

Abstract
Tauopathies, including Alzheimer’s disease, are characterized by progressive accumulation of hyperphosphorylated and pathologic tau protein in association with onset of cognitive and behavioral impairment. Tau pathology is also associated with increased susceptibility to seizures and epilepsy, with tau−/− mice showing seizure resistance in some epilepsy models. To better understand how tau pathology is related to neuronal excitability, we performed whole-cell patch clamp electrophysiology in dentate gyrus granule cells of tau−/− and human-tau expressing, htau mice. The htau mouse is unique from other transgenic tau models in that the endogenous murine tau gene has been and replaced with readily phosphorylated human tau. We assessed several measures of neuronal excitability, including evoked action potential frequency and excitatory synaptic responses in dentate granule cells from tau−/−, htau, and non-transgenic control mice at 1.5, 4, and 9 months of age. Compared to age matched controls, dentate granule cells from both tau−/− and htau mice had a lower peak frequency of evoked action potentials and greater paired pulse facilitation, suggesting reduced neuronal excitability. Our results suggest that neuronal excitability is more strongly influenced by the absence of functional tau than by the presence of pathologic tau. These results also suggest that tau’s effect on neuronal excitability is more complex than previously understood.
Keywords: Microtubule associated protein tau, tauopathy, htau, tau−/−, hippocampus, dentate granule cell, EPSC
Introduction
Microtubule dynamics are critical for central nervous system (CNS) function. Disruptions in microtubule homeostasis are associated with a variety of CNS dysfunctions including Alzheimer’s disease and other tauopathies, epilepsy, Parkinson’s disease, congenital brain malformations, psychiatric disorders, and autism spectrum disorder (Chang et al., 2018; Gardiner and Marc, 2010; Goncalves et al., 2018; Marchisella et al., 2016; Pellegrini et al., 2017; Saha and Sen, 2019). The microtubule-associated protein tau (MAPT, tau) plays an important role in the assembly and stabilization of microtubules. To allow normal microtubule dynamics, phosphorylated tau has reduced affinity for microtubules, promoting their dismantling (Lindwall and Cole, 1984). The equilibrium of tau phosphorylation, which is tightly regulated under physiological conditions (Martin et al., 2013a; Martin et al., 2013b), becomes disrupted in Alzheimer’s disease and other tauopathies, to allow accumulation and aggregation of hyperphosphorylated tau (pTau) (Castellani and Perry, 2019). Tau solubility decreases as hyperphosphorylation increases, eventually forming insoluble neurofibrillary tangles, which are a hallmark of Alzheimer’s disease and other tauopathies (Castellani and Perry, 2019).
Hyperphosphorylated tau promotes increased neuronal excitability. In mouse models expressing mutant human tau protein, principal neurons in the frontal cortex and hippocampus exhibit depolarized neuronal resting membrane potentials (Crimins et al., 2012; Crimins et al., 2011; Rocher et al., 2010), increased evoked action potential firing and spontaneous excitatory postsynaptic currents (sEPSCs) (Crimins et al., 2012; Crimins et al., 2011; Rocher et al., 2010), increased glutamate release and decreased glutamate reuptake (Decker et al., 2016; Hunsberger et al., 2015), and abnormal neuron morphology and synaptic organization (Crimins et al., 2012; Crimins et al., 2011; Rocher et al., 2010; Yoshiyama et al., 2007). Consistent with this increased neuronal excitability, mice expressing mutant human tau protein exhibit increased vulnerability to induced epilepsy (Garcia-Cabrero et al., 2013; Liu et al., 2017), a finding consistent with studies demonstrating increased seizure prevalence in patients with Alzheimer’s disease (Pandis and Scarmeas, 2012; Vossel et al., 2013; Vossel et al., 2016).
In addition to establishing a correlation between tau hyperphosphorylation and neuronal hyperactivity in animals, previous work has investigated the effect of removing or reducing tau expression on seizures. Disruption of Mapt expression reduces seizure burden and improves survival in some genetic models of epilepsy (Gheyara et al., 2014; Holth et al., 2013), confers resistance to chemically induced seizures (DeVos et al., 2013; Li et al., 2014), and prevents glutamate excitotoxicity in cultured neurons (Miyamoto et al., 2017). Furthermore, reduction of tau phosphorylation by administration of the protein phosphatase 2A (PP2A) activator, sodium selenate, reduced seizure burden and promoted survival in multiple seizure models (Jones et al., 2012; Liu et al., 2016). Taken together, these studies suggest involvement of tau hyperphosphorylation in epilepsy due to its effects on neuronal excitability.
While previous studies have outlined an important role for tau in promoting neuronal hyperexcitability, the full impact of tau’s effect on neuronal function remains unknown. Age related functional changes in tau knockout mice may impact neuronal activity and animal survival at later ages. Furthermore, the effect of pTau on excitability has been studied in mouse models that express mutant human tau, without suppressing endogenous mouse tau. The presence of two different tau species could thus confound results, since the murine tau may remain functional despite the presence of pathologic human tau. It is unclear whether the presence of wild-type murine tau in most tauopathy models modulates the pathological functions conferred by hyperphosphorylation. This study will address these gaps in current knowledge by measuring neuronal excitability in tau−/− mice and in the htau mouse model, which expresses six isoforms of non-mutant human tau with a concurrent deletion of endogenous murine tau (Andorfer et al., 2003). The htau model recapitulates features of tau pathology and cognitive deficits present in Alzheimer’s disease, which include appearance of hyperphosphorylated tau species as early as 1.5mo of age, deposition of late-stage tangle pathology at 9mo, and presentation of substantial cognitive deficits by 12mo. Given these considerations and previous data demonstrating profound hippocampal deficits in tauopathy models (Abisambra et al., 2010; Abisambra et al., 2013; Fontaine et al., 2017), we tested the hypotheses that pTau promotes hyperexcitability of dentate gyrus granule cells (DGCs) in the absence of functional endogenous tau, and that complete tau ablation reduces neuronal excitability throughout the life of the animal. We chose to study DGCs because they contribute to epileptogenesis and tauopathy-associated cognitive decline (Alcantara-Gonzalez et al., 2021; Boychuk et al., 2016; Hunt et al., 2010; Lee et al., 2012; Martin-Belmonte et al., 2020), and epilepsy-related changes in the dentate gyrus were ameliorated by tau deletion (Gheyara et al 2014). Despite their importance in cognition and disease processes, the electrophysiological effects of modifying tau expression have not been studied extensively in DGCs.
Materials and Methods
Animals
Transgenic B6.Cg-Mapttm1(EGFP)KltTg(MAPT)8cPdav/J mice (male and female) were produced in house from breeders obtained from The Jackson Laboratory (Jax# 005491; Bar Harbor, ME). This mouse line was generated previously by introducing a transgene encoding six isoforms of human tau without disease-associated mutations onto homozygous tau−/− mice (Andorfer et al., 2003). These mice lack any obvious disease phenotype at birth, but develop impairments in Morris Water Maze, spatial learning, and food burrowing with age (Geiszler et al., 2016; Phillips et al., 2011; Polydoro et al., 2009). This mouse strain was originally generated on a hybrid Swiss Webster/B6D2F1 hybrid background but has been backcrossed to C57BL/6J for more than 10 generations. SNP analysis performed by The Jackson Laboratory were consistent with a pure C57BL/6J background, which served as the control strain. All breeding mice were homozygous for a disruption of the murine tau gene. One mouse in each breeding pair was hemizygous for a transgene expressing all six isoforms of non-mutant human tau protein. The offspring are therefore either full tau knockout (tau−/−) or express only human tau (htau).
DNA was extracted from tail snips and genotype was confirmed via PCR according to the protocols supplied by Jackson labs. Disruption of the endogenous murine tau gene was confirmed using the primer pair 5’-CGTTGTGGCTGTTGTAGTTG-3’ and 5’-TCGTGACCACCCTGACCTAC-3’, which amplifies a fragment at 270 bp in both tau−/− and htau mice. Presence of the human-tau transgene was confirmed using the primer pair 5’-CGAAGTGATGGAAGATCACG-3’ and 5’-GTCTTGGTGCATGGTGTAGC-3’, which amplifies a fragment at 79 bp in htau mice. Protein expression was confirmed via western blot of hippocampal homogenate using the H150 antibody (Fig. 1; 1:2000; Santa Cruz Biotechnology). Age matched male C57BL/6J control mice were obtained from Jackson labs (Jax#000664) and allowed to acclimate after delivery for at least one week prior to any experiments. All mice were housed under a 14 hour light / 10 hour dark cycle in an Association for Assessment and Accreditation of Laboratory Animal Care Internal (AALAC) facility. Food and water were available ad libitum. The University of Kentucky Institutional Animal Care and Use Committee approved all procedures.
Figure 1. Membrane voltage response to injected current in DGCs from tau−/− and htau mice compared to non-transgenic control mice.
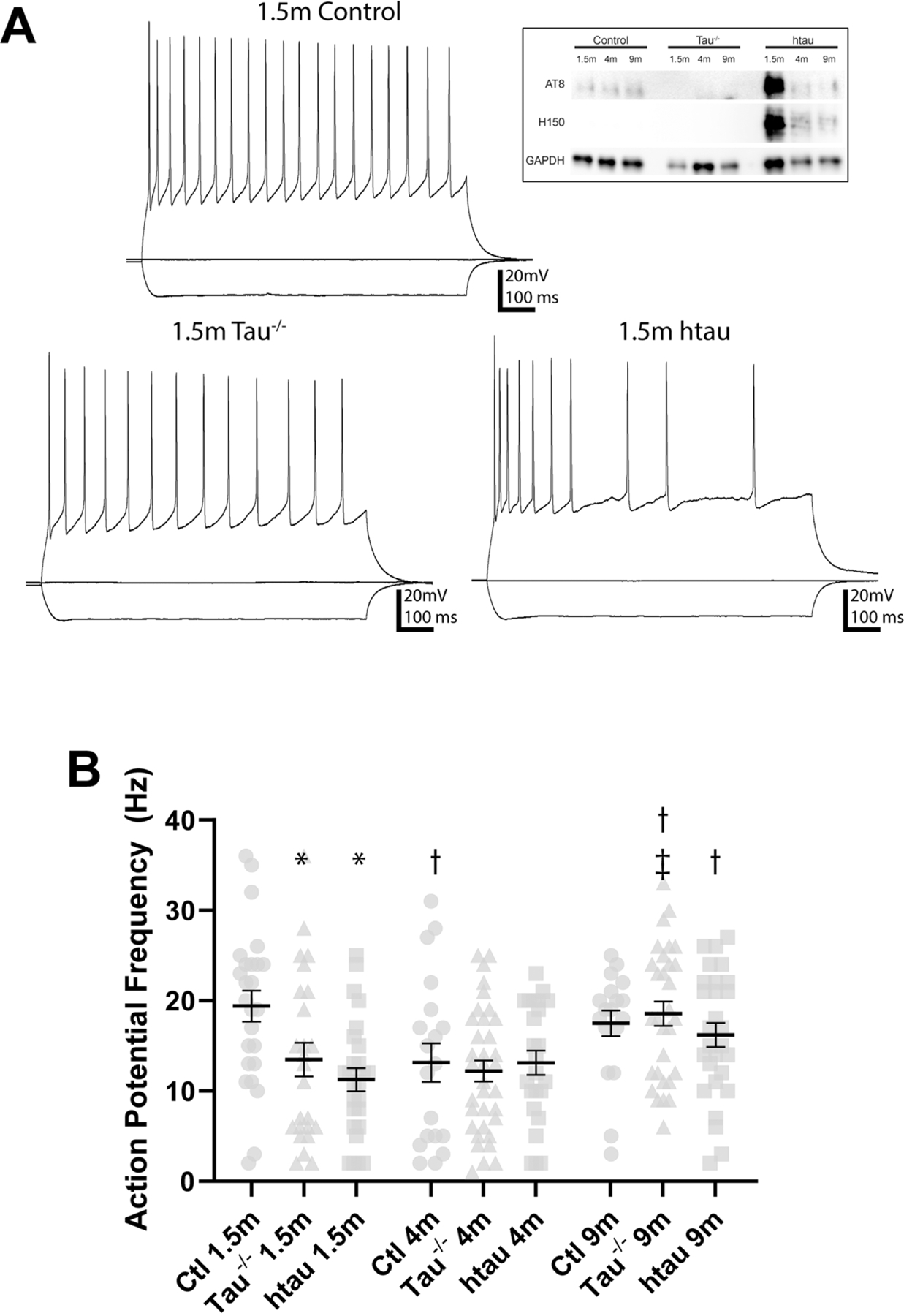
(A) Sample recordings showing voltage response from −100, 0, and 200pA current steps in DGCs from 1.5 month old tau−/−, htau, and control mice. Inset: representative Western blot showing absence of tau protein in tau−/− and presence of human tau protein in htau mice. (B) Comparison of maximum evoked action potential frequency (250pA current injection). At 1.5 months, DGCs from control mice fire more actions potentials than those from tau−/− and htau mice. In tau−/− mice, DGCs from 9 month old mice fire more action potentials than at 1.5 or 4 months. In htau mice, DGCs from 9 month old mice fire more action potentials than at 1.5 months. In control mice, DGCs from 4 month old mice fire more action potentials than at 1.5 months.*: Different from control at same age (p<0.05) †: Different from 1.5 month of same genotype (p<0.05) ǂ: Different from 4 month of same genotype (p<0.05). Error bars indicate SEM. Statistical comparisons were made between all groups by two-way ANOVA with age and genotype as factors (Tukey post hoc).
Hippocampal slice preparation
Mice were deeply anesthetized via inhalation of isoflurane to effect (lack of tail pinch response) and decapitated while anesthetized. The brain was rapidly removed from the skull and immersed in ice-cold oxygenated (95% O2/5% CO2) cutting/holding artificial cerebrospinal fluid (aCSF). The cutting/holding aCSF contained (in mM): 85 NaCl, 75 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4·H2O, 4 MgCl2·6H2O, 0.5 CaCl2·2H2O, and 24 NaHCO3 (pH 7.2–7.4). Coronal sections (300 µm) were cut on a vibrating microtome (Vibratome Series 1000; Technical Products International, St. Louis, MO). Each slice was divided with a midsagittal cut and hippocampi were isolated and transferred to a holding chamber with warmed (30–32° C), oxygenated cutting/holding aCSF and incubated for at least 1 hour before recordings. In a subset of animals, 5–8 hippocampal slices were set aside for protein analysis. Extrahippocampal tissue was removed from these slices. Approximately 30mg of hippocampal tissue was collected and flash frozen in liquid nitrogen. One slice at a time was transferred to a chamber mounted under an upright microscope (BX51WI; Olympus) and was superperfused with warmed (30–32° C) oxygenated recording aCSF. The recording aCSF contained (in mM): 124 NaCl, 3 KCl, 2 CaCl2, 1.3 MgCl, 1.4 NaHCO3, and 11 glucose (pH 7.2–7.4). Each slice was equilibrated in the microscope chamber for >10 minutes before recording.
Electrophysiological recordings
Whole-cell patch-clamp recording were obtained from hippocampal DGCs, identified by location and morphological characteristics. Recording pipettes were pulled from borosilicate glass (open tip resistance 3–5 MΩ; King Precision Glass Co.). The pipette recording solution contained (in mM): 126 K+-gluconate, 4 KCl, 10 HEPES, 4 MgATP, 0.3 NaGTP, and 10 PO-creatine (pH 7.2). Electrophysiological recordings were performed using a Multiclamp 700B amplifier (Molecular Devices), low pass filtered at 2 kHz, digitized at 20 kHz (Digidata 1440A; Molecular Devices), and recorded onto a computer using pClamp 10.2 software (Molecular Devices). Seal resistance was typically 2–5 GΩ. Series resistance was <25 MΩ (mean: 11.4 MΩ ) and was monitored periodically during the recordings. Recordings were discontinued if series resistance changed by more than 20% during the recording.
Spontaneous excitatory post-synaptic currents (sEPSCs) were recorded in voltage-clamp mode at a holding potential of −70 mV. Resting membrane potential and input resistance were measured in current clamp mode. Current steps (−100 pA to 400 pA in 50 pA steps) were injected to record membrane voltage response. The input resistance was calculated from the slope of the linear portion of the resulting current-voltage curve. The resting membrane potential was averaged from the portions of recorded traces between current steps. A platinum-iridium concentric-bipolar electrode (125 µM diameter; FHC) was positioned on the lateral perforant pathway of each slice. A series of 30 pairs of current pulses (10–60 µA; 400 µs; interpulse interval 75 ms; 5 seconds between pulse pairs) were administered to evoke EPSCs. The stimulus intensity was adjusted so that responses occurred after >80% of pulses. Stimulus sweeps that failed to elicit a response with both stimuli were excluded from analysis.
Tissue Homogenization and Western Blot
The hippocampal tissue from each mouse was homogenized as described previously (Koren et al., 2019). Tissue samples were mechanically homogenized in RIPA lysis buffer (VWR) with phosphatase inhibitor cocktails 2 and 3 (Sigma), cOmplete protease inhibitor (Sigma), and PMSF (1 mM final concentration, Roche Diagnostics). Homogenates were centrifuged at 4°C at 13,000 rpm for 30 minutes. The fresh supernatant from each sample was divided into aliquots and stored at −80°C. Protein concentration was quantified using the Pierce BCA kit (ThermoFisher, 23225).
Sample protein concentrations were normalized with lysis buffer and denatured by boiling for 5 minutes in 4x Laemmli buffer (BioRad) plus 10% β-mercaptoethanol (Sigma). The samples were separated on a 10% tris-glycine gel (BioRad) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore). Membranes were blocked in 6% non-fat dry milk (w/v) in 0.01M tris-buffered saline (TBS). All antibodies were diluted in 6% non-fat milk in TBS. Primary antibodies used are as follows: AT8 (pS202/pT205 tau; 1:2000; ThermoFisher), H-150 (human tau; 1:2000; Santa Cruz), GAPDH (1:5000; Cell Signaling Technology). Goat anti-mouse and goat anti-rabbit secondary antibodies conjugated to horseradish peroxidase (Southern Biotech) used were diluted in 6% non-fat dry milk in TBS (w/v). Images were collected on an Amersham Imager 600 (General Electric).
Data Analysis
All electrophysiological measures (evoked action potential frequency, resting membrane potential, input resistance, spontaneous EPSC frequency, and paired pulse responses) were analyzed with MiniAnalysis (Synaptosoft, Fort Lee, NJ). Statistical measures were performed with Prism (GraphPad, San Diego, CA). Data were tested for normality using the Shapiro-Wilk test. Data that were not normally distributed were log-transformed prior to further analysis. Data were normally distributed except where noted. A two-way ANOVA (Tukey posthoc test for multiple comparisons) with age and genotype as factors was used to compare mean values for each electrophysiological value. All data for each electrophysiological measurement were analyzed together. Some control data are presented in multiple figures for presentation clarity. Data were disaggregated by sex and no sex-dependent differences were detected for any measure, so sexes were combined for all analyses. Total cell counts for each electrophysiological measurement are summarized in Table 1. Data are presented as mean ± SEM and statistical significance was set to p<0.05 for all tests.
Table 1.
Summary of Replicates Used for Electrophysiological Measurements
| Age | Genotype | N (cells) | ||
|---|---|---|---|---|
| Induced Action Potential Frequency | Spontaneous EPSC Frequency | Paired Pulse Ratio | ||
| 1.5 months | tau−/− | 26 | 20 | 21 |
| htau | 25 | 16 | 26 | |
| control | 25 | 13 | 22 | |
| 4 months | tau−/− | 24 | 15 | 17 |
| htau | 19 | 11 | 17 | |
| control | 35 | 21 | 16 | |
| 9 months | tau−/− | 28 | 23 | 23 |
| htau | 18 | 16 | 12 | |
| control | 30 | 21 | 19 | |
Results
Resting membrane potential and input resistance in tau−/− and htau mice
Overexpression of hyperphosphorylated tau is associated with a depolarized resting membrane potential in neocortical pyramidal neurons of Tg4510 mutant mice (Crimins et al., 2012; Crimins et al., 2011; Rocher et al., 2010), but the electrophysiological effects of modifying tau expression in DGCs have not been studied in detail. Additionally, the insertion of pathologic human tau in other models (e.g., Tg4510) may compete with endogenous mouse tau to regulate microtubule assembly and function (Alonso Adel et al., 2006). To better understand the contribution of tau to intrinsic properties, resting membrane potential and input resistance were measured in DGCs from non-transgenic control, tau−/−, and htau mice at 1.5, 4, and 9 months of age, which represent time points in which early pTau species are detected, mid-stage pathological processes occur, and insoluble tangle pathology is evident, respectively (Andorfer et al., 2003).
Differences in resting membrane potential and input resistance were analyzed between control, tau−/−, and htau mice at each age by a two-way ANOVA, with age and genotype as factors. The two-way ANOVA found a significant effect of age, but not genotype, on resting membrane potential (F[2, 224]=11.83; p<0.05) and input resistance (F[2, 224]=14.11; p<0.05). In DGCs from tau−/− mice, the resting membrane potential was depolarized compared to those from age-matched control mice at 1.5 months of age (p<0.05; Table 2). No difference in resting membrane potential was detected at 4 months or 9 months of age (p>0.05; Table 2). No difference in resting membrane potential was detected between htau and control mice at any age (p>0.05). Input resistance was also not different between DGCs from control mice and either tau−/− or htau mice at any age (p>0.05; Table 2). With the exception of resting membrane potential differences in young tau −/− mice, differences in passive membrane properties were not detected across genotypes at any age.
Table 2.
Summary of Resting Membrane Potential and Input Resistance
| Age | Genotype | N (cells) | Membrane potential Mean (mV) |
Input Resistance Mean (MΩ) |
|---|---|---|---|---|
| 1.5 months | tau−/− | 25 | −68.8±1.8* | 278.7±28.4MΩ |
| htau | 26 | −71.1±1.8 | 288.5±27.8MΩ | |
| control | 25 | −72.1±2.1 | 265.6±33.1MΩ | |
| 4 months | tau−/− | 35 | −70.1±1.9 | 287.4±22.5MΩ |
| htau | 24 | −70.8±1.7 | 278.4±32.4MΩ | |
| control | 19 | −70.3±2.1 | 276.9±22.1MΩ | |
| 9 months | tau−/− | 30 | −73.9±1.3 †ǂ | 236.4±15.3MΩ †ǂ |
| htau | 28 | −73.2±1.7 | 227.1±17.4MΩ †ǂ | |
| control | 18 | −73.8±1.7ǂ | 234.1±21.5MΩ |
significant difference versus control.
significant difference versus 1.5 month.
significant difference versus 4 month.
The effect of age on resting membrane potential and input resistance was determined within each genotype. Resting membrane potential was hyperpolarized in DGCs from 9 month old tau−/− mice, relative to 1.5 and 4 month old tau−/− mice (p<0.05). Resting membrane potential was also hyperpolarized in DGCs from 9 month old control mice compared to 4 month old control mice (p<0.05). Resting membrane potential did not change with age in DGCs from htau mice (p>0.05), and no age-related difference in input resistance was detected in control mice (p>0.05). Input resistance in DGCs from 9 month old tau−/− mice, however, was lower compared to those from 1.5 or 4 month old tau−/− mice (p<0.05). Similarly, input resistance was lower in DGCs from 9 month old htau mice compared to 1.5 or 4 month old htau mice (p<0.05). Thus, input resistance was lower at the oldest ages examined for DGCs in both tau−/− and htau mice. Input resistance was not different between DGCs from tau−/− and htau mice at any age (p>0.05).
Lower action potential firing frequency in young tau−/− and htau mice
At 4 and 8–9 months of age, neocortical pyramidal cells from the Tg4510 tau mouse model exhibit a higher frequency of evoked action potentials in response to depolarizing current injection, relative to control mice (Crimins et al., 2012; Crimins et al., 2011; Rocher et al., 2010), but lower action potential threshold and reduced action potential frequency was reported in CA1 pyramidal cells from the same mouse strain (Hatch et al., 2017), and age-related effects of tau expression on intrinsic excitability of DGCs has not been assessed. To better understand the role of tau in determining evoked action potential frequency, depolarizing and hyperpolarizing currents were injected into DGCs from non-transgenic control, tau−/−, and htau mice at 1.5, 4, and 9 months of age. The average evoked action potential frequency at the current step resulting in maximum action potential frequency (i.e., 250 pA) was analyzed using a two-way ANOVA, with age and genotype as factors. The two-way ANOVA found a significant effect of interaction between age and genotype on peak action potential frequency (F[4, 221]=2.594; p<0.05).
Dentate granule cells from 1.5 month old tau−/− and htau mice had a lower peak action potential frequency relative to cells from age-matched control mice (p<0.05; Fig. 1B). Peak action potential frequency in these neurons did not differ between tau−/−, htau, and control mice at 4 or 9 months (p>0.05). Thus, versus age-matched controls, lower action potential frequency was observed in DGCs at 1.5 months of age in both tau−/− and htau mice, but not at older ages. No differences in action potential frequency were detected between DGCs from tau−/− and htau mice at any age (p>0.05).
Membrane voltage response to injected current was analyzed at different time points to assess the effect of aging within each genotype. In DGCs from control mice, cells from 4 month old mice had a lower peak frequency of evoked action potentials compared to 1.5 month old mice (p<0.05; Fig. 1B). DGCs from 9 month old tau−/− mice had a higher peak frequency of evoked action potentials compared to 1.5 or 4 month old mice (p<0.05; Fig. 1B). DGCs from 9 month old htau mice had a higher peak frequency of evoked action potentials compared to 1.5 month old mice (p<0.05; Fig. 1B).Thus, higher peak action potential frequency emerged at 9 months of age in both htau and tau −/− mice, but this was not evident in control mice. Together, these results suggest intrinsic properties in both tau−/− and htau mice develop age-related changes that are not observed in control mice. DGCs from both transgenic strains exhibit reduced cellular excitability compared to control early in life, but these differences are abrogated with age. Furthermore, the intrinsic properties of DGCs from tau−/− and htau mice change with age in a similar fashion.
Spontaneous EPSC frequency was not impacted by tau
Excitatory synaptic input to DGCs is thought to contribute to network excitability and seizure generation in several models of acquired epilepsy (Butler et al., 2015; Hunt et al., 2010; Winokur et al., 2004), and changes in sEPSC frequency related to changes in tau expression could contribute to the changes in seizure susceptibility observed by others (Garcia-Cabrero et al., 2013; Gheyara et al., 2014; Holth et al., 2013). To determine if tau expression influenced overall excitatory synaptic input to DGCs, spontaneous EPSC frequency was measured in DGCs from tau−/−, htau, and control mice. A Shapiro-Wilk test found the data were not normally distributed. Since the values were all positive and positively skewed, a log transformation was performed on the raw data. Log-transformed sEPSC frequencies were compared with a two-way ANOVA with age and genotype as factors. No differences were detected between groups at any ages (p>0.05; Fig 3A&B). Overall, there was not a strong influence of age or strain on sEPSC frequency.
Figure 3. Paired pulse ratio in tau−/− mice compared to non-transgenic control mice.
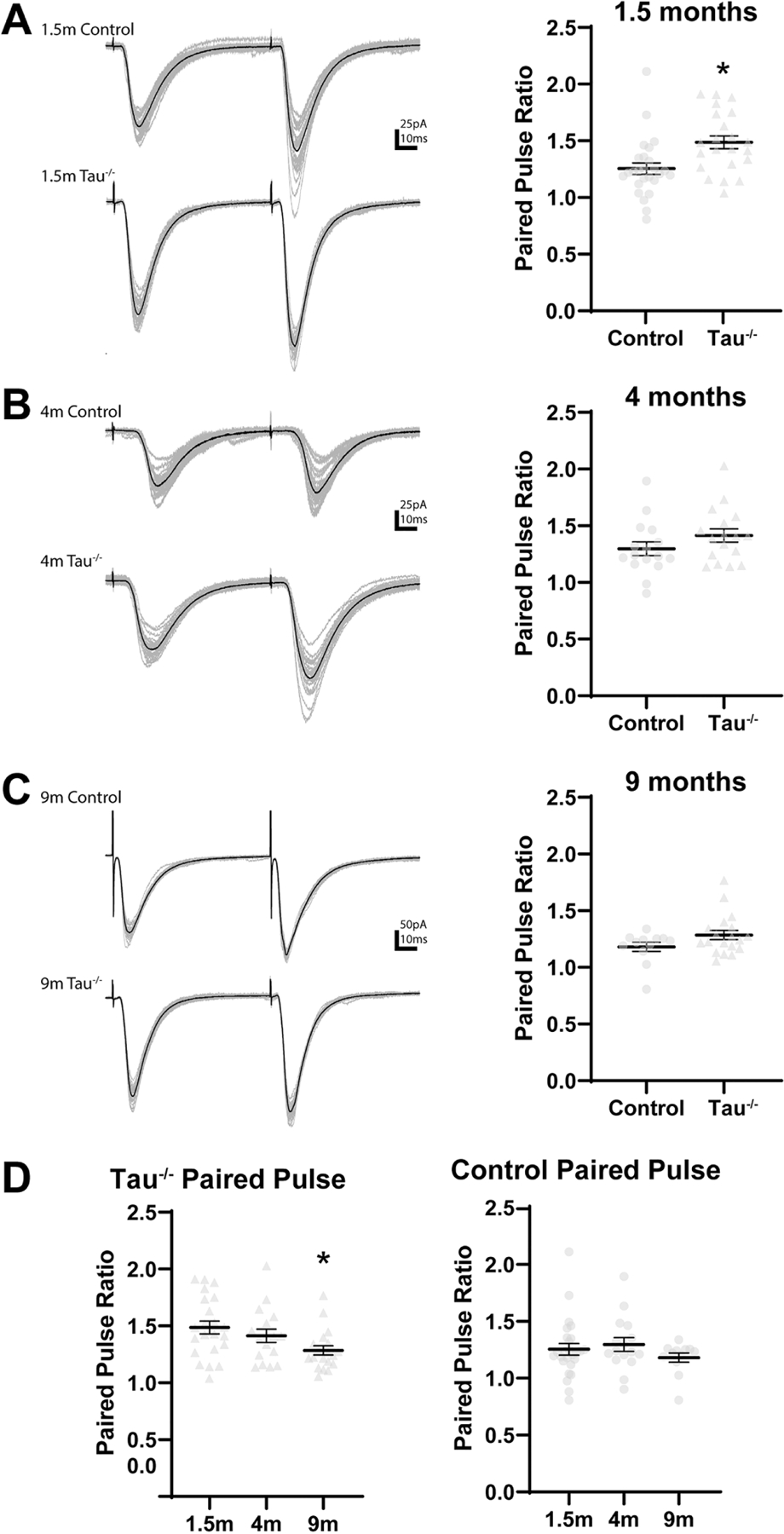
(A) The paired pulse ratio is higher in DGCs from 1.5 month old tau−/− mice compared to 1.5 month old control mice. *: p<0.05 (B, C) No differences in in paired pulse ratio were found between DGCs from tau−/− and control mice at 4 (B) or 9 (C) months old. (D) The paired pulse ratio is lower in DGCs from 9 month old tau−/− mice compared to 1.5 month old tau−/− mice. The paired pulse ratio did not change with age in DGCs from control mice. Statistical comparisons were made between all groups by two-way ANOVA with age and genotype as factors. *: different from 1.5 month (p<0.05) Error bars indicate SEM.
Paired pulse facilitation is enhanced in young tau−/− and htau mice
Altered probability of glutamate release can also influence DGC activity and may be susceptible to tau hyperphosphorylation or mutation (Yoshiyama et al., 2007). To determine effects of tau expression on paired pulse facilitation in the dentate gyrus, pairs of stimuli were administered to the lateral perforant path and evoked EPSCs were recorded in DGCs from tau−/−, htau, and control mice. The paired pulse ratio, which is calculated as the amplitude of the second evoked EPSC divided by the amplitude of the first evoked EPSC, is inversely proportional to the probability of neurotransmitter release from the presynaptic terminal (i.e. a higher paired pulse ratio indicates a lower probability of neurotransmitter release; Graziane and Dong, 2016). The paired pulse ratio of evoked EPSCs was compared between all groups with a two-way ANOVA with age and genotype as factors. The two-way ANOVA found a significant effect of age and genotype, but not their interaction, on paired pulse ratio (Fage[2, 164]=8.924; Fgenotype[2, 164]=6.480; p<0.05).
The paired pulse ratio of evoked EPSCs was significantly greater in DGCs from tau−/−mice compared to age-matched control mice at 1.5 months (control: 1.27±0.10; tau−/−: 1.56±0.16; p=0.002) but did not differ at 4 or 9 months (p>0.05; Fig. 4A-C). The paired pulse ratio was lower in DGCs from 9 month old tau−/− mice compared to 1.5 month old tau−/− mice (1.5 month tau−/−: 1.56±0.16, 9 month tau−/−: 1.28±0.09; Fig. 4D). Similar to results from tau −/− mice, the paired pulse ratio was greater in DGCs from htau mice compared to control mice at 1.5 months (control: 1.27±0.10; htau: 1.43±0.09; p=0.023) but did not differ at 4 or 9 months of age (p>0.05; Fig. 5A-C,). The paired pulse ratio did not change with age in DGCs from htau mice (p>0.05; Fig. 5D). Thus, in both transgenic mouse strains, the paired pulse ratio in DGCs after stimulation of the lateral perforant pathway was greater compared to those from non-transgenic control mice at 1.5 months of age, but not later in life. The paired pulse ratio in DGCs after stimulation of the perforant pathway did not differ between tau−/− and htau animals at any age (p>0.05). The paired pulse ratio decreased with age in tau−/− mice. This contrasted with results from htau mice and non-transgenic control mice, in which age-related changes in paired pulse ratio were not detected (p>0.05). The amplitudes of evoked EPSCs varied significantly across all cells but were not different between any groups (data not shown).
Figure 4. Paired pulse ratio in htau mice compared to non-transgenic control mice.
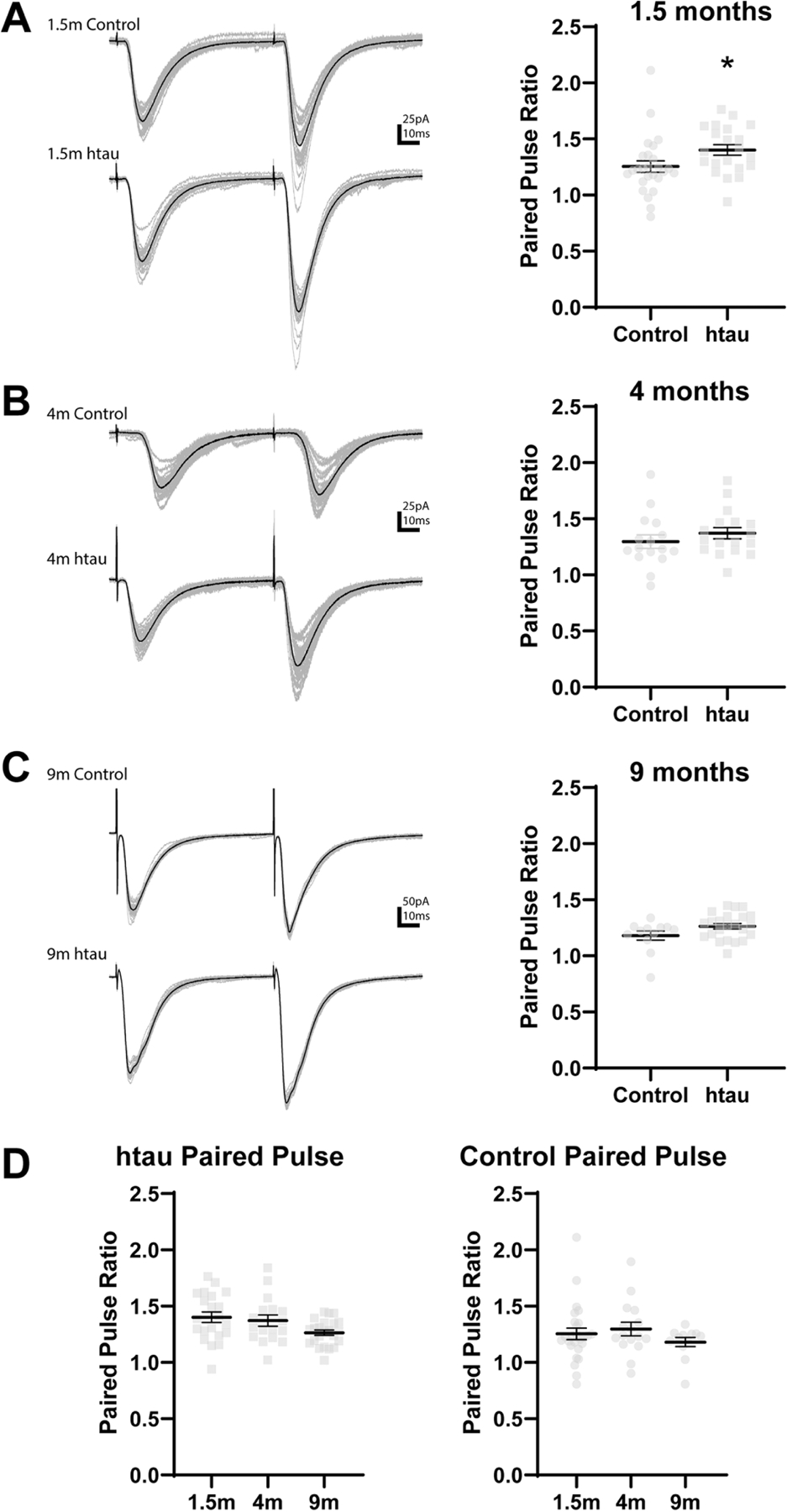
(A) The paired pulse ratio is higher in DGCs from 1.5 month old htau mice compared to 1.5 month old control mice. *: p<0.05 (B, C) No differences in in paired pulse ratio were found between DGCs from htau and control mice at 4 (B) or 9 (C) months old. (D) The paired pulse ratio in DGCs from htau mice compared to 1.5 month old htau mice. The paired pulse ratio did not change with age in DGCs from control mice. Statistical comparisons were made between all groups by two-way ANOVA with age and genotype as factors. *: different from 1.5 month (p<0.05) Error bars indicate SEM.
Discussion
Tau pathology is associated with greater excitability and increased susceptibility to seizures in epilepsy models (Garcia-Cabrero et al., 2013; Liu et al., 2017). Similarly, loss of tau through genetic deletion or suppression with antisense oligonucleotides is associated with decreased mossy fiber sprouting in the dentate gyrus, reduced seizure burden, and improved cognition and survival in models of epilepsy (DeVos et al., 2013; Gheyara et al., 2014; Holth et al., 2013) and ameliorates the increased susceptibility to seizures associated with some models of Alzheimer’s disease (Ittner et al., 2010; Roberson et al., 2007). This study measured several intrinsic and synaptic membrane properties in tau−/− and htau mice to provide better understanding of tau’s role in neuronal excitability, which has been hypothesized to play a role in genetic epilepsies and, possibly, seizure disorders associated with neurodegenerative disease (Decker et al., 2016; Garcia-Cabrero et al., 2013; Gheyara et al., 2014; Holth et al., 2013; Ittner et al., 2010; Roberson et al., 2011; Roberson et al., 2007).
Although other studies have measured the influence of tau expression and hyperphosphorylation on neuronal properties, most have used forms of tau with disease-related mutations such as P301L (Crimins et al., 2012; Crimins et al., 2011; Hatch et al., 2017; Hunsberger et al., 2015; Liu et al., 2017; Rocher et al., 2010), P301S (Yoshiyama et al., 2007), and A152T (Decker et al., 2016). In contrast, the htau mouse used in this study expresses six isoforms of human tau without any disease-related mutations (Andorfer et al., 2003). This model was selected for two reasons. First, the non-mutated human tau expressed in this model results in slower development of pathology that reasonably resembles human aging than in more aggressive pathologic tau models. The htau mouse exhibits progressive tau pathology, developing pTau by 1.5 months, somatodendritic redistribution of tau around 3 months, and neurofibrillary tangles around 9 months (Andorfer et al., 2003). Additionally, the endogenous murine tau gene is deleted in the htau mouse, setting it apart from other commonly used tauopathy models. The deletion of endogenous murine tau is important because tau hyperphosphorylation leads to concurrent loss of normal tau function and consequently increased pathology due to the increase in soluble pTau. Tau hyperphosphorylation reduces the pool of functional tau available to stabilize microtubules, resulting in a loss of physiologic function (Lindwall and Cole, 1984). At the same time, soluble pTau itself is neurotoxic through several mechanisms, including mislocalization of pathologic tau, inhibition of protein translation, and abnormal interaction with other cellular components (Flach et al., 2012; Fulga et al., 2007; Hoover et al., 2010; Koren et al., 2019; Meier et al., 2015; Meier et al., 2016; Thies and Mandelkow, 2007; Tian et al., 2013). Because tauopathy results in loss of function as well as development of neurotoxicity, the presence of normal murine tau expression in other tau mouse models confounds the interpretation of outcomes based on tau hyperphosphorylation. Some inconsistency between the results across previous studies in different tau models, along with the similarities found between the htau and tau−/− animals in the current study, demonstrate the importance of separating the effects of removing tau and adding hyperphosphorylated tau in interpreting electrophysiological outcomes.
While previous work suggests an important role for tau in the electrophysiological function of the neuron, the direct effects of tau deletion on neuronal excitability have not been extensively studied without the presence of additional pathology. Previous studies on the electrophysiological effects of hyperphosphorylated tau have primarily used the rTg4510 transgenic mouse, which expresses 4R0N human tau with the pathogenic P301L mutation with endogenous murine tau expression intact (Santacruz et al., 2005). The major effects on the intrinsic properties of frontal cortex pyramidal neurons in these studies were depolarized resting membrane potential and increased frequency of evoked action potentials (Crimins et al., 2012; Crimins et al., 2011; Rocher et al., 2010). A decrease in paired pulse facilitation in the hippocampus has also been reported in other transgenic tau models (Maeda et al., 2016; Roberson et al., 2011; Sydow et al., 2011; Yoshiyama et al., 2007). Based on these studies, we hypothesized that similar results would be detected in DGCs from htau mice, but this hypothesis was not supported. Instead we found no changes in resting membrane potential in the htau mice, and reduced evoked action potential firing and increased paired pulse facilitation was detected in both htau and tau −/− mice, but only at 1.5 months of age. Thus, deletion of murine tau tended to reduce intrinsic and synaptic excitability of DGCs early, but not later in life, and replacing murine tau with human tau did not reinstate normal excitability.
Our results in htau mice might be linked to pathogenic mechanisms of Alzheimer’s disease. Unlike rTg4510 mice, which overexpress 13x more human mutant 4R0N P301L tau in the forebrain, htau mice express all six splice variants of non-mutant human tau (Andorfer et al., 2003; Santacruz et al., 2005). The P301L mutation is associated with frontotemporal dementia (Hutton et al., 1998); meanwhile, tau pathology in Alzheimer’s brains has accumulation of the six splice variants of non-mutant human tau. The difference in tau pathogenicity between the htau and rTg4510 mice may explain why no change in resting membrane potential was detected in DGCs from htau mice. Other factors, including the endogenous properties of the different neuron types studied (i.e., pyramidal cells versus DGCs) or the continued expression of endogenous mouse tau in the rTg4510 mice, could also contribute substantially to these outcomes. However, the differences in tau species, or even cell type examined, do not adequately explain the patterns of evoked action potentials or paired pulse facilitation observed in each mouse model: A difference in relative pathogenicity could result in a smaller magnitude of difference between these measures, but it does not fully explain the opposite direction of effects (i.e., action potential frequency decreased in htau but increased in rTg4510 and paired pulse ratio increased in htau but decreased in rTg4510).
Because the htau mice lack endogenous tau, physiological tau function is likely reduced because the transgenically expressed human tau may not function normally. This notion is supported by the similar patterns of evoked action potentials observed in dentate granule cells from htau and tau−/− mice early in life, when human tau may not function normally in the htau mouse. Similar to studies demonstrating that the pro-excitatory effects of amyloid-beta rely on the presence of functional tau (Roberson et al., 2011; Shipton et al., 2011), our results suggest that excitatory effects associated with pathologic tau in other models requires a pool of functional tau as well, which is not expressed in either strain used here. Any potential pro-excitatory effects may therefore have been abrogated by the absence of functional tau.
This study found important similarities between dentate granule cell responses in tau−/−and htau mice, consistent with the hypothesis that the substitution of tau species in the htau mouse contributes to the loss of normal tau function. In both tau−/− and htau mice, peak evoked action potential frequency in DGCs was reduced relative to age-matched controls early in life, but not at later ages. Similarly, peak evoked action potential frequency in DGCs increased with age in both tau−/− and htau mice, becoming comparable to that seen in controls. These results together indicate that absence of functional tau, due to hyperphosphorylation and/or gene deletion, has an effect on intrinsic neuronal function early in life that is not evident as the animals age. Tau deletion reduces excitability in both young tau−/− and htau mice, but this does not persist in older mice.
Tau’s ability to affect synaptic function, either on its own or in conjunction with other Alzheimer’s disease pathology, has been established in several transgenic animal models. The major effect of pathologic tau on synaptic function is impairment of long term potentiation (LTP) and a decrease in the paired pulse ratio at several hippocampal synapses (Decker et al., 2016; Maeda et al., 2016; Roberson et al., 2011; Shipton et al., 2011; Sydow et al., 2011; Yoshiyama et al., 2007). The impairment of LTP likely contributes to deficits in learning and memory in these models. The decrease in paired pulse ratio suggests an increased probability of neurotransmitter release and may at least partially explain the hyperexcitability and susceptibility to seizures observed in these models. Previous studies have found that removal of tau rescues synaptic deficits associated with Alzheimer’s disease, particularly amyloid-β induced impairment of LTP (Roberson et al., 2011; Shipton et al., 2011). These studies reported minimal differences between tau−/− and non-transgenic mice in the absence of additional pathology. In the current study we measured the frequency of spontaneous excitatory post-synaptic currents (sEPSCs) and found no differences between genotypes at any age. Conversely, changes in presynaptic release associated with absence of functional murine tau protein were observed in both transgenic strains used herein. We measured the dentate granule cell response after stimulation of the lateral perforant path to assess synaptic release probability, based on paired pulse response ratios. DGCs from both tau−/− and htau mice exhibited greater paired pulse facilitation (suggesting lower probability of synaptic glutamate release) compared to age-matched, non-transgenic control mice early in life, but the paired pulse ratio was not different from controls in either tau−/− or htau mice later in life. The paired pulse ratio decreased significantly with age in tau−/− mice, whereas it did not change with age in htau or non-transgenic mice. Our study found similar presynaptic neurotransmitter release in both transgenic strains, suggesting a loss of tau function in both groups.
Taken together, the reduced frequency of evoked action potentials and increased paired pulse facilitation in young tau−/− mice provide a basis for the seizure resistance in genetic epilepsy models observed in tau−/− mice at similar ages (Gheyara et al., 2014; Holth et al., 2013). One limitation of those studies is the relatively young age at which the mice were, necessarily, assessed. The results of the present study suggest that the anti-epileptic and anti-SUDEP effect of tau deletion evident in mutant mice, which express channelopathies that underlie seizures, may not fully translate to epilepsies with a later age of onset. These effects may be lost or reduced with age as excitability increases. Previous work found changes in expression of other microtubule-associated proteins in tau−/− mice (Harada et al., 1994; Ma et al., 2014). More work is required to understand how these changes in expression might affect electrophysiological function in tau−/− mice.
Conclusions
This study sheds new light on the role of tau in promoting neuronal excitability. We found that DGCs from mice lacking tau protein or in which endogenous mouse tau is replaced by human tau, which has previously been shown to become hyperphosphorylated early in life (Andorfer et al., 2003), exhibited reduced measures of excitability in the form of reduced peak evoked action frequency and increased paired pulse facilitation that were significant in young animals but were abrogated with age. The loss of excitability in the htau mouse contrasts with previous work showing a pro-excitatory role of pathologic tau in pyramidal neurons from other tauopathy models, but this may be due to differences in tau or other pathogenicity, endogenous tau expression in these models, or in the characteristics of the types of neurons and networks examined. Finally, our results suggest that, while early changes in tau expression may influence neuronal excitability and seizures in young mice, other compensatory mechanisms may participate in stabilizing neuronal circuits later in life. Identifying additional links between tau phosphorylation and neuronal network function may help resolve the influence of tauopathy on disease progression.
Figure 2. Average spontaneous EPSC frequency in tau−/−, htau, and non-transgenic control mice.
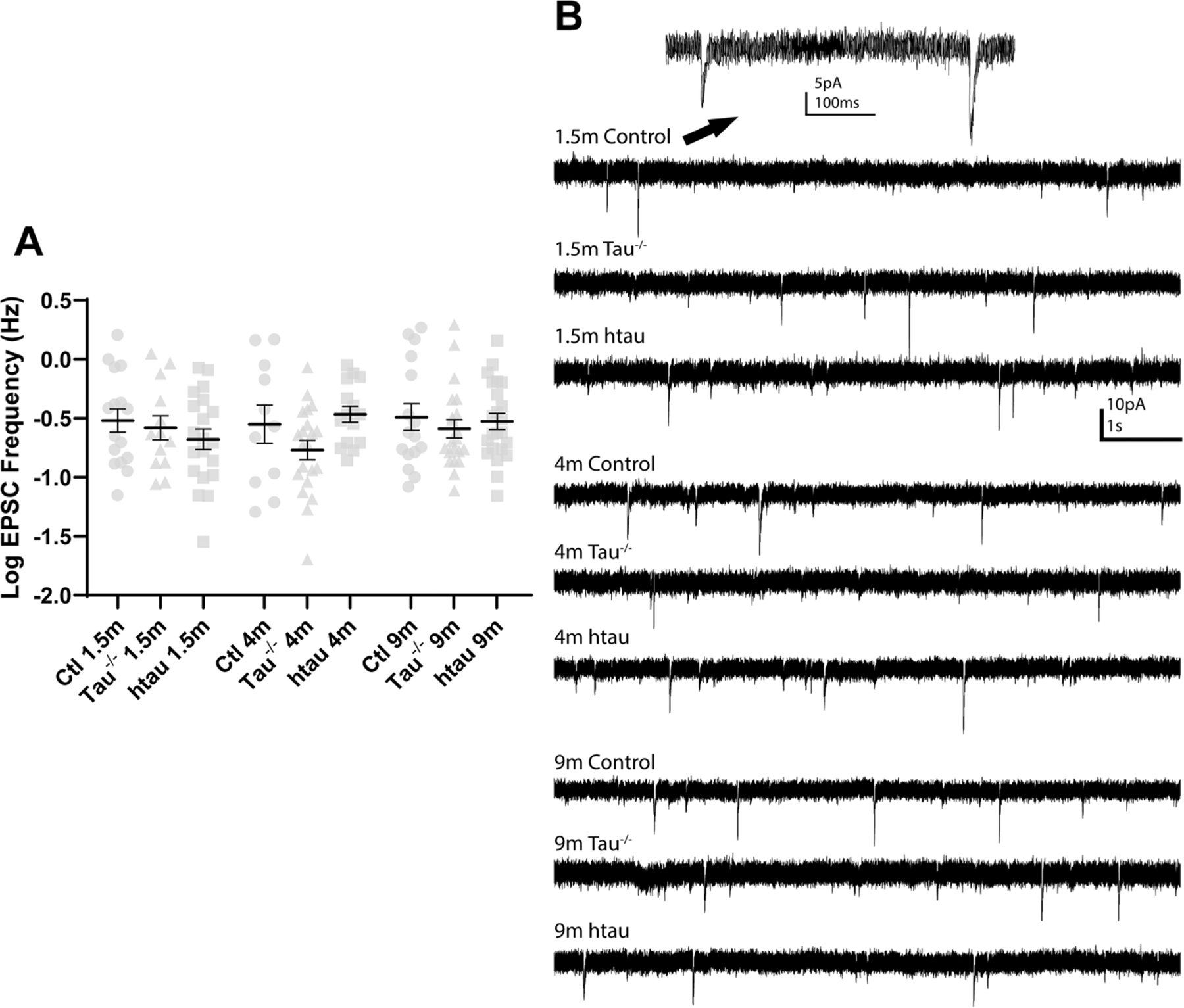
No differences sEPSC frequency were found between DGCs from tau−/− or htau mice and control mice at any age. sEPSC frequency did not change with age in DGCs from tau−/−, htau, or control mice (p>0.05). A Shapiro-Wilk test found the data were not normally distributed because they were were positive and right-skewed, so raw data were log transformed prior to further analysis. Statistical comparisons were made between all groups by two-way ANOVA with age and genotype as factors. (C) Representative traces of sEPSCs in DGCs from tau−/−, htau, or control mice at 1.5, 4, and 9 months. Error bars indicate SEM.
Highlights.
Tau loss is associated with decreased excitability in dentate gyrus in young mice.
Dentate granule cells from tau−/− and htau mice are electrophysiologically similar.
Loss of tau function affects excitability more than addition of pathologic tau.
Acknowledgments
Funding sources
This work was funded by National Institutes on Health [NINDS 1R01NS092552, NINDS 5R01NS091329, NCATS TL1TR001997, and NIGMS 1T32GM118292] and the Department of Defense [W81XWH-15–1-0551].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors have no competing interests to declare.
References
- Abisambra JF, Blair LJ, Hill SE, Jones JR, Kraft C, Rogers J, Koren J 3rd, Jinwal UK, Lawson L, Johnson AG, Wilcock D, O’Leary JC, Jansen-West K, Muschol M, Golde TE, Weeber EJ, Banko J, Dickey CA, 2010. Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci 30, 15374–15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abisambra JF, Jinwal UK, Blair LJ, O’Leary JC 3rd, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, Cockman M, Suntharalingham A, Li P, Jin Y, Atkins CA, Dickey CA, 2013. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci 33, 9498–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara-Gonzalez D, Chartampila E, Criscuolo C, Scharfman HE, 2021. Early changes in synaptic and intrinsic properties of dentate gyrus granule cells in a mouse model of Alzheimer’s disease neuropathology and atypical effects of the cholinergic antagonist atropine. Neurobiol Dis, 105274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso Adel C, Li B, Grundke-Iqbal I, Iqbal K, 2006. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A 103, 8864–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, De Silva R, Tucker KL, Barde Y-A, Duff K, Davies P, 2003. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. Journal of Neurochemistry 86, 582–590. [DOI] [PubMed] [Google Scholar]
- Boychuk JA, Butler CR, Halmos KC, Smith BN, 2016. Enduring changes in tonic GABAA receptor signaling in dentate granule cells after controlled cortical impact brain injury in mice. Exp Neurol 277, 178–189. [DOI] [PubMed] [Google Scholar]
- Butler CR, Boychuk JA, Smith BN, 2015. Effects of Rapamycin Treatment on Neurogenesis and Synaptic Reorganization in the Dentate Gyrus after Controlled Cortical Impact Injury in Mice. Front Syst Neurosci 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Perry G, 2019. Tau Biology, Tauopathy, Traumatic Brain Injury, and Diagnostic Challenges. J Alzheimers Dis 67, 447–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Yang H, Wang M, Wei H, Hu F, 2018. Role of Microtubule-Associated Protein in Autism Spectrum Disorder. Neurosci Bull 34, 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimins JL, Rocher AB, Luebke JI, 2012. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol 124, 777–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimins JL, Rocher AB, Peters A, Shultz P, Lewis J, Luebke JI, 2011. Homeostatic responses by surviving cortical pyramidal cells in neurodegenerative tauopathy. Acta Neuropathol 122, 551–564. [DOI] [PubMed] [Google Scholar]
- Decker JM, Kruger L, Sydow A, Dennissen FJ, Siskova Z, Mandelkow E, Mandelkow EM, 2016. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep 17, 552–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, Bennett CF, Cirrito JR, Holtzman DM, Miller TM, 2013. Antisense reduction of tau in adult mice protects against seizures. J Neurosci 33, 12887–12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach K, Hilbrich I, Schiffmann A, Gartner U, Kruger M, Leonhardt M, Waschipky H, Wick L, Arendt T, Holzer M, 2012. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem 287, 43223–43233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine SN, Ingram A, Cloyd RA, Meier SE, Miller E, Lyons D, Nation GK, Mechas E, Weiss B, Lanzillotta C, Di Domenico F, Schmitt F, Powell DK, Vandsburger M, Abisambra JF, 2017. Identification of changes in neuronal function as a consequence of aging and tauopathic neurodegeneration using a novel and sensitive magnetic resonance imaging approach. Neurobiol Aging 56, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB, 2007. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol 9, 139–148. [DOI] [PubMed] [Google Scholar]
- Garcia-Cabrero AM, Guerrero-Lopez R, Giraldez BG, Llorens-Martin M, Avila J, Serratosa JM, Sanchez MP, 2013. Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol Dis 58, 200–208. [DOI] [PubMed] [Google Scholar]
- Gardiner J, Marc J, 2010. Disruption of normal cytoskeletal dynamics may play a key role in the pathogenesis of epilepsy. Neuroscientist 16, 28–39. [DOI] [PubMed] [Google Scholar]
- Geiszler PC, Barron MR, Pardon MC, 2016. Impaired burrowing is the most prominent behavioral deficit of aging htau mice. Neuroscience 329, 98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane MM, Sanchez PE, Mucke L, 2014. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol 76, 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves FG, Freddi TAL, Taranath A, Lakshmanan R, Goetti R, Feltrin FS, Mankad K, Teixeira SR, Hanagandi PB, Arrigoni F, 2018. Tubulinopathies. Top Magn Reson Imaging 27, 395–408. [DOI] [PubMed] [Google Scholar]
- Graziane N, Dong Y, 2016. 11.Measuring Presynaptic Release Probability, in: Dong Y, Graziane N (Eds.), Electrophysiological Analysis of Synaptic Transmission Humana Press, New York, NY, pp. 133–143. [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N, 1994. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369, 488–491. [DOI] [PubMed] [Google Scholar]
- Hatch RJ, Wei Y, Xia D, Gotz J, 2017. Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol 133, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, Noebels JL, 2013. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci 33, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D, 2010. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger HC, Rudy CC, Batten SR, Gerhardt GA, Reed MN, 2015. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J Neurochem 132, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN, 2010. Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J Neurophysiol 103, 1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P, 1998. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J, 2010. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. [DOI] [PubMed] [Google Scholar]
- Jones NC, Nguyen T, Corcoran NM, Velakoulis D, Chen T, Grundy R, O’Brien TJ, Hovens CM, 2012. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol Dis 45, 897–901. [DOI] [PubMed] [Google Scholar]
- Koren SA, Hamm MJ, Meier SE, Weiss BE, Nation GK, Chishti EA, Arango JP, Chen J, Zhu H, Blalock EM, Abisambra JF, 2019. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol 137, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Kim KR, Ryu SY, Son S, Hong HS, Mook-Jung I, Lee SH, Ho WK, 2012. Impaired short-term plasticity in mossy fiber synapses caused by mitochondrial dysfunction of dentate granule cells is the earliest synaptic deficit in a mouse model of Alzheimer’s disease. J Neurosci 32, 5953–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hall AM, Kelinske M, Roberson ED, 2014. Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol Aging 35, 2617–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindwall G, Cole RD, 1984. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259, 5301–5305. [PubMed] [Google Scholar]
- Liu S, Shen Y, Shultz SR, Nguyen A, Hovens C, Adlard PA, Bush AI, Chan J, Kwan P, O’Brien TJ, Jones NC, 2017. Accelerated kindling epileptogenesis in Tg4510 tau transgenic mice, but not in tau knockout mice. Epilepsia 58, e136–e141. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zheng P, Wright DK, Dezsi G, Braine E, Nguyen T, Corcoran NM, Johnston LA, Hovens CM, Mayo JN, Hudson M, Shultz SR, Jones NC, O’Brien TJ, 2016. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 139, 1919–1938. [DOI] [PubMed] [Google Scholar]
- Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M, Kiosea NC, Nazari S, Chen PP, Nothias F, Chan P, Teng E, Frautschy SA, Cole GM, 2014. Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris Water Maze with aging. J Neurosci 34, 7124–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Djukic B, Taneja P, Yu GQ, Lo I, Davis A, Craft R, Guo W, Wang X, Kim D, Ponnusamy R, Gill TM, Masliah E, Mucke L, 2016. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep 17, 530–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchisella F, Coffey ET, Hollos P, 2016. Microtubule and microtubule associated protein anomalies in psychiatric disease. Cytoskeleton (Hoboken) 73, 596–611. [DOI] [PubMed] [Google Scholar]
- Martin-Belmonte A, Aguado C, Alfaro-Ruiz R, Moreno-Martinez AE, de la Ossa L, Martinez-Hernandez J, Buisson A, Shigemoto R, Fukazawa Y, Lujan R, 2020. Density of GABAB Receptors Is Reduced in Granule Cells of the Hippocampus in a Mouse Model of Alzheimer’s Disease. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Terro F, 2013a. Tau protein phosphatases in Alzheimer’s disease: the leading role of PP2A. Ageing Res Rev 12, 39–49. [DOI] [PubMed] [Google Scholar]
- Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C, Terro F, 2013b. Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res Rev 12, 289–309. [DOI] [PubMed] [Google Scholar]
- Meier S, Bell M, Lyons DN, Ingram A, Chen J, Gensel JC, Zhu H, Nelson PT, Abisambra JF, 2015. Identification of Novel Tau Interactions with Endoplasmic Reticulum Proteins in Alzheimer’s Disease Brain. J Alzheimers Dis 48, 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, Mechas E, Chen J, Wolozin B, LeVine H 3rd, Zhu H, Abisambra JF, 2016. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J Neurosci 36, 1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Stein L, Thomas R, Djukic B, Taneja P, Knox J, Vossel K, Mucke L, 2017. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol Neurodegener 12, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandis D, Scarmeas N, 2012. Seizures in Alzheimer disease: clinical and epidemiological data. Epilepsy Curr 12, 184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Wetzel A, Granno S, Heaton G, Harvey K, 2017. Back to the tubule: microtubule dynamics in Parkinson’s disease. Cell Mol Life Sci 74, 409–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M, Boman E, Osterman H, Willhite D, Laska M, 2011. Olfactory and visuospatial learning and memory performance in two strains of Alzheimer’s disease model mice--a longitudinal study. PLoS One 6, e19567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydoro M, Acker CM, Duff K, Castillo PE, Davies P, 2009. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci 29, 10741–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L, 2011. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci 31, 700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L, 2007. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, Luebke JI, 2010. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol 223, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha P, Sen N, 2019. Tauopathy: A common mechanism for neurodegeneration and brain aging. Mech Ageing Dev 178, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH, 2005. Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, Vargas-Caballero M, 2011. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J Neurosci 31, 1688–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D’Hooge R, Alzheimer C, Mandelkow EM, 2011. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci 31, 2511–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies E, Mandelkow EM, 2007. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci 27, 2896–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Davidowitz E, Lopez P, Emadi S, Moe J, Sierks M, 2013. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int J Cell Biol 2013, 260787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, Mucke L, 2013. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 70, 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, Karydas AM, Coppola G, Roberson ED, Miller BL, Garcia PA, Kirsch HE, Mucke L, Nagarajan SS, 2016. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann Neurol 80, 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winokur RS, Kubal T, Liu D, Davis SF, Smith BN, 2004. Recurrent excitation in the dentate gyrus of a murine model of temporal lobe epilepsy. Epilepsy Res 58, 93–105. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM, 2007. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]