

Summary
Proteasomes are multisubunit complexes that catalyze the majority of protein degradation in mammalian cells to maintain protein homeostasis and influence the regulation of most cellular processes. The proteasome, a multicatalytic protease complex, is a ring-like structure with a narrow pore that exhibits regulated gating, enabling the selective degradation of target proteins into peptide fragments. This process of removing proteins is essential for eliminating proteins that are no longer wanted, such as unfolded or aggregated proteins. This is important for preserving cellular function relevant to brain health and disease. Recently, in the nervous system, specialized proteasomes have been shown to generate peptides with important cellular functions. These discoveries challenge the prevailing notion that proteasomes primarily operate to eliminate proteins and identify signaling-competent proteasomes. This review focuses on the structure, function and regulation of proteasomes and sheds light on emerging areas of investigation regarding the role of proteasomes in the nervous system.
Graphical Abstract
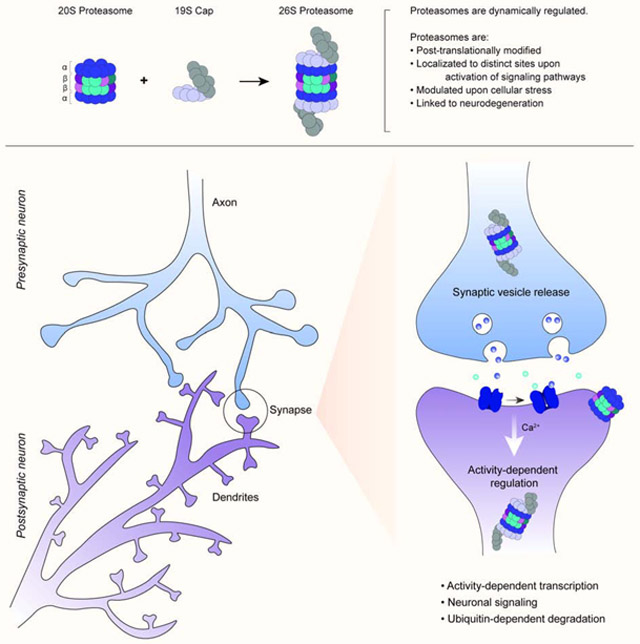
eTOC Blurb:
Proteasome-dependent proteolysis is a tightly regulated process that is essential in all eukaryotic cells, largely due to its central role in regulating protein levels. In this review, Türker et al cover details related to proteasome structure and regulation, particularly focusing on the role of the proteasome in the nervous system.
Introduction
In 1977, Etlinger and Goldberg proposed the presence of a non-lysosomal, soluble, and energy-dependent protein degradation machinery in reticulocytes (Etlinger and Goldberg, 1977). Following this groundbreaking discovery, other groups started to purify and analyze this novel proteolytic complex in other cell types. In 1980, Wilk et al. purified the proteasome, then called the “cation-sensitive neutral endopeptidase,” from bovine pituitary glands. They demonstrated that the complex is composed of distinct subunits with mainly chymotrypsin-like proteolytic activity, which depends on the intact complex, since the dissociated complex, i.e., individual subunits, exhibits no proteolytic function (Wilk and Orlowski, 1980, 1983a, b).
In the last four decades, there has been a tremendous improvement in our understanding of the mechanism and function of the proteasome. The catalytic chamber of the proteasome complex is the 20S core particle (Coux et al., 1996). When this chamber is associated with a regulatory cap, 19S, the final structure is called the 26S proteasome, the major component of the Ubiquitin-Proteasome System (UPS) (Beck et al., 2012; Coux et al., 1996; Huang et al., 2016; Kohler et al., 2001; Matyskiela et al., 2013; Snoberger et al., 2017). While the natural behavior of 26S proteasomes is to mediate ATP-dependent degradation of ubiquitylated proteins, the 20S does not require ubiquitin or ATP and primarily degrades unfolded, intrinsically disordered, and oxidized proteins (Ben-Nissan and Sharon, 2014; Ciechanover, 1998; Ciechanover and Schwartz, 1998). All 20S subunits are essential in mammalian cells and cannot be knocked out. Thus, potent and specific inhibitors of 20S catalytic activity have proven to be very valuable research tools and have helped to establish proteasomes as critical for many cellular functions, including neuronal signaling, immune regulation, and cell death, to name a few. Proteasome inhibitors have also been used as therapeutic agents that have prolonged the lives of many cancer patients (Goldberg, 2012; Goldberg and Rock, 2002; Kisselev and Goldberg, 2001). Despite these tremendous advances, proteasome inhibitors do not easily distinguish between distinct proteasome complexes, therefore, making it challenging to study specific proteasomes and their role in various biological processes.
In the nervous system, the addition of proteasome inhibitors alters synapse biology (Ehlers, 2003; Willeumier et al., 2006) and physiology (Bingol and Schuman, 2006; Cai et al., 2010; Djakovic et al., 2009; Dong et al., 2008; Fonseca et al., 2006; Hegde et al., 1997; Karpova et al., 2006; Rinetti and Schweizer, 2010; Speese et al., 2003). Interestingly, acute addition of a pan-proteasome inhibitor to neurons rapidly attenuates neuronal transmission (Bingol and Schuman, 2006; Cai et al., 2010; Djakovic et al., 2009; Dong et al., 2008; Ehlers, 2003; Karpova et al., 2006; Rinetti and Schweizer, 2010). More recently, a specialized neuronal proteasome was discovered that degrades intracellular proteins into extracellular peptides that mediate rapid changes in neuronal signaling (Ramachandran and Margolis, 2017). This discovery shed new light on the role for proteasomes in the nervous system and will be discussed throughout the review.
In human studies an association between defects in the protein degradation machinery and cognitive dysfunction have been documented (Schmidt and Finley, 2014; Tai and Schuman, 2008). Moreover, many studies have confirmed that dysregulation of protein degradation machinery is likely one of the major contributors to the initiation or progression of neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), or Huntington’s disease (HD) (Ciechanover and Kwon, 2015). Whether the disruption of proper protein degradation is the cause or consequence of these neurological diseases is still an active area of investigation.
In this review, we discuss these emerging areas of study related to proteasomes. We focus on providing background for proteasome structure and regulation along with a current understanding of targeted degradation in the nervous system and relevance to neurodegenerative disease.
Structure of the Proteasome
20S Core Particle
The catalytic core particle (CP or 20S) of the proteasome is composed of four heptameric rings (α7β7β7α7) from seven structurally similar α and β subunits, forming a cylindrical structure with dimensions of approximately 150 Å by 115 Å and a narrow (13 Å) entrance pore (Groll et al., 1997; Harshbarger et al., 2015; Huang et al., 2016) (Figure 1). The multicatalytic protease activity of the CP is achieved by three distinct β subunits: β1 (caspase-like), β2 (trypsin-like), and β5 (chymotrypsin-like), which cleave peptide bonds at the C-terminal side of acidic, basic, and hydrophobic amino acid residues, respectively (Figure 1). The catalytic activity of these β subunits is dependent on threonine residues at their N-termini, which classifies the proteasome as a threonine protease, separating it from other well-studied proteases (serine, cysteine, or carboxyl protease families). Due to the sequestration of the catalytic subunits inside the 20S core particle, the proteasome is selective for those molecules that can enter the chamber . Entry into the core proteasome is in part regulated by the hydrophobic N-terminal tails of the 20S α subunits that extend through the chamber to form a gate and prevent nonselective degradation (Bajorek et al., 2003; Groll et al., 2000).
Figure 1. Structure of the 20S proteasome and regulatory caps.

Cryo-EM structure of the human 20S proteasome with a resolution of 3.50 Å (PDB:5GJR) (Huang et al., 2016). The 20S proteasome has a barrel-shaped structure formed by 4 heptameric rings (α7β7β7α7) with a width of 12 nm and height of 15 nm. β1 (pink), β2 (gold), and β5 (purple) subunits have caspase-like, trypsin-like, and chymotrypsin-like catalytic activity, respectively. The 20S proteasome degrades intrinsically disordered or modified proteins independent of ATP or ubiquitin. The human 26S proteasome, a major player in the ubiquitin-proteasome system (UPS), is formed with the association of the 19S regulatory particle with the 20S. 19S is composed of 1) non-ATPase regulatory subunits (dark gray), which are critical for ubiquitinated substrate recognition and deubiquitylation of the substrate, and 2) ATPase subunits (light gray), which are important for unfolding/translocation of substrates and 20S gate opening. Structure of the mouse PA28αβ cap obtained via X-ray diffraction with a resolution of 2.90 Å (PDB:5MX5)(Huber and Groll, 2017). PA28αβ, an alternative cap for the 20S proteasome, forms a heteroheptameric ring with two distinct subunits: α (light pink) and β (dark pink). PA28αβ activates the proteasome in an ATP- and ubiquitin-independent manner. Cryo-EM structure of the human PA200 cap with a resolution of 3.75 Å (PDB:6KWX) (Guan et al., 2020). PA200 (gold) activates the proteasome for the degradation of short peptides or highly unstructured proteins. Structures adapted from PyMOL (Schrodinger, 2015).
19S Regulatory Particle
The 19S regulatory particle (RP) associates with one or both ends of the 20S CP and is a major component of ubiquitin-dependent protein degradation, which will be discussed later in this review. The 19S RP can be separated biochemically into the base and lid subcomplexes. The lid subcomplex is composed of nine subunits (Rpns 3, 5, 6, 7, 8, 9, 11, 12, 15). The lid subunits are important for structural aspects of the proteasome and deubiqutylation. Rpn11, the intrinsic deubiqutinase (DUB), is a zinc-dependent metalloprotease that cleaves the ubiquitin chain from the substrate (Huang et al., 2016). Additional deubiquitnases associated with the 19S cap include Usp14 and Uch37 (Hu et al., 2002; Hu et al., 2005). The base subcomplex is composed of ten subunits, six AAA+ (ATPases associated with various cellular activities) ATPases (Rpt1-6) and four non-ATPase regulatory subunits (Rpns 1, 2, 10, 13). The non-ATPase regulatory subunits are located at the interface of the base and the lid and are important for ubiquitin recognition and binding in part through their ubiquitin-interacting motif (UIM) (Deveraux et al., 1994; Elsasser et al., 2004; Fu et al., 2001). The AAA+ ATPase subunits are important for substrate unfolding and act as motors to induce translocation of the unfolded polypeptide into the catalytic chamber (Bard et al., 2018; Collins and Goldberg, 2017; Dong et al., 2019; Huang et al., 2016). Simultaneously, the AAA+ ATPases, which bind the 20S CP α subunits through a C-terminal HbYX (Hydrophobic-Tyrosine-X) motif, mediate conformational change in the α subunits’ N-termini and enable gate opening to allow for translocation of the unfolded protein (Rabl et al., 2008; Smith et al., 2007; Yu et al., 2010) (Figure 1).
Alternative Regulatory Particles
In addition to the 19S cap, the 20S CP can be associated with alternative caps such as the 11S regulator (REG; PA28αβ, PA28γ) (Chu-Ping et al., 1992) or Blm10/PA200 (Ortega et al., 2005). PA28 complexes can be composed of distinct but structurally similar subunits: α, β, or γ (Huber and Groll, 2017)(Figure 1). While PA28α and PA28β assemble into heteroheptameric complexes and are located mainly in the cytoplasm (Huber and Groll, 2017), PA28γ is exclusively homoheptameric and located in the nucleus. PA28αβ activates the proteasome in an ATP- and ubiquitin-independent manner and plays a major role in the immunoproteasome function (Dubiel et al., 1992). In the nervous system, it has been shown that the expression level of PA28αβ is upregulated upon stress, such as mitochondrial dysfunction, oxidative stress, or cytokine treatment, suggesting a protective role of PA28 against protein aggregation in neurons (McNaught et al., 2010; Shanley et al., 2020). However, the exact mechanism or function of the PA28 in the nervous system remains an active area of research. PA200, similar to PA28, does not require ATP or ubiquitin to activate proteasome-dependent protein degradation (Guan et al., 2020) (Figure 1). PA200 induces degradation of small peptides or highly unstructured proteins like tau in the absence of ATP in vitro (Dange et al., 2011; Schmidt et al., 2005). The role of the PA200-associated proteasome in the nervous system is still unclear, and in vivo experiments are needed to confirm the results from in vitro studies.
Another endogenous regulator of the proteasome, Proteasome Inhibitor of 31 kDa (PI31), was first identified as a proteasome-interacting protein and inhibitor of the 20S proteasome in vitro. However, in vivo studies indicated that PI31 depletion leads to defects in proteasome-dependent protein degradation as opposed to elevated proteasome activity (Liu et al., 2019, Minis et al., 2019). A recent study from Liu et al. suggests that PI31 acts as an adaptor between the proteasome and dynein light chain, a cellular motor responsible for axonal transport, resulting in proteasome trafficking in neurons in Drosophila and mice. Loss of PI31 leads to disruption in axonal proteasome movement and accumulation of poly-ubiquitylated proteins in the axons and nerve terminals, suggesting insufficient proteasome activity in those sites due to low abundance of the proteasome. The dysregulation of proteasome trafficking results in structural defects in synapses (Liu et al., 2019). A follow-up study from the same group demonstrated that a PI31 conditional knock-out in spinal motor neurons and cerebellar Purkinje cells in mice leads to axonal degeneration, cell death, and motor dysfunction, suggesting essential roles for PI31 and regulated proteasome localization in neuronal survival and function (Minis et al., 2019).
Post-translational Modification and Localization of the Proteasome
Post-translational modifications (PTMs) affect the function, structure, and half-life of many proteins, and the proteasome is no exception. One of the best-studied PTMs is phosphorylation, and there are around 300 phosphorylation sites across the proteasome subunits (Feng et al., 2001; Iwafune et al., 2002; Mason et al., 1996; VerPlank and Goldberg, 2018; VerPlank et al., 2020). Several reports have suggested an increase in proteasome activity upon phosphorylation of the 19S subunit Rpn6 at serine 14 by Protein Kinase A (PKA). PKA is activated upon an increase in cyclic AMP (cAMP) levels, leading to the release of active PKA catalytic subunits from cAMP-bound regulatory subunits. PKA can be activated by pharmacological agents such as forskolin which activates adenylate cyclase, the enzyme responsible for converting ATP into cAMP, or rolipram which blocks hydrolysis of cAMP through inhibition of the phosphodiesterase PDE4. By tracing half-lives of short-lived/misfolded proteins involved in neurodegenerative disease progression, several groups provided evidence that PKA activation through addition of forskolin or rolipram promotes an increase in degradation of toxic protein load (Lokireddy et al., 2015; VerPlank and Goldberg, 2018). Moreover, in vivo, it has been shown that enhancing cAMP-PKA signaling by rolipram or by pure PKA treatment increased proteasome activity, significantly decreased the accumulation of phospho-tau, and improved cognitive performance in a mouse model of tauopathy (rTg4510 mouse, expressing a pathogenic tau mutation (P301L)) (Myeku et al., 2016).
In addition to phosphorylation by PKA, VerPlank et al. demonstrated that cGMP-dependent protein kinase (PKG) activation upon cGMP elevation induced 26S phosphorylation, leading to increased 26S proteolytic activity, reduced toxic protein load, and dimished cell death in a zebrafish model of neurodegenerative disease (VerPlank et al., 2020). Unfortunately, the precise sites of phosphorylation on proteasomes by PKG remain unknown, making it hard to perform proof of concept experiments and understand the exact mechanism of PKG-induced activation of the proteasome. Interestingly, in hippocampal neurons, it has been shown that calcium influx following synaptic depolarization leads to activation of calcium/calmodulin-dependent protein kinase II (CaMKII) and phosphorylation of serine 120 on Rpt6, which in turn increased 26S proteasome activity (Djakovic et al., 2009). This study is especially important since it suggests a potential feedback mechanism between neuronal activation, protein degradation, and synaptic remodeling. In a review by VerPlank and Goldberg, they summarize the effects and mechanisms of 26S proteasome phosphorylation (VerPlank and Goldberg, 2017). While we have some insight into the function of proteasome phosphorylation, there are other PTMs of the proteasome with diverse functions such as O-linked N-acetylglucosamine (O-GlcNAC) (Zhang et al., 2003), ADP-ribosylation (Cho-Park and Steller, 2013), acetylation (Gomes et al., 2006), and even ubiquitylation (Besche et al., 2014; Marshall et al., 2016).
Across different mammalian tissues proteasomes have been observed to be localized to various subcellular sites (Baumeister et al., 1998) such as the nucleus (Adori et al., 2006; Franic et al., 2021; Rivett et al., 1992; Scharf et al., 2007), cytosol (Palmer et al., 1996; Schipper-Krom et al., 2019), plasma membrane (Ramachandran et al., 2018; Ramachandran and Margolis, 2017), and more recently have been demonstrated to be extracellular (Dekel et al., 2021; Dwivedi et al., 2021; Kulichkova et al., 2017; Lavabre-Bertrand et al., 2001; Zoeger et al., 2006). In neurons, due to their highly complex morphology, it is not hard to imagine that the spatial distribution of the proteasome has profound effects on cellular function. The localization of the proteasome is dynamic and regulated by neuronal activity. Bingol and Schuman demonstrated that neuronal activity-induced calcium-dependent signaling results in movement and sequestration of the 26S proteasome into dendritic spines within minutes (Bingol and Schuman, 2006). Moreover, this rapid translocation of the 26S proteasome to the dendritic spine was critical for activity-dependent synapse formation (Hamilton et al., 2012). More recently, the Margolis group reported on their discovery of a 20S proteasome in neurons that localizes to the plasma membrane (Ramachandran et al., 2018; Ramachandran and Margolis, 2017).
Targeted Protein Degradation
Ubiquitin-dependent Degradation
One of the most prominent and well-studied functions of ubiquitin is targeting substrates to the 26S proteasome for degradation, a process that occurs in a highly regulated manner. All proteins have a half-life, generally ranging from minutes to days, and most cellular proteins are ultimately degraded through ubiquitin-dependent degradation (Craiu et al., 1997). This process is also responsible for degrading misfolded/damaged proteins (including defective ribosome products, DRiPs), and orphaned subunits of multimeric complexes (Schubert et al., 2000).
Ubiquitin is a highly conserved 76 amino acid protein that can be conjugated to other proteins as a post-translational modification. The process of adding ubiquitin moieties (ubiquitylation) involves a cascade of E1-E2-E3 enzymes (Figure 2). Ubiquitylation begins with a ubiquitin-activating enzyme (E1), which creates a thiol-ester linkage between the enzyme and the ubiquitin polypeptide in an ATP-dependent manner. Next, the ubiquitin moiety is transferred from the E1 to a ubiquitin-conjugating enzyme (E2), generating another thio-ester linkage. Lastly, the ubiquitin is transferred to a lysine residue of a target protein via an isopeptide bond with the help of a ubiquitin ligase enzyme (E3) (Scheffner et al., 1995).
Figure 2. Ubiquitin-dependent and ubiquitin-independent protein degradation by the proteasome.

(A) The ubiquitin-proteasome system (UPS). The ubiquitin-activating enzyme (E1, cyan) hydrolyzes ATP (orange) and binds to ubiquitin (Ub, gray), activating the C-terminus of ubiquitin for nucleophilic attack. Activated ubiquitin is next transferred to the ubiquitin-conjugating enzyme (E2, purple), which then transfers the ubiquitin to the ubiquitin ligase (E3, dark blue). E3 facilitates target selectivity due to its direct binding to the substrate (dark red). As a result of this E1-E2-E3 enzymatic cascade, ubiquitin attaches to the substrate lysine residue via an isopeptide bond. Upon the formation of ubiquitin chains (at least four ubiquitin) on the substrate, it is delivered to the 26S proteasome. The 19S regulatory particle (19S RP) recognizes the ubiquitin chain, deubiquitylates and unfolds the target protein, and opens the 20S gate, driving translocation of the substrate into the 20S catalytic particle (20S CP). (B) The composition of the proteasome is highly dynamic. The 26S proteasome can be decapped under certain circumstances, such as oxidative stress, leading to the accumulation of the 20S proteasome. However, some of the decapped proteasomes can be recapped quickly, leading to an optimal balance of capped and uncapped proteasomes. (C) Ubiquitin-independent degradation by the 20S proteasome. The 20S proteasome, in the absence of any caps, degrades substrates in an ATP- and ubiquitin-independent manner. Proteins modified as a result of oxidative stress (pink) are susceptible to 20S proteasome degradation, potentially due to their exposed hydrophobic patches. Intrinsically-disordered proteins (IDP, teal) comprise the majority of 20S proteasome substrates due to their flexible structure that enables entry into the narrow pore of the 20S catalytic chamber. In neurons, the 20S proteasome localizes to the plasma membrane and degrades ribosome-associated nascent polypeptide chains (purple).
In humans, there are 2 known E1 enzymes, ~30-50 E2 enzymes, and ~600 E3 enzymes (George et al., 2018). E3 enzymes account for much of the specificity of ubiquitylation, as they recognize target substrate proteins. There are multiple types of E3 ubiquitin ligases, classified by the organization of their catalytic domains and their mechanism of ubiquitin transfer. Various aspects of E3 ligase structure, function, and regulation have been reviewed extensively elsewhere (Buetow and Huang, 2016; Dove and Klevit, 2017; Metzger et al., 2014; Sluimer and Distel, 2018; Wang et al., 2020; Zheng and Shabek, 2017).
Specific ubiquitin-linkages can alter the fate of a protein. The most abundant of these linkages are K48-linked ubiquitin chains, which primarily signal for proteasomal degradation and increase rapidly in response to proteasome inhibition (Jacobson et al., 2009; Xu et al., 2009). Even the structure of ubiquitin chains can influence degradation, such as K48/K11 branched chains that target substrates for degradation with high efficiency. K11, K29, and K63-linked ubiquitin chains promote proteasomal degradation to a lesser extent and may serve alternative functions besides degradation (French et al., 2021; Komander and Rape, 2012).
The polyubiquitinated substrate can then be recognized by proteins containing ubiquitin-binding domains (UBDs). These proteins may be proteasome shuttling factors such as Rad23A/B, UBQLNs, and DDI1/2, which have ubiquitin-associated (UBA) domains to bind ubiquitin chains and ubiquitin-like (UBL) domains to interact with the proteasome (Zientara-Rytter and Subramani, 2019). Ubiquitinated substrates may interact directly with the proteasome, as the 19S cap of the 26S proteasome has three ubiquitin receptors, Rpn1, Rpn10 and Rpn13 (Dikic, 2017; Husnjak et al., 2008; Schreiner et al., 2008; Shi et al., 2016). Together these receptors allow the proteasome to recognize substrates with a variety of ubiquitin chain topologies (Martinez-Fonts et al., 2020).
Ubiquitin-dependent degradation is crucial for proper function of the nervous system and involved in the regulation of various aspects of neuronal development, including neurogenesis, differentiation, and synapse formation (Upadhyay et al., 2017). To this end, many E3 ubiquitin ligases have been discovered to be mutated or dysregulated in neurological diseases. In fact, ~13% of known E3 ligases are mutated in neurological disease (George et al., 2018). Some prominent examples include UBE3A, which is mutated in Angelman Syndrome, and Parkin, which is mutated in inherited instances of Parkinson’s disease (Kishino et al., 1997; Kitada et al., 1998). Further, nearly a dozen E3 ligases have been found mutated in individuals with autism or autism spectrum disorders (ASDs) (George et al., 2018). Understanding the role of E3 ligases in contributing to neurological diseases lies in identifying the substrates of a particular ligase and elucidating the functional consequence of the ubiquitin moieties placed on the substrate. This is a highly active area of research to identify new therapeutic targets for treating neurological conditions related to disruptions in the UPS.
Ubiquitin-independent Degradation
While we have a broad understanding of protein degradation by UPS, proteolysis by the free 20S proteasome has been mostly accepted as the latent form of degradation. Most of the proteasome inhibitors target the catalytic sites found on the 20S core particle, making it difficult to distinguish between the free 20S versus 26S proteasome. However, recent advances in the 20S proteasome field demonstrated that the 20S proteasome largely contributes to overall protein degradation and removal of damaged/oxidized proteins in an ATP-/ubiquitin-independent manner (Baugh et al., 2009; Fabre et al., 2014; Inai and Nishikimi, 2002; Kumar Deshmukh et al., 2019; Pickering et al., 2010; Silva et al., 2012; Wang et al., 2010a)(Figure 2). Additional targets for 20S proteasomes are proteins with intrinsically disordered domains (such as p53 (Asher et al., 2005), c-Fos (Bossis et al., 2003), p21 (Li et al., 2007), tau (David et al., 2002), alpha-synuclein (Tofaris et al., 2001)) with partially unfolded structures. A thorough review of the comparison between the 20S proteolytic pathways was published recently (Raynes et al., 2016).
As mentioned above, among the main targets for the free 20S proteasome are oxidatively damaged proteins produced by oxidative stress. The proteasome is responsible for the degradation of ~90% of oxidized proteins (Jung et al., 2006). Due to conformational changes upon chemical modifications, oxidized proteins expose their hydrophobic patches, enabling recognition/binding by the 20S proteasome (Carrard et al., 2002; Giulivi et al., 1994; Grune et al., 1997; Kisselev et al., 2002; Korovila et al., 2017; Pajares et al., 2015; Rabl et al., 2008). Several groups showed that the 26S proteasome and E1-E2-E3 enzymatic cascade are transiently inactivated upon oxidative insult (Grune et al., 2011; Reinheckel et al., 1998). Interestingly, the affinity of 19S for the 20S proteasome decreases upon oxidative stress, resulting in dissociation of 26S proteasome and accumulation of free 20S proteasome. ECM29, a proteasome adaptor and scaffold protein, is the key modulator in this dissociation event, enabling the disassembly of the 26S proteasome by binding to and removing the 19S cap from the 26S complex (Haratake et al., 2016; Wang et al., 2010b) (Figure 2).
Proteins containing partial or complete disordered regions under physiological conditions are called intrinsically disordered proteins (IDPs), and they lack a well-defined 3D structure. Strikingly, 44% of the entire human protein pool contains disordered regions of more than 30 amino acids in length (Oates et al., 2013). IDPs have a signature amino acid composition, enriched in polar groups, and short in hydrophobic or aromatic residues (Dyson, 2016). Many proteins containing an intrinsically disordered region (IDR) are prone to ubiquitin-independent 20S proteasome degradation. The major question in the field is the targeting mechanism of IDPs to the 20S proteasome. Since it has been shown that IDPs can be degraded by the 20S proteasome that lacks the ubiquitin-recognition particles (19S subunits), there is a massive effort in identifying the targeting mechanism of IDP to the 20S proteasome. Biran et al. recently demonstrated that the 20S proteasome subunit, α3, has a high affinity for and interacts with some IDPs (p21, c-Fos, p53) in vitro (Biran et al., 2017). Interestingly, some proteins with known IDR, such as tau, are involved in neurodegenerative diseases by inducing aggregate formation through interactions between their IDRs. Further investigation revealed that tau is degraded by the 20S proteasome and that degradation kinetics are affected by the phosphorylation state of tau in vitro, indicating a post-translational mechanism that could control targeting of substrates to the 20S proteasome for degradation (Ahmadi et al., 2019; Ukmar-Godec et al., 2020). Due to the strong link between the degradation of IDPs and neurodegenerative diseases, it is critical to study this unique mechanism of ubiquitin-independent degradation in vivo.
Recently, research in our laboratory discovered a novel proteasome complex that is specifically expressed in the nervous system, makes up 40% of all neuronal proteasomes, and is critical for neuronal activity-dependent signaling (Ramachandran and Margolis, 2017). This proteasome complex is made up of a 20S proteasome core and is localized at the neuronal plasma membrane in a manner that allows ubiquitin-independent degradation of intracellular proteins into peptides that are released directly into the extracellular space. The mechanism for this ubiquitin-independent targeting to the neuronal membrane proteasome remains to be determined and is an active area of investigation. One clue lies in the previous findings that ribosomes and proteasomes appear to interact in neurons and may lead to direct co-translational degradation (Ramachandran et al., 2018).
Proteasomes in the Nervous System
Neuronal Activity Regulates Proteasome Function
Neuronal activation leads to dramatic changes in various essential cellular processes, including gene expression, protein translation, and protein degradation. Alteration of neuronal activity affects the level of proteasome-dependent degradation. In rat hippocampal cultures, blockade of action potential firing by a voltage-gated sodium channel antagonist, tetrodotoxin, or elevation of neuronal activity by a GABAA receptor antagonist, bicuculline, leads to 26S proteasome-dependent degradation of different pools of postsynaptic density (PSD) proteins (Ehlers, 2003). In addition, this study showed that bicuculline-induced neuronal signaling is blocked by the addition of the proteasome inhibitor MG132, suggesting that the proteasome has a significant role in synaptic signaling. Moreover, the proteasome localizes to the dendritic spines upon neuronal activation, supporting the idea of proteasomes’ rapid role in modulating synaptic composition (Bingol and Schuman, 2006) (Figure 3). Furthermore, as mentioned above, calcium influx following neuronal excitation activates CaMKII, which enhanced proteasome activity through phosphorylation of the Rpt6 subunit, suggesting that neuronal activation has profound effects on proteolysis (Djakovic et al., 2009). Consistent with this, in 2009, Banerjee et al. showed that neuronal activation through addition of N-methyl-D-Aspartate (NMDA) or potassium chloride to neuronal cultures induced degradation of a synaptic translational repressor, Mov10, by the proteasome, leading to increased levels of some synaptic proteins such as α-CamKII and LimKI. This study indicated the importance of proteasome-dependent protein degradation in regulating activity-dependent protein synthesis (Banerjee et al., 2009).
Figure 3. The function of the proteasome in the nervous system.

(A) Schematic of a hippocampal pyramidal neuron. Dendrites/postsynaptic site (purple), axon/presynaptic site (blue) and nucleus/soma (yellow) are shown. (B) In the nucleus, upon induction of long-term potentiation (LTP), the proteasome degrades transcriptional repressors (orange), leading to the CREB (green)-dependent transcription of genes critical for LTP maintenance. (C) Proteasome-dependent protein degradation mediates axonal maturation by reducing ribosome levels and regulates synaptic vesicle release by balancing the levels of vesicle priming/release proteins (yellow) at the presynaptic site. (D) Upon neuronal activation through NMDAR (dark blue) activation, the neuronal membrane proteasome (NMP) produces more signaling peptides (orange), potentially leading to regulation of the postsynaptic signaling. Moreover, signaling through NMDARs (dark blue) leads to localization and sequestration of the proteasome into dendritic spines, remodeling the postsynaptic protein composition.
Another example of neuronal activity-regulated proteasomes is the aforementioned neuronal membrane proteasome (NMP). A cell impermeable, potent proteasome inhibitor, biotin-epoxomicin, has been synthesized to study the function of the NMP (Ramachandran et al., 2018; Ramachandran and Margolis, 2017). Acute and specific inhibition of the NMP prevents extracellular peptide production and potently reduces activity-induced calcium signaling in cortical neurons. It has been shown that the NMP released more peptides into the extracellular space upon neuronal stimulation by depolarization buffer compared to unstimulated, control buffer-treated mouse cortical neurons. It is still unclear whether this increase in peptide release is due to an increase in proteasome catalytic activity, proteasome localization to the plasma membrane, or availability of the proteasome substrates upon neuronal activation. Release of these peptides might be physiologically important for activity-dependent neuronal function, as the addition of purified peptides derived from the NMP to naive cortical neurons can rapidly and robustly induce calcium signaling through N-methyl-D-Aspartate receptor (NMDAR) activation (Ramachandran and Margolis, 2017). The rise in NMP peptide production upon neuronal activation and the ability of NMP peptides to induce neuronal stimulation suggest a potential feedback mechanism to control the intensity of activity-dependent neuronal signaling (Figure 3).
Proteasome Inhibition in the Nervous System
Since its discovery in 1966, the term long-term potentiation (LTP) has been used to describe the strengthening of synapses based on persistent synaptic activity between two neurons, while long-term depression (LTD) is the weakening of synapses due to repetitive low frequency (0.5-3 Hz) stimulation (LFS) (Dudek and Bear, 1992). LTP is considered the underlying mechanism in learning and memory, making the concept an invaluable tool in the neuroscience field. LTP is induced by short duration, high frequency (over 100 Hz) stimulation of glutamatergic synapses that are made up of NMDA and AMPA receptors. There are several potential mechanisms for LTP expression, including changes in synaptic vesicle release properties and changes in AMPAR conductance or membrane insertion (Bliss and Collingridge, 2013). There are different phases of LTP: early phase-LTP (Ep-LTP) and late phase-LTP (L-LTP). Calcium influx, leading to activation of specific kinases (CaMKII, PKA, PKC), is required to insert more AMPAR at the postsynaptic site and is sufficient to induce Ep-LTP. In contrast, L-LTP maintenance requires gene expression and translation, leading to major morphological changes, such as growth of new dendritic spines or pre-existing spines (Abraham, 2003; Yuste and Bonhoeffer, 2001). On the other hand, LTD is induced by NMDAR-dependent calcium influx upon LFS, leading to activation of protein phosphatase pathways, especially PP1, resulting in AMPAR dephosphorylation (S845). Dephosphorylation of AMPAR decreases its open channel probability and increases its internalization, weakening synapses.
It is generally accepted that proteasome degradation plays a critical role in this synaptic plasticity (Dong et al., 2008; Hegde, 2017; Speese et al., 2003; Widagdo et al., 2015). However, very little is known about the mechanism responsible for this effect. Hegde and colleagues showed that inhibition of the proteasome has opposite effects on induction of LTP versus maintenance of LTP (Dong et al., 2014). Proteasome inhibition enhances the induction of L-LTP by preventing the turnover of dendritic proteins. Consistent with this, they revealed that proteasome inhibition in the presence of protein synthesis inhibitors blocked enhanced induction of L-LTP. Considering the major excitatory receptors, NMDAR and AMPAR, are targets of the UPS, the group proposes that proteasome inhibition stabilizes the receptors. Higher receptor levels leads to an increase in calcium influx and activation of the downstream signaling pathways, enhancing the induction of L-LTP even with a weak, subthreshold stimulation (2x100 Hz) (Dong et al., 2014; Dong et al., 2008; Vashisht et al., 2018). On the other hand, proteasome inhibition following stimulation (subthreshold L-LTP induction via the theta-burst protocol) blocks the maintenance of L-LTP. This is achieved by reducing the transcription of CREB-inducible genes due to stabilization of ATF4 (activating transcription factor 4), a CREB repressor, and by stabilization of translational repressors (e.g., Paip2, 4E-BP2) in dendrites (Dong et al., 2014; Dong et al., 2008) (Figure 3).
On the other hand, Fonseca et al. showed that inhibition of either protein synthesis or degradation weakens the L-LTP. Moreover, they showed that inhibition of both protein synthesis and degradation simultaneously recovers L-LTP, suggesting that the balance between degradation and synthesis is the crucial step in the formation of L-LTP (Fonseca et al., 2006). This finding builds on previous observations that disturbing one of these pathways alone leads to an accumulation or reduction of critical synaptic plasticity proteins (e.g., Homer1a, Dunc-13) (Ageta et al., 2001a; Ageta et al., 2001b; Speese et al., 2003), which likely disrupts proper L-LTP maintenance, suggesting that interfering with both pathways does not have any detrimental effect on L-LTP due to balancing of the levels of synaptic plasticity proteins (Fonseca et al., 2006).
Regardless of which model is more accurate, it is clear that neuronal activity-dependent protein turnover is critical for postsynaptic remodeling and signaling. The UPS regulates key synaptic proteins such as PSD-95, Shank, GKAP, AKAP, SPAR, RIM-1, and GRIP1 (Colledge et al., 2003; Ehlers, 2003; Pak and Sheng, 2003; Yao et al., 2007). Synaptic activity elevated ubiquitin conjugation of PSD proteins in mouse hippocampal cultures (such as postsynaptic scaffolds: Shank, GKAP, AKAP79/150) (Ehlers, 2003). Upon NMDAR receptor activation, PSD-95, an essential anchoring factor for AMPAR at the PSD, was ubiquitinated by Mdm2 and degraded by the proteasome. This NMDAR-induced decrease in PSD-95 levels led to NMDAR-induced AMPAR endocytosis. Additionally, inhibition of proteasome activity blocks AMPAR recycling through the stabilization of PSD-95, leading to attenuation of LTD (Colledge et al., 2003).
The effects of the proteasome in the synaptic composition are not restricted to postsynaptic sites. During axonal maturation, axons transition from a high ribosome and local translation state, required for axonal elongation, to a reduced necessity for ribosomes in axons due to the formation of stable connections that might be disrupted by excess local translation. Costa et al. showed that synapse formation induces a reduction in ribosome levels through clearance of excess ribosomes via proteasome-dependent degradation in mature axons (Costa et al., 2019) (Figure 3). Additionally, DmeI\unc-13, Dunc13, a critical protein for presynaptic vesicle exocytosis in Drosophila neuromuscular junctions, is degraded by the proteasome. Speese et al. reported that inhibition of the proteasome (lactacystin or epoxomicin) results in stabilization of Dunc-13 and increased synaptic transmission due to more efficient priming and release of synaptic vesicles (Speese et al., 2003). At presynaptic sites, a mammalian ubiquitin ligase, SCRAPPER, ubiquitinates Rab3-interacting molecule 1 (RIM1), a major player in the vesicle priming step, to regulate synaptic vesicle release (Yao et al., 2007) (Figure 3).
Proteasome inhibition has been shown to have detrimental effects, not only in the central nervous system (CNS) but also in the peripheral nervous system (PNS). In 2003, bortezomib, a reversible proteasome inhibitor, was approved as a treatment for multiple myeloma, accounting for 1% of all cancer cases, due to its efficiency in driving myeloma cells into apoptosis. The major limitation of this line of therapy is the induction of peripheral neuropathy, due to major damage to the peripheral nerves, observed in up to 50% of bortezomib-treated multiple myeloma patients (Argyriou et al., 2008). Bortezomib-induced peripheral neuropathy (BIPN) could be derived from inflammation, oxidative stress, or mitochondrial changes. Interestingly, treatment with another proteasome inhibitor, carfilzomib, yields a lower incidence of peripheral neuropathy due to its high selectivity for the proteasome β5 subunit, low off-target effects, and irreversible binding to the proteasome (Mushtaq et al., 2018). Understanding and testing a variety of proteasome inhibitors is a major focus in the field of cancer therapy. The mechanism of action of proteasome inhibitors and neuroprotective strategies following treatment have been recently reviewed (Malacrida et al., 2019; Velasco et al., 2019).
Protein Degradation in Neurodegenerative Diseases
Disruption of protein homeostasis is a common hallmark of aging and neurodegenerative diseases (ND) (Limanaqi et al., 2020). Proteasome activity declines with age, triggering the onset of some NDs (Keller et al., 2000). Accumulation of aggregated proteins, the foundation of many NDs, is commonly age-dependent and due to stress-induced defects. One common idea about why aggregate accumulation is a hallmark of most NDs is the potential resistance of proteins with repetitive amino acids to degradation by the proteasome (e.g., polyglutamine in Huntington’s disease, polyalanine in muscular dystrophy, dipeptide repeats in amyotrophic lateral sclerosis), leading to toxic aggregate formation inside the cell. Another idea is that the aggregates act as a competitive inhibitor for the proteasome, leading to the accumulation of proteasome substrates. While both could be occurring simultaneously, the timing of these two harmful mechanisms is still unclear. An enormous amount of work has been done to look at proteolysis deficits in neurodegeneration, for the purposes of illustration we chose to focus our attention to AD and HD.
Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of dementia, leading to progressive neuronal death, shrinkage of the temporal and frontal lobes, and subsequent decline in cognition and memory. Two main aggregate forms have been accepted as the leading risk factors for developing AD: amyloid-beta (Aβ) and tau aggregates. The mechanism of proteasome disruption in AD is still an active area of investigation. Gregori et al. suggest that Aβ enters the catalytic chamber and inhibits the activity of the β5 subunit. However, previous findings show that aggregated Aβ acts as a competitive inhibitor without having a direct impact on the proteolytic activity, restricting entry of the natural proteasome substrates into the catalytic chamber of the proteasome and leading to impairment of proper degradation of the cellular protein pool (Almeida et al., 2006; Cecarini et al., 2008; Gregori et al., 1997; Oh et al., 2005; Thibaudeau et al., 2018; Tseng et al., 2008; Zhao and Yang, 2010). Moreover, hyperphosphorylated insoluble tau is more resistant to proteolysis than its soluble counterpart, and ubiquitinated and hyperphosphorylated tau have been shown to impair proteasome activity both in vivo and in vitro (Myeku et al., 2016).
Another hypothesis for the disruption of proteasome function in AD suggests the involvement of ubiquitin, which has been used as a marker for brain pathology since the late 1980s. A mutant form of ubiquitin (UbB+1) resulting from the replacement of a C-terminal residue of Ub (G76) with an additional 20 residues has been detected in the brains of AD patients (Van Leeuwen et al., 1998a; Van Leeuwen et al., 1998b). Ubiquitin chains bearing UbB+1 on the terminal ends (UbB+1-capped chains) are resistant to deubiquitylation due to the absence of the G76 site. The accumulation of UbB+1 induces proteasome dysfunction by directly binding to the ubiquitin recognition motifs on the 19S proteasome and competing with the polyubiquitinated substrates for proteasome binding (Lam et al., 2000). The UPS plays a significant role in the clearance of tau and Aβ under normal circumstances, so the disruption of proteasome function leads to the accumulation of Aβ and tau, enabling the formation of more Aβ plaques and tau aggregates, eventually causing neuronal loss. In addition to ubiquitin, E3 ubiquitin ligases, TRIM15 and UBR5, have been accepted as potential genetic markers in AD patients through Genome-Wide Association Studies (GWAS) (Shi et al., 2010; Hu et al., 2011). There are many more E3 ubiquitin ligases and deubiquitinases that have been shown to be involved in AD, supporting the idea that the UPS plays a critical role in AD (Harris et al., 2020). . Several reviews are available that discuss the link between the proteolytic pathways and AD (Cheng et al., 2018; Ciechanover and Kwon, 2015; Hong et al., 2014).
Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant disease caused by a mutation in the huntingtin gene leading to an expansion of the polyglutamine repeat at the N-terminus of the huntingtin protein (HTT), making it prone to misfolding and aggregation. The mutant HTT (mHTT) forms aggregates and eventually leads to atrophy and cell death. Since the function of the HTT protein is still unclear, it is challenging to understand the mechanism of the disease. However, several reports suggest that increasing the degradation kinetics of the mutant protein might be an efficient line of therapy as suggested by experiments performed with HD patient-derived cell lines using PROteolysis TArgeting Chimera (PROTAC)-based approaches for drug design, even though there is still no clinical success on this avenue (Harding and Tong, 2018; Ottis and Crews, 2017; Tomoshige et al., 2018). mHTT aggregates, which cannot be fully degraded by the proteasome, colocalize with UPS components and reduce proteasome activity. Holmberg et al. investigated the potential mechanism of UPS disruption by mHTT aggregates (Holmberg et al., 2004). They showed that mHTT aggregates enter the proteasome, are incompletely degraded, and are kinetically trapped, thus obstructing the proteasome active site and leading to proteotoxicity.
Interestingly, free filamentous mHTT aggregates extracted from inclusion bodies isolated from a transgenic mouse model of HD (Tet/HD94) result in impairment of the 26S proteasome activity in a non-competitive manner (Díaz-Hernández et al., 2006). Several reports contradict this finding, since aggregates of synthetic poly-Q peptides have been shown to not change the proteasome activity in vitro (Bennett et al., 2005). However, these studies use different protocols for the source of aggregates (isolated from the mouse brain versus recombinant, synthetic peptides), which might explain the discrepancies in the effects of these filaments on proteasome function. Aggregates isolated from the mouse brain have been processed and bear modifications, like ubiquitylation, that could easily change the interaction of the aggregates with the proteasome (Gandhi et al., 2019; Harding and Tong, 2018; Rai et al., 2019).
Conclusions
Protein degradation is mediated by multiple mechanisms in eukaryotic cells, including autophagy and the proteasome. The proteasome is responsible for the turnover of most of the cellular protein pool, making it an essential mechanism for cellular health. The proteasome-ubiquitin pathway has been studied widely and is accepted as the main degradation machinery. However, the role of the uncapped, free 20S proteasome in ubiquitin and ATP-independent degradation has only recently begun to be revealed. While targeting of substrates to the 26S proteasome is well characterized and ubiquitin-mediated, the exact mechanism of substrate recognition by the 20S proteasome is an active area of research. It is critical to distinguish the functions of the UPS and the free 20S proteasome to gain better insight into the regulation of protein degradation through different mechanisms, potentially under different conditions.
Proteasome activity is regulated by a variety of PTMs, especially phosphorylation. Identification of the exact sites of modification on the proteasome and the conditions under which this occurs will allow us to study the dynamics of proteasome catalytic activity and potentially the signaling pathways involved in modulating proteasome-dependent protein degradation. In addition to PTMs, localization of the proteasome is important for various cellular functions, including regulation of the cell cycle, synaptic signaling, and gene expression. The proteasome can localize to the nucleus, cytoplasm, and even the plasma membrane in neurons. However, treatment with pharmacological proteasome inhibitors or knocking out individual proteasome subunits are not specific for distinct compartments, making it challenging to study the proteasomes’ compartment-specific roles. Understanding the mechanisms related to proteasome compartmentalization will provide new avenues for understanding target selection and cell type-specific functions. Different cell types have distinct morphologies, and proteasome localization to unique domains might dictate cell-specific proteasome function, suggesting that further research in this area could reveal novel roles for the proteasome across eukaryotic tissues in health and disease.
Recent investigation into the link between the proteasome and healthy neuronal function suggests that neuronal activity regulates proteasomes. We now know that neuronal activity is involved in modulation of the proteasome catalytic activity and localization in neuronal cells. Understanding the consequences of aberrant neuronal activation on proteasome-dependent degradation will help us to uncover proteasome-dependent pathways responsible for the initiation/progression of neurological disorders. However, much still remains to be done to decipher the collective mechanisms of proteasome-dependent protein degradation in the nervous system to dissect the disease-relevant pathways. Genetic manipulation of the 20S core subunits is limited in mammalian cells since they are essential proteins and are not suitable for knock-out experiments, making it difficult to test for necessity. Future discovery of reagents for detecting and manipulating the proteasome will enhance our understanding of the underlying mechanisms linking disruption of the proteasome system and neurological disorders.
Lastly, the presence of a neuronal proteasome that mediates the production of signaling peptides provides insight into a new and potentially interesting area of proteasome studies that relate to cellular signaling. Such a mechanism is akin to what we observe in the immune system, which has a specialized immunoproteasome that produces peptides relevant to immunological response(Goldberg and Rock, 1992; McCarthy and Weinberg, 2015; Rock et al., 1994; Zerfas et al., 2020). Discovery of these specialized proteasomes has led to new reagents to control their specific activities independent of other cytosolic constitutive proteasomes. Taken together, this new knowledge indicates the possibility of other tissue-specific proteasomes whose composition and regulation could be further explored for the benefit of fundamental understanding and the possibility of developing tissue-specific targeted-degradation therapies.
Highlights:
Proteasomes are a multisubunit catalytic complex.
Proteasomes are dynamically regulated.
Proteasomes in the nervous system are important for many aspects of neuronal function.
Proteasomes dependent degradation is disrupted in neurodegenerative disease.
Significance.
Proteasome-dependent proteolysis is a tightly regulated process that is essential in all eukaryotic cells, largely due to its central role in regulating protein levels. In the nervous system, proteasome function regulates many neuronal processes essential to brain function in health and disease. This review covers details related to proteasome structure and regulation. It also provides insight into the many studies that look at the role of the proteasome in the nervous system.
Acknowledgments
The authors thank Jordan Barrows and Caitlin Seluzicki for helpful discussions and review of the manusript. The research of S.S.M. is supported by grants from National Institute of Neurological Disorders and Stroke (R01NS110754).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
F.T., E.K.C., and S.S.M. are employees of The Johns Hopkins University School of Medicine. S.S.M. is an inventor on patents related to the neuronal membrane proteasome.
References
- Abraham WC (2003). How long will long-term potentiation last? Philosophical Transactions of the Royal Society B: Biological Sciences 358, 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adori C, Low P, Moszkovkin G, Bagdy G, Laszlo L, and Kovacs GG (2006). Subcellular distribution of components of the ubiquitin-proteasome system in non-diseased human and rat brain. J Histochem Cytochem 54, 263–267. [DOI] [PubMed] [Google Scholar]
- Ageta H, Kato A, Fukazawa Y, Inokuchi K, and Sugiyama H (2001a). Effects of proteasome inhibitors on the synaptic localization of Vesl-1S/Homer-1a proteins. Brain Res Mol Brain Res 97, 186–189. [DOI] [PubMed] [Google Scholar]
- Ageta H, Kato A, Hatakeyama S, Nakayama K, Isojima Y, and Sugiyama H (2001b). Regulation of the level of Vesl-1S/Homer-1a proteins by ubiquitin-proteasome proteolytic systems. J Biol Chem 276, 15893–15897. [DOI] [PubMed] [Google Scholar]
- Ahmadi S, Zhu S, Sharma R, Wu B, Soong R, Dutta Majumdar R, Wilson DJ, Simpson AJ, and Kraatz HB (2019). Aggregation of Microtubule Binding Repeats of Tau Protein is Promoted by Cu 2+. ACS Omega 4, 5356–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CG, Takahashi RH, and Gouras GK (2006). Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci 26, 4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyriou AA, Iconomou G, and Kalofonos HP (2008). Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood 112, 1593–1599. [DOI] [PubMed] [Google Scholar]
- Asher G, Tsvetkov P, and Kahana C (2005). Proteasomal Degradation of the Tumor Suppressors P53 and P73. Genes & Development 1, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajorek M, Finley D, and Glickman MH (2003). Proteasome disassembly and downregulation is correlated with viability during stationary phase. Current Biology 13, 1140–1144. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Neveu P, and Kosik KS (2009). A Coordinated Local Translational Control Point at the Synapse Involving Relief from Silencing and MOV10 Degradation. Neuron 64, 871–884. [DOI] [PubMed] [Google Scholar]
- Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, and Martin A (2018). Structure and Function of the 26S Proteasome. Annual Review of Biochemistry 87, 697–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh JM, Viktorova EG, and Pilipenko EV (2009). Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J Mol Biol 386, 814–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister W, Walz J, Zühl F, and Seemüller E (1998). The proteasome: Paradigm of a self-compartmentalizing protease. Cell 92, 367–380. [DOI] [PubMed] [Google Scholar]
- Beck F, Unverdorben P, Bohn S, Schweitzer A, Pfeifer G, Sakata E, Nickell S, Plitzko JM, Villa E, Baumeister W, et al. (2012). Near-atomic resolution structural model of the yeast 26S proteasome. Proc Natl Acad Sci U S A 109, 14870–14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Nissan G, and Sharon M (2014). Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4, 862–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EJ, Bence NF, Jayakumar R, and Kopito RR (2005). Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Molecular Cell 17, 351–365. [DOI] [PubMed] [Google Scholar]
- Besche HC, Sha Z, Kukushkin VN, Peth A, Hock EM, Kim W, Gygi S, Gutierrez JA, Liao H, Dick L, et al. (2014). Autoubiquitination of the 26S Proteasome on Rpn13 Regulates Breakdown of Ubiquitin Conjugates. EMBO Journal 33, 1159–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, and Schuman EM (2006). Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441, 1144–1148. [DOI] [PubMed] [Google Scholar]
- Biran A, Myers N, Adler J, Broennimann K, Reuven N, and Shaul Y (2017). A 20S proteasome receptor for degradation of intrinsically disordered proteins. bioRxiv, 210898. [Google Scholar]
- Bliss TVP, and Collingridge GL (2013). Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Molecular brain 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossis G, Ferrara P, Acquaviva C, Jariel-Encontre I, and Piechaczyk M (2003). c-Fos Proto-Oncoprotein Is Degraded by the Proteasome Independently of Its Own Ubiquitinylation In Vivo. Molecular and Cellular Biology 23, 7425–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow L, and Huang DT (2016). Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nature Reviews Molecular Cell Biology 17, 626–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai F, Frey JU, Sanna PP, and Behnisch T (2010). Protein degradation by the proteasome is required for synaptic tagging and the heterosynaptic stabilization of hippocampal late-phase long-term potentiation. Neuroscience 169, 1520–1526. [DOI] [PubMed] [Google Scholar]
- Carrard G, Bulteau AL, Petropoulos I, and Friguet B (2002). Impairment of proteasome structure and function in aging. International Journal of Biochemistry and Cell Biology 34, 1461–1474. [DOI] [PubMed] [Google Scholar]
- Cecarini V, Bonfili L, Amici M, Angeletti M, Keller JN, and Eleuteri AM (2008). Amyloid peptides in different assembly states and related effects on isolated and cellular proteasomes. Brain Res 1209, 8–18. [DOI] [PubMed] [Google Scholar]
- Cheng J, North BJ, Zhang T, Dai X, Tao K, Guo J, and Wei W (2018). The emerging roles of protein homeostasis-governing pathways in Alzheimer's disease. Aging Cell 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho-Park PF, and Steller H (2013). Proteasome regulation by ADP-ribosylation. Cell 153, 614–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu-Ping M, Slaughter CA, and DeMartino GN (1992). Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). Journal of Biological Chemistry 267, 10515–10523. [PubMed] [Google Scholar]
- Ciechanover A (1998). The ubiquitin-proteasome pathway: on protein death and cell life. Embo J 17, 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, and Kwon YT (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Experimental & molecular medicine 47, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, and Schwartz AL (1998). The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proceedings of the National Academy of Sciences of the United States of America 95, 2727–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, and Scott JD (2003). Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40, 595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GA, and Goldberg AL (2017). The Logic of the 26S Proteasome. Cell 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RO, Martins H, Martins LF, Cwetsch AW, Mele M, Pedro JR, Tome D, Jeon NL, Cancedda L, Jaffrey SR, et al. (2019). Synaptogenesis Stimulates a Proteasome-Mediated Ribosome Reduction in Axons. Cell Rep 28, 864–876 e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coux O, Tanaka K, and Goldberg AL (1996). Structure and functions of the 20S and 26S proteasomes. Annual Review of Biochemistry 65, 801–847. [DOI] [PubMed] [Google Scholar]
- Craiu A, Gaczynska M, Akopian T, Gramm CF, Fenteany G, Goldberg AL, and Rock KL (1997). Lactacystin andclasto-Lactacystin β-Lactone Modify Multiple Proteasome β-Subunits and Inhibit Intracellular Protein Degradation and Major Histocompatibility Complex Class I Antigen Presentation. Journal of Biological Chemistry 272, 13437–13445. [DOI] [PubMed] [Google Scholar]
- Dange T, Smith D, Noy T, Rommel PC, Jurzitza L, Cordero RJB, Legendre A, Finley D, Goldberg AL, and Schmidt M (2011). Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. Journal of Biological Chemistry 286, 42830–42839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, and Spillantini MG (2002). Proteasomal degradation of tau protein. Journal of Neurochemistry 83, 176–185. [DOI] [PubMed] [Google Scholar]
- Dekel E, Yaffe D, Rosenhek-Goldian I, Ben-Nissan G, Ofir-Birin Y, Morandi MI, Ziv T, Sisquella X, Pimentel MA, Nebl T, et al. (2021). 20S proteasomes secreted by the malaria parasite promote its growth. Nat Commun 12, 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux Q, Ustrell V, Pickart C, and Rechsteiner M (1994). A 26 S protease subunit that binds ubiquitin conjugates. Journal of Biological Chemistry 269, 7059–7061. [PubMed] [Google Scholar]
- Díaz-Hernández M, Valera AG, Morán MA, Gómez-Ramos P, Alvarez-Castelao B, Castaño JG, Hernández F, and Lucas JJ (2006). Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. Journal of Neurochemistry 98, 1585–1596. [DOI] [PubMed] [Google Scholar]
- Dikic I (2017). Proteasomal and Autophagic Degradation Systems. Annu Rev Biochem 86, 193–224. [DOI] [PubMed] [Google Scholar]
- Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, and Patrick GN (2009). Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J Biol Chem 284, 26655–26665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Bach VS, Haynes KA, and Hegde AN (2014). Proteasome modulates positive and negative translational regulators in long-term synaptic plasticity. Journal of Neuroscience 34, 3171–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Upadhya SC, Ding L, Smith TK, and Hegde AN (2008). Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn Mem 15, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Zhang S, Wu Z, Li X, Wang WL, Zhu Y, Stoilova-McPhie S, Lu Y, Finley D, and Mao Y (2019). Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove KK, and Klevit RE (2017). RING-Between-RING E3 Ligases: Emerging Themes amid the Variations. J Mol Biol 429, 3363–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiel W, Pratt G, Ferrell K, and Rechsteiner M (1992). Purification of an 11 S regulator of the multicatalytic protease. Journal of Biological Chemistry 267, 22369–22377. [PubMed] [Google Scholar]
- Dudek SM, and Bear MF (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proceedings of the National Academy of Sciences of the United States of America 89, 4363–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi V, Yaniv K, and Sharon M (2021). Beyond cells: The extracellular circulating 20S proteasomes. Biochim Biophys Acta Mol Basis Dis 1867, 166041. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6, 231–242. [DOI] [PubMed] [Google Scholar]
- Elsasser S, Chandler-Mitilello D, Müller B, Hanna J, and Finley D (2004). Rad23 and Rpn10 serve as alternate ubiquitin receptors for the proteasome. Journal of Biological Chemistry 279, 26817–26822. [DOI] [PubMed] [Google Scholar]
- Etlinger JD, and Goldberg AL (1977). A soluble ATP dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proceedings of the National Academy of Sciences of the United States of America 74, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre B, Lambour T, Garrigues L, Ducoux-Petit M, Amalric F, Monsarrat B, Burlet-Schiltz O, and Bousquet-Dubouch MP (2014). Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. Journal of Proteome Research 13, 3027–3037. [DOI] [PubMed] [Google Scholar]
- Feng Y, Longo DL, and Ferris DK (2001). Polo-like kinase interacts with proteasomes and regulates their activity. Cell Growth and Differentiation 12, 29–37. [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, and Nagerl UV (2006). A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52, 239–245. [DOI] [PubMed] [Google Scholar]
- Franic D, Zubcic K, and Boban M (2021). Nuclear Ubiquitin-Proteasome Pathways in Proteostasis Maintenance. Biomolecules 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French ME, Koehler CF, and Hunter T (2021). Emerging functions of branched ubiquitin chains. Cell Discov 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Reis N, Lee Y, Glickman MH, and Vierstra RD (2001). Subunit interaction maps for the regulatory particle of the 26S proteasome and the COP9 signalosome. EMBO Journal 20, 7096–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi J, Antonelli AC, Afridi A, Vatsia S, Joshi G, Romanov V, Murray IVJ, and Khan SA (2019). Protein misfolding and aggregation in neurodegenerative diseases: A review of pathogeneses, novel detection strategies, and potential therapeutics. Reviews in the Neurosciences 30, 339–358. [DOI] [PubMed] [Google Scholar]
- George AJ, Hoffiz YC, Charles AJ, Zhu Y, and Mabb AM (2018). A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front Genet 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulivi C, Pacifici RE, and Davies KJ (1994). Exposure of hydrophobic moieties promotes the selective degradation of hydrogen peroxide-modified hemoglobin by the multicatalytic proteinase complex, proteasome. Arch Biochem Biophys 311, 329–341. [DOI] [PubMed] [Google Scholar]
- Goldberg AL (2012). Development of proteasome inhibitors as research tools and cancer drugs. J Cell Biol 199, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL, and Rock K (2002). Not just research tools--proteasome inhibitors offer therapeutic promise. Nat Med 8, 338–340. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, and Rock KL (1992). Proteolysis, proteasomes and antigen presentation. Nature 357, 375–379. [DOI] [PubMed] [Google Scholar]
- Gomes VA, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, Jones RC, Thyparambil S, Wang GW, Qiao X, et al. (2006). Mapping the murine cardiac 26S proteasome complexes. Circulation Research 99, 362–371. [DOI] [PubMed] [Google Scholar]
- Gregori L, Hainfeld JF, Simon MN, and Goldgaber D (1997). Binding of amyloid beta protein to the 20 S proteasome. J Biol Chem 272, 58–62. [DOI] [PubMed] [Google Scholar]
- Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R, Glickman MH, and Finley D (2000). A gated channel into the proteasome core particle. Nature Structural Biology 7, 1062–1067. [DOI] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, and Huber R (1997). Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature 386, 463–471. [DOI] [PubMed] [Google Scholar]
- Grune T, Catalgol B, Licht A, Ermak G, Pickering AM, Ngo JK, and Davies KJA (2011). HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radical Biology and Medicine 51, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grune T, Reinheckel T, and Davies KJ (1997). Degradation of oxidized proteins in mammalian cells. FASEB J 11, 526–534. [PubMed] [Google Scholar]
- Guan H, Wang Y, Yu T, Huang Y, Li M, Saeed AFUH, Perculija V, Li D, Xiao J, Wang D, et al. (2020). Cryo-EM structures of the human PA200 and PA200-20S complex reveal regulation of proteasome gate opening and two PA200 apertures. PLoS Biology 18, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AM, Oh WC, Vega-Ramirez H, Stein IS, Hell JW, Patrick GN, and Zito K (2012). Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 74, 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haratake K, Sato A, Tsuruta F, and Chiba T (2016). KIAA0368-deficiency affects disassembly of 26S proteasome under oxidative stress condition. J Biochem 159, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding RJ, and Tong YF (2018). Proteostasis in Huntington's disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacologica Sinica 39, 754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshbarger W, Miller C, Diedrich C, and Sacchettini J (2015). Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 23, 418–424. [DOI] [PubMed] [Google Scholar]
- Hegde AN (2017). Proteolysis, synaptic plasticity and memory. Neurobiol Learn Mem 138, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, and Schwartz JH (1997). Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89, 115–126. [DOI] [PubMed] [Google Scholar]
- Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, and Morimoto RI (2004). Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO Journal 23, 4307–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L, Huang HC, and Jiang ZF (2014). Relationship between amyloid-beta and the ubiquitin-proteasome system in alzheimer's disease. Neurological Research 36, 276–282. [DOI] [PubMed] [Google Scholar]
- Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, and Shi Y (2002). Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111, 1041–1054. [DOI] [PubMed] [Google Scholar]
- Hu M, Li P, Song L, Jeffrey PD, Chenova TA, Wilkinson KD, Cohen RE, and Shi Y (2005). Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO Journal 24, 3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Luan B, Wu J, and Shi Y (2016). An atomic structure of the human 26S proteasome. Nature Structural and Molecular Biology 23, 778–785. [DOI] [PubMed] [Google Scholar]
- Huber EM, and Groll M (2017). The Mammalian Proteasome Activator PA28 Forms an Asymmetric alpha4beta3 Complex. Structure 25, 1473–1480 e1473. [DOI] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, and Dikic I (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inai Y, and Nishikimi M (2002). Increased degradation of oxidized proteins in yeast defective in 26 S proteasome assembly. Arch Biochem Biophys 404, 279–284. [DOI] [PubMed] [Google Scholar]
- Iwafune Y, Kawasaki H, and Hirano H (2002). Electrophoretic analysis of phosphorylation of the yeast 20S proteasome. Electrophoresis 23, 329–338. [DOI] [PubMed] [Google Scholar]
- Jacobson AD, Zhang NY, Xu P, Han KJ, Noone S, Peng J, and Liu CW (2009). The lysine 48 and lysine 63 ubiquitin conjugates are processed differently by the 26 s proteasome. J Biol Chem 284, 35485–35494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung T, Engels M, Kaiser B, Poppek D, and Grune T (2006). Intracellular distribution of oxidized proteins and proteasome in HT22 cells during oxidative stress. Free Radic Biol Med 40, 1303–1312. [DOI] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Thomas U, Knopfel T, and Behnisch T (2006). Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J Neurosci 26, 4949–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Huang FF, and Markesbery WR (2000). Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 98, 149–156. [DOI] [PubMed] [Google Scholar]
- Kishino T, Lalande M, and Wagstaff J (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15, 70–73. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, and Goldberg AL (2001). Proteasome inhibitors: from research tools to drug candidates. Chem Biol 8, 739–758. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Kaganovich D, and Goldberg AL (2002). Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes. Evidence for peptide-induced channel opening in the α-rings. Journal of Biological Chemistry 277, 22260–22270. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, and Shimizu N (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. [DOI] [PubMed] [Google Scholar]
- Kohler A, Bajorek M, Groll M, Moroder L, Rubin DM, Huber R, Glickman MH, and Finley D (2001). The substrate translocation channel of the proteasome. Biochimie 83, 325–332. [DOI] [PubMed] [Google Scholar]
- Komander D, and Rape M (2012). The ubiquitin code. Annu Rev Biochem 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Korovila I, Hugo M, Castro JP, Weber D, Hohn A, Grune T, and Jung T (2017). Proteostasis, oxidative stress and aging. Redox Biol 13, 550–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulichkova VA, Artamonova TO, Lyublinskaya OG, Khodorkovskii MA, Tomilin AN, and Tsimokha AS (2017). Proteomic analysis of affinity-purified extracellular proteasomes reveals exclusively 20S complexes. Oncotarget 8, 102134–102149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Deshmukh F, Yaffe D, Olshina MA, Ben-Nissan G, and Sharon M (2019). The Contribution of the 20S Proteasome to Proteostasis. Biomolecules 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YA, Pickart CM, Alban A, Landon M, Jamieson C, Ramage R, Mayer RJ, and Layfield R (2000). Inhibition of the ubiquitin-proteasome system in Alzheimer ’ s disease. 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavabre-Bertrand T, Henry L, Carillo S, Guiraud I, Ouali A, Dutaud D, Aubry L, Rossi JF, and Bureau JP (2001). Plasma proteasome level is a potential marker in patients with solid tumors and hemopoietic malignancies. Cancer 92, 2493–2500. [DOI] [PubMed] [Google Scholar]
- Li X, Amazit L, Long W, Lonard DM, Monaco JJ, and O'Malley BW (2007). Ubiquitin- and ATP-Independent Proteolytic Turnover of p21 by the REGγ-Proteasome Pathway. Molecular Cell 26, 831–842. [DOI] [PubMed] [Google Scholar]
- Limanaqi F, Biagioni F, Gambardella S, Familiari P, Frati A, and Fornai F (2020). Promiscuous roles of autophagy and proteasome in neurodegenerative proteinopathies. International Journal of Molecular Sciences 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Jones S, Minis A, Rodriguez J, Molina H, and Steller H (2019). PI31 Is an Adaptor Protein for Proteasome Transport in Axons and Required for Synaptic Development. Developmental Cell 50, 509–524.e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokireddy S, Kukushkin NV, and Goldberg AL (2015). cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proceedings of the National Academy of Sciences of the United States of America 112, E7176–E7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malacrida A, Meregalli C, Rodriguez-Menendez V, and Nicolini G (2019). Chemotherapy-induced peripheral neuropathy and changes in cytoskeleton. International Journal of Molecular Sciences 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RS, McLoughlin F, and Vierstra RD (2016). Autophagic Turnover of Inactive 26S Proteasomes in Yeast Is Directed by the Ubiquitin Receptor Cue5 and the Hsp42 Chaperone. Cell Reports 16, 1717–1732. [DOI] [PubMed] [Google Scholar]
- Martinez-Fonts K, Davis C, Tomita T, Elsasser S, Nager AR, Shi Y, Finley D, and Matouschek A (2020). The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat Commun 11, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason GGF, Hendil KB, and Rivett AJ (1996). Phosphorylation of Proteasomes in Mammalian Cells. Identification of Two Phosphorylated Subunits and the Effect of Phosphorylation on Activity. European Journal of Biochemistry 238, 453–462. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Lander GC, and Martin A (2013). Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol 20, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MK, and Weinberg JB (2015). The immunoproteasome and viral infection: A complex regulator of inflammation. Frontiers in Microbiology 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Pruneda JN, Klevit RE, and Weissman AM (2014). RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta 1843, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minis A, Rodriguez JA, Levin A, Liu K, Govek EE, Hatten ME, and Steller H (2019). The proteasome regulator PI31 is required for protein homeostasis, synapse maintenance, and neuronal survival in mice. Proceedings of the National Academy of Sciences of the United States of America 116, 24639–24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushtaq A, Kapoor V, Latif A, Iftikhar A, Zahid U, McBride A, Abraham I, Riaz BI, and Anwer F (2018). Efficacy and toxicity profile of carfilzomib based regimens for treatment of multiple myeloma: A systematic review. Critical Reviews in Oncology/Hematology 125, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeku N, Clelland CL, Emrani S, Kukushkin VN, Yu WH, Goldberg AL, and Duff KE (2016). Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nature Medicine 22, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, et al. (2013). D2P2: Database of disordered protein predictions. Nucleic Acids Research 41, D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, and Mook-Jung I (2005). Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech Ageing Dev 126, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Ortega J, Heymann JB, Kajava VA, Ustrell V, Rechsteiner M, and Steven AC (2005). The axial channel of the 20 S proteasome opens upon binding of the PA200 activator. Journal of Molecular Biology 346, 1221–1227. [DOI] [PubMed] [Google Scholar]
- Ottis P, and Crews CM (2017). Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chemical Biology 12, 892–898. [DOI] [PubMed] [Google Scholar]
- Pajares M, Jimenez-Moreno N, Dias IHK, Debelec B, Vucetic M, Fladmark KE, Basaga H, Ribaric S, Milisav I, and Cuadrado A (2015). Redox control of protein degradation. Redox Biol 6, 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DTS, and Sheng M (2003). Targeted Protein Degradation and Synapse Remodeling by an Inducible Protein Kinase. Science 302, 1368–1373. [DOI] [PubMed] [Google Scholar]
- Palmer A, Rivett AJ, Thomson S, Hendil KB, Butcher GW, Fuertes G, and Knecht E (1996). Subpopulations of proteasomes in rat liver nuclei, microsomes and cytosol. Biochem J 316 (Pt 2), 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering AM, Koop AL, Teoh CY, Ermak G, Grune T, and Davies KJ (2010). The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem J 432, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, and Cheng Y (2008). Mechanism of Gate Opening in the 20S Proteasome by the Proteasomal ATPases. Molecular Cell 30, 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai SN, Singh BK, Rathore AS, Zahra W, Keswani C, Birla H, Singh SS, Dilnashin H, and Singh SP (2019). Quality Control in Huntington’s Disease: a Therapeutic Target. Neurotoxicity Research 36, 612–626. [DOI] [PubMed] [Google Scholar]
- Ramachandran VK, Fu JM, Schaffer TB, Na CH, Delannoy M, and Margolis SS (2018). Activity-Dependent Degradation of the Nascentome by the Neuronal Membrane Proteasome. Molecular Cell 71, 169–177.e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran VK, and Margolis SS (2017). A mammalian nervous-system-specific plasma membrane proteasome complex that modulates neuronal function. Nature Structural and Molecular Biology 24, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynes R, Pomatto LCD, and Davies KJA (2016). Degradation of oxidized proteins by the proteasome: Distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Molecular Aspects of Medicine 50, 41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJA, and Grune T (1998). Comparative resistance of the 20 S and 26 S proteasome to oxidative stress. Biochemical Journal 335, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinetti GV, and Schweizer FE (2010). Ubiquitination acutely regulates presynaptic neurotransmitter release in mammalian neurons. J Neurosci 30, 3157–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivett AJ, Palmer A, and Knecht E (1992). Electron microscopic localization of the multicatalytic proteinase complex in rat liver and in cultured cells. J Histochem Cytochem 40, 1165–1172. [DOI] [PubMed] [Google Scholar]
- Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, and Goldberg AL (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771. [DOI] [PubMed] [Google Scholar]
- Scharf A, Rockel TD, and von Mikecz A (2007). Localization of proteasomes and proteasomal proteolysis in the mammalian interphase cell nucleus by systematic application of immunocytochemistry. Histochem Cell Biol 127, 591–601. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Nuber U, and Huibregtse JM (1995). Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 373, 81–83. [DOI] [PubMed] [Google Scholar]
- Schipper-Krom S, Sanz AS, van Bodegraven EJ, Speijer D, Florea BI, Ovaa H, and Reits EA (2019). Visualizing Proteasome Activity and Intracellular Localization Using Fluorescent Proteins and Activity-Based Probes. Front Mol Biosci 6, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, and Finley D (2014). Regulation of proteasome activity in health and disease. Biochim Biophys Acta 1843, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Haas W, Crosas B, Santamaria PG, Gygi SP, Walz T, and Finley D (2005). The HEAT repeat protein Blm10 regulates the yeast proteasome by capping the core particle. Nature Structural and Molecular Biology 12, 294–303. [DOI] [PubMed] [Google Scholar]
- Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, Elsasser S, Finley D, Dikic I, Walters KJ, and Groll M (2008). Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 453, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC (2015). The PyMOL Molecular Graphics System, Version 2.4.1. [Google Scholar]
- Schubert U, Antón LC, Gibbs J, Norbury CC, Yewdell JW, and Bennink JR (2000). Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774. [DOI] [PubMed] [Google Scholar]
- Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, Lee BH, Zhang N, de Poot SA, Tuebing F, Sun S, et al. (2016). Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva GM, Netto LE, Simoes V, Santos LF, Gozzo FC, Demasi MA, Oliveira CL, Bicev RN, Klitzke CF, Sogayar MC, et al. (2012). Redox control of 20S proteasome gating. Antioxid Redox Signal 16, 1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluimer J, and Distel B (2018). Regulating the human HECT E3 ligases. Cell Mol Life Sci 75, 3121–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DM, Chang SC, Park S, Finley D, Cheng Y, and Goldberg AL (2007). Docking of the Proteasomal ATPases' Carboxyl Termini in the 20S Proteasome's α Ring Opens the Gate for Substrate Entry. Molecular Cell 27, 731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoberger A, Anderson RT, and Smith DM (2017). The Proteasomal ATPases Use a Slow but Highly Processive Strategy to Unfold Proteins. Front Mol Biosci 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, and Broadie K (2003). The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol 13, 899–910. [DOI] [PubMed] [Google Scholar]
- Tai HC, and Schuman EM (2008). Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci 9, 826–838. [DOI] [PubMed] [Google Scholar]
- Thibaudeau TA, Anderson RT, and Smith DM (2018). A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun 9, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, and Spillantini MG (2001). α-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Letters 509, 22–26. [DOI] [PubMed] [Google Scholar]
- Tomoshige S, Nomura S, Ohgane K, Hashimoto Y, and Ishikawa M (2018). Degradation of huntingtin mediated by a hybrid molecule composed of IAP antagonist linked to phenyldiazenyl benzothiazole derivative. Bioorganic and Medicinal Chemistry Letters 28, 707–710. [DOI] [PubMed] [Google Scholar]
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, and LaFerla FM (2008). Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 29, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukmar-Godec T, Fang P, Ibáñez de Opakua A, Henneberg F, Godec A, Pan KT, Cima-Omori MS, Chari A, Mandelkow E, Urlaub H, et al. (2020). Proteasomal degradation of the intrinsically disordered protein tau at single-residue resolution. Science advances 6, eaba3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay A, Joshi V, Amanullah A, Mishra R, Arora N, Prasad A, and Mishra A (2017). E3 Ubiquitin Ligases Neurobiological Mechanisms: Development to Degeneration. Frontiers in Molecular Neuroscience 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen FW, Burbach JPH, Hol EM, and Hol (1998a). Mutations in RNA: A first example of molecular misreading in Alzheimer's disease. Trends in Neurosciences 21, 331–335. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen FW, De Kleijn DPV, Van Den Hurk HH, Neubauer A, Sonnemans MAF, Sluijs JA, Köycü S, Ramdjielal RDJ, Salehi A, Martens GJM, et al. (1998b). Frameshift mutants of β amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science 279, 242–247. [DOI] [PubMed] [Google Scholar]
- Vashisht A, Bach VS, Fetterhoff D, Morgan JW, McGee M, and Hegde AN (2018). Proteasome limits plasticity-related signaling to the nucleus in the hippocampus. Neuroscience Letters 687, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco R, Alberti P, Bruna J, Psimaras D, and Argyriou AA (2019). Bortezomib and other proteosome inhibitors—induced peripheral neurotoxicity: From pathogenesis to treatment. Journal of the Peripheral Nervous System 24, S52–S62. [DOI] [PubMed] [Google Scholar]
- VerPlank JJS, and Goldberg AL (2017). Regulating protein breakdown through proteasome phosphorylation. Biochem J 474, 3355–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VerPlank JJS, and Goldberg AL (2018). Exploring the regulation of proteasome function by subunit phosphorylation. Methods in Molecular Biology 1844, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VerPlank JJS, Tyrkalska SD, Tyrkalska SD, Tyrkalska SD, Fleming A, Fleming A, Fleming A, Rubinsztein DC, and Goldberg AL (2020). CGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proceedings of the National Academy of Sciences of the United States of America 117, 14220–14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Darwin KH, and Li H (2010a). Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol 17, 1352–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yen J, Kaiser P, and Huang L (2010b). Regulation of the 26S proteasome complex during oxidative stress. Science Signaling 3, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Argiles-Castillo D, Kane EI, Zhou A, and Spratt DE (2020). HECT E3 ubiquitin ligases – emerging insights into their biological roles and disease relevance. Journal of Cell Science 133, jcs228072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, and Anggono V (2015). Activity-Dependent Ubiquitination of GluA1 and GluA2 Regulates AMPA Receptor Intracellular Sorting and Degradation. Cell Rep 10, 783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk S, and Orlowski M (1980). Cation-Sensitive Neutral Endopeptidase: Isolation and Specificity of the Bovine Pituitary Enzyme. Journal of Neurochemistry 35, 1172–1182. [DOI] [PubMed] [Google Scholar]
- Wilk S, and Orlowski M (1983a). Evidence that Pituitary Cation-Sensitive Neutral Endopeptidase Is a Multicatalytic Protease Complex. Journal of Neurochemistry 40, 842–849. [DOI] [PubMed] [Google Scholar]
- Wilk S, and Orlowski M (1983b). Inhibition of Rabbit Brain Prolyl Endopeptidase by N-Benzyloxycarbonyl-Prolyl-Prolinal, a Transition State Aldehyde Inhibitor. Journal of Neurochemistry 41, 69–75. [DOI] [PubMed] [Google Scholar]
- Willeumier K, Pulst SM, and Schweizer FE (2006). Proteasome inhibition triggers activity-dependent increase in the size of the recycling vesicle pool in cultured hippocampal neurons. J Neurosci 26, 11333–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, and Peng J (2009). Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, et al. (2007). SCRAPPER-Dependent Ubiquitination of Active Zone Protein RIM1 Regulates Synaptic Vesicle Release. Cell 130, 943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Smith DM, Kim HM, Rodriguez V, Goldberg AL, and Cheng Y (2010). Interactions of PAN's C-termini with archaeal 20S proteasome and implications for the eukaryotic proteasome-ATPase interactions. EMBO Journal 29, 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, and Bonhoeffer T (2001). Morphological Changes in Dendritic Spines Associated with Long-Term Synaptic Plasticity. Annual Review of Neuroscience 24, 1071–1089. [DOI] [PubMed] [Google Scholar]
- Zerfas BL, Maresh ME, and Trader DJ (2020). The Immunoproteasome: An Emerging Target in Cancer and Autoimmune and Neurological Disorders. J Med Chem 63, 1841–1858. [DOI] [PubMed] [Google Scholar]
- Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, and Kudlow JE (2003). O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115, 715–725. [DOI] [PubMed] [Google Scholar]
- Zhao X, and Yang J (2010). Amyloid-beta peptide is a substrate of the human 20S proteasome. ACS Chem Neurosci 1, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, and Shabek N (2017). Ubiquitin Ligases: Structure, Function, and Regulation. Annu Rev Biochem 86, 129–157. [DOI] [PubMed] [Google Scholar]
- Zientara-Rytter K, and Subramani S (2019). The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoeger A, Blau M, Egerer K, Feist E, and Dahlmann B (2006). Circulating proteasomes are functional and have a subtype pattern distinct from 20S proteasomes in major blood cells. Clin Chem 52, 2079–2086. [DOI] [PubMed] [Google Scholar]