

Summary
RAD51 facilitates replication fork reversal and protects reversed forks from nuclease degradation. While potentially a useful replication stress response mechanism, unregulated fork reversal can cause genome instability. Here we show that RADX, a single-strand DNA binding protein that binds to and destabilizes RAD51 nucleofilaments, can either inhibit or promote fork reversal depending on replication stress levels. RADX inhibits fork reversal at elongating forks, thereby preventing fork slowing and collapse. Paradoxically, in the presence of persistent replication stress, RADX localizes to stalled forks to generate reversed fork structures. Consequently, inactivating RADX prevents fork reversal-dependent telomere dysfunction in the absence of RTEL1 and blocks nascent strand degradation when fork protection factors are inactivated. Addition of RADX increases SMARCAL1-dependent fork reversal in conditions where pre-binding RAD51 to a model fork substrate is inhibitory. Thus, RADX directly interacts with RAD51 and single-strand DNA to confine fork reversal to persistently stalled forks.
Highlights and eTOC Blurb
Krishnamoorthy et al., show that RADX prevents replication fork reversal at elongating replication forks, but promotes fork reversal when forks stall. RADX generates these context-dependent effects by binding ssDNA and RAD51 to generate a metastable RAD51 filament at stalled forks when template-strand ssDNA is abundant.
Graphical Abstract
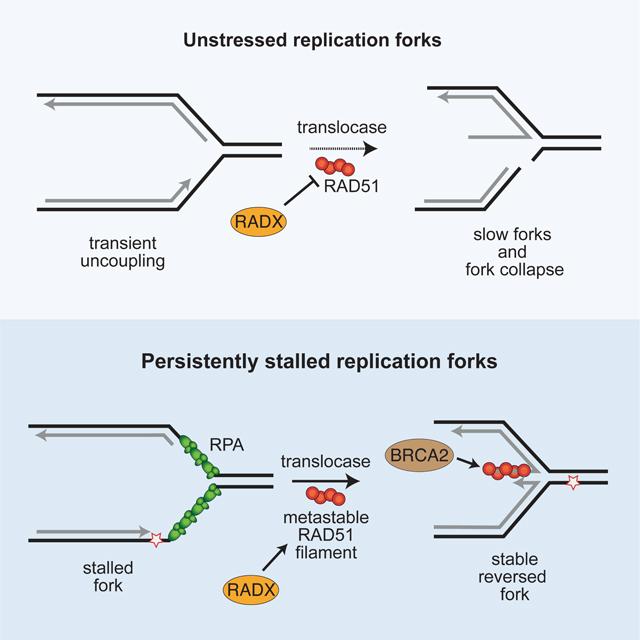
Introduction
Replication fork reversal is a replication stress tolerance mechanism that promotes replication-coupled DNA repair or bypass of DNA damage (Berti et al., 2020a; Cortez, 2019). Multiple proteins regulate the formation and stabilization of reversed forks since unregulated fork reversal can slow replication elongation, increase the frequency of double strand breaks (DSBs), cause extensive degradation of nascent DNA, and result in genome instability.
The recombinase RAD51 is required to promote fork reversal (Zellweger et al., 2015), and protect the nascent DNA from degradation (Hashimoto et al., 2010; Schlacher et al., 2011). The RAD51 reversal function is thought to involve a metastable RAD51 filament (Berti et al., 2020a). In most cases, reversal does not require BRCA2, although it may be involved in circumstances in which RAD51 function is partly compromised (Liu et al., 2020). Instead, other RAD51 regulators including RAD51 paralogs assist fork reversal (Berti et al., 2020b), but exactly how RAD51 promotes reversal is unknown.
A reversed fork is the substrate for nascent strand degradation since inactivating fork reversal enzymes like SMARCAL1, ZRANB3, HLTF, or FBH1 blocks degradation (Higgs et al., 2015; Kolinjivadi et al., 2017; Lemacon et al., 2017; Liu et al., 2020; Mijic et al., 2017; Taglialatela et al., 2017). Fork protection is thought to require RAD51 nucleoprotein filaments that are stabilized by BRCA2. These stable filaments protect the nascent DNA from nucleases including MRE11 and DNA2 (Hashimoto et al., 2010; Schlacher et al., 2011; Thangavel et al., 2015). Many proteins in addition to BRCA2 and RAD51 prevent nascent strand degradation, and they organize into at least two pathways depending on whether the translocases SMARCAL1, ZRANB3 and HLTF or FBH1 cooperate with RAD51 to remodel the fork (Liu et al., 2020). Moreover, different levels of RAD51 function are required for fork reversal and fork protection (Bhat and Cortez, 2018; Bhat et al., 2018). Consistent with this hypothesis, reducing the concentration of RAD51 in cells or reducing its ability to bind DNA using a chemical inhibitor, B02, causes deprotection of reversed forks; whereas, potently silencing RAD51 expression prevents fork reversal and inhibits nascent strand degradation (Bhat et al., 2018; Taglialatela et al., 2017).
RADX is a single-strand DNA (ssDNA) binding protein important for replication fork stability (Dungrawala et al., 2017; Schubert et al., 2017). In the absence of added replication stress, RADX inactivation causes slow replication elongation and increased DSBs (Dungrawala et al., 2017; Schubert et al., 2017). RADX competes with RAD51 for ssDNA, directly interacts with RAD51, destabilizes the RAD51 nucleofilament, and inhibits RAD51-dependent processes in vitro (Adolph et al., 2021). Inactivating RAD51 rescues the slow replication elongation and elevated DSBs observed in RADX-deficient cells (Dungrawala et al., 2017). These data suggest that RADX antagonizes RAD51 to maintain genome stability during DNA replication.
In cells experiencing persistent replication stress caused by hydroxyurea (HU), RADX overexpression reduces the amount of RAD51 at forks and promotes fork degradation (Bhat et al., 2018). In addition, RADX inactivation restores fork protection in BRCA2, BRCA1, or FANCD2-deficient cells treated with HU (Bhat et al., 2018; Dungrawala et al., 2017). Based on the antagonistic relationship between RADX and RAD51 found in biochemical experiments and in unstressed cells, and the observations that RADX inactivation or overexpression increases or reduces RAD51 levels at forks respectively, we proposed that the rescue of fork protection by RADX inactivation could be due to restoration of sufficient RAD51 activity to protect reversed forks in these cells (Bhat and Cortez, 2018; Bhat et al., 2018; Dungrawala et al., 2017). In other words, we hypothesized that removing a negative regulator of RAD51 like RADX may compensate for inactivating positive regulators like BRCA2. However, this hypothesis does not explain why RADX accumulates at stalled forks (Dungrawala et al., 2017; Schubert et al., 2017).
In this study, we find that consistent with our previous hypothesis, RADX inhibits aberrant fork reversal and prevents fork collapse in unstressed cells. However, we now find that RADX promotes the formation of reversed fork structures in cells experiencing persistent replication stress. Our results indicate that RADX has two apparently opposite functions to regulate fork reversal depending on the amount of replication stress. To explain these observations, we propose a unifying model in which the RADX-dependent destabilization of the RAD51 nucleofilament can either inhibit or promote fork reversal depending on whether forks are actively elongating or stalled. These results are reminiscent of N. gonorrhoeae RecX which stimulates functions of the recombinase RecA (the RAD51 orthologue) in cells even though it inhibits its biochemical activities (Gruenig et al., 2010).
Results
RADX inhibits inappropriate fork reversal in the absence of exogenous stress
RADX inactivation causes slow replication elongation and increased fork collapse in the absence of added replication stress (Dungrawala et al., 2017; Schubert et al., 2017). Inactivating the fork reversal enzymes SMARCAL1, ZRANB3, or HLTF using siRNA or CRISPR-Cas9 gene editing rescues the slow fork elongation phenotype caused by RADX silencing in U2OS and hTERT-RPE-1 cells (Figures 1A and S1A–D). Additionally, inhibiting PARP with the small molecule inhibitor Olaparib, which was previously reported to prevent fork reversal by promoting fork restoration (Berti et al., 2013), also rescued the fork elongation defect in RADXΔ cells (Figure 1B). In contrast, silencing MUS81 or inhibiting MRE11 did not rescue fork speeds indicating that the fork elongation defects in the absence of RADX are unlikely to be due to fork cleavage or nuclease degradation (Figures S1E and S1F). Finally, inactivating SMARCAL1, ZRANB3, or HLTF also reduces the frequency of DSBs observed in U2OS cells lacking RADX suggesting these breaks are a consequence of aberrant fork remodeling (Figure 1C and (Dungrawala et al., 2017)).
Figure 1. RADX inhibits inappropriate fork reversal in the absence of replication stress.

(A) U2OS cells transfected with the indicated siRNAs (NT=non-targeting) were labeled with CldU followed by IdU and DNA combing was used to measure elongation rates. A one-way ANOVA with Tukey’s multiple comparison test was used to calculate p values in all DNA fiber experiments. (B) Replication fork elongation was monitored by DNA combing in RADXΔ cells treated with 10μM Olaparib as indicated. (C) DSBs were measured by neutral comet assay in wild-type or HLTFΔ U2OS cells transfected with the indicated siRNAs. A Kruskal-Wallis test was used to calculate p values in all comet assays. All fiber and comet assays are representative experiments of at least n=3 biological replicates. See also Figure S1. (D) Example of a reversed replication fork imaged by EM and the mean+/−SEM percentage of reversed forks from three experiments is shown (Inset, magnified four-way junction at the reversed fork; P, parental strands; D, daughter strands; R, reversed strands). The number of replication intermediates analyzed for each condition is indicated in parenthesis. A Welch’s test was used to calculate p values. See also Table S1 and Figure S2.
These data collectively suggest that RADX prevents inappropriate fork reversal at unchallenged forks that otherwise would impair replication elongation and increase the frequency of fork collapse. To further test this hypothesis, we analyzed replication intermediates using electron microscopy (EM). As predicted, silencing RADX either by siRNA transfection (siRADX) or by CRISPR-Cas9 mediated gene editing (RADXΔ) increased the frequency of reversed forks observed by EM in the absence of added replication stress (Figures 1D and S2 and Table S1). Thus, we conclude that RADX inhibits fork reversal in the absence of replication stress.
RADX inactivation blocks nascent strand degradation at persistently stalled replication forks without restoring RAD51 localization
We previously reported that inactivating RADX in U2OS cells blocks nascent strand degradation when BRCA2 or BRCA1 are also inactivated (Bhat et al., 2018; Dungrawala et al., 2017). We confirmed that this effect was not cell type specific, since RADX silencing by RNA interference also prevents nascent strand degradation in BRCA2-depleted hTERT-RPE-1 cells and BRCA2Δ DLD1 cells (Figures 2A and 2B). We also found that despite causing elevated levels of DSBs when silenced by itself in HU-treated cells, RADX silencing reduces the fork breakage observed in BRCA2-deficient cells, which previously was reported to be dependent on fork reversal (Figures 2C and S3A) (Lemacon et al., 2017).
Figure 2. RADX silencing protects stalled forks from degradation in the absence of BRCA2 without restoring RAD51 localization.

(A and B) Fork protection assays were completed in (A) hTERT-RPE-1 or (B) DLD1 cells with and without BRCA2 after transfection of the indicated siRNAs. (C) Neutral comet assay in siRNA transfected U2OS cells treated with 3mM HU for 5 hours. (D) U2OS cells transfected with siRNA were labeled with 10μM EdU for 20 minutes, treated with 3mM HU for 5h, and stained for RAD51 and EdU. Representative images of RAD51 staining and the number of chromatin-bound RAD51 foci per nucleus is shown (mean +/− 95% confidence interval). Scale bar-10μm. A Mann-Whitney test was used to calculate p-values. See also Figure S3.
Since RADX inactivation increases the amount of RAD51 localized to replication forks (Dungrawala et al., 2017), we previously hypothesized that inactivating RADX at persistently stalled forks prevents nuclease-mediated nascent strand degradation by improving the stability of RAD51 filaments on the reversed forks. This hypothesis predicts that RADX inactivation should restore RAD51 localization to stalled forks in BRCA2-deficient cells. To test this prediction, we performed quantitative immunofluorescence imaging of chromatin-bound RAD51 in S-phase cells. As shown previously, silencing BRCA2 reduces the intensity and number of RAD51 foci while silencing RADX modestly increases the intensity and number of RAD51 foci in HU-treated cells (Figures 2D and S3B) (Dungrawala et al., 2017; Taglialatela et al., 2017). However, contrary to our expectation, silencing RADX in the absence of BRCA2 did not appreciably restore the number or intensity of RAD51 foci. Silencing RADX also did not increase RAD51 localization to stalled forks in BRCA1-deficient cells (Figures 2D and S3B). Thus, the ability of RADX inactivation to prevent nascent strand degradation and fork cleavage when fork protection factors are inactivated may not result from restoration of RAD51 localization to reversed forks.
To further test this idea, we reasoned that if RADX inactivation restores fork protection to reversed forks by stabilizing RAD51 filaments, then loss of RADX should not rescue nascent strand degradation in cases where fork protection is independent of RAD51 filament stability. For example, ABRO1 was reported to protect reversed forks independently of RAD51 (Xu et al., 2017). Thus, if RADX inactivation restores RAD51 filament stability on reversed forks, it should not rescue the nascent strand degradation in ABRO1-deficient cells. In contrast to this prediction, RADX silencing does restore fork protection in ABRO1-depleted cells (Figure S3C). Furthermore, we confirmed that a reversed fork is the substrate for degradation in cells lacking ABRO1 since inactivating the fork reversal proteins ZRANB3 or HLTF also prevents nascent strand degradation when ABRO1 is silenced (Figures S3D and S3E). These observations are inconsistent with the idea that RADX inactivation restores fork protection by increasing the stability of RAD51 filaments on reversed forks.
Partial depletion of RADX causes nascent strand degradation
We did not observe nascent strand degradation in either our RADXΔ cells generated by CRISPR-Cas9 or after transfection with RADX siRNA. However, Schubert et al., reported nascent strand degradation when they silenced RADX with siRNA (Schubert et al., 2017). To further explore how RADX inactivation affects fork stability, we tried to reconcile these observations. RNA interference may not fully inactivate RADX, so we considered the possibility that partial RADX loss might yield a different effect than complete loss of function as we had previously found for RAD51 (Bhat et al., 2018). To test this hypothesis directly, we utilized different concentrations of RADX siRNA to obtain varying levels of silencing. 5nM of RADX siRNA caused efficient knockdown with little RADX protein visible by immunoblotting, while 0.5nM of the same siRNA yielded only partial knockdown (Figure 3A). Strikingly, the partial RADX knockdown cells treated with HU underwent extensive nascent strand degradation even though the cells with more efficient RADX knockdown did not (Figure 3A). The same result was obtained with a second siRNA to RADX (Figure 3A). Partial knockdown of RADX did not yield fork degradation in RADXΔ cells indicating that these are not off-target effects of the siRNAs (Figure S4A). Moreover, nascent strand degradation upon partial RADX inactivation was also observed in hTERT-RPE-1 cells indicating that this effect is not cell-type specific (Figure S4B).
Figure 3. Partial depletion of RADX causes nascent strand degradation.

(A-D) Fork protection assays were performed in wild-type, SMARCAL1Δ, HLTFΔ or ZRANB3Δ U2OS cells transfected with the indicated amount of each siRNA. Cells were treated with (B) 100μM Mirin or (D) 25μM B02 where indicated. A one-way ANOVA with Tukey’s multiple comparison test was used to calculate all p values. All experiments were completed at least twice. See also Figure S4.
The fork degradation caused by partial depletion of RADX siRNA is prevented by treatment with mirin which inhibits MRE11 (Dupre et al., 2008), C5 which inhibits DNA2 (Liu et al., 2016), or by inactivating SMARCAL1, ZRANB3, or HLTF (Figures 3B, 3C, and S4C). Thus, the resection substrate in these cells is likely a reversed fork that is degraded by MRE11 or DNA2.
These results phenocopy the effects previously reported for RAD51 (Bhat et al., 2018). RAD51 partial knockdown by siRNA, inhibition with a small molecule inhibitor, or mutation also yields nascent strand degradation that is dependent on fork reversal (Bhat et al., 2018; Taglialatela et al., 2017; Wang et al., 2015). Strikingly, either efficient or partial knockdown of RADX in cells treated with the RAD51 inhibitor, B02, largely restores fork protection (Figure 3D). A parsimonious explanation for these results is that in the presence of HU, RADX functions like RAD51 to help generate a reversed fork substrate for degradation even though in the absence of added replication stress, it inhibits fork reversal.
RADX is required for fork-reversal dependent telomere catastrophe in cells experiencing added replication stress
We next looked for additional contexts to test the hypothesis that RADX inhibits fork reversal in the absence of added replication stress but is needed to promote fork reversal in the presence of replication stress. One such context is at telomeres where telomerase binding to reversed forks in RTEL1-deficient cells causes telomere catastrophe (Figure 4A) (Margalef et al., 2018). If RADX is needed to promote fork reversal, then we might expect RADX inactivation to rescue the telomere catastrophe in RTEL1-deficient cells. Conversely, if RADX prevents fork reversal, then its inactivation would have no effect or increase telomeric dysfunction. As previously reported, inactivating RTEL1 in HeLa cells with long telomeres increased the frequency of telomere fragility and heterogeneity, and co-depleting the fork reversal enzyme ZRANB3 rescues these phenotypes linking them to fork reversal (Figures 4B-D) (Margalef et al., 2018). Inactivating RADX by itself did not cause a significant increase in telomere heterogeneity or fragility (Figures 4C and 4D). When RADX and RTEL1 were co-depleted, we observed a small but statistically insignificant decrease in fragility and no change in heterogeneity compared to RTEL1 inactivation by itself (Figures 4C and 4D). These data suggest that in the absence of added replication stress agents, loss of RADX has only minor effects on telomere stability and is not required for formation of reversed forks at telomere sequences in RTEL1-deficient cells. We next repeated the experiments in the presence of aphidicolin to increase replication stress levels and generate more persistently stalled forks. As expected, addition of aphidicolin increases telomere catastrophe in RTEL1-deficient cells (Figures S4D and S4E). In this condition, RADX inactivation by itself had no effect on telomere fragility or heterogeneity; but inactivating RADX in the aphidicolin-treated, RTEL1-deficient cells rescued the telomere fragility and heterogeneity similar to inactivating ZRANB3 (Figures 4E and 4F). These results are consistent with the idea that in the presence of added replication stress, RADX is needed to generate the reversed forks that are a source of the telomere instability in RTEL1-deficient cells.
Figure 4. RADX inactivation rescues fork reversal-dependent telomere instability only in cells treated with aphidicolin.

(A) Schematic model indicating that fork reversal causes telomere catastrophe in RTEL1-deficient cells (Margalef et al., 2018). (B) Representative images of normal, fragile, and unequal telomeres. Scale bar-1μm. (C) Quantification of fragile telomeres and (D) telomere heterogeneity in cells transfected with siRNAs. (E) Quantification of fragile telomeres and (F) telomere heterogeneity in cells transfected with siRNAs and treated with 0.2μM aphidicolin for 40h. Each data point represents a metaphase spread. All experiments were completed at least three times. Parentheses indicate the number of total metaphases analyzed. P values were calculated using a Kruskal-Wallis test. See also Figure S4.
RADX promotes fork reversal in the presence of persistent replication stress
The simplest explanation of the fork protection and telomere stability data is that RADX is needed to generate reversed forks in cells experiencing persistent replication stress. To directly test this hypothesis, we utilized EM to observe the frequency of fork reversal in HU-treated cells. As reported previously, treatment with HU yields a high frequency of reversed forks (Figures 5A and 5B, Table S1). Depletion of RADX by siRNA (siRADX) or inactivation by CRISPR-Cas9 (RADXΔ) consistently decreased the percentage of reversed forks although not to the levels of unstressed cells (Figure 5B, Table S1).
Figure 5. RADX inactivation suppresses fork reversal in cells experiencing persistent replication stress.

(A) Example of a reversed fork observed by EM (Inset, magnified four-way junction at the reversed fork; P, parental strands; D, daughter strands; R, reversed strands). (B) Quantitation of fork reversal measured by EM in the indicated U2OS cells treated with 3mM HU for 5h (n=3; Mean +/− SEM). The number of replication intermediates analyzed for each condition is indicated in parenthesis. P values were calculated using a Welch test. See also Table S1 and Figure S2.
RADX binds RAD51 and ssDNA to promote fork reversal in the presence of persistent replication stress
In unstressed cells, the ability of RADX to prevent fork instability is dependent on its direct interactions with RAD51 and ssDNA (Adolph et al., 2021; Dungrawala et al., 2017). To test whether the same mechanisms operate in stressed cells, we analyzed the activities of RADX separation of function mutants with diminished ssDNA binding activity (RADX OB2m, (Dungrawala et al., 2017) or an inability to interact with RAD51 (RADX QVPK, (Adolph et al., 2021)). First, we examined if transient overexpression of these proteins caused nascent strand degradation in HU-treated cells as previously observed for wild type RADX (Dungrawala et al., 2017). In contrast to wild-type RADX, the RADX QVPK or RADX OB2m mutants do not cause nascent strand degradation when overexpressed in U2OS cells (Figures 6A and S5A). The RADX overexpression-induced nascent strand degradation is rescued by overexpressing RAD51 suggesting that it is due to a reduction in the stability of RAD51 filaments on reversed replication forks (Figure S5A).
Figure 6. A direct interaction of RADX and RAD51 is required to maintain fork stability in cells experiencing persistent replication stress.

(A) Fork protection assays with 3mM HU were performed in U2OS cells immediately after infection with lentiviruses to overexpress wild-type (WT) RADX or RADX QVPK. Arrows in the immunoblot indicate the endogenous and GFP-tagged RADX proteins. (B) Wild-type or RADXΔ U2OS cells complemented with WT RADX or RADX QVPK were transfected with non-targeting or BRCA2 siRNA and examined for fork protection. Note that the wild-type RADX but not the RADX QVPK mutant expression in the RADXΔ complemented cells decreases after a few passages as previously reported (Adolph et al., 2021). A one-way ANOVA with Tukey’s multiple comparison test was used to calculate all p values. All experiments were completed at least three times. Extra lanes were removed.
We next examined fork protection in RADXΔ cells complemented with wild-type, QVPK, or OB2m RADX. As we previously reported, expression of the wild-type RADX protein decreases after a few passages to a level that no longer causes fork deprotection and permits nascent strand degradation when BRCA2 is silenced (Figure 6B) (Adolph et al., 2021). In contrast, cells only expressing the RADX QVPK or RADX OB2m proteins have stable forks when BRCA2 is silenced similar to RADXΔ cells consistent with the idea that RADX binding to RAD51 and ssDNA is needed to enable fork reversal in cells experiencing persistent stress (Figure 6B and S5B).
To explain the paradoxical observations for RADX function in unstressed and stressed cells, we reasoned that RADX could either “switch” from an antagonist of RAD51 in the absence of stress to a positive regulator of RAD51 with persistent stress or that it may continue to antagonize RAD51 biochemically in the presence of stress but that this function may help generate the “metastable” RAD51 filament proposed to be needed to reverse forks (Berti et al., 2020a). To test if RADX switched from an antagonist of RAD51 to a positive regulator of RAD51, we purified GFP-Flag-RADX from unstressed and HU-treated 293T cells (Fig. S6A). Both RADX proteins inhibited RAD51-mediated strand exchange and D-loop formation as efficiently as MBP-RADX purified from insect cells while the RADX-QVPK protein did not (Figures S6B and S6C, (Adolph et al., 2021)). Thus, RADX purified from untreated and HU-treated cells have similar biochemical activities. Furthermore, the ability of RAD51 overexpression to rescue the phenotypic effect of RADX overexpression in HU-treated cells (Figure S5A) also suggests it remains a RAD51 antagonist in these conditions.
To directly test whether RADX could promote fork reversal by decreasing the stability of RAD51 filaments, we examined the ability of SMARCAL1 to catalyze fork reversal on model replication fork substrates in the presence of RAD51, RPA and RADX (Figure 7A). Individually, MBP-RADX or RAD51 inhibited fork reversal when added before SMARCAL1 (Figures 7B and 7C) but as previously reported, RPA stimulates SMARCAL1 (Figure 7B; (Betous et al., 2012). As expected, addition of RPA, RAD51 and MBP-RADX together in the absence of SMARCAL1 did not catalyze fork reversal (Figure 7D). Moreover, adding RPA by itself after RAD51 did not improve SMARCAL1-mediated reversal (Figure 7E). In contrast, adding increasing amounts of MBP-RADX in the presence of RPA to a fork substrate with pre-bound RAD51 stimulated fork reversal by SMARCAL1 (Figures 7E). Importantly, adding the MBP-RADX QVPK mutant that cannot bind RAD51 or destabilize RAD51 filaments did not stimulate reversal (Figure 7F). These data suggest that RADX can promote fork reversal in at least some conditions by destabilizing RAD51.
Figure 7. RADX promotes fork reversal by destabilizing RAD51 filaments in vitro.

(A) Schematic of the fork reversal assay. (B). Addition of RPA stimulates SMARCAL1- dependent fork reversal, while addition of RAD51 inhibits fork reversal. (C) Increasing concentrations of RADX by itself inhibits fork reversal by SMARCAL1. (D). Addition of RPA, RADX, and RAD51 do not catalyze fork reversal in the absence of SMARCAL1. (E) Addition of increasing concentrations of RADX in the presence of RPA overcomes the fork reversal inhibition caused by RAD51. Top, quantifications (n=4; mean+/−SEM). Bottom, representative experiment. (F) Unlike wild type RADX, RADX QVPK does not stimulate fork reversal in the presence of RAD51, RPA, and SMARCAL1. Top, quantifications (n=3; mean+/−SEM). A representative experiment is shown. A repeated measures one-way ANOVA with Sidaks multiple comparisons test was used to calculate p values. The line in blots indicate lanes were removed.
Discussion
Our findings indicate that RADX regulates replication fork reversal in a context-dependent manner. In the absence of added replication stress, RADX inhibits fork reversal to prevent fork slowing and fork breakage. However, in the presence of added replication stress, RADX helps to generate reversed forks. Therefore, RADX inactivation blocks nascent strand degradation and telomere dysfunction in fork protection-deficient or RTEL1-deficient cells experiencing persistent replication stress since reversed forks are the substrates for the nuclease processing in these circumstances (Kolinjivadi et al., 2017; Lemacon et al., 2017; Margalef et al., 2018; Mijic et al., 2017; Taglialatela et al., 2017). Both RADX-dependent inhibition of fork reversal in the absence of stress and generation of reversed forks in the presence of stress depend on its ability to interact directly with RAD51 and ssDNA. Based on these findings and our observation that RADX can stimulate fork reversal in vitro in conditions where RAD51 binding a fork substrate is inhibitory, we conclude that RADX likely confines fork reversal to persistently stalled forks by destabilizing RAD51 nucleofilaments. We suggest that the difference in outcome at elongating versus stalled forks is due to the difference in amount of ssDNA.
Our new data also explains the discrepancies in the literature on the stability of nascent DNA when RADX is inactivated. We reported that silencing RADX with RNA interference or knocking out RADX with CRISPR-Cas9 caused fork slowing but did not yield nascent strand degradation in the presence of persistent replication stress (Bhat et al., 2018; Dungrawala et al., 2017). Yet, Schubert et al., reported that RADX siRNA caused nascent strand degradation in HU-treated cells (Schubert et al., 2017). We now find that inefficient RADX silencing causes nascent strand degradation indicating the differences in published results were due to how efficiently RADX was inactivated. In this respect, RADX phenocopies RAD51 since partial silencing of either causes nascent strand degradation but efficient inactivation yields stable forks in HU-treated cells.
Six observations are consistent with the conclusion that RADX prevents fork reversal in the absence of replication stress: 1) RADX is present at actively elongating replication forks placing it where it would need to operate to prevent RAD51-dependent reversal (Dungrawala et al., 2017); 2) RADX inactivation causes an increased abundance of RAD51 at replication forks (Dungrawala et al., 2017); 3) RADX inactivation slows replication elongation and increases fork collapse; 4) both fork slowing and fork collapse in the absence of RADX can be rescued by inactivating the fork reversal enzymes HLTF, ZRANB3, or SMARCAL1 or by silencing RAD51; 5) replication elongation rates in RADX-deficient cells are also rescued by addition of PARP inhibitors which inhibit fork reversal; and 6) direct visualization of replication intermediates by EM shows an accumulation of reversed forks in RADX-deficient cells that are not treated with any replication stress agent.
Six additional observations support the conclusion that RADX helps to generate reversed forks in the presence of replication stress: 1) RADX accumulates at stalled replication forks where it would need to operate to promote RAD51-dependent reversal (Dungrawala et al., 2017; Schubert et al., 2017); 2) RADX silencing or deletion prevents nascent strand degradation when fork protection factors including BRCA2, BRCA1, BOD1L, ABRO1, Fanconi anemia proteins, or DCAF14 are inactivated similar to the rescue observed by inactivating fork reversal enzymes (Bhat et al., 2018; Townsend et al., 2021); 3) the rescue of fork protection in these circumstances is not due to restoration of RAD51 localization to stalled replication forks; 4) RADX inactivation prevents fork reversal-dependent telomere catastrophe in RTEL1-deficient cells treated with aphidicolin to elevate replication stress levels; 5) RADX inactivation prevents reversal-dependent fork breakage that is observed in BRCA2-deficient cells; 6) the percentage of reversed forks observed by EM in HU-treated cells is decreased when RADX is inactivated.
We envision two models to explain the apparent difference in RADX functions in the absence or presence of replication stress (Figure S6). First, RADX could switch functions from a RAD51 antagonist to a RAD51 activator in response to replication stress. A post-translational modification or change in protein binding partners could mediate this change. Second, RADX could retain the same biochemical activity (reducing RAD51 nucleofilament stability) in both stressed and unstressed cells; however, the reduction in RAD51 nucleofilament stability induced by RADX generates the metastable filaments needed for fork reversal in stressed cells, while it prevents inappropriate RAD51 access to elongating forks in unstressed conditions. In this second model, other changes in the replication fork proteome or DNA structures when cells are treated with HU would be needed to yield the apparent difference in outcome. We hypothesize that the difference may be the amount of ssDNA at active versus stalled forks. When there is limited ssDNA for short times, such as what would be present on the lagging template strand at active replication forks or what would be generated by stochastic polymerase speed changes (Graham et al., 2017), RADX-destabilization could prevent RAD51 from accessing the forks to promote reversal. However, when there is persistent fork stalling yielding more ssDNA, then destabilization of RAD51 filaments that form on the template DNA strands could be needed to allow the fork reversal enzymes to reanneal the parental DNA and catalyze fork reversal. In other words, accumulation of RADX at stalled forks could help the reannealing of the parental DNA strands by making the RAD51 nucleofilament more dynamic so it does not act as a road-block to reversal and possibly to allow RPA to remain present where it can stimulate SMARCAL1 (Figure S7).
While both models could explain the apparent switch in RADX function, our data is more consistent with the second model for several reasons. First, RADX function in both stressed and unstressed cells requires a direct interaction with RAD51 and ssDNA. Second, loss of RADX increases RAD51 chromatin localization both in the absence and presence of stress. Third, RADX purified from unstressed or HU-treated cells inhibits RAD51-mediated strand exchange and D-loop activity. Fourth, overexpressing RAD51 overcomes the fork destabilization caused by overexpressing RADX suggesting they retain an antagonistic relationship in the presence of HU. Fifth, RAD51 added to fork reversal assays can inhibit SMARCAL1 presumably by acting as a steric block to enzyme translocation. Adding RADX to these reactions (but not RADX that cannot interact with RAD51) overcomes the RAD51-dependent inhibition when RPA is also present. The same purified RADX protein inhibits RAD51-dependent strand exchange and destabilizes RAD51 nucleofilaments (Adolph et al., 2021). Finally, a “less is more” model for RAD51 activity has precedence in the bacterial literature where the N. gonorrhoeae RecX protein biochemically inhibits N. gonorrhoeae RecA, but actually promotes its function in vivo (Gruenig et al., 2010). Although RADX has no sequence similarity to RecX, the RecA and RAD51 proteins are orthologs that likely require similar regulatory mechanisms. Taken together, we conclude that the RADX may promote fork reversal by making the RAD51 filament metastable, which was previously proposed to be required to generate reversed forks (Berti et al., 2020a).
Inactivating RADX in HU-treated cells reduces the percentage of reversed forks visualized by EM, but it does not fully prevent reversal suggesting either it is only important at a subset of stalled forks or it reduces the time that all stalled forks reside in the reversed state. Fork reversal is a dynamic process with reversed forks being “reset” by fork restoration reactions catalyzed by RECQ1 or by the limited action of nucleases like DNA2 (Berti et al., 2013; Hu et al., 2012; Thangavel et al., 2015). The fork reversal enzymes like SMARCAL1, can also catalyze fork restoration reactions (Betous et al., 2013). Moreover, other fork protection proteins like RAD52 can prevent excessive RAD51 function and engagement of translocases at replication forks to limit fork reversal upon stalling (Malacaria et al., 2019). The stability of the RAD51 filament and whether it is formed on gaps in the template DNA strands, on the nascent DNA of a reversed replication fork, or even its binding to double strand DNA at the fork will also determine the dynamics of the conversion between a three-way to four-way junction. The persistence of the replication block, amount of ssDNA, recruitment of other RAD51 regulators, and the activity of competing pathways all likely combine to yield the static snapshot of reversed fork frequency that is observed by EM. RADX may change the dynamics of RAD51 at HU-stalled forks enough to shift the equilibrium between the 3- and 4- way junctions towards reversal, but not be absolutely essential explaining why some reversed forks remain visible by EM.
Nonetheless, even though RADX inactivation only partially reduces fork reversal in cells exposed to persistent replication stress, it fully prevents nascent strand degradation in HU-treated cells in which fork protection factors are inactivated. Thus, whatever reversal happens in these circumstances is not enough to yield extensive nascent strand degradation. RADX, like RAD51, may be acting in two steps of the fork protection pathway at persistently stalled forks – promoting fork reversal and destabilizing RAD51 filaments on the reversed fork making it more susceptible to nucleases. Finally, nascent strand degradation may require multiple cycles of reversal and degradation. If RADX inactivation reduces this dynamic cycling, then that would also explain why fork degradation is completely dependent on RADX but fork reversal is only partially-dependent on RADX.
In summary, RADX antagonizes RAD51 in unstressed cells to prevent aberrant fork reversal. However, in stressed cells with persistently stalled forks, RADX interacts with RAD51 to promote reversal. In both cases, RADX may operate by destabilizing RAD51 filaments. By confining fork reversal to stalled forks, RADX prevents aberrant processing by nucleases that can generate genome instability.
Limitations of the study
While we used more than one cell line in many experiments, it is possible that the some of the effects we observe by inactivating RADX could be cell type dependent. The in vitro fork reversal assay results are dependent on protein and DNA concentrations. Furthermore, they do not fully mimic the reaction in vivo since only a few proteins are added to simple DNA substrates in these experiments. Recombinant RADX purified from unstressed and HU-treated cells may not retain the appropriate post-translational modifications or the biochemical assays may fail to contain the needed components to visualize a shift in function. Furthermore, RPA and RAD51 are also post-translationally modified in response to replication stress, and the in vitro reactions do not capture the effects of those modifications. Additional biochemical assays using different DNA substrates and additional proteins including other RAD51 regulators will be needed to fully understand how RADX acts.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Cortez (david.cortez@vanderbilt.edu)
Materials availability
No new plasmids or cell lines were generated in this study. All plasmids and cell lines generated in the Cortez laboratory are available upon request. Any plasmids or cell lines obtained from other sources should be requested from those investigators.
Data and code availability
Unprocessed blots, gels, and microscopy images are available on Mendeley Data at http://dx.doi.org/10.17632/pvrw8psnsj.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
U2OS (female) and HEK293T (female) were cultured in DMEM with 7.5% Fetal Bovine Serum (FBS). hTERT-RPE-1 (female) cells were cultured in DMEM F12 with 7.5% FBS and 7.5% Sodium bicarbonate. HeLa1.3 (long telomere) was cultured in DMEM with 10% FBS. DLD-1 (male) was cultured in RPMI1640 with 10% FBS. U2OS RADXΔ, SMARCAL1Δ, ZRANB3Δ, HLTFΔ were generated as described previously (Dungrawala et al., 2017; Liu et al., 2020). U2OS, HEK293T, and hTERT-RPE-1 were purchased from ATCC, tested for mycoplasma, and authentication verified using short tandem repeat profiling. HeLa 1.3 (female) was a gift from Dr. Titia de Lange (Rockefeller University). DLD-1 wild type and DLD-1 BRCA2 knockouts were a gift from Dr. Douglas K. Bishop (University of Chicago). The RADXΔ cells expressing wild-type RADX or RADX QVPK were described previously (Adolph et al., 2021). Plasmid transfections were performed with polyethylenimine for 293T, FugeneHD for U2OS cells and siRNA transfections were performed according to manufacturer’s instructions with Dharmafect 1 (Dharmacon) for U2OS, 293T, and DLD-1 cells, or RNAimax (Thermofisher) for hTERT-RPE-1 and HeLa1.3 cells.
METHOD DETAILS
DNA fiber analysis and DNA combing
Cells were incubated with 20μM CldU and 100μM IdU as indicated in the figures. DNA fiber analysis in which cells were lysed on a glass slide and gravity was used to spread DNA fibers was used in all fork protection experiments and was carried out as described previously (Couch et al., 2013). Unless otherwise indicated, fork speed measurements were made after combing purified genomic DNA on glass coverslips using the Fibercomb Molecular Combing instrument from Genomic Vision. Stained slides and coverslips were imaged using a 40X oil objective (Nikon Eclipse Ti) and lengths measured manually using NIS-Elements software by an investigator blinded to sample identity.
Telomere PNA FISH
Telomeric Peptide Nucleic Acid Fluorescence In situ Hybridization (PNA FISH) was essentially carried out as described previously (Celli and de Lange, 2005). Briefly, cells were treated with 0.1μg/ml colcemid for 2 hours before collecting. Harvested cells were incubated for 20min at 37°C with 75mM KCl to allow the cells to swell, fixed with cold 3:1 methanol:glacial acetic acid, and dropped onto glass slides. Cells were then rehydrated in PBS, fixed with 4% formaldehyde, treated with 1mg/ml pepsin at 37°C, dehydrated sequentially in 70%, 95%, and 100% ethanol, and air dried. Slides were then hybridized with Cy3-TelC (Cy3-CCCTAACCCTAACCCTAA-3’) in hybridization mix (10 mM Tris-HCl pH 7.2, 70% deionized formamide, 0.5% blocking reagent [Roche 11096176001]), washed twice with hybridization wash buffer I (70% formamide, 10 mM Tris-HCl pH 7.2, 0.1% BSA), and washed three times in hybridization wash buffer II (100mM Tris-HCl pH 7.2, 150 mM NaCl, 0.08% Tween-20) with DAPI added to the second wash. Slides were dehydrated with an ethanol series, air dried, and mounted with Prolong Gold. Images were obtained using a 40X oil objective (Nikon Eclipse Ti). For aphidicolin treatment, 0.2μM aphidicolin was added to cells for 40h prior to harvesting.
Electron microscopy
For EM analysis of replication intermediates, 5–10 ×106 cells transfected with the indicated siRNAs were collected immediately after treatment with 3 mM hydroxyurea for 5 hours. Untreated cells were also included. DNA was cross-linked by incubating with 10 μg/mL 4,5’,8-trimethylpsoralen followed by a 3-minute exposure to 366 nm UV light on a precooled metal block, for a total of three rounds. Cells were lysed and genomic DNA was isolated from the nuclei by proteinase K digestion and chloroform-isoamyl alcohol extraction. Genomic DNA was purified by isopropanol precipitation and digested with PvuII HF with the appropriate buffer for 4 hours at 37°C. Replication intermediates were enriched on a benzoylated naphthoylated DEAE-cellulose (Sigma-Aldrich) column. Samples were prepared for visualization by EM by spreading the purified, concentrated DNA on a carbon-coated grid in the presence of benzyl-dimethyl-alkylammonium chloride, followed by platinum rotary shadowing. Images were obtained on a JEOL JEM-1400 electron microscope using a bottom mounted AMT XR401 camera. Analysis was performed using ImageJ software (National Institute of Health). EM analysis allows distinguishing duplex DNA—which is expected to appear as a 10 nm thick fiber after the platinum/carbon coating step necessary for EM visualization—from ssDNA, which has a reduced thickness of 5–7 nm. Criteria used for the assignment of a three-way junction, indicative of a replication fork, include the joining of three DNA fibers into a single junction, with two symmetrical daughter strands and single parental strand. Reversed replication forks consist of four DNA fibers joined at a single junction, consisting of two symmetrical daughter strands, one parental strand and the addition of a typically shorter fourth strand, representative of the reversed arm. The length of the two daughter strands corresponding to the newly replicated duplex should be equal (b=c), whereas the length of the parental arm and the regressed arm can vary (a ≠ b = c ≠ d). Conversely, canonical Holliday junction structures will be characterized by arms of equal length (a = b, c = d). Particular attention is paid to the junction of the reversed replication fork in order to observe the presence of a bubble structure, indicating that the junction is opened up and that it is simply not the result of the occasional crossover of two DNA molecules. These four-way junctions of reversed replication forks may also be collapsed and other indicators such as daughter strand symmetry, presence of single-stranded DNA at the junction or the entire structure itself, all are considered during analysis (Neelsen et al., 2014). The frequency of reversed forks in a sample is computed using the Prism software.
Neutral Comet assay
Trevigen comet assay kit was used to detect DSBs. Tail moment was scored using Opencomet plugin in to ImageJ software.
Immunofluorescence
U2OS cells plated on 96-well plates were treated with 10μM EdU followed by treatment with or without HU. Cells were then pre-extracted with 0.1% Triton X-100 in PBS (PBS-T) prior to fixing with paraformaldehyde/sucrose. Cells were blocked with 10% goat serum in PBS-T and stained with anti-RAD51 antibody. EdU was detected using click chemistry with an Alexa Fluor 488- conjugated azide. Images were acquired in an unbiased manner using ImageXpress Micro (Molecular Devices). Automated intensity analyses were performed using the MetaXpress software.
Cell lysis and immunoblotting
Whole cell lysates were generated using RIPA lysis buffer (50mM Tris-Cl pH 7.4, 0.1% NP-40, 150mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate) containing 1mM sodium vanadate, cOmplete protease inhibitor cocktail (Roche), 1mM PMSF, 1mM DTT, 25 units of Pierce Universal Nuclease, and 1mM MgCl2. Bio-Rad stain-free gels were used to obtain total protein loading controls.
Protein Purification
His-MBP-RADX, was purified from baculovirus infected Sf9 cells. Briefly, cells were lysed in buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 10 mM NaF, 10 mM sodium phosphate monobasic, 10 mM sodium pyrophosphate, 1% Triton X-100, 10% glycerol, 1 mM DTT, and a cOmplete protease inhibitor cocktail tablet (Roche). After high-speed centrifugation, the cleared lysates were incubated with Talon metal affinity resin for 2 hours at 4 °C. The beads were washed once in buffer containing 1% Triton X-100, 500 mM NaCl in PBS and then washed in buffer containing 50 mM Tris (pH 8.0), 300 mM NaCl and increasing amounts of imidazole (5–20 mM). The bound proteins were eluted in 50 mM Tris (pH 8.0), 300 mM NaCl, 10% glycerol, and 1 mM DTT and 200 mM imidazole. The fractions containing protein were then subjected to size exclusion chromatography on a Superdex 200 10/300 Increase GL (GE Healthcare) in elution buffer with added protease inhibitors. GFP-FLAG-RADX was purified from 293T cells in the presence of absence of 3 mM hydroxyurea for 5 hours. Cells were lysed in buffer containing 20 mM Tris (pH 8.0), 150 mM NaCl, 0.5% NP40 and a cOmplete protease inhibitor cocktail tablet (Roche). Clarified lysates were incubated with FLAG M2 Magnetic Beads (Sigma) for 2 hours at 4 °C. The beads were washed twice in lysis buffer, twice in lysis buffer containing 0.3mM LiCl and twice in elution buffer containing 50 mM Tris (pH 8.0), 300 mM NaCl, 10% glycerol and 1 mM DTT. The bound proteins were eluted in elution buffer with 0.25 mg/mL Flag peptide for 90 minutes at 4 °C. FLAG-SMARCAL1 was purified from baculovirus infected Sf9 cells as described previously (Bhat et al., 2015). Human RAD51 was expressed in E. coli and purified using a protocol that exploits its affinity for BRCA2 BRC repeat 4 as previously reported (Bhat et al., 2018).
Strand Exchange Assay
The strand exchange assay was performed as described and conditions adapted from (Bugreev and Mazin, 2004). Briefly, ϕX174 circular ssDNA (30 μM) was incubated with RAD51 (7.5 μM) for 10 minutes at 37 °C in buffer containing 20 mM HEPES pH 7.5, 1 mM MgCl2, 1 mM CaCl2, 2 mM ATP, 1 mM DTT, and 100 mM (NH4)2SO4 added at the time of dsDNA addition. Then, RPA was added to the reaction at a final concentration of 2 μM and further incubated for 10 minutes. The reaction was initiated by the addition of 30 μM ϕX174 dsDNA, linearized by ApaL1, and the reaction proceeded for 180 minutes. The addition of RADX (150 nM) is indicated in the reaction scheme. Time points were taken at 0, 30, and 180 minutes by removal of 7 ul of reaction mixture into 3 μl of 0.5% SDS and 0.5 μg/mL proteinase K, and incubated for 20 minutes at 37 °C. After addition of loading dye, the deproteinized samples were loaded onto a 1.0% agarose gel in 1X TAE buffer and electrophoresed at 20V for 16 hr. The products were visualized after one-hour ethidium bromide staining and quantified using ImageLab software (BioRad) Total percent strand exchange was calculated using the integrated intensities of the dsDNA and product bands, and the formula (JM/1.5)+NC/((JM/1.5)+NC)+ dsDNA (Liu et al., 2011).
Displacement Loop Assay
32P-labeled oligonucleotide D1 (3 μM) which is complementary to positions 1932–2022 of pBluescript SK DNA was incubated with RAD51 (1 μM) in buffer containing 25 mM TrisOAc pH 7.5, 20 mM KCl, 1 mM CaCl2, 1 mM ATP and 1 mM DTT for 5 min at 37 °C. RADX was added to the reactions at the same time as the addition of supercoiled pBluescript SK (35 μM base pairs) to initiate the reaction. Reactions were incubated at 37 °C and at the indicated time point (0, 5, 15 min) an aliquot of the reaction was removed and added to SDS (0.5%) and Proteinase K (5 μg/ml) to deproteinize the reactions, followed by a 20 min incubation at 37 °C. All reaction products were resolved on a 1.0% agarose gel, dried and visualized using a phosphorimager (Typhoon FLA 7000, GE Healthcare) and quantified using ImageLab (BioRad).
In vitro fork reversal assay
Fork reversal substrates were prepared as described earlier (Betous et al., 2013), using the oligonucleotides described in Table S2. Briefly, 5’ 32P-labeled nascent lag82 (135 nM final) was annealed with unlabeled parental lag122 (160 nM final) in 1X saline-sodium citrate (SSC) buffer by heating for 3 minutes at 95 °C and gradually cooled down to room temperature overnight. Unlabeled parental lead122 (175 nM final) was then added and annealed in 25mM Tris acetate pH=7.5, 5mM Magnesium acetate, 0.1mg/ml BSA, 2 mM DTT at 75°C for 3 minutes and then cooled down to room temperature for 2 hours. The DNA substrate was then PAGE purified, vacuum concentrated and stored at −20 °C until further use.
1 nM (molecules) DNA substrate was used for fork reversal assays. The assays were carried out in reaction buffer containing 25 mM Tris- HCl pH 7.5, 5 mM MgCl2, 2 mM DTT, 10mM NaCl, 0.1 mg/ml BSA, and 2.5 mM ATP. Master-mixes were prepared on ice and when indicated, RAD51 was added to the master-mix for 5 minutes. Next, reactions were supplemented with other recombinant proteins (as indicated in figure 7) and incubated for 10 minutes at room temperature. Then, SMARCAL1 was added to the reaction and incubated for an additional 20 minutes at 37 °C before the reactions were terminated by addition of 4 μl stop buffer (1mg/ml Proteinase K, 0.2% SDS [w/v], 30% glycerol, 25mg/ml Ficoll) and incubation for 20 minutes at 37 °C. Samples were loaded onto 10% polyacrylamide (19:1 acrylamide:bisacrylamide) gels and separated for 60 minutes at 80 V at room temperature. The gels were dried, imaged, and quantified using a phosphorimager.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were completed using Prism 9. Details of the individual statistical tests are indicated in figure legends and results. No statistical methods were used to estimate sample size or to include/exclude samples. Investigators were blinded to sample identities and all experiments were repeated at least three times unless otherwise indicated.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-RADX | Novus Biologicals | Cat#NBP2-13887; RRID: AB_2687552 |
| Mouse monoclonal anti-RAD51 (14B4) for immunofluorescence | Abcam | Cat# ab213; RRID: AB_302856 |
| Rabbit polyclonal anti-RAD51 | Abcam | Cat# ab63801; RRID: AB_1142428 |
| Rabbit SMARCAL1 | (Bansbach et al., 2009) | N/A |
| Rabbit monoclonal HLTF | Abcam | Cat# ab183042 |
| Mouse monoclonal BRCA2 | Calbiochem | Cat#OP95; RRID: AB_2067762 |
| Mouse monoclonal BRCA1 | Oncogene | Cat#OP92; RRID: AB_2750876 |
| Rabbit polyclonal RTEL1 | Novus Biotechne | Cat#NBP2-22360; RRID: AB_2722642 |
| Mouse monoclonal MUS81 | Abcam | Cat# ab14387; RRID: AB_301167 |
| Rabbit polyclonal Abro1 | Abcam | Cat# ab83860; RRID: AB_1860658 |
| Mouse monoclonal GAPDH | Chemicon/Millipore | Cat# MAB374; RRID: AB_2107445 |
| Mouse monoclonal Ku70 | Abcam | Cat# ab3114; RRID: AB_2219041 |
| Rabbit polyclonal ZRANB3 | Bethyl | Cat# A303-033A; RRID: AB_10773114 |
| Mouse monoclonal anti BrdU | BD | Cat#347580; RRID:AB_10015219 |
| Rat monoclonal anti BrdU | Abcam | Cat#ab6326; RRID:AB_305426 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Life Technologies | Cat# A32723; RRID: AB_2633275 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Life Technologies | Cat# A11007; RRID:AB_10561522 |
| Goat anti-mouse (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 594 | Life Technologies | Cat# A32742 RRID: AB_2762825 |
| Oligonucleotides | ||
| SMARCAL1 siRNA | Dharmacon | Cat# J-013058-08 |
| ZRANB3 siRNA | Ambion Silencer select | Cat# 4392420 |
| HLTF siRNA | Dharmacon (pool) |
Cat# J-006448-06; J-006448-07; J-006448-08; J-006448-09 |
| BRCA2 siRNA | Qiagen | Cat# S102653595 |
| BRCA1 siRNA | Dharmacon | Cat# D-003461-08 |
| RTEL1 siRNA | Dharmacon | Cat# M-013379-00 |
| RAD51 siRNA | Dharmacon | Cat# J-003530-12 |
| MUS81 siRNA | Dharmacon | Cat# J-016143-09; J-016143-10; J-016143-11; J-016143-12 |
| RADX siRNA | Dharmacon | Cat# J-014634-21 Cat# J-014634-20 |
| Non-targeting (NT) siRNA | Qiagen | Cat# 1027281 |
| DNA oligonucleotides for fork reversal assay (Table S2) | IDT | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CldU | Sigma Aldrich | Cat#C6891 |
| IdU | Sigma Aldrich | Cat#l7125 |
| EdU | VICB synthesis core | N/A |
| Alexa Fluor 488 azide | Thermo Fisher | Cat#10266 |
| Hydroxyurea | Sigma Aldrich | Cat#H8627 |
| Aphidicolin | Sigma Aldrich | Cat#A0781 |
| C5 | Medchemexpress | Cat# HY-128729 |
| Mirin | Selleck | Cat# S8096 |
| Olaparib (AZD2281) | Selleck | Cat# S1060 |
| MBP-RADX | (Adolph et al., 2021) | N/A |
| MBP-RADX QVPK | (Adolph et al., 2021) | N/A |
| RAD51 | Provided by Mauro Modesti | N/A |
| RPA | Provided by Walter Chazin | N/A |
| SMARCAL1 | This study | N/A |
| Critical Commercial Assays | ||
| Trevigen comet assay | Trevigen | Cat# 4250-050-ES |
| Coverslips | MicroSurfaces Inc. | N/A |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | Cat#HTB-96; RRID:CVCL_0042 |
| HEK293T | ATCC | Cat#CRL-11268; RRID:CVCL_1926 |
| hTERT-RPE-1 | ATCC | Cat#CRL-4000; RRID: CVCL_4388 |
| HeLa1.3 | Gift from Dr. Titia De Lange | N/A |
| DLD-1 BRCA2Δ and DLD1-WT | Gift from Dr. Douglas Bishop | N/A |
| U2OS ΔRADX | (Dungrawala et al., 2017) | N/A |
| U2OS ΔSMARCAL1 | (Liu et al., 2020) | N/A |
| U2OS ΔZRANB3 | (Liu et al., 2020) | N/A |
| U2OS ΔHLTF | (Liu et al., 2020) | N/A |
| Recombinant DNA | ||
| Plasmid pDC1272 = pLEGFP-FLAG-RADX | (Dungrawala et al., 2017) | N/A |
| Plasmid pMA32 = pLEGFP-FLAG-RADX QVPK | (Adolph et al., 2021) | N/A |
| Plasmid pKB40 = pLEGFP-FLAG-RADX OB2m | (Dungrawala et al., 2017) | N/A |
| Plasmid pCAGGS-RAD51 | (Schlacher et al., 2011) | N/A |
| Software and Algorithms | ||
| GraphPad Prism8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_000306 |
| ImageLab | BioRad |
http://www.bio-rad.com/en-us/sku/1709690-image-lab-software; RRID:SCR_014210 |
| Fiji/ImageJ | ImageJ software |
https://imagej.nih.gov/ij/ RRID:SCR_003070 |
| Nikon Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Deposited Data | ||
| Unprocessed blots, gels and microscopy images | This paper | http://dx.doi.org/10.17632/pvrw8psnsj.1 |
Highlights.
RADX inhibits inappropriate fork reversal in the absence of replication stress
RADX promotes fork reversal in response to persistent replication stress
Silencing RADX rescues fork reversal-dependent instability at stalled forks
RADX interacts with RAD51 and single-strand DNA to regulate fork reversal
Acknowledgements:
We thank Dr. Courtney Lovejoy for suggesting and assisting with the telomere dysfunction assays. We thank Runxiang Zhao and Dr. Wenpeng Liu for creating knockout cells for HLTF, SMARCAL1, and ZRANB3. We thank Dr. Titia de Lange for the long-telomere HeLa cells and Dr. Douglas Bishop for the DLD1 wild-type and BRCA2Δ cells. We thank Dr. Maria Jasin for the RAD51 plasmid. This research was primarily supported by R01GM116616 from the NIH to D.C. Additional support came from NCI grants R01CA237263 and R01CA248526 to A.V, and a Breast Cancer Research Foundation grant to D.C. The EMB Core of P01CA092584 provided insect cells expressing MBP-RADX and Flag-SMARCAL1 proteins used in this study.
Footnotes
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adolph MB, Mohamed TM, Balakrishnan S, Xue C, Morati F, Modesti M, Greene EC, Chazin WJ, and Cortez D. (2021). RADX controls RAD51 filament dynamics to regulate replication fork stability. Mol Cell 81, 1074–1083 e1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach CE, Betous R, Lovejoy CA, Glick GG, and Cortez D. (2009). The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev 23, 2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Cortez D, and Lopes M. (2020a). The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol 21, 633–651. [DOI] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Teloni F, Mijic S, Ursich S, Fuchs J, Palumbieri MD, Krietsch J, Schmid JA, Garcin EB, Gon S, et al. (2020b). Sequential role of RAD51 paralog complexes in replication fork remodeling and restart. Nature communications 11, 3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Couch FB, Mason AC, Eichman BF, Manosas M, and Cortez D. (2013). Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep 3, 1958–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, and Cortez D. (2012). SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes & development 26, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Betous R, and Cortez D. (2015). High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem 290, 4110–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, and Cortez D. (2018). RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol 25, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Krishnamoorthy A, Dungrawala H, Garcin EB, Modesti M, and Cortez D. (2018). RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep 24, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugreev DV, and Mazin AV (2004). Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci U S A 101, 9988–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli GB, and de Lange T. (2005). DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7, 712–718. [DOI] [PubMed] [Google Scholar]
- Cortez D. (2019). Replication-Coupled DNA Repair. Mol Cell 74, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, and Cortez D. (2017). RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol Cell 67, 374–386 e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, et al. (2008). A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JE, Marians KJ, and Kowalczykowski SC (2017). Independent and Stochastic Action of DNA Polymerases in the Replisome. Cell 169, 1201–1213 e1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenig MC, Stohl EA, Chitteni-Pattu S, Seifert HS, and Cox MM (2010). Less is more: Neisseria gonorrhoeae RecX protein stimulates recombination by inhibiting RecA. J Biol Chem 285, 37188–37197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V. (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, et al. (2015). BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol Cell 59, 462–477. [DOI] [PubMed] [Google Scholar]
- Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, Zhang M, Hu Y, Wang Q, Xu W, et al. (2012). The intra-s phase checkpoint targets dna2 to prevent stalled replication forks from reversing. Cell 149, 1221–1232. [DOI] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881.e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sneeden J, and Heyer WD (2011). In vitro assays for DNA pairing and recombination-associated DNA synthesis. Methods in molecular biology 745, 363–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Krishnamoorthy A, Zhao R, and Cortez D. (2020). Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Science Advances 6, eabc3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Zhou M, Li Z, Li H, Polaczek P, Dai H, Wu Q, Liu C, Karanja KK, Popuri V, et al. (2016). A Selective Small Molecule DNA2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. EBioMedicine 6, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malacaria E, Pugliese GM, Honda M, Marabitti V, Aiello FA, Spies M, Franchitto A, and Pichierri P. (2019). Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat Commun 10, 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalef P, Kotsantis P, Borel V, Bellelli R, Panier S, and Boulton SJ (2018). Stabilization of Reversed Replication Forks by Telomerase Drives Telomere Catastrophe. Cell 172, 439–453 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Chaudhuri AR, Follonier C, Herrador R, and Lopes M. (2014). Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol Biol 1094, 177–208. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M. (2011). Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert L, Ho T, Hoffmann S, Haahr P, Guerillon C, and Mailand N. (2017). RADX interacts with single-stranded DNA to promote replication fork stability. EMBO Rep 18, 1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208, 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend A, Lora G, Engel J, Tirado-Class N, and Dungrawala H. (2021). DCAF14 promotes stalled fork stability to maintain genome integrity. Cell Rep 34, 108669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. (2015). A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell 59, 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Wu X, Wu L, Castillo A, Liu J, Atkinson E, Paul A, Su D, Schlacher K, Komatsu Y, et al. (2017). Abro1 maintains genome stability and limits replication stress by protecting replication fork stability. Genes Dev 31, 1469–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M. (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed blots, gels, and microscopy images are available on Mendeley Data at http://dx.doi.org/10.17632/pvrw8psnsj.1