

Abstract
RNA-binding proteins like human antigen R (HuR) are key regulators in post-transcriptional control of gene expression in several pathophysiological conditions. Diabetes adversely affects monocyte/macrophage biology and function. It is not known whether diabetic milieu affects cellular/exosome-HuR and its implications on cardiac inflammation and fibrosis. Here, we evaluate in vitro and in vivo effects of diabetic milieu on macrophage cellular/exosome-HuR, alterations in intercellular cross talk with fibroblasts, and its impact on cardiac remodeling. Human failing hearts show higher HuR levels. Diabetic milieu activates HuR expression in cardiac- and cultured bone marrow-derived macrophages (BMMØ) and stimulates HuR nuclear-to-cytoplasmic translocation and exosome transfer. Exosomes from macrophages exposed to diabetic milieu (high glucose or db/db mice) significantly increase inflammatory and profibrogenic responses in fibroblast (in vitro) and cardiac fibrosis in mice. Intriguingly, Exo-HuR deficiency (HuR knockdown in macrophage) abrogates the above effects. In diabetic mice, macrophage depletion followed by reconstitution with BMMØ-derived HuR-deficient exosomes inhibits angiotensin II-induced cardiac fibrosis response and preserves left ventricle function as compared to control-exosome administration. To the best of our knowledge, this is the first study to demonstrate that diabetes activates BMMØ HuR expression and its transfer into exosome. The data suggest that HuR might be targeted to alleviate macrophage dysfunction and pathological fibrosis in diabetes.
Keywords: cardiac fibrosis, cardiac inflammation, diabetes, exosome, HuR, intercellular signaling
1 |. INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of mortality in patients with diabetes.1 In the injured myocardium, infiltrating macrophages are the major myeloid cell population that exhibit remarkable phenotypic and functional heterogeneity.2–4 Depending on the tissue milieu, macrophages in the tissue secrete factors that mediate cross talk with other cardiac cell types to either ensure myocardial homeostasis or initiate and propagate adverse cardiac remodeling.4,5 Diabetic milieu has been shown to adversely affect macrophage function in the heart and prolong activation of cardiac myofibroblast and excessive collagen deposition leading to increase in myocardial stiffness and resulting in worse clinical outcomes.6,7 Here we studied the influence of diabetes on paracrine mechanism of macrophage cross talk with fibroblasts and its implications on cardiac remodeling and failure.
RNA-binding proteins (RBPs) regulate gene expression by altering mRNA stability or degradation. Among the RBPs, human antigen R (HuR; encoded by the ELAVL1 gene) is ubiquitously expressed and binds to AU-Rich Elements (ARE) in the 3′-untranslated region (3′UTR) of mRNA and confers its stability.8 HuR regulation of cancer progression and pathology has been extensively studied.9 While the regulatory role of HuR in cardiovascular pathophysiology is slowly emerging, we and other research groups have demonstrated the involvement of HuR in hyperglycemia-mediated human ventricular cardiomyocyte pyroptosis,10 rat myocyte hypertrophic growth,11 inflammation,12 and cell proliferation/death.13,14 Cell-secreted exosomes (Exo) mediate cell-cell interaction and play a crucial role in several biological processes.15,16 However, exosome transfer of RBPs and its role in cellular cross talk and diabetes-mediated cardiac pathology has never been explored so far.
The aim of the present study is to determine the influence of diabetes on macrophage RNA-binding protein (HuR) activity and its paracrine effect via exosomes on cardiac remodeling and function. We hypothesize that diabetes stimulates macrophage HuR expression, activity, and transfer into exosome and Exo-associated HuR promotes cardiac inflammatory/fibrogenic response. In this work, we show that diabetes-mediated increase in macrophage HuR expression, its cytoplasmic translocation, and exosome transfer activates fibrogenesis signaling in vitro and in vivo. Most importantly, under diabetic milieu, intravenous injection of HuR-deficient exosomes inhibits angiotensin II (ANG II)-induced cardiac inflammation and fibrosis and is found to be cardio-protective. To our knowledge, this is the first report to demonstrate that HuR protein is enriched in exosome and its role in BMMØ-fibroblast interaction and diabetes-influenced cardiac remodeling and dysfunction. As fibrotic diseases have significant healthcare implications, for which there are limited treatment options, this work is valuable in demonstrating exosome-associated HuR as a therapeutic target for limiting pathological fibrosis and heart disease.
2 |. METHODS
2.1 |. Vertebrate animals
All animal experiments conform to the protocols approved by the Institutional Animal Care and Use Committee (IACUC) at The University of Alabama at Birmingham (Birmingham, AL). Eight-weeks-old C57BL/6J or db/db (BKS.Cg-Dock7m +/+ Leprdb/J) mice on a same genetic background were procured from Jackson Laboratory (Bar Harbor, ME). The mice were allowed to acclimatize for 10 days under sterile animal management conditions.
2.2 |. Mouse bone marrow macrophage isolation and culture
Mouse bone marrow-derived macrophage (BMMØ) isolation, ex vivo expansion, and culture were performed as previously described.17 Briefly, bone marrow mononuclear cells collected from femur and tibia of mice were differentiated in DMEM (Lonza, Walkersville, MD) supplemented with macrophage colony-stimulating factor (M-CSF) [50% L929-conditioned medium] and 10% FBS. After differentiation for 6 days, cells were maintained in DMEM supplemented with 20% L929-conditioned medium.
2.3 |. Cell culture
RAW 264.7 cells (mouse monocyte/macrophage cell line, ATCC TIB-71) and NIH/3T3 cells (mouse embryo fibroblast, ATCC CRL-1658) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Life technologies, Grand Island, NY) with 10% fetal bovine serum (FBS, ATCC, Manassas, VA) and 1% penicillin-streptomycin (Life Technologies) and were incubated in a humidified chamber at 37°C with 5% CO2. Cells were cultured in either high glucose (HG, 30 mM) or normal glucose (NG, 5 mM) media.
2.4 |. HuR knockdown by shRNA lentiviral transduction
To knockdown HuR, RAW 264.7 cells were infected with short hair-pin expressing lentiviral particle against HuR (Cat# SHCLNV, Sigma, 5 MOI, multiplicity of infection) in the presence of hexadimethrine bromide (Cat# H9268, Sigma; 8 µg/mL). After 24-hour transduction, cells resistant to puromycin (Cat# P9620, Sigma; 6 µg/mL, dose determined by titration) were further cultured. Similarly, BMMØs cultured for seven days were infected for 48 hours. HuR knockdown (HuR KD) and its effects on targets were confirmed by quantitative real-time PCR (qRT-PCR) and/or Western blotting.
2.5 |. Exosomes isolation, characterization, labeling, and target cell uptake
Exosomes were isolated from macrophage-conditioned media using total exosome isolation reagent (Life Technologies, CA) according to the manufacturer’s instructions. Briefly, the condition medium of RAW 264.7 cells cultured in exosome-depleted FBS medium (Cat# A2720801, Life Technologies) with normal glucose (NG; 5 mM) or high glucose (HG; 30 mM) conditions was collected after 72 hours, centrifuged (2000 g for 30 minutes), filtered (0.22 µm), and incubated overnight at 4°C with total exosome isolation reagent. The samples were centrifuged at 10 000 g for 1 hour, and the pellet was dispersed in PBS and further purified using exosome spin columns (Cat# 4484449, Life Technologies). The size, physical characteristics, and exosome-associated protein markers (Alix, HSP70, and Annexin V) were evaluated using dynamic light scattering (DLS, NanoSight NS300; NTA 3.0 software, Malvern), transmission electron microscope (TEM; Tecnai Spirit T12, Thermo Fisher) and Western blotting, and dot blot, respectively. The isolated exosomes from RAW 264.7 cells condition medium were labeled with PKH26 (Cat# MINI26, Sigma-Aldrich) according to the manufacturer’s instructions and incubated with NIH/3T3 cells in four-well chamber slides (30 000 cells/chamber) for 24 hours. The cells were then fixed with 2% paraformaldehyde, permeabilized with 0.5% Triton X-100 for 10 minutes, and then blocked with 5% FBS for 30 minutes. Cells were stained with Flash Phalloidin Green 488 (1:50, Cat# 42420, Biolegend) for intracellular cytoskeleton F-actin. At the end, coverslips were mounted on glass slides with VECTASHIELD antifade mounting medium with 4′,6-diamidino-2-phenylindole-DAPI (Cat# H-1200, Vector Laboratories) and observed under inverted microscope (Cat# IX83, Olympus). Exosomes were also characterized for expression of exosome specific markers using Exo-check exosome antibody array from system bioscience (# Exoray 200A-4).
2.6 |. Macrophage depletion and reconstitution with HuR-deficient exosomes in ANG-II-induced cardiac fibrosis mouse model
Clodronate liposome or control liposome (150 μL; FormuMax Scientific, Sunnyvale, CA, USA) was intraperitoneally injected into 8-week-old C57BL/6J mice, and macrophage depletion was confirmed using FACS analysis (BD LSR II Analyzer). After 48 hours, vehicle (saline) or ANG II (1000 ng·kg−1·min−1; EMD Millipore) was continuously administered using osmotic mini-pumps (ALZET DURECT, Cupertino, CA). Exosomes from control or HuR shRNA-treated BMMØ derived from diabetic (db/db) mice (Exo-db/dbHuR KD vs Exo-db/dbWT) were injected intravenous through tail vein on day 2 and day 4 after pump implantation. Heart function was evaluated by echocardiography (Vevo 2100; VisualSonics, Toronto, Canada). Mice were euthanized on day 28; heart was collected for further analysis.
2.7 |. Echocardiography studies
Transthoracic two-dimensional B-Mode and M-Mode echocardiogram was obtained using Vevo 2100 (VisualSonics, Toronto, Canada) equipped with 30 MHz transducer. Echocardiographic studies were performed after ANG II mini-osmotic pump implantation on mice anesthetized with a mixture of 1.5% isoflurane and oxygen (1 L/min). B-Mode and M-Mode tracings were used to measure left ventricle (LV) percent fractional shortening (%FS) and percent ejection fraction (%EF), respectively, as described earlier.12
2.8 |. Histological analysis
Histopathology studies were performed as described previously.12,18 The hearts were fixed with 4% paraformaldehyde (PFA), embedded in paraffin, and cut into 5-μm-thick slices for Masson’s trichrome staining to detect fibrosis using light microscopy. Immunofluorescent staining for F4/80 and HuR on tissue sections was performed as described previously.12 Tissue sections were permeabilized and stained with anti-F4/80 (1:100, Cat# MA5–16624, Invitrogen) and anti-HuR (1:100, Cat# sc-5261, Santa cruz biotechnology) for macrophage and HuR followed by incubation with respective secondary antibodies (1:500, Cat# A-21208, Code # 715–165-150). Staining without primary antibodies was used as control for non-specific fluorescence. Nuclei were counter-stained with DAPI (1:5000, Sigma-Aldrich, St Louis, MO), and sections were examined under fluorescent microscope (Olympus IX83). Stained heart tissues were used to analyze percent HuR-positive cells, percent perivascular, and percent interstitial fibrosis using NIH-ImageJ software.
2.9 |. RNA isolation and quantitative qRT-PCR
Total cellular and exosome RNA were extracted using a RNA extraction kit (Cat# 217004, Qiagen), and RNA was reverse transcribed to cDNA using qScript cDNA SuperMix (Cat# 95048–025, Quanta Biosciences) according to the manufacturer’s instructions. Quantitative RT-PCR was performed using Quanta bio-studio (Applied Biosystems, Grand Island, NY) using the Fast SYBR Green Master Mix (Cat# 4385612, Applied Biosystems) for detection of gene amplification by following manufacturer’s instructions. Relative mRNA expression of target gene was normalized to the β-Actin or GAPDH gene. The data were represented as fold change versus respective control.
2.10 |. Protein extraction and Western blot analysis
RAW 264.7 cells and exosomes lysates were prepared using cell lysis buffer 1 (Part No.: 890713, R & D Systems). In addition, cells were fractionated into cytosolic and nuclear fractions using a NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Cat# 78833, Thermo Fisher Scientific) according to the manufacturer’s protocol. Equal amounts of proteins from cells, sub cellular fraction, and exosomes were separated by 10% Mini-PROTEAN TGX Stain-Free Protein Gels (Cat# 4568034, Bio-Rad) and blotted on to Trans-Blot Turbo Mini polyvinylidenedifluoride (PVDF) membranes (Cat# 1704156, Bio-Rad). The blots were incubated with antibodies against HuR (1:100, Cat# sc-5261, Santa Cruz Biotechnology Inc); MMP9, MMP13 and vimentin (1:1000, Proteintech); exosomal markers Alix, HSP70 and Annexin V (1:1000, Cat# 7426, Cell Signaling); and GAPDH (1:100, Cat# MA5–15738, Thermo Fisher Scientific) and developed with an enhanced chemiluminescence detection system (Bio-Rad, ChemiDoc Touch Imaging System). The densitometry of the bands from the Western blot was analyzed using NIH-ImageJ software.
2.11 |. HuR subcellular localization
RAW 264.7 cells were cultured in four-well chamber slides (30 000 cells/chamber), using DMEM complete medium with normal glucose (NG; 5 mM), high glucose (HG; 30 mM), and lipopolysaccharide (LPS; 10 ng/mL) conditions. The cultured cells were incubated at 37°C in 5% CO2 for 24 hours. The cells were fixed (2% PFA) for 10 minutes, permeabilized (0.5% Triton X-100) for 10 minutes, and blocked (5% FBS) for 30 minutes at room temperature. Cells were stained with primary antibody HuR (1:100) and secondary antibody FITC-conjugated Donkey anti-mouse IgG (1:100, Cat# 715–095-151, Jackson ImmunoResearch). At the end, coverslips were mounted on glass slides with VECTASHIELD antifade mounting medium with DAPI and observed under inverted microscope.
2.12 |. Statistical analyses
The data were analyzed using Student’s t test or one-way analysis of variance (ANOVA). All the values are presented as mean ± SE. The probability (P) values of ≤0.05 were considered statistically significant.
3 |. RESULTS
3.1 |. HuR expression increases in human failing hearts
HuR binds to mRNA and has been shown to regulate various genes implicated in cancer formation, progression, metastasis, and prognosis.9 Our previous study has shown that anti-inflammatory cytokine (IL-10) administration in mice decreases myocardial HuR expression and attenuates cardiac remodeling and dysfunction.12,19 To determine the effect of heart failure (HF) in humans on myocardial HuR expression and activity, human cardiac biopsies collected from left ventricular free wall of ischemia patients (Temple University School of Medicine, Philadelphia, PA) were subjected to qRT-PCR and immunofluorescence (IF) analyses. HuR mRNA expression is significantly upregulated in the human heart tissue samples from HF patients as compared to patients with non-cardiac-related ailments (Figure 1A, *P < .01). Furthermore, immunofluorescence (IF) analysis shows an increase in the number of myocardial cells positive for HuR protein in patients with heart disease as compared to non-failing hearts (Figure 1B,C, *P < .05; H&E stained section is shown in Figure 1D).
FIGURE 1.
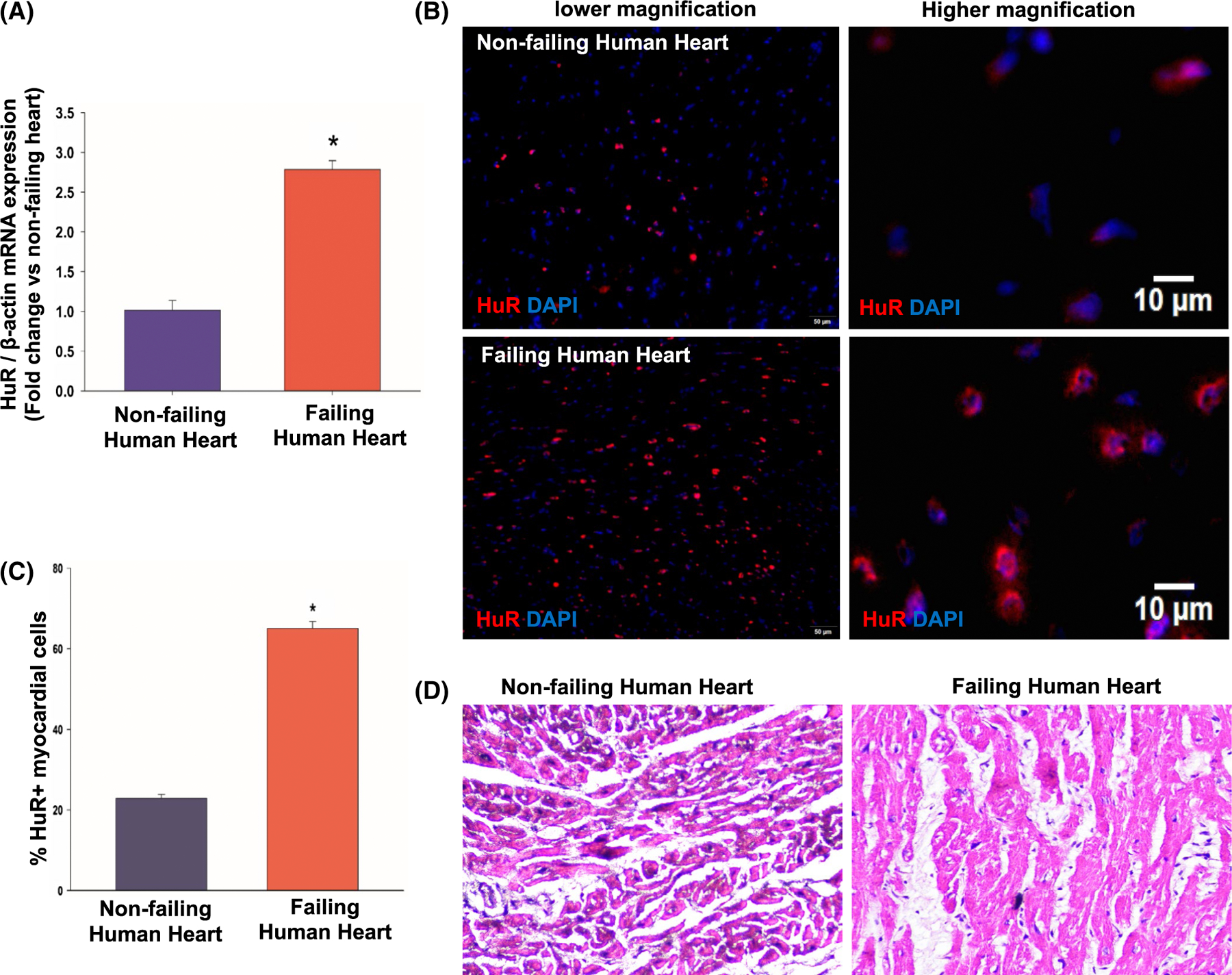
HuR expression increases in ischemic failing human heart. A, qRT-PCR data showing increase in HuR mRNA expression in failing human heart samples as compared to non-failing hearts. Data normalized to β-actin mRNA. n = 3, *P < .01. Immunofluorescence staining of human heart tissue, low and high magnification (B), and bar graph (C) showing increase in HuR+ cells in failing heart (n = 3, *P < .05 vs non-failing hearts). D, H&E staining of non-failing and failing human hearts
3.2 |. Diabetes upregulates HuR expression and activity in mouse cardiac- and bone marrow-derived macrophages
Inflammatory stimuli upregulate HuR expression.20 To determine the influence of diabetes on HuR expression, we performed IF staining on heart sections and cultured BMMØs derived from diabetic (db/db) mouse and compared with non-diabetic (C57BL/6J) mouse. Interestingly, diabetic hearts show a significant increase in HuR-positive myocardial cells as compared to non-diabetic (C57BL/6J) hearts (Figure 2A,B, *P < .01; H&E stained section is shown in Figure 2C). Our previous study has shown that upon myocardial infarction (MI) in mice, HuR expression increases in the myocardium and is localized to border zone of infarct, specifically higher in infiltrating cells.19 Diabetic milieu negatively affects macrophage function.21–23 To determine the influence of diabetes on macrophage HuR activity, we performed co-immunostaining for F4/80 (macrophage marker) and HuR on hearts from db/db and non-diabetic (C57BL/6J) mice. We observed that diabetic (db/db) hearts show higher number of HuR+ F4/80+ macrophages as compared to non-diabetic hearts (Figure 2D,E, *P < .01).
FIGURE 2.
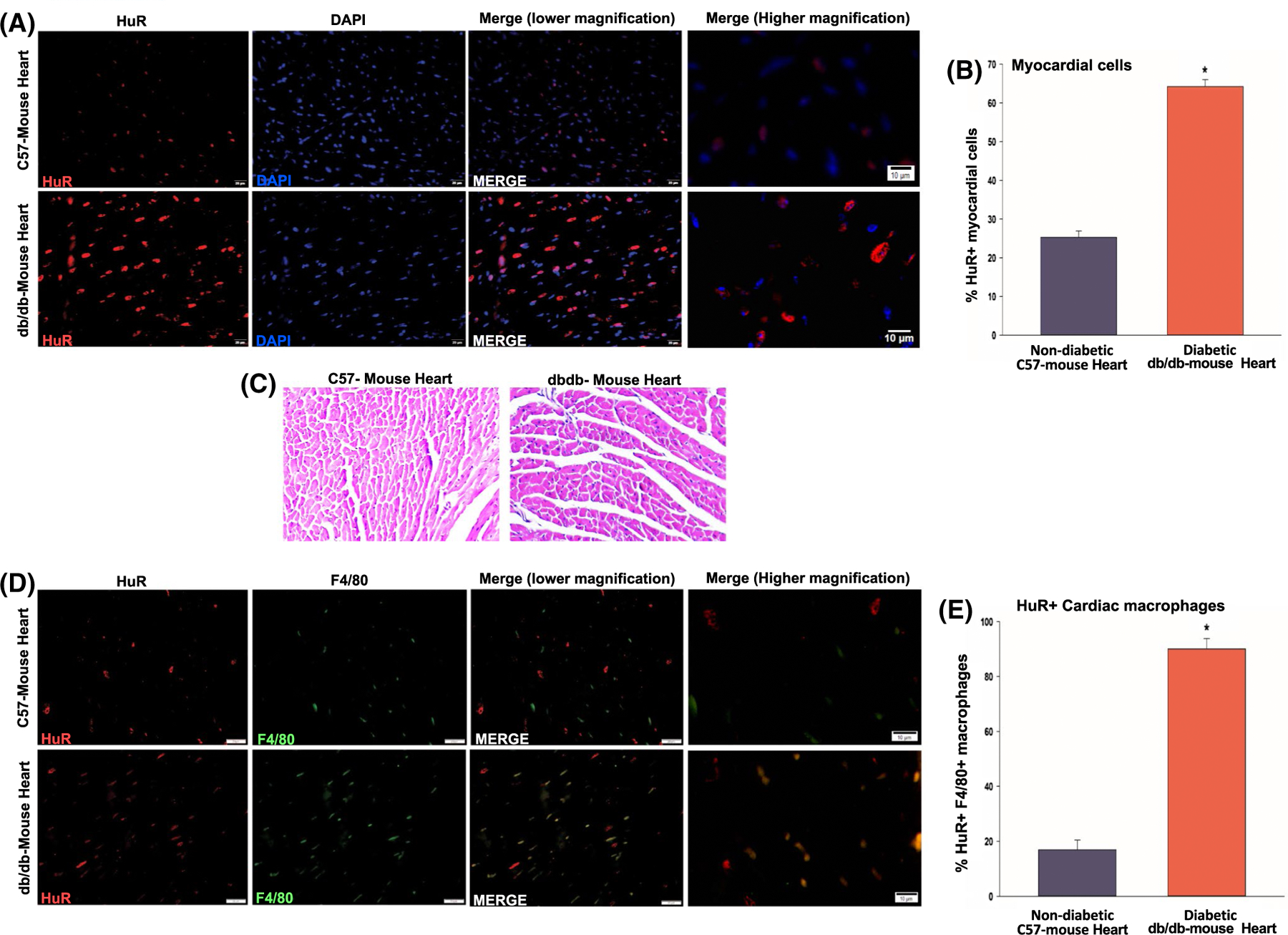
Enhanced HuR levels in myocardial cells and cardiac macrophages in diabetic mouse hearts. A,B, Co-immunofluorescence staining of diabetic (db/db) mouse hearts (low and high magnification) showing increase in HuR+ cells in the myocardial cells. C, H&E staining of WT C57 mice and db/db mice hearts. D,E, Co-immunofluorescence staining showing increased F4/80+ HuR+ cardiac macrophages in db/db mouse heart in comparison with non-diabetic (C57BL/6J) mouse hearts. n = 3, *P < .01
One of the major sources of extracardiac macrophages is bone marrow. Previous reports have shown that HuR activity (mRNA binding and stability) is regulated through nuclear-to-cytoplasmic shuttling in the cell.24 We evaluated the influence of diabetes on HuR expression and activity in ex vivo cultured bone marrow-derived macrophages (BMMØ) from db/db and C57BL/6J mice. Interestingly, IF staining of BMMØ shows both, an increase in nuclear HuR expression and pronounced cytoplasmic translocation in macrophages from diabetic (db/db) mouse as compared to non-diabetic control mouse (Figure 3A). Our data show that HuR is predominantly nuclear in non-diabetic condition while it shuttles to cytoplasm under diabetic milieu. Previous reports have shown that HuR shuttling is regulated through protein kinase C delta (PKCδ).25,26 To determine whether the effect of diabetic milieu on HuR nuclear-to-cytoplasmic translocation is mediated through PKCδ, we exposed mouse BMMØ cells to high glucose (HG, 30 mM) with/without Rottlerin (a widely used selective inhibitor of PKCδ) for 24h and compared to cells exposed to normal glucose (NG, 5 mM) conditions. Intriguingly, Rottlerin treatment (10 μM) limits HG-induced nuclear export of HuR protein (Figure 3B).
FIGURE 3.
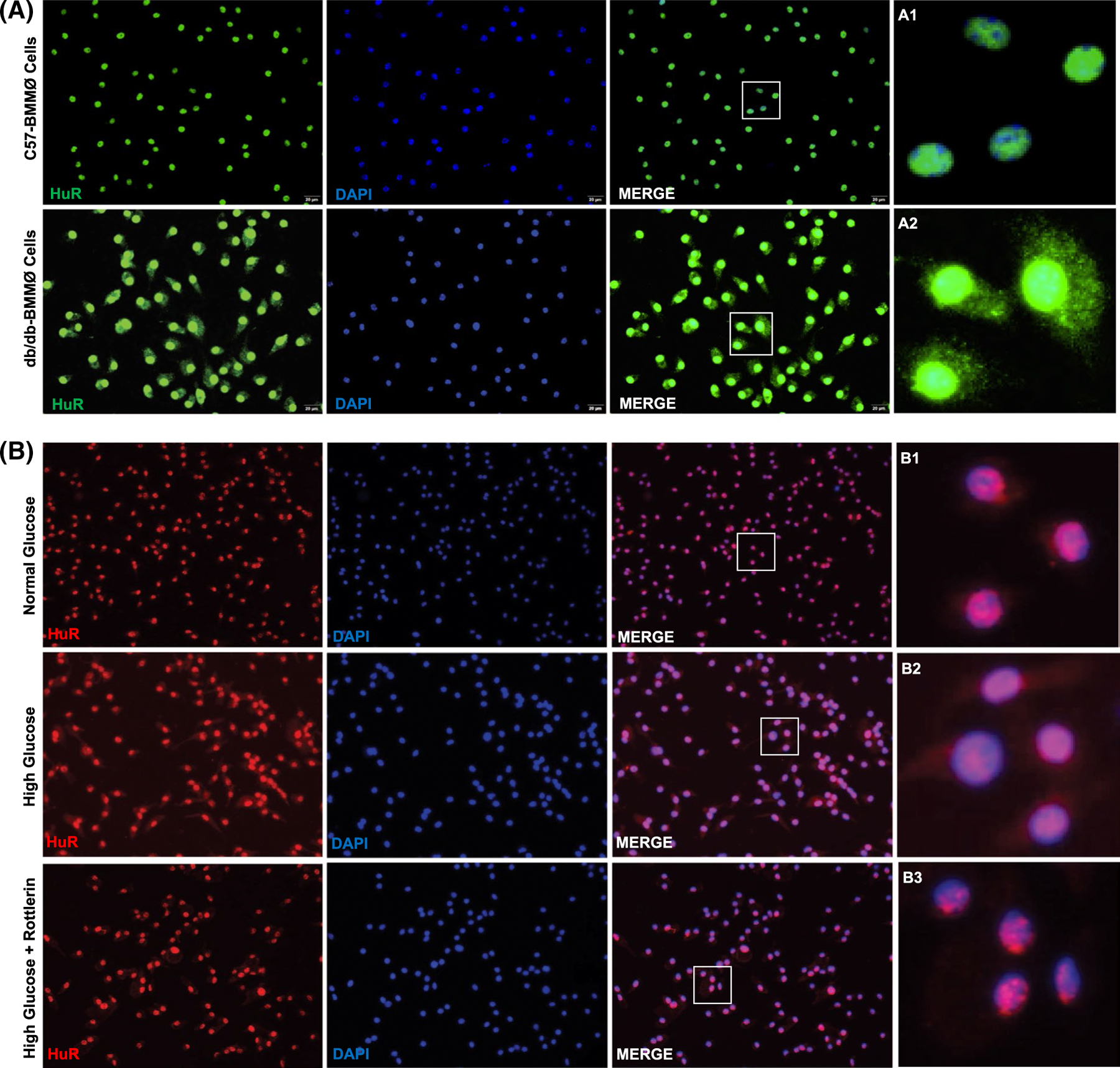
HuR cytoplasmic translocation in mouse bone marrow-derived macrophages. A, Immunofluorescence staining of bone marrow-derived macrophages from diabetic db/db mouse (db/db-BMMØ) showing HuR cytoplasmic translocation, compared with BMMØ from non-diabetic (C57BL/6J) mice. B, Immunofluorescence staining of BMMØ showing nuclear export of HuR in high glucose-treated cells, while PKCδ inhibition (Rottlerin, 10 μM) reverses HuR shuttling. The inset in each panel represents a closer view of the indicated region (A1–2 and B1–3); scale bar, 20 μm
Furthermore, we confirmed the above findings in RAW 264.7 cells (mouse macrophage cell line) treated with HG (30 mM). There was a significant increase in HuR mRNA expression (Figure 4A, *P < .05) and protein levels (Figure 4B,C, *P < .05) in HG vs NG (5 mM)-treated cells. HuR shuttling was evaluated in cellular fractions using Western blotting (Figure 4D,E) and IF staining (Figure 4F). Cells treated with HG show higher cytoplasmic translocation or HuR, in addition to increase in nuclear HuR protein levels (Figure 4D,E, *P < .05; Figure 4F).
FIGURE 4.
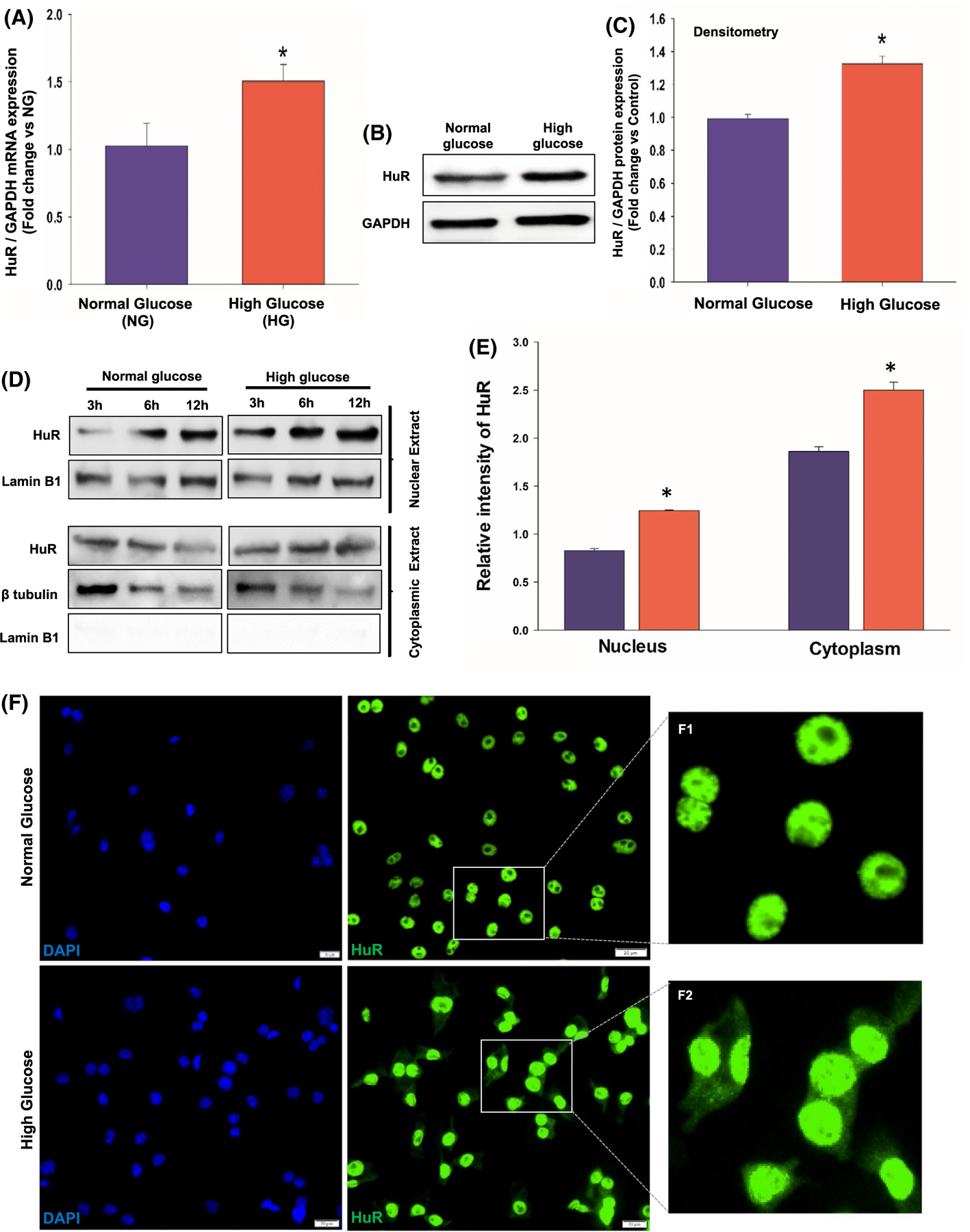
High glucose treatment upregulates HuR expression and cytoplasmic translocation in mouse macrophage cell line (RAW 264.7 cells). qRT-PCR data (A) and Western blotting (B)/densitometry analysis (C) showing increase in HuR mRNA and total cellular HuR protein expression in high glucose-treated RAW 264.7 cells, compared with normal glucose-treated cells. Data normalized to GAPDH expression. n = 3, *P < .05. Western blotting and densitometry analysis (D and E) showing increase in both nuclear HuR protein and translocation into cytoplasmic compartment in high glucose-treated cells. n = 3, *P < .05. F, Immunofluorescence staining confirming HuR cytoplasmic translocation in high glucose-treated cells. The inset in each panel represents close view of indicated region (F1–2); scale bar, 20 μm
3.3 |. Exosomes from high glucose-treated macrophage induce inflammatory and fibrogenesis genes in fibroblasts, in vitro
Chronic inflammation due to macrophage dysfunction and fibroblast activation are characteristics of diabetic heart disease.21,27 To determine whether diabetes-induced HuR increase and cytoplasmic translocation in macrophage contribute to fibrosis through paracrine mechanism, we evaluated the effect of exosomal HuR transfer on inflammation and fibrogenesis response, in vitro. Exosomes were isolated from conditioned media of macrophages exposed 48 hours to HG or NG conditions (Exo-HG or Exo-NG) and transferred to fibroblasts (NIH3/T3 cells), and after 24 hours, inflammatory and fibrogenesis response was assessed. Dynamic light scattering (Figure 5A) and transmission electron microscopy (Figure 5B) demonstrate particle density, size, and cup-shaped morphology that are characteristic of exosomes. Western and dot blot analysis shows expression of exosome markers such as HSP70, Annexin V, CD63, EpCAM, Flot1, and TSG101 (Figure 5C). Most interestingly, qRT-PCR and Western blot analyses of exosomes show transfer of HuR mRNA and proteins into exosomes (Figure 5D,E). Exosomal HuR mRNA expression was significantly higher in exosomes from HG-treated macrophages (Figure 5D, *P < .01). To demonstrate the effect of macrophage exosome on target cell function, we first evaluated exosome uptake by fibroblast cells. Exosomes isolated from conditioned media of macrophages were PKH26-labeled and incubated with fibroblast cells for 24h. Immunofluorescence staining shows uptake of macrophage exosomes (PKH26-labeled; red) by fibroblasts (Flash Phalloidin stained; green) (Figure 5F). Intriguingly, qRT-PCR data show that fibroblast co-cultured with exosomes from HG-treated macrophages (Exo-HG) significantly upregulates mRNA expression of several inflammatory and fibrogenesis signaling molecules such as IL-1β, TGF-β, MMP9, Col1A1, COL1A2, and Col3A1 as compared to fibroblasts treated with Exo-NG (Figure 5G, *P < .01). On the contrary, MMP-2 was not different between the groups (data not shown).
FIGURE 5.
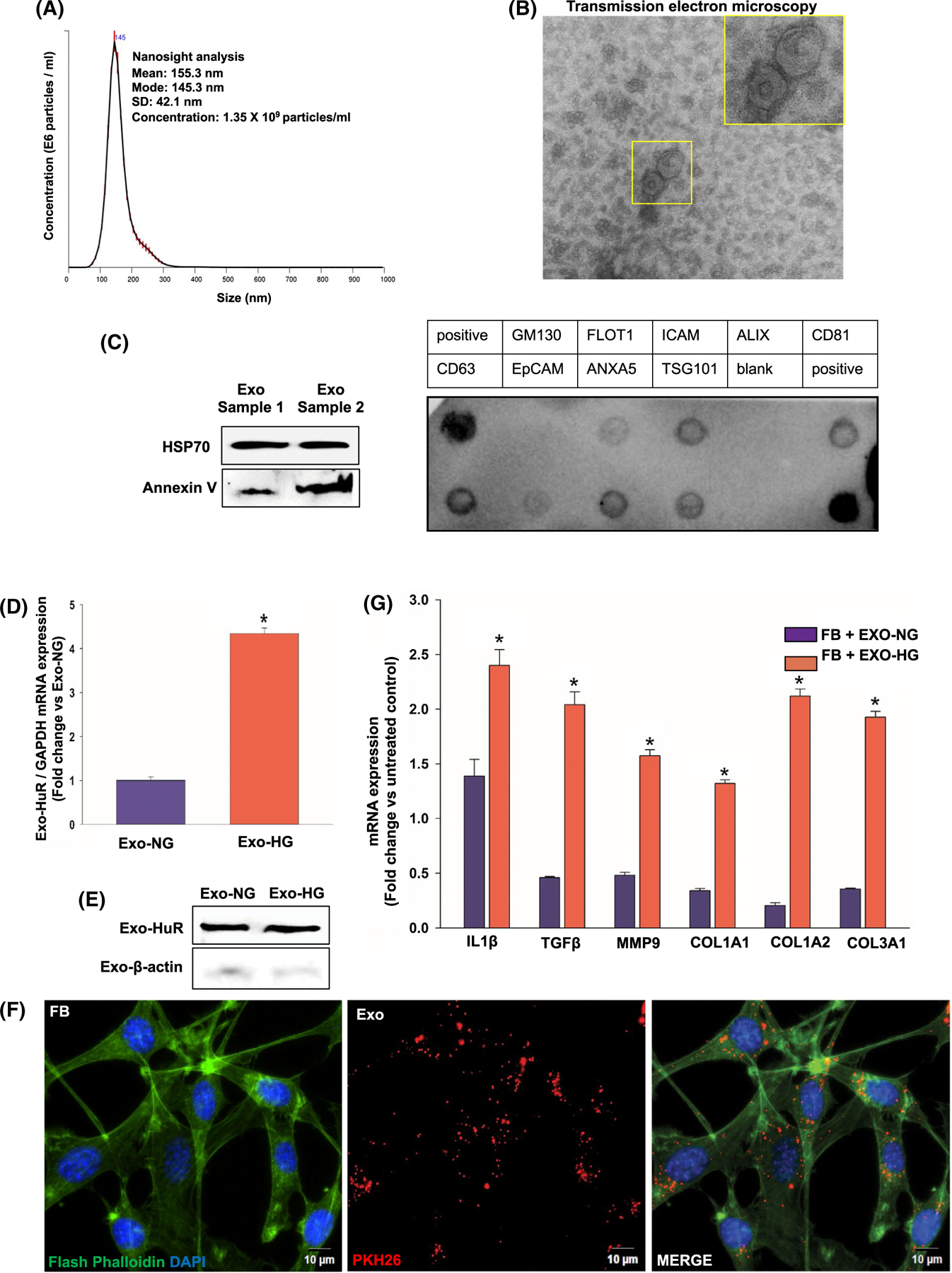
Uptake of exosomes from high glucose-treated macrophages upregulates expression of inflammatory and fibrogenesis genes in fibroblasts. Characterization of exosomes from macrophage cell line (RAW 264.7 cells) by NanoSight dynamic light scattering analysis (A), transmission electron microscopy (B), and Western blotting and dot blot for exosome markers (C). Inset in panel B represents a closer view of the indicated region, scale bar, 100 μm. D, qRT-PCR data showing higher HuR mRNA levels in exosomes of high glucose-treated cells, compared with exosomes from normal glucose-treated cells (normalized to GAPDH, *P < .05). E, Western blot to show presence of HuR protein in macrophage exosome. F, Immunofluorescence staining showing uptake of PKH26-labeled macrophage exosomes (red) by fibroblasts (FB; NIH/3T3 fibroblast cell line; Flash Phalloidin green staining). DAPI to stain nuclei (blue); scale bar, 10 μm. G, qRT-PCR showing increase in mRNA expression of inflammatory and fibrogenesis-related genes in FB co-cultured with exosomes derived from high glucose-treated macrophages (Exo-HG), compared to FB treated with exosomes derived from normal glucose-treated macrophages (Exo-HG). Data normalized to β-actin expression. n = 3, *P < .01
3.4 |. HuR deficiency abrogates Exo-HG-induced inflammatory and fibrogenesis response, in vitro
To confirm the relevance of paracrine mechanism of HuR-mediated effects in target cells, we performed HuR loss-of-function studies. We knockdown HuR in macrophage cell line (RAW 264.7 cells) using lentiviral-based shRNA against HuR, treated with HG, isolated and transferred exosomes to fibroblasts, and evaluated its effects on inflammatory and fibrogenesis signaling. First, we confirmed HuR knockdown in cells treated with shRNA against HuR (HuR KD) in comparison to cells treated with non-specific control shRNA (wild-type, WT), using Western blot analysis (Figure 6A,B, *P < .01) and immunostaining (Figure 6C). Second, we observed that HuR deficiency in exosomes did not alter fibroblast uptake of exosomes (data not shown). Interestingly, as compared to fibroblasts (FB) incubated with HG-treated wild-type macrophage-derived exosomes (ExoWT), fibroblasts incubated with HuR-deficient exosomes from HG-treated macrophages (ExoHuR KD) show reduction in mRNA expression of IL1β, TNFα, TGFβ, MMP9, COL1A1, COL1A2, COL3A1, and αSMA, suggesting lower inflammatory and fibrogenesis response (Figure 6D, *P < .05). Furthermore, Western blot analyses show that FB treated with ExoHuR KD reduced protein expression of MMP13 and MMP9 as compared to FB incubated with ExoWT (Figure 6E,F, *P < .01). These data suggest that diabetes-stimulated HuR function in macrophages and its subsequent transfer into exosomes might alter myocardial inflammation and fibrosis response.
FIGURE 6.
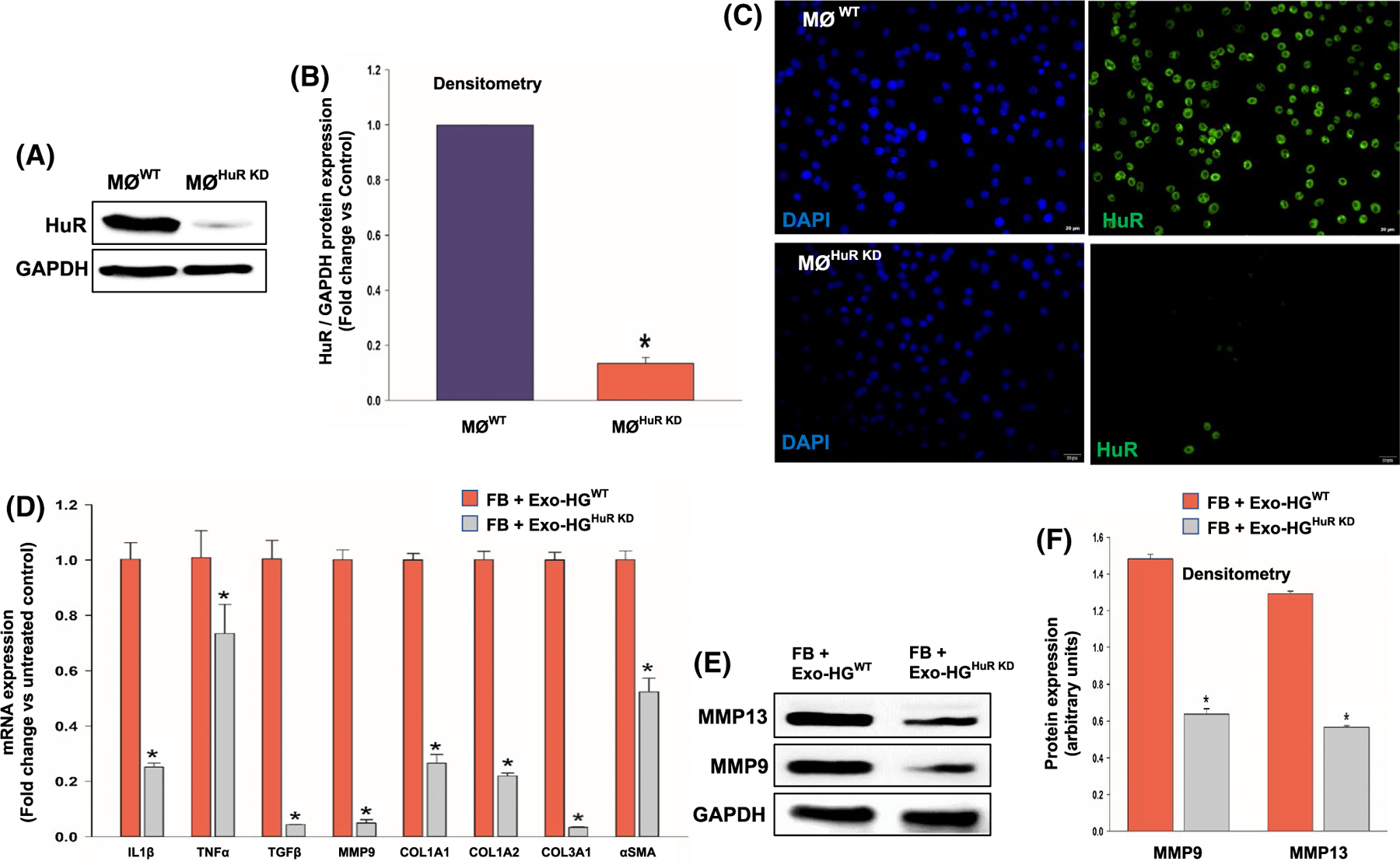
Exosomes from high glucose-treated HuR-deficient macrophage abrogate inflammatory and fibrogenesis response in fibroblasts. Western blotting/densitometry analysis (A and B) and immunofluorescence staining (C) show efficient knockdown of HuR in mouse macrophage cell line (RAW 264.7) after 48 hours of lentiviral-based shRNA transduction. HuR KD, macrophage cells transduced with HuR-specific shRNA; WT, macrophage cells transduced with non-specific control shRNA. Scale bar, 20 μm. D, qRT-PCR analysis showing downregulation of mRNA related to inflammatory and/or fibrogenesis genes in fibroblasts (NIH3T3 cells) when co-cultured with exosomes from high glucose-treated HuR-deficient macrophage cells (HG-Exo-HGHuR KD). Data normalized to β-actin mRNA. (E and F) Western blot and densitometry analysis showing similar trend for MMP-9 and MMP13 proteins, data normalized to GAPDH. n = 3, *P < .01
3.5 |. Injection of BMMØ-exosome from diabetic mice induces cardiac fibrosis and dysfunction, while HuR inhibition and deficiency limit pathology
To demonstrate the direct in vivo effect of exosome-associated HuR on cardiac pathology, we examined the effect of exosomes from diabetic (db/db) mice-derived BMMØ macrophage, with/without HuR (Exo-db/dbHuR KD vs Exo-db/dbWT), on ANG-II-induced cardiac fibrosis and function in non-diabetic (C57BL/6J mice). Western blot analysis confirms that mouse BMMØ cells treated with lentiviral-shRNA against HuR show efficient knockdown (BMMØHuR KD) as compared to non-specific control shRNA-treated cells (BMMØWT; Figure 7A,B, *P < .01; 48h after viral infection). Before injecting db/db-exosomes into the non-diabetic (C57BL/6J) mice, to limit the influence of host macrophages, we depleted macrophages by administering clodronate liposomes. Flow cytometry data confirm in vivo depletion of mouse splenic macrophages (Figure 7C,D, *P < .01). After four weeks of ANG II infusion, Exo-db/dbWT-injected mice display significant interstitial and perivascular fibrosis in the heart; however, interestingly, mice receiving HuR-deficient db/db-Exo (Exo- db/dbHuR KD) show a decrease in cardiac fibrosis (Figure 8A-C, *P < .01). Furthermore, reduced fibrosis is associated with preserved cardiac function (higher %EF and FS%) (Figure 8D,E, *P < .05).
FIGURE 7.
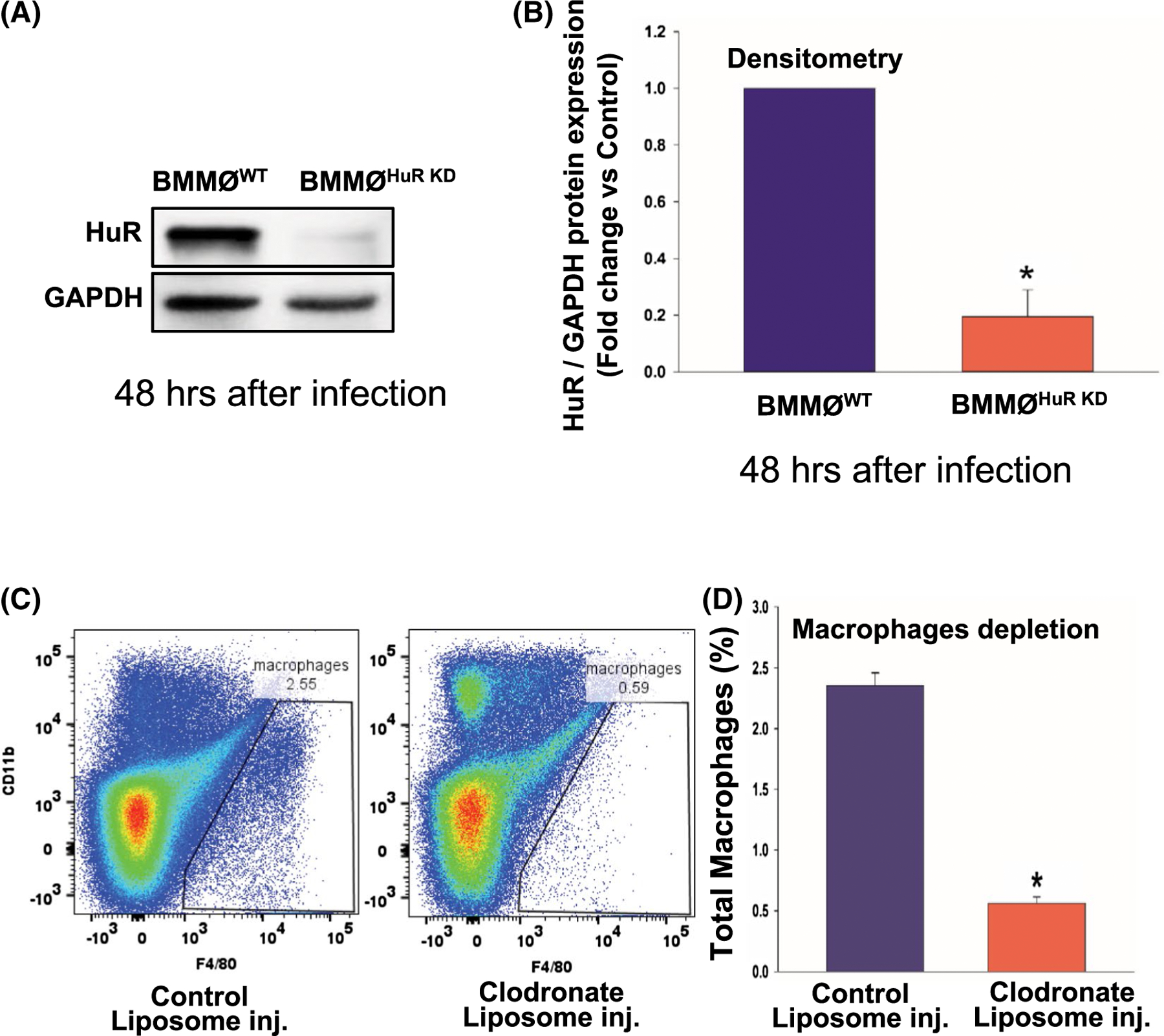
Ex vivo HuR knockdown in mouse BMMØ and in vivo macrophage depletion in mouse. A,B, Western blot and densitometry analysis showing HuR knockdown in mouse bone marrow-derived macrophage cells using lentiviral HuR-specific shRNA (BMMØHuR KD), 48 hours after infection. BMMØWT, macrophages transduced with control non-specific shRNA. Data normalized to GAPDH. C,D, Flow cytometry analysis showing depletion of mouse spleen macrophages after 48 hours of clodronate liposome administration. n = 3, *P < .01
FIGURE 8.
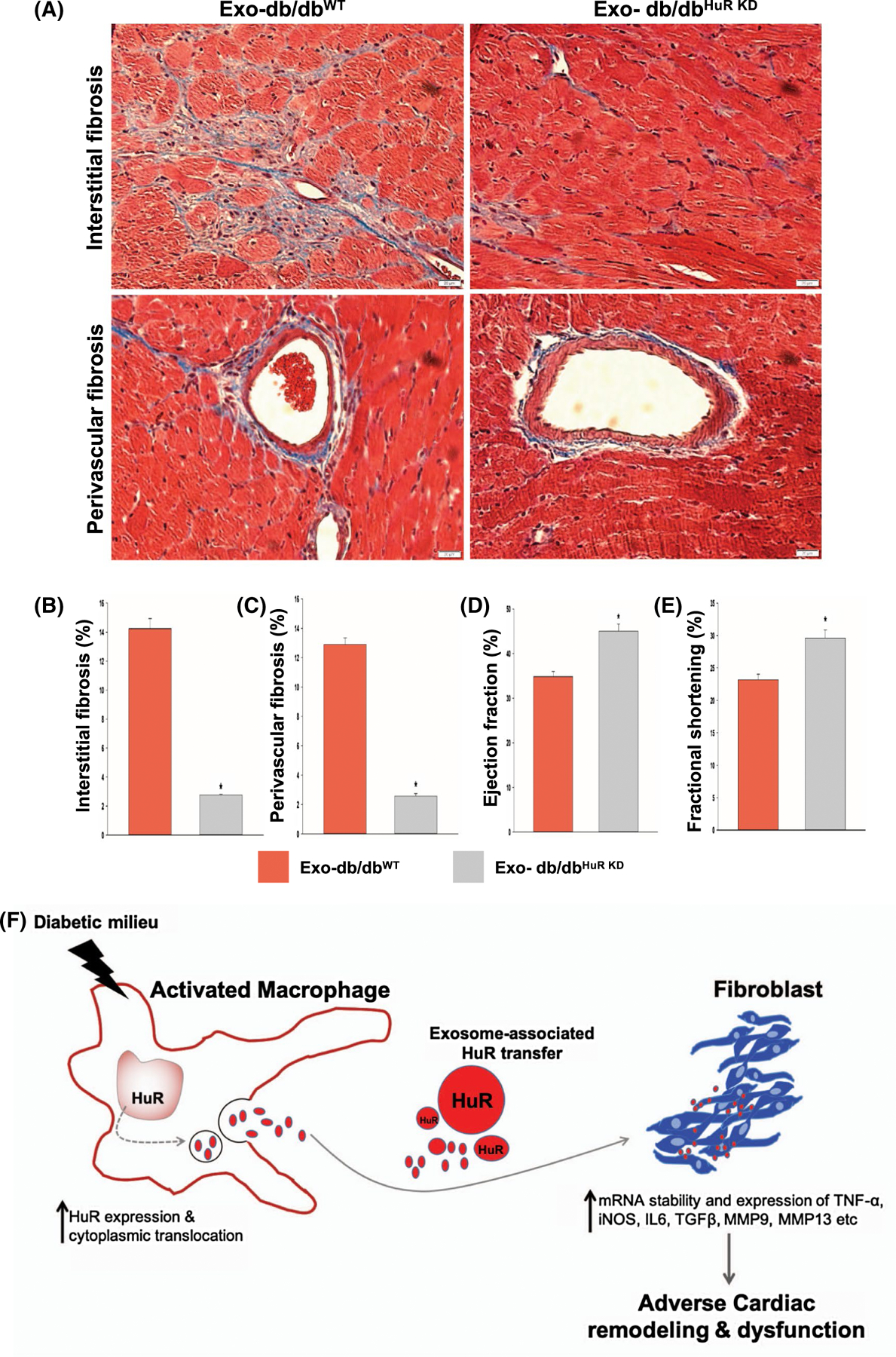
Injection of diabetic (db/db) BMMØ-exosomes with HuR deficiency limits ANG-II-induced cardiac fibrosis in mice. A-C, Masson’s trichrome staining and analysis showing lower perivascular and interstitial fibrosis in hearts or mice receiving diabetic (db/db) BMMØ-exosomes with HuR deficiency (Exo-db/dbHuR KD), as compared to db/db-Exo injected mice. Twenty-eight days post-administration. n = 3, *P < .01; scale bar, 20 μm. D,E, Echocardiography analysis showing improvements in heart function as shown by higher % ejection fraction and % fractional shortening in mice receiving Exo-db/dbHuR KD. n = 3, *P < .05. F, Schematic model depicting Exosome-HuR regulation of fibrogenesis in diabetic heart failure. Diabetes increases HuR expression and cytoplasmic translocation in activated macrophages. The secreted exosome-associated HuR is then transferred into fibroblasts. The macrophage-derived HuR increases mRNA stability and expression of inflammatory and fibrogenesis-related signals thus leading to cardiac fibrosis and dysfunction
4 |. DISCUSSION
Diabetes increases the risk for cardiovascular diseases (CVD), and the underlying causes include aberrant inflammation, loss of cardiomyocytes, microvasculature abnormalities, and adverse remodeling due to excessive fibrosis.21,23 Diabetic milieu adversely affects macrophage function, thus altering the pathogenesis of CVD and its outcome.22 Dysfunctional macrophages not only prolong inflammatory response, but also promote left ventricular fibrotic remodeling possibly via secretion of fibrogenic mediators such as exosomes. However, key molecular players remain elusive. mRNA biogenesis and stability/degradation have been shown to modulate numerous cellular processes involved in cancer, inflammation, cell death, proliferation etc14,28–30 Our previous study has shown that IL-10 (anti-inflammatory cytokine)-mediated suppression of human antigen R (HuR, an mRNA stabilizing protein) attenuates MI-induced cardiac remodeling.12 In this perspective, the influence of diabetic milieu on tissue macrophage and fibroblasts cross talk via exosome transfer of HuR and its implications on cardiac remodeling has never been studied. To our knowledge, this is first such attempt. We hypothesize that diabetes stimulates HuR activity in macrophages and its paracrine transfer via exosomes into fibroblast promotes excessive inflammation and fibrogenesis response leading to cardiac remodeling and dysfunction (Schematic model shown in Figure 8F). In this current study, we made several seminal observations that include (a) elevated HuR levels in ischemic failing human hearts, (b) db/db mice show increase in HuR levels in infiltrating cardiac- and bone marrow-derived macrophages, (c) transfer of exosome-HuR from diabetic milieu-influenced macrophages to fibroblasts activates inflammatory and fibrogenesis signaling, (d) knockdown of HuR in macrophages attenuates diabetic milieu-induced inflammation and fibrogenesis response, both in vitro and in vivo, and (e) intravenous administration of HuR-deficient exosomes attenuates fibrosis and improves left ventricular function in a mouse model of ANG-II-induced cardiac fibrosis and remodeling.
Exosomes have recently emerged as important paracrine mediators of intracellular communication and thus serve as either cause or effect of numerous pathophysiological conditions.31 For example, cardiac fibroblast-derived EV-miR-146a negatively impacts cardiomyocyte contractility.32 However, little is known about how mRNA stabilizing proteins are packaged with in the exosomes or how diabetes might influence the process. Under diabetic milieu, cardiomyocytes have been shown to promote cardiac endothelial cell dysfunction via transfer of exosomal miR-320.33 The present study provides direct evidence that exosome-associated HuR from diabetic macrophages when transferred to fibroblasts promote inflammation and fibrogenesis response, both in vitro and in vivo. Most importantly, HuR knockdown in diabetic macrophages reverses the adverse effects. These data suggest that repression of HuR release from macrophages may provide an anti-inflammatory and anti-fibrotic state and limit adverse cardiac remodeling and dysfunction in diabetes. However, the mode of HuR delivery to the myocardium is not clear, whether exosomes are reaching fibroblasts via infiltrating macrophages or via release of exosomes by circulating monocytes. Despite this limitation, our study might have crucial implications on cancer therapeutics as well, wherein HuR has been shown to be associated with cancer cell proliferation, metastasis, and prognosis.13,14
HuR activity is regulated through nuclear-cytoplasmic shuttling in the cell.24 Increased activity of HuR (mRNA binding and stability) plays critical role in various chronic diseases including cancer and cardiovascular pathophysiology.9,12,19 We have previously shown that myocardial infarction in mice upregulates myocardial HuR expression and IL-10 administration suppresses HuR expression, decreases TNF-α, TGF-β, and MMP-9 mRNA levels, and reduces MI-induced myocardial inflammation and dysfunction.12,19 In addition, HuR has been shown to regulate cardiomyocyte pyroptosis in vitro.10 Consistent with previous reports, we demonstrate an increase in HuR levels in ischemic human hearts. In addition, most importantly, we also show that diabetic milieu stimulates HuR expression and cytoplasmic translocation in BMMØ from db/db mice, in high glucose-treated cells, and in diabetic hearts. These data highlight that HuR expression and activity are a critical mediator of diabetes-induced inflammatory and fibrosis responses.
The cellular source of HuR in vivo has been elusive. We have previously shown that HuR as an independent downstream target of IL-10 and HuR expression increases after myocardial infarction with particularly higher levels in IL-10 knockout mice.19 Furthermore, although HuR expression is ubiquitous, as compared to cardiomyocytes, the expression was prominent in the border zone of myocardial infarct in cells presumably infiltrating cells. In the present study, diabetic (db/db) mice show an increase in the number of HuR+ and F4/80+ cells in the myocardium as compared to non-diabetic mice. Most importantly, in vivo study involving administration of exosomes from HuR-deficient macrophages shows a reduction in fibrosis response in the myocardium. These data suggest the importance of macrophage-associated HuR in mediating diabetes-induced LV remodeling and dysfunction. In conclusion, this study demonstrates a novel regulatory mechanism of diabetic cardiac fibrosis and dysfunction whereby cardiac macrophages influence inflammation and fibrosis signaling via exosome-associated transfer of HuR (Figure 8F), suggesting that targeting HuR might serve as a potential novel strategy to limit cardiac fibrosis in diabetics.
ACKNOWLEDGMENTS
This work is supported, in part, by the National Institutes of Health (NIH) grants HL116729 (to PK), HL138023 (to PK and JZ), HL126186 & HL134608 (to RK), and American Heart Association Grant-in-aid GRNT 25860041 (to PK) and Transformational Project Award 19TPA34850100 (to PK).
Funding information
HHS | National Institutes of Health (NIH), Grant/Award Number: HL116729; HHS | National Institutes of Health (NIH), Grant/Award Number: HL138023; HHS | National Institutes of Health (NIH), Grant/Award Number: HL126186; HHS | National Institutes of Health (NIH), Grant/Award Number: HL134608; American Heart Association (AHA), Grant/Award Number: GRNT25860041; American Heart Association (AHA), Grant/Award Number: 19TPA34850100
Abbreviations:
- ANG II
angiotensin II
- BMMØ
bone marrow macrophage
- Exo
exosomes
- HG
high glucose (30 mM)
- HuR
human antigen R
- KD
knockdown
- NG
normal glucose (5 mM)
- WT
wild-type
Footnotes
CONFLICT OF INTEREST
None. All the authors have reported “nothing to disclose.”
REFERENCES
- 1.Roper NA, Bilous RW, Kelly WF, Unwin NC, Connolly VM. Cause-specific mortality in a population with diabetes: South Tees Diabetes Mortality Study. Diabetes Care 2002;25(1):43–48. [DOI] [PubMed] [Google Scholar]
- 2.Chen B, Frangogiannis NG. Macrophages in the remodeling failing heart. Circ Res 2016;119(7):776–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 2007;204(12):3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hulsmans M, Sager HB, Roh JD, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med 2018;215(2):423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014;71(4):549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honold L, Nahrendorf M. Resident and monocyte-derived macrophages in cardiovascular disease. Circ Res 2018;122(1):113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res 2015;116(7):1269–1276. [DOI] [PubMed] [Google Scholar]
- 8.Suresh Babu S, Joladarashi D, Jeyabal P, Thandavarayan RA, Krishnamurthy P. RNA-stabilizing proteins as molecular targets in cardiovascular pathologies. Trends Cardiovasc Med 2015;25(8):676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotta-Loizou I, Vasilopoulos SN, Coutts RHA, Theocharis S. Current evidence and future perspectives on HuR and breast cancer development, prognosis, and treatment. Neoplasia 2016;18(11):674–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeyabal P, Thandavarayan RA, Joladarashi D, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun 2016;471(4):423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slone S, Anthony SR, Wu X, et al. Activation of HuR downstream of p38 MAPK promotes cardiomyocyte hypertrophy. Cell Signal 2016;28(11):1735–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res 2009;104(2):e9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janakiraman H, House RP, Talwar S, et al. Repression of caspase-3 and RNA-binding protein HuR cleavage by cyclooxygenase-2 promotes drug resistance in oral squamous cell carcinoma. Oncogene 2017;36(22):3137–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wigington CP, Jung J, Rye EA, et al. Post-transcriptional regulation of programmed cell death 4 (PDCD4) mRNA by the RNA-binding proteins human antigen R (HuR) and T-cell intracellular antigen 1 (TIA1). J Biol Chem 2015;290(6):3468–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schorey JS, Cheng Y, Singh PP, Smith VL. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep 2015;16(1):24–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel B, Patel J, Cho J-H, et al. Exosomes mediate the acquisition of the disease phenotypes by cells with normal genome in tuberous sclerosis complex. Oncogene 2016;35(23):3027–3036. [DOI] [PubMed] [Google Scholar]
- 17.Weischenfeldt J, Bone PB. Marrow-derived macrophages (BMM): isolation and applications. CSH Protoc 2008;2008:pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 18.Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008;57(12):3297–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnamurthy P, Lambers E, Verma S, et al. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J 2010;24(7):2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Adamiak W, Huang G, et al. Interaction of RNA-binding protein HuR and miR-466i regulates GM-CSF expression. Sci Rep 2017;7(1):17233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol 2016;90:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parathath S, Grauer L, Huang L-S, et al. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes 2011;60(6):1759–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szuszkiewicz-Garcia MM, Davidson JA. Cardiovascular disease in diabetes mellitus: risk factors and medical therapy. Endocrinol Metab Clin North Am 2014;43(1):25–40. [DOI] [PubMed] [Google Scholar]
- 24.Fan XC, Steitz JA. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci USA 1998;95(26):15293–15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doller A, Schlepckow K, Schwalbe H, Pfeilschifter J, Eberhardt W. Tandem phosphorylation of serines 221 and 318 by protein kinase Cdelta coordinates mRNA binding and nucleocytoplasmic shuttling of HuR. Mol Cell Biol 2010;30(6):1397–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doller A, Akool E-S, Huwiler A, et al. Posttranslational modification of the AU-rich element binding protein HuR by protein kinase Cdelta elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol Cell Biol 2008;28(8):2608–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage phenotypes regulate scar formation and chronic wound healing. Int J Mol Sci 2017;18(7):1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Díaz-Muñoz MD, Turner M. Uncovering the role of RNA-binding proteins in gene expression in the immune system. Front Immunol 2018;9:1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas M, Liu X, Whangbo J, et al. Apoptosis triggers specific, rapid, and global mRNA decay with 3′ uridylated intermediates degraded by DIS3L2. Cell Rep 2015;11(7):1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.García-Martínez J, Delgado-Ramos L, Ayala G, et al. The cellular growth rate controls overall mRNA turnover, and modulates either transcription or degradation rates of particular gene regulons. Nucleic Acids Res 2016;44(8):3643–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathiyalagan P, Adamiak M, Mayourian J, et al. FTO-dependent m6A regulates cardiac function during remodeling and repair. Circulation 2019;139(4):518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh JG, Watanabe S, Lee A, et al. miR-146a suppresses SUMO1 expression and induces cardiac dysfunction in maladaptive hypertrophy. Circ Res 2018;123(6):673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Huang W, Liu G, et al. Cardiomyocytes mediate an-ti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol 2014;74:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]