

Summary
The heterogeneous nature of eukaryotic replication kinetics and the low efficiency of individual initiation sites make mapping the location and timing of replication initiation in human cells difficult. To address this challenge, we have developed Optical Replication Mapping (ORM), a high-throughput single-molecule approach, and used it to map early initiation events in human cells. The single-molecule nature of our data, and a total of over 2000-fold coverage of the human genome on 27 million fibers averaging ~300 kb in length, allow us to identify initiation sites and their firing probability with high confidence. We find that the distribution of human replication initiation is consistent with inefficient, stochastic initiation of heterogeneously distributed potential initiation complexes enriched in accessible chromatin. These observations are consistent with stochastic models of initiation-timing regulation and suggest that stochastic regulation of replication kinetics is a fundamental feature of eukaryotic replication, conserved from yeast to humans.
Graphical Abstract
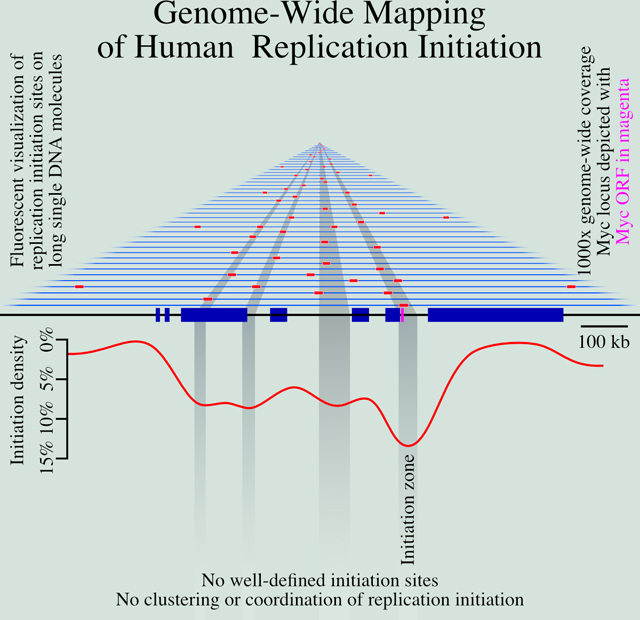
Introduction
The human genome is replicated from approximately 50,000 distinct initiation events in each cell cycle (Masai et al., 2010). However, identifying the location and firing timing of human replication origins is an ongoing challenge (Hyrien, 2015). A major part of this challenge is the inefficient and heterogeneous nature of mammalian origin firing (Berezney et al., 2000; Taylor, 1977). Even in cases where mammalian origins are thought to occur at relatively well-defined sites, they appear to fire with as little as 10% efficiency (Dijkwel et al., 2002); in the more common case where initiation appears to occur within broad initiation zones (IZs), spanning tens of kilobases (Hamlin et al., 2008), they fire with less than 1% efficiency (Demczuk et al., 2012). Therefore, population-based bulk approaches to mapping origins, which average origin-firing behavior across large populations of cells, must contend with low signal-to-noise ratios. As a result, although a number of bulk approaches have been used to map origins in the human genome (Mesner et al., 2013; Besnard et al., 2012; Cayrou et al., 2011; Foulk et al., 2015; Langley et al., 2016; Macheret and Halazonetis, 2018; Dellino et al., 2013; Petryk et al., 2016), there is little concordance among them (Langley et al., 2016; Mesner et al., 2013).
The heterogeneous nature of mammalian origins may be influenced by the non-site-specific nature of the mammalian origin recognition complex (ORC) DNA, which shows only a weak affinity for AT-rich sequences in vitro (Vashee et al., 2003; De Carli et al., 2018) and no discernible sequence preference in vivo (Miotto et al., 2016). G-quadruplex (G4) sequences have been proposed to function as origins (Cayrou et al., 2015; Valton et al., 2014; Prorok et al., 2019), but this hypothesis is controversial (Foulk et al., 2015; Miotto et al., 2016). A simple explanation for the observed patterns of ORC binding and replication initiation is that ORC binds non-specifically to accessible, nucleosome-free DNA (Lubelsky et al., 2011). In particular, ORC binding and replication initiation correlate well with DNase I accessibility across metazoan genomes (Miotto et al., 2016; Gindin et al., 2014b). For these reasons, we will refrain from referring to mammalian sites of replication initiation as origins but rather refer to specific instances of replication initiation as initiation events occurring at initiation sites and to areas enriched in initiation sites as initiation zones.
Although the specific locations of replication initiation are unclear, the general characteristics of mammalian sites of initiation have been described. Initiation is enriched in euchromatic promoter and enhancer regions, consistent with its correlation with accessible chromatin (Ganier et al., 2019; Petryk et al., 2016; Pourkarimi et al., 2016; Cayrou et al., 2015). Although initiation tends to be enriched in euchromatic regions, initiation must also occur in heterochromatin. Initiation in heterochromatin appears to be even more heterogeneous, making heterochromatic initiation sites even less well understood (Petryk et al., 2016; Cayrou et al., 2015).
In budding yeast, where replication origin locations are well defined, origin firing is stochastic, with each origin firing with a specific probability, independent of neighboring origins (Czajkowsky et al., 2008; Yang et al., 2010; de Moura et al., 2010). Such stochastic firing leads to reproducible replication profiles at the population level, because more efficient origins are more likely to fire early and therefore, on average, have early replication times; by contrast, inefficient origins usually fire late or are passively replicated (Rhind et al., 2010). The heterogeneous and inefficient nature of metazoan replication initiation is also consistent with stochastic initiation. Simulations with a stochastic firing model, in which initiation is regulated only by a local-firing-probability function, faithfully reproduce experimental genome-wide replication timing profiles, suggesting that no deterministic timing program is required (Gindin et al., 2014b). Furthermore, if the local initiation rate is predicted by DNase I hypersensitivity, the simulation closely matches experimental results, consistent with the observed correlation between promoters and enhancers, which are DNase I hypersensitive, and initiation frequency (Gindin et al., 2014b). On the other hand, the reproducible replication timing of individual replication domains measured in single cells has led to the suggestion that replication within those domains initiates at defined times in most cells in the population (Dileep and Gilbert, 2018). Furthermore, neighboring initiation sites have been proposed to show both cooperative firing and lateral inhibition (Cayrou et al., 2011; Guilbaud et al., 2011; Löb et al., 2016), neither of which are consistent with strictly stochastic models. Therefore, whether metazoan initiation timing is stochastic or deterministic, or some combination of the two, is still very much an open question (Bechhoefer and Rhind, 2012).
A powerful solution to the problems of heterogeneity and low signal-to-noise ratios is single-molecule analysis, which allows the identification of sites of replication initiation on individual DNA fibers (Técher et al., 2013). Traditional single-molecule approaches—such as fiber autoradiography (Huberman and Riggs, 1968), DNA combing (Herrick and Bensimon, 1999; Anglana et al., 2003; Kaykov et al., 2016) and SMARD (Norio and Schildkraut, 2001)—have provided critical insight into the location and firing kinetics of mammalian replication origins. However, these techniques are restricted to the analysis of at most a few genomic loci. Nanopore-sequencing-based methods offer the potential to map replication genome-wide at high resolution (Müller et al., 2019; Georgieva et al., 2019; Hennion et al., 2020), but the current throughput of nanopore sequencing does not allow genome-wide analysis of metazoan genomes and current average read lengths are on the order of only 30 kb. Optical mapping technologies provide ample throughput on individual long (150 kb to 2 Mb) DNA fibers (Lam et al., 2012) and can be combined with nucleotide analog incorporation to map DNA replication (De Carli et al., 2018). However, the current approach relies on DNA replication in extracts (De Carli et al., 2018), precluding in vivo analysis of replication initiation.
We have developed Optical Replication Mapping (ORM), a single-molecule technique to investigate spatial and temporal distribution of origin firing that combines the Bionano Genomics approach to mapping long individual DNA molecules (Lam et al., 2012) with in vivo fluorescent nucleotide pulse-labeling (Wilson et al., 2016; Panning and Gilbert, 2005) to directly visualize sites of replication initiation within human cells. This approach affords us excellent signal-to-noise characteristics and deep, genome-wide coverage, allowing us to identify initiation sites active in as few as 0.1% of human cells. We have used ORM to identify and analyze human sites of DNA replication initiation. We have further applied ORM to asynchronous human cells to obtain cell-type-specific replication profiles.
Results
Optical Replication Mapping of Early-Firing Human Initiation Sites
We mapped sites of early replication in the human genome by ORM (Figure 1A,B and Methods). Synchronized HeLa cells were electroporated with fluorescent dUTP and released from an aphidicolin block, allowed to complete replication, and harvested for analysis on the Bionano Saphyr platform. As a result, cells incorporate fluorescent nucleotides in replication forks that initiated in early S phase. However, the labeled nucleotides are rapidly depleted, preventing incorporation at later initiation sites (Wilson et al., 2016)
Figure 1. Optical Replication Mapping of Synchronized Cells Identifies Early Initiation Events.

HeLa S3 cells were synchronized by mitotic shake off, arrested in early S-phase with aphidicolin, electroporated with fluorescent dUTP and released from the aphidicolin arrest, allowing fluorescent nucleotide incorporation around early replication initiation events.
A) A representative image of the Bionano data with DNA in blue, Nt.BspQI restriction sites in green and incorporated nucleotides in red. The field of view is 750 kb.
B) Enrichment of ORM signals from the combined 0-minute dataset around early replication timing peaks, those that replicate in the first quarter of S phase.
C) Enrichment around early replication timing peaks of ORM signals from datasets in which cells were labeled between 5 and 90 minutes after release from synchronization.
D, E) Distribution of replication tracks at two previously characterized human replication IZs: Myc (C) and Top1(D). The distribution of replication tracks in shown beneath the HeLa S3 replication timing profile (Hansen et al., 2010) and genes from the region. The distribution of total fibers in shown in grey at 1% the scale of the replication tracks. Discontinuities in the fiber density distribution, such as the one around megabase 127.5 in D, are probably caused by segmental duplication increasing the copy number of the region (see Figure S1E). Therefore, all analyses are normalized to fiber coverage depth.
F) Distribution of replication tracks across the right arm of Chromosome 20, as in D.
G) Distribution of replication timing of replication tracks compared to replication timing of each 50 kb bin in the genome.
N = 4 biological and 11 technical replicates for the 0 minute data and 1 technical replicate for subsequent timepoints.
We collected a dataset of 728 Gb across 2.9 million fibers ranging in length from 150 kb to 1.2 Mb, with an average fiber length of 250 kb and an average genome coverage of 206 fold (Table S1 and Figure S1). On these fibers, we identified fluorescent incorporation signals and mapped them across the genome (Figure 1A, see Methods for details). Technical replicates within our first dataset and three additional biological replicates all show high reproducibility, with the variation dominated by counting noise in infrequently labeled regions of the genome (r = 0.81–0.86, Figures S1E,F). Therefore, we combined the four experiments into one dataset of 11.8 million fibers ranging from 150 kb to 2.2 Mb in length, averaging 285 kb in length and constituting 1,109-fold coverage of the human genome (Table S1). The coverage is uniform, with a coefficient of variation of 12% (Figure S1G).
The fluorescent signals are enriched in replication IZs, as identified in HeLa cells by replication timing peaks (Figure 1B). To test whether the incorporation signals truly reflected initiation events, we modified our protocol to allow for a delay, ranging from 5’ to 90’, after aphidicolin release and before transfection (Table S1). When we aggregate replication signals around early IZs, we see a time-dependent movement of incorporation tracks away from the IZs at an estimated rate of 1.65±0.31 kb per minute (Figure 1C), consistent with prior estimates of mammalian replication fork rates (Jackson and Pombo, 1998; Conti et al., 2007; Chagin et al., 2016). This result is consistent with the signal in our 0’ data representing initiation events and the signals after various release times representing forks moving away from such initiation events.
To facilitate visualization of labeled replication forks, we segmented the fluorescent-nucleotide incorporation signal into discrete replication tracks, identifying replication forks active during our labeling period (see Methods). For fibers on which we identify more than one track, the average distance between track centers is 111±78 kb (Figure S2), consistent with previous measurements (Cayrou et al., 2011; Jackson and Pombo, 1998). We find enrichment of replication tracks around previously identified sites of human replication initiation (Figure 1D,E). In particular, we examined the distribution of replication tracks around the MYC and Top1 loci, both characterized as early-firing origins in the human genome (Tao et al., 2000; Keller et al., 2002), and found a pronounced enrichment at these loci, particularly in the intergenic regions surrounding these genes. More generally, replication tracks are enriched in the earliest replicating parts of the genome, as defined by replication timing profiling (Hansen et al., 2010), as expected for early-initiation events (Figures 1F,G). Interactive display of the ORM data is available on the ORM Browser <http://orm.nucleome.org> (Figure S3, Zhu et al. in prep, personal communication, J. Ma).
We observe 0.97 million initiation events in our combined 0’ dataset. From the median incorporation track length of 75 kb (estimated from our modeling of label-incorporation kinetics, which compensates for the sparseness of labeling, Figure S2B and Methods), we infer that 73 Gb of DNA is labeled, which is ~2% of the 3.36 Tb in the dataset. Therefore, we are labeling the DNA replicated in the first ~2% of S phase and during that time, about 1000 initiation events occur per genome (977,746 initiation events observed in 1,109 genome equivalents).
Determination of Genome-Wide Replication Kinetics in Asynchronous Cells
The experiments described above provide unprecedented single-molecule mapping of initiation events at the onset of S phase. However, most cell types are not amenable to such precise cell-cycle synchronization. To map replication kinetics in unperturbed cells, we labeled asynchronous HeLa cells and H9 human embryonic stem cells, and mapped replication incorporation tracks by ORM (Figure 2A). We identified 412,113 replication tracks in two biologically-independent HeLa replicates totaling 1.4 Tb of data and 299,595 tracks in one H9 dataset totaling 738 Gb of data (Table S1). Using the same analysis as for our synchronous datasets, we infer a similar nucleotide-labeling frequency of 1/1025 and 1/850 thymidines, respectively. The replication tracks average 23.9±35.5 kb in length in the HeLa data and 27.5±40.4 kb in length in the H9 data, which is comparable to the length of tracks in the synchronized data. Thus, forks released from aphidicolin arrest synthesize at about the same rate as untreated replication forks. The tracks are uniformly distributed across the genome, as predicted for asynchronous replication forks, with an average density of 1.3±0.5%. In particular, in contrast to our synchronous dataset, and as expected, we see no enrichment at replication timing peaks in early- or late-replicating regions (Figure 2B).
Figure 2. Optical Replication Mapping of Asynchronous Cells Identifies Cell-Type-Specific Genome-Wide Replication Kinetics.

A) Asynchronous HeLa S3 and H9 human embryonic stem cells were electroporated with fluorescent dUTP, allowing fluorescent nucleotide incorporation into ongoing replication fork.
B) The density of ORM signals from the HeLa asynchronous dataset around replication timing peaks separated into replication timing quartiles, defined by average replication timing (S50, see Methods).
C) Labeled ORM replication forks, with leftward-moving forks colored blue and rightward-moving forks colored red, are depicted below the replication timing profile of the Top1 region and its OK-seq relative fork direction (RFD) data, with blue dots indicating leftward replication, red dots rightward, and the vertical position of the dot, between −1 and 1, indicating the fraction of replication moving in that direction. The polarity of ORM replication forks was determined by calculating their fork direction index (FDI, see Methods).
D) The RFD calculated from ORM and OK-seq data across 7 Mb of Chromosome 1.
E) The enrichment of HeLa fork polarity, as measured by RFD, around early replication timing peaks. F) The enrichment of HeLa and H9 fork polarity around cell-type specific and cell-type general early replication timing peaks.
N = 2 biological and 3 technical replicates for the HeLa data and 1 technical replicate for the H9 data.
In order to use this data to infer replication kinetics, it is necessary to determine the polarity of each incorporation track, so that the direction of each mapped replication fork is known. To determine track polarity, we took advantage of the fact that, as the labeled nucleotides are consumed, the label density diminishes, a strategy that has been used in other fiber-analysis approaches (Müller et al., 2019; Huberman and Riggs, 1968; Hennion et al., 2020). We calculated a fork direction index (FDI) as the ratio of integrated fluorescent signal in the left half of the track to that in the right half, for all tracks with 3 or more signals.
As expected, ORM replication tracks tend to be oriented in the direction of replication inferred from replication timing profiles and Okazaki fragment mapping (Hansen et al., 2010; Petryk et al., 2016) (Figures 2C,D). Furthermore, genome-wide analysis of replication fork polarity around replication timing peaks shows that the polarity signal in ORM data is comparable to that in published OK-seq data (r = 0.59, Figures 2D,E). Importantly, the polarity signal in ORM data is cell-type specific (Figure 2F). These results show that ORM data can be used to characterize replication kinetics in unsynchronized cells, demonstrating its applicability to any cells that can be pulse labeled with fluorescent nucleotides.
Genome-Wide Mapping of Early-Firing Human Initiation Zones
To map the genome-wide distribution of early-firing human initiation sites, we analyzed the incorporation signal in our combined 0’ dataset and identified 4,930 IZs, defined as peaks of replication signal density (Figure 3A, see Methods for details). The IZs are mostly between 20 and 40 kb, with an average length of 32.3±15.9 kb (Figure S2E), comparable to IZs mapped by OK-seq (Petryk et al., 2016). The initiations zones cover about 5.3% of the genome and contain about 19.8% of the ORM signal. Much of the rest of the ORM signal is directly adjacent to defined IZs, but much is distributed elsewhere at lower density, particularly in late-replicating regions on the genome (Figure 3A). Plotting the initiation efficiency of 50 kb genomic bins show that there is a continuous distribution of initiation efficiency between the higher-efficiency regions we define as IZs and lower-efficiency regions, with no obvious break between them (Figure S2F).
Figure 3. Mapping of Human Replication Initiation Zones.

A) A comparison of ORM tracks, ORM IZs, OK-seq RFD (as in Figure 2C), OK-seq IZs (Petryk et al., 2016), SNS-seq (Picard et al., 2014), Ini-seq (Langley et al., 2016) and Orc1 ChIP-seq (Dellino et al., 2013) at the Top1 locus.
B) The correlation between ORM initiation efficiency (fraction of labeled fibers/IZ) in the combined 0-minute dataset and replication timing (S50) in the 4,930 ORM IZs. Spearman rank correlation coefficient between the two datasets is 0.75. Inset is the same data on a semi-log plot fit with an exponential curve.
C) A Venn diagram of the overlap between OK-seq early IZs and 50 kb windows in the genome that have ORM initiation efficiencies of at least 3% or 10%.
D) The estimated minimal number of initiation sites per IZ. The estimate was made by calculating minimum number of replication tracks in each IZ whose centers are more than 15 kb apart. Most of the IZs for which all track centers are within 15 kb of each other are small and contain few replication tracks (Figure S4B).
E) The distribution of the inter-initiation distances in the combined 0’ dataset measured as the distance from the middle of neighboring replications tracks for fibers that have multiple tracks. The average of the distribution is 111 kb and the mode of distribution is 57 kb. Inset: The distribution, plotted on a log y-axis from 60 to 600 kb, fit to an exponential curve (r = 0.99). The exponential distribution of the inter-replication-track distances indicates that the distribution of initiation events on this length scale is random.
F) The correlation between the probability of replication at neighboring IZs. The data is well fit by a model that assumes no correlation between IZs (see Method S1).We also calculated the correlation coefficient among all IZs on each fiber, not just the neighboring ones, and found a coefficient similar to that of neighboring IZs separated by more than 200 kb, consistent with the interpretation that the low level of residual correlation seen at longer distances is a global effect due to the heterogeneity in cellular label uptake.
G) A matrix depicting the correlation between initiation sites predicted by ORM, replication timing (Chen et al., 2010), OK-seq (Petryk et al., 2016), SNS-seq (Picard et al., 2014), Ini-seq (Langley et al., 2016) Orc1 ChIP-seq (Dellino et al., 2013), DNase I hypersensitivity (Bernstein et al., 2012) and G4 motifs (Puig Lombardi et al., 2019).
N = 4 biological replicates and 11 technical replicates for all ORM data. See also Figures S4 and S5.
The median genome-wide inter-initiation-zone distance is 273 kb. However, since our dataset is biased towards early replicating IZs, we suspect that we underestimate the number of IZs in late-replicating parts of the genome. Consistent with that contention, limiting our analysis to the earliest quarter of the genome leads to a median inter-initiation-zone distance of 168 kb. The inter-initiation-zone distances in the other three quarters of the genome are 257, 258 and 337 kb, respectively. We suspect that the lower density of observed IZs in the later-replicating regions of the genome results from having less data rather than fewer initiation events in these regions (Figure 1G).
A major advantage of ORM is that it records the number of fibers that both contain and do not contain replication tracks, allowing for the calculation of the absolute efficiency with which replication initiates at each locus in the genome. Although such an analysis is possible with prior methods, it is considerably more difficult and thus rarely reported (Demczuk et al., 2012). Such an analysis shows that early initiation is quite heterogeneous. Examining the IZs with average replication times in the first quarter of S phase (S50 < 25%), we find that they initiate replication, on average, on only 7.5% of molecules (Figure 3B). Even the 5% earliest IZs are active on only 11% of molecules. We therefore conclude that, in aphidicolin arrested cells, replication initiates in only a stochastically-selected subset of IZs, including only 11% of the most efficient early IZs.
We investigated the frequency of early initiation of replication across the genome, examining all 4,930 ORM IZs. We find that the local efficiency of initiation in these zones is generally below 15%, with occasional efficiencies up to 20% and a median efficiency of 3.7% (Figure 3B). All of the IZs with efficiencies of more than 10%, and 99% of the IZs with efficiencies of more than the median of 3.7%, are in early-replicating regions of the genome (Figure 3B), consistent with the results in Figure 1G. We also find a strong concordance between the IZs identified by ORM and those identified by OK-seq (Petryk et al., 2016). In particular, 68% of the 6,166 early IZs identified by OK-seq show at least 3% initiation efficiency by ORM and 100% of loci that show greater than 10% efficiency by ORM are identified by OK-seq as IZs (Figure 3C).
Visual inspection of the distribution of replication tracks suggests that initiation in human cells is not concentrated at discrete, frequently-used origins. Instead, initiation appears to be distributed across broad IZs, as has been observed at specific loci and in other genome-wide analyses (Petryk et al., 2016; Mesner et al., 2013; Besnard et al., 2012; Hamlin et al., 2008; Dijkwel et al., 2002; Demczuk et al., 2012; Lubelsky et al., 2011; Tao et al., 2000). Even the IZ at the Top1 locus appears to have initiation events distributed over tens of kilobases (Figures 1E, S4A). However, given the sparse labeling of our replication tracks, we cannot identify initiation sites with resolution higher than about 15 kb. Nonetheless, 93.5% of IZs contain at least two incorporation tracks with centers that are more than 15 kb apart, suggesting that these IZs contain at least two, and possibly many more, initiation sites (Figures 3D and S4A,B). Further, the 6.5% of IZs that do not have obviously non-overlapping replication initiation sites tend to be smaller (16 kb v. 33 kb), less efficient (1.2% v. 4.5%) and later replicating (S50 0.55 v. 0.32) than those that do (Figure S4B). Furthermore, a reanalysis of 66 potentially discrete initiation site identified by OK-seq (Petryk et al., 2016) is consistent with the conclusion that there are no isolated, discrete initiation sites in the human genome (Figure S5).
Another way to distinguish between initiations that occur at a single site and those that are distributed across a broad region is to examine the distribution of ORM signals across the IZs, the expectation being that a single initiation site will have narrower distribution than distributed initiation sites. We measured the signal at the edges of IZs as a fraction of the signal at the middle of the IZ and fit to that data two models, one based on a single initiation site and another based on a uniform distribution of initiation sites across the IZ (Figure S4C). For IZs less that 55 kb long, which includes over 90% of IZs, the distribution signal is consistent with a uniform distribution of initiation sites throughout the IZs. Taking all of our data together, we find no evidence of isolated or high-efficiency initiation sites in the human genome.
Initiation Events in Neighboring Initiation Zones are not Clustered
Regardless of how initiation events are locally distributed, it is important to know whether local initiation events are coordinated. With our deep, single-molecule data on long fibers, we can quantitatively test whether initiation events in neighboring IZs are correlated. A simple, unbiased test for correlation between events is to determine whether the spacing between the event deviates from an exponential distribution. Items distributed randomly on a line have an exponential distribution of inter-item distances (Birnbaum, 1954); therefore, correlated clusters of initiation events would deviate from an exponential distribution of inter-initiation distances. We measured 18,275 inter-initiation distances on 13,769 fibers containing multiple initiation events (Figure 3E). We measure few initiation events less than 60 kb apart, due to the facts that the tracks themselves average 40 kb in length and that early IZs have a median distance of 168 kb. However, above 60 kb, there is an exponential distribution of inter-initiation distances (r = 0.99, Figure 3E, inset). This distribution indicates that, on this length scale, initiation events are distributed randomly.
As an orthogonal test of whether initiation events in neighboring IZs are positively or negatively correlated, we compared the frequency of labeling at each IZ with the frequency with which its neighboring IZs were labeled on the same fiber (Figure 3F). Neighboring IZs can be co-labeled as a result of individual initiation in each zone or of initiation in one zone leading to passive incorporation of label in a neighboring zone by the same replication fork. Correlation can also arise because of heterogeneous uptake of labeled nucleotides. Basically, if one cell has a very high level of nucleotide uptake, all IZs (and any two neighboring IZs, in particular) will be more likely to incorporate label, making them appear to be correlated (see Methods for a more detailed explanation). Therefore, we fit a model to our initiation-zone co-labeling data that takes into account the previously determined incorporation track length (Figure S2B) and nucleotide uptake heterogeneity (Figure S1C), and assumes that individual initiation events are independent. This model fits the data well, suggesting there is no measurable positive or negative correlation between initiation in neighboring zones (Figure 3F).
Comparison of Different Origin Mapping Datasets
We compared our optical replication mapping data to four published genome-wide HeLa replication-initiation-mapping datasets: OK-seq (Petryk et al., 2016), SNS-seq (Picard et al., 2014), Ini-seq (Langley et al., 2016) and Orc1 ChIP-seq (Dellino et al., 2013). By visual inspection, replication tracks appear to be enriched around the IZs identified by OK-seq (Figure 3A). Across the genome, we see colocalization of OK-seq IZs and ORM signal. We see similar colocalization of ORM signal with Ini-seq signal, a cell-free initiation mapping approach and, to a lesser extent, Orc1 ChIP-seq peaks and SNS-seq peaks, which maps initiation events by sequencing short nascent strands produced by replication initiation (Figure 3A). To determine whether the apparent co-localization of initiation mapping data is robust, and to quantify its extent, we measured the correlation between the five data sets (Figure 3G). We find that ORM replication tracks correlate well Ini-seq (r = 0.59), to a lesser extent with OK-seq (r = 0.49), and even less well with SNS-seq (r = 0.36) and Orc1 ChIP-seq (r = 0.31). These correlations are further confirmed by ROC analysis (Figure S6A).
Characterization of Initiation Zones Mapped by ORM
To characterize the genomic character and chromatin context of our IZs, we investigated the enrichment of various genomic and epigenomic annotations, all derived from HeLa cells (Figure 4). Comparing our IZs with various chromatin states from the ENCODE project (Bernstein et al., 2012; Ernst and Kellis, 2012), we find that they are enriched in enhancers and low activity intergenic regions and depleted in transcription units and polycomb-repressed chromatin (Figure S7A). Examining specific histone modifications, our IZs are enriched in H2AZ, which has been implicated in ORC binding, and enhancer-specific modifications, such as H3K4me1 and H3K27ac, and they are depleted in, although adjacent to, transcription-elongation-associated modifications, such as H3K79me2 and H3K36me2 (Figures 4 and S7B,D), in agreement with previous studies (Petryk et al., 2016; Long et al., 2020; Pourkarimi et al., 2016). Consistent with the enrichment of initiation events in enhancer regions and their depletion in transcribed regions, IZs are enriched in DNase I hypersensitive sites and depleted in RNA pol II (Figures 4 and S7B,D).
Figure 4. Characterization of Human Initiation Zones.

Enrichment of histone modifications and other genomic features relative to ORM IZs, separated into replication timing quartiles. The upper panels show the genome-normalized relative signal around all IZs. The lower panels show heat maps of the same signal at each IZ. The y-axis scale for the upper panels and the heat-map scale for the lower panels are the same genome-normalized enrichment in relative units.
N = 4 biological replicates and 11 technical replicates for all ORM data. See also Figure S6.
Although metazoan ORC has only a weak affinity for AT-rich sequences in vitro (Vashee et al., 2003; De Carli et al., 2018) and no discernible sequence preference in vivo (Miotto et al., 2016), sequences that form G4 quadruplex structures have been proposed to correlate with initiation sites in vertebrate cells (Cayrou et al., 2015; Langley et al., 2016; Valton et al., 2014; Prorok et al., 2019). We tested whether IZs are enriched in G4 motifs, using a GC-content-adjusted background model to avoid detecting G4 enrichment as a trivial consequence of the GC-rich nature of enhancers. Of the 737,735 G4 sequences computationally identified in the human genome (Puig Lombardi et al., 2019), 68,585 are found in IZs, which is not more than would be expected by chance, given their GC-rich nature (partial correlation r = −0.0513). Moreover, GC-rich regions (with similar GC% as IZs) outside of IZ are no less likely to contain G4 motifs (131,114 out of 225,710, 58.1%) than GC-rich regions within IZs (10,889 out of 18625, 58.4%, Fisher’s Exact Test p = 0.61). Therefore, although we find that IZs are GC rich (Figure S7C), as previously reported (Xu et al., 2012; Cayrou et al., 2015), they appear to contain G4 motifs as a consequence of their GC richness, but do not appear to be enriched for G4 motif DNA sequences, per se.
Early Initiation Events in Late-Replicating Domains
Although 91% of our early initiation events are in early-replicating regions of the genome, 9% occur in late-replicating domains (Figures 1F, 3B). By visual inspection, the replication tracks in late-replicating parts of the genome appear to be in the locally earliest-replicating parts of the late-replicating regions (for instance at 56 and 58 Mb in Figure 1F), suggesting that they reflect the location of the IZs normally used to replicate these regions. To test this suggestion, we calculated the enrichment of replication tracks around IZs identified as replication timing peaks (Chen et al., 2011) grouped into four quartiles, defined by average replication timing (S50). We find significant enrichment of replication tracks IZs in first three quartiles, (but no enrichment in the fourth quartile, which contains only 3% of the genome-wide replication timing peaks), demonstrating that our ORM signal in late-replicating regions is associated with IZs (Figure 5A). Moreover, the signal-to-noise ratios in the first three quartiles is similar (SNR = 3.2–2.8, Figure 5A), demonstrating that, although there is much less signal in the third quartile, it is enriched over background to the same extent as in the earlier quartiles. In contrast, no enrichment is seen in any quartile in asynchronous cells (Figure 2A). These results confirm that the enrichment of early initiation events we see at late-replicating IZs cannot be attributed to contaminating asynchronous background signals and, therefore, presumably results from bona fide early initiation events in IZs that, on average, replicate late in S phase.
Figure 5. Probability of Early Initiation Predicts Replication Kinetics Genome-Wide.
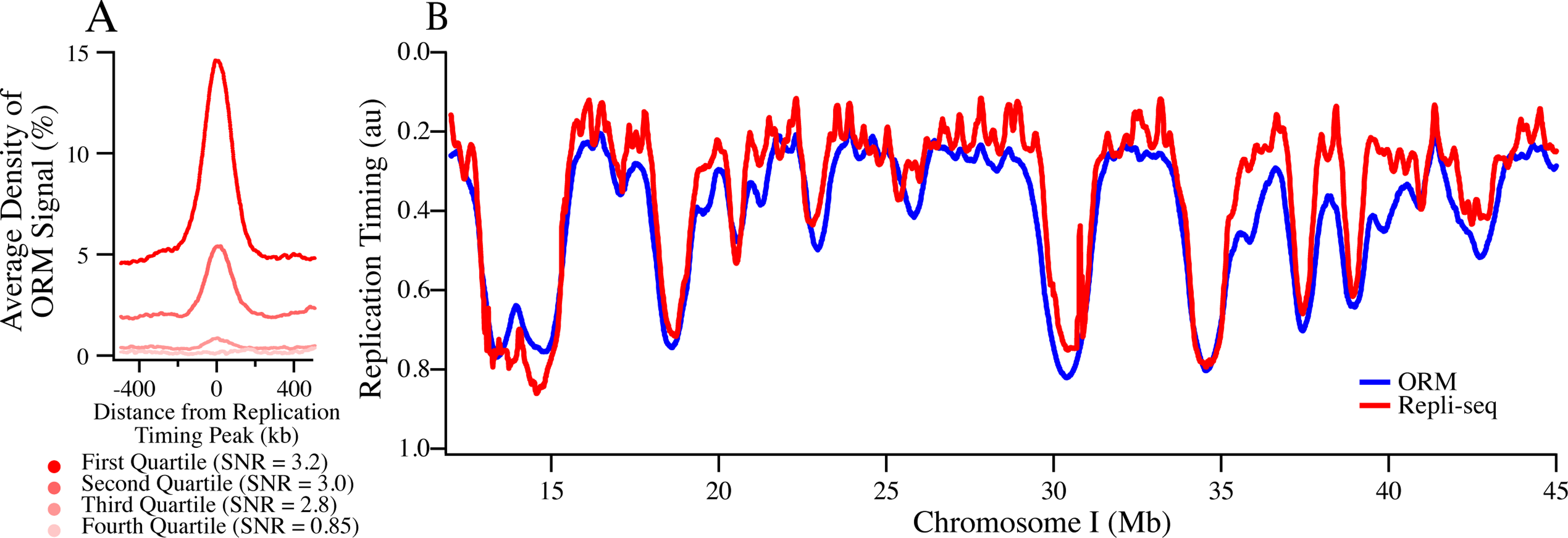
A) Enrichment of ORM signals from the combined 0-minute dataset around replication timing peaks separated into replication timing quartiles. Signal-to-noise ratio (SNR) was calculated as maximum signal over average signal between 400 and 500 kb away.
B) Comparison between experimentally determined replication timing (S50) and replication timing predicted from ORM data using a stochastic model (r = 0.85, Gindin et al., 2014a).
N = 4 biological replicates and 11 technical replicates for all ORM data. See also Figure S6.
Computational Modeling Suggests that Early- and Late-Replicating Initiation Zones are Distinguished Only by Relative Initiation Probability
The distribution of early initiation events we observe—frequent firing in early-replicating IZs and rare firing in late-replicating IZs—is reminiscent of the stochastic regulation of origin-firing timing observed in budding yeast (Czajkowsky et al., 2008; Yang et al., 2010; de Moura et al., 2010). Models of stochastic replication timing posit that the only difference between early- and late-firing origins is their relative probability of firing. More-efficient origins are more likely to fire first and thus, on average, have earlier firing times, but sometimes they still fire late, whereas less-efficient origins are less likely to fire early and thus, on average, have later firing times, but do sometimes fire early (Rhind et al., 2010).
Such models predict that measuring the relative probability of initiation in early-S phase is sufficient to determine the kinetics of initiation for all IZs throughout S phase. If this prediction holds true, we should be able to calculate the complete replication timing profiles of cells, even though we have assayed firing probabilities in only the first 2% of S phase. To test this prediction, we used Replicon, a stochastic replication simulator that calculates replication profiles from the probability of initiation at each point in the genome (Gindin et al., 2014a). We find that we can, indeed, accurately recapitulate genome-wide replication timing profiles with our ORM data (r = 0.85, Figure 5B). Further, our ORM data predicts replication timing better than other initiation data sets, such as, OK-Seq (r = 0.81), Ini-seq (r = 0.69) or SNS-seq (r = 0.48, Figure S6B). ORM predicts replication kinetics equally as well as H3K4me1 (r = 0.85), which marks open enhancer and promoter chromatin, and DNase I hypersensitivity (r = 0.85), which had previously been identified as the best predictor of initiation (Gindin et al., 2014a). These result support the hypothesis that the primary difference between early- and late-replicating IZs is their relative initiation probability, influenced by chromatin accessibility, and not a qualitative difference in their initiation times.
Discussion
We have developed Optical Replication Mapping (ORM), which, for the first time, allows genome-wide, high-throughput, single-molecule replication mapping in human cells. The approach is based on in vivo labeling of replication forks and in vitro detection and genomic mapping of incorporated label on long, single DNA fibers using the Bionano Saphyr platform. Single flow cells using the current mapping approach yielded an average 150-fold coverage of the human genome on fibers from 150 kb to 2 Mb, with an average length of 300 kb (Table S1). Altogether, we have collected 2,550-fold coverage with an average fiber length of 284 kb. Our method is similar to the recently described HOMARD approach, which was used to map DNA replication in Xenopus egg extracts (De Carli et al., 2018).
Direct, single molecule, measurements of initiation frequency
Detection of DNA synthesis on individual DNA fibers is the most direct way to localize sites of DNA replication. Since the preponderance of the literature describes low-frequency, heterogeneous selection of replication initiation sites in mammalian cells, single-molecule measurements may be the only means to accurately map the relative frequencies of initiation events in the human genome. However, to date, single-molecule replication-mapping technologies, such as DNA combing and SMARD, which have throughput of a few hundred fibers, have provided insufficient depth of coverage for metazoan genome-wide analyses. Recently, nanopore sequencing has been used to map replication incorporation tracks genome-wide on single DNA fibers, but the current technology only allows for relatively small datasets of relatively short fibers (Georgieva et al., 2019; Müller et al., 2019; Hennion et al., 2020). The largest dataset from a recent study describing the DNAscent approach to nanopore-based replication mapping in budding yeast was 3.8 Gb (the equivalent of just over 1x coverage of the human genome) with an average fiber length of 32 kb (Müller et al., 2019). Therefore, ORM, for which current typical datasets are 150-times larger, provides a unique opportunity for single-molecule, genome-wide mapping of metazoan replication at high coverage on long fibers.
Using ORM, we have mapped the replication initiation events occurring in the first 2% of S phase in aphidicolin-synchronized HeLa cells. These initiation events, as others have reported (Petryk et al., 2016; Long et al., 2020; Pourkarimi et al., 2016; Ganier et al., 2019; Cayrou et al., 2015), are enriched in the enhancer regions of active genes and are enriched in active chromatin modifications, in particular DNase I HS, H2AZ and H3K4me1 (Figure 4). However, unlike previous reports, we can directly estimate the frequency with which initiation occurs in each IZ because we measure all fibers, both labeled and unlabeled. We find that the distribution of early initiation events is heterogeneous. Although 99% of early IZs predicted by OK-seq analysis are detected in our ORM data, only 6% of them, on average, initiate at the beginning of S phase in any given cell. Furthermore, although most of the initiation events that we observe are in early-replicating IZs, a small but significant minority are in late-replicating IZs, and the frequency of initiation is correlated with the replication timing of the IZ (Figure 3B). This observation is not due to contamination of our synchronized G1/S cells with non-synchronized cells because the signal in asynchronous cells shows no enrichment at specific sites, let alone previously mapped late initiation sites (Figure 2A). These results suggest that, in any given cell, a small number of potential initiation sites are stochastically selected from a large number of potential initiation sites, and that the probability of a site being selected is correlated with the replication time of that site, with early-replicating sites having a higher probability of initiating than late-replicating sites (Figure 3B).
We have mapped replication initiation sites across the genome with over 1,000-fold genome coverage, allowing us to identify initiation sites used in as few as 0.1% of cells. The initiation events that we map appear to be spread over broad IZs, consistent with previous observations (Petryk et al., 2016; Mesner et al., 2013; Besnard et al., 2012; Hamlin et al., 2008; Dijkwel et al., 2002; Demczuk et al., 2012; Lubelsky et al., 2011; Tao et al., 2000; Anglana et al., 2003). However, the sparse labeling and relatively long length (~40 kb) of our incorporation tracks allow us estimate the location of initiation sites at a resolution of only about 15 kb. Nonetheless, in 93.5% of IZs, we observe incorporation track centers that are more than 15 kb apart, suggesting that they initiated from distinct sites (Figures 3D and S4). Furthermore, the distribution of signal across IZs is not consistent with a single initiation site per zone (Figure S4C). We cannot distinguish between frequent, discrete, origins scattered throughout each IZ and diffuse IZs in which initiation can occur almost anywhere. However, we can rule out the hypothesis that human replication initiates at isolated, efficient, well-defined replication origins. In particular, we see no evidence for unique initiation sites at the lamin B or Top1 loci (Figure S4A), which have been proposed to be isolated origins (Keller et al., 2002; Abdurashidova et al., 2000). We suspect that the inefficient nature of human IZs and the low signal-to-noise ratio of previous initiation-mapping technology made it difficult to comprehensively map initiation sites across IZs. However, our data suggests that human IZs contain multiple low-efficiency initiation sites, as has been previously suggested (Langley et al., 2016; Petryk et al., 2016; Mesner et al., 2003; Hamlin et al., 2008).
Human Replication Initiation Events are not Clustered
Our single-molecule data also allows us to directly measure correlations between initiation sites on individual chromosomes. A number of previous reports have claimed that initiations events are clustered, in that initiation events are more likely to occur next to one another on chromosomes than would be expected by chance, and that these clustered events are regularly spaced between 75 and 150 kb apart in human cells (Jackson and Pombo, 1998; Cayrou et al., 2011; Lebofsky et al., 2006; Huberman and Tsai, 1973; Blow et al., 2001; Marheineke and Hyrien, 2004). However, these observations have been largely anecdotal because they have recorded observations only on fibers that have multiple initiation events, and so have not rigorously measured their frequency relative to single initiation events, which is necessary to determine if multiple initiation events happen more often than would be expected by chance. To test whether initiation events tend to occur in clusters, we examined the frequency and distribution of multiple tracks on single fibers. If events on a line are clustered with a characteristic inter-event distance, the distribution of inter-event distances will be bell-shaped, with a peak at the most frequent inter-event distance. If, however, the events are distributed randomly, with no inter-event correlation, the distribution of inter-event distances will be exponential (Birnbaum, 1954). We find that, between 60 and 600 kb, the distribution of inter-initiation-event distances is exponential (r = 0.99, Figure 3E), inconsistent with any significant clustering of initiation.
As an orthogonal test of initiation clustering, we directly examined co-replication of neighboring IZs. We examined every pair of IZs on each fiber we collected and asked whether the frequency with which they were replicated was correlated (Figure 3F). Using a model that takes into account both passive replication predicted by incorporation track lengths (Figures S2B) and nucleotide-uptake heterogeneity (Figure S1C) but otherwise assumes no correlation among initiation events, we can account for the observed frequency of replication in neighboring IZs (Figure 3F), consistent with the conclusion that human replication initiation sites are not clustered in early S phase. Thus, although initiation events with similar replication timing tend to be clustered because regions of similar replication timing are grouped together (Figure 1F), initiation events are no more likely to occur next to one another, or in collinear clusters, than expected by chance (Figures 3E,F).
Stochastic Usage of Initiation Zones in Human Cells
The heterogeneous, uncorrelated nature of replication initiation that we observe is reminiscent of stochastic models that have been proposed to explain the regulation of replication timing in budding yeast. Such models propose that the average timing of initiation in a population is regulated by the probability of a site initiating in an individual cell. In particular, neither hierarchical regulation of initiation timing nor correlation between initiation at neighboring sites is predicted by such models. Previous analyses of human replication kinetics have suggested positive correlations, in which one initiation event, or the forks it produces, would increase the probability of other nearby initiation events (Guilbaud et al., 2011; Löb et al., 2016). However, a more recent, higher resolution replication timing study found no evidence of this so-called “domino model” (Zhao et al., 2020). In any case, we find that a stochastic model assuming independent origin initiation can explain our observed data (Figure 3F).
To test whether our results are compatible with a stochastic model of replication kinetics, we used a computational model of DNA replication that assumes stochastic origin firing (Gindin et al., 2014a). Using our initiation-site mapping data to initialize the model produces predicted replication timing profiles that give an excellent fit (r = 0.85) to experimentally produced profiles (Figure 5B). Since the computational model does not contain any correlation between initiation events, this analysis suggests that any positive or negative correlations between initiation events must be of sufficiently low magnitude or restricted spatial distribution as to be not required to explain observed genome-wide patterns of replication timing. Our work, in combination with work from budding and fission yeast (Kaykov and Nurse, 2015; Yang et al., 2010; Patel et al., 2006; de Moura et al., 2010), supports the hypothesis that stochastic control of initiation timing is a fundamental feature of DNA replication, conserved across eukaryotes.
General Application of ORM to Map Replication Kinetics
In addition to mapping replication initiation sites, we also used ORM to map ongoing replication forks in unperturbed, asynchronous cells. We inferred fork direction from the asymmetric nature of incorporation tracks caused by the reduction of incorporation as labeled nucleotides are depleted, an approach that has been used before for both radio- and fluoro-labeled nucleotides (Müller et al., 2019; Huberman and Riggs, 1968; Hennion et al., 2020). Averaged over our nearly-500-fold fiber coverage of the genome, our inferred fork directions allow us to infer genome-wide replication kinetics that agree with independently determined replication-timing and replication-fork-directionality profiles (Figures 2C,D). In future work, it should be possible to use sequential labeling with nucleotides of different colors to unambiguously determine fork direction, thereby greatly increasing the resolution and sensitivity of asynchronous ORM. In addition to identifying replication initiation sites throughout the S phase of any cell type, sequential labeling will allow investigation of other important aspects of DNA replication, recombination and repair. We envision ORM being used to measure replication fork speed, replication fork arrests and reversal, and sister-chromatid exchange, under both normal growth and replication stress conditions. Such application will make ORM a central technique for studying DNA replication, DNA repair and genome instability.
Limitations of the Study
The primary limitation of current ORM technology is the in vivo pulse-labeling of replication. Other single-fiber DNA-replication-mapping approaches, such as DNA combing (Bensimon et al., 1994) and SMARD (Norio and Schildkraut, 2001), use thymidine analogs to label replicated DNA, allowing kilobase-resolution mapping of replication kinetics in a wide range of cell types and organisms. However, neither the two standard thymidine analogs—BrdU and EdU—are compatible with ORM technology; BrdU because anti-BrdU antibodies only recognize single-stranded DNA, but double-strand DNA is required for fiber stretching in the Bionano optical mapping chips, and EdU because the copper catalyst required to conjugate the EdU to a fluorophore produces hydroxyl radicals, which breaks the DNA fiber into sub-hundred-kb pieces. We have therefore labeled our cells by electroporating them with fluorescently-conjugated dUTP, which limits us to using cell lines. Moreover, the conjugated nucleotides are not efficiently incorporated leading to the sparse labeling and limited resolution that we see. DNA-replication labeling with a number of other nucleoside analogs has been demonstrated recently (Neef and Luedtke, 2014; Rieder and Luedtke, 2014; Mitter et al., 2020). Although none of them are currently compatible with ORM, we predict that improved nucleoside-labeling technology will allow ORM to by applied to most cells, tissues and organisms at kilobase resolution, making ORM a generally applicable technique for replication mapping.
STAR Methods Resource Availability Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nick Rhind (nick.rhind@umassmed.edu).
Materials Availability
No newly generated materials are associated with this paper.
Data and Code Availability
All raw and processed ORM data, including the identified IZs, is available at figshare <https://doi.org/10.6084/m9.figshare.c.5321048>. All ORM analysis code is available at GitHub <https://github.com/cl-chen-lab/ORM>, as are scripts to process ORM data for display on the ORM browser at <https://github.com/ClaireMarchal/ORM2BED> and <https://github.com/nimezhu/ORM>.
Experimental Model and Subject Details
Cell Lines
This study used human HeLa S3 (RRID:CVCL_0058) and H9 cell lines (RRID:CVCL_9773). Both lines are derived from female donors. HeLa S3 cells were grown in DMEM plus 10% Cosmic Calf Serum (GE Life Science SH30087) and Pen/Strep. H9 hESCs were grown in feeder free conditions on Geltrex matrix (Thermo Fisher A14133) coated dishes in StemPro (Thermo Fisher A100701) media according to manufacturer’s specifications.
Method Details Cell Labeling and Sample Preparation
For asynchronous HeLa datasets, cells were grown to ~70% confluency, washed with warm PBS, trypsinized, and electroporated (Lonza, Nucleofection kit SE, HeLa S3 HV program) in the presence of 40 μM Aminoallyl-dUTP-ATTO-647N (Jena Bioscience NU-803–647N). For G1/S synchronized data sets, cells were synchronized at metaphase by incubation with 0.05 μg/ml nocodazole for 4 hours followed by shake off. The percentage metaphase cells was quantified by metaphase spread. Briefly, 3X volumes of ddH20 was added to an aliquot of media from shake off and was incubated at 37°C for 15 minutes. Cells were fixed by adding several drops of ice-cold methanol:glacial acetic acid (3:1). Cells were spun down and pellet were resuspended in ice-cold methanol:glacial acetic acid (3:1) and spotted on to a clean slide. Slides were allowed to dry, spotted with Vectasheild (Vector Laboratories H-1000) containing DAPI and covered with coverslips. Metaphase and interphase cells were counted on a Nikon Eclipse Ti microscope. Metaphase cells were released into fresh media containing 10 μg/ml aphidicolin (Millipore Sigma 178273) for 16 hours. This protocol allows early initiation events but prevents elongation of replication forks from these initiation events and also prevents subsequent initiation events by activation of the intra-S-phase checkpoint (Feijoo et al., 2001). For 0-minute release datasets, cells were washed three times with warm PBS (or PBS plus aphidicolin, in the case of B.0) to remove aphidicolin, trypsinized, and electroporated (Lonza, Nucleofection kit SE, HeLa S3 HV program) in the presence of 40 μM Aminoallyl-dUTP-ATTO-647N (Jena Bioscience NU-803–647N). Cells were recovered in warm media plus aphidicolin for 30 minutes in a 37°C water bath with agitation every 5 minutes to prevent cells from settling in the tube. Cells were then spun down and washed 3 times with warm PBS to remove aphidicolin. For timed release experiments, cells were washed three times with warm PBS to remove aphidicolin, warm media was added, and cells were returned to 37°C/5% CO2 incubator for the indicated times (5, 10, 20, 30, 45, 60, or 90 minutes). Cells were then washed again with PBS, trypsinized, and electroporated in the presence of fluorescent nucleotides as above. In all cases, cells were allowed to recovered overnight in media at 37°C/5% CO2. After recovery, all cells were detached with trypsin and snap frozen in liquid nitrogen. In our initial experiment, 87% of cells were synchronized in mitosis and, after release from aphidicolin, 94% incorporated the fluorescent label, with 100% of labeled cells showing an early-replication pattern of replication foci (Table S1, Figures S1A-C), indicating a high degree of synchrony and <1% of contaminating asynchronous S phase cells.
H9 cells were grown to ~70% confluency, washed with warm PBS, dissociated to single cells with Gentle Cell Dissociation Reagent (StemCell Technologies 07174) and electroporated (Lonza, Nucleofection kit P3, hESC program) in the presence of 40 μM Aminoallyl-dUTP-ATTO-647N (Jena Bioscience NU-803–647N). Cells were recovered overnight in StemPro media plus 10uM Y-27632 dihydrochloride (ROCK inhibitor, Sigma Y0503) at 37°C/5% CO2.
Nucleotide labeling efficiency was assayed by plating a small aliquot of cells on coverslips after electroporation. Following overnight recovery, cells were fixed with 4% formaldehyde for 10 minutes at room temperature. Cells were then washed twice with PBS followed by permeabilization and counterstaining with PBS plus 0.2% Triton X-100 plus DAPI for 5 minutes at room temperature. Coverslips were washed twice with PBS, spotted with Vectasheild, and placed on slides. Images were captured on a DeltaVision (GE Life Sciences) microscope. Replication foci patterns were categorized as previously described (O’Keefe et al., 1992). Briefly, replication foci patterns were categorized as either early, middle, or late replicating. Early foci patterns were characterized as many, small incorporation puncta in the nuclear interior and a distinct lack of incorporation at the nucleolus. Middle foci patterns were characterized as nucleotide incorporation at the nuclear periphery, often in the shape of a ring encircling the entire nuclear interior. Late foci patterns were characterized as large, dense incorporation puncta at either the nuclear interior or the nucleolus.
Optical Replication Mapping
Two Bionano mapping approaches were used: Nick, Label, Repair and Stains (NLRS) and Direct Label and Stain (DLS) (Table S1). For NLRS, cell pellets were thawed on ice, resuspended in cold Cell Buffer (Bionano 80004) and embedded into low-melting point agarose plugs using the CHEF Mammalian Genomic DNA Plug Kit (Bio-Rad #1703591) according to the manufacturer’s instructions, with a target concentration of 1,000,000 cells per plug. Cells immobilized in plugs were lysed and treated with Proteinase K and RNase A. The plugs were then melted and solubilized, and the resulting DNA was further cleaned by drop dialysis. The DNA was fluorescently labeled with green fluorophores at Nt.BspQI sites and counterstained with YOYO-1 using Bionano Genomics’ NLRS labeling kit (Bionano 80001). Throughout the entire process, the samples were shielded from light whenever possible, in order to maintain integrity of the fluorophores. For DLS, DNA was labeled with green fluorophores using the direct labeling enzyme DLE-1 (Bionano Genomics 80005) according to manufacture’s protocol. The rest of processing steps were the same as NLRS labeling.
Labeled DNA samples were loaded onto a Saphyr chip (Bionano Genomics 20319) and run on the corresponding Saphyr instrument (Bionano Genomics IN-011–01), following the manufacturer’s instructions. Briefly, the DNA was coaxed into an array of parallel nanochannels via electrophoresis, thereby elongating the DNA to a uniform contour length, allowing for accurate measurement of distances along the molecules. Molecules were imaged to collect the YOYO-1 DNA signal in a false-color blue channel, the Nt.BspQI or DLE-1 labels in the green channel, and the in vivo replication incorporation tracks in the red channel.
Images were converted to digitized molecules and the positions of the green and red labels on each fiber were determined using Bionano Access. The Nt.BspQI and DLE-1 labels were aligned to human reference genome hg19 by creating an in silico digestion of the reference FASTA file. Alignment of the fibers’ Nt.BspQI and DLE-1 sites allowed mapping of the red replications signal to the genome. The pipeline produces one.bnx file containing the intensity and location of each green and red signal on each fiber and one.xmap file containing the genomic location of each fiber. We observed abnormal ORM signal enrichment in some specific genomic positions (hereafter called hotspots), which only occurred in the samples mapped by NLRS technique but not those mapped by DLS technique. Further investigation confirmed that most (99.36%) these hotspots are located within < 650 bp distance to a BspQI site used for the mapping in the NLRS technique. Detailed analysis showed that these abnormal ORM signals are likely signals of false positive detection due to the strong BspQI signals, since the green signals (for mapping) associated with the hotspot regions have stronger intensity distribution than the green signal outside the hotspot region while the red signals inside the hotspot regions have intensity weaker than the red signals outside of the hotspots. In order to filter these false positive ORM signals, we calculated the ORM signal count (mean = 1.71 and s.d. = 6.35 for all windows containing at least one red signal) within all 300 bp windows by combing all 0’ data, and removed those with >=20 ORM signals (the cutoff was estimated by comparing with the distribution of DLS samples without such hotspots). The removed hotspot signals correspond to 4.7% of total signals in C.0 sample and 3.6% in D.0 sample, and to similar degree (~3–4%) for the other samples from these datasets (i.e. C.5, C.10, D.20, D.30, D.45, D.60 and D.90).
Modeling Signal-Intensity and Inter-Signal-Distance Distributions
The replication-incorporation signal we detect is punctate in nature and uniform in intensity (Figures 1A and S2A). From these observations, we conclude that these incorporation signals consist of individual fluorescent nucleotides. The distributions of signal intensity (Figure S2A) and inter-signal distances (Figure S2B) are consistent with sparse labeling by a pool of fluorescent nucleotides that is depleted by replication, as expected for a one-time bolus of nucleotides delivered by electroporation. The distribution of the signals is predicted by inefficient, random incorporation of fluorescent nucleotides at an initial rate of about 1/900 thymidines that is depleted with a half length of about 75 kb (Figure S2B). Given the lengths of the replication tracks and the sparse nature of the signal within them, we estimate that we can localize initiation events with a resolution of about 15 kb (14.2±0.1 kb). To model the signal-intensity distributions (Figure S2A), we assumed that signals represent individual fluorophores that can be resolved with some finite resolution. We modeled both the photon output and the inter-signal distance (which controls the probability of having two fluors closer together than the resolution of the Saphyr) of each fluor as Poisson processes, resulting in the depicted fit (obtained from the first four terms of Eq. 5, Method S1), which predicts that 20% of the fluors are closer than the resolution of the Saphyr. See Method S1 for details.
To model the distribution of inter-signal distances (Figure S2B), we assume that the incorporation rate is a Poisson process for which the Poisson statistic is determined by the average amount of label taken up by cells. We further assume that there is an exponentially decreasing amount of label over time as it is consumed by replication. Combining these two assumptions results in the depicted fit (Eq. 14, Method S1), with an inferred initial incorporation rate of about 1/900 thymidines and a depletion half length of about 75 kb for the merged 0-minute dataset. See Method S1 for details.
Combining the models in Figures S2A and B, we calculate that 20% of the signals in Figure S2B (corresponding to the 20% of the fluors that we infer from Figure S2A are not individually resolved by the Saphyr) are located within 1.3±0.3 kb of a neighboring fluor, consistent with the reported resolution of the Saphyr and the observation that there are almost no inter-signal distances below 1.3 kb in our datasets.
Identification of Replication Tracks
After mapping the fibers on the human genome, the distances between the genomic mapping signals were recalibrated to match their reference genome positions, compensating for heterogeneity in fiber stretching. We then mapped signals in the red channel between two adjacent genomic mapping signals as nucleotide-incorporation signals. The nucleotide-incorporation signal is discontinuous and appears as clusters of neighboring signals (Figure 1A). To segment the ORM signals into replication-incorporation tracks, we used a two-step process based on Gaussian mixture models. We first fit a Gaussian mixture model using the R package mixtools (https://www.rdocumentation.org/packages/mixtools) with 3-gaussian distributions to the inter-signal distance distribution, with the first peak corresponding to the distances between signals within the same segment, the second peak corresponding to the distances between signals in adjacent segments and the third peak corresponding to signals in non-adjacent segments. The tail of the first Gaussian distribution (16,384 kb) was comparable amongst various data sets and was therefore selected as the cutoff value (cutoff1) to merge adjacent signals into primary segments. We then measured the distance between adjacent primary segments and fit a 2-Gaussian distribution to find the second cutoff value (cutoff2, 32,768 kb), which we used to merge all the adjacent primary segments into final replication tracks.
In our combined 0’ dataset, we observed 977,746 replication tracks averaging 19.5±30.9 kb in length. The observed average track length of 20 kb is shorter than the 75 kb that we estimate is replicated during the labeling period (Figure S2B) due to the sparsity of the labeling. In particular, 58% of our replication tracks are labeled with only one signal. To test whether these solo signals are sparsely labeled replication tracks or spurious background noise, we calculated the enrichment of solo signals at early replication sites. Solo signals are enriched at such sites, demonstrating that they, too, mark regions of replication initiation (Figure S2C), and that noise, if any, is very low with this method. Although many of the tracks are labeled with only one signal, few are not labeled at all. Our modeling of the rate of label incorporation and label depletion suggest that over 90% of initiation events in early S-phase will incorporate at least one label within 15 kb of replication (Figures S2A and B and Method S1 Section 4). Therefore, almost every early-initiation event will be visualized at a resolution of 15 kb.
To estimate the resolution with which we can identify the initiation site of any given replication track, we defined the maximum likelihood distribution of initiation sites for a given replication track and, using numeric simulations, estimated the standard deviation of that distribution as 14.2±0.1 kb. See Method S1 for details.
To determine the direction of replication tracks in our asynchronous data, we defined a fork direction index (FDI): FDI = log2(IntensitySUM_Left / IntensitySUM_Right), where IntensitySUM_Left and IntensitySUM_Right is the total fluorescent signal in the left and right half, respectively, of each ORM track with at least 3 signals. A positive (negative) FDI corresponds to a rightward (leftward) moving replication fork. The mean relative fork direction (RFD) within a given window is defined as (R-L)/(R+L), where R and L is the number of rightward (FDI>0) and leftward (FDI<0) ORM tracks, respectively.
Identification of Initiation Zones
Initiation zones were identified based on the ORM signal density along the genome. We first obtained the ORM signal density by calculating the percentage of fibers containing ORM signal in 10 kb sliding windows with 1 kb steps. LOESS smoothing (with α = 0.75 and the polynomial degree = 2) was then performed in successive 160 kb windows, with 8 kb overlaps to avoid discontinuity of fitting. Within the 8 kb overlaps, the final smoothed values were the averages, weighted by their distance to the corresponding window, of the smoothed values from two adjacent windows. IZs were then defined as the peaks on the smoothed ORM signal density profile. The size of each IZs was defined by the smallest window that containing at least 40% of ORM signals within each peak. The value of 40% was chosen because, when aggregated around IZ peaks, the ORM signal density decreases with distance from the peak at a similar rate until about 40% then decreases much more slowly. This process was performed on all 4 biological replicates of HeLa 0’ data as well as the combined dataset. Only IZs identified in at least 3 replicates and with a relative peak height in the combined dataset (as measured by the difference in signal density between the peak and the edges of the zone) of greater than 0.3% were retained. The precise location of each retained IZ was then determined by analyzing the transition of ORM signals with a K-mean clustering (n=2 if the center cluster < 30 kb, otherwise n=3) on the ΔORM signal between adjacent 1kb windows to identify the region with sharpest ORM density around each IZ center.
To estimate the minimum number of initiation sites in each IZ, we calculated number of replication track centers separated by more than the resolution of our ability to localize initiation sites, which is about 15 kb. We calculated the position of the center of each replication track that overlaps a given IZ, discarding any that are more than 7.5 kb outside of it. We then ranked the centers by position and, starting at one end, counted the number of centers that are more than 15 kb apart.
Comparative Analysis of ORM Data with Other Datasets
The raw Repli-Seq data of HeLa S3 cells were downloaded from the Encode project <http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeUwRepliSeq> (Hansen et al., 2010) and S50 (the fraction of S phase at which 50% of the DNA is replicated in a defined genome region) was computed (Chen et al., 2010) and used to identify replication timing peaks (Chen et al., 2011) as previous described. Published H9 ESCs replication timing and peak calling data were used (Zhao et al., 2020). ORM signal enrichment around timing peaks was calculated in R (version 3.5.1 <https://www.rproject.org>). To compare the ORM IZs and other origin mapping data, we used pROC package to calculate the ROC (Receiver Operating Characteristic) curve and AUC (Area Under the Curve) values by using all 100 kb bins along the genome as example space and the bins overlapped with the ORM IZs as true positive space (Robin et al., 2011).
Correlations between ORM initiation efficiency and published genome-wide HeLa cell replication-initiation-mapping datasets—OK-seq (Petryk et al., 2016), SNS-seq (Picard et al., 2014), Ini-seq (Langley et al., 2016) and Orc1 ChIP-seq (Dellino et al., 2013)—were calculated across the genome in 50 kb adjacent windows. When necessary, genomic coordinates were remapped to hg19 using LiftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). ORM initiation efficiency was calculated as the number of fibers with ORM signal in a 50 kb genomic window divided by the number of fibers covering that window. The OK-seq initiation efficiency was calculated either genome-wide using the previously described origin efficiency metric (OEM) approach (McGuffee et al., 2013) or for each OK-Seq IZ with the ΔRFD between its right and left extremities (Petryk et al., 2016). For a given genomic position, OEM = WL/(WL+CL) – WR/(WR+CR), where WL, CL, WR and CR correspond to the numbers of OK-seq reads mapped, respectively, on the Watson (W) or Crick (C) strand within the 10 kb window on the left (L) or right (R) side of corresponding position. For OK-seq IZs, firing efficiency was calculated by ΔRFD = RFD(R) - RFD(L), where RFD(L) and RFD(R) correspond, respectively, to the RFD values at the left extremity and right extremity of corresponding OK-seq IZ. RFD(L) and RFD(R) were calculated by a linear fit of all 1 kb RFD values within a given OK-seq IZ. Correlations were calculated using Spearman’s correlation coefficient in R.
The genome-segmentation results (ChromHMM) based on ENCODE data were retrieved from UCSC genome browser <https://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeAwgSegmentation>. To compensate for the large size of IZ, relative to many of the ChromHMM segments, we used the IZ’s core regions, defined as the region from center of the IZ to the point of highest ORM signal within that IZ.
Histone modification data for HeLa S3—H2A.Z, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K9me3, H3K27ac, H3K27me3, H3K36me3, H3K79me2 and H4K20me1—was downloaded from ENCODE <https://www.encodeproject.org>. If available, the data was obtained from G1 phase cells. The average of all replicates was use in our analysis. The RNA Pol II-pS2 <https://www.encodeproject.org/experiments/ENCSR000ECT> and DNase I hypersensitivity data (UW DNase I HS data track) for HeLa S3 were also downloaded from ENCODE. The signal enrichment of each feature was re-normalized such that the background regions show zero log2 enrichment ratio. Published G4 sequence locations were retrieved (Puig Lombardi et al., 2019). We analyzed 3 G4 datasets (G4L3, G4L7 and G4L12) with similar results; we therefore used the G4L12 (G3N1–12G3N1–12G3N1–12G3), which contains the largest dataset and give the best correlation with ORM data, in our final analysis. The genome-wide pairwise correlation analysis and enrichment around ORM IZs were performed by deepTools (Ramírez et al., 2016) or with R (version 3.5.1 <https://www.r-project.org>). IZs were classified by timing—early (S50<0.25), mid-early (0.25 ~ 0.5), mid-late (0.5~0.75) and late (S50>0.75)— using the S50 of the IZ center. The partial correlation analysis between ORM initiation efficiency and G4 sequences with controlling the effect of GC percentage (obtained from UCSC <http://hgdownload.soe.ucsc.edu/goldenPath/hg19/gc5Base>) was performed by using the R package ppcor <https://cran.r-project.org/web/packages/ppcor/index.html>. Features were visualized in IGV (Robinson et al., 2011) and the ORM Browser <http://orm.nucleome.org> (Figure S3, Zhu et al. in prep, personal communication, J. Ma <https://github.com/nimezhu/ORM>). All published data sources are complied in the Key Resource Table.
KEY RESOURCES TABLE
Correlation of Initiation in Neighboring Initiation Zones
To calculate the degree of correlation between initiation events in neighboring IZs, we defined an initiation event as any ORM signal in any IZ on a fiber. We then calculated the correlation as the frequency with which we observed initiation in neighboring IZs on the same fiber divided by the product of the frequency of overall initiation in the two IZs. The non-neighbor correlation was calculated in the same manner, but using all pairwise combinations of IZs on each fiber farther that 200 kb apart. To calculate the expected correlation due to passive replication of one IZ due to initiation in a neighboring IZ, we used the observed label-depletion rate and inferred the frequency with which initiation at one IZ would lead to incorporation of label at a neighboring IZ at a given distance away (Figure S2B). To calculate the correlation expected from heterogeneity of label uptake by cells, we used the observed distribution of cellular labeling (Figure S1C). The two calculations were combined to produce the model (Eq. 34, Method S1) shown in Figure 3F. See Method S1 for details.
Replication Timing Simulation
To simulate the replication timing profiles, we used the Replicon simulation code (Gindin et al., 2014a). Replicon uses three sets of user-defined parameters. The first set of parameters is the initiation probability landscape (IPLS), the relative probability of initiating at any point in the genome. In our simulations, we used the ORM the signal distribution or other genomic features with a bin size of 0.5 kb. The second set of parameters control the virtual flow sorter. In agreement with the results from the original paper (Gindin et al., 2014b), we set this to (0.0, 0.17, 0.35, 0.58, 0.92, 0.99, 1.0), although other choices give similar results. The final parameter is the number of forks. Here, we again followed the original paper and set the number of forks equal to 10.24 + x(7.9E-7), where x is the length of the chromosome in base pairs.
Quantification and Statistical Analysis
Statistical analyses were performed in R or Igor Pro. Statistical tests and parameter are reported in the text and figure legends.
Additional Resources
Interactive visualization of the ORM data is available on the ORM Browser <http://orm.nucleome.org>.
Supplementary Material
ORM Dataset Metadata, Related to STAR Methods
Genome-wide, single-molecule optical mapping replication initiation sites
27 million DNA molecules averaging ~300 kb provides 2500-fold genome coverage
Initiation is heterogeneous: initiation zones are active early in > 20% of cells
Contrary to long-held dogma, initiation events are not clustered
Acknowledgements
We are grateful to Feng Yue for his contribution to the collection of the Bionano data, Jian Ma for help developing ORM browser and Peiyao Zhao maintaining the ORM browser server. The work was funded by NIH grants HG010658 to DMG and GM125872 to NR. CLC was supported by the I. Curie YPI program, the ATIP-Avenir program from CNRS, Plan Cancer from INSERM, the CNRS 80|Prime interdisciplinary program, ANR and INCa. WW was supported by a COFUND IC-3i International PhD fellowship.
Footnotes
Declaration of Interests
Alex Hastie is an employee of Bionano Genomics. Other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Wang et al. map early replication initiation sites in human cell using ORM (optical replication mapping).They find that initiation is heterogeneous, inefficient, and that initiation events do not cluster, consistent with stochastic models of replication control.
References
- Abdurashidova G, Deganuto M, Klima R, Riva S, Biamonti G, Giacca M, and Falaschi A. (2000). Start sites of bidirectional DNA synthesis at the human lamin B2 origin. Science 287, 2023–2026. [DOI] [PubMed] [Google Scholar]
- Anglana M, Apiou F, Bensimon A, and Debatisse M. (2003). Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell 114, 385–394. [DOI] [PubMed] [Google Scholar]
- Bechhoefer J, and Rhind N. (2012). Replication timing and its emergence from stochastic processes. Trends Genet 28, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, and Bensimon D. (1994). Alignment and sensitive detection of DNA by a moving interface. Science 265, 2096–2098. [DOI] [PubMed] [Google Scholar]
- Berezney R, Dubey DD, and Huberman JA (2000). Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 108, 471–484. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, and Snyder M. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin JM, and Lemaitre JM (2012). Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol [DOI] [PubMed] [Google Scholar]
- Birnbaum A. (1954). Statistical Methods for Poisson Processes and Exponential Populations. Journal of the American Statistical Association 49, 254–266. [Google Scholar]
- Blow JJ, Gillespie PJ, Francis D, and Jackson DA (2001). Replication origins in Xenopus egg extract are 5–15 kilobases apart and are activated in clusters that fire at different times. J Cell Biol 152, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Ballester B, Peiffer I, Fenouil R, Coulombe P, Andrau JC, van Helden J, and Mechali M. (2015). The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res 25, 1873–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, and Mechali M. (2011). Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res 21, 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagin VO, Casas-Delucchi CS, Reinhart M, Schermelleh L, Markaki Y, Maiser A, Bolius JJ, Bensimon A, Fillies M, Domaing P, Rozanov YM, Leonhardt H, and Cardoso MC (2016). 4D Visualization of replication foci in mammalian cells corresponding to individual replicons. Nat Commun 7, 11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Duquenne L, Audit B, Guilbaud G, Rappailles A, Baker A, Huvet M, d’Aubenton-Carafa Y, Hyrien O, Arneodo A, and Thermes C. (2011). Replication-associated mutational asymmetry in the human genome. Mol Biol Evol 28, 2327–2337. [DOI] [PubMed] [Google Scholar]
- Chen CL, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d’Aubenton-Carafa Y, Arneodo A, Hyrien O, and Thermes C. (2010). Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res 20, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti C, Saccà B, Herrick J, Lalou C, Pommier Y, and Bensimon A. (2007). Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol Biol Cell 18, 3059–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajkowsky DM, Liu J, Hamlin JL, and Shao Z. (2008). DNA Combing Reveals Intrinsic Temporal Disorder in the Replication of Yeast Chromosome VI. J Mol Biol 375, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Carli F, Menezes N, Berrabah W, Barbe V, Genovesio A, and Hyrien O. (2018). High-Throughput Optical Mapping of Replicating DNA. Small Methods 0, 1800146. [Google Scholar]
- de Moura AP, Retkute R, Hawkins M, and Nieduszynski CA (2010). Mathematical modelling of whole chromosome replication. Nucleic Acids Res 38, 5623–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellino GI, Cittaro D, Piccioni R, Luzi L, Banfi S, Segalla S, Cesaroni M, Mendoza-Maldonado R, Giacca M, and Pelicci PG (2013). Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res 23, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demczuk A, Gauthier MG, Veras I, Kosiyatrakul S, Schildkraut CL, Busslinger M, Bechhoefer J, and Norio P. (2012). Regulation of DNA Replication within the Immunoglobulin Heavy-Chain Locus During B Cell Commitment. PLoS Biol 10, e1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkwel PA, Wang S, and Hamlin JL (2002). Initiation sites are distributed at frequent intervals in the Chinese hamster dihydrofolate reductase origin of replication but are used with very different efficiencies. Mol Cell Biol 22, 3053–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dileep V, and Gilbert DM (2018). Single-cell replication profiling to measure stochastic variation in mammalian replication timing. Nat Commun 9, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M. (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, and Smythe C. (2001). Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol 154, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulk MS, Urban JM, Casella C, and Gerbi SA (2015). Characterizing and controlling intrinsic biases of lambda exonuclease in nascent strand sequencing reveals phasing between nucleosomes and G-quadruplex motifs around a subset of human replication origins. Genome Res 25, 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganier O, Prorok P, Akerman I, and Méchali M. (2019). Metazoan DNA replication origins. Curr Opin Cell Biol 58, 134–141. [DOI] [PubMed] [Google Scholar]
- Georgieva D, Liu Q, Wang K, and Egli D. (2019). Detection of Base Analogs Incorporated During DNA Replication by Nanopore Sequencing. bioRxiv 549220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindin Y, Meltzer PS, and Bilke S. (2014a). Replicon: a software to accurately predict DNA replication timing in metazoan cells. Front Genet 5, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindin Y, Valenzuela MS, Aladjem MI, Meltzer PS, and Bilke S. (2014b). A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol Syst Biol 10, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldar A, Marsolier-Kergoat MC, and Hyrien O. (2009). Universal temporal profile of replication origin activation in eukaryotes. PLoS One 4, e5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud G, Rappailles A, Baker A, Chen CL, Arneodo A, Goldar A, d’Aubenton-Carafa Y, Thermes C, Audit B, and Hyrien O. (2011). Evidence for Sequential and Increasing Activation of Replication Origins along Replication Timing Gradients in the Human Genome. PLoS Comput Biol 7, e1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin JL, Mesner LD, Lar O, Torres R, Chodaparambil SV, and Wang L. (2008). A revisionist replicon model for higher eukaryotic genomes. J Cell Biochem 105, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, and Stamatoyannopoulos JA (2010). Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A 107, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennion M, Arbona JM, Lacroix L, Cruaud C, Theulot B, Tallec BL, Proux F, Wu X, Novikova E, Engelen S, Lemainque A, Audit B, and Hyrien O. (2020). FORK-seq: replication landscape of the Saccharomyces cerevisiae genome by nanopore sequencing. Genome Biol 21, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick J, and Bensimon A. (1999). Single molecule analysis of DNA replication. Biochimie 81, 859–871. [DOI] [PubMed] [Google Scholar]
- Huberman JA, and Riggs AD (1968). On the mechanism of DNA replication in mammalian chromosomes. J Mol Biol 32, 327–341. [DOI] [PubMed] [Google Scholar]
- Huberman JA, and Tsai A. (1973). Direction of DNA replication in mammalian cells. J Mol Biol 75, 5–12. [DOI] [PubMed] [Google Scholar]
- Hyrien O. (2015). Peaks cloaked in the mist: the landscape of mammalian replication origins. J Cell Biol 208, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, and Pombo A. (1998). Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140, 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaykov A, and Nurse P. (2015). The spatial and temporal organization of origin firing during the S-phase of fission yeast. Genome Res 25, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaykov A, Taillefumier T, Bensimon A, and Nurse P. (2016). Molecular Combing of Single DNA Molecules on the 10 Megabase Scale. Sci Rep 6, 19636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Ladenburger EM, Kremer M, and Knippers R. (2002). The origin recognition complex marks a replication origin in the human TOP1 gene promoter. J Biol Chem 277, 31430–31440. [DOI] [PubMed] [Google Scholar]
- Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, and Kwok PY (2012). Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol 30, 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley AR, Gräf S, Smith JC, and Krude T. (2016). Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq). Nucleic Acids Res 44, 10230–10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebofsky R, Heilig R, Sonnleitner M, Weissenbach J, and Bensimon A. (2006). DNA Replication Origin Interference Increases the Spacing between Initiation Events in Human Cells. Mol Biol Cell 17, 5337–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löb D, Lengert N, Chagin VO, Reinhart M, Casas-Delucchi CS, Cardoso MC, and Drossel B. (2016). 3D replicon distributions arise from stochastic initiation and domino-like DNA replication progression. Nat Commun 7, 11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Zhang L, Lv M, Wen Z, Zhang W, Chen X, Zhang P, Li T, Chang L, Jin C, Wu G, Wang X, Yang F, Pei J, Chen P, Margueron R, Deng H, Zhu M, and Li G. (2020). H2A.Z facilitates licensing and activation of early replication origins. Nature 577, 576–581. [DOI] [PubMed] [Google Scholar]
- Lubelsky Y, Sasaki T, Kuipers MA, Lucas I, Le Beau MM, Carignon S, Debatisse M, Prinz JA, Dennis JH, and Gilbert DM (2011). Pre-replication complex proteins assemble at regions of low nucleosome occupancy within the Chinese hamster dihydrofolate reductase initiation zone. Nucleic Acids Res 39, 3141–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2018). Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marheineke K, and Hyrien O. (2004). Control of replication origin density and firing time in Xenopus egg extracts: role of a caffeine-sensitive, ATR-dependent checkpoint. J Biol Chem 279, 28071–28081. [DOI] [PubMed] [Google Scholar]
- Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, and Oda M. (2010). Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem 79, 89–130. [DOI] [PubMed] [Google Scholar]
- McGuffee SR, Smith DJ, and Whitehouse I. (2013). Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol Cell 50, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner LD, Li X, Dijkwel PA, and Hamlin JL (2003). The dihydrofolate reductase origin of replication does not contain any nonredundant genetic elements required for origin activity. Mol Cell Biol 23, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner LD, Valsakumar V, Cieslik M, Pickin R, Hamlin JL, and Bekiranov S. (2013). Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res 23, 1774–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B, Ji Z, and Struhl K. (2016). Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc Natl Acad Sci U S A 113, E4810–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitter M, Gasser C, Takacs Z, Langer CCH, Tang W, Jessberger G, Beales CT, Neuner E, Ameres SL, Peters JM, Goloborodko A, Micura R, and Gerlich DW (2020). Conformation of sister chromatids in the replicated human genome. Nature 586, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CA, Boemo MA, Spingardi P, Kessler BM, Kriaucionis S, Simpson JT, and Nieduszynski CA (2019). Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads. Nat Methods 442814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef AB, and Luedtke NW (2014). An azide-modified nucleoside for metabolic labeling of DNA. Chembiochem 15, 789–793. [DOI] [PubMed] [Google Scholar]
- Norio P, and Schildkraut CL (2001). Visualization of DNA replication on individual Epstein-Barr virus episomes. Science 294, 2361–2364. [DOI] [PubMed] [Google Scholar]
- O’Keefe RT, Henderson SC, and Spector DL (1992). Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol 116, 1095–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panning MM, and Gilbert DM (2005). Spatio-temporal organization of DNA replication in murine embryonic stem, primary, and immortalized cells. J Cell Biochem 95, 74–82. [DOI] [PubMed] [Google Scholar]
- Patel PK, Arcangioli B, Baker SP, Bensimon A, and Rhind N. (2006). DNA replication origins fire stochastically in fission yeast. Mol Biol Cell 17, 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Kahli M, d’Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen CL, and Hyrien O. (2016). Replication landscape of the human genome. Nat Commun 7, 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Cadoret JC, Audit B, Arneodo A, Alberti A, Battail C, Duret L, and Prioleau MN (2014). The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet 10, e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourkarimi E, Bellush JM, and Whitehouse I. (2016). Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans. Elife 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prorok P, Artufel M, Aze A, Coulombe P, Peiffer I, Lacroix L, Guédin A, Mergny JL, Damaschke J, Schepers A, Cayrou C, Teulade-Fichou MP, Ballester B, and Méchali M. (2019). Involvement of G-quadruplex regions in mammalian replication origin activity. Nat Commun 10, 3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig Lombardi E, Holmes A, Verga D, Teulade-Fichou MP, Nicolas A, and Londoño-Vallejo A. (2019). Thermodynamically stable and genetically unstable G-quadruplexes are depleted in genomes across species. Nucleic Acids Res 47, 6098–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, and Gilbert DM (2013). DNA replication timing. Cold Spring Harb Perspect Biol 5, a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Yang SC-H, and Bechhoefer J. (2010). Reconciling stochastic origin firing with defined replication timing. Chromosome Res 18, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder U, and Luedtke NW (2014). Alkene-Tetrazine Ligation for Imaging Cellular DNA. Angew Chem Int Ed Engl 53, 9168–9172. [DOI] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, and Müller M. (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Dong Z, Leffak M, Zannis-Hadjopoulos M, and Price G. (2000). Major DNA replication initiation sites in the c-myc locus in human cells. J Cell Biochem 78, 442–457. [DOI] [PubMed] [Google Scholar]
- Taylor JH (1977). Increase in DNA replication sites in cells held at the beginning of S phase. Chromosoma 62, 291–300. [DOI] [PubMed] [Google Scholar]
- Técher H, Koundrioukoff S, Azar D, Wilhelm T, Carignon S, Brison O, Debatisse M, and Le Tallec B. (2013). Replication dynamics: biases and robustness of DNA fiber analysis. J Mol Biol 425, 4845–4855. [DOI] [PubMed] [Google Scholar]
- Valton AL, Hassan-Zadeh V, Lema I, Boggetto N, Alberti P, Saintome C, Riou JF, and Prioleau MN (2014). G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J 33, 732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashee S, Cvetic C, Lu W, Simancek P, Kelly TJ, and Walter JC (2003). Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev 17, 1894–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KA, Elefanty AG, Stanley EG, and Gilbert DM (2016). Spatio-temporal re-organization of replication foci accompanies replication domain consolidation during human pluripotent stem cell lineage specification. Cell Cycle 15, 2464–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yanagisawa Y, Tsankov AM, Hart C, Aoki K, Kommajosyula N, Steinmann KE, Bochicchio J, Russ C, Regev A, Rando OJ, Nusbaum C, Niki H, Milos P, Weng Z, and Rhind N. (2012). Genome-wide identification and characterization of replication origins by deep sequencing. Genome Biol 13, R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SC-H, and Bechhoefer J. (2008). How Xenopus laevis embryos replicate reliably: Investigating the random-completion problem. Phys Rev E Stat Nonlin Soft Matter Phys 78, 041917. [DOI] [PubMed] [Google Scholar]
- Yang SC-H, Rhind N, and Bechhoefer J. (2010). Modeling genome-wide replication kinetics reveals a mechanism for regulation of replication timing. Mol Syst Biol 6, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao PA, Sasaki T, and Gilbert DM (2020). High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol 21, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ORM Dataset Metadata, Related to STAR Methods
Data Availability Statement
All raw and processed ORM data, including the identified IZs, is available at figshare <https://doi.org/10.6084/m9.figshare.c.5321048>. All ORM analysis code is available at GitHub <https://github.com/cl-chen-lab/ORM>, as are scripts to process ORM data for display on the ORM browser at <https://github.com/ClaireMarchal/ORM2BED> and <https://github.com/nimezhu/ORM>.