

Abstract
The emergence of multidrug resistant (MDR) HIV strains severely reduces the effectiveness of antiretroviral therapy. Clinical inhibitor darunavir (1) has picomolar binding affinity for HIV-1 protease (PR), however, drug resistant variants like PRS17 show poor inhibition by 1, despite the presence of only two mutated residues in the inhibitor-binding site. Antiviral inhibitors that target MDR proteases like PRS17 would be valuable as therapeutic agents. Inhibitors 2 and 3 derived from 1 through substitutions at P1, P2 and P2ʹ positions exhibit 3.4- to 500-fold better inhibition than clinical inhibitors for PRS17 with the exception of amprenavir. Crystal structures of PRS17/2 and PRS17/3 reveal how these inhibitors target the two active site mutations of PRS17. The substituted methoxy P2 group of 2 forms new interactions with G48V mutation, while the modified bis-fluoro-benzyl P1 group of 3 forms a halogen interaction with V82S mutation, contributing to improved inhibition of PRS17.
Keywords: Drug resistance, HIV protease, Protease inhibitor, X-ray crystallography
1. Introduction
Combination antiretroviral therapy (cART) has played a critical role in the suppression of human immunodeficiency virus (HIV) replication and the improved outcome for HIV-infected patients [1–3]. HIV protease (PR) inhibitors (PIs) are an integral part of cART regimens together with reverse-transcriptase (RTIs) and integrase inhibitors[4,5]. However, successful treatment is hampered by drug toxicity, side effects, and importantly, the emergence of drug resistant HIV-1 variants. PIs have a higher barrier to resistance than RTIs [6]. Currently, 3 of the 9 approved PIs, ritonavir-boosted darunavir (1), lopinavir and atazanavir are recommended in cART because of their high resistance barrier and potency [7]. Inhibitor 1, which was designed to form hydrogen bonds with the main-chain atoms of PR, is extremely potent and possesses the highest resistance barrier among PIs [8–12]. Other favorable traits of 1 include inhibition of precursor autoprocessing and inhibition of PR dimerization [13,14]. However, the emergence of drug resistant mutations to 1 and the prevalence of multidrug resistant (MDR) viral strains underscore the importance of developing more effective drugs [15,16].
The exceptional antiviral activity and picomolar enzyme inhibition of 1 has led to the design of derivatives to extend its potency, especially for poorly accessible reservoirs of virus. GRL-4410 (2) incorporates a substituted alkoxy group at the C4 position of P2 bis-THF in 1 and a methoxy group replaces the amine group in P2′ aniline of 1 [17]. Compound 2 has an excellent inhibition profile with Ki of 2.9 pM and a potent antiviral efficacy with an IC50 value of 2.4 nM as determined by MTT assay [17]. GRL-142 (3) has a 6–5-5 ring fused crown-like tetrahydropyranofuran (crn-THF) as the P2 ligand, bis-fluoro-benzene at P1 and cyclopropylamino-benzothiazole at P2′ [18,19]. Compound 3 exhibits exceptionally potent antiviral activity with an IC50 value of 0.019 nM compared to values of 3.2 to 33 nM for the nine FDA-approved PIs with tested viral variants, including drug-resistant strains [18]. Compound 3 shows around 1000-fold better inhibition of PR dimerization than 1 [19]. Furthermore, 3 shows better CNS penetration in vitro compared to 1 and studies in rats suggest it can effectively block HIV-1 replication in the brain. These traits make 3 an excellent PI for HIV/AIDS and HIV-associated neurocognitive disorder (HAND).
Recently, MDR variant PRS17 was chosen by mean-shift clustering on genotype-phenotype data using a unified encoding of sequence and 3D structure [20,21]. PRS17 has 17 mutations relative to wild-type PR and exhibits 1.5 to 5 orders of magnitude poorer inhibition relative to wild-type PR for 8 clinical inhibitors [22,23]. PRS17 also shows enhanced binding to substrate analogs[24]. NMR spectroscopy and X-ray crystallography studies show that the dynamic equilibrium conformation of PRS17, unlike that of wild-type PR, is shifted toward the open flap conformation in the absence of inhibitor [25]. Other studied MDR variants, PR20 and MDR769, also exhibit wide open flap conformations and poor binding affinity for inhibitors [26,27]. However, unlike PR20 and MDR769, PRS17 has only two mutations in the inhibitor-binding cavity (G48V and V82S). Hence, PRS17 is an excellent prototype to evaluate inhibitors targeting MDR PR variants with minimal alterations in the binding site.
We have determined the inhibitory activity and crystal structures of PRS17 in complex with 2 and 3. The structures are compared to corresponding wild-type PR complexes and PRS17/1 complex. Insights from this analysis will benefit the design of better drugs for MDR variants like PRS17.
2. Materials and methods
2.1. Expression and Purification of PRS17
The synthetic gene derived from genotype data for PRS17 was expressed in E. coli and purified as described in [24]
2.2. Kinetic inhibition measurements
Compounds 2 and 3 (>95% purity by HPLC) were dissolved in 100% DMSO. Inhibition values (Ki) for PRS17 were measured in a spectroscopic assay with FRET-substrate (BACHEM H-2992) at 37°C and pH 5.6 as described in [24].
2.3. Crystallization
PRS17 was mixed with inhibitor at 1:6 molar ratios and incubated on ice for 30 minutes. PRS17 complex at 5 mg/mL was used in hanging drop vapor diffusion crystallization trials at room temperature. PRS17/2 crystallized in 1.15 M sodium chloride, 0.1 M sodium acetate at pH 5.5. PRS17/3 crystallized from 1.2 M sodium chloride, 0.1 M sodium acetate at pH 5.5. The crystals were cryo-cooled in the respective mother liquor and 30% glycerol.
2.4. X-ray data collection and structure determination
X-ray diffraction data were collected at 100 K on beamline 22-ID (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory. The data were integrated and scaled with HKL2000 [28]. Structures were solved using molecular replacement with PHASER [29,30] with PRS17/1 (5T2Z)[26] as the starting model. Structures were refined using REFMAC5.2 [31] and refitted with COOT [32]. Solvent molecules were inserted at stereochemically reasonable positions using 2Fo-Fc and Fo-Fc maps at 1 and 3 sigma levels, respectively. Hydrogen bonds (2.4–3.5 Å) and hydrophobic contacts (3.6–4.2 Å) were inferred from interatomic distances and chemistry. Molecular figures were prepared with PyMOL (http://www.pymol.org). Coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 7MYP for PRS17/2 and 7MYY for PRS17/3.
3. Results
3.1. Compounds 2 and 3 are excellent inhibitors of PRS17
The Ki values of compounds 2 and 3 were 15.8 ± 4.8 and 17 ± 1.3 nM, respectively, for PRS17. These values are 3- to 500-fold better than those of clinical inhibitors (Ki values of 50 – 8400 nM), except for amprenavir (Ki value of 11 nM) [23]. Interestingly, non-hydrolyzable substrate analogs CA-p2 (Ki = 22 nM) and p2-NC (Ki = 514 nM) also show better inhibition than most clinical inhibitors for PRS17 [24]. Compounds 2 and 3 had similar inhibitory activity to CA-p2 and better inhibition than p2-NC analog for PRS17.
3.2. Overall structure
Crystal structures of PRS17 with investigational inhibitors 2 and 3 derived from compound 1 (Figure 1) were determined at 1.65 and 1.50 Å resolution, respectively, and R-factors of 20% (Table 1). The structures were solved in space group P61 with one PRS17 dimer per asymmetric unit. Residues in the two subunits are numbered 1–99 and 1′−99′ (Figure 1D). The inhibitors were observed in two mutually exclusive orientations related by 180° rotation with relative occupancies of 0.55 and 0.45 for PRS17/2 complex and 0.5 each for PRS17/3 complex. Both inhibitors and all mutations were unambiguously modelled in the structures. The two subunits in PRS17/2 and PRS17/3 dimers are essentially identical with low root mean square deviation (RMSD) values of 0.07 and 0.05 Å for 99 Cα atoms, respectively.
Figure 1. Compounds 1, 2, 3 and sites of mutation in PRS17 dimer.

A. Chemical structure of darunavir (1). B. Chemical structure and Fo-Fc omit map of 2 contour d at 3σ. C. Chemical structure and Fo-Fc omit map of 3 contour d at 3σ. D. PRS17 dimer in cartoon representation showing the sites of 17 mutations. The two active site mutations are shown as red spheres and the other mutations are blue spheres. Compound 3 bound at the active site is shown as pink sticks
Table 1.
Crystallographic data and refinement statistics
| PRS17 Complexes | PRS17/2 | PRS17/3 |
|---|---|---|
| Space group | P61 | P61 |
| Cell Dimensions | ||
| a (Å) | 62.94 | 62.89 |
| b (Å) | 62.94 | 62.89 |
| c (Å) | 82.72 | 83.11 |
| Resolution range (Å) | 50.0 – 1.65 | 50.0 – 1.5 |
| Unique reflections | 21295 | 28781 |
| Redundancy | 4.4 (3.8) | 4.9 (3.9) |
| Completeness | 95.2 (71.1)a | 96.4 (81.5) |
| <I/σ(I)> | 21.0 (3.1) | 34.0 (2.7) |
| Rsym (%) | 5.9 (44.0) | 3.9 (42.6) |
| Refinement resolution range (Å) | 50 – 1.65 | 50.0 – 1.50 |
| R(%) | 20.0 | 20.2 |
| Rfree (%) | 24.8 | 24.8 |
| Number of water molecules | 97 | 124 |
| Average B-factor (Å2) | ||
| Main-chain | 28.2 | 25.7 |
| Side-chain | 33.0 | 30.3 |
| Inhibitor | 22.4 | 21.5 |
| Water | 37.4 | 35.1 |
| RMS deviations from ideality | ||
| Bond length (Å) | 0.01 | 0.01 |
| Angles(°) | 1.6 | 1.7 |
Values in parentheses are for the highest resolution shell
3.3. New interaction of 2 with G48V of PRS17 contributes to its improved inhibition over 1
The P2 alkoxy group at the C4-position of bis-THF of 2 was designed to form additional interactions with the flexible flaps of PR [17]. The dimers of PRS17/2 and wild-type PR/2 [17] superposed with a RMSD of 0.8 Å for 198 equivalent Cα atoms, however, PRS17/2 is more similar to PRS17/1 [25] with a low RMSD of 0.17 Å. The protein residues in the active site cavity share similar conformations in the three structures except at the 80′s loop and flaps, where V82S and G48V mutations are located in PRS17. Mutation V82′S substitutes the polar serine for β-branched hydrophobic valine. The main-chain atoms of Thr80′ to Ser82′ in the S1 pocket of PRS17/2 complex shift by about 0.7–1.0 Å towards P1 of the inhibitor compared to the position in the wild-type PR/2 complex (Figure 2A). This shift maintains the van der Waals contacts of the smaller Ser82′ mutation and Pro81′ of PRS17 with P1 Phe of compound 2. A similar shift in the other subunit acts to maintain the hydrophobic contact between P1′ Leu of 2 and Ser82 mutation of PRS17. PRS17/1 complex shows a similar conformational change, which confirms the importance of V82S mutation.
Figure 2. Interactions of P2 group of compound 2 with G48V mutation of PRS17.
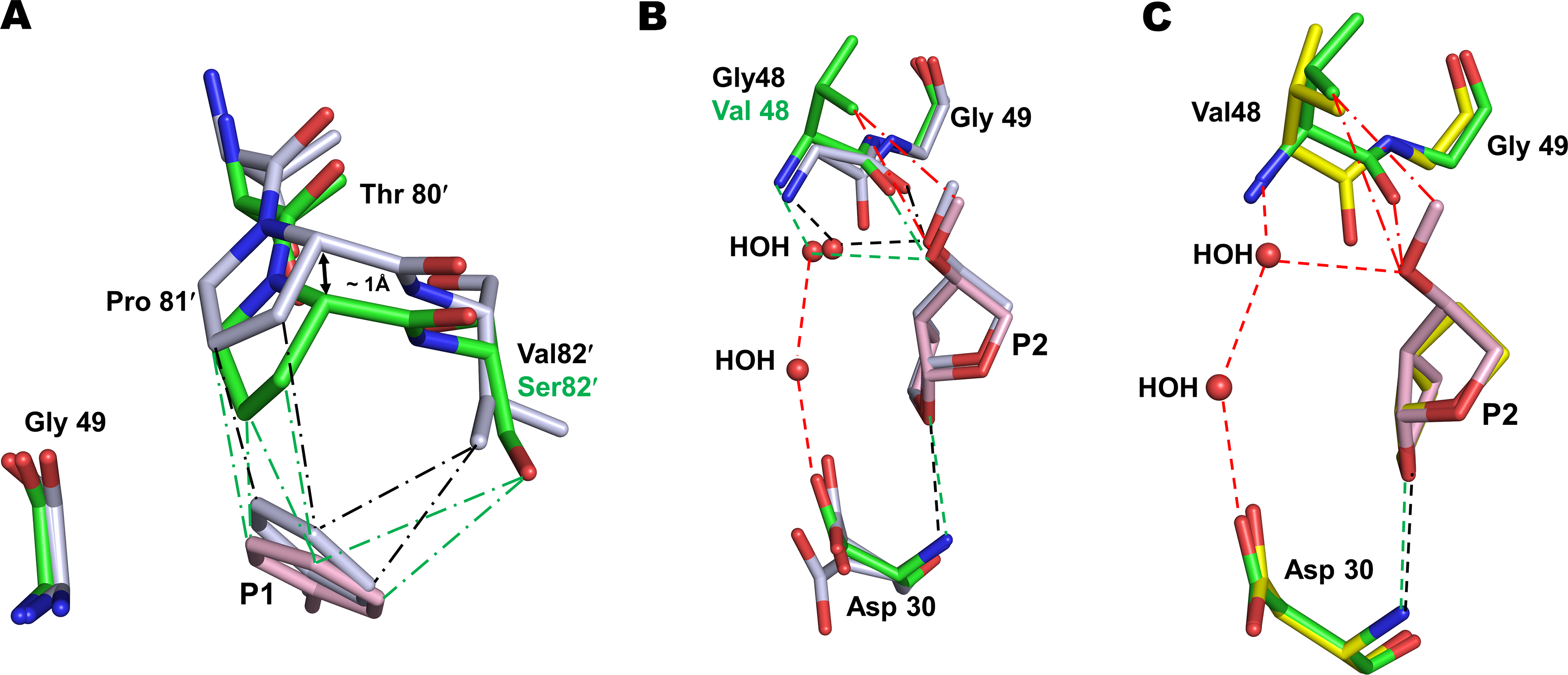
A. The main-chain of residues 80–82 in PRS17/2 complex shifts by ~1 Å due to V82S mutation to maintain van der Waals contacts with P1 group of 2 observed in wild-type PR/2 complex. PR/2 complex is shown as grey sticks colored by element in panels A and B. PRS17/2 amino acids are in green sticks and inhibitor 2 is in pink. Green and black (― - ―) lines represent van der Waals contacts in PRS17/2 and PR/2 complexes, respectively. B. Comparison of P2 methoxy group interaction in the S2 pocket of PRS17/2 and PR/2 complexes. The new interactions of P2 group of 2 are shown in red lines in panels B and C. Green and black (- - -) lines represent hydrogen bonds in mutant and wild-type PR complexes. C. Comparison of interactions at the S2 site of PRS17 by substituted P2 methoxy group of 2 in PRS17/2 complex and bis-THF of 1 in PRS17/1 complex. PRS17/1 is shown as sticks with yellow carbons.
All hydrogen bond interactions between 2 and the main-chain atoms of PR are retained in PRS17/2 complex. The carbonyl group of G48V in PRS17/2 is in a single conformation in contrast to the two conformations in the wild-type PR complex. The substituted methoxy group of P2 bis-THF of 2 forms similar van der Waals contact with the carbonyl oxygen of G48V in PRS17/2 and PR/2 (Figure 2B). The water-mediated hydrogen bond observed between the oxygen of the P2 methoxy group and the amide of Gly48 in PR/2 is conserved in the new PRS17/2 complex. However, the P2 methoxy group forms additional hydrophobic contacts with the side-chain of G48V mutation cannot occur in the wild-type complex. In addition, the P2 group forms water-mediated interactions with Asp30 in PRS17/2 unlike in PR/2. Comparison with PRS17/1 reveals that P2 bis-THF of 1 lacks the water-mediated hydrogen bonds with G48V and Asp30 and has no hydrophobic contacts with G48V (Figure 2C). Thus, the P2 alkoxy group of 2 retains interactions with the main-chain of 48 in wild-type PR/2 and in PRS17/2 complexes. The absence of these interactions in PRS17/1 explains the improved inhibition of compound 2 relative to 1 for PRS17.
3.4. Halogen bond between 3 and V82S confers enhanced inhibition constant for PRS17 over 1
Compound 3 has larger groups compared to 1 with crn-THF as P2-ligand, aminobenzothiazole (Cp-Abt) as P2′-ligand, and bis-fluoro-benzene as P1-ligand. The dimer of PRS17/3 superimposes on wild-type PR/3 with RMSD of 0.79 Å for 198 equivalent Cα atoms. PRS17/3 complex is more similar to PRS17/1 with RMSD of 0.23 Å. PRS17/3 retains all hydrogen bonds observed between 3 and main-chain atoms of protein in previously reported structures of PR/3 and PRS17/1. The crn-THF P2 group of 3 forms similar van der Waals contacts with Ile 47 in the wild-type PR/3 and PRS17/3 structures, while the bis-THF P2 group in PRS17/1 complex has no contacts with Ile47 (Figure 3A). Like in the PR/3 structure, the Cp-Abt at P2′ of PRS17/3 forms two hydrogen bonds with the side-chain of Asp30′. The P2′ cyclopropyl group of 3 in PR/3 and PRS17/3 complexes forms van der Waals interactions with the side-chain of Asp29′. In contrast, the P2′ aminobenzene in PRS17/1 forms a hydrogen bond (3.5 Å) with the side-chain of Asp30′ (Figure 3B). Thus, unlike 1, the large P2ʹ group of 3 makes extensive interactions with Asp29ʹ and Asp30ʹ of PRS17.
Figure 3. Interactions of P1, P2 and P2ʹ groups of 3 with PRS17 in comparison to 1.
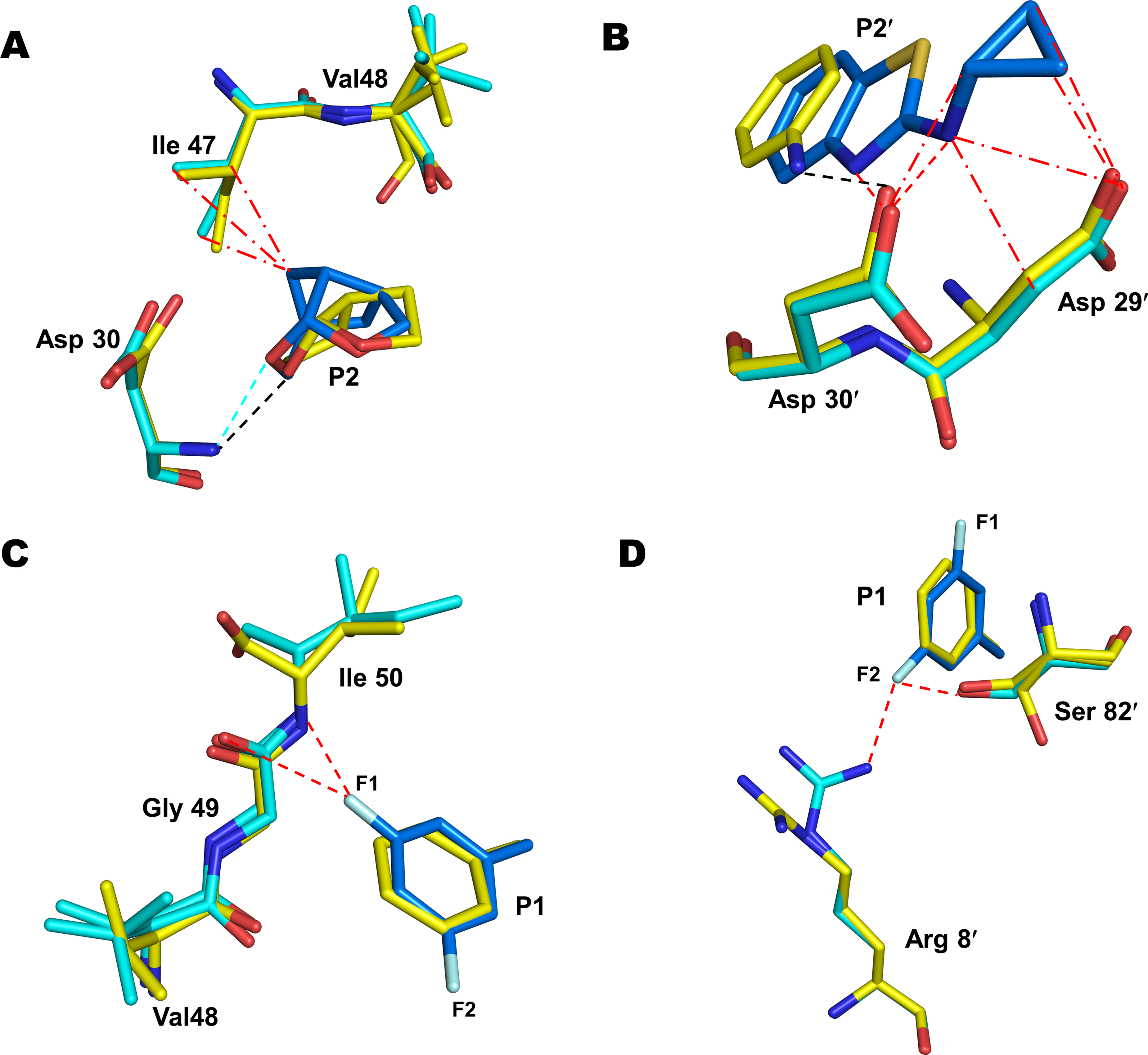
PRS17/1 complex is shown in sticks with yellow carbons. PRS17/3 is shown with cyan carbons for protein and blue carbons for inhibitor. Lines (― - ―) and (- - -) represent van der Waals and hydrogen bond interactions. Black and cyan lines represent interactions observed in PRS17/1 and PRS17/3, respectively. New interactions observed in PRS17/3 are shown as red dashed lines. A. The substituted P2 crown-THF of 3 forms new van der Waals contacts with Ile47 at the S2 pocket of PRS17/3 in comparison to PRS17/1. B. The P2ʹ Cp-Abt of 3 forms 2 new hydrogen bonds with Asp30ʹ and several van der Waals contacts with Asp29ʹ in PRS17/3 compared to PRS17/1. C. one of the fluorine in the P1 bis-fluro-benzyl group of 3 forms two polar interactions with the flap residues Gly49 and Ile50 in PRS17/3 compared to PRS17/1. D. The second fluorine in the P1-ligand of 3 forms polar interactions with Arg8ʹ and the critical active site mutation V82ʹS in PRS17/3 in comparison to PRS17/1.
The fluorine atoms in the P1 bis-fluoro-benzene of 3 play an important role in its binding to PR. One of the fluorine atoms forms a polar interaction (C-F···H-N) to the main-chain amide group of Ile50 in both PR/3 and PRS17/3 complexes. The fluorine also forms an orthogonal multipolar interaction (C-F···C-O) interaction with the main-chain carbonyl of Gly49 in both complexes. Inhibitor 1 lacks these halogen interactions and instead forms weaker van der Waals contacts with the flap residues in PRS17/1 complex (Figure 3C). In the wild-type PR/3, the second fluorine atom forms polar interactions with the guanidinium group of Arg8′. In PRS17/3, the second fluorine retains the polar interaction with Arg8′ in one conformation of 3 while the second conformation forms a water-mediated interaction with Arg8′. The second fluorine also forms a new polar interaction with side-chain of V82′S mutation in PRS17/3 complex (Figure 3D). This interaction is not possible in PR/3 complex with Val82′ nor in PRS17/1 where P1 lacks fluorine atoms. Thus, the new halogen interactions formed by P1 group of 3 with V82ʹS, Arg8ʹ and flap residues Gly49 and Ile50 of PRS17, together with added interactions of substituted P2 and P2′, contribute to its improved inhibition relative to 1 for PRS17.
4. Discussion
Among the 17 mutations, PRS17 has only two mutations, G48V and V82S, in the active site cavity. Drug resistant mutations of Val82 are among the first to emerge in patients undergoing antiviral therapy [33] and are associated with resistance to all clinical drugs except for 1 [34]. Flap mutation G48V is selected by PIs saquinavir, atazanavir, indinavir, lopinavir and nelfinavir [35–37]. Mutations of Gly48 are common in MDR variants [38] like PRS17. Hence, inhibitors that target Gly48 mutations are likely to perform well against MDR PRs. In addition, G48V and V82S mutations were shown to play a vital role in the enhanced binding of substrate analogs CA-p2 and p2-NC to PRS17 thereby contributing to viral fitness [24]. The role of V82 mutations is confirmed by studies of PR with single mutation V82A, which also displays enhanced binding to substrate analogs CA-p2 and p2-NC [24,39]. Amprenavir with the smaller THF at P2 exhibits better inhibition constant for PRS17 compared to other PIs as well as 2 and 3. However, inhibitors 2 and 3 with bigger P2 groups perform better against MDR mutants like PR20 with expanded S2 pockets, whereas amprenavir is a poorer inhibitor of variants with an expanded S2 pocket or active site mutations like V32I or V82I. It is likely that a smaller P2 group may result in improved inhibition profile against MDR PRs with minimal active site mutations such as PRS17. The current study reveals that specific modifications to compound 1 result in better inhibition of MDR PRS17. The substituted P2 moiety of inhibitor 2 targets flap mutation G48V and these interactions contribute to its improved inhibition of PRS17. The modified P1 group of 3 targets V82S mutation through halogen interactions to improve its inhibition of PRS17. These insights will be valuable for the design of improved inhibitors of MDR PRs. A new inhibitor in the scaffold of 1 that combines the P1 and P2 substitutions of 2 and 3 may be more effective for mutants like PRS17.
Clinical inhibitors of HIV-1 protease are ineffective against drug-resistant mutant PRS17
Two new antiviral compounds derived from darunavir show better inhibition of PRS17
Structural analysis reveals new interactions of inhibitors with mutated amino acids in PRS17
Acknowledgments
This research was supported by the National Institute of Health grants AI150461 (ITW) and AI150466 (AKG). We thank the staff at the Southeast Regional Collaborative Access Team (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Funding
This research was supported by the National Institute of Health grants AI150461 (ITW) and AI150466 (AKG) and a Georgia State University Molecular Basis of Disease fellowship.
Footnotes
Conflicts of interest
None declared.
Biochemical and Biophysical Research Communications
Declaration of competing interests
☒ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Please note that all Biochemical and Biophysical Research Communications authors are required to report the following potential conflicts of interest with each submission. If applicable to your manuscript, please provide the necessary declaration in the box above.
(1) All third-party financial support for the work in the submitted manuscript.
(2) All financial relationships with any entities that could be viewed as relevant to the general area of the submitted manuscript.
(3) All sources of revenue with relevance to the submitted work who made payments to you, or to your institution on your behalf, in the 36 months prior to submission.
(4) Any other interactions with the sponsor of outside of the submitted work should also be reported.
(5) Any relevant patents or copyrights (planned, pending, or issued).
(6) Any other relationships or affiliations that may be perceived by readers to have influenced, or give the appearance of potentially influencing, what you wrote in the submitted work. As a general guideline, it is usually better to disclose a relationship than not.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Lederman MM, Connick E, Landay A, Kuritzkes DR, Spritzler J, St Clair M, Kotzin BL, Fox L, Chiozzi MH, Leonard JM, Rousseau F, Wade M, Roe JD, Martinez A, Kessler H, Immunologic responses associated with 12 weeks of combination antiretroviral therapy consisting of zidovudine, lamivudine, and ritonavir: results of AIDS Clinical Trials Group Protocol 315, J Infect Dis 178 (1998) 70–79. 10.1086/515591. [DOI] [PubMed] [Google Scholar]
- [2].Walensky RP, Paltiel AD, Losina E, Mercincavage LM, Schackman BR, Sax PE, Weinstein MC, Freedberg KA, The survival benefits of AIDS treatment in the United States, J Infect Dis 194 (2006) 11–19. 10.1086/505147. [DOI] [PubMed] [Google Scholar]
- [3].Bhaskaran K, Hamouda O, Sannes M, Boufassa F, Johnson AM, Lambert PC, Porter K, Collaboration C, Changes in the risk of death after HIV seroconversion compared with mortality in the general population, JAMA 300 (2008) 51–59. 10.1001/jama.300.1.51. [DOI] [PubMed] [Google Scholar]
- [4].Cihlar T, Fordyce M, Current status and prospects of HIV treatment, Curr Opin Virol 18 (2016) 50–56. 10.1016/j.coviro.2016.03.004. [DOI] [PubMed] [Google Scholar]
- [5].Lv Z, Chu Y, Wang Y, HIV protease inhibitors: a review of molecular selectivity and toxicity, HIV AIDS (Auckl) 7 (2015) 95–104. 10.2147/HIV.S79956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Riddler SA, Haubrich R, DiRienzo AG, Peeples L, Powderly WG, Klingman KL, Garren KW, George T, Rooney JF, Brizz B, Lalloo UG, Murphy RL, Swindells S, Havlir D, Mellors JW, Team ACTGSA, Class-sparing regimens for initial treatment of HIV-1 infection, N Engl J Med 358 (2008) 2095–2106. 10.1056/NEJMoa074609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saag MS, Benson CA, Gandhi RT, Hoy JF, Landovitz RJ, Mugavero MJ, Sax PE, Smith DM, Thompson MA, Buchbinder SP, Del Rio C, Eron JJ Jr., Fatkenheuer G, Gunthard HF, Molina JM, Jacobsen DM, Volberding PA, Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2018 Recommendations of the International Antiviral Society-USA Panel, JAMA 320 (2018) 379–396. 10.1001/jama.2018.8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT, High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains, J Mol Biol 338 (2004) 341–352. 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- [9].Ghosh AK, Chapsal BD, Weber IT, Mitsuya H, Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance, Acc Chem Res 41 (2008) 78–86. 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- [10].Surleraux DL, Tahri A, Verschueren WG, Pille GM, de Kock HA, Jonckers TH, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PB, Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor, J Med Chem 48 (2005) 1813–1822. 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- [11].Dierynck I, De Wit M, Gustin E, Keuleers I, Vandersmissen J, Hallenberger S, Hertogs K, Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier, J Virol 81 (2007) 13845–13851. 10.1128/JVI.01184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].de Meyer S, Vangeneugden T, van Baelen B, de Paepe E, van Marck H, Picchio G, Lefebvre E, de Bethune MP, Resistance profile of darunavir: combined 24-week results from the POWER trials, AIDS Res Hum Retroviruses 24 (2008) 379–388. 10.1089/aid.2007.0173. [DOI] [PubMed] [Google Scholar]
- [13].Louis JM, Aniana A, Weber IT, Sayer JM, Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors, Proc Natl Acad Sci U S A 108 (2011) 9072–9077. 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Huang D, Caflisch A, How Does Darunavir Prevent HIV-1 Protease Dimerization?, J Chem Theory Comput 8 (2012) 1786–1794. 10.1021/ct300032r. [DOI] [PubMed] [Google Scholar]
- [15].El Bouzidi K, White E, Mbisa JL, Sabin CA, Phillips AN, Mackie N, Pozniak AL, Tostevin A, Pillay D, Dunn DT, Database UHDR, and the UKCHIVCSSC, Database UHDR, the UKCHIVCSSC, HIV-1 drug resistance mutations emerging on darunavir therapy in PI-naive and -experienced patients in the UK, J Antimicrob Chemother 71 (2016) 3487–3494. 10.1093/jac/dkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Godfrey C, Thigpen MC, Crawford KW, Jean-Phillippe P, Pillay D, Persaud D, Kuritzkes DR, Wainberg M, Raizes E, Fitzgibbon J, Global HIV Antiretroviral Drug Resistance: A Perspective and Report of a National Institute of Allergy and Infectious Diseases Consultation, J Infect Dis 216 (2017) S798–S800. 10.1093/infdis/jix137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ghosh AK, Martyr CD, Steffey M, Wang YF, Agniswamy J, Amano M, Weber IT, Mitsuya H, Design of substituted bis-Tetrahydrofuran (bis-THF)-derived Potent HIV-1 Protease Inhibitors, Protein-ligand X-ray Structure, and Convenient Syntheses of bis-THF and Substituted bis-THF Ligands, ACS Med Chem Lett 2 (2011) 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aoki M, Hayashi H, Rao KV, Das D, Higashi-Kuwata N, Bulut H, Aoki-Ogata H, Takamatsu Y, Yedidi RS, Davis DA, Hattori SI, Nishida N, Hasegawa K, Takamune N, Nyalapatla PR, Osswald HL, Jono H, Saito H, Yarchoan R, Misumi S, Ghosh AK, Mitsuya H, A novel central nervous system-penetrating protease inhibitor overcomes human immunodeficiency virus 1 resistance with unprecedented aM to pM potency, Elife 6 (2017). 10.7554/eLife.28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ghosh AK, Rao KV, Nyalapatla PR, Kovela S, Brindisi M, Osswald HL, Sekhara Reddy B, Agniswamy J, Wang YF, Aoki M, Hattori SI, Weber IT, Mitsuya H, Design of Highly Potent, Dual-Acting and Central-Nervous-System-Penetrating HIV-1 Protease Inhibitors with Excellent Potency against Multidrug-Resistant HIV-1 Variants, ChemMedChem 13 (2018) 803–815. 10.1002/cmdc.201700824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yu X, Weber IT, Harrison RW, Sparse Representation for Prediction of HIV-1 Protease Drug Resistance, Proc SIAM Int Conf Data Min 2013 (2013) 342–349. 10.1137/1.9781611972832.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yu X, Weber IT, Harrison RW, Prediction of HIV drug resistance from genotype with encoded three-dimensional protein structure, BMC. Genomics Supplement 5 (2014) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yu X, Weber IT, Harrison RW, Identifying representative drug resistant mutants of HIV, BMC. Bioinformatics 16 (2015) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Park JH, Sayer JM, Aniana A, Yu X, Weber IT, Harrison RW, Louis JM, Binding of clinical inhibitors to a model precursor of a rationally selected multidrug resistant HIV-1 protease is significantly weaker than to the released mature enzyme, Biochemistry 55 (2016) 2390–2400. 10.1021/acs.biochem.6b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Agniswamy J, Kneller DW, Brothers R, Wang YF, Harrison RW, Weber IT, Highly Drug-Resistant HIV-1 Protease Mutant PRS17 Shows Enhanced Binding to Substrate Analogues, ACS Omega 4 (2019) 8707–8719. 10.1021/acsomega.9b00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Agniswamy J, Louis JM, Roche J, Harrison RW, Weber IT, Structural Studies of a Rationally Selected Multi-Drug Resistant HIV-1 Protease Reveal Synergistic Effect of Distal Mutations on Flap Dynamics, PLoS One 11 (2016) e0168616. 10.1371/journal.pone.0168616 PONE-D-16–34784 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Agniswamy J, Shen CH, Aniana A, Sayer JM, Louis JM, Weber IT, HIV-1 Protease with 20 Mutations Exhibits Extreme Resistance to Clinical Inhibitors through Coordinated Structural Rearrangements, Biochemistry 51 (2012) 2819–2828. 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Martin P, Vickrey JF, Proteasa G, Jimenez YL, Wawrzak Z, Winters MA, Merigan TC, Kovari LC, “Wide-open” 1.3 A structure of a multidrug-resistant HIV-1 protease as a drug target, Structure 13 (2005) 1887–1895. 10.1016/j.str.2005.11.005. [DOI] [PubMed] [Google Scholar]
- [28].Otwinowski Z, Minor W, Processing of X-ray diffraction data collected in oscillation mode, Method Enzymol 276 (1997) 307–326. [DOI] [PubMed] [Google Scholar]
- [29].Storoni LC, McCoy AJ, Read RJ, Likelihood-enhanced fast rotation functions, Acta Crystallogr D Biol Crystallogr 60 (2004) 432–438. 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- [30].McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ, Likelihood-enhanced fast translation functions, Acta Crystallogr D Biol Crystallogr 61 (2005) 458–464. 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- [31].Murshudov GN, Vagin AA, Dodson EJ, Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr D Biol Crystallogr 53 (1997) 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- [32].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta Crystallogr D Biol Crystallogr 60 (2004) 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- [33].Shafer RW, Stevenson D, Chan B, Human Immunodeficiency Virus Reverse Transcriptase and Protease Sequence Database, Nucleic Acids Res 27 (1999) 348–352. gkc068 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wensing AM, Calvez V, Ceccherini-Silberstein F, Charpentier C, Gunthard HF, Paredes R, Shafer RW, Richman DD, 2019 update of the drug resistance mutations in HIV-1, Top Antivir Med 27 (2019) 111–121. [PMC free article] [PubMed] [Google Scholar]
- [35].Rhee SY, Taylor J, Fessel WJ, Kaufman D, Towner W, Troia P, Ruane P, Hellinger J, Shirvani V, Zolopa A, Shafer RW, HIV-1 protease mutations and protease inhibitor cross-resistance, Antimicrob Agents Chemother 54 (2010) 4253–4261. AAC.00574–10 [pii] 10.1128/AAC.00574-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kantor R, Fessel WJ, Zolopa AR, Israelski D, Shulman N, Montoya JG, Harbour M, Schapiro JM, Shafer RW, Evolution of primary protease inhibitor resistance mutations during protease inhibitor salvage therapy, Antimicrob Agents Chemother 46 (2002) 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Clemente JC, Coman RM, Thiaville MM, Janka LK, Jeung JA, Nukoolkarn S, Govindasamy L, Agbandje-McKenna M, McKenna R, Leelamanit W, Goodenow MM, Dunn BM, Analysis of HIV-1 CRF_01 A/E protease inhibitor resistance: structural determinants for maintaining sensitivity and developing resistance to atazanavir, Biochemistry 45 (2006) 5468–5477. 10.1021/bi051886s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shahriar R, Rhee SY, Liu TF, Fessel WJ, Scarsella A, Towner W, Holmes SP, Zolopa AR, Shafer RW, Nonpolymorphic human immunodeficiency virus type 1 protease and reverse transcriptase treatment-selected mutations, Antimicrob Agents Chemother 53 (2009) 4869–4878. AAC.00592–09 [pii] 10.1128/AAC.00592-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT, Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 angstroms resolution crystal structures of HIV-1 protease mutants with substrate analogs, FEBS J 272 (2005) 5265–5277. 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]