

Abstract
Mouse models of radiation-induced thymic lymphoma are widely used to study the development of radiation-induced blood cancers and to gain insights into the biology of human T-cell lymphoblastic leukemia/lymphoma. Here we aimed to identify key oncogenic drivers for the development of radiation-induced thymic lymphoma by performing whole-exome sequencing using tumors and paired normal tissues from mice with and without irradiation. Thymic lymphomas from irradiated wild-type (WT), p53+/− and KrasLA1 mice were not observed to harbor significantly higher numbers of non-synonymous somatic mutations compared to thymic lymphomas from unirradiated p53−/− mice. However, distinct patterns of recurrent mutations arose in genes that control the Notch1 signaling pathway based on the mutational status of p53. Preferential activation of Notch1 signaling in p53 WT lymphomas was also observed at the RNA and protein level. Reporter mice for activation of Notch1 signaling revealed that total-body irradiation (TBI) enriched Notch1hi CD44+ thymocytes that could propagate in vivo after thymocyte transplantation. Mechanistically, genetic inhibition of Notch1 signaling in immature thymocytes prevented formation of radiation-induced thymic lymphoma in p53 WT mice. Taken together, these results demonstrate a critical role of activated Notch1 signaling in driving multi-step carcinogenesis of thymic lymphoma following TBI in p53 WT mice.
INTRODUCTION
Exposure of a significant part of the bone marrow to ionizing radiation is associated with an excess risk of developing blood cancers, including acute myeloid leukemia and acute lymphoblastic leukemia[1-4]. Experimentally, one of the widely used mouse models to study the carcinogenic effect of total-body irradiation (TBI) on hematopoietic cells is radiation-induced thymic lymphoma[5]. Dr. Henry Kaplan and colleagues first reported that TBI of C57 black mice leads to thymic lymphomas arising from T-cell precursors in the thymus[6, 7]. Lymphoma cells that develop in the thymus can become leukemic and disseminate to other organs including the spleen, liver, kidney and bone marrow[8]. Intriguingly, the incidence of radiation-induced thymic lymphoma after TBI depends on the daily radiation dose, number of fractions, time interval between each fraction, and the accumulated total radiation dose[7]. For example, exposure of C57BL/6 mice to 1.6 to 1.8 Gy TBI once per week for 4 consecutive weeks causes more than 90% of mice to develop thymic lymphoma within 8 months after irradiation. Even though this classic model has been utilized by many laboratories over the past 70 years, mechanisms by which TBI induces thymic lymphoma in wild-type mice are incompletely understood.
The application of high-throughput sequencing provides an opportunity to gain novel insights into mechanisms of radiation-induced carcinogenesis. For example, whole-exome sequencing (WES) studies of mouse solid tumors uncovered mutational signatures enriched in murine radiation-induced solid tumors compared to radiation naïve tumors[9, 10]. In addition, genomic analyses of human cancers reveal specific patterns of single-nucleotide substitutions and DNA deletions among radiation-associated solid cancers[11, 12]. Moreover, results from WES and whole-genome sequencing show distinct mutational patterns in radiation-induced cancers that develop in wild-type mice versus mice with pre-existing germline mutations. For example, using a mouse model of B-cell lymphoma, Patel et al. found that gamma-rays and heavy ion (56Fe) radiation markedly increased the number of insertions and deletions (indels) per tumor only in mice lacking one allele of the mismatch repair gene Mlh1 (Mlh1+/−)[13]. Also, using mice carrying different loss of function alleles of the tumor suppressor p53, Li et al. observed that the genomic landscape of radiation-induced cancers is influenced by radiation quality, germline p53 deficiency and tissue/cell of origin[14]. Collectively, findings from these genomic studies advance our understanding of mutational processes that underlie the development of radiation-induced cancers. However, although high-throughput sequencing studies have identified mutations in many putative driver genes, critical oncogenic pathways that are necessary to drive the development of radiation-induced cancers remain undefined.
Here, we performed WES to identify somatic mutations and copy number alterations of coding genes in thymic lymphoma that develop in mice following 4 weekly fractions of 1.8 Gy TBI. We conducted our study not only in wild-type (C57BL/6J) mice, but also in genetically engineered mice predisposed to thymic lymphomas including KrasLA1 mice[15] and p53+/− mice[16, 17]. As a control, we also analyzed thymic lymphomas in unirradiated mice that developed as a result of germline deletion of both p53 alleles in hematopoietic cells in order to compare the mutational signatures between thymic lymphomas that develop in irradiated mice versus radiation naïve mice. Importantly, utilizing the discoveries from whole-exome sequencing, we performed mechanistic studies to identify Notch1 signaling as a critical oncogenic pathway that drives the formation of radiation-induced thymic lymphoma in wild-type mice.
MATERIALS AND METHODS
Mouse strains and induction of thymic lymphomas
All animal procedures for this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Duke University. Mouse strains used to generate thymic lymphomas with and without irradiation were described previously including wild-type C57BL/6J mice (the Jackson Laboratory #000664), B6.SJL mice (the Jackson Laboratory #002014), KrasLA1 mice on a B6 background[15, 18], p53+/− mice[8, 16], Tie2Cre; p53FL/− mice[19], LckCre mice[20] (the Jackson Laboratory #003802), Rbpj floxed mice[21] and Rbpj-Venus reporter mice[22] (the Jackson Laboratory #020942). To study radiation-induced thymic lymphomas, mice were exposed to 1.8 Gy total-body irradiation (TBI) every week for 4 consecutive weeks (1.8 Gy x 4)[8]. The irradiation experiments were performed using the X-RAD 320 Biological Irradiator (Precision X-ray). Mice were irradiated at 50 cm from the radiation source with a dose rate of 3.18 Gy/min with 320 kVp X-rays, using 12.5 mA and a filter consisting of 2.5 mm Al and 0.1 mm Cu. The dose rate was measured with an ion chamber by members of the Radiation Safety Division at Duke University.
Whole exome sequencing (WES)
To prepare samples for WES, thymic lymphoma specimens stored in RNAlater (Invitrogen) and matched tail that were snap frozen were used for DNA extraction. DNA extraction was performed using the DNeasy Blood and Tissue Kit or the AllPrep DNA/RNA Mini Kit (Qiagen) per the manufacturer’s guidelines. WES was performed using previously described methods[9]. Sequencing libraries were prepared using the Agilent SureSelectXT Mouse All Exon Kit (S0276129). Methods for data analyses including somatic mutation calling, generation of somatic mutation plots, generation of mutational signatures, and assessment of copy number variations (CNVs) were described previously[9], with two exceptions. First, in this study, genes with estimated CNVs of absolute value greater than 0.3 were considered CNVs. Second, for the purposes of inclusion in the heat maps, mean CNV was calculated across all samples, and oncogenes with mean CNV greater than zero and tumor suppressor genes with mean CNV less than zero were included. An additional heat map of CNVs in Notch receptor and ligand genes was also generated, not limited by mean CNV. “Somatic variants were annotated using Ensembl Variant Effect Predictor (VEP)[23]. The impact of somatic variants was defined by the Sequence Ontology (SO) described in the Ensembl database: https://uswest.ensembl.org/info/genome/variation/prediction/predicted_data.html. The SO terms with MODERATE and HIGH impact were categorized as “non-synonymous” mutations.”
Notch1 Exon 1 Deletion PCR
DNA was extracted from thymic tissues using the Quick-DNA Miniprep Plus Kit (Zymo Research) according to the manufacturer’s protocols. PCR was performed using Phusion polymerase (New England Biolabs) using a 90 second elongation for 35 cycles. Primers used were: Notch1 1Δ F 5’- ATGGTGGAATGCCTACTTTGTA-3’, Notch1 1Δ R 5’- CGTTTGGGTAGAAGAGATGCTTTAC-3’ (Annealing Temp = 62.8°C, product size ~500 bp), Notch1 E30E31 F 5’- CACATGCACACATTCCCTAGC-3’, Notch1 E30E31 R 5’- GCTTTGCAGCATCTGAACGA-3’ (Annealing Temp = 64.4°C, Product size ~1000 bp).
qRT-PCR to detect mRNA expression
RNA was isolated from thymic tissues using Direct-zol RNA Miniprep Kit (Zymo Research) according to the manufacturer’s protocols. cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad). Gene expression for Hes1(Mm01342805_m1), Ikzf1(Mm01187878_m1), Notch1 5’(Mm00627192_m1), and Notch1 3’(Mm01326754_g1) were analyzed using Taqman probes and TaqMan™ Universal PCR Master Mix (ThermoFischer) with Gapdh(Mm99999915_g1) as an internal control. Gene expression for Dtx1 (qMmuCID0027483, BioRad), Myc (qMmuCID0006528, BioRad) and Rbpj (Mm.PT.58.7684670, IDT) were analyzed using PrimePCR SYBR Green Assays and SsoAdvanced Universal SYBR Green Supermix (BioRad) with TBP (Fwd: 5’-TGCACAGGAGCCAAGAGTGAA-3’, Rev: 5’-CACATCACAGCTCCCCACCA-3’) as an internal control. Samples were run in triplicate on the QuantStudio 6 Flex Real-Time PCR System.
qRT-PCR to detect DNA copy numbers
Copy number variants determined by WES for NF2 and Ikzf1 were tested using the same genomic DNA. Gene expression was measured using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) in the QuantStudio 6 Flex Real-Time PCR System in triplicate with 10 ng of gDNA per well. We utilized the following primer sequences for the target genes: NF2E12 (Fwd: 5’- GAGGAGGAGGCCAAGCTTTT-3’, Rev: 5’-TCCTCTCTGACTCCTCAGCC-3’); Ikzf1E5 (Fwd: 5’-AATGAATGGCTCCCACAGGG-3’, Rev: 5’-GAACCATGAGCACATTGGGC-3’). qRT-PCR expression results from lymphoma samples were normalized using ΔΔCt analysis, with the TFRC gene and paired tail samples as internal controls. The TFRC primer sequence (Fwd: 5’–TGTAATTGGGTATAGGCTTGCA–3’, Rev: 5’–ATCCTGTACTCAAGAATTGGCT–3’) has been previously described24.
Immunoblotting
Proteins were extracted from cells or tissues using PARIS kit (Invitrogen). The protein concentration was determined using Pierce Rapid Gold BCA Protein Assay (Thermo Scientific) according to the manufacturer’s protocol. Total protein (40ug) was loaded for electrophoresis into 4–20% sodium dodecyl sulfate polyacrylamide gels (Bio-Rad). Separated proteins were transferred to a 0.2 μm nitrocellulose membrane (Bio-Rad). Membranes were blocked with Odyssey Blocking Buffer (Li-Cor) diluted 1:1 with tris buffered saline with 0.1% Tween 20 (TBS-T). Protein levels were detected using antibodies against Intracellular Domain of Notch1 (ICN) (Val1744 Cell Signaling Technology #4147, D3B8, 1:500 dilution) and cylcophylin A (Bio-Rad, VMA00535, 1:1000 dilution) followed by secondary fluorescently-conjugated IRDye 800CW and IRDye 680RD antibodies (Li-Cor, 1:15,000 dilution). Bands were visualized at 700 and 800nm using the Odyssey CLX imaging system (Li-Cor).
Sanger sequencing and targeted sequencing
Lymphoma single nucleotide variants (SNVs) of Notch1, Ikzf1, Pten, Akt1, and p53 determined by WES were validated by Sanger sequencing. Identification of somatic variants in exon 34 of the Notch1 gene in additional lymphoma samples was conducted using targeted sequencing (AMPLICON-EZ, GENEWIZ). The sequences were amplified by PCR using AccuPrime Taq DNA Polymerase (Thermo Fisher Scientific) and performed on the same genomic DNA sent for WES. Primer sequences are described in Table S1. The PCR Protocol for Notch1 exon 34 primers 3 through 5 and Ikzf1 exon 5 was: denaturing stage of 94°C for 2 minutes; 40 cycles of 94°C for 30 seconds, annealing temperature for 30 seconds, and 68°C for 60 seconds; followed by 4°C forever. Notch1 exon 34 primers 1 and 2 differed only in extending the elongation step of 68°C, for 90 seconds instead of 60 seconds. The PCR protocols for Notch1 exons 27 and 26, Ikzf1 exons 6, 8, and 9, TRP53 exon 7, Pten exons 7 and 8, and Akt1 exon 4 utilized the same protocol except for the elongation step, which was 45 seconds. Sequencing was performed by Eton Bioscience, Inc or GENEWIZ. DNA variant analysis was conducted using Mutation Surveyor (Softgenetics).
Mouse and human cell culture
Mouse lymphoma cells were isolated by harvesting the total thymus from mouse in 5 mL HSC buffer: 50 mL FBS, 4 mL of 0.5M EDTA, and 1 L HBSS buffer (Invitrogen). Thymus pieces are homogenized and transferred through cell strainer in 3 mL of HSC buffer. After centrifugation (300 g for 5 minutes), cells are resuspended with 2 mL RBC lysing buffer (Lonza) for 3 minutes, then centrifuged again. Cells were cultured in Opti-MEM (Gibco 11058-021) with 10% FBS, 1x anti-anti, and 1x 2-Mercaptoethanol (Gibco). Mouse lymphoma cells were plated at 1 x 106 cells/mL and treated with either 1 μM DAPT (GSI-IX, LY-374973) (Selleckchem, S2215) or the vehicular control of Dimethylsulfoxide (DMSO) at equal volume. All treatment groups were incubated using standard culture conditions and harvested at 24 or 48 hours post treatment.
Human lymphoblastic leukemia cell line MOLT-4 (ATCC CRL-1582) was purchased from Duke Cell Culture Facility. These cells were plated at 1 x 106 cells/mL in 10 mL of RPMI 1640 (Gibco 11875-093, supplemented with 10% FBS, 1x anti-anti, and 1x Sodium Pyruvate) and harvested 48 hours later for protein extraction. They were passaged for 3 times between thawing and the use in the described experiment. These cells were not tested for mycoplasma nor authenticated by our laboratory.
Flow cytometry
Total thymocytes were isolated from the thymus in HSC buffer (HBSS with Ca2+ and Mg2+, 5% fetal bovine serum, 2 mM EDTA). Red blood cells (RBC) were lysed using ACK lysing buffer (Lonza). 1 x 106 live thymocytes were blocked with a rat anti-mouse CD16/32 antibody (BD Pharmingen) and stained with a combination of the antibodies including PE-Cy5 conjugated anti-mouse CD4 (clone: GK1.5, eBioscience), PE conjugated anti-mouse CD8 (clone: 53-6.7, eBioscience), APC conjugated anti-mouse CD44 (clone: IM7, eBioscience), BV421 conjugated anti-mouse CD25 antibodies (clone: PC61, BioLegend). FITC conjugated anti-mouse CD45.2 (clone: 104, eBioscience), and APC-eFluor780 conjugated anti-mouse CD45.1 (clone: A20, e eBioscience). All antibodies were diluted 1:400. Data were collected from at least 100,000 single cells by FACSCanto (BD Pharmingen) and analyzed by FlowJo (Tree Star, Inc) without knowledge of the genotype or treatment by a single observer (C-LL).
Thymocyte transplantation
Thymocyte transplantation was performed according to a method described previously[24] with slight modifications. Individual recipient mice were exposed to 5 Gy TBI and then received 2 x 107 thymocytes from donor mice through intravenous injection approximately 6 hours after irradiation. Twenty-one days after thymocyte transplantation, thymocytes were harvested from recipient mice for subsequent analysis using flow cytometry.
Statistics
Statistical results presented here were post-hoc analyses. The p-values reported were two-sided and were not adjusted for multiple testing. Pairwise comparisons of quantitative phenotypes were based on the Mann-Whitney U test. Comparisons of three or more groups used the Kruskal-Wallis test. Comparisons of proportions were based on the chi-squared test. Time-to-event analyses were based on the Cox proportional hazards model and reported p-values are from the score test. Kendall’s W was used to test concordance of estimated CNV by qPCR and WES. Inferential analyses were conducted using the R statistical environment[25] and extension packages from the Comprehensive R Archive Network (https://cran.r-project.org/) and the Bioconductor project[26]. Box-and-whisker plots presented in the figures were constructed as previously described[9]. For all experiments, data are presented as mean ± SE and each data point represents one mouse or tumor sample.
Data availability
The knitr package[27] was used to generate dynamic reports and conduct the analyses pertaining to WES results, in accordance with the principles of reproducible analysis. Duke’s GitLab was used for source code management (https://gitlab.oit.duke.edu), and the code for replicating the figures and statistical analyses pertaining to WES results presented here has been made available through a public source code repository (https://gitlab.oit.duke.edu/dcibioinformatics/pubs/kirsch-lee_lymphoma). The sequencing data along with the called mutations in vcf format have been deposited into the National Center for Biotechnology Information Sequence Read Archive under project ID PRJNA627412.
RESULTS
Somatic mutation analysis of thymic lymphomas from irradiated and unirradiated mice
Thymic lymphomas were generated in irradiated and unirradiated mice on four distinct genotypes (Figure S1). Radiation-induced thymic lymphomas were generated in wild-type (WT) (C57BL/6J) mice, KrasLA1 mice[15] or p53+/− mice[16, 17] that were exposed to 1.8 Gy total-body irradiation (TBI) per week for 4 consecutive weeks[7, 8]. Unirradiated Tie2Cre; p53FL/− mice (Non-IR p53−/−) in which both alleles of the p53 gene are deleted in endothelial cells and hematopoietic cells[19] were used to generate thymic lymphomas in the absence of irradiation. Clonality analyses that assess TCR rearrangement have shown that radiation-induced thymic lymphomas and Non-IR p53−/− thymic lymphomas are mostly oligoclonal[8, 28]. The median time for the formation of radiation-induced thymic lymphoma in WT mice was approximately 150 days after irradiation. The latency of thymic lymphomas in KrasLA1 mice and p53+/− mice was reduced to approximately 100 days post-irradiation because these genetically engineered mice are predisposed to develop thymic lymphoma[15, 17]. The latency of radiation-induced thymic lymphomas is comparable to time of spontaneous thymic lymphoma development in Non-IR p53−/− mice (Figure S1).
Thymic lymphomas and paired normal tissues were subjected to WES to identify somatic mutations that occurred specifically in tumors (Figure 1 and Table S2). This analysis showed that while the total number of called somatic mutations per tumor differed between thymic lymphomas induced by TBI alone, TBI plus genetic predisposition (KrasLA1 and p53+/−), and the loss of both alleles of p53 in the absence of TBI (Kruskal-Wallis p-value= 0.022), the number of called nonsynonymous somatic mutations was not significantly different among different lymphoma groups (Kruskal-Wallis p-value=0.068, Figure 1, a and b). Notably, our data revealed that the median total called mutations and called nonsynonymous mutations were the lowest in IR-induced KrasLA1 lymphomas and the highest in Non-IR p53−/− lymphomas (Figure 1, a and b). In addition, we observed that two radiation-induced lymphomas, tumor numbers 5015 (WT) and 5020 (KrasLA1), exhibited substantially higher numbers of called somatic mutations compared to other lymphomas (Figure 1a). Examination of the proportion of indels and SNVs did not show statistically significant difference among different lymphoma groups (Figure 1, c to e and Figure S2, a and b). In addition, unsupervised hierarchical clustering of thymic lymphomas based on data from SNVs did not differentiate radiation-induced thymic lymphomas from spontaneous tumors that arose in Non-IR p53−/− mice (Figure 1f). However, this analysis revealed a distinct cluster comprised tumor numbers 5015 and 5020, two radiation-induced thymic lymphomas with the highest number of somatic mutations (Figure 1a).
Figure 1.
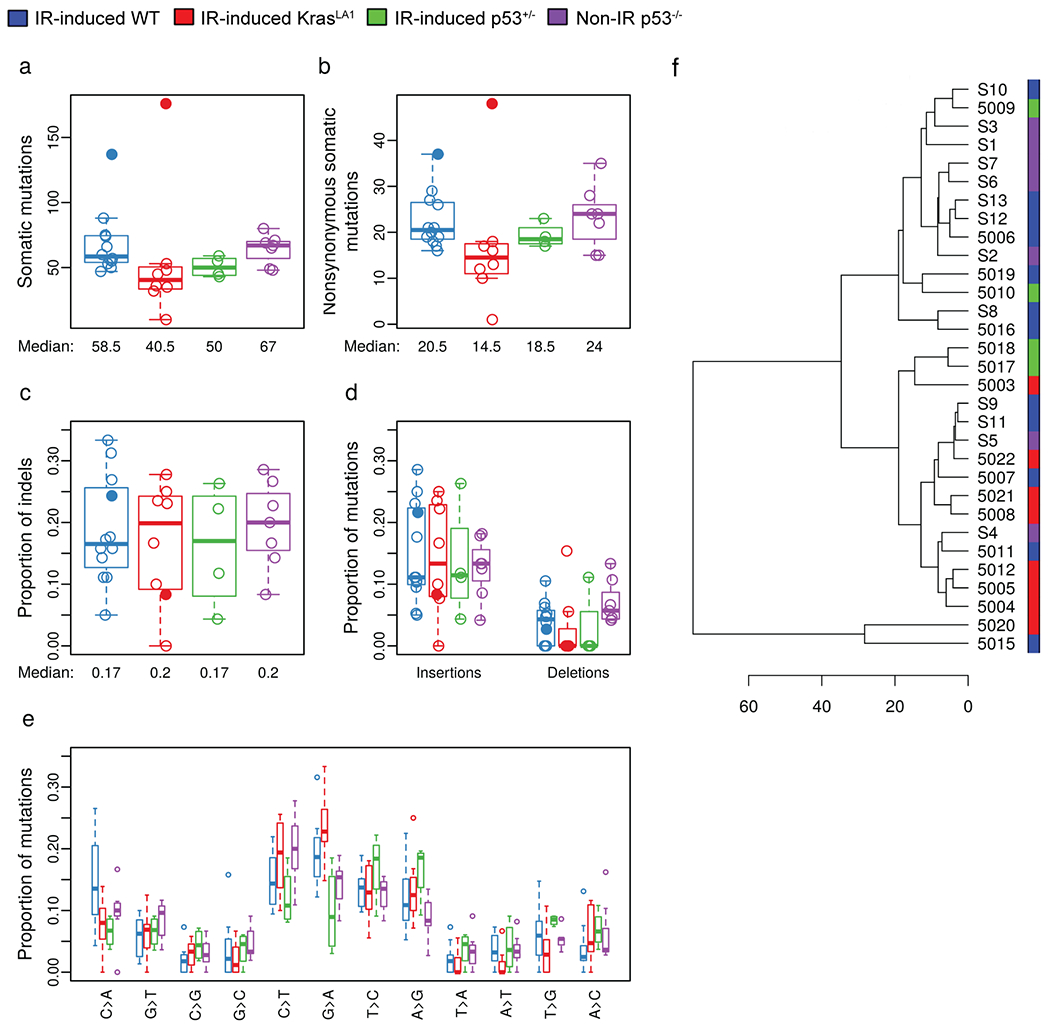
Somatic mutation analysis of murine thymic lymphomas. Thymic lymphomas were harvested from the following cohorts of mice: IR-induced WT (blue), IR-induced KrasLA1 (red), IR-induced p53+/− (green) and Non-IR p53−/− (purple). a, The number of total somatic mutations per tumor. b, The number of somatic nonsynonymous mutations per tumor. c, The proportion of insertion-deletions (indels) within nonsynonymous mutations. d, The proportion of insertions or deletions within nonsynonymous mutations. e, The proportions of different single nucleotide variants. f, Unsupervised hierarchical clustering of thymic lymphomas based on data from single nucleotide variations. All panels illustrate the data for n=31 tumors. Data from lymphomas that had substantially higher number of somatic mutations (5015 and 5020) were labeled or denoted by closed circles.
To generate mutational signatures common to each lymphoma genotype, we performed a signature analysis as described by Alexandrov, et al.[29] and compared mutational signatures of mouse lymphomas with the Catalogue Of Somatic Mutations In Cancer (COSMIC) mutational signatures of human cancers (https://cancer.sanger.ac.uk/cosmic/signatures) (Figure S3). Among radiation-induced thymic lymphomas, all three genotypes exhibited cosine-similarity with two groups of COSMIC mutational signatures of human cancers (Figure S3, a to c): defective DNA mismatch repair (Signatures 6, 15 and 26) and a signature with unknown etiology that has been found in all cancer types (Signature 5). Remarkably, the same groups of signatures were also found in Non-IR p53−/− thymic lymphomas (Figure S3d). In addition, spontaneous deamination of 5-methylcytosine (Signature 1) was found in IR-induced p53 WT, IR-induced KrasLA1 and Non-IR p53−/− lymphomas, while a signature of defects in polymerase-eta (Signature 9) was observed in IR-induced p53+/− lymphomas. Together, our results from the analyses of somatic mutations indicate that murine radiation-induced thymic lymphomas do not typically harbor higher number of somatic mutations and distinct mutational signatures compared to thymic lymphomas driven by the loss of functional p53.
Copy number variations analysis of murine thymic lymphomas
WES data was evaluated using CODEX2 for CNVs[30]. Somatic deletions and amplifications were detected across all 19 mouse chromosomes in the thymic lymphomas (Figure 2a and Table S3). Among the four lymphoma cohorts, IR-induced KrasLA1 and IR-induced WT lymphomas had a generally low number of genes with detected CNVs. Interestingly, tumor numbers 5015 and 5020, which exhibited a higher number of somatic mutations compared to other lymphomas, also had the highest numbers of CNVs detected, compared to other tumors within the same cohort (Figure 2, b to d). In contrast, the number of genes with CNVs detected was significantly higher in IR-induced p53+/− lymphomas compared to IR-induced KrasLA1 and IR-induced WT lymphomas (pairwise Mann-Whitney U p-values 0.004 and 0.001, respectively, Figure 2b). While lymphomas with the greatest number of genes affected by CNVs were initiated in p53+/− mice by TBI with loss of p53 function, the number of genes with detected CNVs was also significantly higher in Non-IR p53−/− lymphomas compared to IR-induced KrasLA1 and IR-induced WT lymphomas (pairwise Mann-Whitney U p-values 0.014 and 0.028, respectively, Figure 2b). In sum, our results suggest that in mouse models of thymic lymphomas, TBI cooperates with loss of p53 to increase the number of CNVs.
Figure 2.
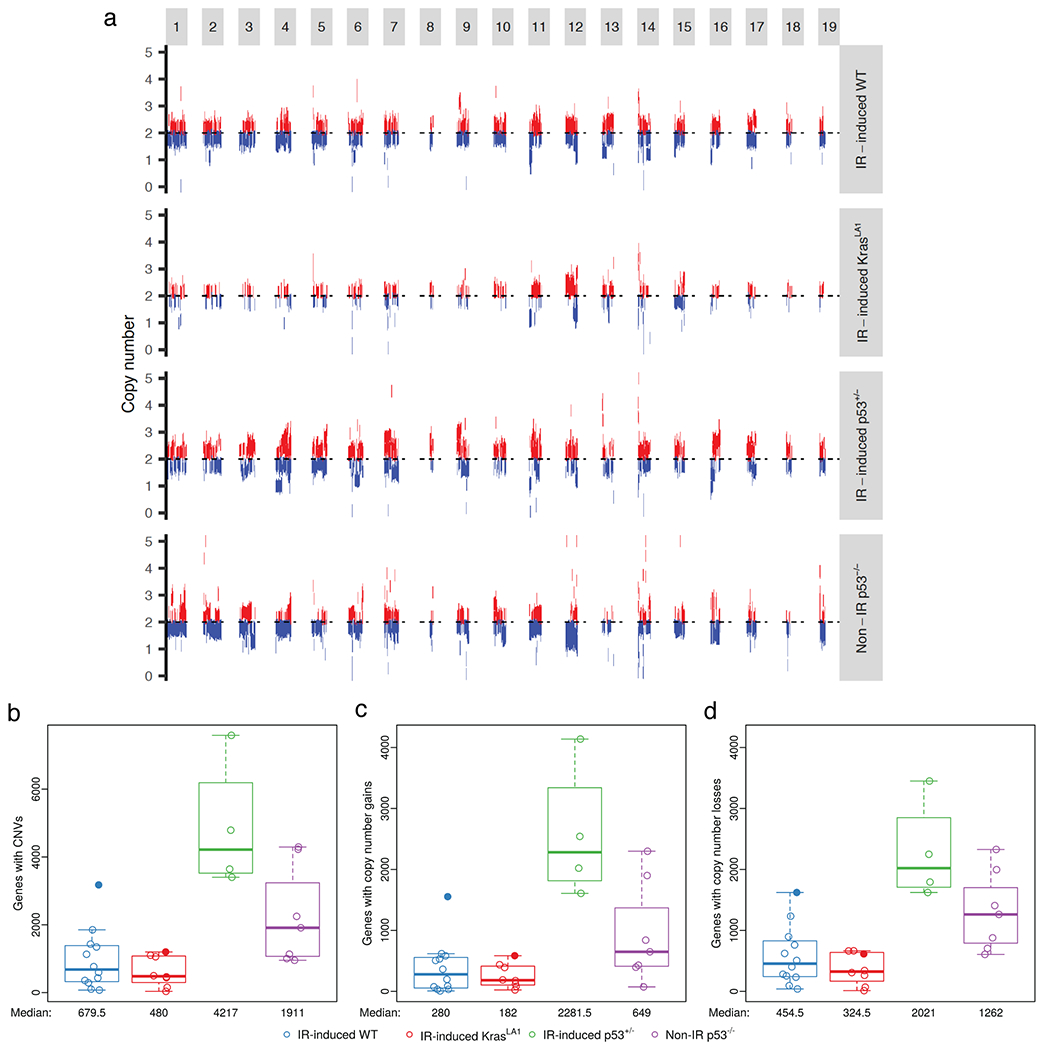
Somatic copy number variations (CNVs) in thymic lymphomas. a, Schematic of CNVs across the 19 mouse chromosomes. Results represent pooled data from lymphomas of the same cohort. DNA deletions and amplifications are labeled by blue and red, respectively. b, The number of genes affected by CNVs. c, The number of genes with copy number gains. d, The number of genes with copy number losses. Panels illustrate the data for n=31 tumors. Data from lymphomas that had substantially higher mutational loads (5015 and 5020) were denoted with closed circles.
Identification of putative genetic drivers underlying T-lymphomagenesis
To investigate genetic alterations that potentially drive oncogenesis of thymic lymphomas, we identified genes from the COSMIC database that were impacted by somatic mutations and CNVs. In IR-induced WT lymphomas, nonsynonymous mutations were found in 1 to 5 COSMIC genes (median = 3) per tumor (Figure 3a). Even fewer nonsynonymous mutations in COSMIC genes were found in thymic lymphomas from mice that harbored a germline tumor-initiating mutation including irradiated KrasLA1 mice, irradiated p53+/− mice, as well as radiation naïve p53−/− mice (Figure 3a). The number of COSMIC genes altered by CNVs in radiation-induced lymphomas from either WT mice or KrasLA1 mice was significantly lower compared to radiation-induced lymphomas in p53+/− mice (Kruskal-Wallis p-value = 0.001, Figure 3b). The tumors from unirradiated p53−/− mice exhibited an intermediate level of CNVs in COSMIC genes (Figure 3b). Of note, tumor numbers 5015 and 5020 also had the highest number of COSMIC genes affected by CNVs compared to other tumors within the same cohort (Figure 3b). We also quantified the number of nonsynonymous mutations and CNVs in non-COMSIC genes (Figure 3, c and d). The number of nonsynonymous mutations and CNVs are substantially higher in non-COSMIC genes compared to COSMIC genes. Non-IR p53−/− lymphomas and IR-induced p53+/− lymphomas harbored the highest median nonsynonymous mutations and CNVs in non-COSMIC genes, respectively (p-value = 0.033 by Kruskal-Wallis for panel c, and p-value = 0.002 by Kruskal-Wallis for panel d).
Figure 3.
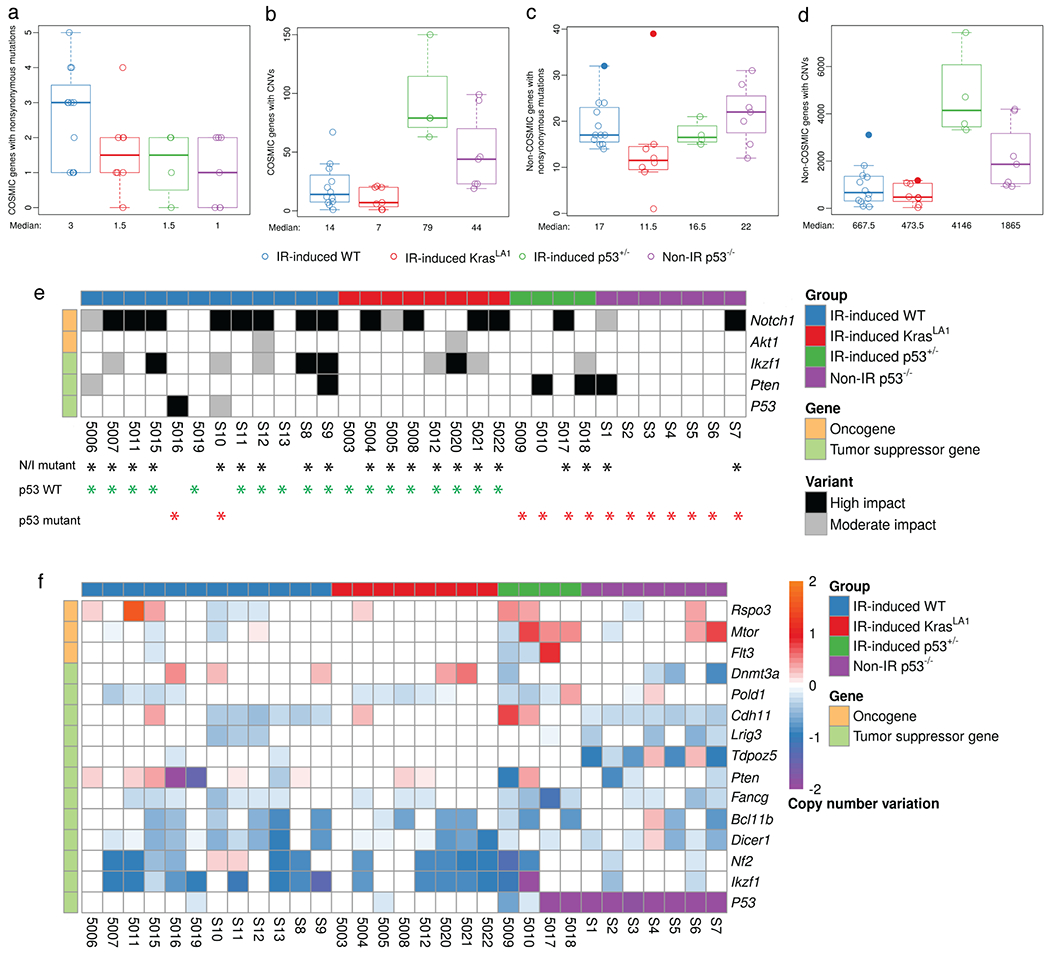
Somatic genetic alterations in COSMIC genes across murine thymic lymphomas. a to d, The number of COSMIC (a-b) and non-COSMIC genes (c-d) affected by nonsynonymous mutations and CNVs per tumor, respectively. Data from lymphomas that had substantially higher mutational loads (5015 and 5020) were denoted with closed circles. e, Recurrent mutations in COSMIC genes across thymic lymphomas. Black asterisk: tumors with either Notch1 and/or Ikzf1 mutations (I/N mutant); Green asterisk: tumors with wild-type p53 (n=18); Red asterisk: tumors with p53 mutations or harvested from p53+/− or p53−/− mice (n=13). f, COSMIC oncogenes that show a mean copy number gain and COSMIC tumor suppressor genes that show a mean copy number loss in thymic lymphomas. Genes are ordered within gene type by mean copy number across all samples. All panels illustrate the data for n=31 tumors.
We next examined individual COSMIC genes affected by nonsynonymous somatic mutations and/or CNVs across lymphomas from mice from the four genotypes (Figure 3, e and f). Somatic mutations identified by WES were validated using Sanger Sequencing (Table S4). Our data revealed genotype-specific genetic alterations in signaling pathways, including Notch signaling, PI3K/AKT/mTOR signaling, MEK/ERK signaling, epigenetic modifiers, Hippo pathway, Wnt signaling and the DNA damage response (Table S5). Remarkably, among lymphomas that arose in irradiated WT and KrasLA1 mice, 90% (18/20) of them retained WT p53 (Figure 3e). Only two of these lymphomas harbored mutations in p53: tumors 5016 (missense mutation) and S10 (splicing mutation). Furthermore, we observed that p53 WT and p53 deficient lymphomas harbored distinct patterns of mutations that affect Notch signaling. In lymphomas that retained WT p53, approximately 83% (15/18) of these tumors harbored mutations in Notch1 and/or Ikzf1, a negative regulator of Notch1 signaling (Figure 3e). Three p53 WT lymphomas did not harbor mutations in either Notch1 or Ikzf1: tumors 5019 (mutations in Ptpn11, Stil, Bcl11b and Ptprb), S13 (mutation in Ezh2) and 5003 (Mutation in Kras) (Figure S4). In contrast to p53 WT lymphomas, mutations in Notch1 or Ikzf1 only occurred in around 38% (5/13) of p53 deficient lymphomas. (chi-squared test of proportions p-value=0.028, Figure 3e) Nonsynonymous mutations in Notch1 were exclusively found in exon 27 that encodes the heterodimerization domain or HD (all missense mutations) and exon 34 that encodes the proline, glutamic acid, serine, threonine-rich or PEST domain (missense, stop-gain and/or frameshift mutations) (Figure S5a). Also, nonsynonymous mutations in Ikzf1 were exclusively found in exons 5/6 that encode the DNA binding domain (all missense mutations) and exon 9 (stop-gain or frameshift mutations) (Figure S5b). These mutations recapitulate hotspot mutations in human NOTCH1 and IKZF1 observed in acute lymphoblastic leukemia patients[31, 32]. Given the small sample size of IR-induced p53+/− lymphomas that we analyzed by WES, and the lack of IR-induced p53−/− lymphomas, we performed targeted sequencing to detect somatic variants in exon 27 and exon 34 of the Notch1 gene in an additional five IR-induced p53+/− and 2 IR-induced p53−/− thymic lymphomas. Nonsynonymous mutations in Notch1 were detected in two out five IR-induced p53+/− lymphomas and neither of the two IR-induced p53−/− lymphomas (Table S6)
Similar to our findings from somatic mutational analysis of COSMIC genes, we observed a notable difference in CNVs of Ikzf1 between p53 WT and p53 deficient lymphomas (Figure 3f). While approximately 72% (13/18) of lymphomas that retained wild-type p53 show loss of Ikzf1, loss of Ikzf1 was found in only 38% (5/13) of p53 deficient lymphomas. CNVs of Ikzf1 were validated using a genomic qPCR assay (Figure S6). In contrast, Mtor, a critical gene that regulates cell growth and proliferation[33], was amplified in a subset of IR-induced p53+/− and Non-IR p53−/− lymphomas (5/11) (Figure 3f). In addition, examination of CNVs in Notch receptors (Notch1, Notch2 and Notch3) and Notch ligands (Dll1, Dll4 and Jag1) revealed that Notch1 was substantially amplified in a Non-IR p53−/− lymphomas (tumor S3) (Figure S7). Collectively, our analyses of somatic mutations and CNVs reveal distinct genetic alterations that occur in thymic lymphomas with and without wild-type p53. In particular, our findings demonstrate that p53 wild-type thymic lymphomas exhibit a high frequency of genetic alterations in Notch1 and Ikzf1, two critical genes that regulate the Notch1 signaling pathway.
Notch1 signaling is dysregulated primarily in thymic lymphomas harboring wild-type p53
Our findings of frequent Notch1 pathway mutations in radiation-induced thymic lymphomas that retained wild-type p53 prompted us to further examine Notch1 signaling among the different lymphoma cohorts. One critical consequence of Notch1 and Ikzf1 mutations is increased expression of the intracellular domain of Notch1 (ICN)[34]. Our data showed that while the expression of 5’ Notch1 mRNA (exon 9 – 10) was not significantly different between thymic lymphomas and normal thymuses from mice of the same genotype (Figure 4a), the expression of 3’ Notch1 mRNA (exon 32 – 33), which encodes for part of the ICN protein, was significantly increased specifically in IR-induced WT and IR-induced KrasLA1 thymic lymphomas compared to normal thymuses (Mann-Whitney U p-values 0.008 and 0.023, respectively, Figure 4b). We previously showed that radiation-induced thymic lymphomas comprise predominately CD4+CD8+ cells[19]. However, we did not observe an increase in the expression of 3’ Notch1 mRNA in CD4+CD8+ thymocytes compared to total thymocytes from unirradiated C57BL/6J mice (Figure S8). These results indicate that the overexpression of 3’Notch1 mRNA in lymphomas is not due to the difference between CD4+CD8+ thymocytes and total thymocytes.
Figure 4.
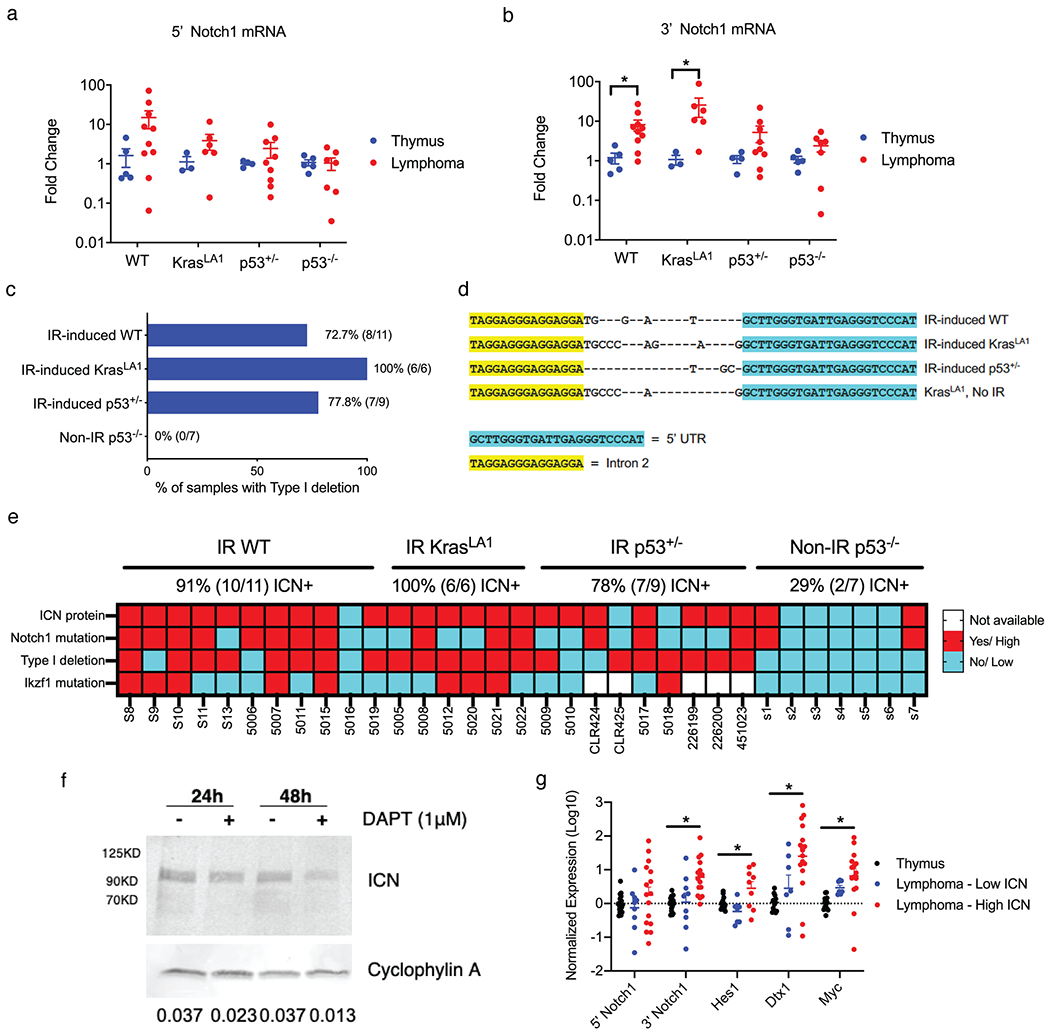
Notch1 signaling is upregulated in thymic lymphomas harboring functional p53. a-b, mRNA expression of 5’ and 3’ region of the Notch1 gene. Results were compared between lymphomas and control thymuses from mice of the same genotype. Data are presented as mean ± SE. Each dot represents one mouse. *P<0.05 by Mann-Whitney U test. c, Detection of Type 1 deletions of the Notch1 gene among thymic lymphomas of different genotypes. d, Sequencing results of the Type 1 deletion PCR product. Sanger sequencing showed that Type 1 deletions resulted from a specific gene recombination between the 5’ UTR and Intron 2. e, Summary of intracellular Notch1 (ICN) protein expression, Notch1 mutations, Notch1 Type 1 deletions and Ikzf1 mutations across thymic lymphomas of different genotypes. f, Expression of ICN protein in IR-induced WT lymphoma cells treated with DMSO or the γ-secretase inhibitor DAPT in vitro. Cyclophylin A was used as a control for protein loading. Quantification of Western blot data is shown as ICN/ Cyclophylin A. g, The mRNA expression of 5’ and 3’ regions of Notch1 as well as transcriptional targets of Notch1. Each dot represents one mouse. *P<0.05 by Kruskal-Wallis test.
It has been shown that high expression of 3’Notch1 mRNA in murine thymic lymphomas can be attributed to deletions within the 5’ region of the Notch1 locus. For example, small deletions of exons 1 and 2 of Notch1, termed Type 1 deletions, can result in overexpression of a truncated 3’ mRNA transcript from a cryptic promoter in exon 25 that is translated into the ICN protein[35] (Figure S9a). To explore this potential mechanism underlying Notch1 overexpression, we performed PCR to detect Type 1 deletions of the Notch1 gene in our tumors (Figure 4c and Figure S9b). We observed that 100% (6 out of 6) of IR-induced KrasLA1 thymic lymphomas harbored Type 1 deletions, while Type 1 deletions were detected in around 70% of IR-induced WT lymphomas (8 out of 11) and IR-induced p53+/− lymphomas (7 out of 9). Interestingly, Type 1 deletions were not detected in any of the Non-IR p53−/− thymic lymphomas that we examined (0 out of 7) (Figure 4c). To further dissect the contribution of irradiation vs. p53 loss to the formation of Type 1 deletion in thymic lymphomas, we examined thymic lymphomas from an independent cohort of unirradiated and irradiated mice. We observed that thymic lymphomas from KrasLA1 mice and LckCre; LSL-KrasG12D mice, in which a mutant KrasG12D is activated specifically in thymocytes[36], all exhibited Type 1 deletions independent of radiation exposure (Figure S9c). The DNA sequence of Type 1 deletion is also conserved among lymphomas that developed in irradiated and unirradiated KrasLA1 mice (Figure 4d). In contrast, the frequency of Type 1 deletions was substantially lower in thymic lymphoma from both unirradiated and irradiated p53−/− mice (Figure S9c). Thus, these results suggest that the absence of Type 1 deletions in Non-IR p53−/− lymphomas is not likely due to the lack of radiation exposure.
We next determined the impact of Notch1 mutations, Notch1 Type 1 deletions and Ikzf1 mutations on the expression ICN protein. Western blot data showed that ICN protein was expressed in 91% of IR-induced WT (10 out of 11), 100% of IR-induced KrasLA1 (6 out of 6) and 78% of IR-induced p53+/− (7 out of 9) thymic lymphomas, whereas ICN protein was only detected in 29% of Non-IR p53−/− (2 out of 7) thymic lymphomas that we examined (Figure 4e and Figure S10). The abundance of ICN protein was reduced in p53 WT thymic lymphoma cells treated with a γ-secretase inhibitor DAPT in vitro (Figure 4f). Of note, thymic lymphomas that expressed ICN protein, expect for one tumor 5010, all harbored either Notch1 mutations, Notch1 Type 1 deletions or both. In contrast, ICN-negative thymic lymphomas, expect for tumor CLR425, were negative for both Notch1 mutations and Notch1 Type 1 deletions (Figure 4e).
To determine the biological effects of ICN protein overexpression on the activation of Notch1 signaling, we quantified the mRNA expression of several transcriptional targets of Notch1. We observed that low-ICN expressing lymphomas, normal thymuses, and lymphomas expressing high ICN at the protein level not only exhibited differential mRNA expression of 3’ Notch1, but also the Notch1 transcriptional targets Hes1, Dtx1 and Myc (Kruskal-Wallis p-values 0.0002, 0.0335, 0.0009, 0.0002, respectively, Figure 4g).
Collectively, our results demonstrate that Notch1-mediated signaling is dysregulated at the DNA, RNA and protein levels in the vast majority of IR-induced WT and IR-induced KrasLA1 thymic lymphomas that we examined. In contrast, only a subset Non-IR p53−/− thymic lymphomas that we studied showed aberrant activation of Notch1. These findings reveal that Notch1 signaling is dysregulated predominately in thymic lymphomas harboring WT p53.
Inhibition of Notch1 signaling prevents the formation of radiation-induced thymic lymphoma in wild-type mice
To identify cells that express activated Notch1 signaling in vivo, we induced thymic lymphomas in Rbpj-Venus reporter mice[22]. Rbpj is a transcriptional co-activator of Notch1[37]. It has been shown that cells positive for Venus in Rbpj-Venus reporter mice exhibit elevated Notch1 activity[22]. We found that the majority of tumor cells were Venus+ in IR-induced WT and IR-induced KrasLA1 thymic lymphomas (Figure S11, a and b). Notably, Venus+ cells formed a distinct population that expressed surface markers CD4 and CD8 as well as CD44. In parallel to the reporter assays, we examined thymocyte populations that can propagate in vivo using a thymocyte transplantation assay[24]. We transplanted thymocytes harvested from WT mice 16 weeks after TBI and age-matched unirradiated controls into irradiated recipient mice. The engraftment of donor thymocytes were analyzed 21 days after transplantation (Figure 5a). The number of total thymocytes was significantly higher in recipient mice that received cells from irradiated donors (Mann-Whitney U p-value = 0.024, Figure 5b). Moreover, flow cytometry analysis indicated that thymocytes from irradiated donors showed a significantly higher percentage of engraftment into the recipient thymus compared to thymocytes from unirradiated donors (Mann-Whitney U p-value = 0.028, Figure 5c). Analysis of cell surface markers indicated that engrafted thymocytes were almost exclusively CD4+CD8+ and CD44+ (Figure 5, d to f). To determine the activity of Nocth1 signaling in engrafted thymocyte, we performed thymocyte transplantation using thymocytes harvested from Rbpj-Venus reporter mice, backcrossed to a B6.SJL background, 19 weeks after TBI. We observed that the majority of donor-derived thymocytes that engrafted in the recipients were Venus+ (Figure 5g). Further analysis of engrafted Venus+ thymocytes showed a significant enrichment for CD44+ cells (Figure 5h). In addition, we examined the changes in CD44+ cells in paired thymocytes before and after thymocyte transplantation. Thymocyte transplantation was performed using CD4+CD8+ thymocytes isolated from KrasLA1 mice 8 weeks post-1.8 Gy x 4 TBI. We found that in two out of four mice the percentage of CD44+ thymocytes were substantially increased in engrafted thymocytes compared to donor thymocytes before thymocyte transplantation (Figure 5i).
Figure 5.
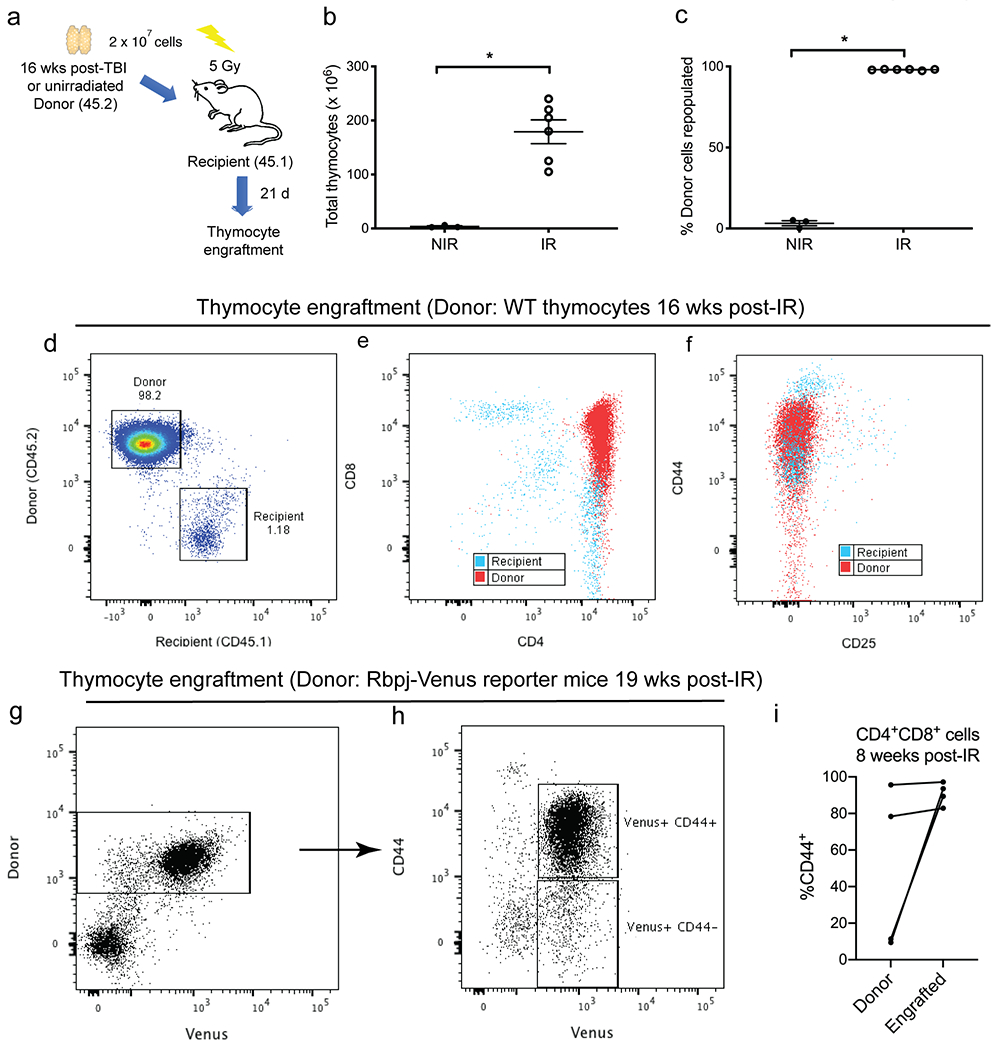
Engrafted thymocytes from irradiated donors are enriched for CD44. a, 2 x 107 donor thymocytes were harvested from age matched C57BL/6J mice (CD45.2) either 16 weeks after 1.8 Gy x 4 TBI (IR) or unirradiated (NIR). Donor thymocytes were injected intravenously into B6.SJL recipients (CD45.1) 6 hours after 5 Gy TBI. Thymocytes were harvested from recipients 21 days after transplantation. b and c, The number of total thymocytes (b) and the engraftment of donor thymocytes (c) were assessed in recipients that received thymocytes from donors that were either unirradiated (NIR, n=3) or previously treated with TBI (IR, n=6). Data are presented as mean ± SE. *P<0.05 by Mann-Whitney U test. d-f, Engraftment of thymocytes harvested from wild-type mice 16 weeks post-irradiation. Representative flow cytometry plots that distinguish thymocytes derived from donor (CD45.2) vs. recipients (CD45.1). g-h, Engraftment of thymocytes harvested from Rbpj-Venus reporter mice 19 weeks post-irradiation. Donor thymocytes were injected intravenously into C57BL/6J recipients (CD45.1) Donor-derived Venus+ thymocytes were predominately CD44+. i, Transplantation of CD4+CD8+ thymocytes sorted from KrasLA1 mice 8 weeks post-irradiation. The percentage of CD44+ cells within the CD4+CD8+ population before (donor) and after transplantation (engrafted) was analyzed by flow cytometry. Each dot represents one mouse.
We next quantified the percentage of Venus+ thymocytes and CD44+ thymocytes from Rbpj-Venus reporter mice without irradiation as well as 8 and 14 weeks after irradiation. Although we did not observe changes in the percentage of Venus+ thymocytes 8 and 14 weeks after irradiation (Figure 6a), the percentage of CD44+ thymocytes were significantly increased at each time point post-irradiation (Mann-Whitney U p-value = 0.004 for No IR vs. 8 weeks and p-value = 0.002 for No IR vs. 14 weeks, Figure 6b). Remarkably, we observed that while only around 30% of Venus+ thymocytes were positive for CD44 in unirradiated mice, the percentage of Venus+ thymocytes that are also CD44+ was increased to more than 80% 8 and 14 weeks after irradiation (Mann-Whitney U p-value = 0.01 for No IR vs. 8 weeks and p-value = 0.003 for No IR vs. 14 weeks, Figure 6c). These data suggest that TBI did not cause an overall increase in Venus+ cells during the latent period of lymphomagenesis. However, the subset of Venus+ thymocytes that are positive for CD44+ thymocytes was significantly enriched by irradiation (Figure 6d).
Figure 6.
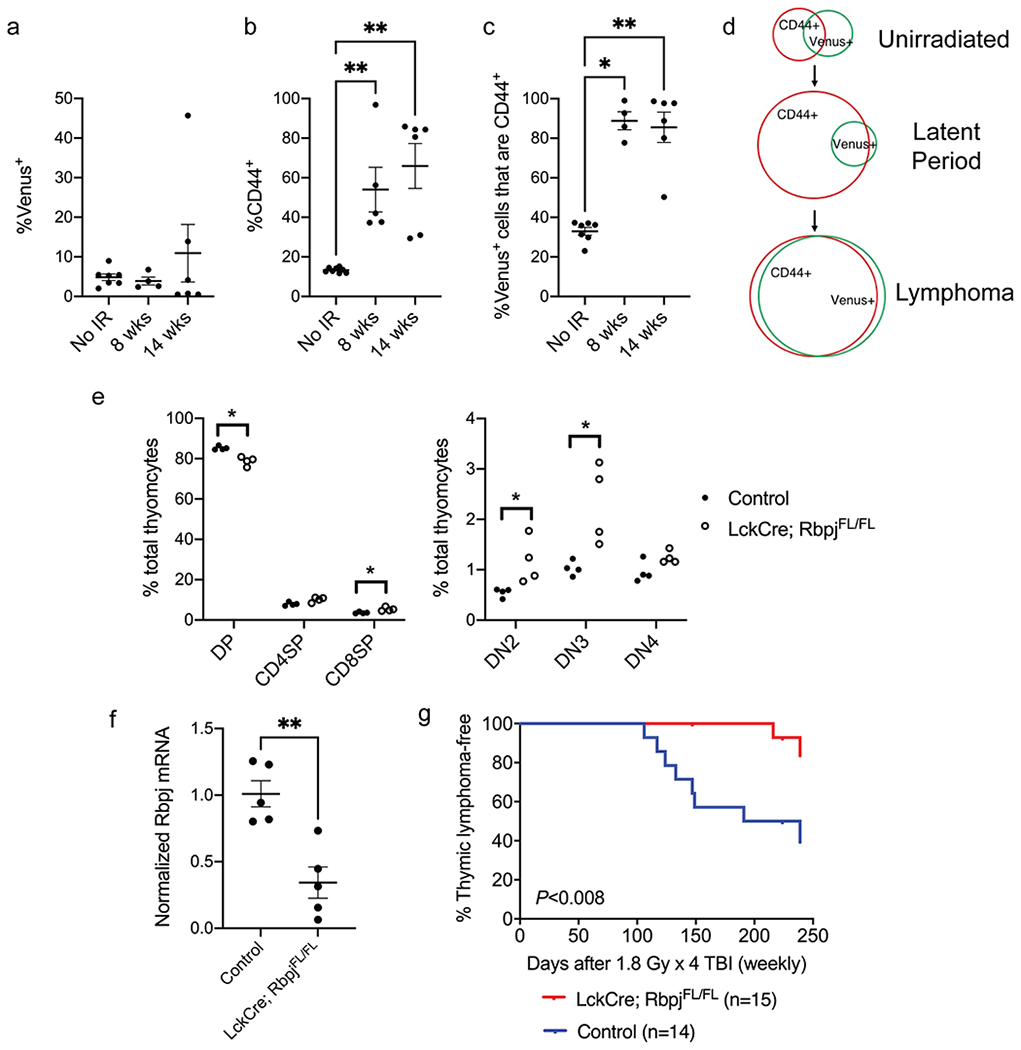
Notch1 signaling is critical for the development of radiation-induced thymic lymphoma. a-b, The percentage of Venus+ cells (a) and CD44+ cells (b) in total thymocytes harvested from Rbpj-Venus reporter mice at various time points after 1.8 Gy x 4 TBI. c, The percentage of Venus+ thymocytes also positive for CD44. Data are presented as mean ± SE. Each dot represents one mouse. *P<0.05 and **P<0.01 by Mann-Whitney U test compared to No IR. d, Schematic diagram showing the changes in Rbpj-Venus+ and CD44+ cells at various stages of radiation-induced lymphomagenesis. e, The frequency of different thymocyte populations in LckCre; RbpjFL/FL mice and littermate controls without Cre. DP (CD4+ CD8+), CD8SP (CD8+), CD4SP (CD4+), DN2 (CD4− CD8− CD25+ CD44+), DN3 (CD4− CD8− CD25+ CD44−), DN4 (CD4− CD8− CD25− CD44−). n=4 mice per genotype. Each dot represents one mouse. *P<0.05 by Mann-Whitney U test. f, The expression of Rbpj mRNA in total thymocytes from LckCre; RbpjFL/FL mice and littermate controls without Cre. Data are presented as mean ± SE. Each dot represents one mouse. **P<0.01 by Mann-Whitney U test. g, Thymic lymphoma-free survival of LckCre; RbpjFL/FL mice and littermate controls that consist of RbpjFL/WT or RbpjFL/FL (No Cre) mice and LckCre; RbpjFL/WT mice that retain the expression of one Rbpj allele. All mice were exposed to 1.8 Gy x 4 TBI. P= 0.008 by log-rank test. Data were collected from at least two independent experiments.
To determine if Notch1-mediated signaling is necessary to drive the formation of radiation-induced thymic lymphoma, we generated LckCre; Rbpj floxed mice in which the Rbpj allele is specifically deleted in immature thymocytes beyond the DN2 stage[20, 21]. Our data showed that deletion of Rbpj by LckCre caused a significant decrease in the frequency of CD4+CD8+ thymocytes and an increase in the frequency of double negative (DN)2 and DN3 thymocytes (Figure 6e). These results are consistent with the findings reported previously[38]. The expression of Rbpj mRNA was significantly decreased in thymocytes harvested from LckCre; RbpjFL/FL mice compared to control mice without Cre (Mann-Whitney U p-value = 0.008, Figure 6f). To elucidate the impact of Rbpj deletion on the development of radiation-induced thymic lymphoma, LckCre; RbpjFL/FL mice and their littermate controls including RbpjFL/FL, RbpjFL/WT mice and LckCre; Rbpj FL/WT mice were exposed to 1.8 Gy x 4 TBI and followed for up to 8 months post-irradiation. Our results showed that the development of radiation-induced thymic lymphoma was significantly inhibited in LckCre; RbpjFL/FL mice (log-rank p-value = 0.008, Figure 6g). Together, our findings from genetic analyses of lymphomas, Rbpj-Venus reporter mice experiments, thymocyte engraftment assays and thymocyte-specific Rbpj knockout mice demonstrate that activated Notch1 signaling drives the development of radiation-induced thymic lymphoma in p53 wild-type mice.
DISCUSSION
In the present study, we conducted a comprehensive analysis of somatic mutations and CNVs in thymic lymphomas that developed in irradiated WT, KrasLA1, p53+/− mice as well as in unirradiated mice lacking both alleles of p53 in hematopoietic cells. Our results reveal that murine radiation-induced thymic lymphomas harbor recurrent mutations in putative driver genes that have been reported in human T-cell acute lymphoblastic leukemia (T-ALL), including Pten, Akt1, Ikzf1 and Notch1[31] (Figure 3). In particular, we found that mutations in the Notch1 oncogene frequently occur in radiation-induced thymic lymphomas harboring wild-type p53. Although the presence of mutations and CNVs that affect the Notch1 signaling pathway in murine radiation-induced thymic lymphomas have been reported previously[39–41], our results from mice lacking functional Rbpj in immature thymocytes, for the first time, demonstrate that activation of Notch1 signaling is necessary to drive radiation-induced thymic lymphomas in p53 WT mice (Figure 6). Our study is distinct from papers that investigate the role of Notch1 in T-cell leukemogenesis through genetic engineering because the development of thymic lymphomas in our model is driven by genetic alterations that spontaneously occur in WT mice following TBI. Together, our findings uncover the mutational landscape of murine radiation-induced thymic lymphomas and define the pivotal role of activated Notch1 signaling in driving tumor development in p53 wild-type mice.
Our results indicate that the overexpression of ICN protein in radiation-induced thymic lymphomas is likely attributed to multiple genetic alterations that affect the Notch1 signaling pathway, including Notch1 mutations, Notch1 Type 1 deletions and Ikzf1 mutations (Figure 4). We noticed that that in a subset of thymic lymphomas, especially the tumors that developed in irradiated WT mice, these genetic alterations are not mutually exclusive. It has been shown in thymic lymphomas generated by viral insertion that Ikzf1 mutation is strongly selected for in tumors that also acquire Notch1 mutations in the intracellular domains[42]. These mutations also represent the vast majority of Notch1 mutations that we observed in radiation-induced thymic lymphomas (Figure S5). The Ikzf1 gene encodes Ikaros, a tumor suppressor in human T-ALL that represses the expression of Notch1 protein and its oncogenic target genes[34, 43]. Loss of Ikaros augments the transcription of Notch1 mRNA from the canonical and alternative promoters in the 5’ and intragenic region of the Notch1 gene by increasing permissive chromatin[44]. As Notch1 Type 1 deletions results in alternative transcription of the Notch1 gene from a cryptic promoter in exon 25[35], it is conceivable that mutations/deletions of Ikzf1 will further increase the expression of 3’ Notch1 mRNA that encodes the ICN protein. These results reveal that Notch1 signaling is activated in radiation-induced thymic lymphomas via a complex genetic and epigenetic network. Further studies are warranted to better understand the order of these oncogenic alterations during the development of radiation-induced thymic lymphomas in WT mice.
Our study also provides novel insights into how TBI selects for Notch1-mutant thymic lymphomas in p53 wild-type mice. Our results from Rbpj-Venus reporter mice indicate that malignant thymocytes expressing high Notch1 activity form a distinct population positive for CD44. In addition, we observed that irradiated thymocytes that can prorogate in vivo are almost exclusively positive for CD44+ (Figure 5). Intriguingly, the results from our time-course study show that TBI does not cause an overall increase in Venus+ cells at 8 and 14 weeks after irradiation, however, the subset of CD44+ thymocytes with activation of the Notch1 pathway as measured by Venus+ reporter expression was significantly enriched by TBI at both time points (Figure 6). The expansion of CD44+ thymocytes after TBI was also observed in an independent study[45]. Moreover, inhibition of CD44 significantly suppresses the engraftment of Notch1-mutant human T-ALL cells[46]. Together, these results suggest that the Notch1-CD44 axis plays an important role in regulating the formation of radiation-induced thymic lymphoma in wild-type mice. Further studies are warranted to investigate how Notch1+CD44+ thymocytes interact with the thymic niches at early stages of radiation-induced T-lymphomagenesis[47].
Our results also demonstrate that alterations in Notch1-mediated signaling occur more frequently in p53 wild-type thymic lymphomas following TBI than in lymphomas that develop in irradiated p53+/− or Non-IR p53−/− mice. Indeed, mutations in Notch1 and/or Ikzf1 were observed in 83% (15/18) lymphomas that retained WT p53 and only in 38% (5/13) of p53 deficient lymphomas (Figure 3). These results are consistent with the findings that Notch1 PEST mutations are detected in approximately 40% of thymic lymphomas that develop in p53+/− mice following a single dose of 4 Gy TBI[48]. The frequency of Notch1 mutations is even lower in radiation naïve thymic lymphomas driven by the loss of p53 (29%) (Figure 3). This inverse relationship between mutations in Notch1 and p53 has also been reported in murine thymic lymphomas generated by viral insertion[42]. In this paper, Uren and colleagues found that mutations that cause overexpression of ICN are almost exclusively detected in tumors harboring wild-type p53 and p19Arf. One possible explanation for this phenomenon is that p53 deficient mice have impaired expression Fbxw7, a transcriptional target of p53 that regulates the turnover of ICN[49, 50]. Indeed, Notch1 PEST domain mutations are not detected in radiation-induced thymic lymphomas of p53+/−; Fbxw7+/− mice[48]. However, as Notch1 and p53 both regulate the expression of numerous transcriptional targets, further investigation is warranted to dissect how Notch1 and p53 signaling pathways interact to regulate the formation of T-cell lymphoblastic lymphoma/leukemia.
Our mutational analyses using whole-exome sequencing data found no statistical evidence of a difference in the number of nonsynonymous somatic mutations per tumor between radiation-induced thymic lymphomas and thymic lymphomas that develop in p53−/− mice without irradiation (Figure 1). Similarly, analyses of mutational signatures by assessing the proportion of indels, the distribution of SNVs and cosine-similarity with COSMIC mutational signatures of human cancers do not show a significant difference between radiation-induced thymic lymphomas and Non-IR p53−/− thymic lymphomas (Figure 1 and Figure S3). While the absence of evidence for these differences may merely be due to the small size of the experimental populations, a study by Wong et al. also reported that therapy-related myeloid neoplasms (t-MNs) and de novo myeloid neoplasms in human patients exhibit a similar number of somatic mutations and single nucleotide substitutions[51]. In contrast to blood cancers, solid tumors that developed in humans and mice following focal irradiation display unique mutational processes compared to radiation naïve solid tumors. For example, radiation-induced sarcomas exhibit a strong oxidative mutation signature as shown by a preference of C to T and the reverse (G to A) single nucleotide substitutions as well as harboring a higher proportion of nonsynonymous deletion[9]sf. However, all of these unique mutational signatures were not observed in radiation-induced thymic lymphomas. Besides somatic mutations, we found that the number of genes affected by CNVs is markedly higher in thymic lymphomas in p53+/− mice treated with four weekly fractions of 1.8 Gy TBI (Figure 2). A similar increase in genomic instability was also reported in thymic lymphomas of p53+/− mice after a single fraction of 4 Gy TBI[52]. Collectively, these genomic studies provide compelling evidence to support a model in humans and mice without germline mutations where the formation of hematologic malignancies following exposure to ionizing radiation occurs through a non-cell-autonomous mechanism[8, 53, 54]. However, for individuals harboring germline mutations in DNA damage response genes, such as p53 and Mlh1, ionizing radiation may cause blood cancers through a more direct cell-autonomous effect by increasing genomic instability in tumor-initiating cells[13, 14, 52].
In sum, the results in the present study reveal the mutational landscape of coding genes and identify key drivers in murine radiation-induced thymic lymphoma, a classic model that has been used to study radiation carcinogenesis for over 70 years. These findings lay the foundation for a better understanding of mechanisms underlying multi-step carcinogenesis of hematologic malignances following total-body irradiation.
Supplementary Material
SIGNIFICANCE.
Findings reveal the mutational landscape and key drivers in murine radiation-induced thymic lymphoma, a classic animal model that has been used to study radiation carcinogenesis for over 70 years.
ACKNOWLEDGMENT
This work was supported by the following grants: National Cancer Institute (R35CA197616 to DGK, K99CA212198 and R00CA212198 to C-LL), Whitehead Scholar Award from Duke University School of Medicine (C-LL), and the Duke Cancer Center Support Grant 5P30CA14236-44.
Footnotes
CONFLICT OF INTEREST
DGK is on the scientific advisory board and owns stock in Lumicell, Inc which is developing intraoperative imaging technology. DGK is a co-founder of X-RAD Therapeutics, which is developing radiosensitizers. DGK reports research support from Merck, Bristol Myers Squibb, Varian, and X-RAD Therapeutics. C-LL reports research support from Janssen R&D. None of these interests present a conflict with the content in this manuscript. The remaining authors have no conflicting financial interests.
REFERENCES
- 1.Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102(1):43–52. [DOI] [PubMed] [Google Scholar]
- 2.Shuryak I, Sachs RK, Hlatky L, Little MP, Hahnfeldt P, Brenner DJ. Radiation-induced leukemia at doses relevant to radiation therapy: modeling mechanisms and estimating risks. J Natl Cancer Inst. 2006;98(24):1794–806. [DOI] [PubMed] [Google Scholar]
- 3.Ozasa K, Shimizu Y, Suyama A, Kasagi F, Soda M, Grant EJ, et al. Studies of the mortality of atomic bomb survivors, Report 14, 1950-2003: an overview of cancer and noncancer diseases. Radiat Res. 2012;177(3):229–43. [DOI] [PubMed] [Google Scholar]
- 4.Hsu WL, Preston DL, Soda M, Sugiyama H, Funamoto S, Kodama K, et al. The incidence of leukemia, lymphoma and multiple myeloma among atomic bomb survivors: 1950-2001. Radiat Res. 2013;179(3):361–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rivina L, Schiestl R. Mouse models of radiation-induced cancers. Advances in genetics. 2013;84:83–122. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan HS. Observations on radiation-induced lymphoid tumors of mice. Cancer Res. 1947;7(3):141–7. [PubMed] [Google Scholar]
- 7.Kaplan HS, Brown MB. A quantitative dose-response study of lymphoid-tumor development in irradiated C 57 black mice. J Natl Cancer Inst. 1952;13(1):185–208. [PubMed] [Google Scholar]
- 8.Lee CL, Castle KD, Moding EJ, Blum JM, Williams N, Luo L, et al. Acute DNA damage activates the tumour suppressor p53 to promote radiation-induced lymphoma. Nat Commun. 2015;6:8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CL, Mowery YM, Daniel AR, Zhang D, Sibley AB, Delaney JR, et al. Mutational landscape in genetically engineered, carcinogen-induced, and radiation-induced mouse sarcoma. JCI Insight. 2019;4(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherborne AL, Davidson PR, Yu K, Nakamura AO, Rashid M, Nakamura JL. Mutational Analysis of Ionizing Radiation Induced Neoplasms. Cell Rep. 2015;12(11):1915–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thibodeau BJ, Lavergne V, Dekhne N, Benitez P, Amin M, Ahmed S, et al. Mutational landscape of radiation-associated angiosarcoma of the breast. Oncotarget. 2018;9(11):10042–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behjati S, Gundem G, Wedge DC, Roberts ND, Tarpey PS, Cooke SL, et al. Mutational signatures of ionizing radiation in second malignancies. Nat Commun. 2016;7:12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel R, Zhang L, Desai A, Hoenerhoff MJ, Kennedy LH, Radivoyevitch T, et al. Mlh1 deficiency increases the risk of hematopoietic malignancy after simulated space radiation exposure. Leukemia. 2019;33(5):1135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose Li Y, Halliwill KD, Adams CJ, Iyer V, Riva L, Mamunur R, et al. Mutational signatures in tumours induced by high and low energy radiation in Trp53 deficient mice. Nat Commun. 2020;11(1):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–6. [DOI] [PubMed] [Google Scholar]
- 16.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4(1):1–7. [DOI] [PubMed] [Google Scholar]
- 17.Kemp CJ, Wheldon T, Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet. 1994;8(1):66–9. [DOI] [PubMed] [Google Scholar]
- 18.Moding EJ, Min HD, Castle KD, Ali M, Woodlief L, Williams N, et al. An extra copy of p53 suppresses development of spontaneous Kras-driven but not radiation-induced cancer. JCI Insight. 2016;1(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee CL, Moding EJ, Cuneo KC, Li Y, Sullivan JM, Mao L, et al. p53 functions in endothelial cells to prevent radiation-induced myocardial injury in mice. Sci Signal. 2012;5(234):ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hennet T, Hagen FK, Tabak LA, Marth JD. T-cell-specific deletion of a polypeptide N-acetylgalactosaminyl-transferase gene by site-directed recombination. Proc Natl Acad Sci U S A. 1995;92(26):12070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanigaki K, Han H, Yamamoto N, Tashiro K, Ikegawa M, Kuroda K, et al. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat Immunol. 2002;3(5):443–50. [DOI] [PubMed] [Google Scholar]
- 22.Nowotschin S, Xenopoulos P, Schrode N, Hadjantonakis AK. A bright single-cell resolution live imaging reporter of Notch signaling in the mouse. BMC Dev Biol. 2013;13:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerby B, Tremblay CS, Tremblay M, Rojas-Sutterlin S, Herblot S, Hebert J, et al. SCL, LMO1 and Notch1 reprogram thymocytes into self-renewing cells. PLoS Genet. 2014;10(12):e1004768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Team RC. R: A language and environment for statistical computing. Vienna, Austria: 2020. [Google Scholar]
- 26.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015;12(2):115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie Y knitr: A Comprehensive Tool for Reproducible Research in R. Implement Reprod Res. 2014;1:20. [Google Scholar]
- 28.Dudgeon C, Chan C, Kang W, Sun Y, Emerson R, Robins H, et al. The evolution of thymic lymphomas in p53 knockout mice. Genes Dev. 2014;28(23):2613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang Y, Wang R, Urrutia E, Anastopoulos IN, Nathanson KL, Zhang NR. CODEX2: full-spectrum copy number variation detection by high-throughput DNA sequencing. Genome Biol. 2018;19(1):202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lana T, de Lorenzo P, Bresolin S, Bronzini I, den Boer ML, Cave H, et al. Refinement of IKZF1 status in pediatric Philadelphia-positive acute lymphoblastic leukemia. Leukemia. 2015;29(10):2107–10. [DOI] [PubMed] [Google Scholar]
- 33.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22. [DOI] [PubMed] [Google Scholar]
- 34.Witkowski MT, Cimmino L, Hu Y, Trimarchi T, Tagoh H, McKenzie MD, et al. Activated Notch counteracts Ikaros tumor suppression in mouse and human T-cell acute lymphoblastic leukemia. Leukemia. 2015;29(6):1301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashworth TD, Pear WS, Chiang MY, Blacklow SC, Mastio J, Xu L, et al. Deletion-based mechanisms of Notch1 activation in T-ALL: key roles for RAG recombinase and a conserved internal translational start site in Notch1. Blood. 2010;116(25):5455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiang MY, Wang Q, Gormley AC, Stein SJ, Xu L, Shestova O, et al. High selective pressure for Notch1 mutations that induce Myc in T-cell acute lymphoblastic leukemia. Blood. 2016;128(18):2229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castel D, Mourikis P, Bartels SJ, Brinkman AB, Tajbakhsh S, Stunnenberg HG. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 2013;27(9):1059–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanigaki K, Tsuji M, Yamamoto N, Han H, Tsukada J, Inoue H, et al. Regulation of alphabeta/gammadelta T cell lineage commitment and peripheral T cell responses by Notch/RBP-J signaling. Immunity. 2004;20(5):611–22. [DOI] [PubMed] [Google Scholar]
- 39.Tsuji H, Ishii-Ohba H, Ukai H, Katsube T, Ogiu T. Radiation-induced deletions in the 5′ end region of Notch1 lead to the formation of truncated proteins and are involved in the development of mouse thymic lymphomas. Carcinogenesis. 2003;24(7):1257–68. [DOI] [PubMed] [Google Scholar]
- 40.Lopez-Nieva P, Santos J, Fernandez-Piqueras J. Defective expression of Notch1 and Notch2 in connection to alterations of c-Myc and Ikaros in gamma-radiation-induced mouse thymic lymphomas. Carcinogenesis. 2004;25(7):1299–304. [DOI] [PubMed] [Google Scholar]
- 41.Ohi H, Mishima Y, Kamimura K, Maruyama M, Sasai K, Kominami R. Multi-step lymphomagenesis deduced from DNA changes in thymic lymphomas and atrophic thymuses at various times after gamma-irradiation. Oncogene. 2007;26(36):5280–9. [DOI] [PubMed] [Google Scholar]
- 42.Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M, et al. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell. 2008;133(4):727–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdulla H, Vo A, Shields BJ, Davies TJ, Jackson JT, Alserihi R, et al. T-ALL can evolve to oncogene independence. Leukemia. 2021. [DOI] [PubMed] [Google Scholar]
- 44.Gomez-del Arco P, Kashiwagi M, Jackson AF, Naito T, Zhang J, Liu F, et al. Alternative promoter usage at the Notch1 locus supports ligand-independent signaling in T cell development and leukemogenesis. Immunity. 2010;33(5):685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sen-Majumdar A, Guidos C, Kina T, Lieberman M, Weissman IL. Characterization of preneoplastic thymocytes and of their neoplastic progression in irradiated C57BL/Ka mice. J Immunol. 1994;153(4):1581–92. [PubMed] [Google Scholar]
- 46.Garcia-Peydro M, Fuentes P, Mosquera M, Garcia-Leon MJ, Alcain J, Rodriguez A, et al. The NOTCH1/CD44 axis drives pathogenesis in a T cell acute lymphoblastic leukemia model. J Clin Invest. 2018;128(7):2802–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasapis S, Caraballo I, Lee CL. Transplantation of Unirradiated Bone Marrow Cells after Total-Body Irradiation Prevents the Development of Thymic Lymphoma in Mice through Niche Competition. Radiation research. 2021;195(3):301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jen KY, Song IY, Banta KL, Wu D, Mao JH, Balmain A. Sequential mutations in Notch1, Fbxw7, and Tp53 in radiation-induced mouse thymic lymphomas. Blood. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432(7018):775–9. [DOI] [PubMed] [Google Scholar]
- 50.Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes & Development. 2008;22(8):986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mao JH, Wu D, Perez-Losada J, Jiang T, Li Q, Neve RM, et al. Crosstalk between Aurora-A and p53: frequent deletion or downregulation of Aurora-A in tumors from p53 null mice. Cancer Cell. 2007;11(2):161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marusyk A, Casas-Selves M, Henry CJ, Zaberezhnyy V, Klawitter J, Christians U, et al. Irradiation alters selection for oncogenic mutations in hematopoietic progenitors. Cancer research. 2009;69(18):7262–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fleenor CJ, Marusyk A, DeGregori J. Ionizing radiation and hematopoietic malignancies: altering the adaptive landscape. Cell Cycle. 2010;9(15):3005–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The knitr package[27] was used to generate dynamic reports and conduct the analyses pertaining to WES results, in accordance with the principles of reproducible analysis. Duke’s GitLab was used for source code management (https://gitlab.oit.duke.edu), and the code for replicating the figures and statistical analyses pertaining to WES results presented here has been made available through a public source code repository (https://gitlab.oit.duke.edu/dcibioinformatics/pubs/kirsch-lee_lymphoma). The sequencing data along with the called mutations in vcf format have been deposited into the National Center for Biotechnology Information Sequence Read Archive under project ID PRJNA627412.