

Abstract
Purpose:
Harnessing the immune-stimulatory effects of radiation by combining it with immunotherapy is a promising new treatment strategy. However, more study characterizing immunotherapy and radiation dose scheduling for the optimal therapeutic effect is essential for designing clinical trials.
Methods and Materials:
The new ablative radiation dosing scheme “personalized ultra-fractionated stereotactic adaptive radiotherapy” (PULSAR) was tested in combination with α-PD-L1 therapy in immune activated and resistant syngeneic immunocompetent mouse models of cancer. Specifically, tumor growth curves comparing immunotherapy and radiotherapy dosing sequencing were evaluated in immunologically “cold” and “hot” tumor models. The response relative to cytotoxic killer T cells was evaluated using an α-CD8 depleting antibody, and immunological memory was tested by tumor re-challenge of cured mice.
Results:
We report that both radiation and immunotherapy sequencing as well as radiotherapy fraction spacing affects the combination treatment response. Better tumor control was achieved by giving α-PD-L1 therapy during or after radiation, and spacing fractions 10 days apart (PULSAR) achieved better tumor control than traditional daily fractions. We showed that CD8+ depleting antibody abrogated tumor control in the PULSAR combination treatment and certain treatment schedules induced immunological memory.
Conclusions:
These results illustrate that radiation therapy dosing and scheduling impacts tumor control in combination with checkpoint blockade therapies. PULSAR styled radiation dosing is more complimentary in combination with single agent immunotherapy than traditional daily fractions in this preclinical model. Pre-clinical investigation could prove helpful in designing clinical trials investigating combination therapy.
Keywords: PULSAR, SAbR, SBRT, Checkpoint-blockade, Scheduling, Immunotherapy
Introduction
For decades, therapeutic radiotherapy for cancer has been delivered using numerous “fractions” of relatively low dose, given daily (except weekends) over several weeks based on a single dosimetry plan prepared at start of therapy. In prior eras of crude delivery and targeting technology, this conventionally fractionated radiotherapy (CFRT) exploited repair differences between targeted tumor and nearby normal tissue in reaction to the small injury caused by the low daily dose. While both tumor and normal tissue ultimately received high cumulative dose at completion of the protracted course, the improved repair of small daily fraction injury within normal tissue compared to tumor allowed a therapeutic gain derived from relative tissue repair biology1.
Delivery technology and imaging improvements in recent decades has facilitated a new radiotherapy allowing deposition of ablative or more destructive dose. The newer technologies permitted dramatic geometric avoidance of surrounding normal tissue thereby reducing the previous requirement to give a low daily dose and rely on a differential repair approach. These newer treatments, called stereotactic ablative radiotherapy (SAbR), stereotactic body radiation therapy (SBRT), stereotactic radiosurgery (SRS) and others, have been used successfully for tumor-only treatments akin to surgery2. While they do not exploit the differential repair between normal tissue and tumor as with CFRT, SAbR treatments still are given daily in practice and have continued to be referred to as “fractions.” Also consistent with CFRT, SAbR is delivered based on a single dosimetry plan prepared at start of therapy.
More recently, further technological innovation has allowed for fast replanning of a course of radiotherapy in response to changes in an individual patient’s situation, including tumor shrinkage (or enlargement) as well as other changing parameters that may occur during a protracted course of therapy. This has been called “adaptive” radiotherapy and is a form of personalized therapy3. SAbR treatments are generally completed in 5 or fewer fractions over 1–2 weeks; hence, there is little time for changes to occur that might justify adaptive replanning. An even newer radiotherapy paradigm being evaluated abandons the classic daily fractions altogether. It employs a limited number of fairly large dose “pulses” in the ablative range that are separated in time by weeks or months. Our group and others4,5 are beginning to use this paradigm clinically which has been coined personalized, ultrafractionated stereotactic adaptive radiotherapy, PULSAR. Separating the individual pulses by longer periods allows for potentially significant changes to be observed both in the tumor, its microenvironment, adjacent tissues as well as systemically. These changes can be interrogated and, if relevant, can trigger an adaptive replanning of the ongoing therapy for a more personalized and, hopefully, more effective overall therapy.
With increasing degrees of freedom, PULSAR potentially provides opportunities not afforded by CFRT or SAbR. But PULSAR has less preclinical basis for justification or to guide its implementation. With tremendous interest in harnessing the immune response against cancer, PULSAR will need to operate in association with immunotherapy6. Herein, we perform early exploration of PULSAR in conjunction with immunotherapy using preclinical vignettes that pertain to clinically pertinent applications. SAbR given daily or every other day has already been combined with immunotherapy in the clinic with mixed but encouraging results. We hypothesized that PULSAR might afford even better opportunities to work synergistically with immunotherapy and set out to test the hypothesis in a murine model.
Methods and Materials
Mice
Female C57BL/6J mice were purchased from Charles River or Jackson Laboratories at 6–8 weeks old. All mice were maintained under specific pathogen-free conditions and all animal procedures were performed in accordance with the animal experimental guidelines set by the Institutional Animal Care and Use Committee of Anonymized for Review.
Cell lines and reagents
Mouse colon carcinoma MC38 cell line was obtained from the American Type Culture Collection. Lewis Lung Carcinoma (LLC) was a kind gift from the Story lab, and was originally derived from lung cancer of the C57BL/6 line. All cells were cultured in 5% CO2 and maintained in vitro in Dulbecco’s Modified Eagle’s Medium supplemented with 10% heat-inactivated fetal bovine serum (all reagents from Sigma-Aldrich), 100 U/ml penicillin, and 100 μg/ml streptomycin. All cell lines are routinely tested for mycoplasma contamination and were confirmed negative prior to this study. For CD8+T cell depletion studies, we used α-CD8 clone 53–6.7, for LLC experiments, and α-CD8 clone 53–5.8 for MC38. We used α-PD-L1 (10F.9G2), and isotype control for α-PD-L1 (clone LTF-2). All antibodies used in this study were purchased from BioXCell.
Local Radiation
Tumor bearing mice were anesthetized using isoflurane and irradiated with either 8 Gy (for MC38) or 10–15Gy (for LLC) according to the schedules listed in the text and figures. MC38 and LLC were given different doses according to their individual in vitro modelling parameters as predicted by the Universal Survival Model7 (Supplementary Figure 6). Local irradiations were conducted on a dedicated X-ray irradiator (X-RAD 32-, Precision X-ray, Inc.). Various sizes of collimators were developed to form the filed-of-view depending on the sizes of the tumors. A tumor bearing mouse was anesthetized using isoflurane and mounted on an acrylic bed equipped with a nose cone. The mouse was positioned such that the source-to-tumor surface distance (SSD) was 20 cm and the tumor was in the center of the X-ray beam. The energy of the X-ray was set to 250 kVp and current was set to 15 mA for the irradiation. The dose rate under this condition was 19.468 Gy/min, which was calibrated using a PTW 31010 ionization chamber and a PTW UnidosE electrometer (PTW North America Corporation, New York, NY) in accord with the AAPM TG-61 protocol.
Tumor growth and treatments
Tumor cells were injected subcutaneously on the right leg of mice. Mice were randomized to treatment groups when tumors reached 150–200mm3 for LLC, and 100–150mm3 for MC38. Tumors were treated with α-PD-L1 or not, then tumor volumes were measured by the length (a), width (b) and height (h) and calculated as tumor volume = abh/2. For the survival curve, if each of length, width or height of tumor is larger than 2cm, the tumor volume is larger than 1500mm3, or the mice had a significant ulceration in the tumor, the mice reached survival endpoint and euthanized for moribundity. For CD8 T cell depletion experiments, 200μg α-CD8 was given intraperitoneally (i.p.) on the same day of first antibody treatment, and every 4 days for a total of 3 weeks. For the experiments in MC38, 25 μg anti–PDL1 or 25μg isotype control was administered i.p. to mice every 2 days for a total of four times starting 1 day before radiation. For the experiments in LLC, 200μg anti–PDL1 or 200μg isotype control was administered i.p. to mice every 2 days for a total of four times starting 2 days before radiation.
Flow Cytometry
Single cell suspensions were obtained from spleen by smashing through 70μM cell strainer. Red blood cells were removed from blood and spleen by 2 minute incubation with ACK buffer (NH4Cl 8,024mg/l, KHCO3 1,001mg/l, EDTANa22H2O 3.722mg/l). Washed cells were incubated with anti-CD16/32 (α-FcγIII/II receptor, clone 2.4G2) for 15 minutes to block non-specific binding and then stained with antibodies α-CD3-PE (clone 145–2C11) α-CD8-AF700 (Clone 53–6.7) α-CD4-BV605 (clone RM4–5). All fluorescently labeled antibodies were purchased from BioLegend. Fixable Viability Dye eFluor™ 506 (eBioscience) was used to exclude dead cells. Data were collected on CytoFLEX (Beckman Coulter, Inc) and analyzed with CytExpert (Beckman Coulter, Inc) or FlowJo (Tree Star Inc., Ashland, OR) software.
Nomenclature
For this paper, the term “fraction” refers to either a single dose or more than 1 dose of radiotherapy given where individual doses are separated by less than 1 week (e.g., daily, every other day, every 4 days, etc). In contrast, a “pulse” refers to more than 1 dose separated by at least a week (7 days or greater). The actual interval between doses (D) will be depicted by DX/Y where X refers to the timing of the first dose and Y the second dose (e.g., D0/1 refers to a one day separation in the two “fractions” while D0/10 refers to a 10 day separation of the two “pulses”). A “cycle” is defined in our context as one round of α-PD-L1 treatment including one dose before radiation, one dose on the day of radiation, and two doses after. No attempt was made in the context of radiation dosing to make the radiation schedules “biologically equivalent.” Instead, the total dose was kept constant for each animal model and comparable set of experiments (e.g., total dose 16 Gy whether given as D0/0, D0/1, D0/10, etc., for MC38).
Statistics
The number of animals and replicates are indicated in each figure legend. Prior to treatment, animals were allocated into groups using stratified randomization by tumor volume. Tumor growth curves are represented as mean ± SEM. For tumor growth curves, repeated measures two-way ANOVA was used to calculate significance between groups and corrected for multiple comparisons using the Tukey method. For survival curves, significance was determined using the log-rank (Mantel-Cox) test. All analysis was performed using GraphPad Prism statistical software (GraphPad Software Inc.). Investigators were blinded to the group allocation during the experiment and drug treatment whenever possible. ns (not significant), *P < 0.05, **P < 0.01, ***P < 0.001.
Results
Sequence of immunotherapy and radiation therapy impacts tumor growth.
To begin to explore the impact of dose and schedule of radiation and immunotherapy on tumor growth, we used a syngeneic immune competent mouse model that is sensitive to immunotherapy. We specifically chose MC38, a colon cancer model in the C57BL/6 background known to have infiltration of CD8+T cells in the untreated tumor microenvironment8, and responds to α-PD-L1 monotherapy9. To understand how sequencing of radiotherapy in combination with immunotherapy affects tumor growth, we implanted MC38 cells subcutaneously (s.c.) in the mouse hind-limb. When tumors reached average of 150mm3 mice were treated with three schedules of a single 16 Gy dose of radiation with different timing of α-PD-L1 antibody as shown in Figure 1A. The three radiation monotherapy (IR + Isotype) schemes had very similar impacts on tumor growth (Supplementary Figure 1A). When all doses of α-PD-L1 were given before radiation, there was no additive effect of α-PD-L1 therapy and radiation (Figure 1B). However, when α-PD-L1 was given concurrent with or after radiation, there was a benefit to the addition of immunotherapy (Figures 1C–D). Furthermore, concurrent immunotherapy resulted in improved overall survival (Supplementary Figure 1B). These experiments defined the optimum combined radio-immunotherapy “cycle”, which we defined as one dose of α-PD-L1 before radiation, one dose on the day of radiation, and two doses after (inset Figure 2A).
Figure 1. Sequence of immunotherapy and radiation therapy within radio-immunotherapy pulses impact tumor growth.
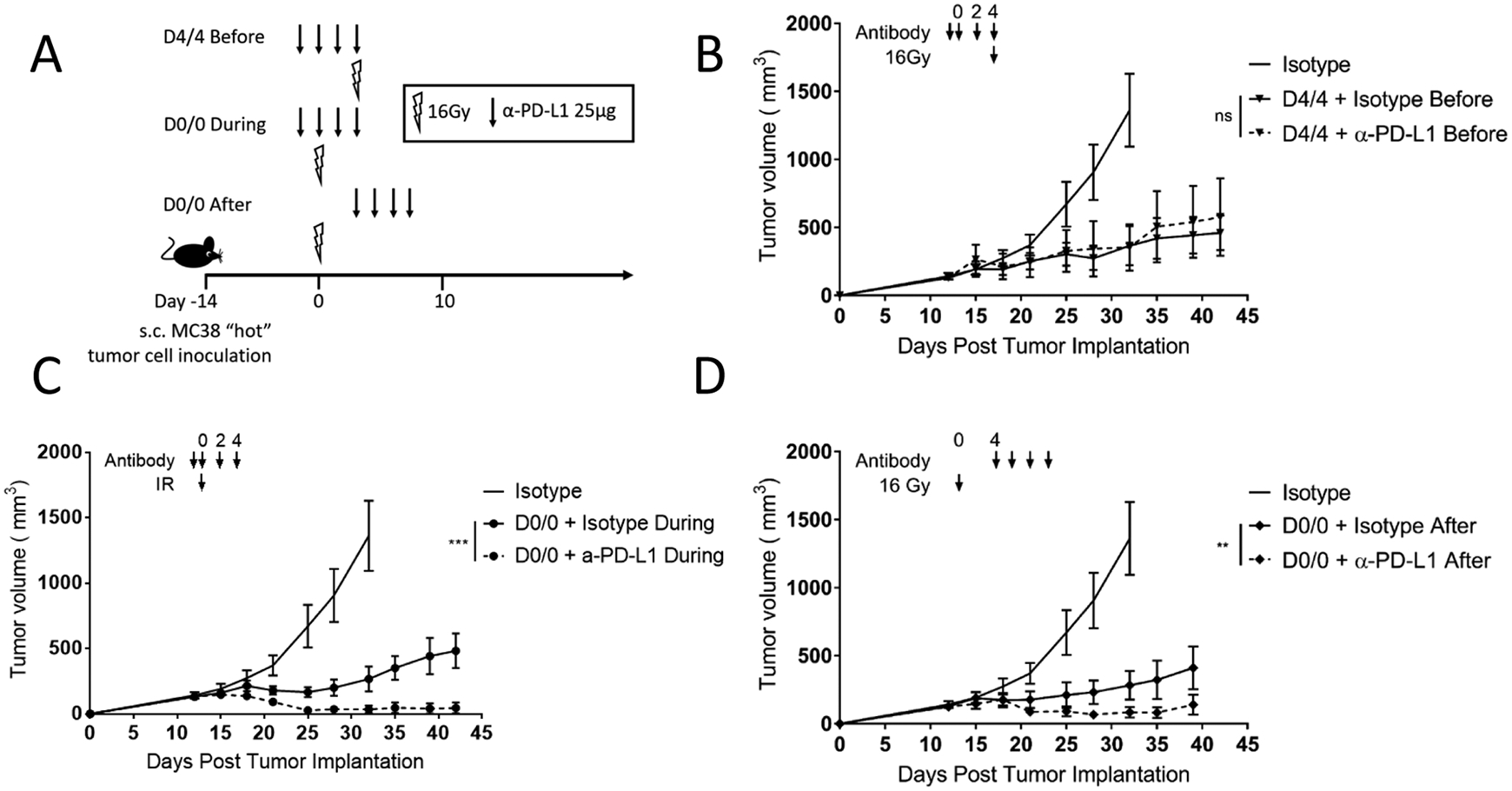
(A) Diagram representing the treatment timeline for panels B–D. 1×106 MC38 cells were injected subcutaneously in the right leg on day −14, and three different combination α-PD-L1 + 16Gy radiation schemes were given as shown. Arrow represents α-PD-L1 or isotype dose, lightning bolt indicates radiation dose.
(B–D) Mice were treated as described in (A) and as shown with arrows above the tumor curves. The same isotype control mice are shown in each panel for relative comparison, as all tumor curves in these panels were collected in the same experiment. Tumor volume was measured, and the results are presented as mean ±SEM, IR irradiation, ns not significant, ** p < 0.01, *** p < 0.001, n=7/group.
Figure 2. Timing of radio-immunotherapy pulses affects tumor growth in a “hot” tumor microenvironment.
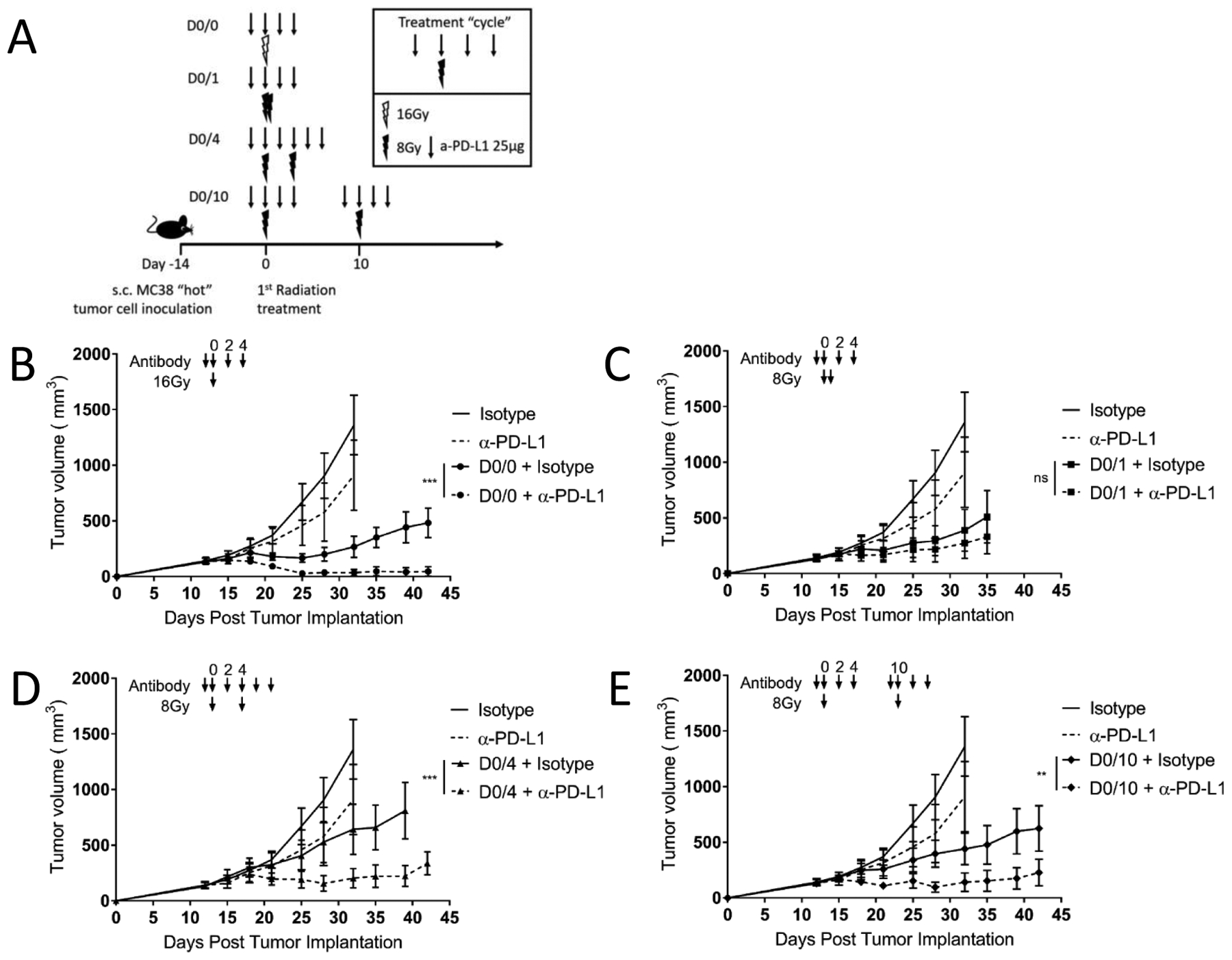
(A) Diagram representing the treatment timeline for panels B–E. 1×106 MC38 cells were injected subcutaneously in the right leg on day −14, and four different radiation schedules of two fractions of 8Gy were given either 0, 1, 4 or 10 days apart starting on day 0. α-PD-L1 (25μg) or isotype control (25μg) was given starting the day before each radiation dose and continuing for 2 doses after radiation. Arrow represents α-PD-L1 or isotype dose, lightning bolt indicates radiation dose.
(B–E) Mice were treated as described in (A) and as shown with arrows above the tumor curves. The same isotype control mice are shown in each panel for relative comparison, as all tumor curves in these panels were collected in the same experiment. Tumor volume was measured, and the results are presented as mean ±SEM, IR irradiation, ns not significant, ** p < 0.01, *** p < 0.001, n=7/group, representative of two independent experiments.
Timing of radio-immunotherapy affects tumor growth in a “hot” tumor microenvironment.
In treatments that are high dose per fraction such as SABR, we hypothesized there would not be a therapeutic penalty by delivering as a pulse, but rather, there could be an advantage from the immune response perspective. Having established the optimum “cycle” schedule for combined radio-immunotherapy using a single fraction of radiation (inset Figure 2A), we set out to define differential responses to fractions or pulses (see definitions in Methods- Nomenclature). We chose four different fraction/pulse schedules, with no spacing (D0/0) single fraction, fractions separated by one day (D0/1), fractions separated by four days (D0/4) and pulses separated by ten days (D0/10). These spacing schemes may interact variably at various time points within the immune response to the first dose of radiation, e.g., with the 10 day pulse occurring just as or after newly primed T cells would be entering the tumor microenvironment from the draining lymph node10. Control animals received isotype antibody or α-PD-L1 monotherapy as shown in the schedule in Supplementary Figure 2A. The four radiation monotherapy schedules (D0/0, D0/1, D0/4 and D0/10 in Figures 2B–E) controlled tumor growth similarly, and tumor growth curves were not significantly different (Supplementary Figure 2B). Additive benefit of α-PD-L1 therapy with radiation depended on radio-immunotherapy spacing. Giving a single fraction (D0/0), two more separated fractions (D0/4) or longer pulse separation of 10 days (D0/10) resulted in significantly improved tumor control (Figure 2B, D, E). However, giving two fractions one day apart (D0/1) prevented the additive effect of α-PD-L1 as it did not improve tumor control over radiation monotherapy (Figure 2C). The most effective treatment regimen giving a single fraction of radiation combined with α-PD-L1 had significantly better tumor control than the combination group with radiation fractions given 1 day apart (Supplementary Figure 2C). However the difference between the two α-PD-L1 and IR combination groups when radiation was split by 1 day vs. 10 days did not reach statistical significance (Supplementary Figure 2C). Finally, overall survival was significantly improved by the addition of α-PD-L1 therapy in the D0/0 and D0/4 radiation schedules (Supplementary Figure 2D).
Response to pulsed radio-immunotherapy depends on CD8+ T cells and induces immunological memory.
It is not known whether a second dose of radiation may stimulate or impair an ongoing immune response. Therefore, we wanted to know whether the benefit of spacing the timing or radiotherapy in identified radio-immunotherapy combinations was immune dependent. We treated MC38 tumor bearing mice with two pulses of radio-immunotherapy 10 days apart (D0/10), a previously identified efficacious combination, as described in Figure 3A. Treated animals were split into two groups, one receiving 200μg α-CD8 depleting antibody on the first day of treatment and continuing every 4 days for 3 weeks. T cell depletion was verified by flow cytometry of peripheral blood lymphocytes (Supplementary Figure 3). In the MC38 model, CD8+T cell depletion reduced the efficacy and survival of pulsed radio-immunotherapy demonstrating that the tumor growth delay from the combination therapy requires CD8+T cells (Figure 3B–C). To understand if the adaptive immune response generated by radio-immunotherapy “pulses” generated immunologic memory, we re-challenged our tumor-free mice with 5x the dose of MC38 cells 60 days after the first tumor inoculation. Tumors were rejected in the D0/0 and D0/10 radio-immunotherapy schedules, while tumors grew rapidly in naïve mice (Figure 3D).
Figure 3. Response to pulsed radio-immunotherapy depends on CD8+ T cells and induces immunological memory.
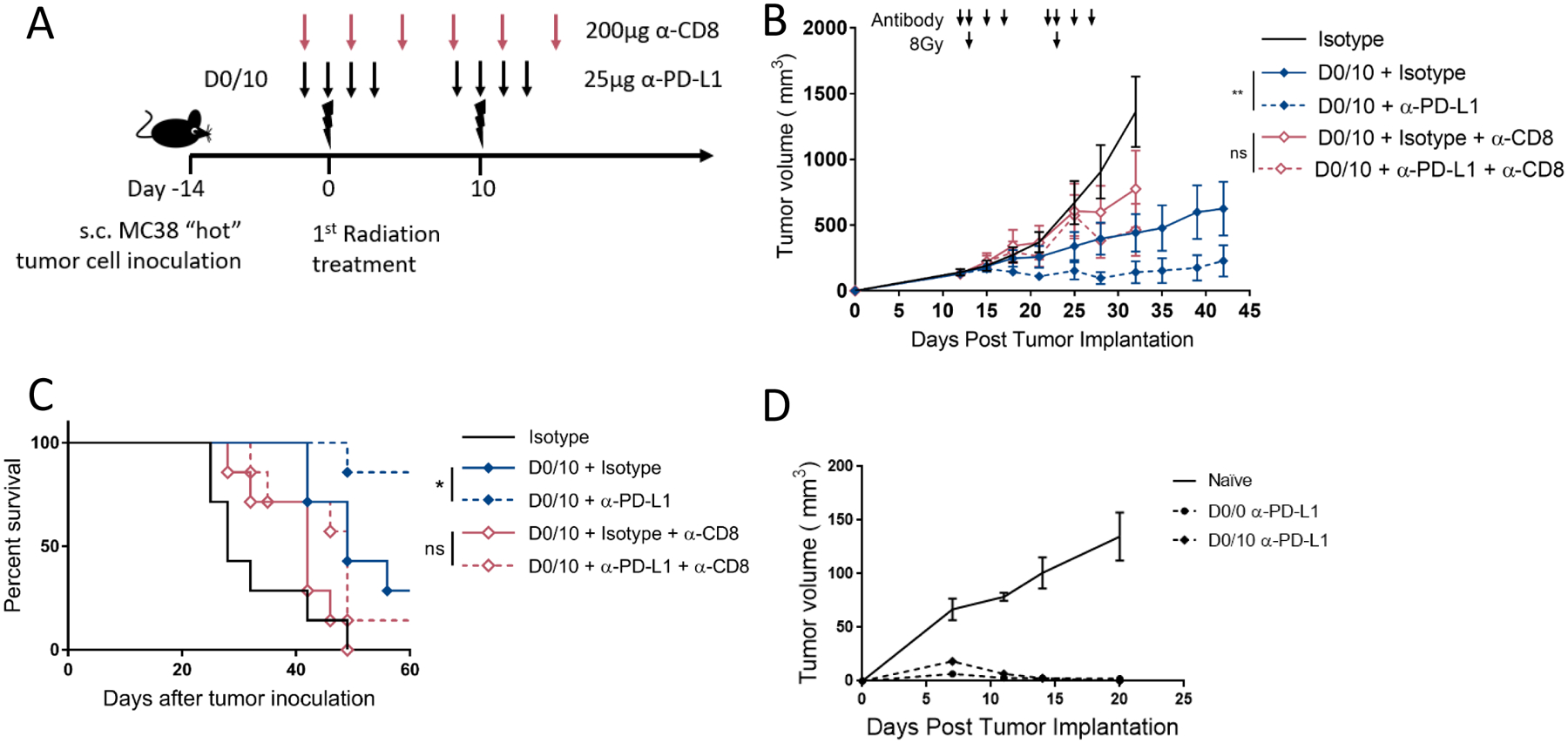
(A) Diagram representing the treatment timeline for panels B and C. 1×106 MC38 cells were injected subcutaneously in the right leg on day −14, and treated with two fractions of 8Gy radiation 10 days apart starting on day 0 after tumor cell implantation. α-PD-L1 (25μg) or isotype control (25μg every 2 days) was given starting two days before each radiation dose and continuing for 2 doses after radiation. α-CD8 depleting antibody (200μg) was given on the first day of treatment and every 4 days for 3 weeks. Lower arrow represents α-PD-L1 or isotype dose, upper arrow represents α-CD8 depleting antibody dose, lightning bolt indicates 8Gy radiation dose.
(B) Mice were treated as described in (A) and as shown with arrows above the tumor curves. Tumor volume was measured, and the results are presented as mean ±SEM, IR irradiation, ns not significant, ** p < 0.01, n=7/group.
(C) Survival curves for mice treated as described in panel B. * p < 0.05, n=7/group.
(D) Naïve or cured mice from Figures 2B and 2E were re-challenged 60 days after first tumor implantation with 5 million MC38 cells s.c. on the opposite flank. Tumor volume was measured, and the results are presented as mean ±SEM.
Radio-immunotherapy combinations show evidence of antitumor immune responses from pulsed (or PULSAR) dosing in “cold” immune-resistant tumors.
After exploring the effect of radio-immunotherapy pulses in an immunogenic mouse tumor model, we wanted to know if the same pulse schedules would similarly impact tumor growth in an immune resistant model. Lewis Lung Carcinoma (LLC) is a syngeneic tumor model in the C57BL/6 background known to be “cold” or non-immunogenic and does not respond to α-PD-L1 monotherapy9. We implanted LLC cells s.c. in the mouse hind-limb and when tumors reached an average of 150mm3, the mice received one of several radio-immunotherapy schedules as shown in Figure 4A. There was no response to high dose α-PD-L1 monotherapy in LLC tumor bearing mice (Figure 4B). For this tumor, when radio-immunotherapy fractions were given on both D0/1 and D0/4, there was no additive effect of combining α-PD-L1 with radiation (Figure 4C, D). However, when pulses were given 10 days apart, there was a greater antitumor effect using combined radiation and α-PD-L1 therapy compared to radiation alone (Figure 4E).
Figure 4. Radio-immunotherapy pulses show synergistic antitumor effects depending on radiation dose and schedule in “cold” immune-resistant tumors.
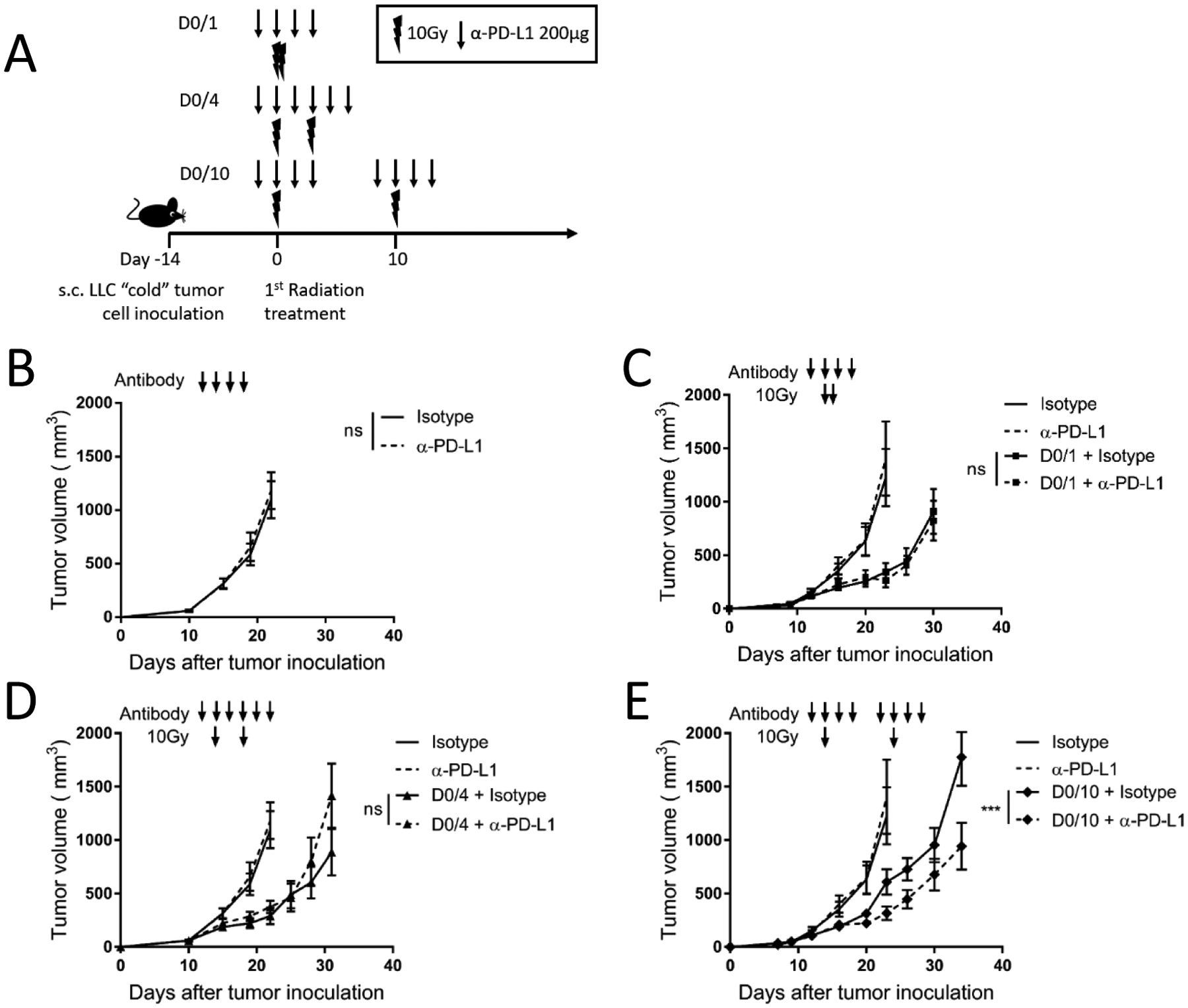
(A) Diagram representing the treatment timeline for panels B–E. 1×106 LLC cells were injected subcutaneously (s.c.) in the right leg on day −14, and four different radiation schedules of two fractions of 10Gy were given either 0, 1, 4 or 10 days apart starting on day 0. α-PD-L1 (200μg) or isotype control (200μg) was given starting the day before each radiation dose and continuing for 2 doses after radiation. Arrow represents α-PD-L1 or isotype dose, lightning bolt indicates radiation dose. N = 8 or 9/group, one of 2 independent replicates.
(B) C57BL/6 mice (n=8 or 9/group) were inoculated s.c. with 1×106 LLC cells in the right leg and treated with either isotype or α-PD-L1 antibody (200μg) as described by the arrows above the growth curve. Tumor volume was measured, and the results are presented as mean ±SEM, ns not significant.
(C–E) Mice were treated as described in (A) and as shown with arrows above the tumor curves. The same isotype control mice are shown in each panel for relative comparison, as all tumor curves in these panels were collected in the same experiment. Tumor volume was measured, and the results are presented as mean ±SEM, IR irradiation, ns not significant, ** p < 0.01, *** p < 0.001. (B–E) data are representative of two independent experiments.
Only the second dose of α-PD-L1 is required for CD8+ T cell dependent therapeutic efficacy in the non-immunogenic LLC model.
Given that only the pulsed radio-immunotherapy treatment showed therapeutic efficacy in LLC, we evaluated which dose of α-PD-L1 antibody was most effective. Mice were treated with α-PD-L1 immunotherapy during either the first “pulse” or the second. Figure 5B shows a significant benefit to giving α-PD-L1 therapy with the second pulse, but not when only given with the first pulse. This shows the first dose of α-PD-L1 is not required for therapeutic efficacy, but the second dose is required for the therapeutic effect of the D0/10 radio-immunotherapy pulse schedule. Finally, to determine if the D0/10 radio-immunotherapy pulse spacing in the non-immunogenic LLC is also T cell dependent, we performed CD8+T cell depletion experiments. The experiment was performed as described with the MC38 model, T cell depletion was confirmed in the spleen, and the combination effect was lost in treated groups receiving α-CD8 depleting antibody (Figure 5D and supplementary 5).
Figure 5. Only the second dose of α-PD-L1 is required for CD8+ T cell dependent therapeutic efficacy in the non-immunogenic LLC model.
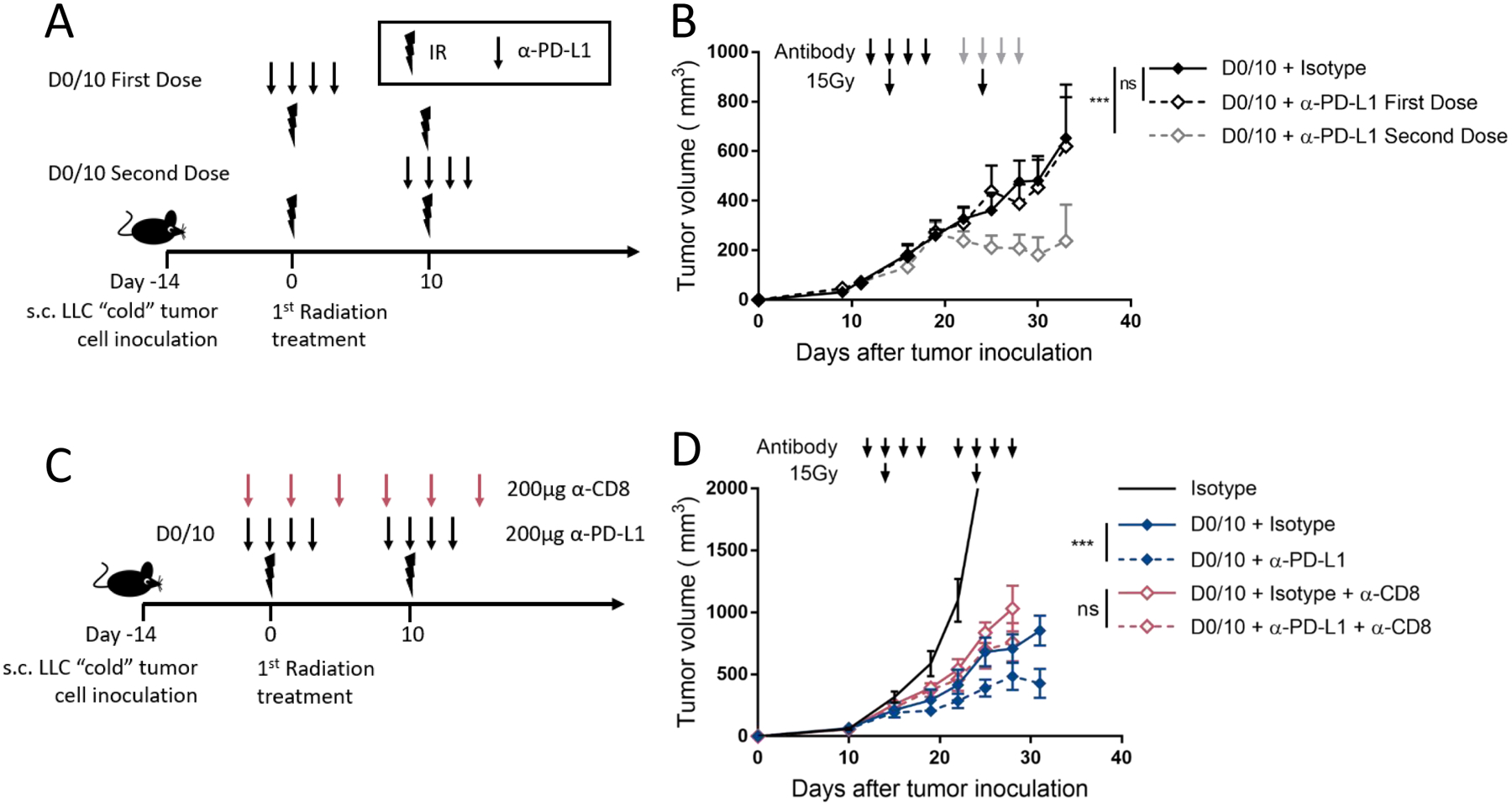
(A)Diagram representing the treatment timeline for panel B. 1×106 LLC cells were injected subcutaneously (s.c.) in the right leg on day −14, and 15Gy radiation was given 10 days apart starting on day 0. α-PD-L1 (200μg) or isotype control (200μg) was given with either the first or second radiation fraction. Arrow represents α-PD-L1 or isotype dose, lightning bolt indicates radiation dose.
(B) Mice were treated as described in (A) and as shown with arrows above the tumor curves. The solid black line represents control mice only receiving two doses of 15Gy and isotype control antibody. The dashed black group represents mice treated with two doses of 15Gy with α-PD-L1 given during the first dose of radiation (black arrows). The dashed grey group represents mice treated with two doses of 15Gy with α-PD-L1 given during the second dose of radiation (grey arrows). Tumor volume was measured, and the results are presented as mean ±SEM, IR irradiation, ns not significant, ** p < 0.01, n=8 or 9/group.
(C) Diagram representing the treatment timeline for panel D. 1×106 LLC cells were injected s.c. in the right leg on day −14, and treated with two fractions of 15Gy radiation 10 days apart starting on day 0 after tumor cell implantation. α-PD-L1 (200μg) or isotype control (200μg) was given starting two days before each radiation dose and continuing for 2 doses after radiation. α-CD8 depleting antibody (200μg) was given on the first day of treatment and every 4 days for 3 weeks. Lower arrow represents α-PD-L1 or isotype dose, upper arrow represents α-CD8 depleting antibody dose, lightning bolt indicates 15Gy radiation dose.
(D) Mice were treated as described in (C) and as shown with arrows above the tumor curves. Tumor volume was measured, and the results are presented as mean ±SEM, IR irradiation, ns not significant, ** p < 0.01, n=8 or 9/group.
Discussion
It is well described in preclinical models that radiotherapy has immune mediated antitumor effects that can synergize with checkpoint blockade11. It is no surprise that in the last two years the number of clinical trials combining radiation therapy with immunotherapies targeting checkpoint inhibitors and cytokines has increased 5 and 16 fold respectively12. However, there have been few systematic preclinical investigations to guide clinical treatment schedules combining radiation and immunotherapy. We sought to better understand the interactions between combination personalized, ultrafractionated stereotactic adaptive radiotherapy (PULSAR) and single agent immune checkpoint blockade. PULSAR is a new treatment paradigm in which radiation “pulses” in the ablative range are separated in time by weeks or months. We first describe the optimum sequencing of single fraction radiotherapy and immunotherapy in ablative ranges, and show that checkpoint blockade given during or after radiation is better than before. We further describe the impact of timing and spacing when combining immunotherapy with SAbR radiation dosing in both a “hot” immunogenic and “cold” immunotherapy-resistant syngeneic tumor models. For both “hot” and “cold” preclinical tumor models, single agent immune checkpoint blockade did not improve response over SAbR daily dose fractions alone. But enhancement was observed when combining the same therapies where the radiation was given in the PULSAR-style. And finally, we demonstrate that our optimized sequencing is CD8+ T cell dependent and able to induce immunological memory.
In this experience, we specifically test PULSAR-styled SAbR dosing in which pulses are separated by 7 or more days. Most preclinical models exploring combination radiation and α-PD-L1 therapy use SAbR doses of 10–24 Gy in 1–3 daily or every other day fractions, and begin PD1/PD-L1 checkpoint blockade therapy within a day of radiation13. One of the more clinically influential dosing regimens for combination checkpoint blockade and SBRT was described by Vanpouille-Box et al. in 2017 using pre-clinical models and included 3 daily SAbR fractions of 8 Gy followed by α-CTLA4 treatment beginning on the day of the last fraction14–16. We show using α-PD-L1 and SAbR that spacing radiation only 24 hours apart is not optimal for combination therapy. While Vanpouille-Box et al. used a different single agent checkpoint blockade, we wonder if even better results could have been achieved using PULSAR-styled SAbR in combination with α-PD-L1. Further, while some reports show no additive benefit of α-PD-L1 therapy when given 6 days after a single dose of radiation17, others support our results and show a clear benefit of α-PD-L1 given even 21 days after radiation18. Yet all are in agreement that α-PD-L1 has additive benefit when given concomitantly with radiation. We believe these differences are likely model specific. Finally, pre-clinical models dose immunotherapy differently than in the clinic. In humans, dosing schemes take into account antibody half-life in the serum, and aim to achieve relatively steady concentrations over months to years. Because this cannot be modeled pre-clinically in mice whose tumors are lethal within months, dosing in the literature has adapted to 1–4 doses given within a week, which is well within the biological half-life. In the cycles we define here, the D0/0 and D0/1 mice receive identical doses of α-PD-L1, however given the spacing of D0/4 fractions and D0/10 pulses, these animals receive additional doses. While this antibody treatment scheme slightly increases the total dose with increasing fraction/pulse spacing, we still believe the interaction between IR schedule and immunotherapy is relevant. Further investigation is certainly needed to optimize immunotherapy dosing for combination with PULSAR in the clinic as it will be very different from dosing in pre-clinical models.
In line with our findings showing that daily radiation scheduling is not enhanced in combination with immune checkpoint blockade, Filatenkov et al. showed that tumor growth after ablative radiotherapy is accelerated by subsequent 3Gy doses given daily, and survival is decreased19. They use similar pre-clinical models and further show mechanistically that CD8+ T cell infiltration into tumors is abrogated after repeated 3Gy daily doses, an important predictor of immunotherapy response in humans20. Mechanistically, Arina et al. showed that tumor resident T cells are resistant to radiation compared to those residing in lymph nodes or spleen21. However, independent from these resident T-cells, radiation potentially recruits circulating T cells from the blood into the tumor microenvironment. These newly arrived T cells may be more sensitive to subsequent doses of radiation. Further, it is not known how subsequent doses of radiation may affect other leukocytes, such as neutrophils and macrophages, which have been shown in pre-clinical models to be important for α-PD-L1 therapy response9,22,23. We therefore hypothesize that increased time between fractions (i.e., pulses) is more likely to preserve the proliferation and function of tumor fighting immune cells in the tumor microenvironment than the typical daily or every other day dosing14.
The two opposing tumor models used in this study lend insight to the importance of radiation timing and potential heterogeneity between tumors. We show the most optimized treatment regimen for the “hot” T cell inflamed MC38 colon cancer model is an up-front dose of radiation combined with α-PD-L1 therapy. However the “cold” LLC model responds best to irradiation doses spaced by 10 days, and only the second dose of radiation is required for therapeutic effect. Previous work has shown that in similar “hot” murine tumor models, pre-existing tumor immunity is required for response to combined radiation and checkpoint blockade therapy24. It may be that in such cases of strong pre-existing immunity, a single dose of radiation is optimal for synergy with immunotherapy because initial priming has already occurred. However, in more immune-resistant tumors similar to the LLC murine model and most human tumors, an initial priming dose with radiation is required which will later be amplified by α-PD-L1 therapy.
Another important advantage of PULSAR styled radiation is the adaptability and personalization available to optimize treatments for each patient. We saw using our MC38 tumor model that giving radiation in a single pulse up front in combination with immunotherapy was significantly better than daily fractions, whereas 10 day spacing did not reach statistical significance when compared to daily fractions. This may be because the tumors were actively regressing when the second dose of radiation was given, and it hindered the ongoing immune response. More investigation is needed to determine the importance of this finding in mouse models and how it translates to the clinical setting. Regardless, PULSAR has a clear advantage – if re-imaging shows progressive tumor shrinkage, clinicians can adapt and postpone further ablative doses until complete tumor control or relapse.
Clinical trials assessing radiotherapy sequencing when combined with immunotherapy have not been conducted. However, Luke et al. in 2018 reported outcomes of multi-site SBRT in combination with pembrolizumab to treat advanced solid tumors25. A favorable overall objective response rate of 13.2% was seen when pembrolizumab was initiated after completion of SBRT. The PEMBRO-RT phase 2 clinical trial in NSCLC demonstrated a doubling in overall response when patients were treated with 3×8Gy immediately followed by pembrolizumab26. When the results of the PEMBRO-RT trial were pooled with phase I/II trials from MD Anderson (MDACC) in which radiation was given concurrently with pembrolizumab, the clinical benefit of combination treatment became significant. Furthermore, the PACIFIC trial showed a benefit for patients with chemoradiotherapy refractory non-small cell lung cancer subsequently treated with durvalumab27. These clinical trials agree with the additive benefit we see when α-PD-L1 therapy is given concurrently or after radiotherapy in the pre-clinical setting. While neither of these trials used pulsed radiotherapy, both show a benefit to additional PD-1/PD-L1 pathway blockade after radiotherapy.
Mouse models have several limitations for exploring long time dependent responses because most syngeneic mouse tumors grow significantly faster than human tumors, and are generally more radio-resistant. Therefore, by the time a new immune response is generated by radiation, the tumor itself has grown and is more difficult to cure. Despite these difficulties, we show impressive tumor growth control by spacing radiotherapy pulses 10 days apart, which is superior to the conventional daily spacing when combined with single agent checkpoint blockade. Growth delay as impacted by direct radiation killing with the various radiation schemes used in our study was not strictly controlled between experiments as we did not use biologically equivalent doses (BED) in the experiments depicted in individual figures. The more common BED modeling formalisms do not account for proliferation between doses which would be important for doses separated by more than a few days. Facing this reality, we chose to use equivalent total doses. The prevailing understanding of radiotherapy interaction with immunotherapy views the radiation as a potential act of in situ, anti-tumor vaccination by stimulating the c-GAS-STING cytosolic DNA sensing pathway10. In turn, the checkpoint inhibition serves to undo mechanisms by which this vaccination effect might be suppressed to avoid autoimmunity. What would be ideal in the context of our work would be to equate the relative success of radiation in effectively vaccinating against tumor, a vaccination equivalent dose (VED), more so than a BED. Either way, this was a shortcoming of our methods. In the end, though, our results suggest that PULSAR may be a more optimal method of radiation dosing of SAbR for synergy with immunotherapy.
Conclusions
Our work confirms what has been long hypothesized in the field – that radiation dosing and scheduling impact its synergy with immunotherapy. We demonstrate that α-PD-L1 therapy given before a single dose of ablative radiation is inferior to giving immunotherapy after radiation. Further, we show that spacing radiation therapy by 10 days (PULSAR) has synergistic effects with α-PD-L1 therapy and giving two fractions separated by only 24 hours does not. Most pre-clinical models and many new clinical trials are designed with ablative fractions separated by 24–48 hours. Thus, our data could point toward a shortcoming in design when adding immunotherapy to the common daily radiation treatment paradigm. We hope our experience leads to more investigation, both observational and mechanistically, of PULSAR dosing schedules for optimum immune preservation and stimulation.
Supplementary Material
Acknowledgements
This work was funded by a grant from the Once Upon a Time Foundation to R.T., R01CA233594 to B.P.C., and RP180725 from CPRIT to Y.X.F.
Footnotes
Conflict of Interest
Dr. Robert Timmerman holds a pending patent for Personalized ultra-fractionated stereotactic adaptive radiotherapy in conjunction with certain immunotherapy. Dr. Saha has a patent for personalized ultrafractionated stereotactic adaptive radiotherapy in combination with some immunotherapy drugs pending. Dr. Story has a patent personalized ultra-fractionated stereotactic adaptive radiotherapy pending.
Data Availability
Research data are stored at the institution and will be shared upon request to the corresponding author.
References
- 1.Williams JP, Newhauser W. Normal tissue damage: its importance, history and challenges for the future. The British journal of radiology. 2019;92(1093):20180048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Timmerman RD, Kavanagh BD, Cho LC, Papiez L, Xing L. Stereotactic body radiation therapy in multiple organ sites. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25(8):947–952. [DOI] [PubMed] [Google Scholar]
- 3.Hall WA, Straza MW, Chen X, et al. Initial clinical experience of Stereotactic Body Radiation Therapy (SBRT) for liver metastases, primary liver malignancy, and pancreatic cancer with 4D-MRI based online adaptation and real-time MRI monitoring using a 1.5 Tesla MR-Linac. PloS one. 2020;15(8):e0236570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jain S, Poon I, Soliman H, et al. Lung stereotactic body radiation therapy (SBRT) delivered over 4 or 11 days: a comparison of acute toxicity and quality of life. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2013;108(2):320–325. [DOI] [PubMed] [Google Scholar]
- 5.Quon HC, Ong A, Cheung P, et al. Once-weekly versus every-other-day stereotactic body radiotherapy in patients with prostate cancer (PATRIOT): A phase 2 randomized trial. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2018;127(2):206–212. [DOI] [PubMed] [Google Scholar]
- 6.Sharabi AB, Tran PT, Lim M, Drake CG, Deweese TL. Stereotactic radiation therapy combined with immunotherapy: augmenting the role of radiation in local and systemic treatment. Oncology (Williston Park). 2015;29(5):331–340. [PMC free article] [PubMed] [Google Scholar]
- 7.Park C, Papiez L, Zhang S, Story M, Timmerman RD. Universal survival curve and single fraction equivalent dose: useful tools in understanding potency of ablative radiotherapy. International journal of radiation oncology, biology, physics. 2008;70(3):847–852. [DOI] [PubMed] [Google Scholar]
- 8.Tang H, Wang Y, Chlewicki LK, et al. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell. 2016;30(3):500. [DOI] [PubMed] [Google Scholar]
- 9.Lin H, Wei S, Hurt EM, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest. 2018;128(4):1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng L, Liang H, Xu M, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41(5):843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutiontov SI, Pitroda SP, Chmura SJ, Arina A, Weichselbaum RR. Cytoreduction and the Optimization Of Immune Checkpoint Inhibition with Radiation Therapy. International journal of radiation oncology, biology, physics. 2020;108(1):17–26. [DOI] [PubMed] [Google Scholar]
- 12.Jagodinsky JC, Harari PM, Morris ZS. The Promise of Combining Radiation Therapy With Immunotherapy. International journal of radiation oncology, biology, physics. 2020;108(1):6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong J, Le TQ, Massarelli E, Hendifar AE, Tuli R. Radiation therapy and PD-1/PD-L1 blockade: the clinical development of an evolving anticancer combination. J Immunother Cancer. 2018;6(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(17):5379–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanpouille-Box C, Alard A, Aryankalayil MJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nature communications. 2017;8:15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamazaki T, Kirchmair A, Sato A, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. 2020;21(10):1160–1171. [DOI] [PubMed] [Google Scholar]
- 17.Azad A, Yin Lim S, D’Costa Z, et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med. 2017;9(2):167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang H, Deng L, Chmura S, et al. Radiation-induced equilibrium is a balance between tumor cell proliferation and T cell-mediated killing. J Immunol. 2013;190(11):5874–5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filatenkov A, Baker J, Mueller AM, et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(16):3727–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75(11):2139–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arina A, Beckett M, Fernandez C, et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nature communications. 2019;10(1):3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang H, Liang Y, Anders RA, et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest. 2018;128(2):580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng Q, Qiu X, Zhang Z, et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nature communications. 2020;11(1):4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crittenden MR, Zebertavage L, Kramer G, et al. Tumor cure by radiation therapy and checkpoint inhibitors depends on pre-existing immunity. Sci Rep. 2018;8(1):7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luke JJ, Lemons JM, Karrison TG, et al. Safety and Clinical Activity of Pembrolizumab and Multisite Stereotactic Body Radiotherapy in Patients With Advanced Solid Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2018;36(16):1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theelen W, Peulen HMU, Lalezari F, et al. Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced Non-Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial. JAMA oncology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antonia SJ, Villegas A, Daniel D, et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. The New England journal of medicine. 2018;379(24):2342–2350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.