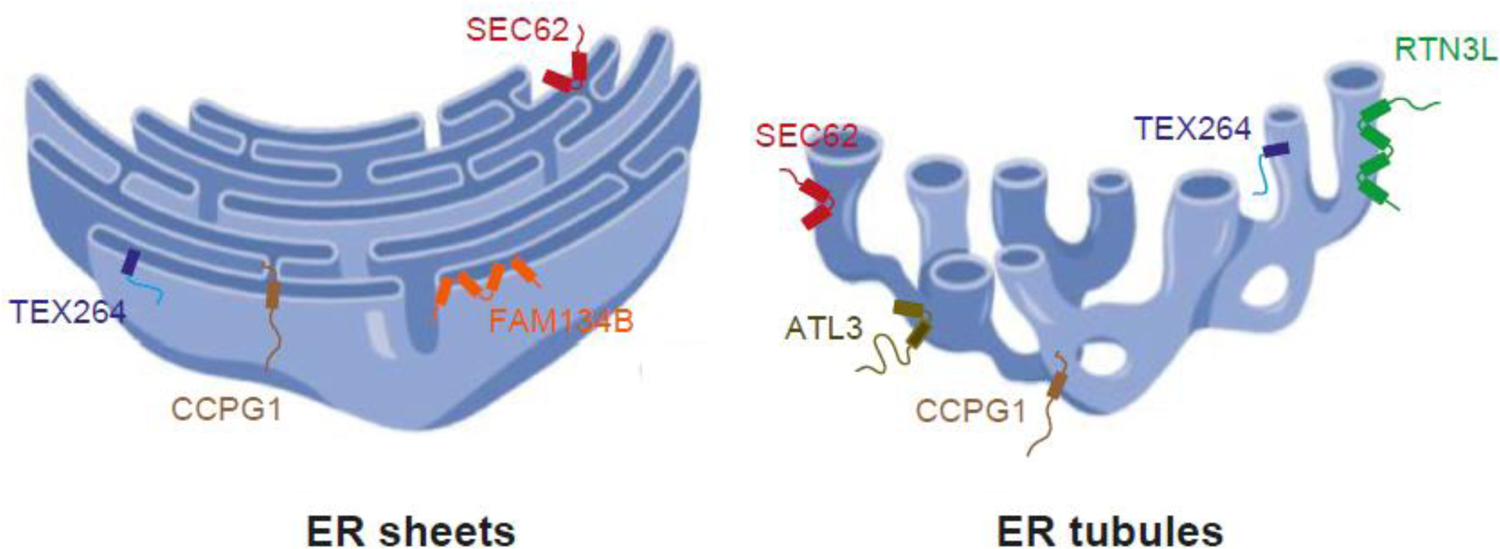
A cross section of an area of sheets (left) and tubules (right) is shown to illustrate the localization of the 6 known mammalian ER membrane ER-phagy receptors. RTN3L localizes to tubules, ATL3 is present at the three-way junctions of the ER tubules, and FAM134B resides on the curved edges of the sheets. The other receptors, i.e., SEC62, CCPG1 and TEX264, are found on flat ER sheets and tubules. Figure created in Biorender.
Figure 4. Macro-ER-phagy in yeast.

Yeast Atg40 has a domain structure similar to mammalian FAM134B, yet it localizes to the tubular ER, like RTN3L. In yeast, the sheets and tubules in the cytoplasm, and at the cell cortex, are referred to as the cortical ER. Atg40 is largely present on the cortical ER, while Atg39 is principally localized in the nuclear envelope. Atg40, which contains a reticulon-like domain, recruits Atg8 via a LIR motif to initiate cortical macro-ER-phagy. Autophagosome-driven fragmentation of the cortical ER occurs at ER-phagy sites (ERPHS) with the aid of the Lst1-Sec23 complex. In contrast, transport vesicles bud from the ER at ER exit sites (ERES), where the COPII coat complex is assembled. Soluble secretory cargo proteins are loaded into vesicles via a receptor-mediated process, while transmembrane cargo proteins interact with the COPII coat subunits or specific adaptors.
In addition to the membrane ER-phagy receptors, two soluble mammalian proteins, p62 and CALCOCO1, were identified as ER-phagy receptors, and one soluble receptor, Epr1, was recently reported in Schizosaccharomyces pombe (24–27). Another soluble receptor, C53, was also recently identified in plants and mammals (28). Interestingly, C53 is recruited to autophagosomes when the ER is stressed. It binds to Atg8 family members via a “shuffled” recognition motif, and is a component of an ER membrane complex that includes an enzyme required for substrate UFMylation, a ubiquitin-like modification that precludes Atg8 binding. Recent reports have confirmed that UFMylation may be a key regulator of some ER-phagy pathways (29). In contrast, p62 appears to promote the degradation of damaged ER sub-domains marked by ubiquitin since it acts with the E3 ligase, TRIM13, to elicit N-degron ubiquitin-dependent ER-phagy (24) (also see below).
As mentioned above, although ER-phagy is commonly induced by amino acid starvation or drugs such as rapamycin or Torin which mimic starvation, ER-phagy can also be induced by other cellular and environmental cues, such as the UPR (5–7). For example, some receptors like TEX264 largely induce macro-ER-phagy by acting in response to nutrient deprivation, while SEC62 (see below) only triggers micro-ER-phagy in response to the resolution of the UPR (19–21). Interestingly, the compound Loperamide, which upregulates the transcription factor ATF4 and other ER stress markers in glioblastoma cells, induces FAM134B-dependent macro-ER-phagy, and to a lesser extent TEX264-dependent ER-phagy (30). Although the precise mechanism by which ER-phagy receptors are activated is largely unknown, their enhanced expression and oligomerization appears to be part of the mechanism by which they are activated (16, 20, 31, 32) (also see below).
ER-phagy, nutrient deprivation, and ER fragmenting activity
Amino acid starvation inhibits the TORC1 kinase, which induces autophagosome biogenesis by de-repressing the ULK complex (33). The response of FAM134B, RTN3L, ATL3, CCPG1, and TEX264 to starvation is consistent with the observation that these receptors mediate macro-ER-phagy pathways that employ autophagosomes (5–7).
TEX264, which resides in ER sheets and tubules (Figure 2), facilitates ~50% of the observed autophagic flux during amino acid starvation (Figure 3) (20, 21). Under nutrient rich conditions, TEX264 is found on punctate structures on the ER membrane that enlarge during starvation. These enlarged structures colocalize with LC3 and other ATG marker proteins. TEX264 associates with LC3 at three-way junctions of ER tubules, suggesting it is packaged into autophagosomes at these locations. Interestingly, TEX264 contains a gyrase inhibitor-like domain and a long intrinsically disordered region (IDR) that is required for its function as an ER-phagy receptor. The IDR, which bridges the ER to the growing autophagosomal membrane, is present in most of the ER-phagy receptors (5). Unexpectedly, recent studies revealed a role for TEX264 that appears to be independent of its activity as an LC3-binding protein and ER-phagy receptor (34). In combination with the ATPase p97 and SPRTN, TEX264 localizes to the nuclear envelope to repair DNA (34).
Figure 3. Cellular roles of ER-phagy and the contributions of metazoan ER-phagy receptors.
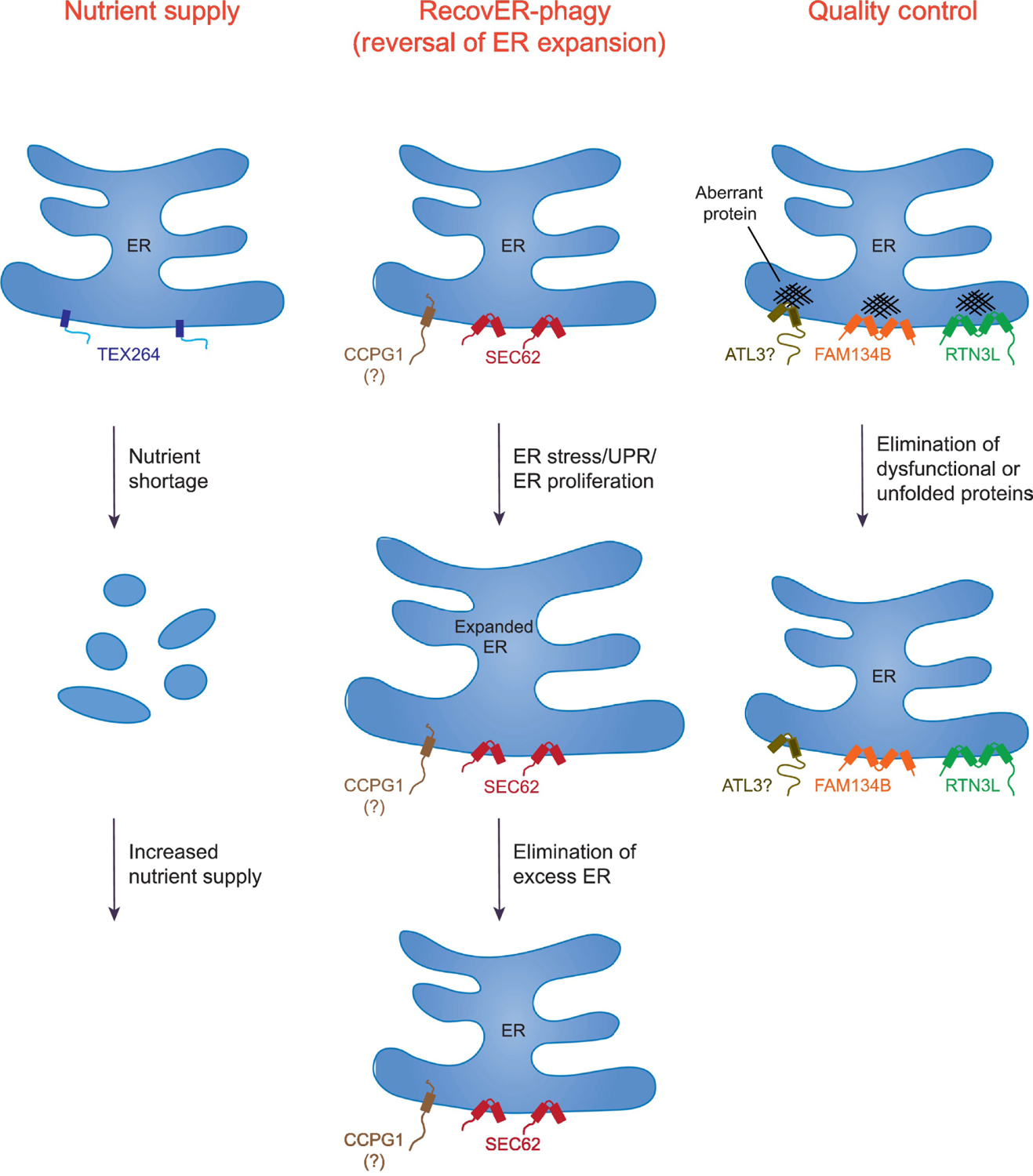
Shown are the general pathways in which ER-phagy is utilized and the contributing receptors. These pathways include: Nutrient Supply (TEX264), RecovER-phagy (Reversal of ER Expansion) (SEC62 and possibly CCPG1), and Quality Control (FAM134B, RTN3L, and possibly ATL3). Not all the listed pathways are limited to the indicated ER-phagy receptors. For example, while TEX264 contributes to ~50% of autophagic flux during amino acid deprivation, other ER receptors, such as FAM134B, can also participate in starvation-induced ER-phagy, albeit far less efficiently. Note that a significant fraction of the ER is fragmented in the Nutrient Supply pathway to illustrate that TEX264 promotes degradation of a large portion of this organelle during amino acid starvation. SEC62 mediates RecovER-phagy. Although CCPG1 function appears to be regulated by the unfolded protein response (UPR), it remains to be determined whether it also definitively participates in RecovER-phagy. In Quality Control, FAM134B facilitates the removal and degradation of misfolded procollagen (shown) and disease-causing mutant NPC1, whereas misfolded pro-insulin (Akita), proopiomelanocortin (POMC) and arginine vasopressin (pro)-AVP utilize RTN3L. There are no known misfolded proteins that specifically utilize ATL3. Also note that FAM134B and RTN3L are inserted into the lipid bilayer via reticulon domains, whereas the other receptors shown possess bona fide transmembrane segments.
During ER-phagy, a portion of the ER must fragment. While TEX264 lacks ER fragmenting activity, the overexpression of FAM134B and RTN3L drives ER fragmentation, as does the multimerization of RTN3L (17, 18). FAM134B and RTN3L, as well as the closely related yeast Atg40 protein, contain reticulon homology domains (RHDs) (16–18). RHDs consist of two hairpin-like domains that each possess 28–36 hydrophobic amino acids separated by a stretch of 60–70 hydrophilic amino acids (1). The RHDs generate membrane curvature by inserting into the cytoplasmic surface of the membrane. FAM134B also contains two amphiphatic helices that may contribute to membrane curvature (35). The other mammalian ER membrane receptors lack RHDs. Instead, these receptors could use another mechanism to fragment the ER, or they might work with FAM134B or RTN3L (5).
ER-phagy restores ER homeostasis.
If, as we suggest above, ER-phagy acts as a general regulator of ER homeostasis, then the UPR and ER-phagy should be linked. Consistent with this hypothesis, the accumulation of misfolded proteins in the ER induces the UPR, ER expansion, and ER-phagy (36). UPR-generated ER expansion dilutes the concentration of misfolded proteins while the concentration of chaperones in the ER increases (36). Autophagy can reestablish pre-stress levels of the ER (37, 38). This notion is consistent with an earlier observation that autophagy destroys excess ER after administered barbiturates are removed (39). The translocon component, SEC62, plays a critical role in the removal of UPR-generated excess ER by virtue of a C-terminal LIR domain that recruits LC3 (19). Because overexpressed SEC62, which is not associated with the translocon, is sufficient to trigger this process—known as “RecovER-phagy”—a signal that releases SEC62 from the translocon must be generated after the UPR has been resolved (Figure 3). The nature of this signal is unknown. In a study where ER-phagy was induced by inhibiting the ER Ca2+ pump, the turnover of excess ER via RecovER-phagy was found to be via micro-ER-phagy (9). This further distinguishes SEC62-mediated RecovER-phagy from macro-ER-phagy, which employs the other five ER-phagy membrane receptors.
CCPG1 function is also linked to the UPR, but unlike SEC62, this protein is induced by the UPR. In addition, CCPG1-mediated ER-phagy utilizes the macroautophagy machinery. Indeed, CCPG1 binds through independent motifs to both GABARAP and FIP200 (40). In the exocrine pancreas of CCPG1 hypomorphic mice, insoluble secreted proteins accumulate in the ER, the ER becomes distended, and ER stress and apoptotic markers are induced. The global increase in insoluble ER proteins may be an indirect secondary consequence of the accumulation of unfolded proteins, or due to the loss of a specific ER function that occurs when CCPG1 activity is compromised. An interesting aspect to explore in the future will be to determine whether CCPG1 also contributes to RecovER-phagy and, if so, whether it participates through a micro- or a macro-ER-phagy process. We suggest that this analysis will permit an understanding of whether the UPR, depending on a specific cue, can engage different ER-phagic programs. In this context, yeast appears to use both Atg40-dependent macro-ER-phagy and ESCRT-dependent micro-autophagy when the UPR is triggered with tunicamycin (41). Notably, the UPR can also be induced by lipid bilayer stress (42–46), but it is unclear whether SEC62- and CCPG1-dependent lysosomal delivery of the ER is associated with this stress. How excess ER is ultimately turned over under these stress conditions, and whether SEC62 or CCPG1 are directly involved, requires further investigation.
ER-phagy and ER quality control.
ER-phagy also prevents disease manifestations and maintains cellular homeostasis by degrading deleterious proteins that are not removed by other ER disposal pathways (Figure 3). Examples include the disease-causing form of human α1-antitrypsin Z variant (ATZ), which fails to induce the UPR, ERAD-resistant misfolded procollagen, and the ERAD-resistant mutant prohormone Akita discussed below (3, 24, 47). ER-phagy receptors that regulate ER quality control likely work in concert with chaperones (3, 48, 49) (also see below), and possibly additional machinery, to mark the site on the ER targeted for degradation. For example, the N-degron ER-phagy pathway, which contributes to the clearance of ATZ aggregates (24) (Figure 3), may target sites on the ER that are marked with ubiquitin. ATZ can also be degraded by an LC3-mediated ER-to-lysosome-associated degradation (ERLAD) pathway that is proposed to be vesicle-mediated (48), suggesting redundant action between different pathways to detoxify the ER. It is worth noting that the term ERLAD has also been used to group all ER-lysosome transport pathways (48).
In contrast to what has been observed in mammalian cells, when ATZ is heterologously expressed in yeast, ATZ turnover is enhanced by Atg40-mediated ER-phagy (4, 50). ATZ overexpression enhances Atg40 expression and upregulates macro-ER-phagy in the absence of the UPR (4). ATZ degradation also requires Lst1 (4), a COPII coat subunit that complexes with Sec23. On the secretory pathway, Lst1-Sec23 packages correctly folded proteins into ER-to-Golgi COPII-coated transport vesicles (4, 51). In either atg40Δ or lst1Δ mutant cells, ATZ aggregates in the ER (4). When ER-phagy is induced, Lst1-Sec23 binds Atg40 to mark specific subdomains on the ER, named ER-phagy sites (ERPHS) (4) (Figure 4). ERPHS are targeted to and sequestered into autophagosomes. ERPHS appear to be distinct from the ER exit sites (ERES) that bud ER-derived COPII-coated vesicles that traffic correctly folded proteins on to the secretory pathway. Formation of the ERPHS may require ER network rearrangements. For example, ERPHS do not form in cells lacking LNP1, the gene that encodes the yeast homologue of LUNAPARK (4, 52). Lnp1 stabilizes nascent ER junctions that arise when two tubules fuse with one another (53). The role of Lnp1 in ERPHS formation may be medically important as mutations in human LNPK lead to a complex neurodevelopmental syndrome (54), although it is unclear whether this syndrome arises directly from a defect in ER-phagy. In addition to the components mentioned above, the lipid transporter, Vps13, and Atg40 oligomerization may participate in the formation and/or function of ERPHS (55, 56). Exactly how ERPHS form on the ER is currently unknown. It also remains to be addressed if misfolded proteins accumulate in the ERPHS during ER-stress. However, ERPHS formation may be conserved in mammalian cells as the Lst1 homologue, SEC24C, is required for the degradation of ER sheets and tubules in Torin-treated U2OS cells (4). Interestingly, recent studies have implicated SEC24C in the maintenance of neuronal homeostasis (57).
As discussed above, some receptors that contribute to ER quality control also bind molecular chaperones. FAM134B binds calnexin, and RTN3L co-purifies with two J domain proteins, DNAJB12 and DNAJB14 (3, 48, 49). Recent studies have shown that RTN3L facilitates the degradation of several aggregation-prone mutant prohormones, including variants of proinsulin (Akita), pro-opiomelanocortin (POMC), and pro-arginine-vasopressin (Pro-AVP) (Figure 3) (8). In the case of Akita, it was shown that low molecular weight oligomers are removed from the ER by ERAD, whereas larger high molecular weight oligomers are only cleared by RTN3-mediated ER-phagy (10). Interestingly, mutations in FAM134B and ATL3 are associated with hereditary sensory and autonomic neuropathy (HSAN), which leads to the loss of myelinated and unmyelinated fibers (58–61). These disorders primarily affect the peripheral nervous system (58–61). RTN3L, which is abundant in neurons, has additionally been linked to Alzheimer’s disease (62). Together, these observations suggest that the ER-phagy pathways that participate in ER quality control may be particularly important in cells, like neurons, that do not actively divide.
FAM134B is also required for the degradation of the transmembrane Niemann-Pick disease type C1 protein (NPC1), as well as misfolded and mutant forms of procollagen by ER-phagy (3, 63). Like Akita, procollagen is degraded by ERAD, but some forms of procollagen are resistant to degradation by ERAD and are degraded by ER-phagy instead (11). Procollagen is an abundant, large fibrillar trimeric protein that traffics through the secretory pathway in enlarged COPII-coated secretory vesicles (51). A significant fraction of procollagen is unable to fold correctly, and ER-phagy prevents ER accumulation. Two distinct mechanisms have been described for the degradation of misfolded and mutant procollagen by ER-phagy (3, 47). One report showed that FAM134B delivers procollagen to lysosomes via macro-ER-phagy (3). In this context, procollagen binds calnexin, which in turn interacts with FAM134B to trigger lysosomal delivery by ER-phagy (3). In another study, procollagen accumulated in COPII-coated ER exit sites that were directly engulfed into lysosomes via micro-ER-phagy which appears to employ p62 and ubiquitin (47). It is currently unclear why procollagen can be degraded by both macro-ER-phagy or micro-ER-phagy, although these two mechanisms may function redundantly to avoid procollagen accumulation in the ER, which would be cytotoxic.
Concluding remarks
Since the identification of the first ER-phagy receptors five years ago, many questions have emerged about the contribution of ER-phagy to cellular homeostasis and the roles that the different ER-phagy receptors play (see Outstanding Questions). Here, we have outlined the major functions of ER-phagy and discussed the growing connection between these processes and disease. Examples include mutations in ER-phagy receptors linked to neurodegenerative disorders that have also been shown to disrupt ER-phagy (17, 22). Additionally, certain mutant proteins that are known ER-phagy substrates, such as proinsulin Akita and vasopression, lead to autosomal dominant forms of diabetes (8). ER-phagy has also recently been implicated in the regulation of metabolism as well as cellular differentiation and development. Specifically, the induction of macro-ER-phagy in chondrocytes was shown to be triggered by FGF signaling, which in turn increased the levels of FAM134B and macro-ER-phagy via the TFEB transcriptional factor (32).
Outstanding questions:
What is the spectrum of endoplasmic reticulum (ER)-phagy receptors and binding partners, and what are their molecular roles?
What are the role of post-translational modifications in ER-phagy?
How are different ER-phagy pathways selected for different substrates?
Do ER-phagy receptors function together, synergistically, or play redundant roles?
While we have outlined three main roles for ER-phagy (Figure 3), some autophagy receptors, like FAM134B, exhibit other ER-phagy-associated functions. For example, an ER stress-dependent increase in cytosolic calcium initiates FAM134B phosphorylation and macro-ER-phagy (31). FAM134B has also been implicated in degradation at ER-mitochondria contact sites (64), and it limits viral replication by favoring the turnover of specific viral glycoproteins (65, 66). These observations raise an important question. How can one autophagy receptor affect so many pathways? Studies in yeast provide a potential answer to this question (4). Atg40 largely resides on and degrades the cortical ER, yet a minor fraction of Atg40 resides on the nuclear membrane where it also participates in nucleophagy (16). However, when Atg40 functions with Lst1-Sec23, it acts exclusively in cortical ER-phagy, and not nucleophagy (4). Likewise, FAM134B may only contribute to ER quality control when bound to calnexin (3, 48). We propose that the interaction of an ER-phagy receptor with a specific binding partner defines the pathway in which it functions. Identifying and understanding how these binding partners interact with ER-phagy receptors is an important goal for future studies.
As noted in this article, a growing body of evidence indicates that ER-phagy degrades ERAD-resistant forms of ERAD substrates. As a component of ER proteostasis, the relative roles of ER-phagy versus ERAD are unclear because direct measurements of substrate flux through these pathways have not been studied. The contribution of each pathway is possibly controlled by a myriad of ER stress response transducers, or simply the levels of different aggregation-prone or ERAD-resistant proteins. Similarly, we only have a rough understanding of the substrate specific decisions that direct a protein to a specific pathway. In ERAD, glycan- and chaperone-based selection play critical roles (67). In turn, while the chaperone-like protein calnexin has been implicated in the selection of macro-ER-phagy substrates (see above), we suggest that other chaperone-like proteins function similarly.
In summation, these and many other questions remain unanswered, including how specific domains of the ER are targeted for ER-phagy. We propose that posttranslational modifications, such as ubiquitinylation and UFMylation, play a central role in this process. Based on the findings discussed above, we also suggest that multiple second messengers regulate ER-phagy. Currently, it is also unclear how the decision to utilize different ER-phagy pathways is made, and whether multiple receptors work with one another in these processes. In addition to shedding light on the multi-faceted tasks of ER-phagy, future studies could provide insight into the development of therapeutic approaches that may delay the onset or cure the growing number of diseases associated with ER-phagy.
Highlights:
The selective degradation of the endoplasmic reticulum (ER), called ER-phagy or reticulophagy, has multiple physiological functions
Many different types of cell stress can induce ER-phagy, including starvation and misfolded protein accumulation
ER-phagy pathways participate in ER homeostasis and ER quality control
ER-phagy performs functions that are independent of the unfolded protein response (UPR) and ER associated degradation (ERAD)
ER-phagy pathways, which play a role in ER quality control, are cytoprotective
Acknowledgments
Research in the lab of SF-N was supported by grants R35GM131681 and R01NS117440 from the National Institutes of Health and the Alpha-1 Foundation. Research in the lab of FR was supported by ZonMW TOP (91217002), ENW Open Program (OPENW.KLEIN.118), ALW Open Programme (ALWOP.310), Marie Skłodowska-Curie Cofund (713660) and Marie Skłodowska Curie ETN (765912) grants. Research in the lab of JLB was supported by grants R35 GM131732 and P30 DK079307 from the National Institutes of Health.
Glossary terms
- Autophagy
a term describing a “self-eating” process in which cellular content is degraded after delivery to the vacuole (in yeasts/fungi) or to the lysosome in (higher organisms); the autophagy pathway can remove malfunctioning or aggregated proteins, aberrant or unwanted protein complexes, damaged or excess organelles, and pathogens to maintain homeostasis, and macro-autophagy, micro-autophagy, and chaperone-mediated autophagy are three mechanistically distinct types of autophagy (only macro-autophagy and micro-autophagy are discussed in this article)
- Cortical ER
a term used to describe peripheral and cytoplasmic endoplasmic reticulum (ER) in yeast; cortical ER contains ER sheets and tubules, but not nuclear ER membranes
- ER Associated Degradation (ERAD)
a pathway that rids the ER of misfolded proteins, unassembled subunits of multimeric complexes, or proteins that fail to acquire proper post-translational modifications, and also regulates the steady-state levels of metabolic enzymes in the ER; after selection, ERAD substrates are “retrotranslocated” or “dislocated” into the cytoplasm, ubiquitinated, and degraded by the proteasome
- ER expansion
an event that accompanies the application of select compounds or stresses that either need to be metabolized (e.g., barbiturates) or that induce the UPR; expansion requires an increase in lipid synthesis, and in some cases the expression of ER resident proteins also rises
- ER-to-lysosome-associated degradation (ERLAD)
a term used to describe the spectrum of pathways by which ER-to-lysosome transport degrades cargo molecules
- N-degron ubiquitin-dependent ER-phagy
a pathway in which ER-resident cargo proteins containing destabilizing N-terminal amino acids are delivered into lysosomes; this pathway requires recognition of the N-terminal amino acid by an autophagy receptor, as well as its ubiquitination
- Nucleophagy
a selective type of autophagy in which a part of the nucleus is targeted for degradation
- ULK complex
a protein complex that contains the ULK1 or ULK2 kinase as well as FIP200, ATG13, and ATG101; the complex functions as the most upstream component of the autophagy machinery and responds to various cellular stimuli to repress or initiate autophagy by acting on downstream components of the autophagy machinery
- Unfolded Protein Response (UPR)
an inducible response that is mediated by ER membrane proteins that responds to an increase in misfolded protein levels in the ER, lipid disequilibrium, and alterations in the ER lumenal ion composition and protein glycosylation; downstream effects of the UPR include induction of ER-phagy/autophagy, ERAD, secretory pathway functions, and the ER protein folding machinery, as well as reduced protein translation and in specific cases apoptosis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen S, Novick P, Ferro-Novick S, ER structure and function. Curr Opin Cell Biol 25, 428–433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun Z, Brodsky JL, Protein quality control in the secretory pathway. J Cell Biol 218, 3171–3187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forrester A et al. , A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J 38, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui Y et al. , A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science 365, 53–60 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chino H, Mizushima N, ER-phagy: Quality control and turnover of endoplasmic reticulum. Trends Cell Biol 30, 384–398 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Hübner CA, Dikic I, ER-phagy and human diseases. Cell Death Differ 27, 833–842 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilkinson S, Emerging Principles of Selective ER Autophagy. J Mol Biol 432, 185–205 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunningham CN et al. , Cells Deploy a Two-Pronged Strategy to Rectify Misfolded Proinsulin Aggregates. Mol Cell 75, 442–456.e444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loi M, Raimondi A, Morone D, Molinari M, ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat Commun 10, 5058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cunningham CN et al. , Cells deploy a two-pronged strategy to rectify misfolded proinsulin aggregates. Mol Cell 75, 442–456 e444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishida Y et al. , Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell 20, 2744–2754 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turco E et al. , FIP200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol Cell 74, 330–346 e311 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravenhill BJ et al. , The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol-invading bacteria. Mol Cell 74, 320–329 e326 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vargas JNS et al. , Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell 74, 347–362 e346 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith MD et al. , CCPG1 Is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev Cell 44, 217–232 e211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mochida K et al. , Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522, 359–362 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Khaminets A et al. , Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Grumati P et al. , Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fumagalli F et al. , Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol 18, 1173–1184 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Chino H, Hatta T, Natsume T, Mizushima N, Intrinsically disordered protein TEX264 mediates ER-phagy. Mol Cell 74, 909–921 e906 (2019). [DOI] [PubMed] [Google Scholar]
- 21.An H et al. , TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol Cell 74, 891–908 e810 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Q et al. , ATL3 Is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr Biol 29, 846–855 e846 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Liang JR, Lingeman E, Ahmed S, Corn JE, Atlastins remodel the endoplasmic reticulum for selective autophagy. J Cell Biol 217, 3354–3367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji CH et al. , The N-Degron Pathway Mediates ER-phagy. Mol Cell 75, 1058–1072.e1059 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Stefely JA et al. , Mass spectrometry proteomics reveals a function for mammalian CALCOCO1 in MTOR-regulated selective autophagy. Autophagy, 1–19 (2020). [DOI] [PMC free article] [PubMed]
- 26.Nthiga TM et al. , CALCOCO1 acts with VAMP-associated proteins to mediate ER-phagy. EMBO J 39, e103649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao D et al. , A UPR-Induced Soluble ER-Phagy Receptor Acts with VAPs to Confer ER Stress Resistance. Mol Cell, (2020). [DOI] [PubMed]
- 28.Stephani M et al. , A cross-kingdom conserved ER-phagy receptor maintains endoplasmic reticulum homeostasis during stress. Elife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang JR et al. , A Genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 180, 1160–1177 e1120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zielke S et al. , ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy, 1–17 (2020). [DOI] [PMC free article] [PubMed]
- 31.Jiang X et al. , FAM134B oligomerization drives endoplasmic reticulum membrane scission for ER-phagy. EMBO J 39, e102608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cinque L et al. , MiT/TFE factors control ER-phagy via transcriptional regulation of FAM134B. EMBO J 39, e105696 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizushima N, Yoshimori T, Ohsumi Y, The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27, 107–132 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Fielden J et al. , TEX264 coordinates p97- and SPRTN-mediated resolution of topoisomerase 1-DNA adducts. Nat Commun 11, 1274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhaskara RM et al. , Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat Commun 10, 2370 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schuck S, Prinz WA, Thorn KS, Voss C, Walter P, Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol 187, 525–536 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogata M et al. , Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26, 9220–9231 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernales S, McDonald KL, Walter P, Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4, e423 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolender RP, Weibel ER, A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of threatment. J Cell Biol 56, 746–761 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith MD et al. , CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev Cell 44, 217–232.e211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schafer JA et al. , ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast. EMBO J 39, e102586 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Promlek T et al. , Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell 22, 3520–3532 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hou NS et al. , Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc Natl Acad Sci U S A 111, E2271–2280 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koh JH, Wang L, Beaudoin-Chabot C, Thibault G, Lipid bilayer stress-activated IRE-1 modulates autophagy during endoplasmic reticulum stress. J Cell Sci 131, (2018). [DOI] [PubMed] [Google Scholar]
- 45.Tam AB et al. , The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev Cell 46, 327–343.e327 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Volmer R, van der Ploeg K, Ron D, Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A 110, 4628–4633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Omari S et al. , Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc Natl Acad Sci U S A 115, E10099–E10108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fregno I et al. , ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J 37, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen YJ, Williams JM, Arvan P, Tsai B, Reticulon protects the integrity of the ER membrane during ER escape of large macromolecular protein complexes. J Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kruse KB, Brodsky JL, McCracken AA, Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human a-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell 17, 203–212 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gomez-Navarro N, Miller E, Protein sorting at the ER-Golgi interface. J Cell Biol 215, 769–778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen S, Cui Y, Parashar S, Novick PJ, Ferro-Novick S, ER-phagy requires Lnp1, a protein that stabilizes rearrangements of the ER network. Proc Natl Acad Sci U S A 115, E6237–E6244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S et al. , Lunapark stabilizes nascent three-way junctions in the endoplasmic reticulum. Proc Natl Acad Sci U S A 112, 418–423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breuss MW et al. , Mutations in LNPK, Encoding the Endoplasmic Reticulum Junction Stabilizer Lunapark, Cause a Recessive Neurodevelopmental Syndrome. Am J Hum Genet 103, 296–304 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen S et al. , Vps13 is required for the packaging of the ER into autophagosomes during ER-phagy. Proc Natl Acad Sci U S A 117, 18530–18539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mochida K et al. , Super-assembly of ER-phagy receptor Atg40 induces local ER remodeling at contacts with forming autophagosomal membranes. Nat Commun 11, 3306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang B et al. , The COPII cargo adapter SEC24C is essential for neuronal homeostasis. J Clin Invest 128, 3319–3332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurth I et al. , Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet 41, 1179–1181 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Xu H et al. , ATL3 gene mutation in a Chinese family with hereditary sensory neuropathy type 1F. J Peripher Nerv Syst 24, 150–155 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Kornak U et al. , Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain 137, 683–692 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Krols M et al. , Sensory neuropathy-causing mutations in ATL3 affect ER-mitochondria contact sites and impair axonal mitochondrial distribution. Hum Mol Genet 28, 615–627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi Q, Ge Y, He W, Hu X, Yan R, RTN1 and RTN3 protein are differentially associated with senile plaques in Alzheimer’s brains. Sci Rep 7, 6145 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schultz ML et al. , Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat Commun 9, 3671 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mookherjee D et al. , RETREG1/FAM134B mediated autophagosomal degradation of AMFR/GP78 and OPA1 -a dual organellar turnover mechanism. Autophagy, 1–24 (2020). [DOI] [PMC free article] [PubMed]
- 65.Chiramel AI, Dougherty JD, Nair V, Robertson SJ, Best SM, FAM134B, the Selective Autophagy Receptor for Endoplasmic Reticulum Turnover, Inhibits Replication of Ebola Virus Strains Makona and Mayinga. J Infect Dis 214, S319–S325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lennemann NJ, Coyne CB, Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 13, 322–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oikonomou C, Hendershot LM, Disposing of misfolded ER proteins: A troubled substrate’s way out of the ER. Mol Cell Endocrinol 500, 110630 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]