

Abstract
Background and Objectives
Equilibrative nucleoside transporter (ENT) 1 is a widely-expressed drug transporter, handling nucleoside analogues as well as endogenous nucleosides. ENT1 has been postulated to be inhibited by some marketed tyrosine kinase inhibitors (TKIs). To obtain insights into this point, the interactions of 24 TKIs with ENT1 activity have been analyzed.
Methods
Inhibition of ENT1 activity was investigated in vitro through quantifying the decrease of [3H]-uridine uptake caused by TKIs in HAP1 ENT2-knockout cells, exhibiting selective ENT1 expression. TKI effects towards ENT1-mediated transport were additionally characterized in terms of their in vivo relevance and of their relationship to TKI molecular descriptors. Putative transport of the TKI lorlatinib by ENT1/ENT2 was analyzed by LC-MS/MS.
Results
Of 24 TKIs, 12 of them, each used at 10 µM, were found to behave as moderate or strong inhibitors of ENT1, i.e., they decreased ENT1 activity by at least 35%. This inhibition was concentration-dependent for at least the strongest ones (IC50 less than 10 µM) and was correlated with some molecular descriptors, especially with atom-type E-state indices. Lorlatinib was notably a potent in vitro inhibitor of ENT1/ENT2 (IC50 values around 1.0–2.5 µM) and was predicted to inhibit these nucleoside transporters at relevant clinical concentrations, without, however, being a substrate for them.
Conclusion
Our data unambiguously add ENT1 to the list of drug transporters inhibited by TKIs, especially by lorlatinib. This point likely merits attention in terms of possible drug–drug interactions, notably for nucleoside analogues, whose ENT1-mediated uptake into their target cells may be hampered by co-administrated TKIs such as lorlatinib.
Key Points
| Inhibition of equilibrative nucleoside transporter (ENT1) activity by 24 marketed or under clinical development tyrosine kinase inhibitors (TKIs) was investigated in vitro using cells selectively expressing ENT1. |
| Of 24 TKIs, 12 of them, used at 10 µM, behaved as moderate or strong inhibitors of ENT1-mediated uridine transport, with notably one (lorlatinib) exerting potent in vitro inhibition (IC50 = 2.5 µM) and predicted to inhibit ENT1 activity at clinically achievable plasma concentrations, without, however, being an ENT1 substrate. |
| These data indicate that TKIs, especially lorlatinib, may have to be considered for possible ENT1-based drug–drug interactions (DDIs), notably with known ENT1 substrates like nucleoside analogues. |
Introduction
Equilibrative nucleoside transporter (ENT) 1 (SLC29A1) is a major plasma membrane transporter of nucleosides, widely expressed in various cell types [1]. It mediates facilitated diffusion of both purine (adenosine, guanosine, and inosine) and pyrimidine (cytidine, thymidine, and uridine) nucleosides into cells, in a sodium-independent manner [2]. Such substrates are shared by ENT2 (SLC29A2), another member of the ENT subfamily, also ubiquitously expressed, but with an average expression level lower than that of ENT1, and exhibiting low sensitivity to the potent ENT1 inhibitor, nitrobenzylmercaptopurine riboside (NBMPR) [3].
Besides physiological nucleosides, ENT1 transports nucleosides-derived drugs, used for the treatment of cancers, inflammatory diseases, or viral infections [4]. In this way, ENT1 is directly implicated in the uptake of anticancer or antiviral drugs into target cells, and, consequently, in their pharmacological effects. ENT1 activity is thus a key determinant of the anticancer activity of gemcitabine in pancreatic tumor cells [5]. Decreased expression and/or activity of ENT1 can therefore result in cellular resistance to cytotoxic nucleoside analogues through reduction of their intracellular accumulation [6, 7]. ENT1 may also be implicated in pharmacokinetics, notably at the hepatic level, and at the placental and blood–testis barriers [8–10]. ENT1 additionally deserves recent attention in the context of coronavirus disease 2019 (COVID-19). Indeed, it may transport nucleoside analogue candidates for inhibiting Severe Acute Respiratory Syndrome CoronaVirus-2 (SARS-CoV-2) replication [11]. Moreover, it may be considered as a therapeutic target in COVID-19-related thrombosis [12], because inhibition of its activity enhances extracellular levels of the platelet activation inhibitor, adenosine, likely through decreasing ENT1-mediated re-uptake of this compound in red blood cells, as already demonstrated for the platelet inhibitor, ticagrelor [13].
ENT1 activity can be inhibited by various compounds, including marketed drugs, such as the vasodilatators, dilazep and dipyridamole, and the reverse transcriptase inhibitor, nevirapine [1, 14]. Such ENT1 inhibitors can potentially cause drug–drug interactions (DDIs), through inhibiting the cellular uptake of anticancer nucleoside drugs handled by ENT1, thereby causing resistance of tumoral cells. For this reason, ENT1 has been postulated to be an emergent transporter of clinical importance, that has to be studied during the development of new molecular entities with nucleoside-like structures according to the International Transporter Consortium [15]. In this context, it is noteworthy that various tyrosine kinase inhibitors (TKIs), such as the bcr-abl inhibitors nilotinib, ponatinib, bosutinib, dasatinib, and imatinib, the EGF receptor inhibitors erlotinib and gefitinib, and the multi-kinase inhibitors axitinib, pazopanib, sunitinib, and vandetanib, have been shown to inhibit ENT1 activity in vitro [16–19]. Whether other TKIs can also behave as inhibitors for ENT1 is, however, unknown, but likely deserves interest owing to the current development of TKIs as a leading pharmacological class for various therapeutic indications, and to the potential co-administration of nucleoside analogues and TKIs, notably for the treatment of cancers. To obtain insights into this point in the present study, we have analyzed the inhibition of ENT1 activity by 24 TKIs, targeting various tyrosine kinases and regulatory-approved (n = 22) or under clinical development (n = 2).
Materials and Methods
Chemicals
The 22 marketed TKIs, abemaciclib, acalabrutinib, afatinib, alectinib, brigatinib, cabozantinib, ceritinib, crizotinib, dacomitinib, entrectinib, ibrutinib, lapatinib, lenvatinib, lorlatinib, neratinib, nintedanib, osimertinib, regorafenib, ribociclib, ruxolitinib, tofacitinib, and vemurafenib, as well as itacitinib and pacritinib, 2 TKIs under clinical phase 3 development, were provided by MedChemtronica (Sollentuna, Sweden). These TKIs target various kinases, as indicated in Table 1. Stock solutions of TKIs were initially prepared using dimethylsulfoxide as solvent. NBMPR was from Sigma-Aldrich (Saint-Quentin Fallavier, France) whereas [5,6-3H]-uridine (specific activity = 30 Ci/mmol) was from Perkin-Elmer (Villebon-sur-Yvette, France).
Table 1.
Effect of 10 µM TKIs on ENT1 activity in HAP1-ENT2 KO cells.
| TKI | Main targeted kinase | % inhibition of ENT1 activity (mean ± SD) |
|---|---|---|
| Abemaciclib | CDK4/6 | 25.0 ± 7.9** |
| Acalabrutinib | BTK | 42.6 ± 10.4*** |
| Afatinib | EGFR | 54.5 ± 2.4*** |
| Alectinib | ALK | 54.0 ± 4.9*** |
| Brigatinib | ALK | 52.5 ± 11.1*** |
| Cabozantinib | VEGFR2, c-Met, c-Kit, Axl and Flt3 | 48.8 ± 2.5*** |
| Ceritinib | ALK | 26.3 ± 1.7** |
| Crizotinib | ALK | 34.7 ± 2.7*** |
| Dacomitinib | EGFR | 43.9 ± 2.3*** |
| Entrectinib | Pan-TRK, ROS1, and ALK | 22.9 ± 5.2* |
| Ibrutinib | BTK | 72.7 ± 1.1*** |
| Itacitinib | JAK1 | 21.9 ± 13.3* |
| Lapatinib | ErbB-2 and EGFR | 25.7 ± 15.2** |
| Lenvatinib | VEGFR1-3, FGFR1-4, PDGFR, Kit, and Ret | 63.1 ± 5.9*** |
| Lorlatinib | ROS1 and ALK | 86.6 ± 6.4*** |
| Neratinib | ErbB-2 and EGFR | 61.0 ± 8.5*** |
| Nintedanib | VEGFR, FGFR and PDGFR | 46.9 ± 9.6*** |
| Osimertinib | EGFR | 32.3 ± 11.3*** |
| Pacritinib | JAK2 and Flt3 | 61.6 ± 8.1*** |
| Regorafenib | VEGFR1/2/3, PDGFRβ, Kit, Ret and Raf-1 | 21.4 ± 2.7* |
| Ribociclib | CDK4/6 | 17.8 ± 10.2 |
| Ruxolitinib | JAKs | 34.1 ± 13.2*** |
| Tofacitinib | JAKs | 4.8 ± 8.3 |
| Vemurafenib | B-Raf | 27.3 ± 10.9** |
Experimental data are the means ± SD of three independent assays. ENT1 activity in control HAP1-ENT2 KO cells not exposed to TKIs cells is 2.1 ± 0.2 pmol uridine/mg protein/5 min
ALK anaplastic lymphoma kinase, BTK Bruton’s tyrosine kinase, c-Met hepatocyte growth factor receptor (HGFR), CDK cyclin-dependent kinase, EFGR epidermal growth factor receptor (also known as ErbB-1 or HER1), ErbB-2 human epidermal growth factor receptor-2 (also known as HER2), FGFR fibroblast growth factor receptor, Flt3 Fms-like tyrosine kinase 3, JAK Janus kinase, PDGFR platelet-derived growth factor, TRK tropomyosin receptor kinase (also known as NTRK, neurotrophic tyrosine receptor kinase), VEGFR vascular endothelial growth factor, TKIs tyrosine kinase inhibitors
*p < 0.05, **p < 0.01, and ***p < 0.001, when compared to control cells not exposed to TKIs
Cell Culture
HAP1 wild-type cells (reference C631) and CRISPR-Cas9-edited HAP1 knockout (KO) cells deficient for ENT1 (HAP1 ENT1-KO cells; reference HZGHC000785c004) or for ENT2 (HAP1 ENT2-KO cells; reference HZGHC005532c010) were from Horizon Discovery (Waterbeach, UK). HAP1 wild-type cell line is a human near-haploid cell line derived from the male chronic myelogenous leukemia cell line KBM-7 [20]. HAP1 wild-type and KO cells were routinely cultured in Iscove’s modified Dulbecco’s medium (Thermo Fisher Scientific) supplemented with 10% (vol/vol) fetal calf serum, 20 IU/mL penicillin, and 20 μg/mL streptomycin. Lung A549 cells were routinely cultured in F-12K medium, supplemented with 2 mM l-glutamine, 10% fetal calf serum (vol/vol), 20 IU/mL penicillin, and 20 μg/mL streptomycin, as previously described [21]; the A549 cell line is a human cancerous alveolar basal epithelial cell line, established from a pulmonary adenocarcinoma of a 58-year-old Caucasian male [22].
Nucleoside Transport Assays
Cells previously seeded in 96-well plates were incubated with 8.33 nM [3H]-uridine for 5 min at 37 °C, in a defined sodium-free transport assay buffer, consisting of 5.3 mM KCl, 1.1 mM KH2PO4, 0.8 mm MgSO4, 1.8 mM CaCl2, 11 mM d-glucose, 10 mM Hepes, and 136 mM N-methyl-glucamine, and adjusted to pH 7.4, in the absence or presence of NBMPR (used at 0.1 or 100 µM for inhibiting ENT1 or ENT1/ENT2, respectively) or TKI, as previously described [23]. After two washes in ice-cold phosphate-buffered saline (PBS), cells were lysed using distilled water, and cellular uptake of radiolabeled uridine (U) was determined through scintillation counting and normalized to total protein content, determined by the Bradford method [24]. Nucleoside transporter activities in the absence of TKIs were next determined using the following equations (Eqs. 1 and 2):
| 1 |
| 2 |
with Ucontrol is the uptake of [3H]-uridine in the absence of NPMPR, U0.1 µM NBMPR is the uptake of [3H]-uridine in the presence of 0.1 µM NBMPR and U100 µM NBMPR = uptake of [3H]-uridine in the presence of 100 µM NBMPR.
ENT1 activity in HAP1 ENT2-KO cells in the presence of TKI was expressed as % of ENT1 activity found in control cells or as % of ENT1 activity inhibition according to the following equations (Eqs. 3 and 4):
| 3 |
| 4 |
with UTKI is the [3H]-uridine uptake in the presence of TKI.
ENT2 activity in the presence of TKI in HAP1 ENT1-KO cells was expressed as % of ENT2 activity found in control cells, as described in the Eq. (5):
| 5 |
ENT activity in the presence of lorlatinib in A549 cells was expressed as % of total ENT activity found in control cells using the following equation:
| 6 |
Half-maximal inhibitory concentration (IC50) for TKIs towards ENT1, ENT2, or total ENT activity was calculated using Prism 8.4.2 software (GraphPad Software, La Jolla, CA, USA), through nonlinear regression based on the following four-parameter logistic equation (Eq. 7):
| 7 |
with A is the percentage of transporter activity for a given concentration of TKI, determined as described in Eqs. (3), (5), or (6), [I] is the TKI concentration in the medium, and Hill slope = a coefficient describing the steepness of the curve.
Prediction of In vivo Inhibition of ENT Activity by TKIs
From in vitro IC50 values, in vivo inhibition of ENT activity by TKIs was predicted through applying criteria defined by the US Food and Drug Administration (FDA) guidance on in vitro drug interaction studies [25] to ENT1 or ENT2, i.e., a transporter-related DDI can be expected at a systemic level if (Eq. 8):
| 8 |
with Imax,u is the maximum unbound plasma concentration of the TKI.
Lorlatinib Accumulation Assay
HAP1 wild-type, ENT1-KO, and ENT2-KO cells were incubated with 10 µM lorlatinib, in the absence or presence of 0.1 or 100 µM NBMPR, for 5 min at 37 °C. Cells were then washed twice in ice-cold PBS, lysed in distilled water, and acetonitrile-based extraction of cell lysates was performed. Lorlatinib quantification was next performed through liquid chromatography-tandem mass spectrometry (LC–MS/MS), based on a high-performance liquid chromatography Aria system (Agilent, Les Ulis, France), equipped with a Poroshell 120 C18 (4.6 × 100 mm) column (Agilent) and coupled to a tandem mass spectrometry TSQ Quantum (Thermo Fisher Scientific, Villebon sur Yvette, France), fitted with an electrospray ionization source (ESI+). Monitored ion transitions were at 407 > 228.1 m/z. Data were finally normalized to protein content.
TKI Molecular Descriptor Generation
The total of 829 molecular descriptors belonging to the blocks “constitutional indices” (n = 47), “ring descriptors” (n = 32), “topological indices” (n = 75), “walk and path counts” (n = 46), “connectivity indices” (n = 37), “information indices” (n = 50), “geometrical descriptors” (n = 38), “functional group counts” (n = 154), “atom-centred-fragments” (n = 115), “atom-type E-state indices” (n = 172), “charge descriptors” (n = 15), “molecular properties” (n = 20), and “drug-like indices” (n = 28), were determined for all TKIs using the Dragon 7.0 software (Kode Chemoinformatics, Pisa, Italy) (See https://chm.kode-solutions.net/products_dragon_descriptors.php for a complete list of these descriptors). These compounds, initially expressed in SMILES format, were converted to 3D format using the MarvinView software (ChemAxon, Budapest, Hungary) before processing by Dragon 7.0 software to obtain molecular descriptors, as previously described [26].
Statistical Analysis
Experimental data were routinely expressed as means ± SD of at least three independent assays, each usually being performed in triplicate. Data were statistically analyzed through analysis of variance followed by the Dunnett’s post hoc test or through Spearman’s rank correlation test. The criterion of significance was p < 0.05.
Results
ENT activity was first investigated via [3H]-uridine uptake in HAP1 wild-type, HAP1-ENT1-KO, and HAP1-ENT2-KO cells. As shown in Fig. 1, HAP1 wild-type cells exhibited ENT1 activity higher than that of ENT2. HAP1-ENT2-KO cells exhibited similar levels of ENT1-mediated [3H]-uridine uptake compared to HAP1 wild-type cells, but failed to exhibit notable ENT2 activity (Fig. 1). By contrast, HAP1 ENT1-KO cells displayed similar ENT2 activity compared to HAP1 wild-type counterparts, but ENT1 activity was fully suppressed in these KO cells (Fig. 1). Taken together, these data indicate that HAP1 ENT-KO cells were fully functional, HAP1 ENT2-KO cells exhibited only ENT1 activity and can thus be used for specifically studying ENT1, whereas HAP1 ENT1-KO cells exhibited only ENT2 activity cells and can therefore be retained for specifically investigating ENT2 activity.
Fig. 1.
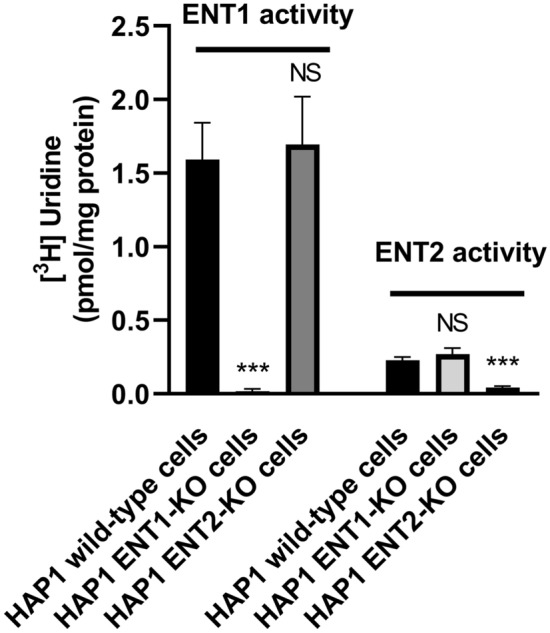
ENT activities in HAP1 wild-type, ENT1-KO, and ENT2-KO cells. Cells were incubated with [3H]-uridine for 5 min at 37 °C, in the absence or presence of 0.1 µM or 100 µM NBMPR. Intracellular uptake of [3H]-uridine was next determined by scintillation counting and normalized to protein content. Data are expressed as ENT1 or ENT2 activity, according to Eqs. (1) and (2) reported in “Materials and Methods” and are the means ± SD of at least three independent assays. ***p < 0.001, NS not statistically significant, when compared to HAP1 wild-type cells. ENT equilibrative nucleoside transporter, KO knockout, NBMPR nitrobenzylmercaptopurine riboside
The effects of 10 µM TKIs towards ENT1-mediated transport of [3H]-uridine were next studied in HAP1 ENT2-KO cells. As shown in Table 1, 2/24 TKIs, i.e., ribociclib and tofacitinib, failed to significantly alter ENT1 activity. Among the 22/24 TKIs which inhibited ENT1, some of them (10/24 TKIs), i.e., abemaciclib, ceritinib, crizotinib, entractinib, itacitinib, lapatinib, osimertinib, regoferanib, ruxolitinib, and vemurafenib, decreased ENT1 activity by less than 35% and can thus be considered as weak ENT1 inhibitors. Other TKIs (7/24 TKIs), i.e., acalabrutinib, afatinib, alectinib, brigatinib, cabozantinib, dacomitinib, and nindetanib, were found to inhibit ENT1 activity by more than 35% but less than 60% (Table 1), and thus appear as moderate inhibitors of the transporter. Finally, 5/24 TKIs, i.e., ibrutinib, lenvatinib, lorlatinib, neratinib, and pacritinib were found to inhibit ENT1 by more than 60% (Table 1), and are consequently potent inhibitors of ENT1 activity. These five strong ENT1 inhibitors were next found to inhibit ENT1-mediated transport of [3H]-uridine in HEK ENT2-KO cells in a concentration-dependent manner, with IC50 values ranging from 2.5 µM (for lorlatinib) to 8.5 µM (for neratinib) (Fig. 2).
Fig. 2.

Concentration-dependent inhibition of ENT1 activity by TKIs. HAP1 ENT2-KO cells were incubated with [3H]-uridine for 5 min at 37 °C, in the absence or presence of various concentrations of TKIs. Intracellular uptake of radiolabeled uridine was next determined by scintillation counting and normalized to protein content. Data are expressed as % of ENT1 activity found in control cells, arbitrarily set at 100%, and are the means ± SD of three independent assays. TKI IC50 values are indicated on the top of the graphs. ENT equilibrative nucleoside transporter, KO knockout, TKIs tyrosine kinase inhibitors
In order to investigate the physico-chemical structural requirements for ENT1 inhibition by TKIs, the percentages of inhibition of ENT1 activity by the 24 TKIs were next compared to values of 829 TKI molecular descriptors, through Spearman’s rank correlation analysis, and 20/829 molecular descriptors were found to be significantly either positively (n = 13) or negatively (n = 7) correlated with ENT1 inhibition (Table 2). Among these correlated descriptors, the most significant belonged to the blocks “constitutional indices” (Mi, p = 0.005), “functional group counts” (nArOR, p = 0.006) and “Atom-Type E-state indices” (SssO, p = 0.0003, and NssO, p = 0.0006) (Table 2). The molecular descriptors not correlated with ENT1 activity inhibition included lipophilicity-related parameters such as the Moriguchi octanol–water partition coefficient and the Ghose–Crippen octanol–water partition, as well as the topological polar surface area (data not shown).
Table 2.
TKI molecular descriptors correlated with ENT1 activity inhibition percentage by 10 μM TKIs.
| Molecular descriptor | Correlation with ENT1 inhibition % | ||
|---|---|---|---|
| Block | Name | Spearman coefficient ( ) | Significance (p value) |
| Constitutional indices | Mean first ionization potential (scaled on Carbon atom) (Mi) | – 0.56 | 0.005 |
| Ring descriptors | Number of 10-membered rings (nR10) | 0.47 | 0.020 |
| Distance/detour ring index of order 10 (D/Dtr10) | 0.43 | 0.034 | |
| Geometrical descriptors | Length-to-breadth ratio by WHIM (L/Bw) | – 0.42 | 0.039 |
| Functional group counts | Number of total tertiary C(sp3) (nCt) | – 0.42 | 0.041 |
| Number of ring tertiary C(sp3) (nCrt) | – 0.42 | 0.041 | |
| Number of aliphatic secondary C(sp2) (nR=Cs) | 0.45 | 0.027 | |
| Number of nitriles (aromatic) (nArCN) | 0.45 | 0.029 | |
| Number of ethers (aromatic) (nArOR) | 0.54 | 0.006 | |
| Atom-centred fragments | =CHR (C-016) | 0.45 | 0.027 |
| H attached to alpha-C (H-051) | – 0.45 | 0.028 | |
|
Al–O–Ar / Ar–O–Ar / R..O..R/R–O–C=X (O-060) |
0.52 | 0.01 | |
| Atom-type E-state indices | Sum of dsCH E-states (SdsCH) | 0.45 | 0.026 |
| Sum of sNH2 E-states (SsNH2) | 0.44 | 0.031 | |
| Sum of ssO E-states (SssO) | 0.67 | 0.0003 | |
| Number of atoms of type dsCH (NdsCH) | 0.45 | 0.027 | |
| Number of atoms of type sNH2 (NsNH2) | 0.47 | 0.021 | |
| Number of atoms of type ssO (NssO) | 0.65 | 0.0006 | |
| Charge descriptors | Maximum positive charge (qpmax) | – 0.52 | 0.010 |
| Submolecular polarity parameter (SPP) | – 0.43 | 0.037 | |
ENT equilibrative nucleoside transporter, TKIs tyrosine kinase inhibitors
The prediction of in vivo ENT1 inhibition was next performed for TKIs defined above as strong inhibitors, i.e., ibrutinib, lenvatinib, lorlatinib, neratinib, and pacritinib. It was done using the criteria defined by the FDA for systemic inhibition of transporters and based on the ratio Imax,u/IC50 [25]. As indicated in Table 3, only lorlatinib was predicted to inhibit in vivo ENT1 activity at clinically achievable concentrations. We therefore further characterized this interaction of lorlatinib with ENT1. We notably determined whether lorlatinib-mediated inhibition of ENT1 activity described above in engineered HAP1 ENT2-KO cells may also occur in human tumoral cells. For this, we used lung cancer A549 cells, which exhibited a high level of [3H]-uridine uptake inhibited by a low concentration (0.1 µM) of NBMPR, representing 88.9 ± 0.5% of total [3H]-uridine uptake (Fig. 3A). This demonstrates that ENT1 is the preponderant functional ENT isoform in lung A549 cells. Lorlatinib used at 10 µM markedly decreased [3H]-uridine accumulation in A549 cells by 82.5 ± 1.1%. This inhibitory effect of the TKI in A549 cells was demonstrated to be concentration-dependent, with an IC50 of 2.0 µM (Fig. 3B), close to the value reported above for HAP1 ENT2-KO cells. The effects of lorlatinib towards ENT2 activity was next studied in HAP1 ENT1-KO cells. As shown in Fig. 4, lorlatinib potently inhibited ENT2 activity in a concentration-dependent manner, with an IC50 (1.0 µM) even lower than the one measured on ENT1 activity in HAP1 ENT2-KO cells (2.5 µM). With a lorlatinib Imax,u of 485 nM (Table 3), the ratio Imax,u/IC50 for ENT2 inhibition by lorlatinib was consequently 0.48, greater than the threshold of 0.1 given by the FDA guidance on DDIs [24]. This means that this interaction with ENT2 should also be clinically relevant, as predicted above for that with ENT1. Whether lorlatinib may be a substrate for ENT1/ENT2 was finally investigated through comparing the cellular accumulation of the TKI in HAP1 wild-type, ENT1-KO, and ENT2-KO cells, in the absence or presence of 0.1 or 100 µM NBMPR. Lorlatinib was found to similarly accumulate in HAP1 wild-type, ENT1-KO, and ENT2-KO cells, and the presence of 0.1 µM or 100 µM NBMPR failed to reduce cellular uptake of lorlatinib (Fig. 5).
Table 3.
Prediction of in vivo ENT1 inhibition by TKIs.
| TKI | Cmax | fu | Imax,u | IC50 | Imax, u/IC50 | Predicted in vivo inhibitiona | Reference |
|---|---|---|---|---|---|---|---|
| Ibrutinib | 354 nM | 0.027 | 9.56 nM | 4.5 µM | 0.002 | No | [43] |
| Lenvatinib | 761 nM | 0.016 | 12.02 nM | 7.4 µM | 0.002 | No | [44] |
| Lorlatinib | 1466 nM | 0.34 | 485 nM | 2.5 µM | 0.194 | Yes | [45] |
| Neratinib | 146 nM | 0.01 | 1.46 nM | 8.5 µM | <0.001 | No | [46] |
| Pacritinib | 22500 nM | 0.011 | 247.5 nM | 3.7 µM | 0.067 | No | [47] |
Cmax maximum plasma concentration, fu unbound fraction, Imax,u maximum unbound plasma concentration, ENT equilibrative nucleoside transporter, TKIs tyrosine kinase inhibitors
aAn in vivo inhibition can be predicted if the ratio Imax,u/IC50 ≥ 0.1 according to the US FDA guidance criteria [25] applied to ENT1
Fig. 3.
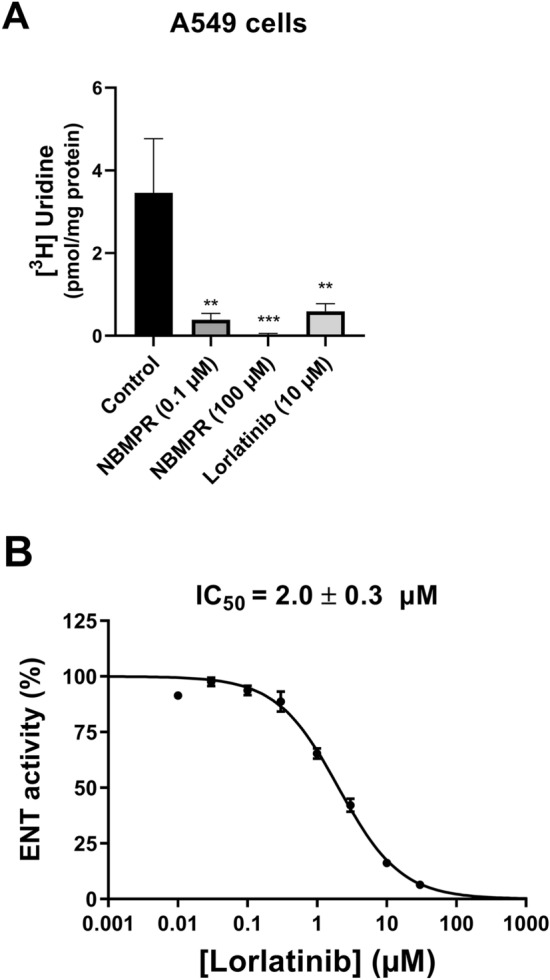
Inhibition of ENT activity by lorlatinib in human lung A549 cells incubated with [3H]-uridine for 5 min at 37 °C, in the absence (control) or presence of A 0.1 µM NBMPR, 100 µM NBMPR, or 10 µM lorlatinib, or B various concentrations of lorlatinib. Intracellular accumulation of [3H]-uridine was next determined by scintillation counting and normalized to protein content. Data are expressed as A [3H]-uridine uptake or B % of ENT activity found in control cells, arbitrarily set at 100%, and are the means ± SD of three independent assays. A **p < 0.01 and ***p < 0.001, when compared to control cells. B Lorlatinib IC50 value is indicated on the top of the graph. ENT equilibrative nucleoside transporter, NBMPR nitrobenzylmercaptopurine riboside
Fig. 4.

Concentration-dependent inhibition of ENT2 activity by lorlatinib. HAP1 ENT1-KO cells were incubated with [3H]-uridine for 5 min at 37 °C, in the absence (control) or presence of various concentrations of lorlatinib. Intracellular uptake of [3H]-uridine was next determined by scintillation counting and normalized to protein content. Data are expressed as % of ENT2 activity found in control cells, arbitrarily set at 100%, and are the means ± SD of three independent assays. Lorlatinib IC50 value is indicated on the top of the graph. ENT equilibrative nucleoside transporter, KO knockout
Fig. 5.
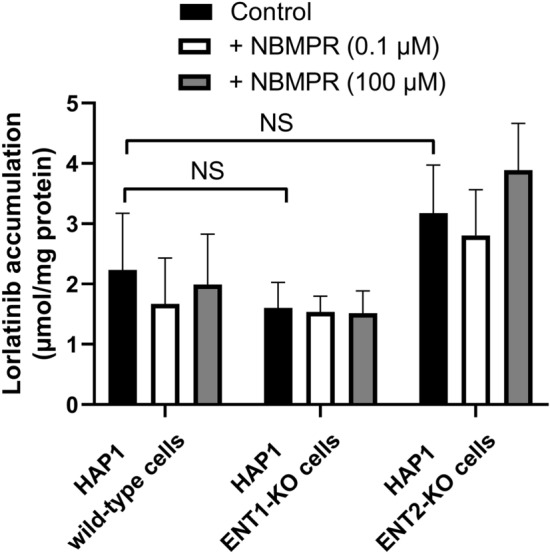
Accumulation of lorlatinib in HAP1 wild-type, ENT1-KO and ENT2-KO cells. HAP1 wild-type, ENT1-KO, and ENT2-KO cells were incubated with 10 µM lorlatinib for 5 min at 37 °C, in the absence (control) or presence of 0.1 µM NBMPR or 100 µM NBMPR. Intracellular accumulation of lorlatinib was next determined by LC-MS/MS and normalized to protein content. Data are the means ± SD of four independent assays. NS not statistically significant, ENT equilibrative nucleoside transporter, KO knockout, NBMPR nitrobenzylmercaptopurine riboside
Discussion
The present study fully confirms that TKIs can interact with ENT1 activity. Indeed, only 2/24 TKIs were devoid of any inhibitory effect towards ENT1 when used at 10 µM, whereas 12/24 TKIs were characterized as moderate (35–60% inhibition of ENT1 activity) or strong (more than 60% inhibition of ENT1 activity) inhibitors of ENT1 and 10/24 TKIs as weak inhibitors (less than 35% of ENT1 inhibition). In particular, ibrutinib, lenvatinib, lorlatinib, neratinib, and pacritinib were found to potently inhibit ENT1 with relative low IC50 values (ranging from 2.5 to 8.5 µM) and can be unambiguously added to the list of TKIs blocking ENT1 activity, which already comprises 11 other TKIs, including the Bcr-Abl inhibitors, imatinib and nilotinib, and the epidermal growth factor receptor inhibitors, gefitinib and erlotinib [17, 18]. In addition to ENT1, TKIs have already been shown to inhibit various SLCs, handling organic cations, like organic cation transporter (OCT) 1 (SLC22A1), OCT2 (SLC22A2), OCT3 (SLC22A3), multidrug and toxin extrusion protein (MATE) 1 (SLC47A1) and MATE2-K (SLC47A2) [27, 28], or organic anion transporters, like organic anion-transporting polypeptide (OATP) 1B1 (SLCO1B1) [29]. Sodium-dependent concentrative nucleoside transporters (CNTs) are also inhibited by some TKIs, including CNT2 (SLC28A2) blocked by imatinib [17] and CNT3 (SLC28A3) by erlotinib [18]. In this context, it is noteworthy that the nature of the inhibited SLCs depends on the TKIs. For example, the Janus kinase inhibitor (JAK) 1/2 ruxolitinib rather potently inhibits OCT2, MATE1, MATE2-K, and the organic anion transporter (OAT) 3 (SLC22A8) (with IC50 values less than 10 µM), whereas it only weakly decreases ENT1 activity and fails to alter that of OCT1 [30]. In the same way, ceritinib and crizotinib are potent inhibitors of OCT3 (with IC50 values in the range 30–40 nM) [28], but only modestly decrease ENT1 activity when used at 10 µM (Table 1). Such data indicate that any general, unspecific and uniform inhibition of SLC transporters by TKIs can be discarded.
The exact mechanism by which TKIs block activity of ENT1 remains to be determined. A possible implication of tyrosine kinase inhibition has to be considered, because such a mechanism is thought to contribute to SLC activity inhibition by TKIs. Indeed, inhibition of OATP1B1 by various TKIs involves repression of the Src kinase LYN-mediated OATP1B1 phosphorylation [29], whereas those of OCT1 and OCT2 have been linked to inhibition of the kinase Yes1 [31, 32]. A major contribution of kinase activity inhibition to TKIs-mediated ENT1 inhibition is nevertheless rather unlikely, owing to (1) the time of incubation with TKIs (5 min), which may be shorter to induce changes in transporter phosphorylation status, and (2) the diverse nature of kinases targeted by TKIs inhibiting ENT1. Indeed, for example, lorlatinib blocks the kinase ROS1 and the anaplasic lymphoma kinase (ALK) [33], whereas ibrutinib represses activity of the Bruton’s tyrosine kinase (BTK) [34] and pacritinib those of JAK2 and Fms-like tyrosine kinase (Flt3) [35]. TKIs may consequently inhibit ENT1 through directly interacting with this transporter, as already reported for the ENT1 inhibitors, NBMPR and dilazep, for which binding sites on ENT1 have recently been characterized [36].
Various molecular descriptors have been associated with drug-mediated inhibition of SLCs acting as transporters. This notably concerns OATPs, OATs, OCTs, and MATEs, but not, or only poorly, ENT1 [27, 37–39]. In the present study, we have brought data about this nucleoside transporter, through identifying several physical/chemical parameters significantly correlated with ENT1 inhibition by TKIs. Among them, the atom-type E-state SssO and NssO exhibit the highest significant correlations; they correspond to electro-topological state indices, reflecting the combinations of electronic, topological, and valence state information [40]. The constitutional index, Mi, reflecting mean first ionization potential (scaled on carbon atom), as well as the functional group count, nArOR, corresponding to the number of ethers (aromatic), were also highly associated with TKI-mediated inhibition of ENT1 activity. Such parameters may consequently represent important descriptors to consider for establishing quantitative structure–activity relationship-based models, in order to predict ENT1 inhibition by drugs, notably TKIs.
Among five strong ENT1 inhibitors identified in the present study, only lorlatinib is predicted to in vivo inhibit ENT1, according to the criteria defined by the FDA guidance on DDIs [25]. This point may merit consideration in terms of possible DDIs and/or adverse effects. Indeed, even if ENT1 is presently not considered as a priority transporter according to the FDA [25], it has been postulated to be a transporter of emerging importance by the International Transporter Consortium, because its inhibition is susceptible to decrease the cellular uptake, and, in this way, the pharmacological effects, of anticancer nucleoside analogues, such as gemcitabine or cytarabine [15]. This has likely to be taken into account for lung cancer cells, which constitute pharmacological targets for lorlatinib [41]. Indeed, ENT1 activity is susceptible to be markedly inhibited by the TKI in these tumoral cells, as demonstrated in lung cancer A549 cells, which may result in a decreased ENT1-mediated accumulation of anticancer nucleoside analogues, and, possibly, in reduced anticancer activity of these drugs, if they are co-administrated with lorlatinib. Moreover, lorlatinib is predicted to also in vivo inhibit ENT2 activity, thus suggesting that this TKI may fully abrogate facilitated transport of nucleoside analogues into target cells and, by this way, may cause potential DDIs. Lorlatinib may also be suspected to reduce intestinal absorption of ENT1 substrates, through blocking their ENT1-related efflux at the basolateral membrane of enterocytes [42]; it may additionally inhibit ENT1-mediated drug transport at the hepatic and/or placental level. Nevertheless, ENT activity inhibition may have beneficial consequences, i.e., it may be hypothesized to contribute to the antiproliferative effects of lorlatinib, through reducing cellular uptake of nucleosides in cancer cells, in a kinase-independent manner. In addition, it may exert antithrombotic effects, through enhancing extracellular levels of the platelet repressor, adenosine, as already described for other ENT1 inhibitors [13]. Finally, it is noteworthy that lorlatinib is not a substrate for either ENT1 or ENT2, since (1) HAP1 wild-type, ENT1-KO, and ENT2-KO cells similarly accumulated the TKI, and (2) the ENT inhibitor, NBMPR, was without effect on lorlatinib uptake in these parental or ENT-KO cells, whenever it was used at 0.1 µM (inhibiting ENT1) or 100 µM (inhibiting both ENT1 and ENT2). ENT1 and ENT2 are therefore unlikely to play a role in lorlatinib pharmacokinetics or to contribute to its accumulation in target cells.
Inhibitory effects of TKIs towards ENT activity have been studied using functional knock-out cells, deficient in either ENT1 or ENT2 activity. The use of such cells for specifically investigating ENT1 or ENT2 activity therefore appears as a convenient alternative to that of transfected transporter-overexpressing cells, notably because native cells often display both ENT1 and ENT2 activities, even if, in the case of HAP1 wild-type cells, that of ENT1 is the main one. This conclusion is fully supported by the recent use of CRISPR/Cas9-generated HeLa cell variants, deficient in either ENT1 or ENT2, for characterizing ENT–drug interactions [14].
Conclusion
Various TKIs (12/24) used at 10 µM have been demonstrated to behave as moderate or strong inhibitors of ENT1, thus unambiguously adding ENT1 to the list of SLCs targeted by TKIs. As regards ALK-targeting lorlatinib, relevant clinical concentrations were even predicted to inhibit ENT1 activity in vivo, as well as that of ENT2. Lorlatinib was, however, not transported by ENT1 or ENT2, thus discarding any role of these nucleoside transporters in its cellular or general pharmacokinetics. Lorlatinib may thus appear as a potent inhibitor of ENT1, which likely merits attention if this TKI is co-administrated with known ENT1 substrates, like anticancer nucleoside analogues.
Declarations
Funding
The authors received no specific funding for this work.
Conflict of Interest
All the authors have no conflict of interest to declare.
References
- 1.Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004;447:735–743. doi: 10.1007/s00424-003-1103-2. [DOI] [PubMed] [Google Scholar]
- 2.Molina-Arcas M, Casado FJ, Pastor-Anglada M. Nucleoside transporter proteins. Curr Vasc Pharmacol. 2009;7:426–434. doi: 10.2174/157016109789043892. [DOI] [PubMed] [Google Scholar]
- 3.Naes SM, Ab-Rahim S, Mazlan M, Abdul RA. Equilibrative Nucleoside Transporter 2: Properties and Physiological Roles. Biomed Res Int. 2020;2020:5197626. doi: 10.1155/2020/5197626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pastor-Anglada M, Pérez-Torras S. Nucleoside transporter proteins as biomarkers of drug responsiveness and drug targets. Front Pharmacol. 2015;6:13. doi: 10.3389/fphar.2015.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, Abrams R, Benson AB, Macdonald J, Cass CE, Dicker AP, Mackey JR. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136:187–195. doi: 10.1053/j.gastro.2008.09.067. [DOI] [PubMed] [Google Scholar]
- 6.Galmarini CM, Thomas X, Calvo F, Rousselot P, Rabilloud M, El Jaffari A, Cros E, Dumontet C. In vivo mechanisms of resistance to cytarabine in acute myeloid leukaemia. Br J Haematol. 2002;117:860–868. doi: 10.1046/j.1365-2141.2002.03538.x. [DOI] [PubMed] [Google Scholar]
- 7.Spratlin J, Sangha R, Glubrecht D, Dabbagh L, Young JD, Dumontet C, Cass C, Lai R, Mackey JR. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res. 2004;10:6956–6961. doi: 10.1158/1078-0432.CCR-04-0224. [DOI] [PubMed] [Google Scholar]
- 8.Endres CJ, Moss AM, Ishida K, Govindarajan R, Unadkat JD. The role of the equilibrative nucleoside transporter 1 on tissue and fetal distribution of ribavirin in the mouse. Biopharm Drug Dispos. 2016;37:336–344. doi: 10.1002/bdd.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishimura T, Sano Y, Takahashi Y, Noguchi S, Uchida Y, Takagi A, Tanaka T, Katakura S, Nakashima E, Tachikawa M, Maruyama T, Terasaki T, Tomi M. Quantification of ENT1 and ENT2 proteins at the placental barrier and contribution of these transporters to ribavirin uptake. J Pharm Sci. 2019;108:3917–3922. doi: 10.1016/j.xphs.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Klein DM, Evans KK, Hardwick RN, Dantzler WH, Wright SH, Cherrington NJ. Basolateral uptake of nucleosides by sertoli cells is mediated primarily by equilibrative nucleoside transporter 1. J Pharmacol Exp Therap. 2013;346:121–129. doi: 10.1124/jpet.113.203265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borbone N, Piccialli G, Roviello GN, Oliviero G. Nucleoside analogs and nucleoside precursors as drugs in the fight against SARS-CoV-2 and other coronaviruses. Molecules. 2021;26:986. doi: 10.3390/molecules26040986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geiger JD, Khan N, Murugan M, Boison D. Possible role of adenosine in COVID-19 pathogenesis and therapeutic opportunities. Front Pharmacol. 2020;11:594487. doi: 10.3389/fphar.2020.594487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armstrong D, Summers C, Ewart L, Nylander S, Sidaway JE, van Giezen JJ. Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. J Cardiovasc Pharmacol Ther. 2014;19:209–219. doi: 10.1177/1074248413511693. [DOI] [PubMed] [Google Scholar]
- 14.Miller SR, Zhang X, Hau RK, Jilek JL, Jennings EQ, Galligan JJ, Foil DH, Zorn KM, Ekins S, Wright SH, Cherrington NJ. Predicting drug interactions with human equilibrative nucleoside transporters 1 and 2 using functional knockout cell lines and bayesian modeling. Mol Pharmacol. 2021;99:147–162. doi: 10.1124/molpharm.120.000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, Zhang L. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther. 2013;94:52–63. doi: 10.1038/clpt.2013.74. [DOI] [PubMed] [Google Scholar]
- 16.Huang M, Wang Y, Cogut SB, Mitchell BS, Graves LM. Inhibition of nucleoside transport by protein kinase inhibitors. J Pharmacol Exp Ther. 2003;304:753–760. doi: 10.1124/jpet.102.044214. [DOI] [PubMed] [Google Scholar]
- 17.Damaraju VL, Weber D, Kuzma M, Cass CE, Sawyer MB. Selective Inhibition of Human Equilibrative and Concentrative Nucleoside Transporters by BCR-ABL Kinase Inhibitors: identification of key hENT1 Amino Acid Residues For Interaction With BCR-ABL kinase inhibitors. J Biol Chem. 2016;291:18809–18817. doi: 10.1074/jbc.M116.741074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Damaraju VL, Scriver T, Mowles D, Kuzma M, Ryan AJ, Cass CE, Sawyer MB. Erlotinib, gefitinib, and vandetanib inhibit human nucleoside transporters and protect cancer cells from gemcitabine cytotoxicity. Clin Cancer Res. 2014;20:176–186. doi: 10.1158/1078-0432.CCR-13-2293. [DOI] [PubMed] [Google Scholar]
- 19.Damaraju VL, Kuzma M, Mowles D, Cass CE, Sawyer MB. Interactions of multitargeted kinase inhibitors and nucleoside drugs: achilles heel of combination therapy? Mol Cancer Ther. 2015;14:236–245. doi: 10.1158/1535-7163.MCT-14-0337. [DOI] [PubMed] [Google Scholar]
- 20.Essletzbichler P, Konopka T, Santoro F, Chen D, Gapp BV, Kralovics R, Brummelkamp TR, Nijman SM, Bürckstümmer T. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res. 2014;24:2059–2065. doi: 10.1101/gr.177220.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Vée M, Jouan E, Fardel O. Carrier-mediated uptake of clonidine in cultured human lung cells. Naunyn Schmiedebergs Arch Pharmacol. 2018;391:361–369. doi: 10.1007/s00210-018-1467-7. [DOI] [PubMed] [Google Scholar]
- 22.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 23.Mayati A, Moreau A, Jouan E, Febvre-James M, Denizot C, Parmentier Y, Fardel O. mRNA expression and activity of nucleoside transporters in human hepatoma HepaRG cells. Pharmaceutics. 2018;10:246. doi: 10.3390/pharmaceutics10040246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry, US Food and Drug Administration. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed 20 Sept 2020.
- 26.Chedik L, Bruyere A, Le Vee M, Stieger B, Denizot C, Parmentier Y, Potin S, Fardel O. Inhibition of human drug transporter activities by the pyrethroid pesticides allethrin and tetramethrin. PLoS ONE. 2017;12:e0169480. doi: 10.1371/journal.pone.0169480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minematsu T, Giacomini KM. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther. 2011;10:531–539. doi: 10.1158/1535-7163.MCT-10-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alim K, Moreau A, Bruyère A, Jouan E, Denizot C, Nies AT, Parmentier Y, Fardel O. Inhibition of organic cation transporter 3 activity by tyrosine kinase inhibitors. Fundam Clin Pharmacol. 2021 doi: 10.1111/fcp.12657. [DOI] [PubMed] [Google Scholar]
- 29.Hayden E, Chen M, Pasquariello KZ, Gibson AA, Petti JJ, Shen S, Qu J, Ong SS, Chen T, Jin Y, Uddin ME, Huang KM, Paz A, Sparreboom A, Hu S, Sprowl JA. Regulation of OATP1B1 function by tyrosine kinase-mediated phosphorylation. Clin Cancer Res. 2021 doi: 10.1158/1078-0432.CCR-21-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruyère A, Le Vée M, Jouan E, Molez S, Nies AT, Fardel O. Differential in vitro interactions of the Janus kinase inhibitor ruxolitinib with human SLC drug transporters. Xenobiotica. 2021;51:467–478. doi: 10.1080/00498254.2021.1875516. [DOI] [PubMed] [Google Scholar]
- 31.Sprowl JA, Ong SS, Gibson AA, Hu S, Du G, Lin W, Li L, Bharill S, Ness RA, Stecula A, Offer SM, Diasio RB, Nies AT, Schwab M, Cavaletti G, Schlatter E, Ciarimboli G, Schellens JHM, Isacoff EY, Sali A, Chen T, Baker SD, Sparreboom A, Pabla N. A phosphotyrosine switch regulates organic cation transporters. Nat Commun. 2016;7:10880. doi: 10.1038/ncomms10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uddin ME, Garrison DA, Kim K, Jin Y, Eisenmann ED, Huang KM, Gibson AA, Hu Z, Sparreboom A, Hu S. Influence of yes1 kinase and tyrosine phosphorylation on the activity of OCT1. Front Pharmacol. 2021;12:644342. doi: 10.3389/fphar.2021.644342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, Cui JJ, Deal JG, Deng YL, Dinh D, Engstrom LD, He M, Hoffman J, Hoffman RL, Huang Q, Kania RS, Kath JC, Lam H, Lam JL, Le PT, Lingardo L, Liu W, McTigue M, Palmer CL, Sach NW, Smeal T, Smith GL, Stewart AE, Timofeevski S, Zhu H, Zhu J, Zou HY, Edwards MP. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57:4720–4744. doi: 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- 34.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, Goh KC, Ong WC, Goh SK, Hart S, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW. Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011;54:4638–4658. doi: 10.1021/jm200326p. [DOI] [PubMed] [Google Scholar]
- 36.Wright NJ, Lee SY. Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol. 2019;26:599–606. doi: 10.1038/s41594-019-0245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Bruyn T, van Westen GJ, Ijzerman AP, Stieger B, de Witte P, Augustijns PF, Annaert PP. Structure-based identification of OATP1B1/3 inhibitors. Mol Pharmacol. 2013;83:1257–1267. doi: 10.1124/mol.112.084152. [DOI] [PubMed] [Google Scholar]
- 38.Liu HC, Goldenberg A, Chen Y, Lun C, Wu W, Bush KT, Balac N, Rodriguez P, Abagyan R, Nigam SK. Molecular properties of drugs interacting with SLC22 transporters OAT1, OAT3, OCT1, and OCT2: A machine-learning approach. J Pharmacol Exp Ther. 2016;359:215–229. doi: 10.1124/jpet.116.232660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wittwer MB, Zur AA, Khuri N, Kido Y, Kosaka A, Zhang X, Morrissey KM, Sali A, Huang Y, Giacomini KM. Discovery of potent, selective multidrug and toxin extrusion transporter 1 (MATE1, SLC47A1) inhibitors through prescription drug profiling and computational modeling. J Med Chem. 2013;56:781–795. doi: 10.1021/jm301302s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall LH, Kier LB. Electrotopological state indexes for atom types—a novel combination of electronic, topological, and valence state information. J Chem Inf Comput Sci. 1995;35:1039–1045. doi: 10.1021/ci00028a014. [DOI] [Google Scholar]
- 41.Shaw AT, Bauer TM, de Marinis F, Felip E, Goto Y, Liu G, Mazieres J, Kim DW, Mok T, Polli A, Thurm H, Calella AM, Peltz G, Solomon BJ. First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N Engl J Med. 2020;383:2018–2029. doi: 10.1056/NEJMoa2027187. [DOI] [PubMed] [Google Scholar]
- 42.Yuasa H, Yasujima T, Inoue K. Current understanding of the intestinal absorption of nucleobases and analogs. Biol Pharm Bull. 2020;43:1293–1300. doi: 10.1248/bpb.b20-00342. [DOI] [PubMed] [Google Scholar]
- 43.Ibrutinib. Pharmacology review, Center for Drug Evaluation and Research, US Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205552Orig2s000ClinPharmR.pdf. Accessed 15 Feb 2021.
- 44.Lenvatinib. Pharmacology review, Center for Drug Evaluation and Research, US Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206947Orig1s000ClinPharmR.pdf. Accessed 15 Feb 2021.
- 45.Lorlatinib. Pharmacology review, Center for Drug Evaluation and Research, US Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210868Orig1s000MultidisciplineR.pdf. Accessed 14 Feb 2021.
- 46.Neratinib. Label, Center for Drug Evaluation and Research, US Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208051Orig1s000MultidisciplineR.pdf. Accessed 26 Feb 2021.
- 47.Pacritinib. Assessment report, European Medecines Agency. https://www.ema.europa.eu/en/documents/withdrawal-report/withdrawal-assessment-report-enpaxiq_en.pdf. Accessed 16 Feb 2021.