

Abstract
Ubiquitylation, a highly regulated post-translational modification, controls many cellular pathways that are critical to cell homeostasis. Ubiquitin ligases recruit substrates and promote ubiquitin transfer onto targets, inducing proteasomal degradation or non-degradative signaling. Accumulating evidence highlight the critical role of dysregulated ubiquitin ligases in processes associated with the initiation and progression of cancer. Depending on the substrate specificity and biological context, an ubiquitin ligase can act either as a tumor promoter or as a tumor suppressor. In this review, we focus on the regulatory roles of ubiquitin ligases and how perturbations of their functions contribute to cancer pathogenesis. We also briefly discuss current strategies for targeting or exploiting ubiquitin ligases for cancer therapy.
Graphical Abstract
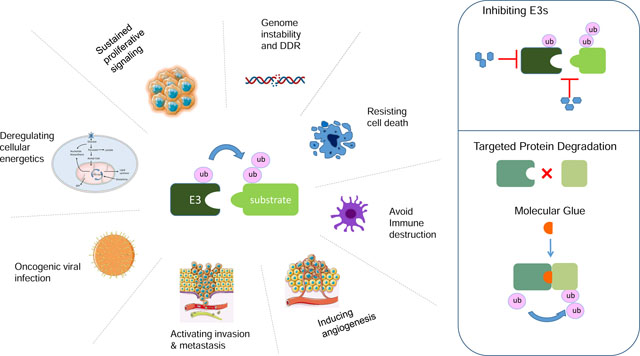
Ubiquitin ligases specifically recognize and mediate the ubiquitylation of critical, regulatory proteins. In this review, Duan and Pagano highlight recent findings about ubiquitin ligases in the regulation of cancer-relevant pathways. They also discuss strategies to exploit ubiquitin ligases for cancer therapy.
Introduction
Ubiquitylation refers to the enzymatic post-translational modification in which the ubiquitin protein is covalently attached to cellular proteins (Hershko and Ciechanover, 1998). The core enzymes driving this process are the ubiquitin activating enzyme (UAE or E1), the ubiquitin-conjugating enzyme (UBC or E2), and the ubiquitin ligase (E3).
E3s recruit substrates and thus determine the overall specificity for ubiquitylation. They constitute a wide class of proteins, with the human genome encoding for more than 600 putative E3s (Li et al., 2008), further subdivided into several families based on their conserved structural domains (e.g., RING, HECT, UBOX or RBR domains) (Buetow and Huang, 2016; Metzger et al., 2012). Amongst these, RING finger ubiquitin ligases comprise the largest family, with members able to function as monomers, dimers, or multi-subunit complexes (Lipkowitz and Weissman, 2011; Petroski and Deshaies, 2005). Of the latter, the best characterized subfamily is formed by ~230 E3s, known as cullin-RING ligases (CRLs). CRLs are modular complexes that contain a cullin scaffold, a RING finger protein (RBX1 or RBX2) that serves as the site for E2 binding, and a variable/exchangeable substrate receptor (SR) subunit (Lydeard et al., 2013; Skaar et al., 2013). Mammals possess eight cullin proteins (CUL1, CUL2, CUL3, CUL4A, CUL4B, CUL5, CUL7, and CUL9), each of which interacts with a unique family of SRs that provides the substrate specificity (Cardozo and Pagano, 2004; Petroski and Deshaies, 2005; Skaar et al., 2013). F-box proteins, VHL/BC-box proteins, BTB proteins, DCAF proteins, and SOCS box proteins are the SRs of CRL1, CRL2, CRL3, CRL4A/B, and CRL5 complexes, respectively. CRL7 uses only one SR (i.e., FBXW8), and it is not clear to date whether CRL9 binds any SR. CRL1 complexes are also known as SCF (SKP1, CUL1, F-box protein) complexes.
The large number of E3s and their targeted substrates connects the ubiquitin-proteasome system to diverse biological processes and human diseases, particularly cancer. Studies during the last 20 years have shown that deregulated E3s play a critical role in the development, progression, and therapeutic response of human cancers. Depending on their substrates, E3s themselves can play tumor-suppressive or pro-oncogenic roles, serving as therapeutic targets for anti-tumor drugs (Senft et al., 2018; Wang et al., 2014). This review will provide an overview of current understanding about the functions of E3s in cancers, and discuss perspectives on cancer therapies that depend on inhibition or activation of ubiquitylation of target proteins.
Role of E3s in tumor development
The development of cancer is a multistep process through which normal cells evolve progressively into a neoplastic state and acquire features that Bill Hanahan and Bob Weinberg defined as the “hallmarks of cancers”, including increased capacity for proliferation and survival, invasion and metastasis, and the ability to evade immunosurveillance and destruction (Hanahan and Weinberg, 2011). Such a complex process requires the reprogramming of cellular signaling networks to meet the needs for malignant transformation. By controlling protein abundance and activity in a timely and specific manner, E3s serve as central regulatory nodes for many signaling pathways. It is therefore no surprise to observe that E3s and their substrates are frequently deregulated in human cancers (Qi and Ronai, 2015). Aberrant regulation of E3s can occur at the genetic, epigenetic, or post-translational levels (Figure 1). These alterations in E3s can either convert proto-oncoproteins into oncoproteins or inactivate tumor suppressors. Moreover, some oncogenic viruses can alter or hijack E3s activity to modulate the abundance of cellular substrates for the benefit of viral replication, ultimately promoting tumor transformation.
Figure 1. Deregulation of E3-mediated ubiquitylation in cancer.
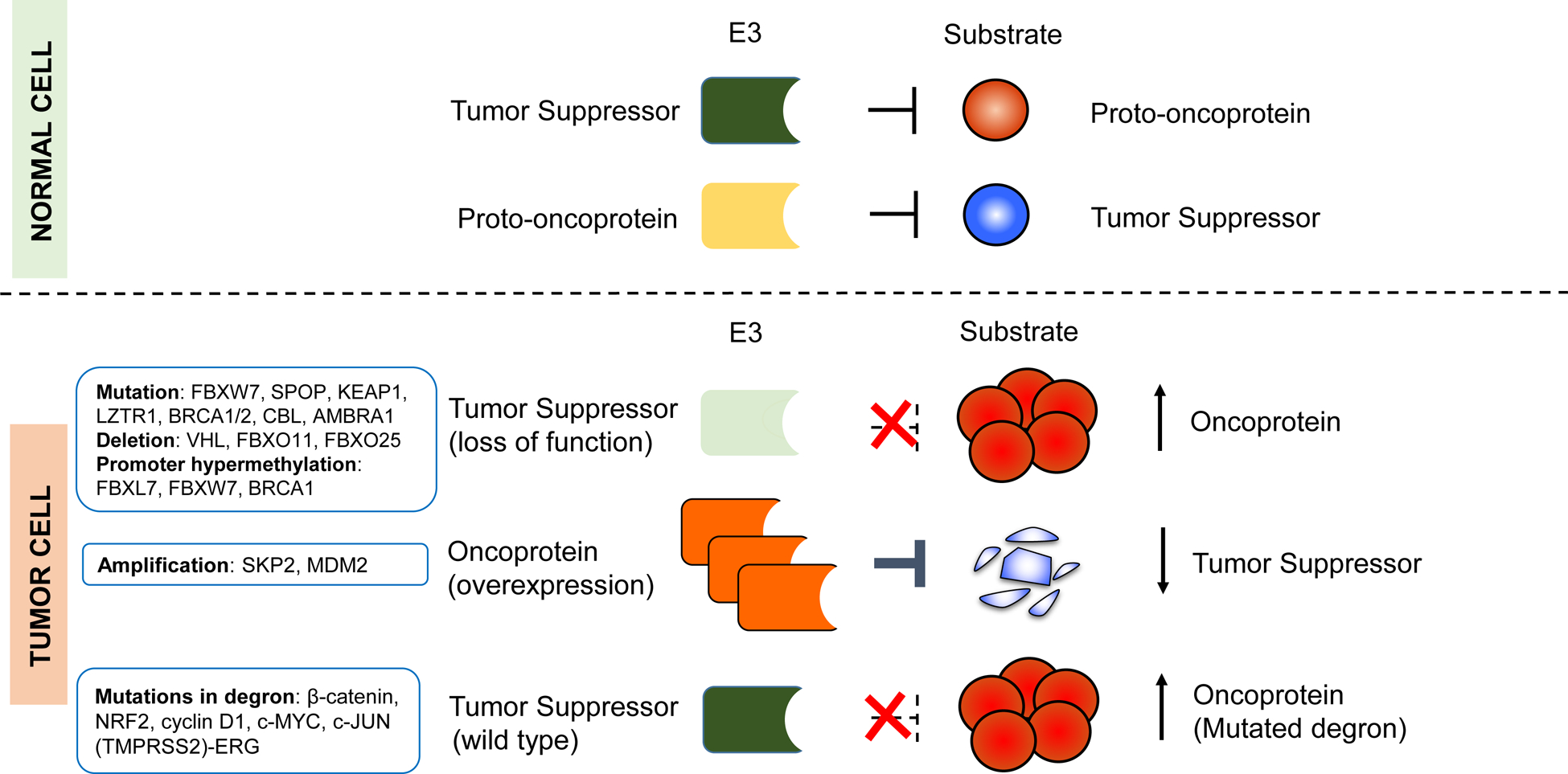
The illustration shows different modes of E3 deregulation, with well-established examples listed. Most deregulations in E3s arise as a consequence of genetic or epigenetic alterations, which can affect the abundance and/or activity of their substrates. For example, deletions, mutations, or promoter methylations can inactivate E3s that normally function as tumor suppressors, and lead to the overexpression of oncoprotein substrates (e.g., c-Myc, cyclin E, and ERG). Alternatively, overexpression of E3s (e.g., through gene amplification of MDM2 and SKP2 loci) that target tumor suppressors (p53 and p27, respectively) can promote tumor formation. In addition, mutations in the substrates, which enable them to escape the recognition by E3s can lead to their accumulation.
In the following sections, we will discuss the critical roles of E3s in processes associated with the initiation and progression of cancers (Figure 2) and give examples on how deregulation of E3s affect these biological processes.
Figure 2. E3s in the regulation of the hallmarks of cancer.
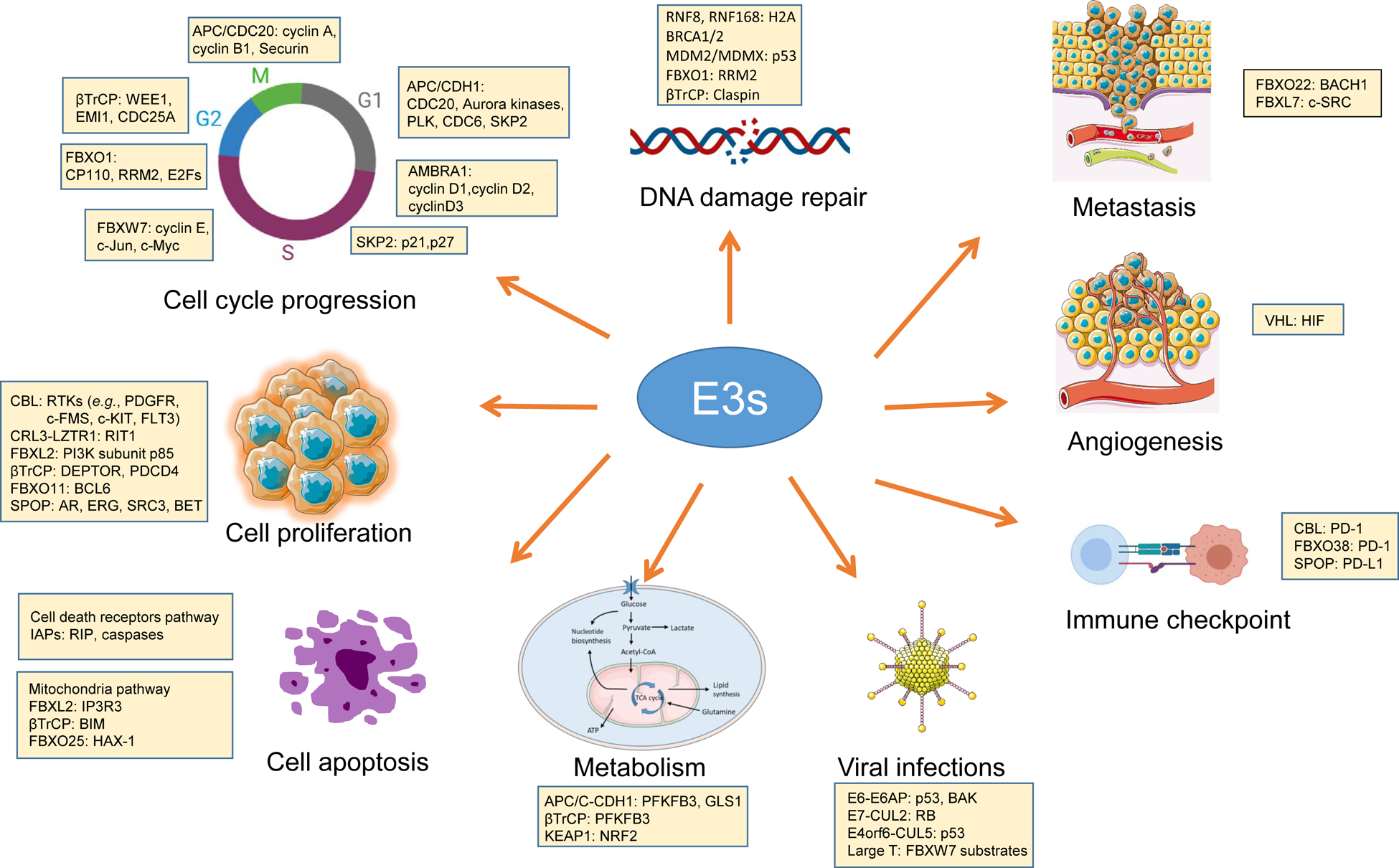
E3s are hubs of many cellular processes, including those playing key roles in cell proliferation, cell cycle, the DNA damage response, apoptosis, metabolism, metastasis, angiogenesis, immune checkpoint, and viral infections. Representative E3s with their best-defined substrates in these biological processes are listed.
E3s and cell proliferative signaling
One of the most fundamental traits of cancer cells is its ability to sustain constant proliferation. Under normal conditions, cellular proliferation signaling pathways are tightly controlled by the limited availability of growth factors and nutrients, contact inhibition, as well as various feedback mechanisms (Sever and Brugge, 2015). Cancer cells, by deregulating these signals, become masters of their own destinies and no longer depend on external stimuli to proliferate.
E3s modulate the abundance of regulatory proteins that participate in major hubs of the proliferative signaling. Thus, dysregulations of E3s may have profound effects on both upstream regulators and downstream effectors. As it is impossible to cover all the signaling molecules involved, we will focus primarily on major proliferative signaling pathways to illustrate the functions of E3s in monitoring essential signal transducers to ensure proper signaling dynamics. Constitutive hyperactivation of these signaling pathways can lead to excessive proliferation and ultimately tumorigenesis (Lavoie et al., 2020; Manning and Toker, 2017).
E3s mediates the ubiquitylation and downregulation of RTKs
Receptor tyrosine kinases (RTKs) function as entry points for many extracellular cues, initiating intracellular signaling cascades. Inappropriate activation of RTKs is associated with a large number of malignancies (Lemmon and Schlessinger, 2010). Among other mechanisms, escaping from ubiquitylation-mediated negative regulation is a central event in RTK deregulation. Activated RTKs are downregulated through endocytosis and subsequent intracellular degradation, and ubiquitylation either serves as a sorting signal for targeting RTKs to clathrin-coated pits for endocytosis, or mediates the degradation of RTKs (Marmor and Yarden, 2004; Tomas et al., 2014). A well-documented example is the epidermal growth factor receptor (EGFR). Following ligand binding and receptor activation by phosphorylation, EGFR is modified by the E3 c-CBL, leading to its internalization by endocytosis and subsequent degradation via the lysosome (Levkowitz et al., 1999; Thien and Langdon, 2001). The CBL family of E3s also target other receptors, including PDGFR, c-Fms/CSF-1R, c-Kit, and Met (Peschard and Park, 2003). Mutations in CBL or RTKs can impede their ubiquitylation and contribute to their hyperactivation. CBL mutations have been found in ~5% of a wide variety of myeloid neoplasms, and many of the mutations are missense mutations affecting its E3 activity (Kales et al., 2010). Moreover, RTK mutants impaired in their ability to bind c-CBL, exhibit prolonged protein stability and enhanced activity (Grandal et al., 2007; Peschard and Park, 2003).
E3s regulate the RAS-MAPK and PI3K-AKT signaling
Ligand-bound RTKs transduce the signaling by activating downstream signaling pathways, including the RAS-MAPK and the PI3K-AKT-mTOR signaling cascades, to regulate various cytoplasmic and nuclear substrates that are responsible for cell growth and proliferation. RTKs recruit adaptor proteins, such as SOS, a RAS-specific guanine nucleotide exchange factor, to the plasma membrane. SOS enhances the exchange of GDP for GTP by the RAS small GTPase, resulting in RAS activation (Chardin et al., 1993; Lemmon and Schlessinger, 2010). The RAS superfamily of GTPases are key signal transducers that activates kinase cascades including the mitogen-activated protein kinases (MAPKs) to regulate key pathways promoting cell proliferation. Ubiquitylation on several members of the RAS family have been observed, leading to their altered cellular localization and activity (de la Vega et al., 2011). LZTR1, the SR of a CRL3, has been proposed to mediate the ubiquitylation of RAS to either control its activity or its degradation (Abe et al., 2020; Bigenzahn et al., 2018; Steklov et al., 2018). Mutations in LZTR1 have been identified in human cancers and developmental diseases such as Noonan syndrome (NS), a genetic disorder caused by germline mutations in genes involved in the RAS-MAPK pathway (Motta et al., 2019; Rauen, 2013). However, the role of LZTR1 in RAS ubiquitylation has not been confirmed by others, and an elegant study instead showed that CRL3LZTR1 targets RIT1, a different small GTPase and proto-oncoprotein, for ubiquitin- and proteasome-mediated degradation (Castel et al., 2019). Pathogenic mutations in LZTR1 affects its interaction with RIT1, leading to RIT1 accumulation and subsequent dysregulation of growth factor signaling responses (Castel et al., 2019), in agreement with what observed in human patients with LZTR1 mutations.
Another major downstream effector of RTKs is the phosphoinositide 3-kinase (PI3K), which catalyzes the production of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) at the plasma membrane, resulting in the activation of AKT kinases (Hoxhaj and Manning, 2020). AKT activates numerous downstream effectors, including mTOR kinase and forkhead box O (FOXO) family of transcription factors, to promote protein synthesis and cell proliferation. The PI3K-AKT signaling is regulated in multiple ways, including via E3-mediated ubiquitylation. PI3K is a dimeric enzyme composed of a catalytic subunit (p110α, p110β, or p110γ) and a regulatory subunit (p85α, p85β, p55α, p55γ, or p50α). Ubiquitylation on both subunits have been reported, with various effects on PI3K signaling. For example, FBXL2, a CRL1 SR, was shown to mediate the degradation of free p85 to prevent its competition with active PI3K (composed of p85–p110 heterodimers) for phospho-Tyr docking sites at the cell membrane (Kuchay et al., 2013). Via this mechanism, FBXL2 ensures efficient activation of the PI3K signaling cascade to promote cell proliferation and survival.
mTOR belongs to the PI3K-related kinases family and exists in two distinct complexes called complex 1 (mTORC1) and complex 2 (mTORC2). Both complexes respond to numerous environmental cues, especially those related to nutrient, energy, and oxygen levels, to modulate cell growth and survival (Liu and Sabatini, 2020). Within the mTORC complex, the DEPTOR protein binds mTOR and inhibits mTORC activity (Peterson et al., 2009). Upon mitogenic stimulation, CRL1βTrCP cooperates with mTOR and CK1 to induce the degradation of DEPTOR, generating an auto-amplification loop that promotes the full activation of mTOR (Duan et al., 2011; Gao et al., 2011; Zhao et al., 2011). A key function of activated mTOR is to regulate mRNA translation and promote protein synthesis, which is an energy costly process. Under nutrient deprivation conditions, PDCD4 (a substrate of the mTOR pathway) inhibits the translation initiation factor eIF4A (Yang et al., 2003). Activated mTOR leads to the phosphorylation of PDCD4, promoting its interaction with βTrCP and leading to its degradation (Dorrello et al., 2006). This event relieves PDCD4 repression of eIF-4A, allowing efficient protein translation and cell growth. Hence, βTrCP coordinates increased protein synthesis with pro-survival signaling.
Other E3s linked to cell proliferation and cancer
E3s can of course regulate numerous other signaling pathways involved in cell proliferation. Notably, a number of E3s regulating cell proliferation are deregulated in specific types of cancers. For example, SPOP (Speckle-type POZ protein) is a CRL3 SR whose corresponding gene is mutated in 5–15% of prostate cancer (Barbieri et al., 2012) and endometrial cancer (Le Gallo et al., 2012). CRL3-SPOP has been implicated in the regulation of key pro-oncogenic signaling proteins, including the androgen receptor, SRC3 (also known as NCOA3), ERG, and BET proteins (BRD2, BRD3, and BRD4) (Clark and Burleson, 2020). In prostate cancer, SPOP mutations clustered within the N-terminal MATH domain, a region responsible for substrate recognition and ubiquitin transfer, thus, leading to impaired E3 activity. Intriguingly, mutations in endometrial cancer occur in a distinct, uncharacterized region of the MATH domain, and have the opposite effect in potentiating the degradation of BET proteins (Janouskova et al., 2017). Moreover, many SPOP substrates contain a SPOP-binding consensus motif (Zhuang et al., 2009), and mutations in this motif can disrupt their interaction with SPOP. For example, prostate cancer tumors harbor SPOP mutations and TMPRSS2–ERG gene fusions were found to occur in a mutually exclusive manner, with both alterations inhibiting CRL3SPOP-dependent degradation of ERG (An et al., 2015; Gan et al., 2015).
Like SPOP, KLHL6 (Kelch-like protein 6) is a CRL3 SR, which is encoded by a gene mutated in human Diffuse large B-cell lymphoma (DLBCL) (Choi et al., 2018). Mutations in KLHL6 inhibit its ligase activity by disrupting the interaction with CUL3, leading to the stabilization of its substrate, the mRNA decay factor roquin-2, which subsequently promotes tumor cell growth.
Deletions and mutations in the gene encoding the F-box protein FBXO11 have also been observed in ~10% of patients with DLBCL, leading to stabilization of the BCL6 oncoprotein (Duan et al., 2012). BCL6 functions as a transcription factor in B cell development, differentiation and activation (Ci et al., 2008). Thus, increased BCL6 expression drives the development of DLBCL.
E3s and cell cycle progression
The transduction of mitogenic signals stimulates cell cycle progression to promote proliferation. The eukaryotic cell cycle progression is driven by oscillations in the activity of cyclin-dependent kinases (CDKs), which are activated by cyclins and inhibited by CDK inhibitors (CKIs) (Bassermann et al., 2014; King et al., 1996). Proteasomal degradation of cyclins and CKIs regulates CDKs activity and its dysregulation contributes to the sustained proliferation observed in cancer cells.
E3s in different phases of cell cycle
Two multi-subunit families of E3s are crucial to cell cycle progression, the CRL1 complexes and the APC/C (anaphase-promoting complex, also known as the cyclosome, which binds to one of its co-activators, CDC20 and CDH1) (Nakayama and Nakayama, 2006; Skaar and Pagano, 2009). APC/CCDC20 and APC/CCDH1 are active in mitosis and G1, respectively, while CRL1 complexes are active throughout the cell cycle. APC/C targets substrates with short linear sequence motifs, referred to as degradation motifs or degrons (e.g., D-box, KEN-box, and ABBA motifs) (Davey and Morgan, 2016). Some of the CRL1 SRs, such as β-TRCP and FBXW7, trigger the degradation of cell cycle regulatory proteins through conserved degron motif only when these are phosphorylated.
Upon mitogen stimulation, cells commit to cell division and pass a restriction checkpoint in G1. Mitogenic factors activate intracellular signaling cascades to induce the expression of D-type cyclins (D1, D2, and D3) that activate CDK4 and CDK6. A well-known target of CDK4/6-cyclin D is the retinoblastoma tumor suppressor protein (RB), which binds some members of the E2F family of DNA-binding transcription factors (E2F) to block their transcriptional activity (Harbour and Dean, 2000). RB phosphorylation promotes its dissociation from E2Fs, releases the E2F transcription factors, and drives the expression of E2F-target genes, including CCNE that encodes cyclin E. In turn, activation of CDK2-cyclin E drives cell cycle progression from G1 into S phase, during which DNA replication occurs (Duronio and Xiong, 2013; Otto and Sicinski, 2017). Upon successful S-phase entry, cyclin D1 is phosphorylated and degraded by CRL4AMBRA1(Chaikovsky et al., 2021; Simoneschi et al., 2021). CDK2-cyclin E and CDK2-cyclin A become main CDK complexes in S phase, promoting the initiation of DNA and centrosome duplication. The rising of CDK2 activity also requires removal of inhibition of the associated CDK inhibitors, such as p21 or p27. CRL1SKP2 mediates the ubiquitylation and degradation of p21 and p27, liberating CDK2 from CKI inhibition. Finally, during G2, E2F activity is turned off via the degradation of E2F1/2/3A mediated by FBXO1 (also known as cyclin F) (Burdova et al., 2019; Clijsters et al., 2019; Emanuele et al., 2020).
During S and G2, it is essential for the cell to assure accurate DNA replication and chromosomes integrity. CRL1s are key players in DNA surveillance mechanisms. Upon detection of DNA damage, CRL1βTrCP in cooperation with the kinase CHK1 targets the CDK1/2 activating phosphatase, CDC25A, for degradation (Busino et al., 2003), resulting in an attenuated CDK activity and consequent halt in cell cycle progression. βTrCP also controls recovery from this checkpoint. Claspin, a checkpoint mediator, is phosphorylated by PLK1 during recovery from genotoxic stress and subsequently degraded in a βTrCP-dependent manner (Mailand et al., 2006; Peschiaroli et al., 2006).
CRLs also monitor the process of centrosome duplication to prevent chromosome aberrations. CP110, a protein essential for centrosome duplication, is targeted by FBXO1 on the centrioles during G2 (D’Angiolella et al., 2010). Thus, FBXO1 ensures that centrosomes are replicated only once during the cell cycle, avoiding centrosome overduplication.
As cells progress through G2 and prepares to enter mitosis, CDK2 gradually steps down from the stage. CDK1 now takes the role to phosphorylate downstream substrates, thereby shaping the mitotic environment. FBXW7 mediates the degradation of cyclin E (Strohmaier et al., 2001), resulting in attenuation of CDK2 activity, whereas β-TrCP promotes activation of CDK1 by mediating the degradation of WEE1, a CDK1 inhibitory kinase (Watanabe et al., 2004).
Starting from metaphase, APC/C becomes a key player in promoting mitotic progression and, eventually, mitotic exit (Skaar and Pagano, 2009). After inactivation of the spindle assembly checkpoint, APC/CCDC20 targets securin for degradation, leading to sister chromatid separation. APC/CCDC20 also targets cyclin B1 to induce mitotic exit, so, this E3 coordinates the segregation of replicated genetic material with the end of mitosis. At the end of anaphase, CDH1 replaces CDC20 within the APC/C complex, and promotes further degradation of mitotic proteins, including PLK1, Aurora kinases, and CDC20 itself. βTrCP mediates degradation of the APC/CCDH1 inhibitor EMI1, thus, contributing to the activation of APC/C (Guardavaccaro et al., 2003). After exiting from mitosis, or during withdrawal from the cell cycle, APC/CCDH1 contributes to maintenance of the G1 state or establishment of a stable G0, respectively. APC/CCDH1 targets drivers of DNA replication and mitosis (e.g., CDC6, cyclin A, and E2F), as well as SKP2, thus promoting the stabilization of CKIs.
Deregulated ubiquitylation affects cell cycle progression
Selective degradation of cell cycle regulators ensures the order and timing of cell cycle events. The functions of theE3s mentioned above in coordinating cell cycle progression give them protooncogenic or tumor suppressor properties, as supported by their frequent alterations in many tumors.
In addition to cyclin E, CRL1FBXW7 targets several well-known oncoproteins that regulates cell proliferation, including c-MYC, c-JUN, and NOTCH (Welcker and Clurman, 2008). FBXW7 is a haploinsufficient tumor suppressor that is frequently subjected to mutations and deletions. Mutations of FBXW7 has been identified in many human cancers, with the highest rate (approximately 30%) in cholangiocarcinoma and T-cell acute lymphoblastic leukemias (T-ALL) (Welcker and Clurman, 2008). A majority of the FBXW7 mutations are point mutations that occurs at key residues that form the substrate-binding interface. Thus, FBXW7 mutations associated with cancer appear to disrupt substrate recognition. Most of the remaining mutations are nonsense codons that lead to premature termination of FBXW7 translation. Studies leveraging mouse models showed that loss-of-function mutations in FBXW7 promoted hematopoietic or solid organ tumor formation (Mao et al., 2004; Matsuoka et al., 2008), supporting a tumor suppressor role for FBXW7. However, the function of FBXW7 is context dependent, as studies have also shown that FBXW7α functions as a pro-survival gene in multiple myeloma by constitutively targeting the NFκB inhibitor p100 (Busino et al., 2012), and these tumors do not harbor FBXW7 mutations.
As an important regulator of cell cycle progression, cyclin D1 is frequently overexpressed in several cancer tissues, including breast cancer, lung cancer, prostate cancer, melanoma, oral squamous cell carcinomas, and colorectal cancer (Alao, 2007; Musgrove et al., 2011). Besides gene amplification, defects in cyclin D1 degradation accounts for its overexpression. The literature suggests several CRL1 SRs and APC/C are the E3s for cyclin D1. However, these studies were not confirmed by genetic studies (Kanie et al., 2012; Qie and Diehl, 2016). Two recent papers also were unable to support a role for CRL1s in cyclin D1 degradation and, instead, they demonstrate that CRL4AMBRA1 targets all three D-type cyclins for degradation (Chaikovsky et al., 2021; Simoneschi et al., 2021). AMBRA1 is frequently mutated in human cancers, resulting in the accumulation of these pro-oncogenic cyclins. Moreover, cancer patients often display somatic mutations in the degron of all three D-type cyclins, also resulting in their accumulation. Finally, mouse models confirm a role of AMBRA1 in the degradation of D-type cyclins both allowing proper embryogenesis and in promoting oncogenesis.
Overexpression of SKP2 levels is pro-oncogenic, due to the enhanced degradation of p27 and the resulting overactivation of CDK1/2. Indeed, amplification of SKP2 is found in multiple human cancers, including lymphomas, prostate cancer, colorectal cancer, melanoma, gastric cancer, pancreatic cancer, and breast cancer (Frescas and Pagano, 2008; Gstaiger et al., 2001).
By virtue of its ability to regulate broad cell cycle regulators, β-TRCP can function as either a tumor suppressor or an oncogenic protein, and may function as an oncoprotein in some tissue types despite having tumor-suppressive effects in others (Frescas and Pagano, 2008; Wang et al., 2014). Overexpression of β-TRCP has been reported in colorectal cancer, hepatoblastoma, and breast cancers (Fuchs et al., 2004; Koch et al., 2005; Ougolkov et al., 2004). Somatic mutations in β-TRCP which may affects its E3 activity, although rare, were observed in certain human cancers, such as gastric and prostate cancers (Gerstein et al., 2002; Kim et al., 2007a; Saitoh and Katoh, 2001). Moreover, aberrant degradation of β-TrCP substrates can be caused by mutations in the substrates themselves or their upstream regulators. Therefore, the roles of β-TRCP in cancer need to be evaluated in a tissue-specific or cellular context-dependent manner.
E3s and cell death
Programmed cell death by apoptosis serves as a natural barrier to cancer development. Apoptosis can be mainly initiated through one of two pathways, the intrinsic and the extrinsic pathway (Fulda and Debatin, 2006; Igney and Krammer, 2002). Both pathways are regulated by a number of proapoptotic and anti-apoptotic regulators, whose levels need to be carefully controlled, as malregultion can introduce defects in cell death signaling and confer a survival advantage to cancer cells. E3-mediated ubiquitylation acts at many levels in regulating the apoptotic machinery (Jesenberger and Jentsch, 2002).
Regulation of the extrinsic apoptosis pathway
The extrinsic apoptosis pathway is initiated at the plasma membrane by death receptor ligation. Stimulation of death receptors [e.g., the tumor necrosis factor (TNF) receptor superfamily (CD95/APO-1/Fas) and TNF-related apoptosis-inducing ligand (TRAIL) receptors] by CD95 ligand or TRAIL results in receptor aggregation and recruitment of the death-inducing stimulating complex (DISC) and caspase-8. Upon recruitment, caspase-8 is activated to initiate apoptosis by direct cleavage of downstream effector caspases. The inhibitors of apoptosis (IAPs) family of proteins are RING-domain E3s that suppress apoptosis. For example, cIAP1 and cIAP2 induce the ubiquitylation of RIP1, an adaptor subunit of DISC, thus promoting cancer cell survival (Bertrand et al., 2008). Recurrent up-regulation of IAP expression have been implicated in cancer development by suppressing apoptosis (Wong, 2011). c-IAP2 is highly expressed in pancreatic cancer and its level correlates to chemotherapy resistance (Lopes et al., 2007). Another IAP, livin, was observed to be highly expressed in human melanoma and other cancers (Vucic et al., 2000).
Regulation of the intrinsic apoptosis pathway
The intrinsic apoptosis signaling is initiated by stress signals through the release of proapoptotic factors, such as cytochrome c or the IAP antagonist Smac/DIABLO, from the mitochondrial intermembrane space, an event that ultimately leads to caspase activation and apoptosis. E3s plays important roles in stress sensing, signaling, and activation of this apoptotic pathway. For instance, overload of Ca2+ into mitochondria can trigger apoptosis (Orrenius et al., 2003). CRL1FBXL2 binds the IP3 (inositol 1,4,5-trisphosphate) receptor 3 (IP3R3) and targets it for degradation to limit Ca2+ influx into mitochondria (Kuchay et al., 2017). Disruption of FBXL2-mediated regulation of IP3R3 leads to increased cytosolic Ca2+ release from the endoplasmic reticulum and sensitizes cells to Ca2+-dependent apoptotic stimuli.
The BCL-2 family of proteins control cell death largely by regulating mitochondrial outer membrane permeabilization (MOMP) and releasing intermembrane space pro-apoptotic proteins. The BCL-2 family is divided into anti-apoptotic proteins (such as BCL-2 and BCL-XL) and pro-apoptotic proteins (such as BAX, BAK, BIM, NOXA, and PUMA) (Kale et al., 2018). Hyperactivation of pro-survival signaling (e.g., RAS-ERK and PI3K-AKT) in cancer cells, together with E3s, can disrupt the balance between the pro-apoptotic and anti-apoptotic proteins and promote cell survival. For example, CRL1βTrCP targets BIM for degradation, following its ERK- and RSK-mediated phosphorylation of BIM1 (Dehan et al., 2009). Consequently, knockdown of either β-TRCP or RSK sensitizes non-small-cell lung cancer (NSCLC) cell lines to gefitinib, a tyrosine kinase inhibitor that induces apoptosis through BIM1.
HCLS1-associated protein X-1 (HAX-1) is a BCL-2-family-related protein that is required to suppress apoptosis in lymphocytes and neurons (Chao et al., 2008). HAX-1 is targeted for degradation by CRL1FBXO25 (Baumann et al., 2014). FBXO25 is located at chromosome 8p23.3, a region that is frequently deleted in human mantle cell lymphoma (up to 29%) and other malignancies, such as DLBCL (~8%). In these patients, deletion of FBXO25, and therefore disruption of the FBXO25–mediated HAX-1 degradation, function as means for promoting lymphomagenesis.
E3s and DNA damage response
Living organisms are continuously exposed to several endogenous and exogenous DNA damaging agents (Jackson and Bartek, 2009). To avoid deleterious mutations, the cell needs to rapidly detect DNA damage, identify the nature of the lesion, and recruit the appropriate repair machinery. Defects in the DNA damage repair (DDR) mechanisms can cause gross chromosomal abnormalities that may contribute to tumorigenesis. Ubiquitylation of histones and DDR factors play an important role in recruiting proteins to damaged DNA sites and initiate the repair process.
As an early event of the double-strand break repair pathway, the RING-type RNF8 E3 binds to the damaged sites and assembles Lys-63-linked ubiquitin chains onto histones H1, H2A and H2AX, in cooperation with the E2 enzyme UBC13 (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007; Thorslund et al., 2015; Wang and Elledge, 2007). This recruits a second RING-E3 ligase, RNF168, which recognizes RNF8 ubiquitylation products and catalyzes the monoubiquitylation of histone H2A on K13 and K15 (Mattiroli et al., 2012). RNF168 could also bind the ubiquitylation products of its own activity, thus enabling it to spread the histone ubiquitylation signals at the DNA damage sites. These ubiquitin tags provide a crucial signaling platform for the assembly of the late effector complex, which contains the E3 BRCA1 along with its binding partner BARD1 to form a BRCA1-BARD1 RING heterodimeric E3 (Jackson and Durocher, 2013; Lukas et al., 2011). The RAP80 protein contains a tandem ubiquitin-interacting motifs (UIMs), which bind preferentially to Lys-63-linked ubiquitin chains, is required for recruitment of the BRCA1 complex and the repair of damaged DNA (Kim et al., 2007b). Mutations in the BRCA1 gene leading to the expression of a truncated or inactive BRCA1 protein are strongly linked to development of ovarian and breast cancer (Kuchenbaecker et al., 2017), although more work is needed to clearly define the functions of BRCA1-BARD1 E3 activity.
E3-mediated degradation is also required in DNA damage detection to enable cells to orchestrate subsequent cell cycle checkpoints and repair. A classic example is the interplay between MDM2, a RING finger E3, and the tumor suppressor p53. p53, known as the “guardian of the genome”, transcriptionally controls a number of genes involved in the regulation of cell cycle, apoptosis, DNA repair, senescence, and angiogenesis (Levine and Oren, 2009; Vousden and Prives, 2009). p53 induces the transcription of MDM2, while MDM2 mediates the ubiquitylation of p53, thus forming a negative feedback loop (Manfredi, 2010; Wade et al., 2010). MDM2 as a homodimer or a heterodimer with the related E3 MDMX/HDMX, binds to and ubiquitylate p53. This results in the proteasomal degradation of p53, keeping p53 levels in check under normal conditions. In response to genotoxic stress, however, various modifications on p53, MDM2, and their regulators inhibit p53 ubiquitylation and degradation, promoting p53 transcriptional activity. The human MDM2 gene is located on chromosome 12 (12q14–15) and its amplification is observed in various types of cancers, particularly in sarcomas (Wade et al., 2013). High MDM2 expression levels negatively correlate with p53 protein levels and activity, resulting in poor survival and prognosis (Mccann et al., 1995; Quesnel et al., 1994). Thus, amplification and/or overexpression of MDM2 represents alternative means to mutations in p53 for escaping growth control in cancer.
The DNA repair process requires building blocks, the deoxynucleotide triphosphates (dNTPs). CRL1FBXO1 participates in genome integrity and DDR by mediating the degradation of RRM2, a subunit of the ribonucleotide reductase that catalyzes the conversion of ribonucleotides to deoxyribonucleotides for both replicative and repair DNA synthesis (D’Angiolella et al., 2012). In response to a variety of genotoxic stimuli, FBXO1 is degraded, allowing the accumulation of RRM2 within the nucleus to enhance dNTPs production and facilitate DNA repair. Frequent cancer mutations in CCNF (which encodes FBXO1) are thought to contribute to genome instability (D’Angiolella et al., 2013).
E3s and cancer metabolism
To fuel cell growth and proliferation, cancer cells display fundamental changes in energy metabolism to enable increased nutrient acquisition and macromolecular precursor biosynthesis (Pavlova and Thompson, 2016). E3s contributes to the reprogramming of metabolism in cancer cells by regulating components of diverse metabolic processes.
One of the most common metabolic alteration in cancer cells is increased glucose uptake. Through the aerobic glycolysis pathway, cancer cells metabolize glucose at higher rates than normal cells, and convert it to lactate, a phenomenon known as the Warburg effect. Some cancer cells also have increased glutamine metabolism, which exceeds the metabolic use of other nonessential amino acids (Wise and Thompson, 2010). Two E3 complexes, APC/CCDH1 and CRL1βTrCP control the metabolism of glucose and glutamine at specific phases of the cell cycle in cancer cells (Colombo et al., 2011; Duan and Pagano, 2011). APC/CCDH1 mediates the degradation of PFKFB3 (6-phosphfructo-2-kinase/fructose-2,6-bisphosphatase, isoform 3) and GLS1 (glutaminase 1), both of which play key roles in the glycolysis and glutaminolysis pathways, as cells exit mitosis and enter G1. CRL1βTrCP specifically targets PFKFB3 during S phase. The oscillation in protein levels of these two enzymes coincides with their respective metabolic activities, in terms of lactic acid generation and glutamine utilization.
To adapt to the high reactive oxygen species (ROS) production due to elevated proliferation and metabolic activity, tumor cells leverage an increased antioxidant program to optimize ROS-driven proliferation and avoid ROS-triggered senescence or apoptosis. The transcriptional factor NRF2 regulates a network of genes with antioxidant functions that mitigate oxidative damage via detoxification of ROS and xenobiotics. NRF2 also regulates metabolic enzymes to rewire cellular metabolism to support antioxidant response (Romero et al., 2017; Wu and Papagiannakopoulos, 2020). Just as NRF2 protects normal cells, studies have shown that aberrant accumulation of NRF2 provides a survival advantage to cancer cells. Indeed, NRF2 overexpression is associated with a poor prognosis in patients with various types of cancer (Jaramillo and Zhang, 2013). At the protein level, NRF2 is regulated by the E3 CRL3KEAP1 (Furukawa and Xiong, 2005; Itoh et al., 1999; Zhang et al., 2004). Inactivating mutations in KEAP1 are found in ~20% NSCLCs, impairing its ability to target NRF2 for degradation (Singh et al., 2006; Wu and Papagiannakopoulos, 2020). While somatic mutations in KEAP1 occur throughout the protein, NRF2 activating mutations predominantly occur within either the DLG or the ETGE degron, which mediate the interaction between NRF2 and KEAP1 (McMahon et al., 2006; Tong et al., 2006). The high mutation frequency of KEAP1 and NRF2 suggests a critical role for this pathway in lung tumor development, as confirmed by numerous mouse models. Genetic alterations in CUL3 (which encodes the backbone protein of the CRL3KEAP1 E3 complex) have also been found to be significantly mutated in human cancers (Armenia et al., 2018; Campbell et al., 2016; Tokheim et al., 2021). Interestingly, at least 3 other CRL3 SRs (i.e., KLHL6, LZTR1 and SPOP) play a tumor suppressor role (see above), therefore, mutations in CUL3 are not expected provide a growth advantage by affecting only KEAP1 activity.
E3s and metastasis
Metastasis describes the process through which malignant cells develop the ability to invade tissues beyond their normal boundaries and seed new tumors at secondary sites (Gupta and Massague, 2006; Lambert et al., 2017). For successful colonization, tumor cells need to disseminate from the primary site, enter circulation, and adapt to foreign tissue microenvironments. Metastasis is a complex process which requires a coordinated network of cellular regulators, including kinases, transcription factors, and E3s. Multiple E3s are reported to either positively or negatively regulate the cellular processes involved metastasis, and some of these E3s are deregulated in human cancers.
CRL1FBXO22 has been shown to suppress metastasis by mediating the degradation of BACH1, a pro-metastatic transcription factor (Lignitto et al., 2019). Heme, the catabolic target of Heme Oxygenase 1 (HO1) enzyme, enhances the interaction between FBXO22 and BACH1. NRF2 regulates HO1 expression, thus, NSCLCs bearing NRF2 stabilizing mutations (see above) experience higher induction of HO1, leading to prominent catabolic processing of Heme. As a result, BACH1 interacts less with FBXO22, accumulates, and promotes metastatic events.
CRL1FBXL7 ubiquitylates c-SRC (Moro et al., 2020), a proto-oncoprotein that has been strongly implicated in cancer development and metastasis formation (Summy and Gallick, 2003). In prostate and pancreatic cancer models, downregulation of FBXL7 promotes metastatic progression by increasing cell invasion and migration in a c-SRC-dependent manner. Importantly, the promoter of the gene encoding FBXL7 is hypermethylated in advanced prostate and pancreatic cancers, correlating with decreased FBXL7 mRNA and protein levels (Moro et al., 2020).
E3s and tumor angiogenesis
Cancer cells hijack agiogenesis, the growth of new blood vessels from pre-existing blood vessels, to supply nutrients and oxygen for the rapid expansion of the tumor mass (Potente et al., 2011). In response to hypoxia, tumor tissues produce and release pro-angiogenic factors, such as VEGF (vasculo-endothelial growth factor) and FGFs (fibroblast growth factors), to activate endothelial cells of pre-existing blood vessels (Niu and Chen, 2010). Vessels also need to adjust their morphology and function to meet changing tissue oxygen demands.
A master regulator of hypoxia-induced angiogenesis is the transcription factor HIF (hypoxia inducible factor) (Semenza, 2012). HIF controls the transcription of multiple genes that promotes oxygen delivery to tissues by inducing angiogenesis, including VEGF, erythropoietin, the angiopoietin growth factors, stromal-derived factor 1, and glucose transporter 1 (Koh and Powis, 2012; Krock et al., 2011). HIF contains a labile, oxygen-sensitive alpha subunit (i.e., HIF1α or HIF2α) and a constitutively expressed beta subunit (HIF1β or Aryl). In the presence of high oxygen levels, HIF alpha subunit are hydroxylated and targeted for degradation by VHL (von Hippel–Lindau), a SR of the CRL2 complex (Kaelin, 2017; Maxwell et al., 1999). Under hypoxic conditions, HIF alpha subunit escape VHL-mediated degradation and activate hypoxia-inducible genes. Depletion or mutations in VHL allows expression of HIF target genes even under normoxic conditions, which supports vascularization and growth of tumors. Inactivation of VHL is causative of the von Hippel–Lindau syndrome, a familiar predisposition to develop tumors in multiple organs (Lonser et al., 2003). Mutations in the VHL protein are frequently found in familial and sporadic clear cell carcinomas of the kidney, hemangioblastomas of the retina and central nervous system, pancreatic cysts and tumors, as well as pheochromocytomas, underscoring its tumor suppressor function in the pathogenesis of these tumors (Kaelin, 2017). Neoplasms caused by VHL inactivation are highly vascularized due to constitutive activation of HIF target genes (Kaelin, 2002). HIF activation is an early event in the evolution of neoplastic kidney lesions in VHL patients, and increased expression of certain HIF target genes is associated with disease progression (Mandriota et al., 2002). These observations also suggest the therapeutic strategy of inhibiting HIF or its target genes in VHL-defective carcinogenesis. For example, clear cell renal carcinomas bear high VEGF levels due to loss of pVHL, and are highly sensitive to single agents directed against VEGF (Choueiri and Motzer, 2017). Moreover, an HIF2α inhibitor is showing promise for treatment of clear cell renal carcinoma (Courtney et al., 2018). These findings demonstrate that targeting the VHL-HIF pathway could be an effective strategy for the treatment of certain angiogenic tumors, notably kidney cancer.
E3s and oncogenic viruses
A few human cancers are triggered by viruses that promote tumorigenesis through activation of oncogenic pathways and/or inactivation of tumor suppressors. The versatility of the ubiquitin system in regulating multiple cellular pathways makes it an attractive target for oncogenic viruses. Viruses may utilize E3s in several ways to promote the host cell proliferation: some viral proteins inhibit E3s activities, others hijack host E3s to target new substrate proteins.
Human oncogenic papillomavirus (HPV) infection plays an etiologic role in a subset of cervical and head and neck cancers (Burd, 2003; Tumban, 2019). The major oncoproteins of HPV are encoded by the viral E6 and E7 genes, both of which exploit the ubiquitin-system to inactivate cellular tumor suppressor proteins and facilitate HPV-induced carcinogenesis (McLaughlin-Drubin and Munger, 2009; Munger and Howley, 2002; Scheffner and Whitaker, 2003). E6 binds through its N-terminus to the E6-AP E3 ligase, the founding member of the HECT family of E3s, thereby promoting binding, ubiquitylation, and consequent degradation of the tumor suppressor p53. Of note, in HPV-negative cells, E6-AP has no role in p53 degradation (BeerRomero et al., 1997; Scheffner, 1998), showing that E6 utilizes a cellular E3 to inactive this tumor suppressor. E6-AP was also shown to target the pro-apoptotic protein BAK for degradation, thus inhibiting apoptosis (Thomas and Banks, 1998). As for E7, an important aspect in its oncogenic activity is the inactivation of the retinoblastoma (RB) tumor suppressor. Analysis of E7-associated proteins uncovered CUL2 as an interactor of E7, and subsequently showed to promote degradation of RB in a E7-dependnet manner (Huh et al., 2007). Similar mechanism has been employed by other viruses. For example, the adenoviral E4orf6 protein hijacks CRL5 to target p53 for degradation (Querido et al., 2001).
In addition to hijacking host E3s to target tumor suppressors, viral protein may also inhibit E3s to stabilize oncoproteins. The large tumor antigen (LTAg) encoded by Simian virus 40 (SV40) is a powerful oncoprotein that is capable of transforming a variety of cell types (Sullivan and Pipas, 2002). LTAg was found to inhibit FBXW7-driven degradation of proto-oncoproteins such as cyclin E (Welcker and Clurman, 2005). LTAg binds FBXW7 through its phospho-epitope within the C-terminus that closely mimics the consensus phospho-degron for FBXW7. LTAg is not degraded, but instead acts as a decoy functioning as a competitive inhibitor for FBXW7, thereby reducing the turnover of cellular substrates. LTAg also associated with CUL 7 (Ali et al., 2004), and mutants with CUL7 binding deficiency exhibit a decreased ability to support anchorage-independent growth of mouse embryonic fibroblasts (Hartmann et al., 2014). Further studies regarding the link between LTAg–CUL7 interaction and cell transformation will be necessary to better understand the mechanisms by which this viral protein leads to tumorigenesis.
E3s in Cancer Immunotherapy
The immune system monitors and eradicates the formation and progression of neoplastic tissues in a process called immunosurveillance (Kim et al., 2007c). Often, tumors hijack immune suppression mechanisms to avoid detection by the immune system. In recent years, great achievements have been made on multiple cancers by blocking immune inhibitory signals, enabling patients to produce an effective anti-tumor response (Mahoney et al., 2015). Immunotherapies that consist of antibodies targeting immune checkpoint proteins, such as PD-1 (programmed cell death protein 1) on T cells, PD-L1 (programmed cell death 1 ligand 1) on antigen presenting cells and tumor cells, CTLA-4 (cytotoxic T lymphocyte antigen 4), have revolutionized cancer treatment (Boussiotis, 2016; Dong et al., 2002).
Emerging evidences suggest that E3s contribute to the regulation of tumor immunosurveillance, and can be exploited for enhancing antitumor immunity (Fujita et al., 2019). Large-scale screens revealed that several E3s, most notably CBL-B, are involved in T cell activation (Shifrut et al., 2018; Zhou et al., 2014). CBL-B negatively regulates T cell activation (Paolino and Penninger, 2010), and cblb−/− T cells are less susceptible to PD-1-mediated inhibition (Fujiwara et al., 2016). Therefore, CBL-B has become a focus for understanding the mechanisms of antitumor immunity, and its activity could serve as a an indicator of responsiveness to PD-1/PD-L1 immunotherapies.
Other studies have suggested to a role of E3s in directly regulating the levels of checkpoint molecules. For example, CRL1FBXO38 targets PD-1 for proteasomal degradation, thereby promoting T cell anti-tumor immunity (Meng et al., 2018). Moreover, FBXO38 transcription is downregulated in CD8+PD-1+ tumor-infiltrating T cells in colorectal and hepatocellular carcinoma patients, and its levels negatively correlated with cell-surface expression of PD-1. Another study showed that PD-L1 protein abundance is regulated by CRL3SPOP (Zhang et al., 2018). Loss-of-function mutations in SPOP compromise PD-L1 degradation, leading to accumulation of PD-L1 levels and reduced numbers of tumor-infiltrating lymphocytes in prostate murine tumors. Further understanding of these biochemical events will provide insight into the mechanisms contributing to the efficacy of tumor immunotherapy and the development of resistance.
E3s as targets in cancer therapy
The critical roles played by E3s in the regulation of fundamental cellular processes and cancer development suggest that they could be key targets for therapeutic strategies. In the following section, we will describe strategies of targeting E3s for cancer therapies (Figure 3).
Figure 3: Therapeutic strategies targeting E3s.
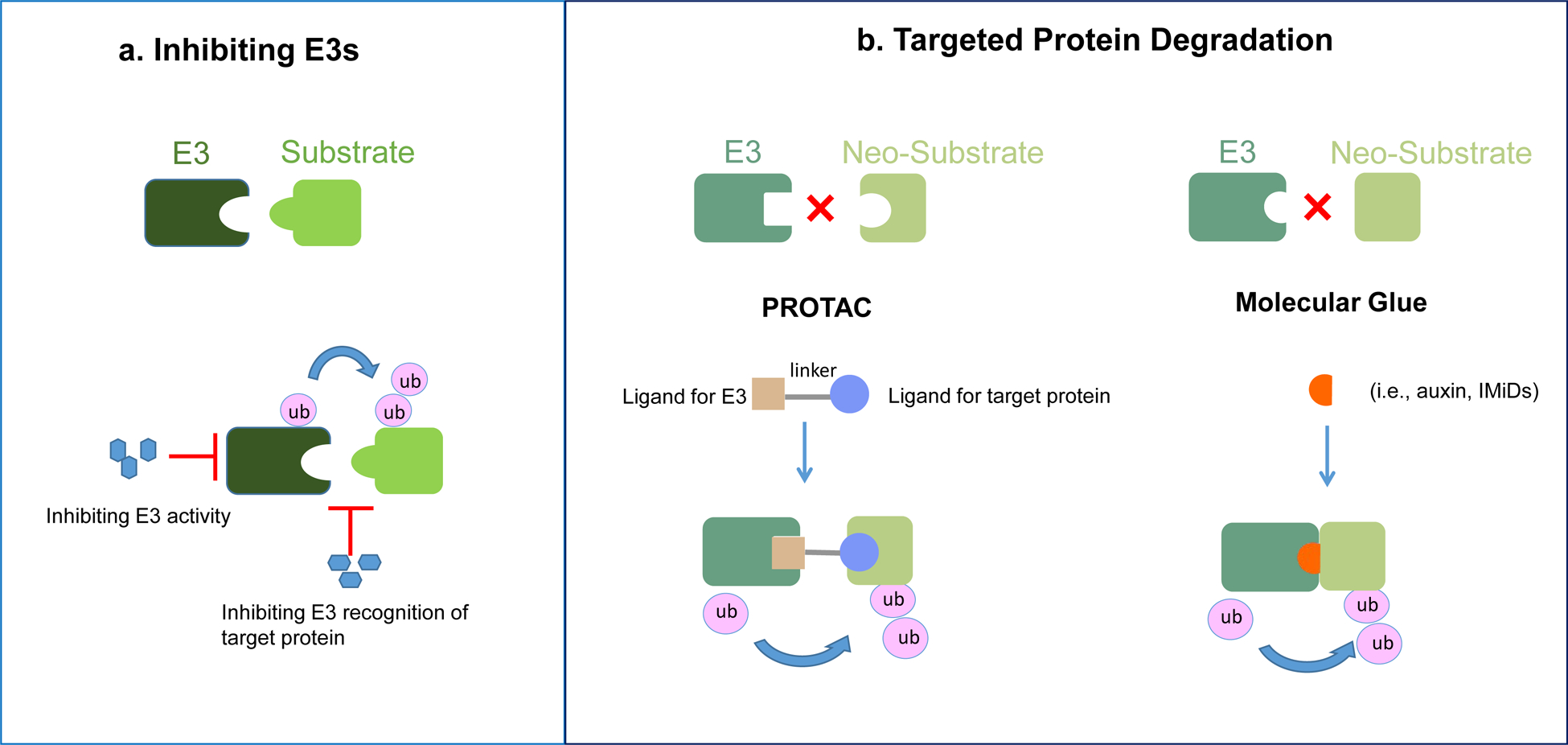
a. E3 inhibitors. Small molecules that either inhibit the activity of E3, or block its interaction with substrates.
b. Targeted protein degradation. PROteolysis TArgeting Chimeras (PROTACs) are bifunctional molecules simultaneously engage a protein of interest and a E3, to form a ternary complex and enable the E3 to mediate the ubiquitylation and subsequent degradation of the neo-substrate. Molecule glues are small compounds that redirect E3s to neo-substrates. They can be either natural compounds (for example, plant hormones), or synthetic compounds (such as IMiDs and indisulam). The induced proximity by the small molecule leads to substrate ubiquitylation by E3s and subsequent proteasomal degradation. Molecule glues do not require high affinity on both the E3 and the neo-substrate, eliminating the need for at least one of the two ligand-binding pockets required for PROTACs
The success of proteasome inhibitors in cancer treatment is one of the reasons that makes targeting the ubiquitin-proteasome system an attractive therapeutic modality. Despite the effectiveness of proteasome inhibitors (e.g., bortezomib, ixazomib, and carfilzomib) in the treatment of certain cancers, especially hematological malignancies, undesirable side effects are observed presumably due to the non-specific inhibition of the proteasome-dependent degradation of many cellular substrates. Inhibitors targeting specific, oncogenic E3s provides the rational for the development of more selective drugs with fewer side effects. Specifically, inhibitors may target the E3 levels or their catalytic activity, block its interaction with substrates, or alter its subcellular localization (Nalepa et al., 2006; Skaar et al., 2014).
One obvious target for cancer therapy is MDM2, which targets the tumor suppressor p53 and is overexpressed in several types of human tumors. Small molecules that designed to disrupt MDM2-p53 interactions, inhibit MDM2 expression, or its activity are expected to activate the p53 pathway, leading to cell cycle arrest and apoptosis. Intensive research efforts in the past decades resulted in several small-molecule compounds, such as nutlins, that made their way into human clinical trials demonstrating antitumor effects (Tisato et al., 2017; Vassilev et al., 2004; Wang et al., 2017). However, drug-related toxicities and acquired resistance caused by the emergence of p53 mutations, are two major concerns arising from these clinical trials. Nevertheless, these discoveries are paving the way for the design of new MDM2 inhibitors with higher potency and selectivity, leading to better pharmacokinetics.
SKP2 is another E3 with oncogenic potentials and is overexpressed in certain human cancers (see above). Different classes of SKP2 inhibitors are designed, either to suppress the expression of SKP2 (Jiang et al., 2020), to interrupt its interaction with other subunits of the CRL complex (Chan et al., 2013), or to block the interaction with its substrates (Wu et al., 2012). CRL1SKP2 and its cofactor CKS1 recognize a phospho-degron in the cell cycle inhibitor p27, and the structure analysis revealed a pocket between SKP2 and CKS1 that is flanked by residues required for p27 binding (Carrano et al., 1999; Hao et al., 2005). Compounds designed to target the binding interface for p27 were shown to selectively inhibit SKP2-mediated p27 degradation, leading to p27 accumulation and cell cycle arrest. These inhibitors showed some activity in cancer cell lines, as well as in mouse cancer models (Pavlides et al., 2013; Rodriguez et al., 2020; Wu et al., 2012).
Overall, despite huge efforts both in academia and industry, the strategy of targeting oncogenic E3s did not produce the effects predicted two decades ago (Garber, 2005). The hurdles could come from the fact that, E3s are not conventional enzymes with most of them lacking well-defined activity pockets to be targeted by small molecules. So, inhibition of these enzymes would require blocking protein-protein interactions (e.g., those between the E3 and the substarte or between the SR subunitcore subunits of the E3), with all the associated challenges.
Targeting E3s with tumor suppressor activity requires different approaches, such as restoring the E3 activity or exploiting the concept of synthetic lethality. For example, as described above, FBXL7 is hypermethylated in advanced prostate and pancreatic cancers, correlating with poor prognosis (Moro et al., 2020). Treatment with decitabine, a DNA-demethylating agent, recovers FBXL7 expression, promoting c-SRC degradation and reducing cell invasion and metastasis formation. The notion of targeting synthetic lethal vulnerabilities in cancer has been validated clinically through the effectiveness of poly(ADP)-ribose polymerase (PARP) inhibitors in breast and ovarian cancers with defect in BRCA1 and BRCA2 (Ashworth and Lord, 2018). Identifying novel E3 synthetic-lethal interactions will shed light on more discovery of targetable vulnerabilities in cancers harboring deficiencies or loss-of-function mutations in tumor suppressor E3s.
Controlled Proteostasis Targeted Protein Degradation
In recent years, huge efforts have been made to hijack E3s to promote the degradation of oncogenic substrates by proteolysis-targeting chimaera (PROTAC) technology or compounds that act as molecular glues. Controlled proteostasis provides for the opportunity to target the proteome once considered as “undruggable” by the traditional modality of drug discovery.
PROTAC technology utilizes bifunctional molecules to guide an E3 towards the recognition and consequent degradation of oncogenic substrates (Lai and Crews, 2017). Such molecules bind the target protein on one end and an E3 on the other. The PROTAC does not require directly binding to the active site. Compared with catalytic inhibition, degradation could lead to more potent effects on cell proliferation and a robust downstream signaling response. In fact, PROTACs often work at lower concentrations than small-molecule inhibitors, thanks to their catalytic mechanism of action (i.e., the target protein is eliminated and the PROTAC re-used to form new E3-PROTAC-substrate trimeric complexes). Our expanding knowledge of E3s and their substrates keeps adding more to the molecular toolbox for the PTOTAC design. A variety of E3s including cereblon (CRBN), VHL, β-TrCP, MDM2, and cIAP have been successfully exploited by this strategy, and the approach was demonstrated for targets that include hormone receptors (ER and AR), kinases, and bromodomain-containing proteins (Cromm and Crews, 2017). ARV-110, which targets the androgen receptor (AR), is the first PROTAC that has advanced into the clinic (Mullard, 2019). These trials will provide important insight into the efficacy of these degrader compounds in vivo.
However, there are associated challenges for the PROTAC technology, including the requirement of a bi-functional molecule with high affinity on both ends, and a molecular weight often above 500 Da, which may limit cell availability. Another emerging strategy for targeted proteolysis utilizes the so-called “molecular glues”, which similarly induces a neo-interaction between an E3 and a protein of interest. Two advantages of molecular glues include the use of non-bivalent molecules that are small enough to be drug-like compounds, and the lack of requirement for high affinity on both the E3 and the neo-substrate. The latter eliminates the need for at least one of the two ligand-binding pockets required for PROTACs, thus, molecular glues can target not only undruggable substrates but also “unligandable” proteins.
The idea of molecular glues in targeted degradation is partly inspired by the finding that auxin, a plant hormone, binds to the plant E3 CRL1TIR1, creating a molecular surface that favors substrates binding (Tan et al., 2007). A similar mechanism was found in the jasmonate signaling. Jasmonates (JAs) are a family of plant hormones that regulates plant growth, development, and response to stress. An amino acid-conjugated form of JA, (3R,7S)-jasmonoyl-L-isoleucine, mediates jasmonate signaling by promoting binding of the F-box protein COI1 to the transcriptional repressor Jasmonate ZIM domain (JAZ) proteins, thereby inhibiting transcription of JA-responsive genes (Katsir et al., 2008; Sheard et al., 2010).
Although molecular glues have been first carachterized in plants, their relevance as a therapeutic modality was first introduced by the discovery of the mechanism of action for immunomodulatory drugs (IMiDs). IMiDs such as thalidomide and its analogues lenalidomide and pomalidomide are used as anticancer agents (Bartlett et al., 2004), but the biochemical basis for their antineoplastic activity was not fully understood until recently. IMiDs bind to CRBN, a CRL4 SR (Ito et al., 2010), redirecting CRL4CRBN to neo-substrates, including the lymphoid transcription factors Ikaros and Aiolos (also known as IKZF1 and IKZF3) (Gandhi et al., 2014; Kronke et al., 2014; Lu et al., 2014), casein kinase 1α (CK1α) (Kronke et al., 2015), and the translation termination factor GSPT1 (Matyskiela et al., 2016). Ikaros proteins are essential for the survival of multiple myeloma cells, accounting for the clinical efficacy of IMiDs. CK1α and GSPT1 are the main targets of IMiDs in the treatment of 5q-deletion-associated myelodysplastic syndrome and acute myeloid leukemia, respectively. Structure analysis showed that lenalidomide binds the substrate-binding domain of CRBN, blocking the interaction with (and ubiquitylation of) MEIS2, an endogenous substrate of CRL4CRBN (Fischer et al., 2014). Lenalidomide and CRBN jointly provide the binding interface for neo-substrates (Matyskiela et al., 2016; Schafer et al., 2018). Notably, Ikaros proteins, CK1α, and GSPT1 share no obvious sequence homology; instead, they are recruited to CRBN through a β-hairpin containing a glycine residue, suggesting a “structural degron” determining CRBN-IMiDs selectivity. Moreover, recruitment specificity of neo-substrates varies among these different thalidomide derivatives. For instance, CK1α is recruited by lenalidomide, but not pomalidomide or thalidomide. Thus, structure and biochemistry insights into the specific interactions between thalidomide analogues with particular substrate will lead to the discovery of more potent compounds. Finally, current efforts are dedicated to the identification of new thalidomide derivatives that may target CRBN to different neo-substrates with additional beneficial effects.
Following these remarkable findings, the aryl-sulfonamide drug indisulam was also shown to redirect CRL4DCAF15 to neo-substrates. Indisulam is a potential anticancer agent originally discovered by screening sulfonamides for inhibition of cancer cell proliferation (Owa et al., 1999). Structural and biochemical studies revealed that indisulam, and its related compounds, E7820 and tasisulum, induce the binding of the RNA splicing factor RBM39 to DCAF15, promoting its degradation and producing anti-proliferative effects (Bussiere et al., 2020; Du et al., 2019; Faust et al., 2020; Han et al., 2017; Uehara et al., 2017).
Continued efforts in the field is leading toward the discovery of an increasing number of compounds acting as molecular glues with therapeutic potentials (Chamberlain and Hamann, 2019). Three different orthogonal approaches revealed CR8 and analogous compounds as molecular glue degraders (Lv et al., 2020; Mayor-Ruiz et al., 2020; Slabicki et al., 2020a). These molecules direct the CDK12–cyclin K complex to DDB1, a core subunit of the CRL4 complex, thereby inducing cyclin K degradation and exerting toxicity for cancer cells. Another screening looking at compounds that mediate the degradation of the oncogenic transcription factor BCL6 identified BI-3802, which binds to the BTB domain of BCL6 and promote its protein aggregation and degradation (Kerres et al., 2017; Slabicki et al., 2020b).
This accelerated progress on PROTACs and molecular glues provide new strategies on targeting what otherwise would be considered as undruggable, and even unligandable, protein targets. It also opens up the possibility for the development of pharmaceutical agents to restore E3-mediated ubiquitylation of substrates that are impaired by genetic alterations on either the E3s or themselves.
Conclusions
E3s act as molecular switches that control the cellular abundance of key regulatory components of signaling pathways. They are often deregulated in cancer through diverse mechanisms, resulting in altered expression and activity of their target proteins. A better appreciation of the mechanisms underlying deregulation of E3s would help to understand the initiation and progression of cancer, and may provide critical breakthroughs for targeted therapy. In addition to inhibit E3 activities, controlled proteostasis as a therapeutic modality has attracted substantial interest and investments, and made great progress in just the last few years. However, the mechanism of action of drugs that function as molecular glues has been identified a posteriori in most cases. How to identify de novo molecular glues that repurpose E3s for a specific target remains an open, fundamental question in the field of ubiquitin-mediated degradation. Moreover, how to ensure that molecular glues induce productive ubiquitylation, rather tha just proximity, remains a major challenge. As the field advances, more rational and systematic approaches are being leveraged, and we anticipate seeing many further clinical successes in this area.
Acknowledgements
We apologize to authors whose relevant publications were not cited due to space limitations. The authors thank Tania J. González -Robles for critical reading of the manuscript and discussion. M.P. is an investigator with the Howard Hughes Medical Institute and his laboratory is funded by grants from the National Institute of Health (R01-CA76584 and R35-GM136250). S.D. was supported by 2T32CA009161-41 Training Program in Molecular Oncology and Tumor Immunology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
M.P. is a consultant for and has financial interests in Coho Therapeutics, CullGen, Kymera Therapeutics, and SEED Therapeutics. M.P. is a cofounder of Coho Therapeutics, is on the SAB of CullGen and Kymera Therapeutics, and is a consultant for Santi Therapeutics. S.D. declares no competing interests.
References
- Abe T, Umeki I, Kanno SI, Inoue SI, Niihori T, and Aoki Y (2020). LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ 27, 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alao JP (2007). The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SH, Kasper JS, Arai T, and DeCaprio JA (2004). Cu17/p185/p193 binding to simian virus 40 large T antigen has a role in cellular transformation. Journal of Virology 78, 2749–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Ren S, Murphy SJ, Dalangood S, Chang C, Pang X, Cui Y, Wang L, Pan Y, Zhang X, et al. (2015). Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol Cell 59, 904–916. [DOI] [PubMed] [Google Scholar]
- Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I, et al. (2018). The long tail of oncogenic drivers in prostate cancer. Nat Genet 50, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth A, and Lord CJ (2018). Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat Rev Clin Oncol 15, 564–576. [DOI] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. (2012). Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 44, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett JB, Dredge K, and Dalgleish AG (2004). Timeline - The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nature Reviews Cancer 4, 314–322. [DOI] [PubMed] [Google Scholar]
- Bassermann F, Eichner R, and Pagano M (2014). The ubiquitin proteasome system - Implications for cell cycle control and the targeted treatment of cancer. Bba-Mol Cell Res 1843, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann U, Fernandez-Saiz V, Rudelius M, Lemeer S, Rad R, Knorn AM, Slawska J, Engel K, Jeremias I, Li Z, et al. (2014). Disruption of the PRKCD-FBXO25-HAX-1 axis attenuates the apoptotic response and drives lymphomagenesis. Nat Med 20, 1401–1409. [DOI] [PubMed] [Google Scholar]
- BeerRomero P, Glass S, and Rolfe M (1997). Antisense targeting of E6AP elevates p53 in HPV-infected cells but not in normal cells. Oncogene 14, 595–602. [DOI] [PubMed] [Google Scholar]
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, and Barker PA (2008). cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30, 689–700. [DOI] [PubMed] [Google Scholar]
- Bigenzahn JW, Collie GM, Kartnig F, Pieraks M, Vladimer GI, Heinez LX, Sedlyarov V, Schischlik F, Fauster A, Rebsamen M, et al. (2018). LZTR1 is a regulator of RAS ubiquitination and signaling. Science 362, 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussiotis VA (2016). Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med 375, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow L, and Huang DT (2016). Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Bio 17, 626–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd EM (2003). Human papillomavirus and cervical cancer. Clin Microbiol Rev 16, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdova K, Yang H, Faedda R, Hume S, Chauhan J, Ebner D, Kessler BM, Vendrell I, Drewry DH, Wells CI, et al. (2019). E2F1 proteolysis via SCF-cyclin F underlies synthetic lethality between cyclin F loss and Chk1 inhibition. EMBO J 38, e101443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, and Draetta GF (2003). Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature 426, 87–91. [DOI] [PubMed] [Google Scholar]
- Busino L, Millman SE, Scotto L, Kyratsous CA, Basrur V, O’Connor O, Hoffmann A, Elenitoba-Johnson KS, and Pagano M (2012). Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol 14, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere DE, Xie L, Srinivas H, Shu W, Burke A, Be C, Zhao J, Godbole A, King D, Karki RG, et al. (2020). Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat Chem Biol 16, 15–23. [DOI] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et al. (2016). Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T, and Pagano M (2004). The SCF ubiquitin ligase: Insights into a molecular machine. Nat Rev Mol Cell Bio 5, 739–751. [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, and Pagano M (1999). SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature Cell Biology 1, 193–199. [DOI] [PubMed] [Google Scholar]
- Castel P, Cheng A, Cuevas-Navarro A, Everman DB, Papageorge AG, Simanshu DK, Tankka A, Galeas J, Urisman A, and McCormick F (2019). RIT1 oncoproteins escape LZTR1-mediated proteolysis. Science 363, 1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikovsky AC, Li C, Jeng EE, Loebell S, Lee MC, Murray CW, Cheng R, Demeter J, Swaney DL, Chen S-H, et al. (2021). The AMBRA1 E3 ligase adaptor regulates Cyclin D protein stability. Nature In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain PP, and Hamann LG (2019). Development of targeted protein degradation therapeutics. Nat Chem Biol 15, 937–944. [DOI] [PubMed] [Google Scholar]
- Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, et al. (2013). Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 154, 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao JR, Parganas E, Boyd K, Hong CY, Opferman JT, and Ihle JN (2008). Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 452, 98–102. [DOI] [PubMed] [Google Scholar]
- Chardin P, Camonis JH, Gale NW, Vanaelst L, Schlessinger J, Wigler MH, and Barsagi D (1993). Human Sos1 - a Guanine-Nucleotide Exchange Factor for Ras That Binds to Grb2. Science 260, 1338–1343. [DOI] [PubMed] [Google Scholar]
- Choi J, Lee K, Ingvarsdottir K, Bonasio R, Saraf A, Florens L, Washburn MP, Tadros S, Green MR, and Busino L (2018). Loss of KLHL6 promotes diffuse large B-cell lymphoma growth and survival by stabilizing the mRNA decay factor roquin2. Nature Cell Biology 20, 586–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choueiri TK, and Motzer RJ (2017). Systemic Therapy for Metastatic Renal-Cell Carcinoma. N Engl J Med 376, 354–366. [DOI] [PubMed] [Google Scholar]
- Ci WM, Polo JM, and Melnick A (2008). B-cell lymphoma 6 and the molecular pathogenesis of diffuse large B-cell lymphoma. Curr Opin Hematol 15, 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A, and Burleson M (2020). SPOP and cancer: a systematic review. Am J Cancer Res 10, 704–726. [PMC free article] [PubMed] [Google Scholar]
- Clijsters L, Hoencamp C, Calis JJA, Marzio A, Handgraaf SM, Cuitino MC, Rosenberg BR, Leone G, and Pagano M (2019). Cyclin F Controls Cell-Cycle Transcriptional Outputs by Directing the Degradation of the Three Activator E2 Fs. Mol Cell 74, 1264–1277 e1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo SL, Palacios-Callender M, Frakich N, Carcamo S, Kovacs I, Tudzarova S, and Moncada S (2011). Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. P Natl Acad Sci USA 108, 21069–21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, Zojwalla NJ, Lowe AM, Wang K, Wallace EM, et al. (2018). Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J Clin Oncol 36, 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromm PM, and Crews CM (2017). Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem Biol 24, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, and Pagano M (2012). Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 149, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, Dynlacht B, and Pagano M (2010). SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 466, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Esencay M, and Pagano M (2013). A cyclin without cyclin-dependent kinases: cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol 23, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, and Morgan DO (2016). Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol Cell 64, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Vega M, Burrows JF, and Johnston JA (2011). Ubiquitination: Added complexity in Ras and Rho family GTPase function. Small GTPases 2, 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, Dustin M, Huang DC, Taunton J, and Pagano M (2009). betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell 33, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8, 793–800. [DOI] [PubMed] [Google Scholar]
- Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, and Pagano M (2006). S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471. [DOI] [PubMed] [Google Scholar]
- Du X, Volkov OA, Czerwinski RM, Tan H, Huerta C, Morton ER, Rizzi JP, Wehn PM, Xu R, Nijhawan D, et al. (2019). Structural Basis and Kinetic Pathway of RBM39 Recruitment to DCAF15 by a Sulfonamide Molecular Glue E7820. Structure 27, 1625–1633 e1623. [DOI] [PubMed] [Google Scholar]
- Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, Chapuy B, Shipp M, Chiarle R, and Pagano M (2012). FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature 481, 90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, and Pagano M (2011). Linking metabolism and cell cycle progression via the APC/CCdh1 and SCFbetaTrCP ubiquitin ligases. Proc Natl Acad Sci U S A 108, 20857–20858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Skaar JR, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y, and Pagano M (2011). mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol Cell 44, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, and Xiong Y (2013). Signaling Pathways that Control Cell Proliferation. Csh Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele MJ, Enrico TP, Mouery RD, Wasserman D, Nachum S, and Tzur A (2020). Complex Cartography: Regulation of E2F Transcription Factors by Cyclin F and Ubiquitin. Trends Cell Biol 30, 640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust TB, Yoon H, Nowak RP, Donovan KA, Li Z, Cai Q, Eleuteri NA, Zhang T, Gray NS, and Fischer ES (2020). Structural complementarity facilitates E7820-mediated degradation of RBM39 by DCAF15. Nat Chem Biol 16, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, et al. (2014). Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D, and Pagano M (2008). Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer 8, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SY, Spiegelman VS, and Kumar KGS (2004). The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene 23, 2028–2036. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Tinoco R, Li Y, Senft D, and Ronai ZA (2019). Ubiquitin Ligases in Cancer Immunotherapy - Balancing Antitumor and Autoimmunity. Trends Mol Med 25, 428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Anstadt EJ, and Clark RB (2016). Cbl-b deficiency renders T cells resistant to PD-L1/PD-1 mediated suppression. J Immunol 196. [Google Scholar]
- Fulda S, and Debatin KM (2006). Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811. [DOI] [PubMed] [Google Scholar]
- Furukawa M, and Xiong Y (2005). BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol 25, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, Varmeh S, Zhang J, Cheng L, Sun Y, et al. (2015). SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol Cell 59, 917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, Ito T, Ando H, Waldman MF, Thakurta A, et al. (2014). Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol 164, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, et al. (2011). mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell 44, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber K (2005). Missing the target: ubiquitin ligase drugs stall. J Natl Cancer Inst 97, 166–167. [DOI] [PubMed] [Google Scholar]
- Gerstein AV, Almeida TA, Zhao GJ, Chess E, Shih IM, Buhler K, Pienta K, Rubin MA, Vessella R, and Papadopoulos N (2002). APC/CTNNB1 (beta-catenin) pathway alterations in human prostate cancers. Gene Chromosome Canc 34, 9–16. [DOI] [PubMed] [Google Scholar]
- Grandal MV, Zandi R, Pedersen MW, Willumsen BM, van Deurs B, and Poulsen HS (2007). EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 28, 1408–1417. [DOI] [PubMed] [Google Scholar]
- Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, and Krek W (2001). Skp2 is oncogenic and overexpressed in human cancers. P Natl Acad Sci USA 98, 5043–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, Margottin-Goguet F, Jackson PK, Yamasaki L, and Pagano M (2003). Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell 4, 799–812. [DOI] [PubMed] [Google Scholar]
- Gupta GP, and Massague J (2006). Cancer metastasis: building a framework. Cell 127, 679–695. [DOI] [PubMed] [Google Scholar]
- Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, and Nijhawan D (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, and Pavletich NP (2005). Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCFSkp2 ubipuitin ligase. Mol Cell 20, 9–19. [DOI] [PubMed] [Google Scholar]
- Harbour JW, and Dean DC (2000). The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 14, 2393–2409. [DOI] [PubMed] [Google Scholar]
- Hartmann T, Xu XS, Kronast M, Muehlich S, Meyer K, Zimmermann W, Hurwitz J, Pan ZQ, Engelhardt S, and Sarikas A (2014). Inhibition of Cullin-RING E3 ubiquitin ligase 7 by simian virus 40 large T antigen. P Natl Acad Sci USA 111, 3371–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, and Ciechanover A (1998). The ubiquitin system. Annu Rev Biochem 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hoxhaj G, and Manning BD (2020). The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nature Reviews Cancer 20, 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, and Chen J (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131, 901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh K, Zhou X, Hayakawa H, Cho JY, Libermann TA, Jin J, Harper JW, and Munger K (2007). Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol 81, 9737–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igney FH, and Krammer PH (2002). Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2, 277–288. [DOI] [PubMed] [Google Scholar]
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, and Handa H (2010). Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, and Yamamoto M (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Gene Dev 13, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Durocher D (2013). Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell 49, 795–807. [DOI] [PubMed] [Google Scholar]
- Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, Bernasocchi T, Civenni G, Losa M, Svinkina T, et al. (2017). Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med 23, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo MC, and Zhang DD (2013). The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 27, 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesenberger V, and Jentsch S (2002). Deadly encounter: ubiquitin meets apoptosis. Nat Rev Mol Cell Biol 3, 112–121. [DOI] [PubMed] [Google Scholar]
- Jiang WX, Lin M, and Wang ZW (2020). Dioscin: A new potential inhibitor of Skp2 for cancer therapy. Ebiomedicine 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG Jr. (2002). Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2, 673–682. [DOI] [PubMed] [Google Scholar]
- Kaelin WG Jr. (2017). The VHL Tumor Suppressor Gene: Insights into Oxygen Sensing and Cancer. Trans Am Clin Climatol Assoc 128, 298–307. [PMC free article] [PubMed] [Google Scholar]
- Kale J, Osterlund EJ, and Andrews DW (2018). BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kales SC, Ryan PE, Nau MM, and Lipkowitz S (2010). Cbl and human myeloid neoplasms: the Cbl oncogene comes of age. Cancer Res 70, 4789–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanie T, Onoyama I, Matsumoto A, Yamada M, Nakatsumi H, Tateishi Y, Yamamura S, Tsunematsu R, Matsumoto M, and Nakayama KI (2012). Genetic reevaluation of the role of F-box proteins in cyclin D1 degradation. Mol Cell Biol 32, 590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsir L, Schilmiller AL, Staswick PE, He SY, and Howe GA (2008). COI1 is a critical component of a receptor for jasmonate and the bacterial virulence factor coronatine. P Natl Acad Sci USA 105, 7100–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerres N, Steurer S, Schlager S, Bader G, Berger H, Caligiuri M, Dank C, Engen JR, Ettmayer P, Fischerauer B, et al. (2017). Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Rep 20, 2860–2875. [DOI] [PubMed] [Google Scholar]
- Kim CJ, Song JH, Cho YG, Kim YS, Kim SY, Nam SW, Yoo NJ, Lee JY, and Park WS (2007a). Somatic mutations of the beta-TrCP gene in gastric cancer. APMIS 115, 127–133. [DOI] [PubMed] [Google Scholar]
- Kim H, Chen J, and Yu X (2007b). Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316, 1202–1205. [DOI] [PubMed] [Google Scholar]
- Kim R, Emi M, and Tanabe K (2007c). Cancer immunoediting from immune surveillance to immune escape. Immunology 121, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters JM, and Kirschner MW (1996). How proteolysis drives the cell cycle. Science 274, 1652–1659. [DOI] [PubMed] [Google Scholar]
- Koch A, Waha A, Hartmann W, Hrychyk A, Schuller U, Waha A, Wharton KA, Fuchs SY, von Schweinitz D, and Pietsch T (2005). Elevated expression of Wnt antagonists is a common event in hepatoblastomas. Clin Cancer Res 11, 4295–4304. [DOI] [PubMed] [Google Scholar]
- Koh MY, and Powis G (2012). Passing the baton: the HIF switch. Trends Biochem Sci 37, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock BL, Skuli N, and Simon MC (2011). Hypoxia-induced angiogenesis: good and evil. Genes Cancer 2, 1117–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, et al. (2015). Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 523, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchay S, Duan S, Schenkein E, Peschiaroli A, Saraf A, Florens L, Washburn MP, and Pagano M (2013). FBXL2- and PTPL1-mediated degradation of p110-free p85beta regulatory subunit controls the PI(3)K signalling cascade. Nat Cell Biol 15, 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, Florens L, Washburn MP, Collazo-Lorduy A, Castillo-Martin M, et al. (2017). PTEN counteracts FBXL2 to promote IP3R3- and Ca(2+)-mediated apoptosis limiting tumour growth. Nature 546, 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, et al. (2017). Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 317, 2402–2416. [DOI] [PubMed] [Google Scholar]
- Lai AC, and Crews CM (2017). Induced protein degradation: an emerging drug discovery paradigm. Nature Reviews Drug Discovery 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging Biological Principles of Metastasis. Cell 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Gagnon J, and Therrien M (2020). ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol 21, 607–632. [DOI] [PubMed] [Google Scholar]
- Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, Price JC, Zhang S, England BM, Godwin AK, et al. (2012). Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet 44, 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, and Schlessinger J (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, and Oren M (2009). The first 30 years of p53: growing ever more complex. Nature Reviews Cancer 9, 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, et al. (1999). Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell 4, 1029–1040. [DOI] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, and Joazeiro CA (2008). Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS One 3, e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, Pass HI, Bhutkar AJ, Tsirigos A, Ueberheide B, et al. (2019). Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 178, 316–329 e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkowitz S, and Weissman AM (2011). RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer 11, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, and Sabatini DM (2020). mTOR at the nexus of nutrition, growth, ageing and disease (vol 29, pg 145, 2020). Nat Rev Mol Cell Bio 21, 246–246. [DOI] [PubMed] [Google Scholar]
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, and Oldfield EH (2003). von Hippel-Lindau disease. Lancet 361, 2059–2067. [DOI] [PubMed] [Google Scholar]
- Lopes RB, Gangeswaran R, McNeish IA, Wang YH, and Lemoine NR (2007). Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int J Cancer 120, 2344–2352. [DOI] [PubMed] [Google Scholar]
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, and Kaelin WG Jr. (2014). The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J, Lukas C, and Bartek J (2011). More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 13, 1161–1169. [DOI] [PubMed] [Google Scholar]
- Lv L, Chen P, Cao L, Li Y, Zeng Z, Cui Y, Wu Q, Li J, Wang JH, Dong MQ, et al. (2020). Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Schulman BA, and Harper JW (2013). Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep 14, 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney KM, Rennert PD, and Freeman GJ (2015). Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 14, 561–584. [DOI] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Bartek J, and Lukas J (2006). Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 23, 307–318. [DOI] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, and Lukas J (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, Wykoff CC, Maher ER, Harris AL, Ratcliffe PJ, et al. (2002). HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 1, 459–468. [DOI] [PubMed] [Google Scholar]
- Manfredi JJ (2010). The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Gene Dev 24, 1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, and Toker A (2017). AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao JH, Perez-Iosada J, Wu D, DelRosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, and Balmain A (2004). Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 432, 775–779. [DOI] [PubMed] [Google Scholar]
- Marmor MD, and Yarden Y (2004). Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 23, 2057–2070. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al. (2008). Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Gene Dev 22, 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, and Sixma TK (2012). RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, Fang W, Wang NY, Nguyen D, Houston J, et al. (2016). A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 535, 252.−+. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, and Ratcliffe PJ (1999). The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275. [DOI] [PubMed] [Google Scholar]
- Mayor-Ruiz C, Bauer S, Brand M, Kozicka Z, Siklos M, Imrichova H, Kaltheuner IH, Hahn E, Seiler K, Koren A, et al. (2020). Rational discovery of molecular glue degraders via scalable chemical profiling. Nat Chem Biol 16, 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccann AH, Kirley A, Carney DN, Corbally N, Magee HM, Keating G, and Dervan PA (1995). Amplification of the Mdm2 Gene in Human Breast-Cancer and Its Association with Mdm2 and P53 Protein Status. Brit J Cancer 71, 981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, and Munger K (2009). The human papillomavirus E7 oncoprotein. Virology 384, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M, Thomas N, Itoh K, Yamamoto M, and Hayes JD (2006). Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem 281, 24756–24768. [DOI] [PubMed] [Google Scholar]
- Meng XB, Liu XW, Guo XD, Jiang ST, Chen TT, Hu ZQ, Liu HF, Bai YB, Xue MM, Hu RG, et al. (2018). FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature 564, 130.-+. [DOI] [PubMed] [Google Scholar]
- Metzger MB, Hristova VA, and Weissman AM (2012). HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci 125, 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro L, Simoneschi D, Kurz E, Arbini AA, Jang S, Guaragnella N, Giannattasio S, Wang W, Chen YA, Pires G, et al. (2020). Epigenetic silencing of the ubiquitin ligase subunit FBXL7 impairs c-SRC degradation and promotes epithelial-to-mesenchymal transition and metastasis. Nat Cell Biol 22, 1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta M, Fidan M, Bellacchio E, Pantaleoni F, Schneider-Heieck K, Coppola S, Borck G, Salviati L, Zenker M, Cirstea IC, et al. (2019). Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum Mol Genet 28, 1007–1022. [DOI] [PubMed] [Google Scholar]
- Mullard A (2019). First targeted protein degrader hits the clinic. Nat Rev Drug Discov. [DOI] [PubMed] [Google Scholar]
- Munger K, and Howley PM (2002). Human papillomavirus immortalization and transformation functions. Virus Res 89, 213–228. [DOI] [PubMed] [Google Scholar]
- Musgrove EA, Caldon CE, Barraclough J, Stone A, and Sutherland RL (2011). Cyclin D as a therapeutic target in cancer. Nature Reviews Cancer 11, 558–572. [DOI] [PubMed] [Google Scholar]
- Nakayama KI, and Nakayama K (2006). Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 6, 369–381. [DOI] [PubMed] [Google Scholar]
- Nalepa G, Rolfe M, and Harper JW (2006). Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov 5, 596–613. [DOI] [PubMed] [Google Scholar]
- Niu G, and Chen XY (2010). Vascular Endothelial Growth Factor as an Anti-Angiogenic Target for Cancer Therapy. Curr Drug Targets 11, 1000–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, and Nicotera P (2003). Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4, 552–565. [DOI] [PubMed] [Google Scholar]
- Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nature Reviews Cancer 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ougolkov A, Zhang B, Yamashita K, Bilim V, Mai M, Fuchs SY, and Minamoto T (2004). Associations among beta-TrCP, an E3 ubiquitin ligase receptor, beta-catenin, and NF-kappa B in colorectal cancer. Jnci-J Natl Cancer I 96, 1161–1170. [DOI] [PubMed] [Google Scholar]
- Owa T, Yoshino H, Okauchi T, Yoshimatsu K, Ozawa Y, Sugi NH, Nagasu T, Koyanagi N, and Kitoh K (1999). Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J Med Chem 42, 3789–3799. [DOI] [PubMed] [Google Scholar]
- Paolino M, and Penninger JM (2010). Cbl-b in T-cell activation. Semin Immunopathol 32, 137–148. [DOI] [PubMed] [Google Scholar]
- Pavlides SC, Huang KT, Reid DA, Wu L, Blank SV, Mittal K, Guo L, Rothenberg E, Rueda B, Cardozo T, et al. (2013). Inhibitors of SCF-Skp2/Cks1 E3 ligase block estrogen-induced growth stimulation and degradation of nuclear p27kip1: therapeutic potential for endometrial cancer. Endocrinology 154, 4030–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschard P, and Park M (2003). Escape from Cbl-mediated downregulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 3, 519–523. [DOI] [PubMed] [Google Scholar]
- Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, and Pagano M (2006). SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell 23, 319–329. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, and Sabatini DM (2009). DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 137, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski MD, and Deshaies RJ (2005). Function and regulation of Cullin-RING ubiquitin ligases. Nat Rev Mol Cell Bio 6, 9–20. [DOI] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, and Carmeliet P (2011). Basic and Therapeutic Aspects of Angiogenesis. Cell 146, 873–887. [DOI] [PubMed] [Google Scholar]
- Qi J, and Ronai ZA (2015). Dysregulation of ubiquitin ligases in cancer. Drug Resist Updat 23, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qie S, and Diehl JA (2016). Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med 94, 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, and Branton PE (2001). Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev 15, 3104–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel B, Preudhomme C, Fournier J, Fenaux P, and Peyrat JP (1994). Mdm2 Gene Amplification in Human Breast-Cancer. Eur J Cancer 30a, 982–984. [DOI] [PubMed] [Google Scholar]
- Rauen KA (2013). The RASopathies. Annu Rev Genom Hum G 14, 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez S, Abundis C, Boccalatte F, Mehrotra P, Chiang MY, Yui MA, Wang L, Zhang HJ, Zollman A, Bonfim-Silva R, et al. (2020). Therapeutic targeting of the E3 ubiquitin ligase SKP2 in T-ALL. Leukemia 34, 1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nature Medicine 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, and Katoh M (2001). Expression profiles of beta TRCP1 and beta TRCP2, and mutation analysis of beta TRCP2 in gastric cancer. International Journal of Oncology 18, 959–964. [PubMed] [Google Scholar]
- Schafer PH, Ye Y, Wu L, Kosek J, Ringheim G, Yang Z, Liu L, Thomas M, Palmisano M, and Chopra R (2018). Cereblon modulator iberdomide induces degradation of the transcription factors Ikaros and Aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann Rheum Dis 77, 1516–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M (1998). Ubiquitin, E6-AP, and their role in p53 inactivation. Pharmacol Therapeut 78, 129–139. [DOI] [PubMed] [Google Scholar]
- Scheffner M, and Whitaker NJ (2003). Human papillomavirus-induced carcinogenesis and the ubiquitin-proteasome system. Semin Cancer Biol 13, 59–67. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2012). Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senft D, Qi J, and Ronai ZA (2018). Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer 18, 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever R, and Brugge JS (2015). Signal transduction in cancer. Cold Spring Harb Perspect Med 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheard LB, Tan X, Mao H, Withers J, Ben-Nissan G, Hinds TR, Kobayashi Y, Hsu FF, Sharon M, Browse J, et al. (2010). Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature 468, 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971 e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoneschi D, Rona G, Zhou N, Jeong Y, Milletti G, Jiang S, Arbini AA, O’sullivan A, Wang AA, Nithikasem S, et al. (2021). CRL4-AMBRA1 is a master regulator of D-type cyclins. Nature (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, et al. (2006). Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. Plos Med 3, 1865–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, and Pagano M (2013). Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol 14, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, and Pagano M (2014). SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov 13, 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, and Pagano M (2009). Control of cell growth by the SCF and APC/C ubiquitin ligases. Curr Opin Cell Biol 21, 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabicki M, Kozicka Z, Petzold G, Li YD, Manojkumar M, Bunker RD, Donovan KA, Sievers QL, Koeppel J, Suchyta D, et al. (2020a). The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabicki M, Yoon H, Koeppel J, Nitsch L, Roy Burman SS, Di Genua C, Donovan KA, Sperling AS, Hunkeler M, Tsai JM, et al. (2020b). Small-molecule-induced polymerization triggers degradation of BCL6. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steklov M, Pandolfi S, Baietti MF, Batiuk A, Carai P, Najm P, Zhang M, Jang H, Renzi F, Cai Y, et al. (2018). Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 362, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, and Reed SI (2001). Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 413, 316–322. [DOI] [PubMed] [Google Scholar]
- Sullivan CS, and Pipas JM (2002). T antigens of Simian virus 40: Molecular chaperones for viral replication and turnorigenesis. Microbiol Mol Biol R 66, 179.-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summy JM, and Gallick GE (2003). Src family kinases in tumor progression and metastasis. Cancer Metast Rev 22, 337–358. [DOI] [PubMed] [Google Scholar]
- Tan X, Calderon-Villalobos LI, Sharon M, Zheng C, Robinson CV, Estelle M, and Zheng N (2007). Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645. [DOI] [PubMed] [Google Scholar]
- Thien CB, and Langdon WY (2001). Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol 2, 294–307. [DOI] [PubMed] [Google Scholar]
- Thomas M, and Banks L (1998). Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 17, 2943–2954. [DOI] [PubMed] [Google Scholar]
- Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, et al. (2015). Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527, 389–393. [DOI] [PubMed] [Google Scholar]
- Tisato V, Voltan R, Gonelli A, Secchiero P, and Zauli G (2017). MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol 10, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokheim C, Wang X, Timms RT, Zhang B, Mena EL, Wang B, Chen C, Ge J, Chu J, Zhang W, et al. (2021). Systematic characterization of mutations altering protein degradation in human cancers. Mol Cell 81, 1292–1308 e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas A, Futter CE, and Eden ER (2014). EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol 24, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Kobayashi A, Katsuoka F, and Yamamoto M (2006). Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem 387, 1311–1320. [DOI] [PubMed] [Google Scholar]
- Tumban E (2019). A Current Update on Human Papillomavirus-Associated Head and Neck Cancers. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Minoshima Y, Sagane K, Sugi NH, Mitsuhashi KO, Yamamoto N, Kamiyama H, Takahashi K, Kotake Y, Uesugi M, et al. (2017). Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat Chem Biol 13, 675–680. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Vousden KH, and Prives C (2009). Blinded by the Light: The Growing Complexity of p53. Cell 137, 413–431. [DOI] [PubMed] [Google Scholar]
- Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, and Dixit VM (2000). ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol 10, 1359–1366. [DOI] [PubMed] [Google Scholar]
- Wade M, Li YC, and Wahl GM (2013). MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M, Wang YV, and Wahl GM (2010). The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 20, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, and Elledge SJ (2007). Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci U S A 104, 20759–20763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhao Y, Aguilar A, Bernard D, and Yang CY (2017). Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu P, Inuzuka H, and Wei W (2014). Roles of F-box proteins in cancer. Nat Rev Cancer 14, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, and Osada H (2004). M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A 101, 4419–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, and Clurman BE (2005). The SV40 large T antigen contains a decoy phosphodegron that mediates its interactions with Fbw7/hCdc4. J Biol Chem 280, 7654–7658. [DOI] [PubMed] [Google Scholar]
- Welcker M, and Clurman BE (2008). FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nature Reviews Cancer 8, 83–93. [DOI] [PubMed] [Google Scholar]
- Wise DR, and Thompson CB (2010). Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35, 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RS (2011). Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 30, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, and Cardozo TJ (2012). Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol 19, 1515–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WL, and Papagiannakopoulos T (2020). The Pleiotropic Role of the KEAP1/NRE2 Pathway in Cancer. Annu Rev Canc Biol 4, 413–435. [Google Scholar]
- Yang HS, Jansen AP, Komar AA, Zheng XJ, Merrick WC, Costes S, Lockett SJ, Sonenberg N, and Colburn NH (2003). The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol 23, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, and Hannink M (2004). Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 24, 10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JF, Bu X, Wang HZ, Zhu YS, Geng Y, Nihira NT, Tan YY, Ci YP, Wu F, Dai XP, et al. (2018). Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91.-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YC, Xiong XF, and Sun Y (2011). DEPTOR, an mTOR Inhibitor, Is a Physiological Substrate of SCF beta TrCP E3 Ubiquitin Ligase and Regulates Survival and Autophagy. Mol Cell 44, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, Cremasco V, Dougan SK, Cowley GS, Elpek K, et al. (2014). In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 506, 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, et al. (2009). Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell 36, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]