

Abstract
Objective
This study aimed to compare the pharmacokinetic, pharmacodynamic and safety profiles of HLX01 (a rituximab biosimilar) and reference rituximab sourced from China (MabThera®; rituximab-CN).
Methods
Here we report the results of two phase 1 studies. In the phase 1a, open-label, dose-escalation study (NCT03218072, CTR20140400), eligible patients received 250, 375 and 500 mg/m2 HLX01 sequentially at 7-day intervals, after confirming no dose-limiting toxicity (DLT). In the phase 1b, double-blind study (NCT02584920, CTR20140764), eligible patients were given a single dose of 375 mg/m2 HLX01 or rituximab-CN. The primary endpoints included safety and tolerability parameters for the phase 1a and the area under the plasma concentration-time curve from time zero to day 91 (AUC0−91 d) for the phase 1b study. Equivalence was concluded if 90% confidence interval (90% CI) for the geometric least squares mean ratio (GLSMR) fell in the pre-specified equivalence criteria (80%−125%).
Results
Between June 20, 2014 and January 5, 2015, 12 patients were enrolled in the phase 1a study. The pharmacokinetics of HLX01 showed dose proportionality and accumulation to steady state. HLX01 was well tolerated, with no serious adverse events (AEs), discontinuations or DLTs. Between November 8, 2014 and August 13, 2015, 87 eligible patients were enrolled in the phase 1b study, including 43 who received HLX01 and 44 who were treated with rituximab-CN. The equivalence endpoint was met with GLSMR for AUC0−91 d being 89.6% (90% CI: 80.4%−99.8%). AEs, anti-drug antibodies, and CD19+ and CD20+ B lymphocyte counts were similar between the HLX01 and rituximab-CN treatment groups.
Conclusions
Treatment with HLX01 was safe and well tolerated in Chinese patients with B-cell lymphoma. HLX01 and rituximab-CN have similar pharmacokinetic, pharmacodynamic and safety profiles.
Keywords: Biosimilar, lymphoma, monoclonal antibody, pharmacokinetics, safety
Introduction
Non-Hodgkin lymphoma (NHL) is the most common adult hematological cancer worldwide, with 85% of cases derived from B lymphocytes (1-3). As the most prevalent form of NHL, diffuse large B-cell lymphoma (DLBCL) accounts for approximately one third of lymphoid malignancies in Western countries and in China (4-6).
Rituximab is a chimeric mouse/human anti-CD20 monoclonal antibody, which has been approved for the treatment of B-cell malignancies for over two decades. CD20 is a valuable therapeutic target in B-cell NHL, since it is expressed as a B-cell-specific cell surface antigen of pre-B and mature B lymphocytes, occurring in more than 90% of B-cell lymphomas (7).
Improved clinical outcomes for patients with B-cell malignancies treated with rituximab have been confirmed in practice, and this treatment option represents a significant advance in the treatment of B-cell NHL, both as monotherapy and in combination with chemotherapy regimens (7-10). Rituximab maintenance therapy also provides survival benefits to patients with follicular lymphoma and mantle cell lymphoma (11,12).
Whilst rituximab provides clear clinical benefit, patients’ access to this product is restricted due to its high costs in China (13). HLX01, the first biosimilar product manufactured in China, was approved by the National Medical Products Administration (NMPA) on February 22, 2019 (14,15). It was developed in accordance with the NMPA and the World Health Organization similar bio-therapeutic product development guidelines. Similarities between HLX01 and reference rituximab in their physicochemical properties and biological activities have previously been demonstrated (16).
An approval of biosimilar products requires demonstration of similar efficacy and safety between biosimilar and reference drug (17). It is necessary to carefully select endpoints to ensure that differences between biosimilar and reference drug are the main focus, while evaluating the impacts of other factors such as tumor burden, previous treatment and underlying clinical conditions. The stepwise approach of biosimilar development and demonstration of similar efficacy and safety requires demonstration of equivalence in pharmacokinetics (PK) and pharmacodynamics (PD) in clinical trials.
Here we report the findings of two phase 1 (1a and 1b) studies of HLX01. Phase 1a study assessed PK, PD, safety and tolerability with escalating and then multiple doses of HLX01. Phase 1b study was a single-dose, multicenter, randomized, double-blind, parallel-controlled study that compared PK, PD, safety and immunogenicity profiles between HLX01 and reference rituximab sourced from China (MabThera®; rituximab-CN). It was the first PK/PD study of reference rituximab in Chinese patients.
Materials and methods
HLX01 and rituximab-CN
Reference rituximab (MabThera®) was purchased from Shanghai Roche Pharmaceuticals Ltd, China. HLX01 [(Han-Li-Kang®) Rituximab Injection] was manufactured by Shanghai Henlius Biotech, Inc., China.
Study design and ethics
Phase 1a study (NCT03218072, CTR20140400) was an open-label, dose-escalation study conducted at two sites in China. Patients were enrolled sequentially into one of three dose-escalating cohorts (250, 375 and 500 mg/m2). Following the initial infusion of HLX01, patients were monitored for seven days through the recording of vital signs, laboratory tests and safety indicators. Enrolment into the sequential, dose-escalating cohort was only permitted if the incidence of dose-limiting toxicities (DLTs) during the monitoring period occurred in less than one-third of participants. In the absence of DLTs, patients in each cohort received further three doses of HLX01, administered weekly. The total observation period was 13 weeks from the initial dose. The primary objective of the phase 1a study was to evaluate the safety and tolerability of single and multiple escalating intravenous (IV) doses of HLX01.
The phase 1b study (NCT02584920, CTR20140764) was a randomized, double-blind, parallel-controlled study conducted at 12 sites in China (Supplementary Table S1 ). Patients were randomized 1:1 to receive a single IV dose of HLX01 (375 mg/m2) or rituximab-CN (375 mg/m2) and were observed for up to 13 weeks (91±3 d). The primary endpoint was the area under the plasma concentration-time curve (AUC) from time zero to day 91 (AUC0−91 d). Equivalence was achieved if 90% confidence interval (90% CI) of the geometric least square mean ratio (GLSMR) of AUC0−91 d fell in the pre-specified equivalence margins of 80%−125%. Secondary endpoints included AUC from time zero to week 1, 2, 4 and 8, AUC from time zero to infinity (AUC0−inf) and maximum drug concentration (Cmax). Other objectives were to compare the PD parameters of HLX01 and rituximab-CN, and evaluate the safety and immunogenicity of HLX01 maintenance therapy.
Table S1. Phase 1b study sites.
| No. | Study sites |
| 1 | National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College |
| 2 | Peking University Third Hospital |
| 3 | Peking University People’s Hospital |
| 4 | Harbin Medical University Cancer Hospital |
| 5 | Tang Du Hospital of the Fourth Military Medical University |
| 6 | Fudan University Shanghai Cancer Centre |
| 7 | The First Affiliated Hospital, College of Medicine, Zhejiang University |
| 8 | The Second Affiliated Hospital, College of Medicine, Zhejiang University |
| 9 | The 307th Hospital of the Chinese People’s Liberation Army |
| 10 | Jiangsu Cancer Hospital |
| 11 | The First Affiliated Hospital of Soochow University |
| 12 | The Second Affiliated Hospital of Soochow University |
The study was conducted in accordance with the Declaration of Helsinki, and applicable national and local regulations for clinical trials. It received approval from independent ethics committees at each participating center, and written informed consent was obtained from all patients who participated in this study prior to screening.
Patients
Eligible patients were adults aged 18−65 years, with histologically diagnosed CD20+ B-cell lymphoma. Patients in phase 1a study had relapsed/refractory B-cell lymphoma requiring consolidation therapy, or chronic lymphocytic leukemia with a peripheral leukocyte count ≤50×109/L. Patients who had achieved complete remission (CR) or unconfirmed CR (CRu) after prior treatment, had peripheral leukocyte counts ≤5×109/L and were considered by the Investigator as likely to benefit from further maintenance treatment, were enrolled in the phase 1b study. Key exclusion criteria for both studies were primary or secondary immunodeficiency, and use of hematopoietic growth factors within one week of enrollment or live vaccines within four weeks of enrollment. The cut-off periods for patients who had received prior rituximab were two years (phase 1a) or four months (phase 1b) before study enrollment. For the phase 1b study, a threshold for residual serum rituximab ≤24 µg/mL was applied at screening (full inclusion and exclusion criteria are detailed in Supplementary Table S2 ).
Table S2. Inclusion and exclusion criteria of phase 1a study and phase 1b study.
| Phase 1a
study |
Phase 1b
study |
Criteria |
| CLL, chronic lymphocytic leukemia; ECOG, Eastern Cooperative Oncology Group; PS, performance status; ULN, upper limit of normal; HBV, hepatitis B virus, DNA, deoxyribonucleic acid; HIV, human immunodeficiency virus; mAb, monoclonal antibody; ADA, anti-drug antibody. | ||
| Inclusion | ||
| Yes | No | Subjects with relapsed or refractory B-cell lymphoma, diagnosed through histopathology, requiring consolidation therapy; CLL subjects with peripheral leukocyte count ≤50×109/L. |
| Yes | Yes | Subjects with CD20+ Bcell lymphoma who had reached CR/CRu after treatment, and could further benefit from continuing treatment from the discretion of the Investigator; Peripheral blood lymphocyte count <5×10 9/L. |
| Yes | Yes | Histologically diagnosed with CD20+ B-cell lymphoma. |
| Yes | Yes | ECOG PS ≤1 at the time of enrolment, life expectancy >3 months. |
| Yes | Yes | Aged ≥18 and ≤65 years |
| Yes | Yes | Signed informed consent and voluntary participation |
| Exclusion | ||
| Yes | Yes | Diagnosed in the last 5 years with squamous cell carcinoma or cutaneous basal cell carcinoma, surgically treated malignant melanoma, or history of malignant tumors other than cervical carcinoma in situ. |
| Yes | No | Used rituximab or other anti-CD20 mAb within 2 years prior to enrolment. |
| Yes | Yes | Used hematopoietic cytokines, such as granulocyte colony-stimulating factor, within 1 week prior to enrolment. |
| Yes | Yes | Inoculation of (attenuated) live vaccine within 4 weeks prior to enrolment. |
| Yes | Yes | Major surgery in the past 8 weeks (excluding diagnostic surgical operation). |
| Yes | Yes | Peripheral nervous system or central nervous system diseases; history of shingles with sequelae or latent infections; other severe diseases possibly limiting the subject’s participation in this study (e.g., progressive infection, uncontrolled diabetes, severe cardiac insufficiency or angina, gastric ulcer, active autoimmune disease). |
| Yes | Yes | Prior to the study, a hematology test matched any of the following conditions: white blood cell <3×10 9/L; absolute neutrophil (segmented and band cell) count <1.5×10 9/L; platelets <100×10 9/L; hemoglobin <90 g/L. Subjects with bone marrow involvement with absolute neutrophil (segmented and band cell) count <1.0×10 9/L; platelets <75×10 9/L; hemoglobin <80 g/L. |
| No | Yes | Residual concentration of rituximab in the peripheral blood >24 μg/mL at screening; (Depending on subject willingness, the Investigator could recommend a time for a subject that failed screening to be re-screened at the hospital based on t 1/2=22 days). |
| Yes | Yes | Prior to the study, liver function test matched any of the following conditions: total bilirubin >1.5× normal value; alanine aminotransferase or aspartate aminotransferase >2.0×ULN; alkaline phosphatase >3×ULN; renal disorder (serum creatinine >1.5× normal value). |
| Yes | Yes | Subjects with thyroid dysfunction (thyroid stimulating hormone below lower limit of normal or above ULN and identified as clinically significant by the Investigator). |
| Yes | Yes | Positive for:
Serum HIV antigens or antibodies; Hepatitis B surface antigen or hepatitis B core antibody-positive and HBV-DNA >ULN; Serum hepatitis C virus antibodies. |
| Yes | Yes | Pregnant or lactating female subjects, or subjects unwilling to adopt contraception during the study period. |
| Yes | Yes | Allergic constitution, known allergic reaction to rituximab, or other anti-CD20 mAb drug components. |
| Yes | Yes | Subjects with a history of alcohol or drug abuse. |
| Yes | No | Subjects who participated in other clinical studies within 3 months prior to the start of this study. |
| Yes | Yes | Subjects deemed unsuitable by the Investigator. |
| No | Yes | Used chemotherapy drugs within one month prior to enrolment. |
| No | Yes | Used rituximab or other anti-CD20 mAb within 4 months prior to enrolment. |
| No | Yes | Used rituximab or other anti-CD20 mAb within 4 months prior to enrolment and ADA positive. |
Randomization and masking (phase 1b study)
Randomization was performed by an independent statistician using the PROC PLAN process (SAS software; Version 9.2; SAS Institute Inc., Cary, NC, USA). Patients were stratified into two subgroups according to the history of rituximab use. Independent personnel were responsible for completing the blind packaging of each vial of study medication with randomization codes. Double-blind masking was achieved by a dispensing nurse, who was involved in the drug administration and preparation of the medication in identical infusion containers.
Safety and tolerability
In both studies, the safety analysis set comprised all patients who received ≥1 dose of study drug. Safety and tolerability were assessed by physical examinations, laboratory measurements and occurrence of adverse events (AEs), serious AEs (SAEs) and adverse drug reactions (ADRs). AEs, including infusion-related reactions (IRRs), were classified using Medical Dictionary for Regulatory Activities Terminology (Version 16.1) and graded according to the National Cancer Institute Common Terminology Criteria for AEs (Version 4.03). All AEs were assessed for their relationship to the study drug; events that were very likely related or possibly related were defined as ADRs. DLT was defined as grade ≥4 hematologic toxicity or grade ≥3 non-hematologic toxicity, excluding IRRs.
PK evaluation
Serum concentrations of HLX01 and rituximab-CN were evaluated by validated enzyme-linked immunosorbent assay. In the phase 1a study, blood samples were collected before and immediately after each HLX01 and rituximab-CN infusion, with further samples collected following the first and fourth doses (at 2, 24, 72 and 120 h). In addition, samples were taken at 7, 14±1, 38±2 and 68±3 d after the fourth dose.
The PK analysis population for the phase 1b study comprised patients in the safety analysis set for whom AUC0−91 d could be reliably calculated and who had no major protocol deviations. Blood samples for analysis were collected within 1 h prior to study drug administration, immediately after study drug infusion and at 6, 24, 72 and 168±1 h. Further samples were collected at 14±1, 28±2, 56±3 and 91±3 d after study drug administration. The subgroup of patients that had received prior reference rituximab treatment also had samples collected at screening.
PD evaluation
The proportions and absolute counts of circulating lymphocytes were determined by flow cytometry. In the phase 1a study, testing was performed prior to study drug administration and at 72 and 168 h after the first dose of study drug. Other time points were immediately after administration of the fourth dose, and at 38±2 and 68±3 d thereafter. In the phase 1b study, samples were taken prior to study drug administration, and at 5 min, 24 h, 14±1, 56±3 and 91±3 d after study drug infusion.
Immunogenicity evaluation
The presence of anti-drug antibodies (ADAs) was assessed by electrochemiluminescent immunoassay. In the phase 1a study, ADAs were analyzed prior to the first and third doses of HLX01, immediately after the fourth dose, and at 38±2 and 68±3 d thereafter. In the phase 1b study, testing was performed prior to study drug administration and at 14, 28±2, 56±3 and 91±3 d after study drug infusion.
Efficacy evaluation
No efficacy objective was defined in the phase 1b study because the patients had achieved CR/CRu prior to inclusion. However, the study design included evaluation of efficacy endpoints, CR, and CRu to determine whether the patients’ disease was stable. Fisher’s exact test was used at week 8 post study drug infusion to assess the overall efficacy rate. Computerized tomography (CT) scans were evaluated by the Investigator according to International Working Group 1999 and 2007 to assess treatment efficacy and response.
Statistical analysis
Sample size for the phase 1b study was estimated based on equivalence of primary endpoint of AUC. The smallest sample size was estimated to be 78 (n=39 per group), aiming for the equivalence margins of 80%−125%, with a two one-sided alpha (α) of 0.05, power of 90%, coefficient variation of 0.3 and the AUC ratio (study group: control group) of 1.0.
Statistical analyses were performed using the SAS software (Version 9.2; SAS Institute Inc., Cary, NC, USA). PK parameters, including AUC from time zero to last sampling time point (AUC0−t), AUC0−inf and AUC0−91 d, were assessed via non-compartmental analysis using Phoenix WinNonlin® software (Version 6.3; Pharsight Corporation, Mountain View, California, United States). Cmax concentrations are reported as actual values. Descriptive statistics are reported for both studies and include mean, standard deviation, median and range (minimum and maximum values). AEs and ADA positivity are reported using frequency and percentages. For the phase 1b study, subgroup analyses were performed for immunogenicity, PK and PD parameters according to prior rituximab use. A sensitivity analysis of PK parameters by ADA status was also performed and the equivalence assessment was conducted in the ADA-negative and the ADA-positive (consecutive) subgroups. Inter-group comparisons of pre-treatment indicators and measurements were analyzed by t-test or Wilcoxon rank sum test, and enumeration data were analyzed by Fisher’s exact test. A P value <0.05 was considered statistically significant.
Results
Patient demographics
Between June 20, 2014 and January 5, 2015, a total of 12 patients, with relapsed/refractory B-cell lymphoma, were enrolled in the phase 1a study (Figure 1A ). All patients received the first single dose and the subsequent four doses of HLX01. Patient baseline characteristics confirmed that all cohorts had a similar distribution of Eastern Cooperative Oncology Group (ECOG) performance status (PS), tumor type and number of prior lines of chemotherapy (Table 1 ). Most patients were male (66.7%) and the median age was 51.5 (range: 31.5−64.3) years. Patients in the 375 mg/m2 cohort (n=6; 50.0% male) were younger (median age: 42.5 years; range: 31.5−51.5 years), with lower body weight compared with patients in the 250 mg/m2 and 500 mg/m2 cohorts (Table 1 ).
Figure 1.

Patient flow diagrams for (A) phase 1a study and (B) phase 1b study. AE, adverse event; GCP, Good Clinical Practice; PK, pharmacokinetics.
Table 1. Patients’ demographics and baseline characteristics (phase 1a study and phase1b study) in safety analysis set.
| Characteristics | Phase 1a study | Phase 1b study (375 mg/m2) | ||||
| 250 mg/m2 (n=3) | 375 mg/m2 (n=6) | 500 mg/m2 (n=3) | HLX01 (n=43) | Reference rituximab* (n=43) | ||
| ECOG, Eastern Cooperative Oncology Group; PS, performance status; FL, follicular lymphoma; CLL, chronic lymphocytic leukemia; SLL, small lymphocytic lymphoma; LPHL, Hodgkin’s lymphoma, classic, lymphocyte-predominant; cHL, classical Hodgkin lymphoma; DLBCL, diffuse large B cell lymphoma; MALT, mucosa-associated lymphoid tissue lymphoma; U-BCL, unclassified B cell lymphoma; U-DLBCL/BL, unclassified B cell lymphoma with feature intermediate between diffuse large B-cell lymphoma and Burkitt’s lymphoma; MCL, mantel cell lymphoma; MZL, marginal zone lymphoma; NA, not applicable; *, Reference rituximab was sourced from China (MabThera®; rituximab-CN). | ||||||
| Age [median (range)] (year) | 57.2 (51.6−59.2) | 42.5 (31.5−51.5) | 57.0 (32.1−64.3) | 51.0 (28.6−63.6) | 51.0 (24.2−65.1) | |
| Male [n (%)] | 2 (66.7) | 3 (50.0) | 3 (100) | 21 (48.8) | 21 (48.8) | |
Weight (
 ) (kg)
) (kg)
|
75.0±13.1 | 65.7±19.4 | 78.7±27.1 | 66.2±11.0 | 64.4±10.2 | |
Body surface area (
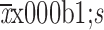 ) (m2)
) (m2)
|
1.8±0.2 | 1.7±0.3 | 1.9±0.3 | 1.7±0.2 | 1.7±0.2 | |
| ECOG PS [n (%)] | ||||||
| 0 | 3 (100) | 5 (83.3) | 3 (100) | 28 (65.1) | 29 (67.4) | |
| 1 | 0 | 1 (16.7) | 0 | 15 (34.9) | 14 (32.6) | |
| Primary tumor type [n (%)] | ||||||
| FL | 0 | 2 (33.3) | 0 | 12 (27.9) | 9 (20.9) | |
| CLL/SLL | 1 (33.3) | 0 | 0 | 2 (4.7) | 2 (4.7) | |
| LPHL | 0 | 1 (16.7) | 0 | 0 | 0 | |
| cHL | 0 | 0 | 1 (33.3) | 0 | 0 | |
| Systemic plasma-cell type CD | 0 | 1 (16.7) | 0 | 0 | 0 | |
| DLBCL | 1 (33.3) | 2 (33.3) | 1 (33.3) | 22 (51.2) | 26 (60.5) | |
| Gastric MALT | 0 | 0 | 1 (33.3) | 0 | 0 | |
| MALT | 1 (33.3) | 0 | 0 | 0 | 0 | |
| U-BCL | 0 | 0 | 0 | 2 (4.7) | 3 (7.0) | |
| U-DLBCL/BL | 0 | 0 | 0 | 0 | 1 (2.3) | |
| MCL | 0 | 0 | 0 | 2 (4.7) | 1 (2.3) | |
| MZL | 0 | 0 | 0 | 3 (7.0) | 1 (2.3) | |
| Tumor stage [n (%)] | ||||||
| I−II | 0 | 4 (66.7) | 1 (33.3) | 20 (46.5) | 13 (30.2) | |
| III−IV | 3 (100) | 2 (33.3) | 2 (66.7) | 23 (53.5) | 30 (69.8) | |
| Duration of disease [median (range)] (year) | NA | NA | NA | 0.9 (0.3−4.3) | 1.3 (0.5−6.5) | |
| Number of chemotherapy regimens [median (range)] | 2.0 (1−4) | 1.5 (0−7) | 2.0 (1−3) | NA | NA | |
| Prior chemotherapy [n (%)] | NA | NA | NA | 34 (79.1) | 34 (79.1) | |
| Prior rituximab [n (%)] | NA | NA | NA | 22 (51.2) | 21 (48.8) | |
A total of 87 patients were enrolled in the phase 1b study between November 8, 2014 and August 13, 2015, including 43 patients in the HLX01 group and 44 patients in the rituximab-CN group (Figure 1B ). Two patients discontinued prior to study completion; one patient in the HLX01 group withdrew consent and one patient in the rituximab-CN group experienced a treatment-emergent adverse event (TEAE). As one patient was excluded from the rituximab-CN group due to deviation from Good Clinical Practice guidelines, the safety analysis set comprised 86 patients. Of these patients, 81 were included in the PK analysis, 40 in the HLX01 group, and 41 in the rituximab-CN group. Five patients were excluded from the PK analysis; one had incomplete infusion data due to an IRR, three were ADA-positive prior to administration and one patient had an incomplete sample set due to withdrawn informed consent. Patients in both study groups were similar in terms of demographics and baseline disease characteristics, except that patients in the rituximab-CN arm had a slightly longer median duration of the disease compared with those in the HLX01 arm (1.3 years vs. 0.9 years) (Table 1 ). Half of the patients in the safety analysis set had received prior rituximab treatment for B-cell lymphoma.
PK
The PK characteristics of HLX01 (phase 1a) were assessed after single and multiple doses in each dose cohort (Supplementary Figure S1 , Table S3 ). The AUC0−t and Cmax showed linear correlation with increasing administered dose (Supplementary Table S3 ). The mean terminal half-life (t1/2) after a single dose of HLX01 was 124.5 h across the dose ranges. Therefore, steady state would be anticipated at approximately 498.0−622.5 h. Consistently, the mean Cmax of HLX01 after multiple doses in each cohort was higher than the mean Cmax observed after a single dose, indicating drug accumulation to steady state (data not shown).
Figure S1.

Serum concentration-time curves (
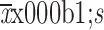 ) following (A) single dose and (B) multiple doses of HLX01 (250, 375 and 500 mg/m2) (phase 1a study).
) following (A) single dose and (B) multiple doses of HLX01 (250, 375 and 500 mg/m2) (phase 1a study).
Table S3. Summary of PK parameters after single infusion of HLX01 (phase 1a study).
| Parameters | 250 mg/m2 | 375 mg/m2 | 500 mg/m2 |
| PK, pharmacokinetic; AUC0−t, area under the plasma concentration-time curve (from time zero to last sampling time point); Cmax, maximum drug concentration; Tmax, time to maximum drug concentration; t1/2, terminal half-life. | |||
AUC0−t (
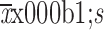 ) (×105 ng·h/mL)
) (×105 ng·h/mL)
|
113.3±8.1 | 183.9±33.8 | 254.7±30.8 |
Cmax (
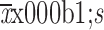 ) (×105 ng/mL)
) (×105 ng/mL)
|
1.4±0.3 | 1.8±0.3 | 2.5±0.5 |
| Tmax (median) (h) | 2 | 2 | 2 |
t1/2 (
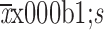 ) (h)
) (h)
|
120.2±40.0 | 136.4±24.8 | 116.9±42.0 |
In phase 1b study, 22 patients in the HLX01 group and 21 patients in the rituximab-CN group had received prior rituximab treatment. The mean exposure to HLX01 and rituximab-CN was 648.7 mg and 624.7 mg in each treatment group (n=43 per treatment group), respectively. PK parameters following single-dose treatment were analyzed using a non-compartmental approach and are shown in Supplementary Table S4 . The geometric mean AUC and AUC0−91 d were similar following single doses (375 mg/m2) of HLX01 and rituximab-CN (Figure 2 , Table 2 ). The primary endpoint was met, as the GLSMR of AUC0−91 d (HLX01: rituximab-CN; 89.6 %) and 90% CI (80.4%−99.8%) fell in the pre-specified equivalence margins of 80%−125% (Table 2 ). The Cmax for HLX01 and rituximab-CN were similar (GLSMR: 102.6%; 90% CI: 95.2%−110.6%). The GLSMR for AUC0−inf was 87.0% (90% CI: 77.7%−97.5%) (Table 2 ), with the upper margin of 90% CI contained within and the lower margin falling outside the pre-specified equivalence margins. The sensitivity analysis of PK parameters by ADA status showed consistent results (Table 2 ).
Table S4. Summary of PK parameters of HLX01 and reference rituximaba (phase 1b study) .
| Parameters | HLX01 | Reference rituximab |
| PK, pharmacokinetic; AUC0−91 d, area under the plasma concentration-time curve (from time zero to day 91); AUC0–x week, area under the plasma concentration time curve (from time 0 to week x) the time point was 91±3 days; AUC0–inf, area under the plasma concentration-time curve (from time zero to infinity); Cmax, maximum drug concentration; t1/2, terminal half-life; a, The reference rituximab was sourced from China (MabThera®; rituximab-CN); b, AUC0−91 d is the primary PK endpoint, to compare equivalence of HLX01 and reference rituximab; c, AUC0−13 week is a secondary endpoint PK parameter where the time point was 91±3 d. | ||
AUC0−91 d (
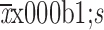 ) (×105 ng·h/mL)b
) (×105 ng·h/mL)b
|
670.9±219.7 | 728.8±179.5 |
Cmax (
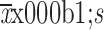 ) (×105 ng/mL)
) (×105 ng/mL)
|
2.2±0.6 | 2.1±0.4 |
AUC0−1 week (
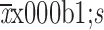 ) (×105 ng·h/mL)
) (×105 ng·h/mL)
|
185.2±32.9 | 186.8±26.1 |
AUC0−2 week (
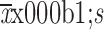 ) (×105 ng·h/mL)
) (×105 ng·h/mL)
|
279.2±52.7 | 285.8±40.2 |
AUC0−4 week (
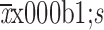 ) (×105 ng·h/mL)
) (×105 ng·h/mL)
|
432.3±95.1 | 452.0±71.8 |
AUC0−8 week (
 ) (×105 ng·h/mL)
) (×105 ng·h/mL)
|
593.1±161.3 | 636.1±129.9 |
AUC0−13 week (
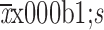 ) (×105 ng·h/mL)c
) (×105 ng·h/mL)c
|
674.8±214.4 | 732.8±176.5 |
AUC0−inf (
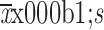 ) (×105 ng·h/mL)
) (×105 ng·h/mL)
|
705.4±231.4 | 794.2±223.7 |
t1/2 (
 ) (h)
) (h)
|
527.0±193.9 | 562.3±134.5 |
Clearance (
 ) (L/h)
) (L/h)
|
0.010±0.004 | 0.009±0.003 |
Figure 2.

Serum concentration-time curves (
 ) following single doses of HLX01 and reference rituximab (phase 1b study). The reference rituximab was sourced from China (MabThera®; rituximab-CN), and 375 mg/m2 of HLX01 or reference rituximab was administered.
) following single doses of HLX01 and reference rituximab (phase 1b study). The reference rituximab was sourced from China (MabThera®; rituximab-CN), and 375 mg/m2 of HLX01 or reference rituximab was administered.
Table 2. Sensitivity analysis and evaluation of equivalence of PK parameters between HLX01 and reference rituximaba (375 mg/m2; phase 1b study) .
| Parameters | Patients | N | Dose corrected geometric mean (mg) | Geometric mean ratiob HLX01/reference rituximab (%) (90% CI) | |
| HLX01 | Reference rituximab | ||||
| PK, pharmacokinetic; PKAS, pharmacokinetics analysis set; ADA, anti-drug antibody; AUC0−91 d, area under the plasma concentration-time curve (from time zero to day 91); AUC0−inf, area under the plasma concentration-time curve (from time zero to infinity); 90% CI, 90% confidence interval; Cmax, maximum drug concentration; N, number of subjects in analysis set; a, Reference rituximab was sourced from China (MabThera®; rituximab-CN); b, Determined using WinNonlin analysis; c, Six patients were excluded from the sensitivity analysis due to ADA positivity after baseline; d, Two patients were excluded from the sensitivity analysis due to ADA positivity at consecutive time points; e, Patients with ADA positivity at baseline were included in the sensitivity analysis; f, A patient with extrapolated area as a percentage of total AUC0−inf (% AUCextra) >20% was excluded from the sensitivity analysis of AUC 0–inf; g, Two patients with baseline ADA positivity were included in the sensitivity analysis of AUC0−inf, one patient had % AUCextra >20% and was excluded from the sensitivity analysis of AUC 0−inf. | |||||
| AUC0−91 d
(×105 ng·h/mL) |
PKAS | 81 | 635.3 | 709.3 | 89.6 (80.4−99.8) |
| PKAS excluding patients with ADA positivityc | 75 | 660.2 | 728.7 | 90.6 (81.7−100.5) | |
| PKAS excluding patients with consecutive ADA positivityd | 79 | 649.1 | 715.0 | 90.8 (81.9−100.7) | |
| PKAS and patients with baseline ADA positivitye | 84 | 628.4 | 703.6 | 89.3 (80.3−99.3) | |
| AUC0−inf
(×105 ng·h/mL) |
PKASf | 80 | 667.3 | 767.1 | 87.0 (77.7−97.5) |
| PKAS excluding patients with ADA positivityf | 74 | 694.2 | 791.0 | 87.8 (78.7−97.9) | |
| PKAS excluding patients with consecutive ADA positivityf | 78 | 682.6 | 773.9 | 88.2 (79.1−98.4) | |
| PKAS and patients with baseline ADA positivityf,g | 82 | 660.1 | 767.1 | 86.1 (77.0−96.2) | |
| Cmax
(×105 ng/mL) |
PKAS | 81 | 2.1 | 2.1 | 102.6 (95.2−110.6) |
| PKAS excluding patients with ADA positivityc | 75 | 2.1 | 2.1 | 103.0 (95.1−111.5) | |
| PKAS excluding patients with consecutive ADA positivityd | 79 | 2.1 | 2.1 | 103.3 (95.8−111.5) | |
| PKAS and patients with baseline ADA positivitye | 84 | 2.1 | 2.1 | 101.6 (94.4−109.3) | |
Safety and tolerability
In the phase 1a study, no deaths, SAEs, AEs leading to treatment discontinuation, or DLTs were observed following single-dose administration of the three tested doses of HLX01. Mean drug exposures in the dose-escalation cohorts (250, 375 and 500 mg/m2) were 461.0, 649.3 and 933.3 mg, respectively, after first infusion. Two patients experienced AEs after receiving single doses of 250 mg/m2 HLX01 (Supplementary Table S5 ).
Table S5. TEAEs following single and multiple administrations of HLX01 (phase 1a study).
| TEAEa | n (%) | |||||||
| 250 mg/m2 (N=3) | 375 mg/m2 (N=6) | 500 mg/m2 (N=3) | ||||||
| TEAE, treatment-emergent adverse event; ADR, adverse drug reaction; SAE, serious adverse event; WBC, white blood cell; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Ig, immunoglobulin; GGT, gamma-glutamyl transpeptidase; LDH, lactic acid dehydrogenase. a, Excluding infusion-related reactions; b, Where possible causal relationship to drug, device or administration; c, The patient has experienced upper respiratory tract infection twice; d, The patient has experienced sinus bradycardia for three times. | ||||||||
| Single dose | ||||||||
| Patients with ≥1 TEAE | 2 (66.7) | 0 | 0 | |||||
| Patients with ADRb | 1 (33.3) | 0 | 0 | |||||
| Patients with significant TEAE | 1 (33.3) | 0 | 0 | |||||
| Patients with SAE | 0 | 0 | 0 | |||||
| Patient death due to TEAE | 0 | 0 | 0 | |||||
| Patient discontinuation due to TEAE | 0 | 0 | 0 | |||||
| Grade of TEAE with single dose | Grade 1 | Grade 2 | Grade 1 | Grade 2 | Grade 3 | Grade 1 | Grade 3 | |
| Fever | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | |
| Chills | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | |
| Upper respiratory infection | 0 | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | |
| Multiple doses | ||||||||
| Patients with ≥1 TEAE | 3 (100) | 4 (66.7) | 3 (100) | |||||
| Patients with ADRb | 2 (66.7) | 3 (50.0) | 2 (66.7) | |||||
| Patients with significant TEAE | 1 (33.3) | 1 (16.7) | 0 | |||||
| Patients with SAE | 0 | 0 | 1 (33.0) | |||||
| Patient death due to TEAE | 0 | 0 | 0 | |||||
| Patient discontinuation due to TEAE | 0 | 0 | 0 | |||||
| Grade of TEAE with multiple doses | Grade 1 | Grade 2 | Grade 3 | Grade 1 | Grade 2 | Grade 3 | Grade 1 | Grade 3 |
| WBC decreased | 2 (66.7) | 0 | 0 | 0 | 2 (33.3) | 0 | 0 | 0 |
| ALT increased | 1 (33.3) | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 |
| AST increased | 1 (33.3) | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 |
| Blood calcium decreased | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Chills | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fever | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Protein albumin ratio decreased | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| IgG decreased | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| IgM decreased | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Blood potassium decreased | 1 (33.3) | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 |
| Neutrophil count decreased | 0 | 0 | 1 (33.3) | 0 | 1 (16.7) | 0 | 0 | 0 |
| Palpitations | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Upper respiratory tract infection | 0 | 1 (33.3)c | 0 | 0 | 0 | 0 | 1 (33.3) | 0 |
| Sinus bradycardia | 0 | 0 | 0 | 1 (16.7)d | 0 | 0 | 0 | 0 |
| Dizziness | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 |
| GGT increased | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 |
| LDH increased | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 |
| Platelets decreased | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 |
| Blood uric acid increased | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 |
| Ventricular arrhythmia | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33.3) | 0 |
| Elevated blood pressure | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33.3) | 0 |
| Herpes virus infection | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 |
| Anal fistula | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33.3) |
No deaths or TEAEs leading to treatment discontinuation after multiple-dose administration of HLX01 (250, 375 and 500 mg/m2) were observed. One patient reported an SAE (grade 3 anal fistula) in the 500 mg/m2 HLX01 cohort, which was considered unrelated to treatment by the Investigator (Supplementary Table S5 ). In total, 35 TEAEs were recorded following administration of multiple doses of HLX01, including fever, chills and upper respiratory infection reported by ten patients (83.3%) in both the single- and multiple-dose periods (Supplementary Table S5 ) and 19 TEAEs experienced by seven patients (58.3%) that were considered very likely/possibly related to treatment (Supplementary Table S6 ). Of these 19 TEAEs, most were grade 1 (13 events) or grade 2 (5 events). Only one grade 3 recovering/resolving TEAE (elevated alanine aminotransferase) was considered likely/possibly related to treatment. A few changes in vital signs and physical examination relative to baseline were observed during the study.
Table S6. Adverse drug reactions following multiple administrations of HLX01 (phase 1a study).
| Adverse drug reactions | n (%) | ||||||
| 250 mg/m2 (N=3) | 375 mg/m2 (N=6) | 500 mg/m2 (N=3) | |||||
| Grade 1 | Grade 1 | Grade 2 | Grade 3 | Grade 1 | |||
| WBC, white blood cell count; Ig, immunoglobulin; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma-glutamyl transpeptidase. a, Where possible causal relationship to drug, device or administration. | |||||||
| General disorders and administration site conditions | |||||||
| Fever | 1 (33.3) | 0 | 0 | 0 | 0 | ||
| Chills | 1 (33.3) | 0 | 0 | 0 | 0 | ||
| Investigations | |||||||
| WBC decreased | 1 (33.3) | 0 | 2 (33.3) | 0 | 0 | ||
| Neutrophil count decreased | 0 | 0 | 1 (16.7) | 0 | 0 | ||
| Decreased platelet count | 0 | 1 (16.7) | 0 | 0 | 0 | ||
| Elevated blood pressure | 0 | 0 | 0 | 0 | 1 (33.3) | ||
| Decreased serum IgG | 1 (33.3) | 0 | 0 | 0 | 0 | ||
| Decreased serum IgM | 1 (33.3) | 0 | 0 | 0 | 0 | ||
| Protein albumin ratio decreased | 1 (33.3) | 0 | 0 | 0 | 0 | ||
| Blood potassium decreased | 0 | 1 (16.7) | 0 | 0 | 0 | ||
| Elevated AST | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | ||
| Elevated ALT | 0 | 0 | 0 | 1 (16.7) | 0 | ||
| Elevated GGT | 0 | 1 (16.7) | 0 | 0 | 0 | ||
| Infections | |||||||
| Upper respiratory tract infection | 0 | 0 | 0 | 0 | 1 (33.3) | ||
| Herpes virus infection | 0 | 0 | 1 (16.7) | 0 | 0 | ||
| Cardiac disorders | |||||||
| Ventricular arrhythmia | 0 | 0 | 0 | 0 | 1 (33.3) | ||
In the phase 1b study, only one patient in the rituximab-CN group discontinued the single-dose infusion due to an IRR (a grade 1 scalp itchiness and rash) (Table 3 ). IRRs were reported in similar proportions of patients in the treatment groups (20.9% per HLX01 vs. 23.3% per rituximab-CN). No deaths or SAEs were reported following single-dose treatment with HLX01 or rituximab-CN.
Table 3. TEAEs following single administration of HLX01 or reference rituximaba (phase 1b study) .
| TEAEb | n (%) | |||||
| HLX01 (N=43) | Reference rituximab (N=43) | |||||
| TEAE, treatment-emergent adverse event; ADR, adverse drug reaction; SAE, serious adverse event; WBC, white blood cell; a, The reference rituximab was sourced from China (MabThera®; rituximab-CN); b, Excluding infusion-related reactions; c, This includes events of chills, pyrexia and upper respiratory infection that started during the single dose phase; d, Where possible causal relationship to drug, device or administration. | ||||||
| Patients with ≥1 TEAEc | 24 (55.8) | 29 (67.4) | ||||
| Patients with ADRd | 14 (32.6) | 21 (48.8) | ||||
| Patients with significant TEAE | 12 (27.9) | 16 (37.2) | ||||
| Patients with SAE | 0 | 0 | ||||
| Patient death due to TEAE | 0 | 0 | ||||
| Patient discontinuation due to TEAE | 0 | 1 (2.3) | ||||
| Grade | Grade 1 | Grade 2 | Grade 3 | Grade 1 | Grade 2 | Grade 3 |
| Patients with ≥1 TEAE | 14 (32.6) | 10 (23.3) | 0 | 15 (34.9) | 12 (28.0) | 2 (4.7) |
| TEAEs ≥2 events by preferred term | ||||||
| Urinary tract infection | 2 (4.7) | 0 | 0 | 1 (2.3) | 0 | 0 |
| Upper respiratory tract infection | 0 | 2 (4.7) | 0 | 1 (2.3) | 3 (7.0) | 0 |
| WBC decreased | 1 (2.3) | 4 (9.3) | 0 | 1 (2.3) | 1 (2.3) | 1 (2.3) |
| Neutrophil count decreased | 1 (2.3) | 4 (9.3) | 0 | 1 (2.3) | 2 (4.7) | 0 |
| Blood immunoglobulin M decreased | 0 | 0 | 0 | 2 (4.7) | 0 | 0 |
| Blood glucose increased | 0 | 0 | 0 | 2 (4.7) | 0 | 0 |
| Arthralgia | 1 (2.3) | 2 (4.7) | 0 | 0 | 0 | 0 |
| Pruritus | 3 (7.0) | 0 | 0 | 3 (7.0) | 0 | 0 |
| Headache | 0 | 0 | 0 | 2 (4.7) | 0 | 0 |
| Cough | 2 (4.7) | 0 | 0 | 0 | 2 (4.7) | 0 |
| Nausea | 3 (7.0) | 0 | 0 | 0 | 0 | 0 |
| Rash | 1 (2.3) | 0 | 0 | 4 (9.3) | 3 (7.0) | 0 |
| Dry mouth | 1 (2.3) | 0 | 0 | 2 (4.7) | 0 | 0 |
During the phase 1b study, 115 AEs were reported (49 AEs in the HLX01 group and 66 AEs in the rituximab-CN group) (Table 3 ). Of the 49 AEs reported in the HLX01 group during the study, 23 were classified as ADRs (Supplementary Table S7 ). Of the 66 AEs reported in the rituximab-CN group, 34 were considered ADRs (Supplementary Table S7 ). When looking for clustering among these ADRs, there were multiple events of neutropenia, itchy skin/rashes and respiratory events. Most ADRs were resolved during the study. Most AEs were grade 1−2, with two patients in the rituximab-CN group experiencing grade 3 AEs during the study. No grade 3 AEs were reported in the HLX01 group during the study. There were no grade ≥4 AEs recorded over the observation period.
Table S7. Adverse drug reactions following of HLX01 or reference rituximaba (phase 1b study) .
| Adverse drug reactions | 375 mg/m2 [n (%)] | ||||||
| HLX01 (N=43) | Reference rituximab (N=43) | ||||||
| Grade 1 | Grade 2 | Grade 3 | Grade 1 | Grade 2 | Grade 3 | ||
| WBC, white blood cell count; Ig, immunoglobulin. a, The reference rituximab was sourced from China (MabThera®; rituximab-CN); b, Where possible causal relationship to drug, device or administration. | |||||||
| Patients with ≥1 ADRb | 12 (27.9) | 2 (4.7) | 0 | 13 (30.2) | 6 (14.0) | 2 (4.7) | |
| Metabolism and nutrition disorder | |||||||
| Hypoalbuminemia | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | |
| Investigations | |||||||
| WBC decreased | 1 (2.3) | 2 (4.7) | 0 | 0 | 1 (2.3) | 1 (2.3) | |
| Decreased globulin | 1 (2.3) | 0 | 0 | 1 (2.3) | 0 | 0 | |
| Neutrophil count decreased | 1 (2.3) | 2 (4.7) | 0 | 1 (2.3) | 2 (4.7) | 0 | |
| Decreased blood pressure | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | |
| Abnormal liver function | 0 | 0 | 0 | 0 | 1 (2.3) | 0 | |
| Decreased immunoglobulin | 0 | 0 | 0 | 2 (4.7) | 0 | 0 | |
| Decreased serum IgA | 0 | 0 | 0 | 1 (2.3) | 0 | 0 | |
| Decreased serum IgG | 0 | 0 | 0 | 1 (2.3) | 0 | 0 | |
| Decreased serum IgM | 0 | 0 | 0 | 2 (4.7) | 0 | 0 | |
| Hemoglobin decreased | 0 | 0 | 0 | 0 | 1 (2.3) | 0 | |
| Decreased platelet count | 0 | 0 | 0 | 1 (2.3) | 0 | 0 | |
| Nervous system disorders | |||||||
| Dizziness | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | |
| Headache | 0 | 0 | 0 | 1 (2.3) | 0 | 0 | |
| Respiratory/thoracic disorders | |||||||
| Cough | 1 (2.3) | 0 | 0 | 0 | 1 (2.3) | 0 | |
| Nasal congestion | 0 | 0 | 0 | 0 | 1 (2.3) | 0 | |
| Oropharyngeal discomfort | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | |
| Sneezing | 0 | 0 | 0 | 0 | 1 (2.3) | 0 | |
| Immune system disorders | |||||||
| Hypersensitivity | 0 | 0 | 0 | 0 | 1 (2.3) | 0 | |
| Skin and subcutaneous tissue disorders | |||||||
| Rash | 1 (2.3) | 0 | 0 | 4 (9.3) | 3 (7.0) | 0 | |
| Pruritus | 3 (7.0) | 0 | 0 | 3 (7.0) | 0 | 0 | |
| General disorders and administration site conditions | |||||||
| Asthenia | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | |
| Fever | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | |
| Gastrointestinal disorders | |||||||
| Diarrhea | 0 | 0 | 0 | 1 (2.3) | 0 | 0 | |
| Dry mouth | 1 (2.3) | 0 | 0 | 1 (2.3) | 0 | 0 | |
| Vomiting | 0 | 0 | 0 | 1 (2.3) | 0 | 0 | |
| Nausea | 2 (4.7) | 0 | 0 | 0 | 0 | 0 | |
| Vascular and lymphatic diseases | 0 | ||||||
| Hypotension | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | |
| Flushing | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | |
| Blood and lymphatic system disorders | |||||||
| Febrile neutropenia | 0 | 0 | 0 | 0 | 0 | 1 (2.3) | |
Immunogenicity
In the phase 1a study, serum samples were tested for the development of anti-HLX01 antibodies up to 90 d post first HLX01 infusion. One patient tested positive for ADA during the screening assessment and, therefore, did not enter the study. All other samples were ADA-negative.
In the phase 1b study, at baseline, two patients tested positive for anti-HLX01 antibodies (neither had prior rituximab use) and one patient tested positive for anti-rituximab antibodies. Two HLX01-treated patients and four rituximab-CN-treated patients tested positive for ADAs at ≥1 time points. There were no significant differences between treatment groups in ADA positivity. Of the five ADA-positive patients treated with rituximab-CN, two had used rituximab previously.
PD
In the phase 1a study, CD19+ and CD20+ B lymphocyte counts were reduced 72 h after the HLX01 infusion and remained at these lower levels at subsequent time points. CD4+ and CD8+ T cell counts (median) were replenished by day 90.
In the phase 1b study, depletion of CD19+ and CD20+ B lymphocyte counts occurred within 5 min of study drug administration and was maintained throughout the study to a similar extent for both HLX01 and rituximab-CN groups (intergroup difference at all time points: P>0.05) (Table 4 ). Similarly, within 5 min of study drug administration, the CD4+ and CD8+ T cell counts decreased from baseline in both groups, and returned to close to baseline values within 24 h and remained stable over the study duration (Supplementary Table S8 ). Inter-group comparison supported similarity between HLX01 and rituximab-CN, as P-values were >0.05, except for the 24-h CD4+ cell count (P=0.04).
Table 4. Mean changes in peripheral CD19+ and CD20+ B lymphocyte following HLX01 and reference rituximab administration (phase 1b study).
| Time point | Statistics | CD19+ B cell (count/µL) | CD20+ B cell (count/µL) | |||
| HLX01 (N=43) | Reference rituximab (N=43) | HLX01 (N=43) | Reference rituximab (N=43) | |||
| Reference rituximab was sourced from China (MabThera®; rituximab-CN); 375 mg/m2 of HLX01 or reference rituximab was administered. | ||||||
| Baseline | n (miss) | 43 (0) | 42 (1) | 43 (0) | 42 (1) | |
| Mean | 114.7 | 132.8 | 112.0 | 132.7 | ||
| Median | 55.0 | 94.6 | 54.0 | 96.5 | ||
| Min, Max | 0.1, 591.1 | 0.1, 632.7 | 0, 522.9 | 0, 629.1 | ||
| 5 min after infusion | n (miss) | 43 (0) | 42 (1) | 43 (0) | 42 (1) | |
| Mean | 1.8 | 0.9 | 0.1 | 0.1 | ||
| Median | 0.3 | 0.7 | 0.0 | 0.1 | ||
| Min, Max | 0, 32.3 | 0, 4.1 | 0, 0.7 | 0, 0.6 | ||
| 24 h after infusion | n (miss) | 43 (0) | 42 (1) | 43 (0) | 42 (1) | |
| Mean | 1.5 | 1.4 | 0.1 | 0.1 | ||
| Median | 0.6 | 1.0 | 0.0 | 0.0 | ||
| Min, Max | 0, 17.5 | 0, 6.4 | 0, 0.3 | 0, 0.7 | ||
| 14 d after infusion | n (miss) | 42 (1) | 42 (1) | 42 (1) | 42 (1) | |
| Mean | 0.6 | 0.6 | 0.1 | 0.1 | ||
| Median | 0.4 | 0.3 | 0.0 | 0.1 | ||
| Min, Max | 0, 4.6 | 0, 5.5 | 0, 0.7 | 0, 0.8 | ||
| 56 d after infusion | n (miss) | 42 (1) | 42 (1) | 42 (1) | 42 (1) | |
| Mean | 2.0 | 0.5 | 1.7 | 0.3 | ||
| Median | 0.4 | 0.3 | 0.2 | 0.2 | ||
| Min, Max | 0, 58.4 | 0, 2.9 | 0, 59.0 | 0, 1.1 | ||
| 91 d after infusion | n (miss) | 42 (1) | 42 (1) | 42 (1) | 42 (1) | |
| Mean | 4.2 | 1.2 | 4.1 | 1.0 | ||
| Median | 0.3 | 0.3 | 0.3 | 0.2 | ||
| Min, Max | 0, 142.4 | 0, 34.1 | 0, 142.4 | 0, 34.1 | ||
Table S8. Mean changes of circulating CD4+ and CD8+ T cell pre- and post-treatment (phase 1b study).
| Time point | Statistics | CD4+ T cell (count/µL) | CD8+ T cell (count/µL) | |||
| HLX01 (N=43) | Reference rituximab (N=43) | HLX01 (N=43) | Reference rituximab (N=43) | |||
| These results were collected from safety analysis set. | ||||||
| Baseline | n (miss) | 43 (0) | 42 (1) | 43 (0) | 42 (1) | |
| Mean | 428.8 | 421.1 | 467.3 | 468.0 | ||
| Median | 409.9 | 376.9 | 388.8 | 375.8 | ||
| Min, Max | 138.2−1,026.8 | 130.3−978.3 | 99.6−1,023.5 | 196.4−1,455.2 | ||
| 5 min after infusion | n (miss) | 43 (0) | 42 (1) | 43 (0) | 42 (1) | |
| Mean | 100.8 | 132.8 | 139.8 | 185.8 | ||
| Median | 80.9 | 83.4 | 101.9 | 144.1 | ||
| Min, Max | 26.1−520.0 | 32.0−780.1 | 21.7−719.0 | 36.9−666.1 | ||
| 24 h after infusion | n (miss) | 43 (0) | 42 (1) | 43 (0) | 42 (1) | |
| Mean | 433.1 | 480.1 | 372.5 | 438.3 | ||
| Median | 399.8 | 429.1 | 283.0 | 374.0 | ||
| Min, Max | 112.8−1,111.9 | 124.2−876.3 | 86.2−927.7 | 197.4−1,504.9 | ||
| 14 d after infusion | n (miss) | 42 (1) | 42 (1) | 42 (1) | 42 (1) | |
| Mean | 464.4 | 456.8 | 523.7 | 509.2 | ||
| Median | 427.2 | 439.7 | 417.2 | 427.5 | ||
| Min, Max | 136.8−1,130.1 | 165.4−1,004.9 | 99.7−1,466.5 | 147.8−1,299.8 | ||
| 56 d after infusion | n (miss) | 42 (1) | 42 (1) | 42 (1) | 42 (1) | |
| Mean | 431.0 | 448.8 | 486.1 | 523.1 | ||
| Median | 400.7 | 414.1 | 415.7 | 460.9 | ||
| Min, Max | 130.8−1,022.5 | 124.3−983.5 | 99.8−1,155.2 | 178.3−2,263.3 | ||
| 91 d after infusion | n (miss) | 42 (1) | 42 (1) | 42 (1) | 42 (1) | |
| Mean | 436.8 | 450.6 | 487.3 | 496.7 | ||
| Median | 407.6 | 425.9 | 410.2 | 405.2 | ||
| Min, Max | 139.1−1,160.2 | 145.1−1,000 | 90.7−1,334.6 | 162.2−1,708.8 | ||
Efficacy
All patients enrolled in the phase 1b study who had achieved CR or CRu were evaluable for disease progression following a single dose of HLX01 or rituximab-CN treatment. As of eight weeks after study drug infusion (the final assessment point), the percentage of evaluable patients who remained at CR or CRu was similar in both groups (97.7% in HLX01 group, 95.3% in rituximab-CN group, P=1.00).
Discussion
Biologics are complex therapeutic products, requiring careful manufacturing to ensure homogeneity. Developing biosimilar biologics requires rigorous evaluations to confirm structural and functional similarity to originator products. Controlled clinical trials, using commercially available reference products, are therefore invaluable for confirmation of equivalence of proposed biosimilars and reference products in terms of PK, PD, immunogenicity, safety and efficacy.
HLX01 is the first biosimilar product approved by the NMPA in China. The application for approval was supported by the evidence demonstrating similarity between HLX01 and reference rituximab in terms of their physicochemical properties, biological activities, PK, clinical efficacy and safety (16,18). In the absence of China-specific guidelines prior to 2015, the early clinical development program for HLX01 followed the principles and requirements of the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) regulatory guidelines for developing and evaluating biosimilar products. The NMPA guidelines that have since been developed are largely consistent with those from the EMA and FDA, and have been adhered to in this study (14,17,19). AUC0−t rather than AUC0−inf was chosen as the primary endpoint in the phase 1b study, as it was calculated from actual measured values and was highly recommended by the regulatory authority in China at the time. AUC0−inf was set as a key secondary endpoint in this study.
This initial phase 1a study demonstrated the safety and tolerability of HLX01 in 12 patients. There were no DLTs, and escalating doses of HLX01 were safe and well tolerated, including the recommended therapeutic dose for rituximab (375 mg/m2). PK profiles of escalating doses of HLX01 displayed an expected linear increase and, in agreement with the early phase PK data for reference rituximab and other anti-CD20 monoclonal antibodies, study drug accumulation was observed upon multiple-dose treatment to steady state (20,21). Population PK of the phase 1b study and the data from a phase 3 clinical study (data not shown), have confirmed that age, weight and gender do not have a significant impact on the PK of HLX01. Therefore, the observed differences between baseline characteristics of different groups in the phase 1a study are not anticipated to impact the outcomes. These outcomes provided scientific basis and dose suggestion for patients with NHL in phase 1b study.
The phase 1b study was a comparative PK and safety study of HLX01 and rituximab-CN. The primary (AUC0−91 d) and secondary (Cmax) endpoints met the equivalence criteria and demonstrated PK equivalence of HLX01 and rituximab-CN, although the lower 90% CI margin of GLSMR for AUC0−inf (77.7%) was slightly outside the pre-specified equivalence margins (80%−125%). The sensitivity analysis, stratified by the ADA status, further demonstrated PK equivalence of HLX01 and rituximab-CN.
In both studies, HLX01 was safe and well tolerated. Overall, HLX01-related toxicities were as expected from the observations of prior studies of reference rituximab and rituximab biosimilar products, and no new safety concerns were identified (20-24). Most AEs, including IRRs, hematological toxicities and infections were mild (grade ≤2) and did not affect study completion. Fewer ADRs of fever were reported in this study than in a previous phase 1 reference rituximab trial, and no grade 4 (or above) events were reported in this trial (21). Interestingly, the rate of infections in this phase 1b study was relatively low (11.6%) compared to other rituximab biosimilar studies such as GP2013 (21.5%) (25) and PF-05280586 (31.1%) (26), possibly related to the status of enrolled patients. The rates of infections in the HLX01 and rituximab-CN groups were comparable, supporting the conclusion of similarity between the two drugs in terms of safety.
Immunogenicity to biologics may result in reduced treatment efficacy or AEs (27). While ADA development has been previously reported following rituximab therapy (23), all patients in phase 1a study tested negative for ADA over the study duration. Consistent with previous studies (24,27), the incidence of ADA was low and similar in the HLX01 and rituximab-CN groups in the phase 1b trial.
Furthermore, the PD profiles of HLX01 and rituximab-CN were similar with respect to counts of CD19+, CD20+ B lymphocytes, and CD4+ and CD8+ T-cells. There was no difference between the two groups in disease progression at week 8 (final assessment point).
Conclusions
Results of the phase 1a and phase 1b studies demonstrate similar PK, PD, safety and immunogenicity profiles between HLX01 and rituximab-CN. HLX01 was safe and well tolerated in Chinese patients with B-cell lymphoma.
Footnote
Conflicts of Interest: Xiaodong Dong is an employee of Henlius. Other authors have no conflicts of interest to declare.
Acknowledgements
This study was funded by Shanghai Henlius Biotech, Inc., Science and Technology Commission of Shanghai Municipality (No. 14431908500), and China National Major Project for New Drug Innovation (No. 2012ZX09303012).
References
- 1.Armitage JO, Gascoyne RD, Lunning MA, et al Non-Hodgkin lymphoma. Lancet. 2017;390:298–310. doi: 10.1016/S0140-6736(16)32407-2. [DOI] [PubMed] [Google Scholar]
- 2.Mohammed R, Milne A, Kayani K, et al How the discovery of rituximab impacted the treatment of B-cell non-Hodgkin’s lymphomas. J Blood Med. 2019;10:71–84. doi: 10.2147/JBM.S190784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. International Agency for Research on Cancer: Non-Hodkin lymphoma; Source Globocan 2018. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/34-Non-hodgkin-lymphoma-fact-sheet.pdf
- 4.Chihara D, Nastoupil LJ, Williams JN, et al New insights into the epidemiology of non-Hodgkin lymphoma and implications for therapy. Expert Rev Anticancer Ther. 2015;15:531–44. doi: 10.1586/14737140.2015.1023712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun J, Yang Q, Lu Z, et al Distribution of lymphoid neoplasms in China: analysis of 4,638 cases according to the World Health Organization classification. Am J Clin Pathol. 2012;138:429–34. doi: 10.1309/AJCP7YLTQPUSDQ5C. [DOI] [PubMed] [Google Scholar]
- 6.Chinese guidelines for diagnosis and treatment of malignant lymphoma 2018 (English version). Chin J Cancer Res 2019;31:557-77.
- 7.McLaughlin P, Grillo-López AJ, Link BK, et al Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16:2825–33. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 8.Coiffier B, Lepage E, Briere J, et al CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:235–42. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 9.Habermann TM, Weller EA, Morrison VA, et al Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol. 2006;24:3121–7. doi: 10.1200/JCO.2005.05.1003. [DOI] [PubMed] [Google Scholar]
- 10.Pfreundschuh M, Trümper L, Osterborg A, et al CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol. 2006;7:379–91. doi: 10.1016/S1470-2045(06)70664-7. [DOI] [PubMed] [Google Scholar]
- 11.Hilal T, Wang Z, Almader-Douglas D, et al Rituximab maintenance therapy for mantle cell lymphoma: A systematic review and meta-analysis. Am J Hematol. 2018;93:1220–6. doi: 10.1002/ajh.25226. [DOI] [PubMed] [Google Scholar]
- 12.Salles G, Seymour JF, Offner F, et al Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011;377:42–51. doi: 10.1016/S0140-6736(10)62175-7. [DOI] [PubMed] [Google Scholar]
- 13.Yu-Wen H, Mei-Bian Z, Xiang X, et al Socioeconomic inequality in the use of rituximab therapy among non-Hodgkin lymphoma patients in Chinese public hospitals. Asia Pac J Public Health. 2014;26:203–14. doi: 10.1177/1010539512464648. [DOI] [PubMed] [Google Scholar]
- 14.National Medical Products Administration. Technical guideline for the development and evaluation of biosimilars (Interim). Available online: https://www.fdanews.com/ext/resources/files/03-15/03-15-China-Biosimilars.pdf?1425486653
- 15.World Health Organization. Guidelines on evaluation of monoclonal antibodies as similar biotherapeutic products (SBPs). Available online: https://www.who.int/biologicals/expert_committee/mAb_SBP_GL-ECBS_review_adoption-2016.10.26-11.7post_ECBS-Clean_Version.pdf?ua=1
- 16.Xu Y, Xie L, Zhang E, et al Physicochemical and functional assessments demonstrating analytical similarity between rituximab biosimilar HLX01 and the MabThera(R) MAbs. 2019;11:606–20. doi: 10.1080/19420862.2019.1578147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf
- 18.Shi Y, Song Y, Qin Y, et al A phase 3 study of rituximab biosimilar HLX01 in patients with diffuse large B-cell lymphoma. J Hematol Oncol. 2020;13:38. doi: 10.1186/s13045-020-00871-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.U.S. Food and Drug Administration. Considerations in Demonstrating Interchangeability With a Reference Product Guidance for Industry. Available online: https://www.fda.gov/media/124907/download
- 20.Gui L, Han X, He X, et al Phase I study of chimeric anti-CD20 monoclonal antibody in Chinese patients with CD20-positive non-Hodgkin’s lymphoma. Chin J Cancer Res. 2016;28:197–208. doi: 10.21147/j.issn.1000-9604.2016.02.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maloney DG, Grillo-López AJ, Bodkin DJ, et al IDEC-C2B8: results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. J Clin Oncol. 1997;15:3266–74. doi: 10.1200/JCO.1997.15.10.3266. [DOI] [PubMed] [Google Scholar]
- 22.Jiang B, Qi JY, Sun MY, et al Tolerance and pharmacodynamics phase clinical trial study of chimeric anti-CD20 monoclonal antibody IBI301 in Chinese patients with CD20-positive non-Hodgkin’s lymphoma. Zhonghua Xue Ye Xue Za Zhi. 2018;39:320–4. doi: 10.3760/cma.j.issn.0253-2727.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoo DH, Suh CH, Shim SC, et al A multicentre randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76:566–70. doi: 10.1136/annrheumdis-2016-209540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jurczak W, Moreira I, Kanakasetty GB, et al Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol. 2017;4:e350–e361. doi: 10.1016/s2352-3026(17)30106-0. [DOI] [PubMed] [Google Scholar]
- 25.Smolen JS, Cohen SB, Tony HP, et al A randomised, double-blind trial to demonstrate bioequivalence of GP2013 and reference rituximab combined with methotrexate in patients with active rheumatoid arthritis. Ann Rheum Dis. 2017;76:1598–602. doi: 10.1136/annrheumdis-2017-211281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen S, Emery P, Greenwald M, et al A phase I pharmacokinetics trial comparing PF-05280586 (a potential biosimilar) and rituximab in patients with active rheumatoid arthritis. Br J Clin Pharmacol. 2016;82:129–38. doi: 10.1111/bcp.12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Einarsson JT, Evert M, Geborek P, et al Rituximab in clinical practice: dosage, drug adherence, Ig levels, infections, and drug antibodies. Clin Rheumatol. 2017;36:2743–50. doi: 10.1007/s10067-017-3848-6. [DOI] [PMC free article] [PubMed] [Google Scholar]