

Abstract
Although prostate cancer is known to have a strong genetic basis and is influenced by both common and rare variants, the ability to investigate the combined effect of such genetic risk factors has been limited to date. We conducted an investigation of 81 094 men from the UK Biobank, including 3568 prostate cancer cases, to examine the combined effect of rare pathogenic/likely pathogenic/deleterious (P/LP/D) germline variants and common prostate cancer risk variants, measured using a polygenic risk score (PRS), on prostate cancer risk. The absolute risk of prostate cancer for HOXB13, BRCA2, ATM, and CHEK2 P/LP/D carriers ranged from 9% to 56%, and the absolute risk in noncarriers ranged from 2% to 31%, by age 85 yr, for men in the lowest and highest PRS decile, respectively. The high-penetrant HOXB13 G84E prostate cancer risk variant was most common in cases in the lowest PRS quintile (4.4%) and least common in cases in the highest PRS quintile (0.5%; p = 0.005), whereas there was no statistically significant difference in frequencies by PRS in controls. While rare and common variants strongly and distinctly influence prostate cancer onset, consideration of rare and common variants in conjunction will lead to more precise estimates of a man’s lifetime risk of prostate cancer.
Keywords: Biobank, Common variants, Exome sequencing, Genetics, Genomics, HOXB13, Polygenic risk score, Prostate cancer, Rare variants
Patient summary:
We found that the risk of prostate cancer conveyed by rare variants could vary depending on an individual’s genetic profile of common risk variants. This implies that in order to comprehensively assess genetic risk of prostate cancer, it is important to consider both rare and common variants.
Prostate cancer (PCa) is a leading cause of death, with high heritability and risk among family members suggesting a strong genetic basis of this disease [1,2]. Rare germline genetic variants have been shown to increase PCa risk [3,4], as have common variants in aggregate as measured by polygenic risk scores (PRSs), with men in the highest PRS decile having approximately four-fold increased odds of PCa than men in the average 40–60% PRS category [5]. Until recently, the ability to investigate the combined influence of rare and common variants has been limited. Recent studies have shown that common variants modify the influence of rare variants on breast cancer, colorectal cancer, and coronary artery disease risk [6,7], and the influence of rare BRCA2 and HOXB13 variants on PCa risk has been shown to vary by common variants [8–10]. Here, we investigated the combined effect of rare and common germline variants on PCa risk using whole-exome sequencing and genome-wide genotype data in a large sample of 81 094 European ancestry men from the UK Biobank (October 2020 release of 200K whole-exome sequences), including 3568 PCa cases and 77 526 controls.
We first performed exome-wide gene-based analyses to determine whether novel PCa risk genes could be identified from rare pathogenic/likely pathogenic/deleterious (P/LP/D) variants. Across 14 905 tested genes, only HOXB13 (odds ratio [OR] = 4.63, 95% confidence interval [CI] = 3.26–6.59, p = 1.4 × 10−17) and CHEK2 (OR = 2.06, 95% CI = 1.51–2.80, p = 4.9 × 10−6) reached genome-wide significance (Supplementary Fig. 1). Limiting to 151 DNA repair genes, which have been implicated in PCa risk [4,11], only CHEK2 (see above) and BRCA2 (OR = 2.15, 95% CI = 1.40–3.28, p = 4.2 × 10−4) were significantly associated with PCa risk after multiple testing adjustment (Supplementary Fig. 2). By testing individual exome-wide P/LP/D variants, two significant associations were identified: known rs138213197 (G84E) in HOXB13 (control carrier frequency = 0.31%, case carrier frequency = 1.29%, p = 6.9 × 10−18) and novel rs769540160 in MYO3A (control carrier frequency = 0.004%, case carrier frequency = 0.11%, p = 1.1 × 10−7; Supplementary Fig. 3). Although MYO3A was not genome-wide significant in gene-based tests, results were suggestive of carriers having 1.67-fold increased odds of PCa (95% CI = 1.06–2.65, p = 0.027). The carrier frequency of rs769540160 in 32 330 cancer-free European ancestry individuals in gnomAD was 0.009% [12], while it was four-fold more common in a whole-exome sequencing study of 5545 European ancestry men with aggressive and nonaggressive PCa, with a carrier frequency of 0.04% (carried by two men both of whom died due to PCa and had Gleason scores ≥8) [4]. Given the extreme rarity of this variant, additional large-scale PCa sequencing studies are necessary to further validate this novel association.
Combined rare and common variant analyses focused on carrier status of P/LP/D variants in known PCa risk genes (HOXB13 and DNA repair genes BRCA2, BRCA1, PALB2, ATM, CHEK2, NBN, and MSH2) [4,11,13] and our recently developed multiancestry PRS [5]. HOXB13, BRCA2, ATM, and CHEK2 had sufficient numbers of P/LP/D carriers for analyses and were consequently the focus of our investigation. Analyses jointly evaluating the PRS and carrier status excluded HOXB13 G84E and/or CHEK2 1100delC from the PRS when carrier status included either of these variants. Of the total 1576 carriers of P/LP/D alleles in these four genes, 19 men carried two P/LP/D alleles (including two cases) and the remaining 1557 men carried one P/LP/D allele (including 143 cases). As expected, these four genes showed strong associations with PCa risk, as did the multiancestry PRS, which had stronger effects than a previously developed European ancestry PRS [14] (Supplementary Tables 1 and 2). In aggregate, P/LP/D carriers had 2.52-fold increased odds of PCa (2.10–3.04, p = 1.40 × 10−22) and 4.73-fold increased odds of dying due to PCa (95% CI = 2.82–7.94, p = 4.1 × 10−9). Although we had insufficient clinical data to further evaluate aggressive or lethal disease (220 men died due to PCa, of whom 16 carried P/LP/D alleles in these genes), we previously reported that P/LP/D variants in ATM and BRCA2 were more common in men with aggressive (and lethal) disease than in men with nonaggressive PCa [4]. Aggregate effects of P/LP/D variants in these genes did not differ significantly in men with and without a first-degree family history of PCa or in men ≤60 or >60 yr of age (Supplementary Tables 3 and 4). PRS effects also did not differ significantly by family history; however, the PRS had significantly larger effects in younger than in older men (Supplementary Tables 5 and 6), consistent with previous findings [5].
Relative to noncarriers in the average 40–60% PRS category, ORs ranged from 0.28 (95% CI = 0.22–0.36) to 4.34 (95% CI = 3.87–4.87) for noncarriers and from 1.06 (95% CI = 0.43–2.64) to 10.21 (95% CI = 6.53–15.96) for carriers in the lowest and highest PRS decile, respectively (Fig. 1A). The absolute risk of PCa by age 85 yr ranged from 2% to 31% for noncarriers and from 9% to 56% for carriers in the lowest and highest PRS decile, respectively (Fig. 1B). The absolute risk for carriers in the 90–100% PRS category (56%) was similar to the 55% absolute risk for men in the 99–100% PRS category (independent of carrier status) and two-fold higher than the 26% absolute risk for carriers (independent of PRS). The absolute risk for carriers in the 0–10% PRS category (9%) was similar to the 11% absolute risk for noncarriers (independent of PRS; Fig. 1C). Effects and absolute risks were slightly weaker when excluding HOXB13 G84E from carrier status (Supplementary Fig. 4). Evaluation of the four genes separately revealed similar findings (Supplementary Fig. 5–8), with HOXB13 G84E carriers having notably increased PCa risk compared with noncarriers across PRS quintiles (used instead of deciles, given the smaller numbers of carriers within individual genes). Across PRS quintiles, ORs for HOXB13 G84E carriers ranged from 2.96 (95% CI = 1.19–7.34) to 10.10 (95% CI = 5.03–20.28; relative to HOXB13 G84E noncarriers in the average 40–60% PRS category), while absolute risks for HOXB13 G84E carriers ranged from 23% to 56% by age 85 yr (Supplementary Fig. 5). We observed a statistically significant interaction between the continuous PRS and carrier status for HOXB13 (p = 0.041), but not for the other genes separately or in aggregate (p ≥ 0.14; Supplementary Table 1).
Fig. 1 -.
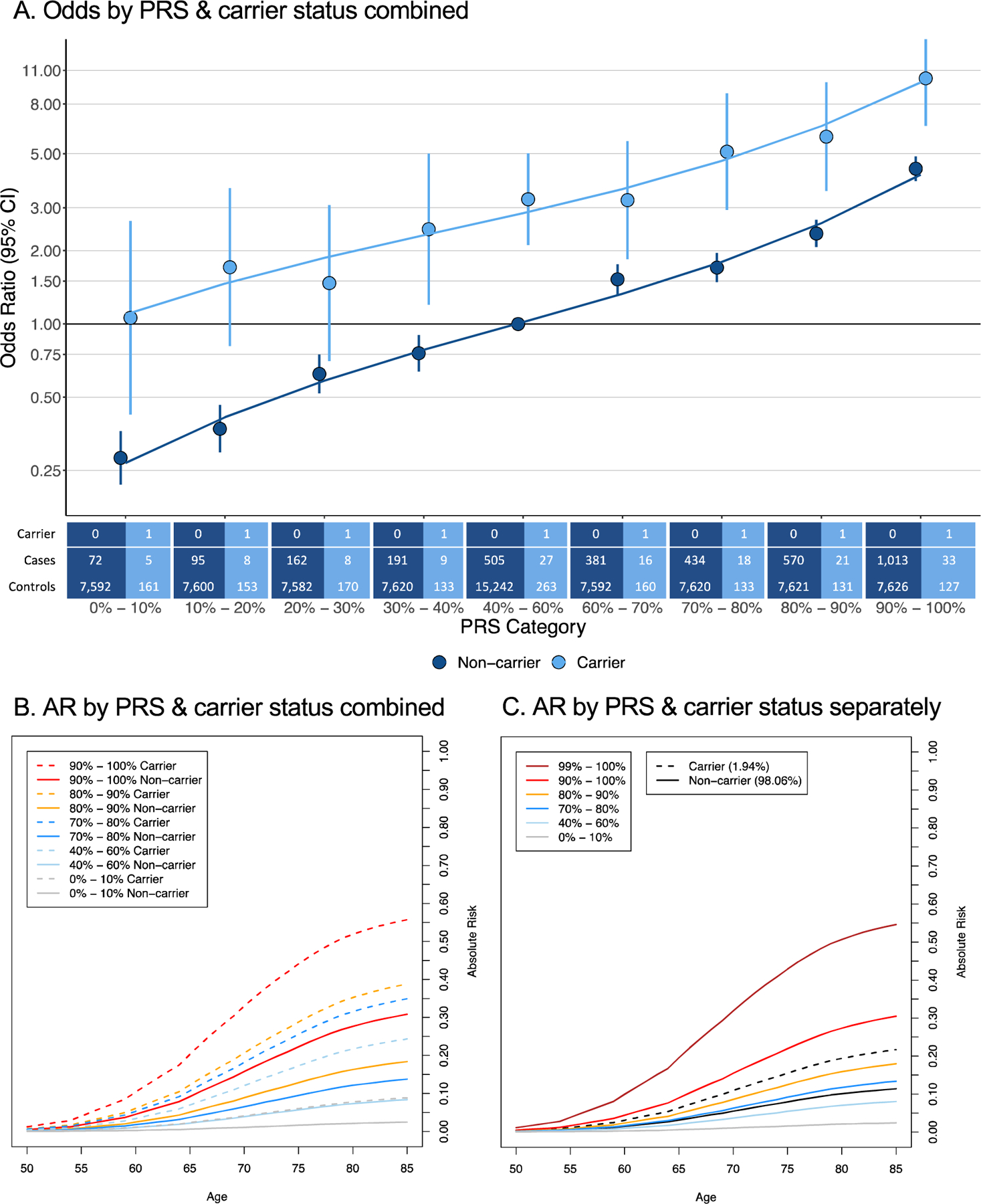
Aggregate effect of P/LP/D variants in ATM, BRCA2, CHEK2, and HOXB13 and a polygenic risk score (PRS) on prostate cancer risk. (A) Odds of prostate cancer by PRS category and carrier status. ORs are calculated with respect to the referent noncarrier 40–60% PRS category. Percentage of total cases are annotated for each effect estimate, and sample sizes of carriers and noncarriers by case status and PRS category are indicated below the figure. In the 40–60% PRS category, 0.76% and 14.15% of total cases are carriers and noncarriers, respectively. ORs are plotted on a log scale. (B) Absolute risk (AR) of prostate cancer by age and the combination of carrier status and PRS category. The 40–60% PRS noncarrier line estimates baseline AR by age (8.4% lifetime AR). (C) AR of prostate cancer by age and carrier status (independent of PRS) and PRS category (independent of carrier status). The 40–60% PRS line (8.1% lifetime AR) and noncarrier line (11.3% lifetime AR) estimate baseline AR by age. CI = confidence interval; OR = odds ratio.
Interestingly, among cases, HOXB13 G84E was most common in the lowest PRS quintile (4.4%) and least common in the highest PRS quintile (0.5%), whereas control carrier frequencies across PRS quintiles were consistently 0.3% (Fig. 2A). Accordingly, the average PRS was higher in HOXB13 G84E noncarriers than in carriers among cases (p = 0.005) and did not significantly differ by carrier status among controls (p = 0.3; Fig. 2B). The frequency of CHEK2 P/LP/D carriers was also most common in cases in the lowest PRS quintile (2.3%) and least common in the highest PRS quintile (1.3%; Supplementary Fig. 9); however, PRS did not differ significantly by carrier status (p = 0.3; Supplementary Fig. 10). The HOXB13 G84E finding was validated using genome-wide association study (GWAS) data in an independent sample of 5197 cases and 115 796 controls in the UK Biobank, with cases having a carrier frequency of 3.2% in the lowest PRS quintile and 1.2% in the highest PRS quintile (Supplementary Fig. 11). Similar results were observed in the full UK Biobank sample (8765 cases and 193 322 controls; Supplementary Fig. 12). This finding suggests that HOXB13 G84E may account for more PCa in men with low versus high PRS. While carrying rare or common risk variants could serve as independent pathways to PCa onset, our results suggest that carrying rare variants in these genes and having high PRS compound PCa risk.
Fig. 2 -.
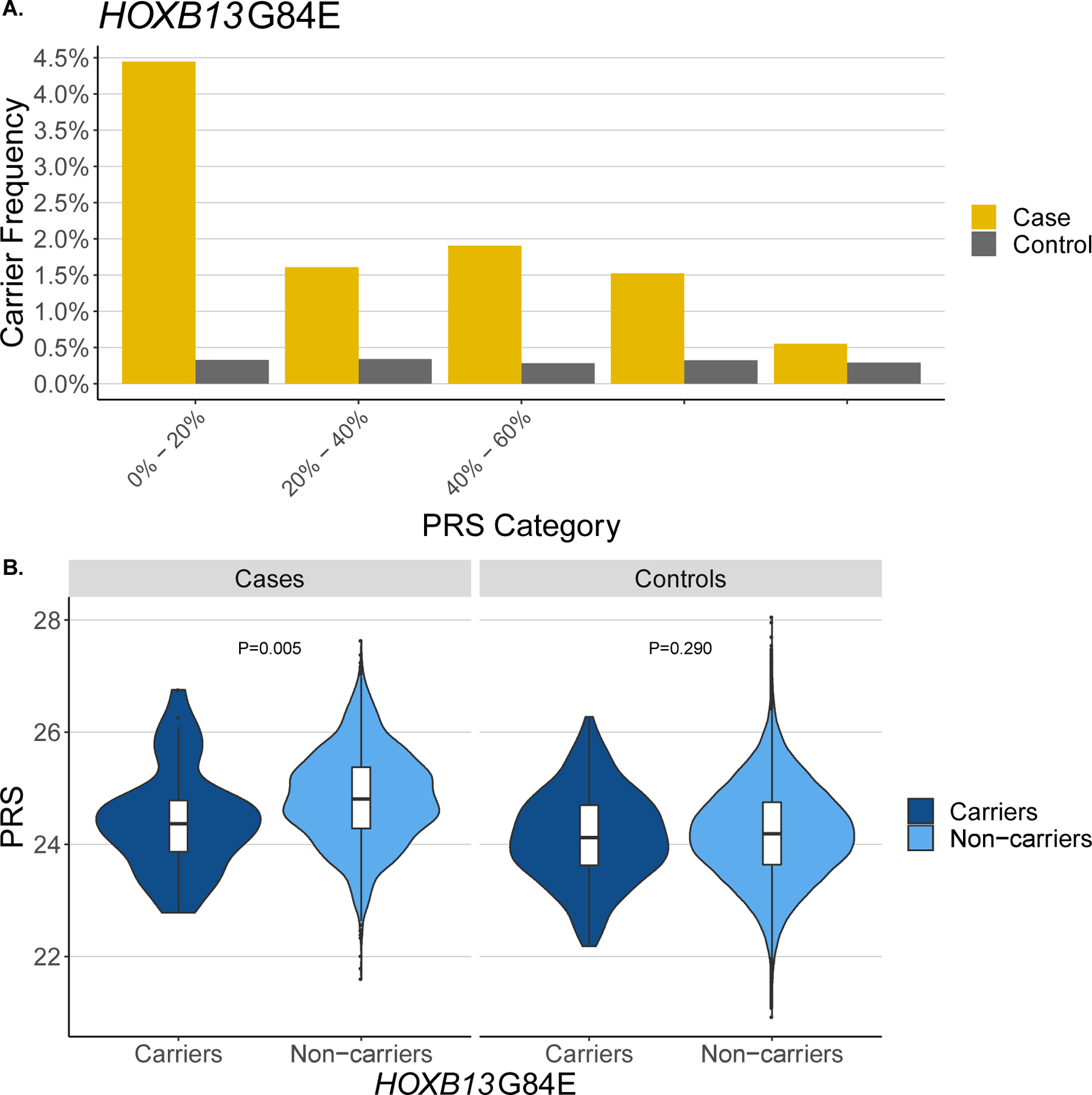
Polygenic risk score (PRS) distribution of HOXB13 G84E carriers. (A) HOXB13 G84E carrier frequency by PRS category and prostate cancer status. (B) PRS distribution by HOXB13 G84E carrier status and prostate cancer status. PRS differences between carriers and noncarriers are calculated using a two-sided t test.
Findings from this investigation suggest that PCa risk may vary depending on an individual’s genetic profile of common risk variants, measured by PRS, and carrier status for rare P/LP/D variants in HOXB13 and BRCA2, with novel evidence for variants in CHEK2 and ATM. In particular, men in the top PRS decile had a higher absolute risk of PCa than carriers (31% vs 25%); however, considering the PRS and carrier status jointly, the absolute risk for noncarriers in the top PRS decile was 31%, while it increased to 56% for carriers in the top PRS decile. This is supported by previous findings of rare and common variants collectively improving discriminative ability of PCa risk models [15] and could have important clinical implications, such as informing decisions regarding PCa screening, with P/LP/D carriers and/or men with a high PRS potentially benefiting from earlier and more frequent screening. Further studies are underway and needed to evaluate the impact of such clinical implementations. Consistent with studies of other diseases [16], our findings also suggest that rare and common variants could independently lead to PCa onset, with low PRS cases being more likely to carry HOXB13 G84E, for example. Whole-genome sequencing efforts could have improved the power to identify additional moderate- to high-penetrant rare PCa risk variants by prioritizing low PRS cases, as extreme sampling has been shown to improve the power to detect rare variants [17]. It will be important to extend this to clinical investigations to determine whether PRS in conjunction with carrier status for rare P/LP/D variants could better discern aggressive PCa, which we were unable to investigate in this study. Further, similar investigations in non-European ancestry men will be critical, particularly in men of African ancestry, given the established genetic contribution to high PCa incidence rates in this population [5].
Supplementary Material
Acknowledgments
Funding/Support and role of the sponsor: This work was supported by the National Cancer Institute at the National Institutes of Health grant (K99 CA246063, BFD and R01 CA196931, CAH) and an award from the Achievement Rewards for College Scientists Foundation Los Angeles Founder Chapter (BFD). This research has been conducted using the UK Biobank Resource under application number 42195.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial disclosures: Burcu F. Darst certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
References
- [1].Hjelmborg JB, Scheike T, Holst K, et al. The heritability of prostate cancer in the Nordic Twin Study of Cancer. Cancer Epidemiol Biomarkers Prev 2014;23:2303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hemminki K. Familial risk and familial survival in prostate cancer. World J Urol 2012;30:143–8. [DOI] [PubMed] [Google Scholar]
- [3].Mancuso N, Rohland N, Rand KA, et al. The contribution of rare variation to prostate cancer heritability. Nat Genet 2016;48:30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Darst BF, Dadaev T, Saunders E, et al. Germline sequencing DNA repair genes in 5,545 men with aggressive and non-aggressive prostate cancer. J Natl Cancer Inst In press. 10.1093/jnci/djaa132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Conti DV, Darst BF, Moss LC, et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet 2021;53:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fahed AC, Wang M, Homburger JR, et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun 2020;11:3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gallagher S, Hughes E, Wagner S, et al. Association of a polygenic risk score with breast cancer among women carriers of high- and moderate-risk breast cancer genes. JAMA Netw Open 2020;3:e208501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Karlsson R, Aly M, Clements M, et al. A population-based assessment of germline HOXB13 G84E mutation and prostate cancer risk. Eur Urol 2014;65:169–76. [DOI] [PubMed] [Google Scholar]
- [9].Kote-Jarai Z, Mikropoulos C, Leongamornlert DA, et al. Prevalence of the HOXB13 G84E germline mutation in British men and correlation with prostate cancer risk, tumour characteristics and clinical outcomes. Ann Oncol 2015;26:756–61. [DOI] [PubMed] [Google Scholar]
- [10].Lecarpentier J, Silvestri V, Kuchenbaecker KB, et al. Prediction of breast and prostate cancer risks in male BRCA1 and BRCA2 mutation carriers using polygenic risk scores. J Clin Oncol 2017;35:2240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016;375:443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Matejcic M, Patel Y, Lilyquist J, et al. Pathogenic variants in cancer predisposition genes and prostate cancer risk in men of African ancestry. JCO Precis Oncol 2020;4:32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schumacher FR, Al Olama AA, Berndt SI, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 2018;50:928–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shi Z, Platz EA, Wei J, et al. Performance of three inherited risk measures for predicting prostate cancer incidence and mortality: a population-based prospective analysis. Eur Urol 2021;79:419–26. [DOI] [PubMed] [Google Scholar]
- [16].Lu T, Zhou S, Wu H, Forgetta V, Greenwood CMT, Richards JB. Individuals with common diseases but with a low polygenic risk score could be prioritized for rare variant screening. Genet Med 2021;23:508–15. [DOI] [PubMed] [Google Scholar]
- [17].Li D, Lewinger JP, Gauderman WJ, Murcray CE, Conti D. Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies. Genet Epidemiol 2011;35:790–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.