

Abstract
Receptor Interacting Protein Kinases (RIPKs) are a family of Ser/Thr/Tyr kinases whose functions, regulation and pathophysiologic roles have remained an enigma for a long time. In recent years, these proteins garnered significant interest due to their roles in regulating a variety of host defense functions including control of inflammatory gene expression, different forms of cell death, and cutaneous and intestinal barrier functions. In addition, there is accumulating evidence that while these kinases seemingly follow typical kinase blueprints, their functioning in cells can take forms that are atypical for protein kinases. Lastly, while these kinases generally belong to distinct areas of innate immune regulation, there are emerging overarching themes that may unify the functions of this kinase family. Our review seeks to discuss the biology of RIPKs, and how typical and atypical features of this family informs the activity of a rapidly growing repertoire of RIPK inhibitors.
Introduction
RIPKs are a seven-member family of Ser/Thr and Tyr TKL (Tyrosine Kinase-Like) kinases (Fig. 1). These proteins display significant homology in the kinase domains, but otherwise share limited similarities. The family includes RIPK1–RIPK7. The latter two family members LRRK (RIPK6)/LRRK2 (RIPK7) are more distant cousins of RIPK1–RIPK5. Furthermore, LRRK2 attracted significant interest through its genetic connections to the pathogenesis of Parkinson’s disease and we direct readers to recent reviews dedicated to LRRK/LRRK2 [1–3]. It is also worth noting that RIPKs belong to the same TKL sub-family as other proteins involved in regulation of parallel mechanisms of immunity, such as IRAK kinases, as well as proteins that have been shown to regulate RIPK1/RIPK3 signaling - TAK1 and pseudokinase MLKL (Fig. 1).
Figure 1.
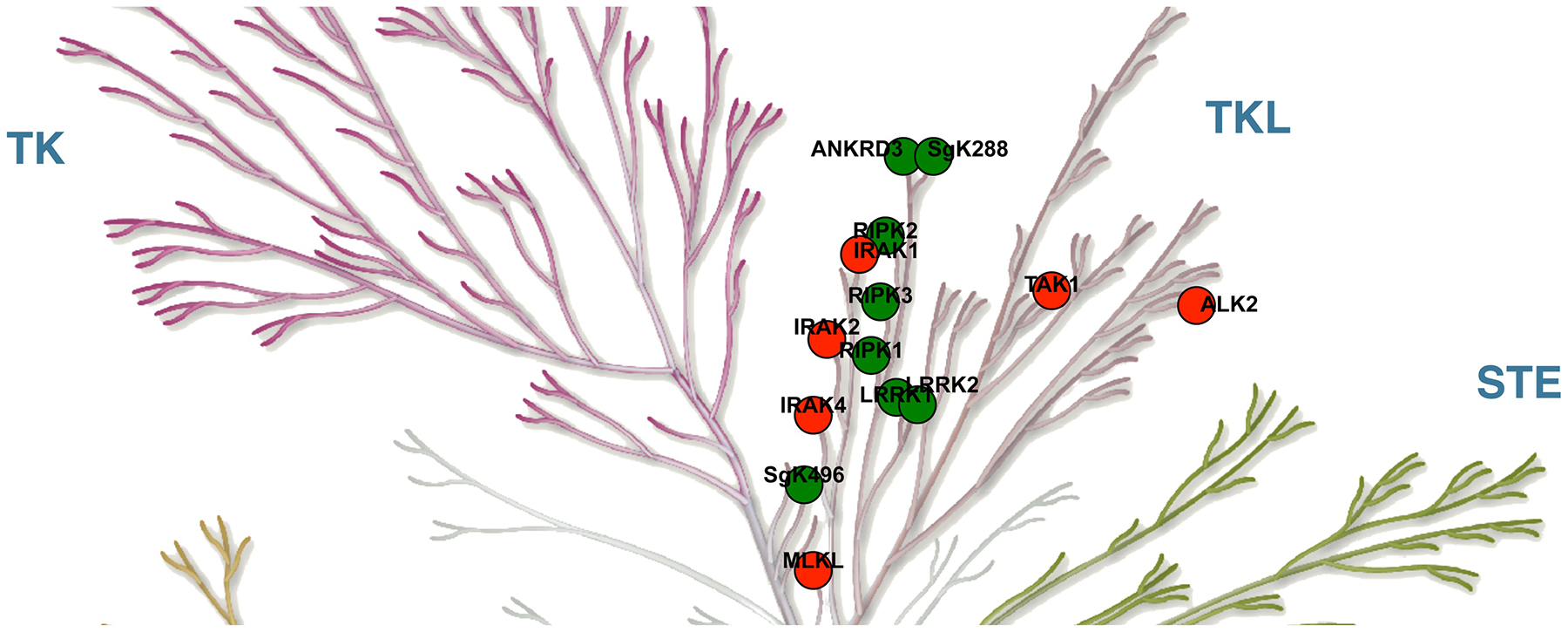
RIPKs belong to the Tyrosine Kinase-Like (TKL) subfamily of human kinases. RIPK family members are shown in green (ANKRD3 is RIPK4, SgK288 or SgK496 is RIPK5, see text for further details). Related TKL kinases are shown in red. Illustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
RIPK1 (RIP, RIP1) was the founding member of the family. It was first described by Stranger et al. in 1995 as an adaptor associated with Fas receptor, which is capable of inducing apoptosis [4]. The induction of death may have been an artifact of protein overexpression, and the main function was quickly recognized to be activation of NF-κB downstream from Tumor Necrosis Factor (TNF) receptor family members [5, 6]. The function of the catalytic activity of RIPK1 remained enigmatic until Holler et al. showed its critical role for the activation of the newly observed Fas ligand (FasL)-dependent regulated necrosis mechanism [7], which we later termed “necroptosis” [8].
RIPK2 and RIPK3 soon followed RIPK1 [9–13]. As with RIPK1, RIPK2 was proposed to have a role in caspase activation [9, 12]. However, as we will discuss below, it may rather reflect regulation of RIPK2 by caspases. On the other hand, its role in the regulation of NF-κBactivation withstood the test of time [10]. Similarly, RIPK3 was initially proposed to induce NF-κBactivation and apoptosis [11, 13]. However, this is no longer considered the main function of this protein [14], once the role of its kinase domain in necroptosis was identified [15–17]. Lastly, NF-κB-activating RIPK4 and RIPK5 were added to the repertoire of RIPKs [18–20], although the function(s) and even identity of RIPK5 remain poorly understood as we will discuss below.
Architecture of RIPK family
RIPKs share homologous kinase domains (Fig. 2A), which contain all the canonical catalytic elements including homologous but not identical P-loops, an ionic pair of catalytic Lys and Glu in the center of αC helix, and an HXD motif in the catalytic loop (Fig. 2B). It should be noted that there is some controversy regarding which kinase represents RIPK5 as both SgK288/ANKK1 and SgK496/RIPK5 have been assigned this role [21, 22]. Based on homology, SgK496 is only slightly closer to other RIPKs and shows greatest homology to RIPK4 (Fig. 2C). On the other hand, SgK288 is most similar to RIPK1. Thus, both kinases could be considered members of the RIPK family. However, further analyses of their physiologic utilities will be needed to establish whether these kinases are functionally related to other RIPKs.
Figure 2.
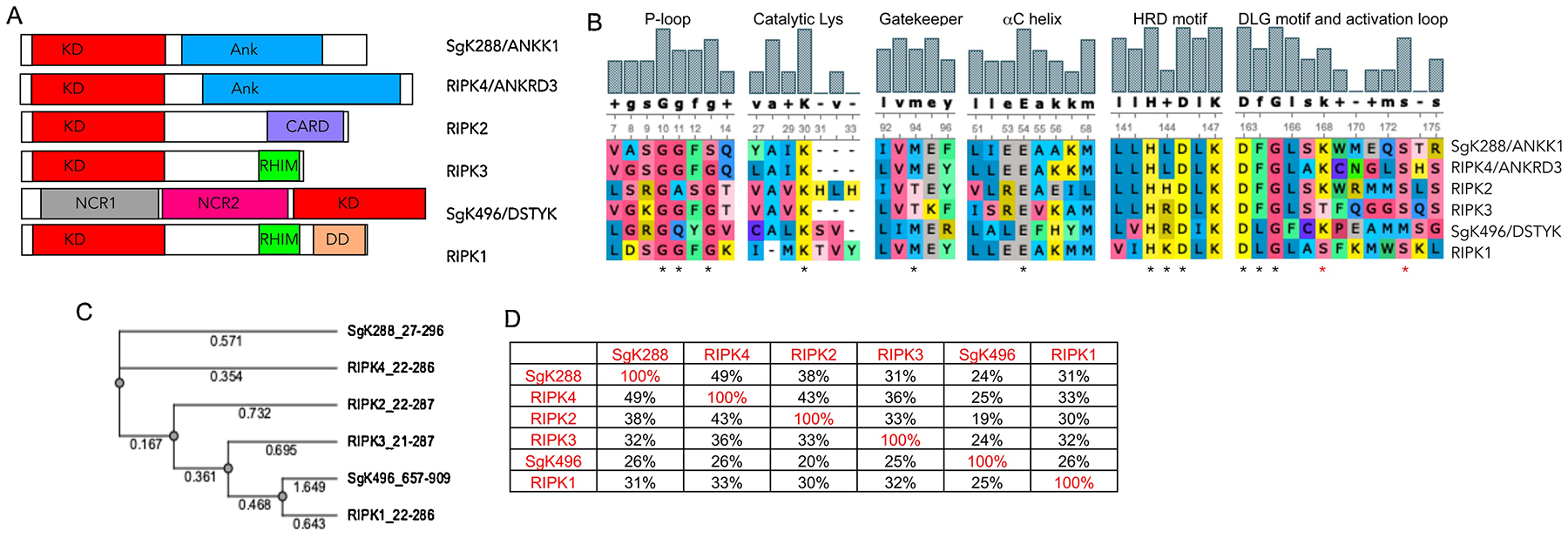
A) Domain architecture of RIPKs. B) Alignment of the critical catalytic elements of RIPKs. * indicate critical residues in each motif, * depict RIPK1 autophosphorylation sites Ser161 and Ser166. C) Phylogenic tree of RIPK kinase domains. Alignments in B and C were generated using UGENE software package and T-Coffee alignment algorithm. D) Degree of similarity between RIPKs was calculated in UGENE using the built-in similarity matrix. Alignment was also performed using ClustalW with similar results.
Along with similarities, there are also notable differences between RIPKs. For example, RIPKs differ in the size of the gatekeeper residue. This residue limits inhibitor access to the allosteric back pocket of kinases (Fig. 3A). RIPK1, RIPK4 and RIPK5 contain bulky Met, while RIPK2 and RIPK3 possess a much smaller and hydrophilic Thr. This has significant implications for the development of allosteric RIPK inhibitors as we showed for RIPK1 [23]. Furthermore, RIPK1 belongs to a subset representing ~15% of the human kinome that contains a non-Phe residue in the middle of the Mg2+-binding DFG motif (Leu157, Fig. 3A), which, as we discuss below, contributes to the unusual flexibility of the back-pocket of RIPK1 [23]. This is a unique feature of RIPK1, which is shared only by SgK288, but no other RIPK family member. In addition, RIPKs display diversity in the central residue of the HXD motif (Fig. 2B). Positively charged Arg in this motif has been shown to interact with a negatively charged phosphate on the residue that undergoes phosphorylation in the activation segment. This stabilizes the active kinase conformation in HRD kinases (Fig. 3B) [24, 25]. Conversely, activation loop phosphorylation may be less important in non-HRD kinases, which lack this interaction. Lastly, it is worth noting that two residues, Ser161 and Ser166, in the activation loop of RIPK1 have been shown to represent RIPK1 autophosphorylation sites, and the latter is used as a marker of RIPK1 activation in cells [26–28]. Notably, N-terminal 160AS161 site of RIPK1 is replaced by either ST in RIPK3 or SK in RIPK2, and SgK288. RIPK4 and Sgk496 lack this Ser residue. Thus, regulation of the activation loop conformation by N-terminal Ser phosphorylation likely significantly differs between RIPKs. In contrast, Ser166 is more highly conserved.
Figure 3.
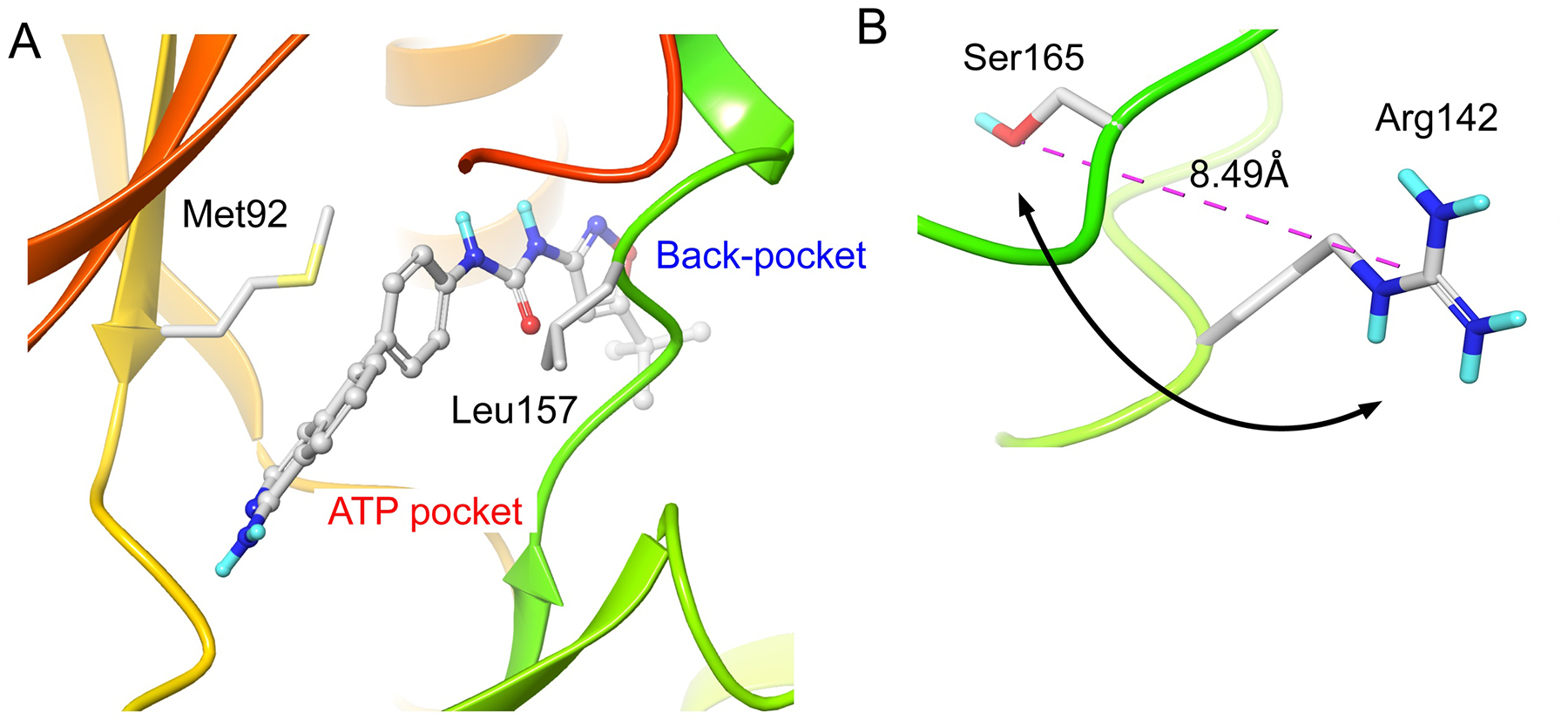
Structural features of RIPKs. A) The size and flexibility of the side chains of Met92 gatekeeper and Leu157 of the DLG define access into the allosteric back-pocket of RIPK1. Based on Glu-in/DLG-out structure 4NEU. Figures were generated using the Schrödinger Maestro software package. B) Arg142 of the HRD motif of RIPK3 is in sufficient proximity to Ser165 in the activation loop. We propose that phosphorylation of Ser165 will result in mutual rotation (arrow) of the two side chains to enable interaction. Based on the active Glu-in/DFG-in structure 4M66.
The C-termini of RIPKs contain a number of protein-protein interaction motifs. RIPK1 contains the Death Domain (DD) required for the assembly of the receptor-proximal complexes on TNF receptor family proteins and recruitment of the critical adaptor FADD [29]. Interestingly, DDs also play a vital role in signaling through MyD88 adaptor downstream from Toll-like receptor (TLR) and IL1 receptor families, which involves recruitment of related IRAK family of kinases (Fig. 1). However, association of RIPK1 with MyD88 signaling platform has not been established. Another interaction motif is RIP Homotypic Interaction Motif (RHIM), present in both RIPK1 and RIPK3, which is critical for the formation of pro-necroptotic RIPK1/RIPK3 necrosomes [30], and is also the main motif for signaling from innate immune sensor Zbp1 and TLR adaptor TRIF [31]. These four proteins are the only RHIM domain-containing factors in mammalian cells and the RHIM domain of RIPK1 may serve as a integrator of both pro-death upstream signal, mediated by TRIF, as well as controller preventing inappropriate RIPK3 activation by Zbp1 [28, 32]. In addition, TRIF and Zbp1 can signal directly through RHIM of RIPK3 [33–35], creating a complex regulatory node. It should also be noted that the intermediate domain of RIPK1 contains a Ser320 residue phosphorylated by TAK1 and MAPKAPK2 kinases [36–38] as well as an Asp324 cleavage site for caspase-8 [39, 40], which are both critical for inhibition of pro-death activity of RIPK1, and a Lys377 poly-ubiquitination site needed for the assembly of NF-κB-activating IKK kinase complex [41, 42].
In contrast to RIPK1/RIPK3, RIPK2 contains a C-terminal Caspase Activation and Recruitment Domain (CARD). However, despite the initial evidence that the CARD of RIPK2 can promote caspase activation, it is currently believed to primarily function in the context of homotypic interactions with the upstream RIPK2 activators Nod1 and Nod2 [43, 44]. Notably, recent work by Goncharov et al. identified C-terminal residues K410 and K538 as sites of poly-ubiquitination of RIPK2 via X-linked Apoptosis Inhibitor Protein (XIAP) in addition to the previously reported K209 in the kinase domain [45–47]. These poly-ubiquitination events are critical for NF-κB activation by RIPK2 as we will discuss below.
Lastly, SgK288 and RIPK4 contain C-terminal Ankyrin repeats. In the case of RIPK4, 11 ankyrin repeats are present and attenuate its ability to activate NF-κB [19]. Recent work hypothesized that the function of Ank repeats may be to interfere with RIPK4 homodimerization, which is critical for its activation [24].
Functional roles of RIPKs
RIPKs have attracted a lot of interest in recent years due to their contributions to physiologic and pathologic inflammatory and cell death responses, and potential connections to a range of human diseases. Multiple in-depth reviews describing molecular mechanisms involving RIPKs have been published and we refer readers to these sources [48–52]. In this review, we are going to only briefly mention some of the major mechanisms and roles of RIPKs.
RIPK1
RIPK1 is a “Swiss Army knife” of innate immune regulation. It has now been linked to multiple innate immune receptors and sensors, including TNFRs, TLRs, IFNAR1, STING, MAVS and others [53]. Downstream from these receptors and associated adaptors, like TRIF, RIPK1 has been connected to the regulation of a multitude of responses including transcription and translation of inflammatory genes [28, 54], necroptosis, apoptosis and pyroptosis [26, 55–57]. RIPK1 utilizes distinct mechanisms for different modes of regulation. This includes kinase-independent functions - activation of NF-κB in the TNFR1-bound Complex I and homeostatic control against inappropriate activation of RIPK3 and caspase-8. Conversely, as a kinase RIPK1 promotes activation of caspase-8 and RIPK3 leading to cell death and inflammation through different versions of secondary Complex II. RIPK1 requires various effectors to exert its kinase-dependent functions, including RIPK3, caspase-8, TBK1, and MAP kinases Erk1/2 and p38 [28, 50, 57–59]. How RIPK1 balances the signals towards these different factors remains incompletely understood.
Not surprisingly, given the range of powerful responses under the control of RIPK1, it has been linked to a diverse range of degenerative, inflammatory and autoimmune peripheral and CNS diseases [50, 60]. Recent work also identified RIPK1 Asp324 mutations, which block inhibitory processing by caspase-8, as a cause of recurring febrile episodes in the affected patients [39, 40]. Additionally, loss of RIPK1 inhibition by TAK1 and TBK1 kinases was recently shown to contribute to aging [61]. To counter these actions of RIPK1, small molecule inhibitors were recently advanced into clinical testing as discussed below.
RIPK3
RIPK3, a partner of RIPK1 in the activation of necroptosis, has a similarly illustrious set of functions. Its main kinase-dependent functions are necroptosis, as well as pro-inflammatory gene expression and sustained translation [15–17, 28, 62]. Similarly to RIPK1, RIPK3 is cleaved by caspase-8 at Asp328, inactivating its kinase-dependent activities [63]. In a kinase-independent manner, it has also been linked to activation of inflammasomes and apoptosis [17, 64–67]. The disease roles of RIPK3 kinase activity are currently difficult to assess because of on-target apoptotic activity of RIPK3 inhibitors [66]. RIPK3 kinase activates necroptosis through a pseudokinase MLKL. Using Mlkl−/− mice as a surrogate for RIPK3 inhibition, Newton et al. showed that it contributes to some, but not all injury paradigms linked to RIPK1 kinase and RIPK3 protein as assessed using Ripk3−/− mice [68]. Thus, RIPK3 may represent a valid therapeutic target but in a more limited subset of conditions compared to RIPK1.
RIPK2
RIPK2’s main recognized function is activation of NF-κB. RIPK2 is stimulated by Nod1 and Nod2 receptors that recognize different peptidoglycan (PGN) chains of Gram-positive and Gram-negative bacteria as recently reviewed by Helm et al. [49]. PGN chains, present on the surface of invading pathogens such as S. flexneri or L. monocytogenes, interact with Leucine Rich Repeat (LRR) domains of Nod1 and Nod2 proteins, respectively [69, 70], leading to a conformational change and Nod oligomerization [71]. Multiple molecules of RIPK2 are recruited through CARD-CARD interactions [44], forming filamentous structures critical for signaling [43, 72]. These filamentous structures appear to be common and potentially even interchangeable [73] signaling intermediates in various innate immune mechanisms including inflammasomes, TLR complexes, necrosomes and caspase-8-activating DISC complexes [30, 74–76]. Notably, RIPK2 was shown to autophosphorylate on Ser176 (activation loop) and Tyr474 (CARD domain) residues. Furthermore, despite being a non-HRD kinase (Fig. 1B), mutations of these residues were shown to reduce RIPK2 signaling in experiments involving protein overexpression [77]. However, more recently the significance of the catalytic activity of RIPK2 for signaling was brought into question. In two separate studies, expression of catalytically inactive RIPK2 has been shown to support Nod responses in cells, even though multiple RIPK2 kinase inhibitors were found to block these responses [45, 78]. Instead, inhibition of Nod signaling by RIPK2 kinase inhibitors was linked to another critical event in the RIPK2 signaling pathway – its binding and ubiquitination by X-linked Inhibitor of Apoptosis Protein (XIAP). XIAP activity is critical for NF-κB activation by RIPK2 through regulation of K63 poly-ubiquitination along with Pellino3, and recruitment of the M1-ubiquitinating LUBAC complex [79, 80]. Notably, this two-step IAP/LUBAC K63/M1 mechanism was also demonstrated for NF-κB activation by RIPK1 [81]. There is no evidence currently that RIPK2 is directly involved in the regulation of cell death despite the presence of CARD domain. On the other hand, recent data suggest that similar to other RIPKs, RIPK2 is cleaved by caspase-1, which is recruited through CARD-CARD-interactions. Thus, the CARD domain may serve to control RIPK2 by caspases and the lack of this cleavage leads to recurring febrile episodes [82].
RIPK4
RIPK4 is also an NF-κB activating kinase, which exerts its functions through phosphorylation of IRF6 on Ser413 and Ser426 [83–85]. Catalytic activity of RIPK4 was shown to play a critical role in normal epidermal differentiation in mice and maintenance of the barrier function of the skin. This phenotype is mediated by IRF6 and another RIPK4 target – plakophilin-1 [85, 86]. Furthermore, in humans mutations in RIPK4 and IRF6 have been found in two highly related epidermal developmental disorders - Bartsocas-Papas syndrome and popliteal pterygium syndrome, respectively [87, 88].
Lastly, very little is currently known about SgK288 and SgK496 kinases and their functional roles.
Emerging common themes in the functions and regulation by RIPKs
The RIPK family was originally assembled based on homology in their kinase domains. However, given the lack of bona fide substrates of these kinases, it is prudent to consider whether there is evidence that RIPKs are indeed related. In this section, we summarize the currently understood overlaps between family members that support this notion.
RIPKs as guardians of barrier integrity
In general, RIPK1–RIPK4 kinases operate in highly related areas of host defense and barrier function [89]. RIPK4 has been shown to contribute, in a kinase-dependent manner, to the integrity of epidermal barriers through regulation of keratinocyte differentiation [85]. RIPK1 in a kinase-independent manner protects epithelial barrier integrity in the gut and epidermis by preventing damage through apoptosis and necroptosis [90, 91]. RIPK3 also has a kinase-independent role as a protector of epithelial barrier integrity by promoting IL-23 and IL-1β mediated repair after severe dextran sodium sulfate (DSS)-induced intestinal injury [65].
Roles and interplay of RIPKs in innate immune responses
RIPK1, RIPK2 and RIPK3 further contribute to distinct, but highly related innate immune processes. RIPK2 has a well-established role in promoting NF-κB-dependent inflammatory responses to invading Gram-positive and Gram-negative bacteria downstream from Nod sensors as we discussed above. RIPK1 and RIPK3 were shown to work in their regulatory functions together as well as separately. For example, RIPK1 works together with RIPK3 and caspase-8 to promote parallel pathways of cell death and inflammation by bacterial pore forming toxins leading to enhanced inflammation in the lungs in vivo [92]. Similarly, RIPK1 and RIPK3 act in concert to restrict neuronal propagation of West Nile virus, although in this case recruitment of immune cells through production of immune mobilizing chemokines, rather than neuronal cell death, has been shown to play the central role [93]. In contrast, RIPK1 has been linked, in a RIPK3-independent manner [94], to the activation of pyroptosis by Y. pseudotuberculosis, which results from inactivation of the proinflammatory TAK1 kinase by the injected bacterial protein YopJ [95]. RIPK1 was also proposed to control Leishmania replication independently from RIPK3 kinase activity [96]. Conversely, RIPK3 has been proposed to mediate necroptosis independent of RIPK1 upon infections with mouse cytomegalovirus (MCMV) [35]. In addition, Influenza-A infection has been shown to activate RIPK3 in lung epithelial cells leading to parallel pathways of kinase-dependent necroptosis and kinase-independent apoptosis [67]. Notably, neither mechanism involves the kinase activity of RIPK1.
It should also be noted that in the context of infection, different sensors are triggered and, thus, multiple innate immune mechanisms are simultaneously induced. This may also apply to RIPK1/3 and RIPK2 pathways, which are triggered by different receptors - TNFR1 or TLRs vs. Nod1/2, respectively. An elegant interplay of RIPK1 and RIPK2 pathways has been recently shown for PGN-induced responses by Stafford et al. [97]. In this case, Nod2/RIPK2/XIAP in a small population of sentinel cells was proposed to initiate TNFα synthesis, triggering the TNFR1/RIPK1/cIAP1 autocrine TNFα loop in other leukocytes, which is required for full PGN response.
Significance of RIPKs for host defense?
At the same time, it remains poorly understood whether there are specific human pathogens for which RIPKs play critical, non-redundant roles in host defense. For example, recent work by Webster et al. questioned the importance of RIPK1 kinase activity and RIPK3 for the control of viral propagation given that infections with vaccinia virus and mouse gammaherpesvirus MHV68 showed comparable rates of viral clearance in WT, RIPK1 kinase dead knock-in (KI) and Ripk3−/− mice [98]. In can also be questioned whether the pathogens that can elicit RIPK responses have already developed means to counter them earlier in their evolution. For example, MCMV virus has been shown to express RHIM-containing M45 protein, which inhibits RIPK3-mediated necroptosis [35]. Similarly, human herpes simplex virus 1 (HSV1) was found to encode necroptosis inhibitor protein ICP6 [99]. Interestingly, an oral Gram-negative bacterial pathogen P. gingivalis and HIV1 virus were both proposed to possess mechanisms leading to proteolytic processing and inactivation of both RIPK1 and RIPK2 [100, 101]. Overall, these data are consistent with pathogens developing mechanisms targeting multiple RIPKs, but also raises questions about the importance of RIPKs for physiologic host defense responses at present.
Mechanistic similarities between RIPKs
Multiple commonalities are also seen between RIPKs on the molecular level. A key function of RIPK1–RIPK4 is activation of the pro-survival, pro-repair, and pro-inflammatory NF-κB pathway. RIPK1 and RIPK2 regulate this function with the help of IAP-dependent poly-ubiquitination as we discussed above. RIPK3 has also been shown to induce NF-κB though a yet to be defined mechanism [65]. Interestingly, activation of NF-κB by these RIPKs does not require kinase activity, and only RIPK4 induces this pathway through phosphorylation of IRF6 [83]. Notably, a Drosophila homologue of RIPKs is an innate immune protein IMD involved in peptidoglycan sensing. It only shows homology to the C-termini of RIPK1 and RIPK2, but not the kinase domains [102]. This gives credence to the hypothesis that kinase-independent activation of NF-κB may have been an initial core property of the RIPK family with different family members developing additional and divergent kinase-dependent functions later in their evolution. Supporting this notion, IMD has been shown to form amyloid fibrils through cryptic RHIM domains [103], which were also shown for RIPK1 and RIPK3 RHIMs [30]. However, in contrast to mammalian counterparts, IMD fiber formation promoted NF-κB activation, but not cell death [103].
Common regulation of RIPKs through ubiquitination and caspase cleavage
It is also striking that all of these RIPKs are the substrates of the same E3 ligases - IAP family (although different RIPKs are likely targeted by particular IAP family members physiologically) and Pellino3 for RIPK1 and RIPK2 [80, 104]. This may suggest a coordinated regulation of different RIPKs. Interestingly, many of the identified ubiquitination sites on these kinases are located on C-terminal domains that share little similarity between RIPKs. So why are the same E3 ligases able to target multiple RIPKs at non-homologous regions? Our recent work identified a binding site for XIAP BIR2 domain as two Arg residues in the β-sheet bundle of RIPK2s’ kinase domain [78]. Given the homology in the kinase domains, an analogous mechanism of kinase domain binding of E3 ligases could be an answer to this question.
Another common regulatory theme for RIPKs is cleavage by caspases: caspase-8 in the case of RIPK1 and RIPK3, and caspase-1 in the case of RIPK2, which limits their kinase-domain dependent functions. The inability to cleave RIPK1 or RIPK2 leads to recurring febrile disease, while consequences of the lack of RIPK3 cleavage are not yet described. Thus, multiple RIPKs not only function in similar pathways, but also appear to be regulated in an analogous manner.
Overlapping pathophysiologic roles of RIPKs
Pathologically, RIPK1, RIPK2 and RIPK3 have been implicated in a range of inflammatory conditions. Furthermore, there are a number of conditions that appear to involve multiple RIPK proteins. One example is multiple sclerosis (MS). Genetic and pharmacologic evidence suggest that both RIPK1/RIPK3 and RIPK2 contribute to pathology in the mouse experimental autoimmune encephalomyelitis (EAE) chronic progressive model of demyelinating disease [105–107]. As with the work by Stafford et al. cited earlier, RIPK1 and RIPK2 may exert their functions in different cell populations with RIPK1 acting in resident CNS cells, while RIPK2 plays a critical in the functions of infiltrating peripheral dendritic cells. Similarly, colitis and Crohn’s disease may involve contributions of RIPK1/RIPK3 and RIPK2. Genetic deletion or inhibition of RIPK2 has been shown to be protective in multiple models of intestinal inflammation [108–110] and recent evidence suggest that RIPK1 may also play a role [111]. Based on the available data, one can envision a mechanism whereby RIPK1 functions in maintaining integrity of the epithelial barrier through controls over various cell death mechanisms, whereas compromised barrier function may lead to chronic inflammation mediated by sentinel cells sequestering invading microbiota and triggering RIPK2 activation.
Small molecule inhibitors of RIPK family
RIPK1 inhibitors
Twenty-five years after RIPK1 was first described, interest in RIPK family member inhibitors is now accelerating. The first reported inhibitor of this kinase family did not emerge from a kinase screen, but rather a phenotypic cell-based screen seeking to identify compounds that would block necroptosis in an assay of TNFα-induced cell death in FADD-deficient Jurkat cells [8]. One of the most promising compounds to emerge from that study was an indole derivative called Necrostatin-1 (Nec-1), which is the product from Edman degradation of tryptophan peptide residues with methyl isocyanate. Optimization of this initial lead molecule resulted in 1 (Nec-1s, Fig. 4A), which demonstrated improved activity and metabolic stability [112]. In 2008, RIPK1 was identified as the responsible molecular target of these compounds [26]. In addition, the surprising selectivity of 1 for RIPK1 from among other kinases was revealed. However, it would be another five years before the binding mode of 1 with RIPK1 was confirmed by co-crystallization [113] as described below. Since these initial findings, several structurally distinct classes of RIPK1 inhibitors have been reported, including benzoxazepinones such as 2 (GSK2982772) [114], brain penetrant 3 [115], as well as 4 [116], the low molecular weight hydroxamic acid 5 (RIPA-56) [117], and dihydropyrazoles 6 (GSK’547) and 7 [118]. Encouragingly, several RIPK1 compounds have now advanced to clinical assessments, including 2 in phase 2 trials for the treatment of ulcerative colitis (NCT02903966), psoriasis (NCT02776033), and rheumatoid arthritis (NCT02858492) by GlaxoSmithKline, and undisclosed compounds in phase 1b trials for amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease by Denali Therapeutics (NCT03757351 and NCT03757325, respectively). For a more comprehensive discussion of the medicinal chemistry development of these and other RIPK1 inhibitors see a recent review by Zhuang and Chen [119].
Figure 4.
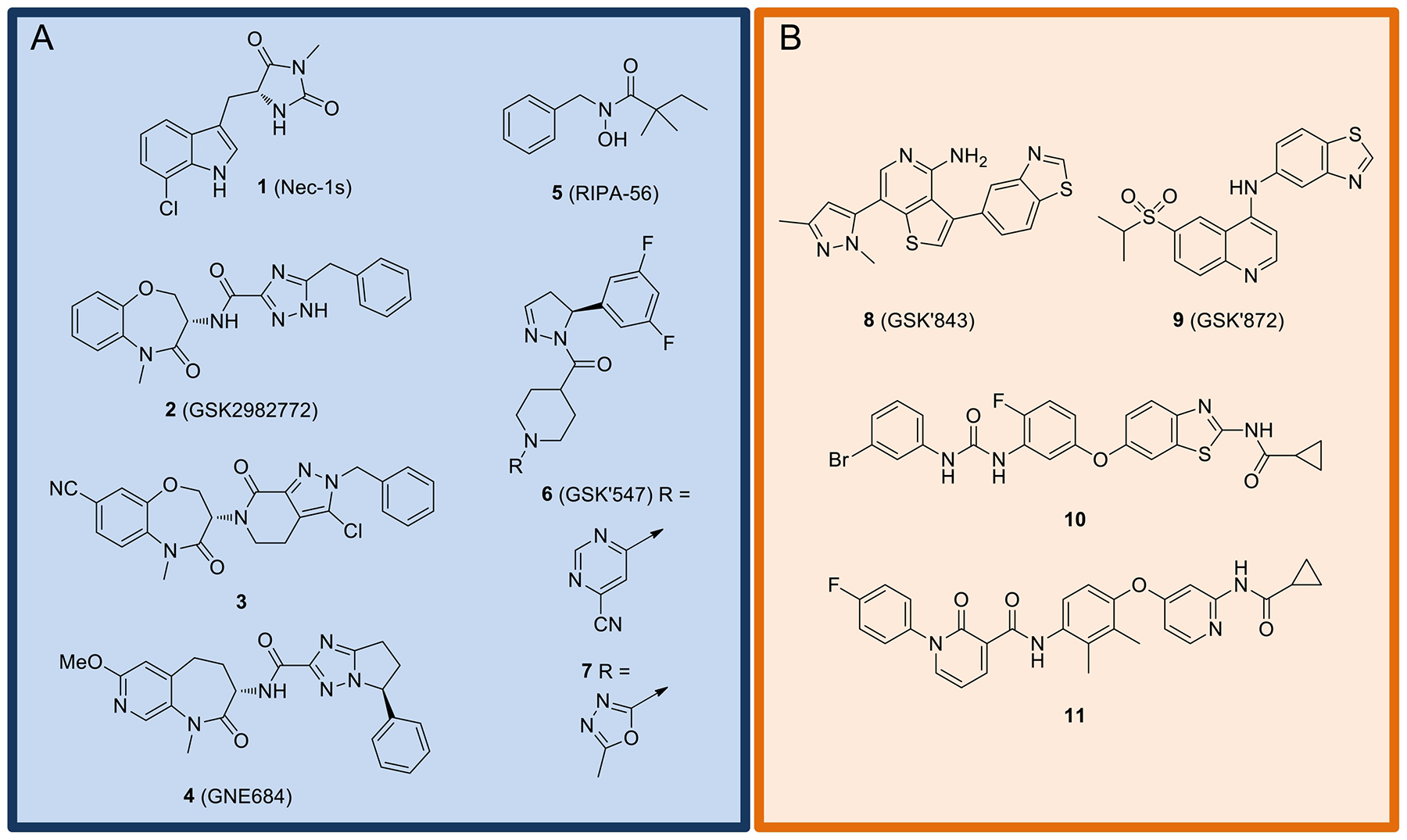
A) Representative examples of RIPK1 inhibitors. B) Representative examples of RIPK3 inhibitors.
RIPK3 inhibitors
The emergence of RIPK1 inhibitors naturally led to interest in the development of inhibitors of its partner in crime RIPK3 (Fig. 4B). The first RIPK3 inhibitors to emerge were 8 (GSK’843) and 9 (GSK’872) [34]. Even though their mode of binding has not yet been revealed, these compounds are structurally related to several RIPK2 inhibitors (15 – 17, see below) that bind to the active form of the kinase. Unexpectedly, these compounds displayed dichotomous activities, including both inhibition of necroptosis as well as RIPK3-dependent induction of apoptosis [66]. Two other RIPK3 inhibitors recently reported are 10 [120] and 11 [121]. Both compounds demonstrate excellent selectivity over RIPK1. Although 10 was presumed to also interact with the hinge region in the kinase’s active conformation, the molecule consists of numerous structural features that would suggest it might bind to the inactive conformation of the kinase. Inhibitor 11 has been confirmed to bind the inactive DFG-out conformation of RIPK3. Unfortunately, nothing has yet been reported regarding RIPK3-dependent toxicities of these molecules.
RIPK2 inhibitors
Some of the first well-characterized RIPK2 inhibitors reported were structurally interesting macrocylic derivatives exemplified by pyrazolo[1,5-a]pyrimidine 12 and imidazo[1,2-b]pyridazine 13 that are ATP-competitive [108] (Fig. 5). Both compounds inhibited RIPK2 tyrosine autophosphorylation as well as NF-κB and MAPK signaling induced by MDP. This was followed by the disclosure of the 1H-pyrazolo[3,4-d]pyrimidine derivative 14 (WEHI-345) that inhibited RIPK2 kinase activity, RIPK2 ubiquitylation, NF-κB activation in vitro, and cytokine production in vivo. Notably, 14 (WEHI-345) reduced experimental autoimmune encephalomyelitis in mice at 20 mg/kg bid [105]. Selective and potent quinoline-based RIPK2 inhibitors were identified through screening of DNA-encoded libraries and optimized to provide 15 (GSK’583) [122]. Further refinement achieved quinazoline derivative 16 (GSK2983559), which advanced into clinical trials as prodrug 17 [109]. However, the trial (NCT03358407) was terminated due to non-clinical toxicology findings and reduced safety margins. In 2017, RIPK2 inhibitor 18, which contains a sulfone and ethers substituted imidazo[1,2-a]pyridine core, was disclosed [123]. Finally, a series of 3,5-diphenyl-2-aminopyridine inhibitors of RIPK2, exemplified by 19 (CSLP37), were reported to antagonize XIAP-binding and XIAP-mediated ubiquitination of RIPK2 [78, 124].
Figure 5.
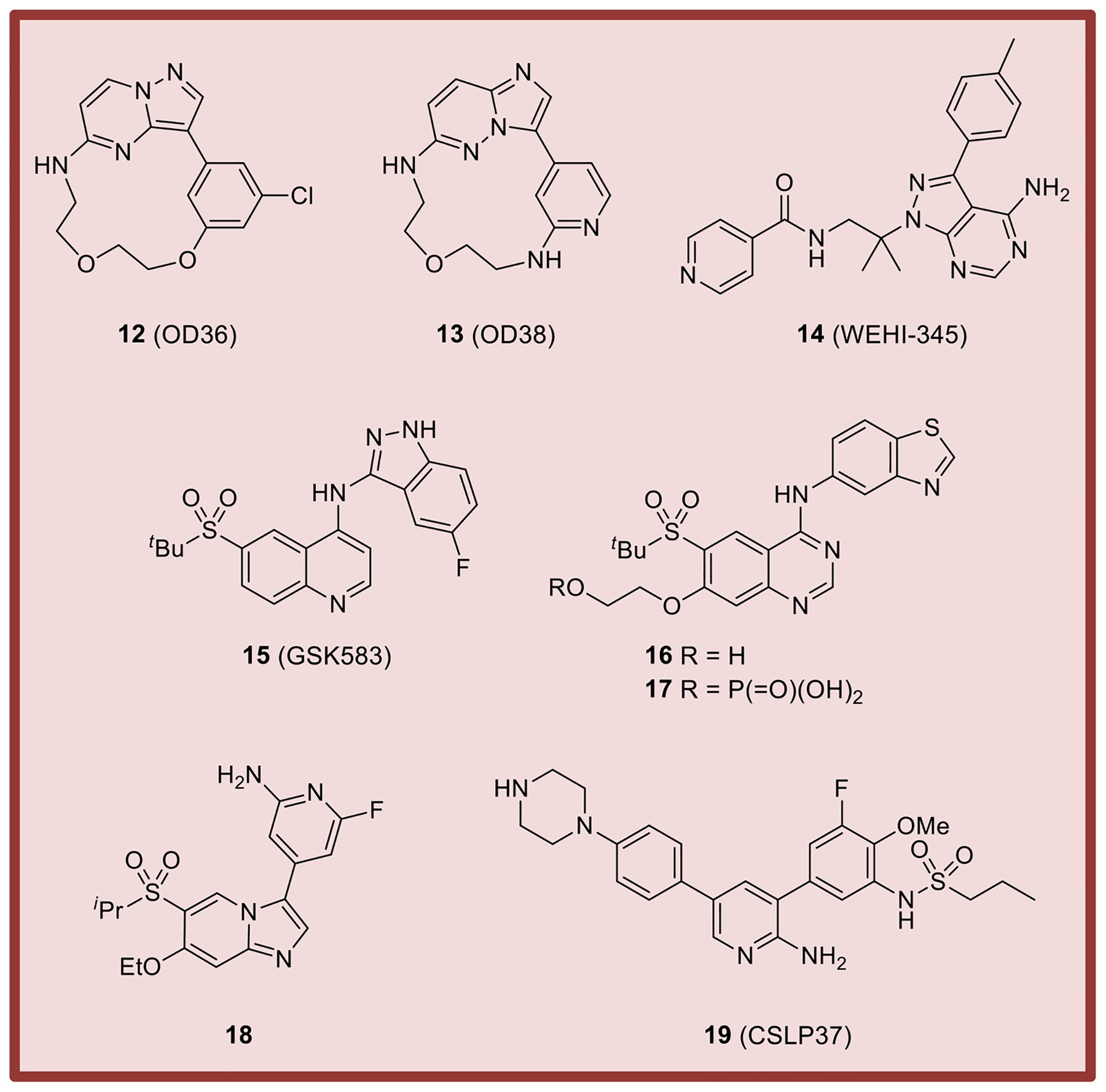
Representative examples of RIPK2 inhibitors.
Importantly, a number of the inhibitors highlighted herein are commercially available allowing the research community to further illuminate the functions of RIPK family members. In addition, further structure-activity relationship (SAR) analyses and in vivo experiments with current and emerging RIPK3 inhibitors will hopefully reveal strategies for RIPK3 modulation in clinically viable settings. No inhibitors of RIPK4 have been reported thus far.
Structural insights into RIPK family
As discussed at the beginning of this review, RIPKs possess all the requisite kinase motifs in the correct places (Fig. 2), thus representing a fairly typical kinase family. However, as we will discuss next, these proteins appear to display a number of atypical features in their structures, mechanisms of action and small molecule inhibition, which we will attempt to link together.
Atypical features of RIPK1 and its inhibitors
At first glance, multiple RIPKs appear to be structurally similar. Kinase active conformation structures of RIPK2, RIPK3 and RIPK4 have been reported [24, 125, 126], and show a canonical kinase fold, typical Glu-in/DFG-in conformation, and a high degree of similarity for all three proteins (Fig. 6A). Structures bound to Type II inhibitors [127] are also available for multiple RIPKs [121, 128–130]. Likewise, these structures reveal typical inactive DFG-out/Glu-in conformations of RIPK1, RIPK2 and RIPK3 kinase domains (Fig. 6B). Furthermore, pan-specific Tyr kinase inhibitor ponatinib has been found to equally and efficiently inhibit all tested RIPKs [23, 131], which is consistent with these proteins not behaving differently from many other human kinases.
Figure 6.
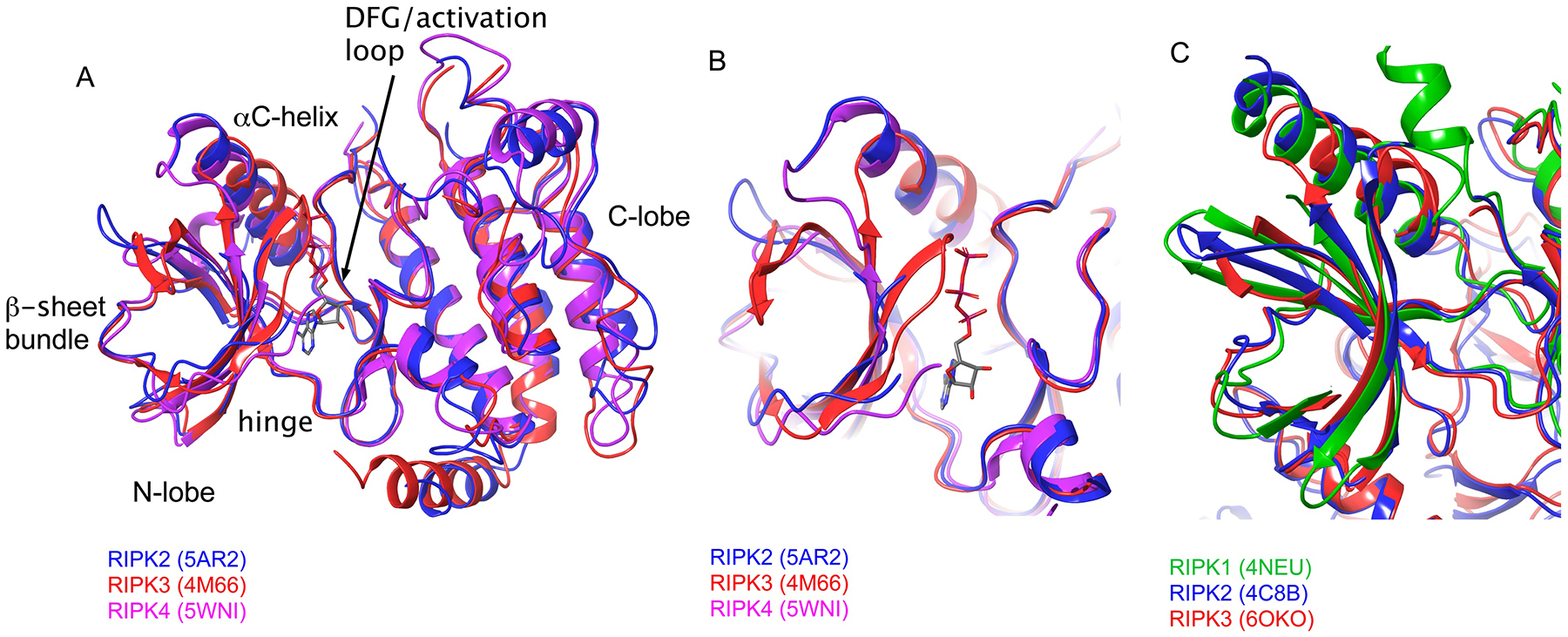
A,B) Alignment of the DFG-in/Glu-in active conformations of RIPKs. A – entire kinase domains, B – ATP-binding pockets. C) Alignment of inactive DFG-out/Glu-in conformations of RIPKs.
However, this conclusion is in opposition to other evidence suggesting that RIPKs are anything but typical kinases. Most strikingly, it does not explain rapid development of multiple RIPK1 inhibitors that show monovalent selectivity for RIPK1 (summarized in [50]). The RIPK1 binding mode was first described for necrostatins, like 1 [113] (Fig. 7A,B), and has since been observed for multiple RIPK1 inhibitors [50]. All of these molecules display a conformation characterized by two inactivating events – movement of the αC-helix away from the active center, resulting in the loss of catalytic Lys coordination by the αC Glu residue, and rotation of the DLG motif into an inactive DLG-out conformation with activation loop blockage of the putative substrate binding site (Fig. 7A). Thus, these inhibitors are termed Type III allosteric inhibitors [132], because they predominantly bind within the kinase back-pocket and lack contacts in the more promiscuous ATP binding pocket. Remarkably, RIPK1 remains to the best of our knowledge the only human kinase for which this “double-inactive” Glu-out/DLG-out conformation characterized by maximal opening of the back-pocket has been described, which explains the amazing selectivity of these molecules.
Figure 7.
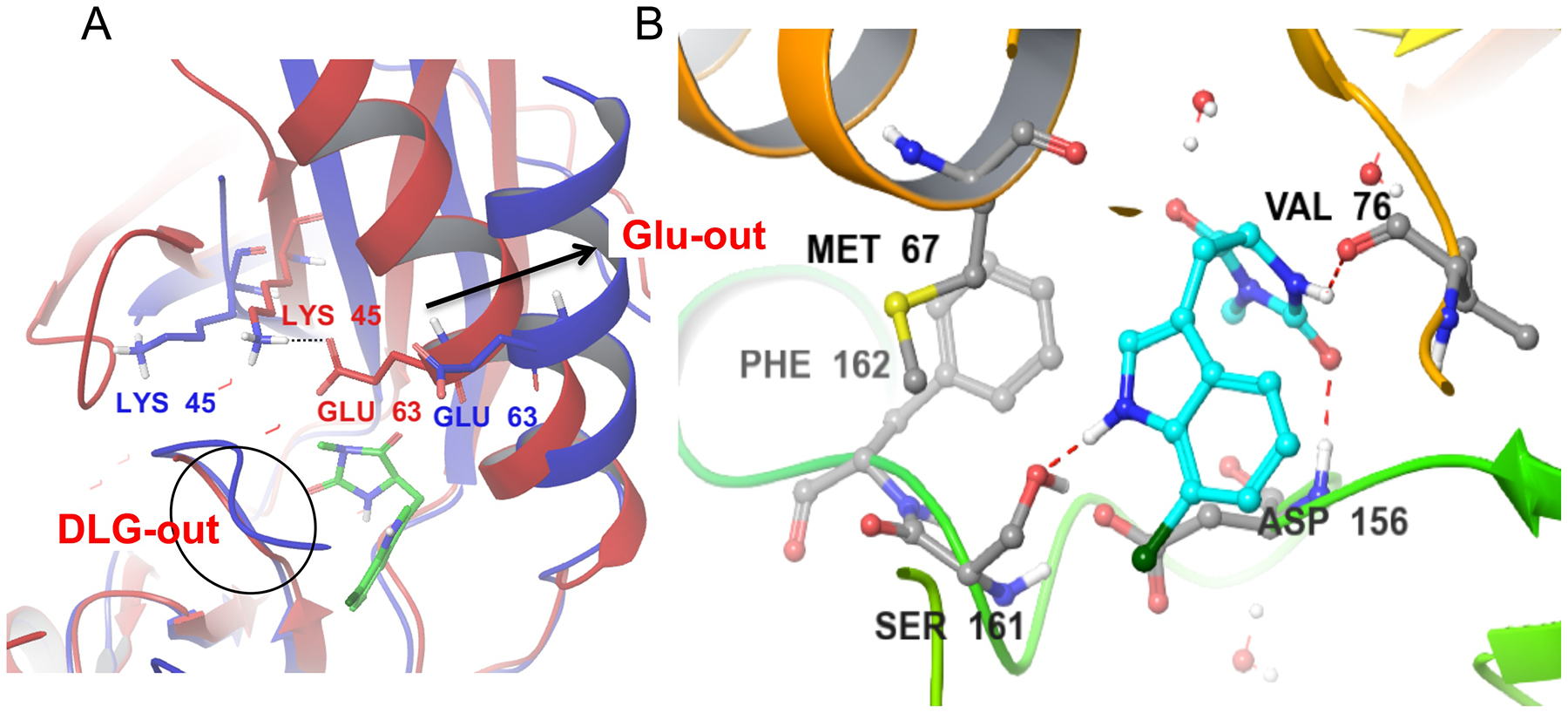
A) Alignment of the Nec-1s bound to RIPK1 in the DLG-out/Glu-out conformation (4ITH, blue) vs. the model of the active RIPK1 conformation, generated using homology modeling (red). Nec-1s binds exclusively in RIPK1 back pocket between activation segment and αC helix. This conformation is associated with the outward movement of the αC helix (arrow), breaking an ionic interaction of Glu63 with catalytic Lys45 and rotation of the DLG motif into the inactive conformation where the side-chain of Asp is not aligned with the ATP pocket. B) Binding of Nec-1s is mediated by three hydrogen bonds, including to the side chain of Ser161, an autophosphorylation site at the N-terminus of the activation loop. It is also stabilized by hydrophobic interactions, especially with the side-chains of Met67 in the αC helix and Phe162 in the activation loop.
Another unexpected feature of RIPK1 inhibitors is the tremendous species selectivity observed for a number of Type III molecules. For example, benzoxazepinone inhibitors, but not others including 1, potently inhibit human, but not a 77% homologous murine RIPK1 [133]. What could explain the ability of this kinase to accommodate such highly selective inhibitors? In our opinion, unusual flexibility of RIPK1’s back-pocket facilitates its opening and ability to bind Type III inhibitors. In part, this is due to RIPK1 being a non-DFG kinase. As we have reported, substitution of the bulkier and more rigid Phe residue with Leu leads to a much more energetically favorable opening of the kinase back-pocket [23]. Work by Harris et al. further showed that not only the opening, but an entire back-pocket of mouse RIPK1 may be unusually flexible, which may explain difficulty for many inhibitors to stay bound to mouse protein. Notably, substitution of residues in the activation loop of mouse RIPK1, which forms part of the back-pocket, with human counterparts restored sensitivity of mouse RIPK1 to “human-only” inhibitors. The decreased flexibility of the activation loop of human kinase was proposed to underlie these changes [133]. Overall, these data paint a striking picture where flexibility of the back-pocket of human RIPK1 is optimal for Type III inhibitor binding without it being excessive and leading to low affinity as in mouse RIPK1.
Interestingly, RIPK1 inhibitors have also been found to possess unexpected activity in cellular experiments. Namely, recent work clearly demonstrated that activation of necroptosis critically depends on RIPK3, while RIPK1 may be less generally critical. This is based on the observations that Ripk1−/− cells were still sensitive to different induction signals, such as TLR3 ligand poly(I:C), which could activate necroptosis by causing direct activation of RIPK3 by TRIF [34, 134]. At the same time, RIPK1 inhibitors blocked necroptosis in the matched wild type cells similarly induced to undergo necroptosis. This creates an apparent paradox. How can small molecule inhibitors of RIPK1 efficiently block necroptosis if RIPK1 kinase is not absolutely required for this form of cell death? Importantly, RIPK1 inhibitors are not active in Ripk1−/− cells confirming that activity of inhibitors is not due to the targeting of some other factor(s) in the pathway [134, 135]. To help explain these data, we suggest considering that RIPK1 inhibitors stabilize a very specific, inactive conformation of the kinase domain. If a specific conformation (in addition to catalytic activity) of the kinase domain of RIPK1 is critical for signaling, then an inhibitor locking the kinase domain in a particular inactive conformation may create a dominant negative form of the protein, which would interfere with downstream RIPK3 activation. Such a conformation-dependent mechanism of signaling may also explain why RIPK1 seems to lack targets besides autophosphorylation sites, which typically control kinase domain conformation. Such a mechanism would also be well suited for activation of innate immune responses through protein-protein interaction-dependent oligomerization [74]. However, a proposed non-canonical scaffolding role of RIPK1 kinase domain in signaling currently remains a hypothesis and awaits experimental validation.
RIPK2 – kinase activity vs. ubiquitination
This hypothesis is potentially buoyed by the recent data generated by us using CSLP inhibitors like 19 of a highly related RIPK2 kinase, which highlighted a greater role of this protein as a scaffold compared to its kinase activity [78]. In this work, we have shown that catalytic activity of RIPK2 is not essential for cellular signaling, a view also shared by Goncharov et al. [45]. Instead, molecules, such as ATP competitive CSLPs, and GSK’583 or Type II ponatinib, efficiently block Nod1/2 responses in cells by extending into a specific sub-pocket in the back of the active center. This site is located proximally to the XIAP BIR2 domain binding site on the surface of the kinase domain of RIPK2. Thereby, occupancy of the sub-pocket interferes with XIAP binding and RIPK2 poly-ubiquitination, explaining why some RIPK2 inhibitors are effective in blocking Nod1/2 responses even though catalytic activity of RIPK2 is dispensable.
Conformational switches in RIPK3 dictate its activity
Unlike RIPK1 and RIPK2 that currently lack targets for phosphorylation, RIPK3 and RIPK4 behave as more canonical kinases, phosphorylating MLKL or IRF6 and Pkp1, respectively, to achieve their signaling goals [83, 86, 136]. RIPK3 and RIPK4 may also share a related mode of activation that requires oligomerization [24, 30]. However, other data suggest that RIPK3 is still far from an ordinary kinase. It assumes an active conformation as a monomer in solution, but binding to substrate, MLKL, leads to movement of the αC helix into the inactive Glu-out state [126]. The reason for this unexpected RIPK3 inactivation upon substrate binding remains an enigma. Furthermore, the kinase dead D161N mutant of RIPK3 as well as several RIPK3 inhibitors have been reported to induce a gain-of-function pathway of RIPK3-mediated apoptosis in cells and in vivo [17, 66]. Strikingly, equally kinase-inactivating D161G, D143N and K51A RIPK3 mutants are non-lethal. This suggests that a specific conformation of the inhibitor-bound or D161N-mutant RIPK3 kinase domain, rather than loss of the catalytic activity per se, may be responsible for this toxicity. It should be noted that the ability of RIPK3 kinase domain to drive apoptosis in a kinase-independent manner is likely a physiologic function of RIPK3, which is selectively enhanced by kinase inhibition [67].
Short synopsis: atypical features of the RIPK family
In summary, RIPKs are ordinary-looking kinases that possess not so ordinary features, including unusual flexibilities of their kinase domains, oligomerization-dependent modes of activation, and unusual conformational roles of the kinase domains extending beyond just the regulation of catalysis. We hypothesize that these additional conformation scaffolding functions may have evolved to provide family members, such as RIPK1 and RIPK3, with the ability to more precisely regulate switching between different cellular responses, which is absolutely critical on the organismal level given the deleterious consequences of inappropriate activation of RIPK1/RIPK3 functions. Scaffolding roles of RIPK kinase domains are not typical for kinases, and, furthermore, each kinase may utilize their kinase domain differently. RIPK inhibitors intersect with these complex mechanisms, creating distinct and unusual activities. Along these lines, necrostatin binding to RIPK1 induces conformational changes that likely globally alter the shape of the kinase domain and, possibly, create a dominant negative protein. However, RIPK2 inhibitors likely cause small local perturbations that specifically affect the area required for XIAP binding. RIPK3 undergoes mysterious inactivating transition during substrate binding, and the conformation of the kinase domain may control the balance between activation of necroptosis and apoptosis. It remains to be seen if additional data will paint a further unifying picture of the regulation of this intriguing kinase family and if they can be harnessed for therapeutic utility.
Acknowledgements
We thank Dr. Soumya Ray (Schrödinger) for help with generating RIPK1 homology model. This work was supported by grants 1R01CA190542, R56AG058642, R21AI124049 and R01AI144400 to G.D.C. and A.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Berwick DC, et al. , LRRK2 Biology from structure to dysfunction: research progresses, but the themes remain the same. Mol Neurodegener, 2019. 14(1): p. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langston RG, Rudenko IN, and Cookson MR, The function of orthologues of the human Parkinson’s disease gene LRRK2 across species: implications for disease modelling in preclinical research. Biochem J, 2016. 473(3): p. 221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor M and Alessi DR, Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr Opin Cell Biol, 2020. 63: p. 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanger BZ, et al. , RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell, 1995. 81(4): p. 513–23. [DOI] [PubMed] [Google Scholar]
- 5.Kelliher MA, et al. , The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity, 1998. 8(3): p. 297–303. [DOI] [PubMed] [Google Scholar]
- 6.Ting AT, Pimentel-Muinos FX, and Seed B, RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J, 1996. 15(22): p. 6189–96. [PMC free article] [PubMed] [Google Scholar]
- 7.Holler N, et al. , Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol, 2000. 1(6): p. 489–95. [DOI] [PubMed] [Google Scholar]
- 8.Degterev A, et al. , Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol, 2005. 1(2): p. 112–9. [DOI] [PubMed] [Google Scholar]
- 9.Inohara N, et al. , RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol Chem, 1998. 273(20): p. 12296–300. [DOI] [PubMed] [Google Scholar]
- 10.McCarthy JV, Ni J, and Dixit VM, RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J Biol Chem, 1998. 273(27): p. 16968–75. [DOI] [PubMed] [Google Scholar]
- 11.Sun X, et al. , RIP3, a novel apoptosis-inducing kinase. J Biol Chem, 1999. 274(24): p. 16871–5. [DOI] [PubMed] [Google Scholar]
- 12.Thome M, et al. , Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol, 1998. 8(15): p. 885–8. [DOI] [PubMed] [Google Scholar]
- 13.Yu PW, et al. , Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkappaB. Curr Biol, 1999. 9(10): p. 539–42. [DOI] [PubMed] [Google Scholar]
- 14.Newton K, Sun X, and Dixit VM, Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol, 2004. 24(4): p. 1464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho YS, et al. , Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell, 2009. 137(6): p. 1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang DW, et al. , RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science, 2009. 325(5938): p. 332–6. [DOI] [PubMed] [Google Scholar]
- 17.Newton K, et al. , Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science, 2014. 343(6177): p. 1357–60. [DOI] [PubMed] [Google Scholar]
- 18.Holland P, et al. , RIP4 is an ankyrin repeat-containing kinase essential for keratinocyte differentiation. Curr Biol, 2002. 12(16): p. 1424–8. [DOI] [PubMed] [Google Scholar]
- 19.Meylan E, et al. , RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-kappa B and is processed during apoptosis. EMBO Rep, 2002. 3(12): p. 1201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zha J, et al. , RIP5 is a RIP-homologous inducer of cell death. Biochem Biophys Res Commun, 2004. 319(2): p. 298–303. [DOI] [PubMed] [Google Scholar]
- 21.Meylan E and Tschopp J, The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci, 2005. 30(3): p. 151–9. [DOI] [PubMed] [Google Scholar]
- 22.Peng J, et al. , Dusty protein kinases: primary structure, gene evolution, tissue specific expression and unique features of the catalytic domain. Biochim Biophys Acta, 2006. 1759(11–12): p. 562–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Najjar M, et al. , Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep, 2015. 10(11): p. 1850–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang CS, et al. , Crystal Structure of Ripk4 Reveals Dimerization-Dependent Kinase Activity. Structure, 2018. 26(5): p. 767–777 e5. [DOI] [PubMed] [Google Scholar]
- 25.de Leon-Boenig G, et al. , The crystal structure of the catalytic domain of the NF-kappaB inducing kinase reveals a narrow but flexible active site. Structure, 2012. 20(10): p. 1704–14. [DOI] [PubMed] [Google Scholar]
- 26.Degterev A, et al. , Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol, 2008. 4(5): p. 313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berger SB, et al. , Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol, 2014. 192(12): p. 5476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Najjar M, et al. , RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity, 2016. 45(1): p. 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bratton SB, et al. , Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis. Exp Cell Res, 2000. 256(1): p. 27–33. [DOI] [PubMed] [Google Scholar]
- 30.Li J, et al. , The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell, 2012. 150(2): p. 339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mocarski ES, et al. , True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J Immunol, 2014. 192(5): p. 2019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newton K, et al. , RIPK1 inhibits ZBP1-driven necroptosis during development. Nature, 2016. 540(7631): p. 129–133. [DOI] [PubMed] [Google Scholar]
- 33.He S, et al. , Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A, 2011. 108(50): p. 20054–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaiser WJ, et al. , Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem, 2013. 288(43): p. 31268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Upton JW, Kaiser WJ, and Mocarski ES, DAI/ZBP1/DLM-1 Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis that Is Targeted by Murine Cytomegalovirus vIRA. Cell Host Microbe, 2019. 26(4): p. 564. [DOI] [PubMed] [Google Scholar]
- 36.Dondelinger Y, et al. , MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol, 2017. 19(10): p. 1237–1247. [DOI] [PubMed] [Google Scholar]
- 37.Dondelinger Y, Vandenabeele P, and Bertrand MJ, Regulation of RIPK1’s cell death function by phosphorylation. Cell Cycle, 2016. 15(1): p. 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng J, et al. , Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun, 2017. 8(1): p. 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao P, et al. , A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature, 2020. 577(7788): p. 109–114. [DOI] [PubMed] [Google Scholar]
- 40.Lalaoui N, et al. , Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature, 2020. 577(7788): p. 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang Y, et al. , K63-linked ubiquitination regulates RIPK1 kinase activity to prevent cell death during embryogenesis and inflammation. Nat Commun, 2019. 10(1): p. 4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, et al. , Ubiquitination of RIPK1 suppresses programmed cell death by regulating RIPK1 kinase activation during embryogenesis. Nat Commun, 2019. 10(1): p. 4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong Q, et al. , Structural basis of RIP2 activation and signaling. Nat Commun, 2018. 9(1): p. 4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maharana J, Pradhan SK, and De S, NOD1CARD Might Be Using Multiple Interfaces for RIP2-Mediated CARD-CARD Interaction: Insights from Molecular Dynamics Simulation. PLoS One, 2017. 12(1): p. e0170232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goncharov T, et al. , Disruption of XIAP-RIP2 Association Blocks NOD2-Mediated Inflammatory Signaling. Mol Cell, 2018. 69(4): p. 551–565 e7. [DOI] [PubMed] [Google Scholar]
- 46.Hasegawa M, et al. , A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J, 2008. 27(2): p. 373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Panda S and Gekara NO, The deubiquitinase MYSM1 dampens NOD2-mediated inflammation and tissue damage by inactivating the RIP2 complex. Nat Commun, 2018. 9(1): p. 4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He S and Wang X, RIP kinases as modulators of inflammation and immunity. Nat Immunol, 2018. 19(9): p. 912–922. [DOI] [PubMed] [Google Scholar]
- 49.Heim VJ, Stafford CA, and Nachbur U, NOD Signaling and Cell Death. Front Cell Dev Biol, 2019. 7: p. 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Degterev A, Ofengeim D, and Yuan J, Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci U S A, 2019. 116(20): p. 9714–9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silke J, Rickard JA, and Gerlic M, The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol, 2015. 16(7): p. 689–97. [DOI] [PubMed] [Google Scholar]
- 52.Humphries F, et al. , RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ, 2015. 22(2): p. 225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vanden Berghe T, Hassannia B, and Vandenabeele P, An outline of necrosome triggers. Cell Mol Life Sci, 2016. 73(11–12): p. 2137–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muendlein HI, et al. , Constitutive Interferon Attenuates RIPK1/3-Mediated Cytokine Translation. Cell Rep, 2020. 30(3): p. 699–713 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amin P, et al. , Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFalpha-mediated apoptosis. Proc Natl Acad Sci U S A, 2018. 115(26): p. E5944–E5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sarhan J, et al. , Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A, 2018. 115(46): p. E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wegner KW, Saleh D, and Degterev A, Complex Pathologic Roles of RIPK1 and RIPK3: Moving Beyond Necroptosis. Trends Pharmacol Sci, 2017. 38(3): p. 202–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu K, et al. , Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis, 2018. 9(5): p. 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saleh D, et al. , Kinase Activities of RIPK1 and RIPK3 Can Direct IFN-beta Synthesis Induced by Lipopolysaccharide. J Immunol, 2017. 198(11): p. 4435–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan J, Amin P, and Ofengeim D, Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci, 2019. 20(1): p. 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu D, et al. , TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell, 2018. 174(6): p. 1477–1491 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Orozco SL, et al. , RIPK3 Activation Leads to Cytokine Synthesis that Continues after Loss of Cell Membrane Integrity. Cell Rep, 2019. 28(9): p. 2275–2287 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng S, et al. , Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal, 2007. 19(10): p. 2056–67. [DOI] [PubMed] [Google Scholar]
- 64.Moriwaki K, et al. , A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J Immunol, 2015. 194(4): p. 1938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moriwaki K, et al. , The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity, 2014. 41(4): p. 567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mandal P, et al. , RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell, 2014. 56(4): p. 481–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nogusa S, et al. , RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe, 2016. 20(1): p. 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Newton K, et al. , RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ, 2016. 23(9): p. 1565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim YG, et al. , The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity, 2008. 28(2): p. 246–57. [DOI] [PubMed] [Google Scholar]
- 70.Girardin SE, et al. , CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep, 2001. 2(8): p. 736–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maekawa S, et al. , Crystal structure of NOD2 and its implications in human disease. Nat Commun, 2016. 7: p. 11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pellegrini E, et al. , RIP2 filament formation is required for NOD2 dependent NF-kappaB signalling. Nat Commun, 2018. 9(1): p. 4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tan Y and Kagan JC, Innate Immune Signaling Organelles Display Natural and Programmable Signaling Flexibility. Cell, 2019. 177(2): p. 384–398 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ferrao R, et al. , Structural insights into the assembly of large oligomeric signalosomes in the Toll-like receptor-interleukin-1 receptor superfamily. Sci Signal, 2012. 5(226): p. re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu A, et al. , Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell, 2014. 156(6): p. 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schleich K, Krammer PH, and Lavrik IN, The chains of death: a new view on caspase-8 activation at the DISC. Cell Cycle, 2013. 12(2): p. 193–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tigno-Aranjuez JT, Asara JM, and Abbott DW, Inhibition of RIP2’s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev, 2010. 24(23): p. 2666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hrdinka M, et al. , Small molecule inhibitors reveal an indispensable scaffolding role of RIPK2 in NOD2 signaling. EMBO J, 2018. 37(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Damgaard RB, et al. , The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell, 2012. 46(6): p. 746–58. [DOI] [PubMed] [Google Scholar]
- 80.Yang S, et al. , Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol, 2013. 14(9): p. 927–36. [DOI] [PubMed] [Google Scholar]
- 81.Dondelinger Y, et al. , Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol Life Sci, 2016. 73(11–12): p. 2165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heymann MC, et al. , Human procaspase-1 variants with decreased enzymatic activity are associated with febrile episodes and may contribute to inflammation via RIP2 and NF-kappaB signaling. J Immunol, 2014. 192(9): p. 4379–85. [DOI] [PubMed] [Google Scholar]
- 83.Kwa MQ, et al. , Receptor-interacting protein kinase 4 and interferon regulatory factor 6 function as a signaling axis to regulate keratinocyte differentiation. J Biol Chem, 2014. 289(45): p. 31077–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kwa MQ, Scholz GM, and Reynolds EC, RIPK4 activates an IRF6-mediated proinflammatory cytokine response in keratinocytes. Cytokine, 2016. 83: p. 19–26. [DOI] [PubMed] [Google Scholar]
- 85.Oberbeck N, et al. , The RIPK4-IRF6 signalling axis safeguards epidermal differentiation and barrier function. Nature, 2019. 574(7777): p. 249–253. [DOI] [PubMed] [Google Scholar]
- 86.Lee P, et al. , Phosphorylation of Pkp1 by RIPK4 regulates epidermal differentiation and skin tumorigenesis. EMBO J, 2017. 36(13): p. 1963–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kondo S, et al. , Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet, 2002. 32(2): p. 285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kalay E, et al. , Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am J Hum Genet, 2012. 90(1): p. 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garcia-Carbonell R, et al. , Dysregulation of Intestinal Epithelial Cell RIPK Pathways Promotes Chronic Inflammation in the IBD Gut. Front Immunol, 2019. 10: p. 1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dannappel M, et al. , RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature, 2014. 513(7516): p. 90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takahashi N, et al. , RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature, 2014. 513(7516): p. 95–9. [DOI] [PubMed] [Google Scholar]
- 92.Gonzalez-Juarbe N, et al. , Bacterial Pore-Forming Toxins Promote the Activation of Caspases in Parallel to Necroptosis to Enhance Alarmin Release and Inflammation During Pneumonia. Sci Rep, 2018. 8(1): p. 5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Daniels BP, et al. , RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell, 2017. 169(2): p. 301–313 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Philip NH, et al. , Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proc Natl Acad Sci U S A, 2014. 111(20): p. 7385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Orning P, et al. , Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science, 2018. 362(6418): p. 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Farias Luz N, et al. , RIPK1 and PGAM5 Control Leishmania Replication through Distinct Mechanisms. J Immunol, 2016. 196(12): p. 5056–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stafford CA, et al. , IAPs Regulate Distinct Innate Immune Pathways to Co-ordinate the Response to Bacterial Peptidoglycans. Cell Rep, 2018. 22(6): p. 1496–1508. [DOI] [PubMed] [Google Scholar]
- 98.Webster JD, et al. , RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J Leukoc Biol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guo H, et al. , Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis, 2018. 9(8): p. 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Madrigal AG, et al. , Pathogen-mediated proteolysis of the cell death regulator RIPK1 and the host defense modulator RIPK2 in human aortic endothelial cells. PLoS Pathog, 2012. 8(6): p. e1002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wagner RN, Reed JC, and Chanda SK, HIV-1 protease cleaves the serine-threonine kinases RIPK1 and RIPK2. Retrovirology, 2015. 12: p. 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dondelinger Y, et al. , An evolutionary perspective on the necroptotic pathway. Trends Cell Biol, 2016. 26(10): p. 721–732. [DOI] [PubMed] [Google Scholar]
- 103.Kleino A, et al. , Peptidoglycan-Sensing Receptors Trigger the Formation of Functional Amyloids of the Adaptor Protein Imd to Initiate Drosophila NF-kappaB Signaling. Immunity, 2017. 47(4): p. 635–647 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang S, et al. , Pellino3 targets RIP1 and regulates the pro-apoptotic effects of TNF-alpha. Nat Commun, 2013. 4: p. 2583. [DOI] [PubMed] [Google Scholar]
- 105.Nachbur U, et al. , A RIPK2 inhibitor delays NOD signalling events yet prevents inflammatory cytokine production. Nat Commun, 2015. 6: p. 6442. [DOI] [PubMed] [Google Scholar]
- 106.Ofengeim D, et al. , Activation of necroptosis in multiple sclerosis. Cell Rep, 2015. 10(11): p. 1836–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shaw PJ, et al. , Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity, 2011. 34(1): p. 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tigno-Aranjuez JT, et al. , In vivo inhibition of RIPK2 kinase alleviates inflammatory disease. J Biol Chem, 2014. 289(43): p. 29651–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Haile PA, et al. , Discovery of a First-in-Class Receptor Interacting Protein 2 (RIP2) Kinase Specific Clinical Candidate, 2-((4-(Benzo[d]thiazol-5-ylamino)-6-(tertbutylsulfonyl)quinazolin-7-yl)oxy)ethyl Dihydrogen Phosphate, for the Treatment of Inflammatory Diseases. J Med Chem, 2019. 62(14): p. 6482–6494. [DOI] [PubMed] [Google Scholar]
- 110.Ermann J, et al. , Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/−.Rag2−/− (TRUC) mice. Proc Natl Acad Sci U S A, 2014. 111(25): p. E2559–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lu H, et al. , RIPK1 inhibitor ameliorates colitis by directly maintaining intestinal barrier homeostasis and regulating following IECs-immuno crosstalk. Biochem Pharmacol, 2020. 172: p. 113751. [DOI] [PubMed] [Google Scholar]
- 112.Teng X, et al. , Structure-activity relationship study of novel necroptosis inhibitors. Bioorg Med Chem Lett, 2005. 15(22): p. 5039–44. [DOI] [PubMed] [Google Scholar]
- 113.Xie T, et al. , Structural basis of RIP1 inhibition by necrostatins. Structure, 2013. 21(3): p. 493–9. [DOI] [PubMed] [Google Scholar]
- 114.Harris PA, et al. , Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J Med Chem, 2017. 60(4): p. 1247–1261. [DOI] [PubMed] [Google Scholar]
- 115.Yoshikawa M, et al. , Discovery of 7-Oxo-2,4,5,7-tetrahydro-6 H-pyrazolo[3,4- c]pyridine Derivatives as Potent, Orally Available, and Brain-Penetrating Receptor Interacting Protein 1 (RIP1) Kinase Inhibitors: Analysis of Structure-Kinetic Relationships. J Med Chem, 2018. 61(6): p. 2384–2409. [DOI] [PubMed] [Google Scholar]
- 116.Patel S, et al. , RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ, 2020. 27(1): p. 161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ren Y, et al. , Discovery of a Highly Potent, Selective, and Metabolically Stable Inhibitor of Receptor-Interacting Protein 1 (RIP1) for the Treatment of Systemic Inflammatory Response Syndrome. J Med Chem, 2017. 60(3): p. 972–986. [DOI] [PubMed] [Google Scholar]
- 118.Harris PA, et al. , Discovery and Lead-Optimization of 4,5-Dihydropyrazoles as Mono-Kinase Selective, Orally Bioavailable and Efficacious Inhibitors of Receptor Interacting Protein 1 (RIP1) Kinase. J Med Chem, 2019. 62(10): p. 5096–5110. [DOI] [PubMed] [Google Scholar]
- 119.Zhuang C and Chen F, Small-Molecule Inhibitors of Necroptosis: Current Status and Perspectives. J Med Chem, 2020. 63(4): p. 1490–1510. [DOI] [PubMed] [Google Scholar]
- 120.Zhang H, et al. , N-(7-Cyano-6-(4-fluoro-3-(2-(3-(trifluoromethyl)phenyl)acetamido)phenoxy)benzo[d] thiazol-2-yl)cyclopropanecarboxamide (TAK-632) Analogues as Novel Necroptosis Inhibitors by Targeting Receptor-Interacting Protein Kinase 3 (RIPK3): Synthesis, Structure-Activity Relationships, and in Vivo Efficacy. J Med Chem, 2019. 62(14): p. 6665–6681. [DOI] [PubMed] [Google Scholar]
- 121.Hart AC, et al. , Identification of RIPK3 Type II Inhibitors Using High-Throughput Mechanistic Studies in Hit Triage. ACS Medicinal Chemistry Letters, 2020. 11(3): p. 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Haile PA, et al. , The Identification and Pharmacological Characterization of 6-(tert-Butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine (GSK583), a Highly Potent and Selective Inhibitor of RIP2 Kinase. J Med Chem, 2016. 59(10): p. 4867–80. [DOI] [PubMed] [Google Scholar]
- 123.He X, et al. , Identification of Potent and Selective RIPK2 Inhibitors for the Treatment of Inflammatory Diseases. ACS Med Chem Lett, 2017. 8(10): p. 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Suebsuwong C, et al. , Receptor-interacting protein kinase 2 (RIPK2) and nucleotide-binding oligomerization domain (NOD) cell signaling inhibitors based on a 3,5-diphenyl-2-aminopyridine scaffold. Eur J Med Chem, 2020. 200: p. 112417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Charnley AK, et al. , Crystal structures of human RIP2 kinase catalytic domain complexed with ATP-competitive inhibitors: Foundations for understanding inhibitor selectivity. Bioorg Med Chem, 2015. 23(21): p. 7000–6. [DOI] [PubMed] [Google Scholar]
- 126.Xie T, et al. , Structural insights into RIP3-mediated necroptotic signaling. Cell Rep, 2013. 5(1): p. 70–8. [DOI] [PubMed] [Google Scholar]
- 127.Zhao Z, et al. , Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem Biol, 2014. 9(6): p. 1230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Harris PA, et al. , Discovery of Small Molecule RIP1 Kinase Inhibitors for the Treatment of Pathologies Associated with Necroptosis. ACS Med Chem Lett, 2013. 4(12): p. 1238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Canning P, et al. , Inflammatory Signaling by NOD-RIPK2 Is Inhibited by Clinically Relevant Type II Kinase Inhibitors. Chem Biol, 2015. 22(9): p. 1174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roskoski R Jr., Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res, 2016. 103: p. 26–48. [DOI] [PubMed] [Google Scholar]
- 131.Fauster A, et al. , A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis, 2015. 6: p. e1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Harris PA, et al. , Identification of a RIP1 Kinase Inhibitor Clinical Candidate (GSK3145095) for the Treatment of Pancreatic Cancer. ACS Med Chem Lett, 2019. 10(6): p. 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Harris PA, et al. , DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J Med Chem, 2016. 59(5): p. 2163–78. [DOI] [PubMed] [Google Scholar]
- 134.Dillon CP, et al. , RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell, 2014. 157(5): p. 1189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kearney CJ, et al. , RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J, 2014. 281(21): p. 4921–34. [DOI] [PubMed] [Google Scholar]
- 136.Sun L, et al. , Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell, 2012. 148(1–2): p. 213–27. [DOI] [PubMed] [Google Scholar]