

Abstract
T-cell activation and expansion in the tumor microenvironment (TME) are critical for antitumor immunity. Neutrophils in the TME acquire a complement-dependent T-cell suppressor phenotype that is characterized by inhibition of T-cell proliferation and activation through mechanisms distinct from those of myeloid-derived suppressor cells. In this study, we used ascites fluid supernatants (ASC) from patients with ovarian cancer as an authentic component of the TME to evaluate the effects of ASC on neutrophil function and mechanisms for neutrophil-driven immune suppression. ASC prolonged neutrophil lifespan, decreased neutrophil density, and induced nuclear hypersegmentation. Mass cytometry analysis showed that ASC induced 15 distinct neutrophil clusters. ASC stimulated complement deposition and signaling in neutrophils resulting in surface mobilization of granule constituents, including NADPH oxidase. NADPH oxidase activation and phosphatidylserine signaling were required for neutrophil suppressor function, although we did not observe a direct role of extracellular reactive oxygen species in inhibiting T-cell proliferation. Post-operative surgical drainage fluid also induced a complement-dependent neutrophil suppressor phenotype, pointing to this effect as a general response to injury. Like circulating lymphocytes, ASC-activated neutrophils caused complement-dependent suppression of tumor-associated lymphocytes. ASC-activated neutrophils adhered to T cells and caused trogocytosis of T-cell membranes. These injury and signaling cues resulted in T-cell immunoparalysis characterized by impaired NFAT translocation, IL2 production, glucose uptake, mitochondrial function, and mTOR activation. Our results demonstrate complement-dependent priming of neutrophil effector functions in the TME inducing a T-cell non-responsiveness distinct from established checkpoint pathways and identify targets for immunotherapy.
Keywords: neutrophil, complement, NADPH oxidase, ovarian cancer, immunosuppression
Introduction
Epithelial ovarian cancer (OC) is the leading cause of death from gynecological malignancies in the United States. Ascites is common in metastatic OC, and both the presence and volume of ascites at diagnosis are negative predictors of outcome (1,2). Ascites is a distinct part of the OC microenvironment that facilitates peritoneal metastasis, mediates resistance to chemotherapy, and impairs antitumor immunity (2,3). Ascites also contains tumor-associated lymphocyte populations that are being explored for cellular therapy (4) and immunosuppressive myeloid cells (5) that can impair antitumor immunity.
Neutrophils and neutrophil-like cells associated with human tumors can arise from expansion of myeloid-derived suppressor cells (MDSC) or polarization of mature neutrophils by tumor-derived products. PMN-MDSC result from disordered granulopoiesis (6), and are characterized by the ability to suppress T-cell activation. Based primarily on mouse data, tumor-associated neutrophils have been divided into N1 (antitumorigenic) and N2 (immunosuppressive and pro-tumorigenic) populations with distinct transcriptional profiles (7). Previously, we observed that normal neutrophils can acquire a suppressor phenotype within OC ascites fluid (8). These cells are completely distinct from PMN-MDSC and N2 neutrophils; we define them as “suppressor neutrophils,” which is a term that we use to describe circulating neutrophils that are modified by malignant effusions or other inflammatory or injurious conditions resulting in acquisition of suppressor function. The acquisition of this suppressor phenotype requires several neutrophil effector functions, including complement signaling, and it can be induced not only by OC ascites fluid, but also by malignant effusions from patients with different metastatic cancers, showing generalizability of these findings (8). Although complement can have pro- or anti-tumorigenic effects (9), these results raise the potential for complement inhibition to abrogate neutrophil-driven suppression in the tumor microenvironment (TME).
In this study, we probed the molecular mechanisms underlying how neutrophils acquire a suppressor phenotype in the TME and the response of T cells to neutrophil-driven injury. We used OC ascites fluid supernatants (ASC) and primary neutrophils and T cells under conditions that mimic as closely as feasible those present in the peritoneal cavity at OC diagnosis. ASC primed complement-dependent degranulation and translocation of NADPH oxidase to neutrophil membranes, and NADPH oxidase and phosphatidylserine signaling were required for the T-cell suppressive function. Suppressor neutrophils induced immunoparalysis in T cells that was characterized by disruption of key signaling and metabolic pathways. A similar neutrophil suppressor phenotype was induced by surgical injury, pointing to suppressor neutrophils as a generalized response to injury. Our work demonstrates that neutrophils in the TME induce a non-responsive state in T cells that is distinct from established checkpoint pathways and identifies these neutrophils as potential therapeutic targets to abrogate cancer-induced immunosuppression.
Methods
Patient samples and normal donor blood:
Participants included normal donors (n=8), patients with cancer and malignant effusions (n=61), patients who underwent primary debulking surgery for newly diagnosed metastatic OC (n=10), and patients without cancer undergoing diagnostic lung surgery (n=2). Normal donors included males and females ages 21–55. From 2015–2020, blood and ascites were collected from patients with newly diagnosed advanced (stage III or IV) OC, as previously described (10). Blood was collected prior to primary surgery, and ascites was collected either by diagnostic paracentesis or in the operating room prior to surgery. In patients with recurrent OC following surgery and chemotherapy, ascites fluid was obtained from therapeutic paracentesis. Ascites fluid was filtered through 300μM filters and then centrifuged (500xg, 10 minutes). Aliquots of supernatants were stored at −80°C until further use. Post-operative drainage fluid (POF) samples consisted of fluid collected 1-day after OC debulking surgery or lung surgery performed at Roswell Park. Samples were processed using the same protocol as OC ascites fluid. In studies of chronic granulomatous disease (CGD), blood was collected in EDTA-coated tubes from CGD patients and normal donors at the National Institutes of Health (Bethesda, MD), shipped overnight at room temperature to Roswell Park, and used immediately upon arrival. Data were provided regarding age, sex, CGD genotype, reactive oxygen species (ROS) production, and use of immunomodulators (Supplementary Table S1). NADPH oxidase activity was assessed by superoxide production and the DHR assay, as previously described (11).
Study Approval:
This study was approved by the Institutional Review Board (IRB) of Roswell Park Comprehensive Cancer Center (Roswell Park), Buffalo, NY, and was in compliance with federal and state requirements. All participants gave written informed consent under an IRB-approved protocol prior to inclusion in the study (protocols i215512 and i188310). Samples were de-identified prior to sharing with collaborators. Blood collection from the NIH was under an NIH-approved protocol, and samples were de-identified prior to shipment. All studies were conducted in compliance with the Declaration of Helsinki.
Cell isolation:
Peripheral blood was collected in EDTA-coated tubes (BD Biosciences). T cells and neutrophils were isolated using negative selection magnetic kits (purity ≥97%) (cat. #130-098-193 and cat. #130-104-434, respectively, Miltenyi Biotec, Inc.), according to the manufacturer’s instructions. Purity was assessed using flow cytometry and gating on CD15+ (clone HI98; cat. #301904, Biolegend) for neutrophils and CD3+ (clone UCHT1; cat. #300446, Biolegend) for T cells. For imaging experiments, erythrocytes were depleted using positive selection magnetic kits according to the manufacturer’s directions (cat. #130-098-196, Miltenyi Biotec, Inc.). All cells were resuspended in RPMI-1640 (cat. #MT-10-040-CV, Mediatech) supplemented with 5mL HEPES (cat. #25-060-CI, Mediatech), 5mL Pen/Strep (cat. #MT 30-001-CI, Mediatech), 5mL MEM non-essential amino acids (cat. #MT 25-025-CI, Mediatech), 5mL sodium pyruvate (cat. #MT 25-000-CI, Mediatech), and 10% heat-inactivated FBS (cat. #10082147, Thermo Fisher Scientific) unless otherwise indicated. In all experiments, ASC was diluted 1:1 in media.
For neutrophil nuclear morphology experiments, heparinized venous blood was obtained from normal donors using protocol 200307026 approved by the IRB at the University of Iowa. Neutrophils were isolated by sequential dextran (cat. # 5510 0500 9007, Pharmacosmos) sedimentation, density gradient separation on Ficoll-Paque Plus (cat. #GE17-1440-03, GE Healthcare), and hypotonic lysis of erythrocytes. Neutrophil purity was assessed by manual counting of PROTOCOL Hema-3 Stat Pack reagent (cat. #22-1222911, Fisher Scientific)-stained cells attached to glass slides by cytocentrifugation. Neutrophils were distinguished from other leukocyte types based on nuclear morphology and appearance of cytoplasmic granules. By this assay, neutrophil purity was routinely 95–97% with eosinophils as the major contaminant.
Flow cytometry:
Flow staining was performed at room temperature and samples were protected from light. All washes were done with FACS buffer (PBS (cat. #21-031-CV, Mediatech) supplemented with 5g BSA (cat. #A7906-100MG, Sigma-Aldrich) and 1mL EDTA (cat. #46-034-CI, Mediatech)) and samples were centrifuged at 500xg for 5 minutes. Cells were pipetted into polypropylene tubes, washed, and Fc blocked with Human TruStain FcX (cat. #422301, Biolegend) for 5 minutes at room temperature before staining with antibodies for 30 minutes at room temperature. Samples were analyzed on a BD LSR Fortessa, LSR II (BD Biosciences) or Canto II (BD Biosciences) and data analysis was performed using WinList (Verity House Software), FCS express 6 (De Novo software), or FlowJo (Tree Star). Cell sorting was performed on a BD FACSAria (BD Biosciences). Gating strategies for neutrophils and T cells are in Supplementary Figure S1.
Neutrophil viability and Bovine Serum Albumin uptake:
Neutrophils were incubated in ASC and viability was assessed at 6, 12, 24, and 54hrs using PI and Annexin V-FITC staining (cat. #88-8005-72, Thermo Fisher Scientific). Briefly, cells were washed and stained in Annexin V binding buffer then analyzed by flow cytometry. To assess extracellular fluid uptake, neutrophils were incubated (5% CO2, 37°C) in ASC or media with Alexa fluor 488 conjugated bovine serum albumin (BSA) (cat. #A13100, Thermo Fisher Scientific) for 21hrs and evaluated by flow cytometry.
Evaluation of neutrophil density:
Peripheral blood was collected in EDTA-coated tubes, diluted in equal volume of PBS and centrifuged at 1,200x g for 20 minutes to isolate the buffy coat. Complete blood count was evaluated in the buffy coat and 1mL (containing ~2.5×106 neutrophils) was plated with equal volume of ASC or media and incubated for 1–3hrs (5% CO2, 37°C). Peripheral blood mononuclear cells (PBMCs) were purified by density centrifugation using lymphocyte separation media (cat. #25-072-CV, Mediatech, Inc.) and SepMate tubes (cat. #85450, Stemcell Technologies) following manufacturer’s protocol. Low-density neutrophils were defined by co-sedimentation with PBMCs (12). Cell count and differential were performed in the clinical Hematology laboratory, and cytospins were stained by Wright-Giemsa.
Effects of ASC on neutrophil nuclear morphology:
Neutrophils in HEPES-buffered RPMI-1640 (cat. # BE12-115F, LONZA) containing 10% heat-inactivated FBS (cat. # S11150: Atlanta Biologicals) were diluted 1:1 with ASC at a final concentration 2×106 cells/mL and were incubated at 37°C for 0–24hrs. At various time points, cells were cytocentrifuged onto glass coverslips, fixed and stained with Hema-3 reagents and mounted onto glass slides as we previously described (13). At least 100 cells per sample in random fields of view were analyzed using light microscopy and nuclear morphology was scored as normal (3–4 lobes), condensed/apoptotic (spherical or 2 lobes), or hypersegmented (5 or more lobes) by manual counting (13). Replicate experiments (n=3) used neutrophils from different donors and ASC from different OC patients. Control samples were incubated in media alone.
Super-resolution confocal microscopy was used to assess nuclear morphology in greater detail. To this end, control neutrophils and cells treated with ASC for 18hrs were attached to pooled human serum-coated, acid washed round #1.5 German glass coverslips, 8mm diameter (cat. #7229608, Electron Microscopy Sciences) and then fixed, permeabilized and blocked using our established methods. In brief, cells were fixed with 10% formalin, permeabilized with cold acetone-methanol (1:1) and then blocked in PBS supplemented with 0.5mg/mL NaN3 (cat. # 71289, Sigma-Aldrich), 5 mg/mL BSA (cat. # A2058, Sigma-Aldrich) and 10% horse serum (cat. # H1270-500ML, Sigma-Aldrich). Cells were stained with antibodies specific for the lamin B receptor (Goat anti-rabbit mAb clone E398L; cat. #ab32535, Abcam) and Dylight 549-conjugated F(ab)’2 secondary antibody (polyclonal; cat. #305-506-047, Jackson ImmunoResearch Laboratories). Cells were mounted to slides using Prolong Glass Antifade mounting medium (cat. #P36984, Invitrogen) and analyzed using a Leica SP8 STED Super Resolution Microscope and LAS X software (Leica Microsystems, Buffalo Grove, IL). A resolution of 40–50nm was obtained after processing using Huygens Professional Software deconvolution wizard (version 19.04) set to a maximum of 40 iterations. Z-stack reconstructions and processing were done using Oxford Bitplane Imaris Software (version 9.2.1). At least 100 cells per condition were analyzed for each donor.
Production of Cp40 or scramble (SCR) peptide:
Cp40 was made by Pierce Custom Peptides (Thermo Fisher Scientific). Cp40: (D-Tyr-Ile-[Cys-Val-1MeTrp-Gln-Asp-Trp-Sar-Ala-His-Arg-Cys]-meIle)[Amide] with a disulfide bond between Cys3-Cys12. SCR: (D-Tyr-Ile-[Ala-Val-1MeTrp-Gln-Asp-Trp-Sar-Ala-His-Arg-Ala]-meIle)[Amide]. Cp40 peptide sequence was as described in (14).
Evaluation of Factor B (FB) inhibitor binding affinity:
Surface Plasmon Resonance (SPR) experiments were performed at 25°C using a Biacore T200 instrument (GE Healthcare). PBS (pH 7.4) supplemented with 0.05% Tween 20 was used as running buffer. Human FB was immobilized covalently to a Series S Sensor Chip CM5 (GE Healthcare) at a flow rate of 10μL/min using an amine coupling protocol. Reagents for the immobilization were purchased from GE Healthcare (Amine Coupling Kit, cat. #BR-1000-50). The sensor chip surface was activated by a 5-minute injection of a 1:1 (v/v) mixture of a 100mM N-hydroxysuccinimide (NHS) solution and a 390mM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) hydrochloride salt solution in water. Both proteins were diluted to 0.05mg/mL in 20mM sodium acetate (pH 5.5) for immobilization onto different flow cells of the chip. After a 5-minute injection of the protein, remaining reactive groups were deactivated by injecting a 1M ethanolamine hydrochloride solution in aqueous NaOH (pH 8.5) for 5 minutes. Different chips were used with immobilization levels ranging between 3,000 and 7,000 response units (RUs). To determine binding kinetic parameters, several independent experiments were run. Single Cycle Kinetics (SCK) measurements were performed to determine the kinetic parameters of the binding interaction: Three-fold serial dilutions of inhibitor were prepared ranging from 1 to 600nM. Five increasing concentrations were injected successively at 40 or 60μL/min for 60 or 120 seconds each without allowing for the dissociation of the protein–ligand complex. Dissociation of the complex was measured once, after the final inhibitor injection. Dissociation time was >10,000 seconds. Data was fitted with Biacore T200 Evaluation Software using a Langmuir single-site binding model.
Evaluation of FB inhibitor function:
Proteolytic function of the FB inhibitor was assessed by measurement of C3a by ELISA. Preparation of recombinant human complement FB was as described in (15). Human CVF:Bb complex (3nM concentration) was incubated with inhibitor at various concentrations for 1hr at room temperature in PBS at pH 7.4, containing 10mM MgCl2 and 0.05 % (w/v) CHAPS (AppliChem, cat. # APA1099.0025). The enzyme reaction was started by addition of C3 diluted in the assay buffer to a final concentration of 1μM. After 1hr incubation time at room temperature, the enzyme reaction was stopped by addition of an excess of various enzyme inhibitors (Roche Complete Inhibitor tablets; cat. #11836170001, Sigma-Aldrich). Generation of C3a as the enzymatic cleavage product was quantified by an enzyme-linked-immunosorbent assay (ELISA). Aliquots of reaction samples were pipetted into 384-well high-capacity protein binding plates (NUNC Maxisorp™; cat. #460518, Thermo Fisher) pre-filled with 97μL/well of coating buffer. After an overnight incubation at 4°C, assay plates were washed with PBS–Tween 20. Remaining free binding capacity was saturated by the addition of Starting Block T20 (cat. #37539, Thermo Fisher) for 5 minutes at room temperature, and assay plates were then washed with PBS–Tween 20. Anti-C3a neoepitope (clone 2991; cat. #HM2074-100UG, Hycult Biotech) was added to each well, followed by incubation for 60 minutes at room temperature and removal of excess antibody by washing with PBS–Tween 20. Then, goat anti-mouse labeled with HRP (0.2μg/well in PBS–Tween 20) was incubated for 60 minutes at room temperature as in the former step, and excess antibody was removed by extensive washing with PBS–Tween 20. HRP activity was measured after 20-minute incubation time with Quantablu fluorogenic peroxidase substrate (100μL) (cat. #15169, Thermo Fisher) at room temperature in a TECAN Safire2 fluorimeter. IC50 values were calculated from percentage of inhibition of FD activity as a function of test compound concentration.
Ability of the FB inhibitor to prevent formation of the membrane attack complex (MAC) was also assessed by ELISA. Serum from healthy donors was obtained under informed consent through the Novartis Basel tissue donor program and approved by the local ethics committee (EKNZ). For activation, zymosan A (cat. #Z-4250, Sigma Aldrich) was suspended at 1% (w/v) in Tris-buffered saline (cat. #12498S, Cell signaling) (TBS, pH 7.6) and heated to 100°C for 1hr using a water bath. The suspension was centrifuged at 4000 rpm for 30 minutes and the pellet containing the zymosan A was sonicated for 30 minutes and then resuspended in TBS buffer at 50mg/mL. Black MaxiSorb™ plates (cat. #460518, Thermo Fisher) were coated with activated zymosan A diluted to 1mg/mL in carbonate buffer (pH 9.5, (cat. #28382 Thermo Fisher) and incubated for overnight at 4°C. Inhibitor was serially diluted in DMSO (cat. # D8148, Sigma) in polypropylene V-bottom plates. Serum was diluted to 50% (v/v) with 2x buffer (0.15mM CaCl2 (cat #21115, Sigma), 141mM NaCl (cat. #71386, Sigma), 4.5mM MgCl2 (cat. #AM9530G, Invitrogen), 4.2mM HEPES (cat. #15630049, Gibco), at pH 7.4) containing 20mM EGTA (cat. # E0396 Sigma-Aldrich) or 20mM EDTA (cat. #15575-038, Invitrogen) and added to the compound containing plate. After a 30-minute incubation on ice, 25μL of the mixture were transferred to the washed zymosan A coated plate to allow activation of the alternative pathway. The reaction plate was incubated at 37°C for 30 minutes. The reaction was terminated by addition of 25μl EDTA buffer, aspirating the supernatant, addition of blocking buffer (cat. #37539, Thermo Fisher Scientific) for 10 minutes and washing, and MAC formation was detected with a mouse anti-human C9 neoepitope (clone aE11; cat #M077701-8, Agilent). The antibody was added to the plate at 0.25μg/mL in PBS-Tween (cat #524653-1EA, Calbiochem) for 1hr at room temperature. After washing, a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1/1000 dilution in TBS-T) (cat. #A0168-1ml, Sigma-Aldrich), was added to each well for 45 minutes at room temperature and, after washing, the reaction was developed using 25μL Quanta blue (cat. #15169, Thermo Fisher) for 20 minutes at room temperature. The plate was read at 325nm excitation and 420nm emission wavelengths in a TECAN Safire2 fluorimeter. The baseline (EDTA-treated serum, maximum inhibition control) and the maximum signal (EGTA-treated serum in the absence of inhibitor) were used to generate percent inhibition values for each of the wells. Graphpad Prism was used to calculate the average ± standard deviation for the duplicate analysis. Inhibition was calculated as: % inhibition = [((Maximum average - Baseline average)-(Test well average − Baseline average)) × 100]/ (Maximum avg − Baseline avg). The IC50 value was calculated using GraphPad Prism software.
T-cell proliferation assays:
Unless otherwise indicated, normal donor neutrophils and T cells were used. T cells were stimulated with anti-CD3/CD28 beads (cat. #11131D, Life Technologies) and cocultured 1:1 with neutrophils in ASC as previously described (8). Cocultures were incubated (5% CO2, 37°C) for 72hrs before addition of [3H] thymidine (1μCi per well) (cat. #NET027×001MC, Perkin Elmer). [3H] thymidine was allowed to incorporate for 16–18hrs and cells were harvested on a Filtermat and counted on a Beta counter (Perkin Elmer or Hidex). Net counts per minute (CPM) was calculated by subtracting the average CPM of unstimulated T cells from that of stimulated T cells. The same methods were used for ASC from patients with recurrent OC and POF samples. For delayed addition of ASC and neutrophils, T cells were stimulated with anti-CD3/CD28, and ASC + neutrophils were added after 0.5, 1, 2hrs. For delayed addition of the C3 inhibitor Cp40, ASC was pretreated with 20μM Cp40 (10μM final concentration) for at least 5 minutes before addition to cocultures. In separate experiments, T-cell proliferation was measured by a flow cytometry based EdU incorporation assay (cat. #C10420, Thermo Fisher Scientific). T cells, neutrophils, and ASC cocultures were prepared as described above, but were scaled for 24-well plates (800μl total volume). After 72hrs, 2μM EdU was added into cocultures and allowed to incorporate for 16–18hrs. Samples were harvested and processed for EdU detection following the manufacturer’s protocol. Briefly, cells were stained with antibodies specific for CD15 (clone HI98; cat. #563872, BD Biosciences), CD4 (clone SK3; cat. #344632, Biolegend), and CD8 (clone SK1; cat. #344714, Biolegend) followed by fixation, permeabilization and staining for EdU detection. Samples were analyzed as described above in Flow cytometry.
Evaluation of neutrophil suppressor function on tumor-associated lymphocytes (TALs):
TALs were harvested from OC ascites at diagnosis and cryopreserved or used fresh. In short, cells were stained with antibodies specific for CD4 (clone SK3; cat. #344632, Biolegend) and CD8 (clone SK1; cat. #344714, Biolegend). CD4+ and CD8+ T cells were sort-purified by flow cytometry from fresh or frozen samples and used in T-cell suppression assays with autologous ASC and normal donor neutrophils. Due to low numbers of recovered T cells, CD4+ and CD8+ T cells were combined before plating in T-cell suppression assays.
Assessment of complement activation and complement products in ASC:
ASC samples were thawed, centrifuged, and kept on ice before use. WIESLAB ELISA kits evaluating the classical pathway (cat. #COMPL CP310), MBL pathway (cat. #COMPL MP320), and alternative pathway (cat. #COMPL AP330) were purchased from Svar Life Science AB and used according to the manufacturer’s protocols. Briefly, samples were diluted 1:101 (classical and MBL) or 1:18 (alternative) before plating. MBL samples were kept at room temperature for at least 15 minutes before use and all samples were used within 60 minutes after thawing. Plates were read at 405nm on a Synergy HT microplate reader (BioTek). OD values were normalized to “Blank” wells containing sample diluent. Percent of complement activity was calculated using the following equation: ((Sample-Negative control)/(Positive control- Negative control)) x100. C3 and C5a levels were quantified using C3 (cat. #HK366-01) and C5a (cat. #HK349-01) ELISAs (Hycult Biotech) following the manufacturer’s protocols and read at 450nm on a Synergy HT microplate reader (BioTek). C3b/c levels were assessed by ELISA as described previously (16) with minor modifications. In brief, plates were coated with 2 μg/mL mAb anti-C3-9 (generated as described in (17)), which binds to a neoepitope expressed on C3b, iC3b and C3c after disruption of the internal thioester (17). Samples were incubated and subsequently detected with polyclonal biotinylated anti-C3c antibodies (generated as described in (17)) followed by peroxidase-labelled streptavidin (cat. #RPN1231, GE Healthcare). Results are expressed in nanomole (nmol) calculated using a calibration curve of aged normal human serum containing 7400nmol C3b/c. C4b/c levels were assessed as described previously (16) with minor modifications. In short, plates were coated with 2μg/mL affinity purified anti-C4-1 mAb (generated as described in (16)) detecting C4b, iC4b, C4c (C4b/c). Samples were incubated and subsequently detected with a biotinylated polyclonal anti-C4 antibody (generated as described in (16)) followed by peroxidase-labelled streptavidin (cat. #RPN1231, GE Healthcare). Results are expressed in nmol calculated using a calibration curve of aged normal human serum containing 1100nmol C4b/c. Plates were read at 450nm on a Synergy 2 microplate reader (BioTek).
Evaluation of classical and alternative pathways of complement activation on neutrophil suppressor function:
To inhibit the classical pathway, ASC was incubated with 2μM of SALO (salivary anti-complement from Lu. longipalpis) for 30 minutes at room temperature before addition into cocultures. SALO (generously provided by Dr. Jesus G. Valenzuela; National Institutes of Health) is a specific classical pathway inhibitor of C1 from the saliva of the sand fly Lutzomyia longipalpis (18). To inhibit the function of properdin, 100μg/mL of 6E11A4 anti-properdin monoclonal antibody was added to cocultures. 6E11A4 was generated as previously described (19). Briefly, the anti-properdin 6E11A4 hybridoma cell line was developed by Genscript USA Inc. using human properdin that was purified as described previously (20). The antibody was purified by Protein G chromatography and the isotype was determined to be IgG1 using a mouse MoAb isotyping test kit (Abd Serotec) (19). Inhibition of both the classical and alternative pathways involved pretreatment with SALO and addition of 6E11A4. The Factor D inhibitor (15,21) and Factor B inhibitor (Patent: WO2013192345) were provided by Novartis. ASC were pretreated with 20μM, 10μM, or 2μM of either inhibitor before being plated into assays (final concentrations were 10μM, 5μM, and 1μM). C5aR was inhibited by pretreating neutrophils with 16.7μM of PMX53 (cat. #5473, R&D Systems Inc.) or control peptide (CTRL) (cat. #5697, R&D Systems Inc.) for 30 minutes at room temperature. Neutrophils were washed and counted before plating.
Evaluation of effect of ASC on neutrophil surface markers, properdin bound to the neutrophil surface, iC3b/C3b deposition, and plasma membrane fusion of secretory vesicles and granules:
Neutrophils were incubated in ASC or media and levels of properdin were measured by flow cytometry after 1.5hrs. Primary staining with anti-Factor P (cat. #A233, Quidel) followed by a secondary antibody (polyclonal Donkey anti-mouse IgG; cat. #A21202, Thermo Fisher Scientific) was used to detect properdin.
Neutrophils were incubated in media or ASC for 1hr before evaluating CD10 (clone HI10a; cat. #312204, Biolegend) or for 1.5hrs before evaluating LOX-1 (clone 15C4; cat. #358610, Biolegend) expression by flow cytometry. PMA (20nM) (cat. #P1585-1MG, Sigma-Aldrich) was used as a positive control to induce LOX-1 expression.
In studies of iC3b/C3b deposition, neutrophils and T cells were incubated (5% CO2, 37°C) in cocultures containing media or 50% ASC for 1.5hrs. Cells were stained with antibodies specific for iC3b/C3b (clone 3E7/C3b; cat. #846104, Biolegend), CD8 (clone SK1; cat. #344714, Biolegend), CD4 (clone SK3; cat. #344632, Biolegend), and CD15 (clone HI98; cat. #563872, BD Biosciences), as well as Live/Dead Fixable Aqua (cat. #L34957, Thermo Fisher Scientific), and analyzed by flow cytometry.
For evaluation of neutrophil secretory vesicle and granule fusion with the plasma membrane, normal donor T cells and neutrophils were cocultured 1:1 in ASC. Cocultures were incubated for 2.5hrs (5% CO2, 37°C) then stained with antibodies specific for CD11b (clone ICRF44; cat.#301324, Biolegend), CD35 (clone E11; cat. #333404, Biolegend), CD66b (clone G10F5; cat. #305126, Biolegend), CD63 (clone H5C6; cat. #353016, Biolegend), CD15 (clone HI98; cat. #563872, BD Biosciences), and flavocytochrome b558 (clone 7D5, cat. #D162-5, MBL International Corporation), as well as Live/Dead fixable Aqua.
Inhibition of extracellular ROS, neutrophil granule proteases, and surface phosphatidylserine:
Superoxide dismutase (SOD) (600units/mL; cat. #S7571-15KU, Sigma-Aldrich), catalase (1000units/mL; cat. #C9322-1G, Sigma-Aldrich), or both were added to cocultures.
Sivelestat (100μg/mL; cat. #17779, Cayman Chemical Co.), Cathepsin G Inhibitor I (5μM; cat. #219372-1MG, Sigma-Aldrich), and 4-aminobenzoic acid hydrazide (4-ABAH 500μM; cat. #14845, Cayman Chemical Co.) were used to inhibit neutrophil elastase, cathepsin G, and MPO, respectively, by addition into cocultures.
To inhibit phosphatidylserine, anti-phosphatidylserine (clone 1H6, 20μg/mL; cat. #05-719, Sigma-Aldrich;) was added into cocultures with mouse IgG1, k isotype (cat. #16-4714-85, Life Technologies) used as a control.
Evaluation of extracellular ROS and peroxynitrite formation:
Extracellular ROS was measured by bioluminescence. Briefly, neutrophils were cultured in 1X Diogenes (cat. #CL-202, National Diagnostics), 1mM 5-amino-2,3-dihydro-1,4-phthalazinedione (Luminol; cat. #123072-2.5G, Sigma-Aldrich). Time zero was taken before ASC (50% total volume) or PMA (20ng/mL) addition. An IVIS Spectrum bioluminescence reader was set to image cells every 5 minutes.
Peroxynitrite formation was measured using a flow cytometry based Peroxynitrite Assay Kit (cat. #AB233470, Abcam) according to the manufacturer’s protocol. Briefly, T cells and neutrophils were cocultured in ASC with peroxynitrite sensor green for 21hrs and fluorescence in T cells was analyzed by flow cytometry. SIN-1 is a nitric oxide donor and was used in coculture with stimulated T cells for a positive control. Sample analysis was performed as described in Flow cytometry.
Mass cytometry (CyTOF) analysis of neutrophils exposed to OC ASC:
CyTOF was performed at the Harvard Medical Area CyTOF core. Normal donor neutrophils were isolated by negative selection and erythrocytes depleted using positive selection as described above in Cell isolation. Neutrophils were plated at 2.5×106 cells in 5mL total volume (50% ASC where applicable). Cells were incubated (5% CO2, 37°C) in ASC, Cp40/SCR treated ASC for 0.5hrs or 3hrs. Cells were harvested and immediately stained for CyTOF. CyTOF staining was performed at room temperature and all washes were by centrifugation of staining plates or tubes at 750 x g for 3 minutes. Cells were stained in cell staining buffer prepared as PBS with 1% BSA (cat. #A3059, Sigma-Aldrich) and 0.05% sodium azide (cat. #71289, Sigma-Aldrich). Isolated neutrophils were suspended in media (RPMI-1640 (cat. #11875-085, Gibco) supplemented with 5% heat-inactivated fetal bovine serum (cat. #16000044, Gibco), 1 mM GlutaMAX (cat. #35050079, Gibco), antibiotic-antimycotic (cat. #15240062, Gibco), 2 mM MEM non-essential amino acids (cat. #11140050, Gibco), 10 mM HEPES (cat. #15630080, Gibco), 2.5 × 10-5 M 2-mercaptoethanol (cat. #M3148, Sigma-Aldrich), and 25 units/mL benzonase nuclease (cat. #E1014, Sigma-Aldrich) and plated in 96-well round-bottom polypropylene plates (Corning life sciences). The cells were spun down and 5μm cisplatin viability staining reagent (cat. #201064, Fluidgm) was added and incubated for 2 minutes. Media was added to dilute cisplatin and cells were then fixed with 0.2% paraformaldehyde (PFA) (cat. #O4042-500, Fisher Scientific) and incubated for 5 minutes. After centrifugation, TruStain FcX Fc receptor blocking reagent (cat. #422302, Biolegend) was added for 10 minutes. The 43-marker neutrophil panel consisting of heavy metal–tagged antibodies was added directly and incubated for 30 minutes. Purified antibodies were obtained from a variety of different suppliers and labeled in house using the MaxPar kit (cat. #201300, Fluidgm) (Supplementary Table S2). Cells were washed with CyTOF cell staining buffer (CSB; calcium/magnesium-free PBS, 0.2% BSA, and 0.05% sodium azide). Media was added, cells were fixed with 4% PFA and then permeabilized with fixation/permeabilize buffer (cat. #00-5523-00, eBioscience) for 30 minutes. Permeabilization buffer (cat. #00-5523-00, eBioscience) was added directly and cells were spun down. Cells were barcoded using a 6 choose 3 palladium barcoding reagent (generated as described in (22)) and incubated for 15 minutes. After washes with CSB were performed to dilute excess barcoding reagent, cells were pooled together at 20 samples per tube. A 4% PFA fix was performed for 10 minutes. 18.75μm of iridium intercalator solution (cat.# 201192B, Fluidgm) was added to the cells and incubated for 20 minutes. Cells were washed with CSB and Cell Acquisition Solution (CAS) (cat. #201239, Fluidgm) and then suspended in CAS with EQ four element calibration beads (cat. #201078, Fluidgm). Compensation data was generated for each of the 43 metal-tagged antibodies used in this study using species appropriate compensation beads as described (23). Samples were then analyzed using a Helios CyTOF mass cytometer (Fluidgm).
CyTOF Pre-Data Analysis:
Data was normalized and then compensated using the catalyst package through R studio as described (23). The data was then debarcoded and uploaded into OMIQ (www.omiq.ai, Santa Clara, CA) for gating and analysis. Debris was gated out using gaussian gating strategy and the data was gated to produce singlet, bead negative, live cells. All other debris and nonessential cells were manually gated out using CD15+ positive and CD45low strategy, leaving only the neutrophil population for analysis. A randomized PCA was run and pre-embedded into the opt-SNE dimensional reduction algorithm. The opt-SNE algorithm was performed on the gated neutrophil data with a perplexity of 50, a theta of 0.5, with a random seed to reduce the high dimensionality data into two dimensions for easier analysis. The PARC analysis algorithm was run using the Euclidean distance metric in order to determine the number of clusters in the data. The results of the PARC were used to run a SPADE on opt-SNE in order to produce different clusters which represented the different neutrophil phenotypes.
Evaluation of trogocytosis of T cells by neutrophils:
Trogocytosis was assessed by live cell imaging, confocal microscopy, and flow cytometry. Live cell imaging was performed at the University at Buffalo Confocal Microscope and Flow Cytometry Facility. T cells were stained with PKH26 (cat. #MINI26, Sigma-Aldrich) according to manufacturer’s protocol and neutrophils were unstained. Before imaging, cells were cultured in IBIDI 8 well glass chambers (cat. #80827, ibidi USA Inc.) in 1:1 ratio in media or 50% ASC. T cells were stimulated with 25μl/mL anti-CD3/CD28 tetramers (cat. #10971, Stemcell Technologies). Three focal spots were selected per well and cells were imaged over the course of 3hrs at 37°C on Leica DMi 8 (63x glycerin immersion). Images were collected in Leica LAS X software. For quantification of neutrophils adhered to T cells and neutrophil morphology, images were de-identified and divided amongst 4 individuals for review.
For confocal imaging, T cells were stained with PKH26 while neutrophils were stained with PKH67 (cat. #MINI67, Sigma-Aldrich). T cells were stimulated with 25μl/mL anti-CD3/CD28 tetramers. Cells were cultured for 3hrs at 37°C on cover slips coated with 0.01% poly-L-lysine (cat. #A-005-C, EMD Millipore). After incubation, cover slips were washed and mounted with Prolong Diamond Mountant (cat. #P36965, Life Technologies). Slides were kept at 4°C and analyzed within 1 week. Samples were imaged using Leica TCS SP8 Laser scanning spectral confocal microscope (63x oil immersion) using Leica LAS X software.
For quantification of trogocytosis, T cells were stained with PKH26 while neutrophils were left unstained. T cells were stimulated with anti-CD3/CD28 tetramers (Stemcell). Cocultures were incubated for 3hrs (5% CO2, 37°C) before analysis by flow cytometry. In CD11b blockade experiments, neutrophils were pretreated with neutralizing anti-CD11b (clone ICRF44; cat. #301362, Biolegend) or mouse IgG1, k isotype for 30 minutes before being used in coculture. In Cp40/SCR treated conditions (see T-cell proliferation assays above), ASC was pretreated with 20μM Cp40/SCR before addition into coculture. CD66b (clone G10F5; cat. #305126, Biolegend) was used to gate on neutrophils.
Additional experiments on trogocytosis were performed as described previously (24). Blood leukocytes were separated based on density by centrifugation over isotonic Percoll (Pharmacia) with a specific density of 1.076 g/mL. The interphase fraction, containing PBMCs, was harvested for isolation of untouched T cells by magnetic-activated cell sorting with the pan T cell isolation kit (cat# 130-096-535, Miltenyi Biotec, Inc.) according to the manufacturer’s instructions. Neutrophils were obtained from the pellet fraction after erythrocyte lysis with hypotonic ammonium chloride solution at 4 °C. T cells were stained with Vybrant DiD (cat. #V22887, Invitrogen) and stimulated with 10ng/mL IL-15 (cat. #247-ILB-005, R&D Systems). Cocultures were incubated as described above and analyzed by flow cytometry.
Activation of NFAT and NF-κB in T cells:
NFAT and NF-κB activation were assessed based on nuclear translocation using the ImageStream platform. Neutrophils and T cells were plated in 24-well plates at 0.75×106 cells/well in 50% ASC, stimulated with anti-CD3/CD28 beads, and incubated for 2hrs (5% CO2, 37°C). Cells were stained with antibodies specific for CD4 (clone RPA-T4; cat. #300519, Biolegend) and CD8 (clone SK1; cat. #344706, Biolegend), then incubated in ice cold 2% formaldehyde for 10 minutes. Cells were washed then incubated with anti-NFAT1 (clone D43B1; cat. #5861, Cell Signaling Technology) or anti-NF-kB p65 (clone D14E12; cat. #8242, Cell Signaling Technology) for 25 minutes in a permeabilization buffer containing 0.1% Triton X in flow staining buffer (1% BSA, 1mM EDTA in PBS). This was followed by secondary antibody (polyclonal Donkey ant-rabbit IgG; cat. #711-605-152, Jackson ImmunoResearch) staining for 25 minutes. DAPI (0.5μg/mL; cat. #422801, Biolegend) was added before analysis on Amnis® Image Stream Mk-205 (Luminex corporation, Austin, TX). Data was analyzed in the IDEAS software where cells were gated on single cells in focus that were CD3+DAPI+. A morphology mask outlining the nucleus (DAPI channel) was created and similarity scores were generated to look for overlap of DAPI and NFAT or NF-κB. Similarity scores ≥ 1 were considered positive for nuclear translocation of NFAT or NF-κB.
Intracellular IL2 staining:
T cells, neutrophils and ASC were plated as described in T-cell suppression assays. Brefeldin A (10μg/mL; cat. #B7651-5MG, Sigma-Aldrich) was added at 4 and 18hrs after incubation and intracellular IL2 was measured 6hrs later (10 and 24hrs, respectively). Cells were stained with antibodies specific for CD4 (clone RPA-T4; cat. #562425, BD Biosciences) and CD8 (clone SK1; cat. #344706, Biolegend). Cells were fixed with 1x IC Fixing buffer (cat. #00-8222-49, eBiosciences) for 10 minutes, washed, resuspended in 1X Permeabilization Buffer (cat. #00-5523-00, eBiosciences), stained with an antibody specific for IL2 (clone MQ1-17H12; cat. #561054, BD Biosciences) for 25 minutes, then washed with 1X Permeabilization Buffer, and analyzed by flow cytometry.
Evaluation of glucose uptake, mitochondrial mass and depolarization, and mTOR activation:
T cells, neutrophils and ASC were plated as described in T-cell suppression assays. Glucose uptake by T cells was evaluated 48hrs after coculture using the fluorescently labeled deoxyglucose analog 2-NBDG (cat. #600470, Cayman Chemical Co.) following the manufacturer’s protocol. Cells were incubated in glucose-free RPMI-1640 (cat. #MT10–043-CV, Mediatech Inc.) with 100μM 2-NBDG at 37°C for 30 minutes, stained with antibodies specific for CD4 (clone RPA-T4; clone 560158, BD Biosciences), CD8 (clone SK1; cat. #344706, Biolegend) and Live/Dead fixable Aqua and analyzed by flow cytometry.
To evaluate mitochondrial mass and membrane potential, cells were cocultured for 48hrs (5% CO2, 37°C) then stained with 30nM MitoTracker Green FM (cat. #M7514, Thermo Fisher Scientific) or MitoTracker Orange CMTMRos (cat. #M7510, Thermo Fisher Scientific) at 37°C, 5%CO2 for 30 minutes. Cells were stained with antibodies specific for CD4 (clone RPA-T4; cat. #300519, Biolegend) or (clone SK3; cat. #344632, Biolegend), and CD8 (clone SK1; cat. #344714, Biolegend) then analyzed by flow cytometry.
Activation of mTOR in T cells was assessed by p70S6k phosphorylation 48hrs after coculture. Cells were stained with antibodies specific for CD4 (clone RPA-T4; cat. #300519, Biolegend) and CD8 (clone SK1; cat. #344706, Biolegend) then fixed and permeabilized with Perm/Wash Buffer I (cat. #557885, BD Biosciences), stained with primary antibody for p-p70S6k (Thr389) (cat. #9205, Cell Signaling Technology) followed by anti-rabbit secondary antibody (polyclonal Donkey anti-Rabbit IgG; cat. #711-605-152, Jackson ImmunoResearch). Samples were analyzed by flow cytometry.
Statistical Analysis:
For CyTOF data, Opt-SNE plots, opt-SNE density plots, PARC clustering, SPADE maps, and heat maps were created and analyzed using OMIQ (Santa Clara). Cluster abundance statistics were exported from OMIQ and analyzed using GraphPad Prism 8 (San Diego). Statistical analyses of thymidine uptake assays and flow cytometry MFI data were performed using the R Studio 4.0.2 statistical computing language. A nominal significance threshold of 0.05 was used. Statistical testing utilized 1-way ANOVA and prespecified interactions were tested within the ANOVA framework. Time course thymidine assays were analyzed using paired Wilcoxon while flow cytometry data with reported percentages and ELISA data were analyzed using two-tailed Mann-Whitney in GraphPad Prism 8.
Results
OC ascites fluid increases neutrophil lifespan, alters neutrophil nuclear morphology, and increases the proportion of low-density neutrophils
OC ascites contains pro-inflammatory cytokines and DAMPs that can activate neutrophils (10,25). For example, ASC from patients with newly diagnosed OC stimulated normal donor neutrophils to generate neutrophil extracellular traps (NETs) (25), a distinct mode of neutrophil death characterized by extracellular release of chromatin and granule constituents. However, inflammatory mediators (e.g., C5a and TNF-α) can also increase neutrophil lifespan. We evaluated the effect of ASC exposure on other neutrophil phenotypes and observed that ASC increased the lifespan of normal donor neutrophils (Supplementary Fig. S2a).
Both PMN-MDSC and N2 neutrophils are suppressive low-density granulocytes (26,27); however, low-density neutrophils are not unique to cancer, and can be induced by a number of stimuli. Incubation of normal donor mature neutrophils in ASC increased the number of low-density neutrophils based on co-sedimentation with PBMCs (Supplementary Fig. S2b). Neutrophils exposed to ASC also had increased uptake of fluorescently tagged BSA, reflecting extracellular protein uptake, which may alter density (Supplementary Fig. S2c). We next evaluated expression of lectin-type oxidized LDL receptor-1 (LOX-1), a recently defined PMN-MDSC marker and also inducible in normal neutrophils exposed to endoplasmic reticulum stress (28), in neutrophils exposed to ASC. Using x to denote fold-reduction in proliferation compared to anti-CD3/CD28 stimulated T cells alone, we defined ASC samples as capable of generating suppressors (x ≥ 1 log10), intermediate suppressors (0.5 log10 ≤ x < 1 log10), or non-suppressors (x < 0.5 log10), as previously described (8). Both suppressor and non-suppressor ASC similarly induced surface expression of LOX-1 (Supplementary Fig. S2d). Together, these findings show that normal mature neutrophils can acquire MDSC-like characteristics in the TME.
At diagnosis, OC ascites fluid neutrophils are morphologically mature and variably have hypersegmented nuclei (8), a feature associated with responses to Helicobacter pylori (13). It was unclear whether ASC induced nuclear hypersegmentation in situ, or whether neutrophils had undergone morphologic changes prior to recruitment into the peritoneal cavity. We therefore evaluated normal donor neutrophil nuclear morphology following exposure to ASC versus media. Based on cytology and STED imaging, ASC induced nuclear hypersegmentation (Supplementary Fig. S2e–f). Together, these results demonstrate that ASC modulates neutrophil lifespan, density, and nuclear morphology in ways that mimic responses to infection and injury.
OC ascites-activated neutrophils suppress tumor-associated lymphocytes via complement-dependent signaling
Purified ascites fluid neutrophils from patients with newly diagnosed OC suppress ex vivo stimulated proliferation of T cells (5). While circulating neutrophils from OC patients or normal donor neutrophils are not intrinsically suppressive of T-cell function, they acquire a suppressor phenotype following ASC exposure (8). This suppressor activity requires several neutrophil effector functions and is fully abrogated by Cp40, a peptide inhibitor of complement activation that works by binding to C3 and interfering with C3 convertase formation (8). The prior studies are based on an ex vivo model of neutrophil–T-cell interactions in the TME using purified circulating neutrophils and T cells at concentrations in the range of those observed in OC ascites fluid.
In this study, we asked whether ASC-activated neutrophils would suppress proliferation of tumor-associated lymphocytes (TALs). TALs from banked frozen or fresh OC ascites were sort-purified and cocultured in autologous ASC with normal donor neutrophils and appropriate controls. Neutrophils + ASC reduced anti-CD3/CD28 stimulated proliferation of TALs to unstimulated levels and suppression was fully abrogated by Cp40 (Fig. 1a). These results further support neutrophil suppression of T-cell function in the TME as an obstacle to the activation of TALs required for durable antitumor immunity and point to the potential of C3 inhibition to overcome this barrier.
Figure 1: Complement activation drives neutrophil-mediated suppression of OC TAL and neutrophil suppressor function induced by surgical injury.
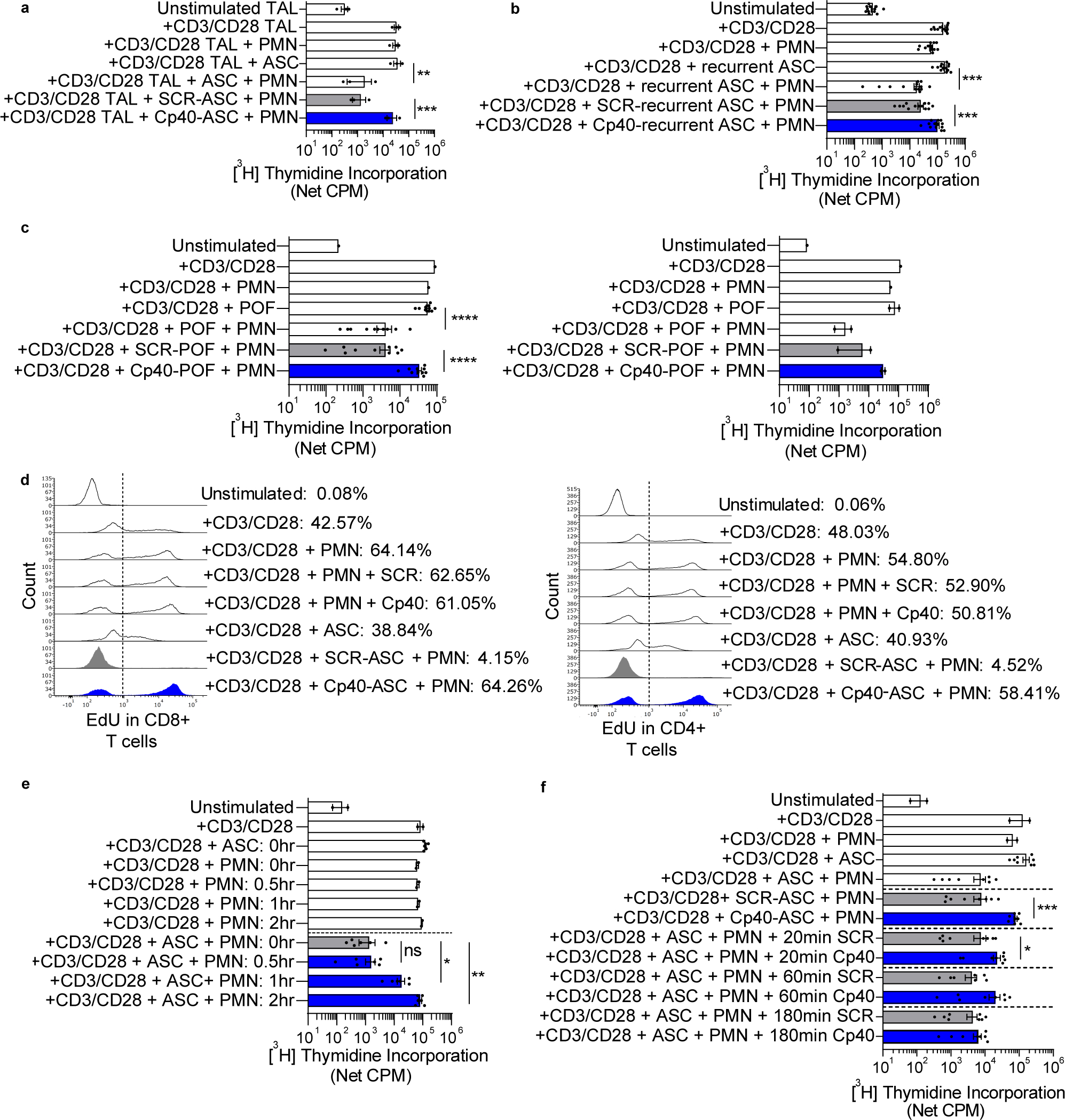
(a) ASC + PMN suppressed anti-CD3/CD28 stimulated TAL proliferation, an effect abrogated by Cp40. TALs (n=3) were incubated with autologous ASC collected at OC diagnosis or media, with Cp40, a peptide inhibitor of C3 activation, or scramble peptide (SCR), with or without normal donor neutrophils (PMN). TAL proliferation was assessed at 96hrs by [3H]-thymidine incorporation. (b-f) Subsequent experiments involved autologous normal donor neutrophils and T cells. (b) ASC from patients with recurrent OC induced C3-dependent neutrophil suppressor function. Anti-CD3/CD28 stimulated T cells were incubated with neutrophils and ASC from patients with recurrent OC (n=13). (c) POF from OC patients or patients without cancer undergoing diagnostic lung surgery induced a complement-dependent neutrophil suppressor phenotype. T cell and neutrophil cocultures were plated as described in panel b, except POF supernatants from OC patients after debulking surgery (n=10, left) or patients undergoing lung surgery (n=2, right) were used. (d) Flow cytometry–based EdU incorporation was used to quantify the effects of neutrophil suppressors and Cp40 on the proportion of CD8+ and CD4+ T cells undergoing proliferation. Anti-CD3/CD28 stimulated T cells were incubated with neutrophils and ASC from patients with newly diagnosed OC or media for 96hrs. Data are representative of two experiments. (e) ASC + PMN suppress T-cell proliferation when added ≤1hr after T-cell stimulation. T cells were stimulated with anti-CD3/CD28. ASC + PMN were added at the same time (0hr) or at 0.5, 1, or 2hrs after T-cell stimulation. Proliferation was assessed at 96hrs. Data pooled from two independent experiments. (f) Addition of Cp40 within 1hr of anti-CD3/CD28 stimulation abrogated neutrophil suppressor function. T cells were cultured with neutrophils and ASC (n=8) and Cp40/SCR was added at 0, 20, 60, or 180 minutes. Data pooled from two independent experiments. Symbols represent individual samples (n) and bars represent ± SEM. Data from a–c were analyzed by ANOVA with Tukey post hoc test with pre-specified groups. Data from e and f were analyzed by paired Wilcoxon (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Recurrent OC ascites fluid and surgical injury induce similar complement-driven neutrophil suppressor function
Metastatic OC cannot be cured and recurrence is expected following surgery and chemotherapy. The results of trials evaluating single-agent checkpoint inhibitors in recurrent OC have been disappointing (29), suggesting that immunosuppression pathways may impair their efficacy. Similar to ASC from patients with newly diagnosed OC, ASC from patients with tumor recurrence induced a C3-dependent neutrophil suppressor phenotype (Fig. 1b). These results point to a persistent complement-dependent neutrophil-driven suppression in OC ascites fluid both at diagnosis and in recurrent disease that may be a barrier to immunotherapy.
To understand if induction of a neutrophil suppressor phenotype occurs as a result of injury, we evaluated post-operative drainage abdominal fluid supernatants from OC patients after primary surgery or patients without cancer undergoing diagnostic lung surgery. Similar to malignant effusions, POF from patients without cancer undergoing diagnostic lung surgery induced a complement-dependent neutrophil suppressor phenotype (Fig. 1c). Although POF after OC debulking could be influenced by the pre-operative TME and microscopic residual tumor, this concern doesn’t apply to non-cancer surgery. These results suggest that neutrophils acquire a suppressor function as a general response to injury rather than this being a distinct feature of the TME.
Neutrophil-mediated suppression occurs early after T-cell stimulation
To determine the effects of neutrophil suppressors and C3 on the proportion of T cells undergoing proliferation, we used a flow cytometry–based EdU incorporation assay. Approximately 40–50% of T cells were EdU+ following anti-CD3/CD28 stimulation. Neutrophils + ASC, but not neutrophils alone or ASC alone, suppressed T-cell proliferation to unstimulated levels, an effect that was abrogated fully by Cp40 (Fig. 1d). Cp40 has no effect on the proliferation of anti-CD3/CD28 stimulated T cells.
To delineate the timeframe for when neutrophils + ASC cause T-cell non-responsiveness, we delayed the addition of neutrophils and ASC after T-cell stimulation (Fig. 1e). The combination of neutrophils + ASC fully suppressed T-cell proliferation when added 30 minutes after anti-CD3/CD28 stimulation, had a partial effect when added at 1hr, and was not suppressive if added at 2hrs. Addition of Cp40 within 1hr after anti-CD3/CD28 stimulation abrogated neutrophil suppressor function, while rescue of T-cell proliferation was lost if Cp40 was added after 3hrs (Fig. 1f). These results point to a critical window in which already activated T cells are rendered non-responsive by suppressor neutrophils and T-cell proliferation is rescued by C3 inhibition.
Complement-induced neutrophil suppressor function is driven by the alternative and classical pathways
Consistent with the results of Bjørge et al. (30), we observed that the majority of ASC samples activated both the alternative and classical pathways, with only 3/38 samples activating the lectin pathway (Supplementary Fig. 3). Activation of the alternative pathway was significantly increased in suppressor ASC versus non-suppressor ASC samples (Supplementary Fig. S3b), but there were no significant differences in activation of classical or lectin pathways between suppressor and non-suppressor ASC. Suppressor ASC contained significantly higher levels of C3 and C5a than non-suppressor ASC (Supplementary Fig. S3c,d). Interestingly, suppressor ASC contained lower levels of C3 and C4 activation products compared with non-suppressor ASC (Supplementary Fig. S3e), whereas paired serum samples from the same patients showed similar levels of C3 and C4 activation products (Supplementary Fig. S3f). These findings raise the possibility that higher levels of intact, undegraded C3 (and therefore lower levels of C3 activation products) in suppressor ASC may result in more C3 available for activation of neutrophils and induction of suppressor function.
SALO, derived from the saliva of the sand fly Lutzomyia longipalpis, is a small peptide inhibitor of the classical pathway (18). Pre-treatment of ASC with SALO partially abrogated neutrophil suppressor activity (Fig. 2a). Properdin stabilizes the C3bBb convertase required for alternative pathway activity (31) and may also serve as a platform for alternative pathway activation. Once bound to the convertase on the cell membrane, properdin increases the half-life of the convertase by 5–10 fold, thus promoting C3b deposition and amplifying alternative pathway activation. Neutralizing anti-properdin monoclonal antibody 6E11A4 (32) significantly abrogated neutrophil suppressor activity, resulting in a ~1-log increase in T-cell proliferation compared with isotype control (Fig. 2a). The combination of SALO and anti-properdin fully abrogated neutrophil-mediated T-cell suppression (Fig. 2a). Factor B binds to C3b and is cleaved by Factor D into the Ba and Bb subunits to form the alternative C3 convertase C3bBb. Consistent with the effect of properdin inhibition, small molecule inhibitors of Factor D (15) and Factor B (Supplementary Fig. S4) abrogated neutrophil suppressor activity (Fig. 2b,c), indicating a major role for the alternative pathway in neutrophil-mediated T-cell suppression.
Figure 2: Complement-induced neutrophil suppressor function is driven by the alternative and classical pathways, and ASC-stimulated membrane expression of CR3 and neutrophil degranulation are dependent on C5aR signaling.
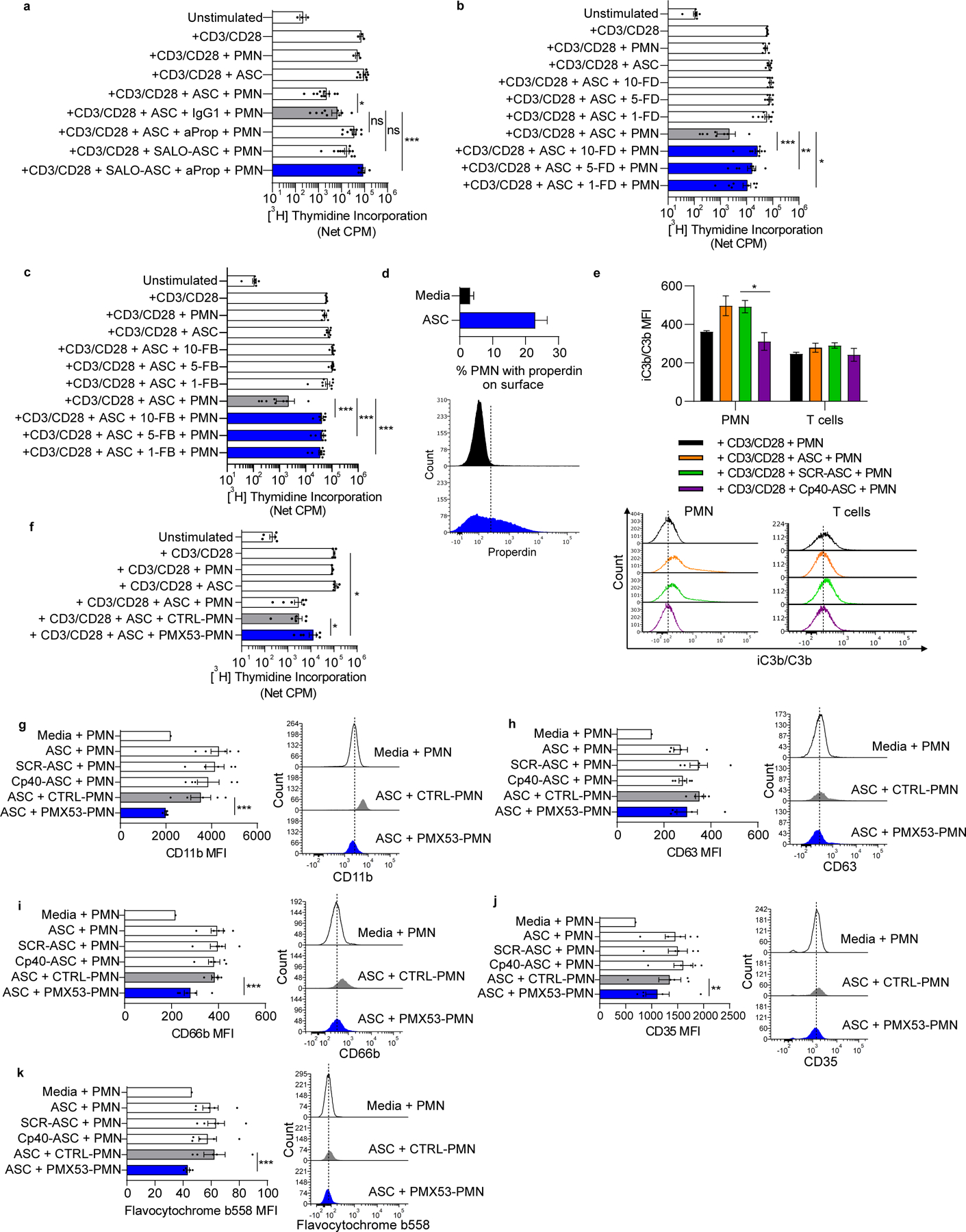
Cocultures consisted of normal donor neutrophils and T cells and ASC from patients with newly diagnosed OC. (a) Inhibition of the classical and alternative pathways abrogated neutrophil suppressor function. SALO, a peptide inhibitor of the classical pathway, and anti-properdin mAb 6E11A4 (aProp) abrogated ASC-induced neutrophil suppressor function. IgG1= isotype control. Data pooled from two independent experiments. (b,c) Inhibition of Factor D (FD) or Factor B (FB) abrogated neutrophil suppressor function. ASC (n=8) was treated with a FD inhibitor or FB inhibitor before addition to cocultures (10μM, 5μM or 1μM final concentration). (d) ASC increased neutrophil surface expression of properdin. Neutrophils were incubated in ASC (blue, n=8) or media (black) for 90 minutes and surface expression of properdin was quantified by flow cytometry. Upper panel is representative of two independent experiments. Bottom panel shows a representative flow plot. (e) ASC increased surface expression of iC3b/C3b on neutrophils, whereas no significant effect was observed on T cells. T cells and neutrophils were incubated with ASC (n=6) or media for 90 minutes with or without Cp40/SCR and surface expression of iC3b/C3b was quantified by flow cytometry. Data representative of two independent experiments. Representative flow overlay is shown below. (f) C5aR inhibition partially abrogated neutrophil suppressor function. Neutrophils were pre-treated with the C5aR inhibitor, PMX53 or peptide control (CTRL), for 30 minutes, washed, then cocultured with T cells. (g-k) ASC increased neutrophil surface expression of CD11b and of granule membrane constituents, an effect that was partially or fully abrogated by C5aR inhibition. Neutrophils were pretreated with PMX53/CTRL, washed, then incubated in ASC or media for 2.5hrs and analyzed by flow cytometry. A representative flow plot is shown on the right side of each panel. Data representative of two independent experiments. Symbols represent individual samples (n) and bars represent ± SEM. Data from a and f were analyzed by ANOVA with Tukey post hoc test while data from b, c, e, and g-k were analyzed by Mann-Whitney (*p<0.05, **p<0.01, ***p<0.001).
Release of properdin from neutrophil secondary granules can promote alternative pathway activation and amplify neutrophil responses, including NADPH oxidase activation (33,34). ASC increased the proportion of neutrophils with surface properdin (likely bound to C3b or a convertase) (Fig. 2d). In addition, ASC increased iC3b/C3b deposition on neutrophil membranes, an effect that was decreased by Cp40 (Fig. 2e). In contrast, iC3b/C3b deposition on T cells was unaffected by Cp40. Thus, ASC can amplify complement activation on neutrophil membranes through augmentation of surface alternative pathway C3 convertase stabilized by properdin, which is required for induction of neutrophil suppressor function.
OC ascites fluid stimulated membrane expression of CR3 and neutrophil degranulation are dependent on C5aR signaling
C5a can prime neutrophils resulting in increased degranulation, NADPH oxidase activation, and CD11b expression (32,35). To evaluate whether C5a receptor 1 (C5aR) on neutrophils contributes to suppression of T-cell proliferation, we pretreated neutrophils with PMX53, a C5aR antagonist, followed by ASC exposure. PMX53 partially rescued T-cell proliferation (Fig. 2f), which was consistent with our previous findings using inhibitors of C5 activation (8).
Activation of neutrophils can increase surface expression of various membrane-bound proteins through fusion of secretory vesicles and granules with the plasma membrane (36,37), a process that enhances neutrophil trafficking and antimicrobial defense. Surface expression of CD11b/CD18 (CR3), which mediates both cell–cell adhesion and binding to iC3b, can increase upon neutrophil activation by infection, microbial products, and pro-inflammatory cytokines and chemokines due to secretory vesicle fusion (36,38). ASC induced mobilization of neutrophil secretory vesicles and granules to the plasma membrane (Fig. 2g–k). Inhibition of C5aR significantly reduced this ASC-induced CD11b surface expression to unstimulated levels (Fig. 2g). These findings suggest a positive feedback loop in the complement cascade in which stimulation of C5aR increases CR3 signaling. Inhibition of C5aR also significantly decreased levels of CD35 and CD66b on the neutrophil surface, but it had no obvious effect on CD63 expression (Fig. 2h–j).
The phagocyte NADPH oxidase is composed of the membrane-bound flavocytochrome b558 heterodimer (gp91phox/p22phox) and cytosolic constituents (p47phox, p67phox, p40phox and Rac2) that translocate to the membrane component following neutrophil activation and are required for NADPH oxidase function. In neutrophils, flavocytochrome b558 is mostly expressed in membranes of secondary granules (39). Neutrophil activation can stimulate fusion of secondary granules with the plasma membrane resulting in redistribution of flavocytochrome b558 to the plasma membrane and increasing extracellular ROS production. ASC increased surface expression of flavocytochrome b558 on neutrophils, and this was blocked by PMX53, indicating a dependence on C5aR signaling (Fig. 2k). In contrast, Cp40 did not significantly alter surface expression of CD11b, granule surface proteins, or flavocytochrome b558 (Fig. 2g–k). Although Cp40 inhibits C3 convertase and downstream new generation of C5a, it does not affect endogenous C5a in ASC that can signal to neutrophils. Thus, these data indicate an important role for C5aR signaling in neutrophil suppressor function, an effect that may be mediated by priming of multiple downstream neutrophil effector functions through increased surface mobilization of CR3 and NADPH oxidase.
Neutrophil suppressor function requires NADPH oxidase activation, but not extracellular ROS
CGD is an inherited disorder of the phagocyte NADPH oxidase characterized by severe bacterial and fungal infections and by excessive inflammation. To delineate the role of NADPH oxidase in neutrophil-mediated T-cell suppression, we evaluated neutrophils from patients with gp91phox-deficient or p47phox-deficient CGD (Supplementary Table S1). Normal donor neutrophils suppressed T-cell proliferation following ASC exposure, whereas similarly treated neutrophils from gp91phox-deficient CGD patients did not suppress anti-CD3/CD28 stimulated T-cell proliferation (Fig. 3a). Neutrophils from p47phox-deficient patients had a modest suppressor effect that did not reach statistical significance. These results indicate a requirement for NADPH oxidase in neutrophil suppressor function.
Figure 3: Neutrophil suppressor function requires NADPH oxidase activation, but not extracellular ROS.
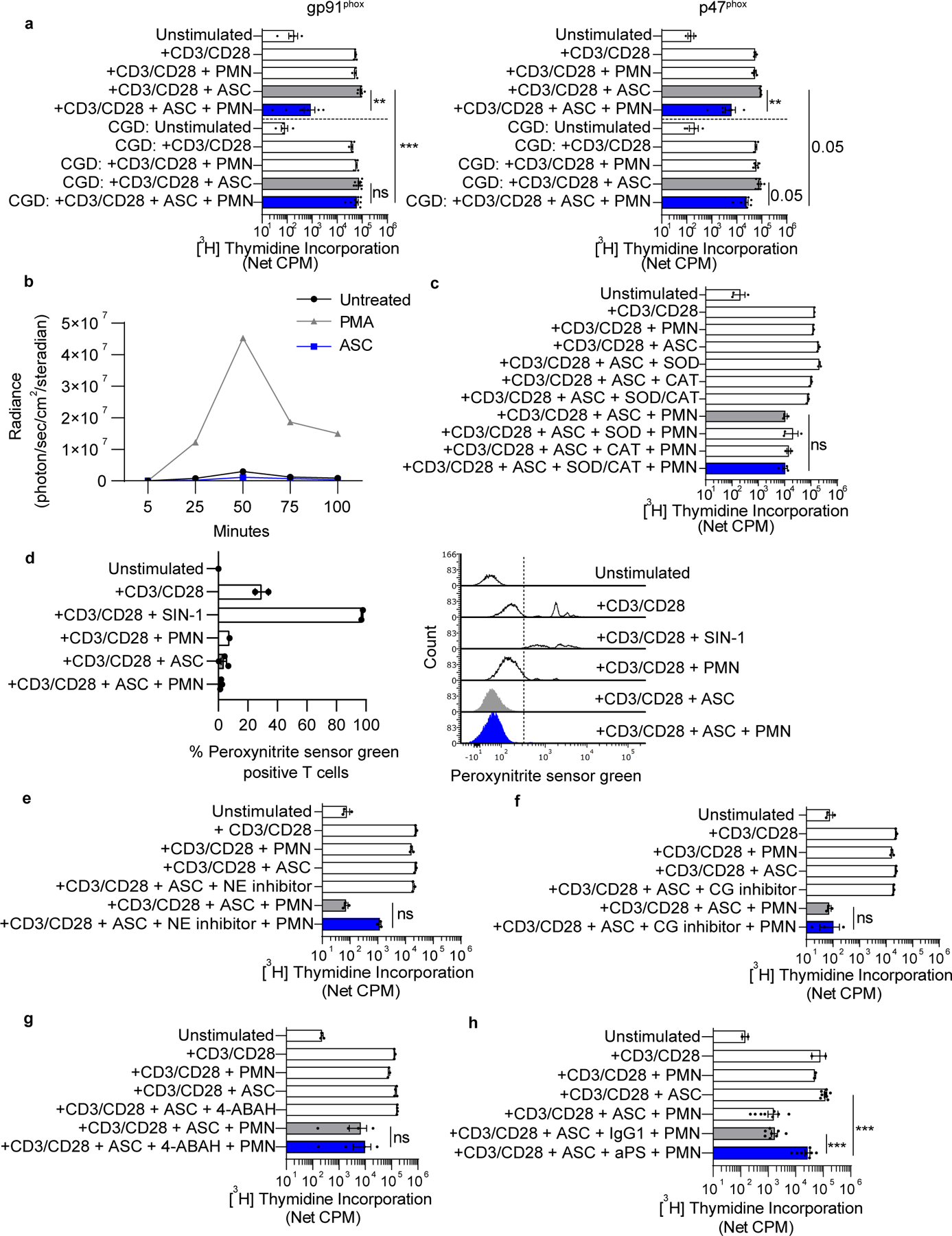
(a) Normal donor neutrophils suppressed T-cell proliferation following ASC exposure, whereas similarly treated neutrophils from gp91phox-deficient patients with CGD did not. Neutrophils from p47phox-deficient CGD patients + ASC had a modest effect on T-cell proliferation that trended to statistical significance (p = 0.05). Autologous T cells and neutrophils from patients with gp91phox-deficient (n=5) or p47phox-deficient (n=4) CGD were used in T-cell proliferation assays and compared to similarly treated cocultures containing normal donor cells. (b) ASC did not induce intracellular ROS generation in neutrophils. Normal donor neutrophils were cultured in media containing Diogenes and Luminol. ASC was added immediately before imaging and luminescence was recorded in 5 minute intervals. PMA was used as a positive control. Data representative of two independent experiments. (c) SOD, CAT, and the combination of SOD + CAT did not abrogate neutrophil suppressor function. Normal donor T cells were cultured with normal donor neutrophils and ASC, super oxide dismutase (SOD), catalase (CAT), or SOD + CAT, and proliferation was measured at 96hrs. (d) ASC-activated neutrophils did not augment peroxynitrite formation on T-cell membranes. Normal donor T cells and neutrophils were incubated with peroxynitrite sensor green for 21hrs before analysis by flow cytometry. Peroxynitrite sensor green reacts with ONOO− to generate a fluorescent product. SIN-1 is a nitric oxide donor that serves as a positive control. Representative flow overlay is shown on the right. (e-g) Neutrophil suppressor function was not dependent on neutrophil elastase (NE), cathepsin G (CG), or myeloperoxidase (MPO). T cells were incubated with neutrophils and ASC in the presence of Sivelestat (NE inhibitor), Cathepsin G Inhibitor I (CG inhibitor), or 4-ABAH (MPO inhibitor). Proliferation was assayed as described previously. (h) Anti-phosphatidylserine monoclonal antibody (aPS, clone 1H6) abrogated neutrophil suppressor function, while IgG1 isotype had no effect (n= 8 ASC samples). Symbols represent individual samples (n) and bars represent ± SEM. Statistical comparisons were by ANOVA with Tukey post hoc test (**p<0.01, ***p<0.001).
We next evaluated whether the suppressive effect of neutrophils + ASC was the result of NADPH oxidase–generated ROS acting directly on T cells. For example, neutrophil NADPH oxidase–generated ROS and downstream metabolites (e.g., peroxynitrite anion) can cause nitration of the TCR of T cells leading to impaired T-cell activation and concomitant loss of antigen recognition (40). We observed that incubation of normal donor neutrophils in ASC did not stimulate extracellular ROS production (Fig. 3b). Furthermore, the addition of SOD, catalase, or SOD + catalase to ASC to neutralize ROS did not abrogate suppression of T-cell proliferation, arguing against a direct role for extracellular ROS in neutrophil suppressor function (Fig. 3c). In addition, we did not observe increased peroxynitrite formation on T cells after coculture with neutrophils + ASC (Fig. 3d). These data suggest that suppression of T-cell proliferation does not occur from a direct effect of NADPH oxidase–generated ROS, and instead may involve intracellular signaling regulated by NADPH oxidase.
Activation of the neutrophil NADPH oxidase results in solubilization and activation of serine granular proteases (e.g., neutrophil elastase and cathepsin G) (41). Small molecule inhibitors of neutrophil elastase, cathepsin G, and myeloperoxidase did not abrogate neutrophil suppressor function (Fig. 3e–g). Externalization of phosphatidylserine (PS) on cell surfaces is known to have broad immunosuppressive properties that limit innate and T-cell responses (42). Neutrophil NADPH oxidase mediates externalization of PS and its catabolite, lysophosphatidylserine (lyso-PS), in both activated live and dying neutrophils (43). We observed that anti-PS abrogated the suppressor function of ASC-activated neutrophils, resulting in increased anti-CD3/CD28 stimulated T-cell proliferation by at least 1-log10 compared with isotype (Fig. 3h).
OC ascites fluid induces distinct neutrophil subpopulations based on surface protein expression
Although the notion of neutrophil heterogeneity has been posited for decades, little is known about neutrophil subpopulations in the human TME. We hypothesized that ASC would induce one or more unique neutrophil subpopulations that were distinct from circulating neutrophils. We used CyTOF to evaluate 43 surface markers on purified normal donor neutrophils after incubation in ASC known to induce suppressor neutrophils. Using opt-SNE analysis, 15 distinct clusters of neutrophil surface marker expression were discovered (Fig. 4a), whose significance was confirmed using multiple t-tests. Comparing neutrophils incubated in media with those incubated in ASC for 0.5 and 3hrs, we observed a significant decrease in the neutrophil population residing in cluster 13 upon treatment with ASC (Fig. 4b–c). Cluster 13 is defined by high expression of CXCR1/2, CD16, CD15, BLT1, CD95, CD88 and CD162, and is suggestive of a circulating neutrophil that can respond to chemoattractants. After only 0.5hr exposure to ASC, neutrophils showed an increased percentage of cells that appeared in clusters 1, 3 and 9, although statistical significance was not reached. Clusters 1, 3 and 9 have high expression of molecules suggestive of neutrophil activation and possible degranulation, such as increased CD10, activated CD11b, CD172a/b, and CD66b. CD10 expression has been linked to a suppressive population of neutrophils (44) and can be increased after C5a exposure (45). We observed by flow cytometry that ASC induced CD10 expression in neutrophils (Supplementary Fig. S5a).
Figure 4: ASC exposure induces distinct neutrophil subpopulations based on CyTOF profiling.
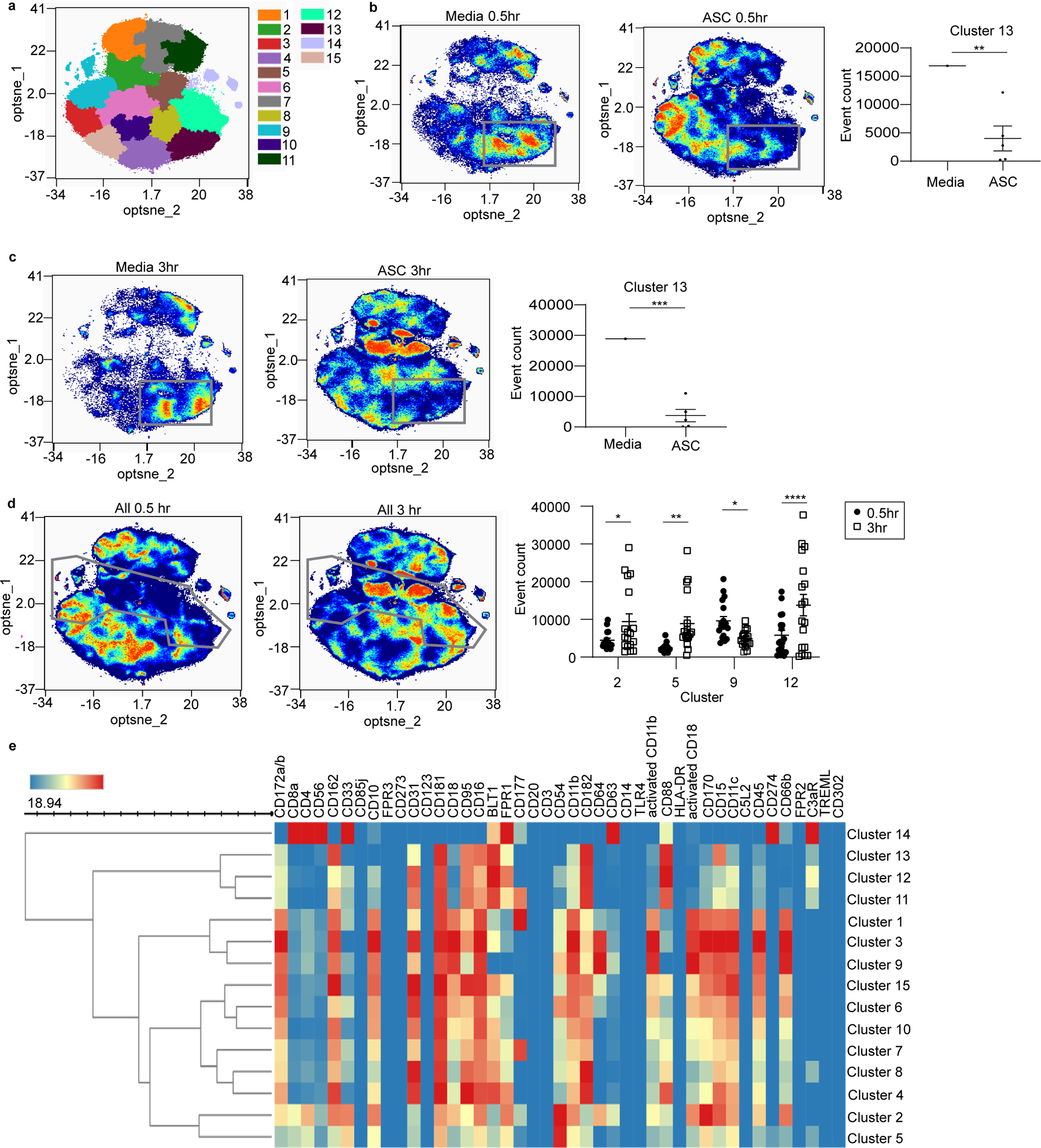
(a) Normal donor neutrophils incubated in ASC or media for 0.5 or 3hrs were surface labeled using heavy metal-conjugated antibodies against 43 surface proteins and analyzed by CyTOF. Clustering of the data using opt-SNE analysis identified 15 distinct neutrophil clusters denoted by the indicated colors. (b,c) Comparison of media vs ASC at 0.5 and 3hr showed a significant decrease in the percentage of neutrophils occupying cluster 13 (outlined by grey boxes). (d) Comparison of ASC samples at 0.5 and 3hr showed significant increases in neutrophil populations within clusters 2, 5, and 12 and a decrease in cluster 9 at 3hr (borders of these clusters are outlined in grey). (e) Heat map indicating marker expression levels that define each individual cluster. Opt-SNE plots, opt-SNE density plots, PARC clustering, SPADE maps, and heat maps were created and analyzed using OMIQ (Santa Clara, CA). Cluster abundance statistics were exported from OMIQ and analyzed using multiple t-tests (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Comparison of all samples at 0.5 and 3hrs showed significant increases in the percentage of cells residing in clusters 2, 5, and 12 and a decrease in cluster 9 at 3hrs (Fig. 4d). The marker expression defining each cluster is shown in Figure 4e. Cluster 2 has high expression of adhesion molecules (CD54, CD162, activated CD11b, and CD18) as well as CD10, CD170, CD11c and CD66b. Cluster 5 has high expression of CD33, CD181 and CD54, but low expression of CD172a/b, CD10, activated CD11b, and CD66b. Cluster 12 has high expression of CD31, CD181, CD95, CD16, BLT1, FPR1, CD182, and CD88. Neutrophils incubated with suppressor ASC treated with Cp40 at 0.5hr showed a significant increase in cluster 12 compared to SCR treatment (Supplementary Fig. S5b). Cluster 12 was also significantly increased after 3hr incubation in non-suppressor ASC (Supplementary Fig. S5c). Incubation of neutrophils with non-suppressor ASC resulted in an absence of cells in clusters 2 and 5 and an increase in clusters 8 and 12. Together, these results indicate that the TME modifies the relative proportion of distinct neutrophil subpopulations.
OC ascites-activated neutrophils damage T cells by trogocytosis of T-cell membranes
Neutrophils can damage extracellular parasites (46) and tumor cells (47) by trogocytosis, a process involving transfer of plasma membrane fragments between conjugated cells. We observed using live cell imaging that upon coculture in ASC, neutrophils and T cells form stable interactions and neutrophils acquired pieces of T-cell membrane (Fig. 5a). Trogocytosis by neutrophils was confirmed by confocal microscopy (Fig. 5b) and quantified by flow cytometry. Membrane transfer occurred in media only, but it was increased by ASC based on MFI of T-cell membrane dye on neutrophils (Fig. 5c) and by the percentage of neutrophils positive for T-cell membrane dye (Fig. 5d–e). Trogocytosis was inhibited by anti-CD11b, but not by C3 inhibition (Fig. 5d–e), suggesting that CD11b/CD18 drives trogocytosis by enabling neutrophil adherence to T cells. Fc-blockade had no effect on trogocytosis, arguing against opsonizing antibodies enhancing trogocytosis. The proportion of neutrophils binding to T cells was similar in ASC and media (Fig. 5f). In media, neutrophils had a predominantly rounded morphology; however, in ASC, neutrophils were more likely to be elongated and flattened and caused stretching of T-cell membranes when bound to T cells. Neutrophil elongation and increased movement lasted about 80 minutes before neutrophils again assumed a rounded morphology that may reflect neutrophil desensitization (Fig. 5g). These results raise the possibility of trogocytosis as a mode of neutrophil-driven T cell injury in the TME and that additional injury or signaling pathways are required to render T cells non-responsive to anti-CD3/CD28 stimulation.
Figure 5: Ascites fluid-activated neutrophils damage T cells by trogocytosis of T-cell membranes.
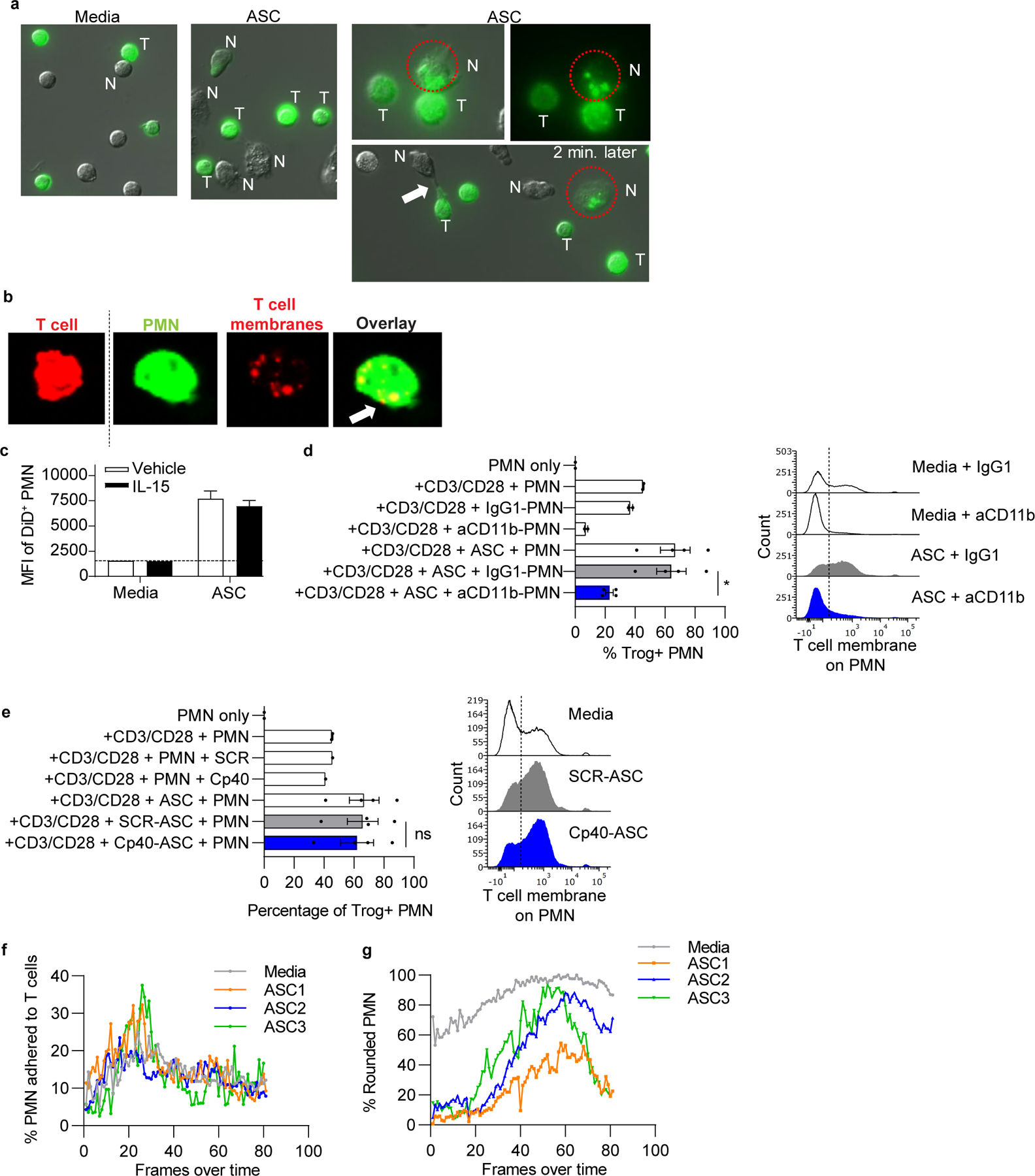
(a) Normal donor T cells were stained with a membrane dye, stimulated with anti-CD3/CD28 tetramers, and incubated with normal donor neutrophils and/or ASC. Cocultures were imaged by fluorescent microscopy over 3hrs. Representative images show cell contact and transfer of labeled T-cell membranes (green) to neutrophils in ASC cocultures. Circle shows neutrophil adherent to a T cell and containing T-cell membrane fragments, then separated from the T cell 2 minutes later. Arrow, neutrophil stretching T-cell membrane. N= neutrophil, T = T cell. (b) Anti-CD3/CD28 stimulated T cells (red) and neutrophils (green) were stained with membrane dyes, cocultured for 3hrs on glass coverslips, and analyzed by confocal microscopy. Representative images (left to right) of an intact T cell and a neutrophil with internalized T-cell membranes as demonstrated in the red channel and overlay image showing overlap of dyes (arrow). (c) ASC augments trogocytosis of T-cell membranes by neutrophils. T cells (n=3 normal donors) were stained with Vybrant DiD, stimulated with IL-15, and incubated with neutrophils in ASC (n=3) for 3hrs. Quantification of membrane transfer was assessed using MFI of Vybrant DiD on neutrophils. Results are from one experiment. (d,e) Neutrophil-mediated trogocytosis of T-cell membranes is dependent on CD11b, but independent of complement C3 activation. T cells and neutrophils were prepared and incubated as mentioned in panel a. Percent neutrophils with T-cell membrane (Trog+ neutrophils) was assessed by flow cytometry. Where indicated, neutrophils were pretreated with blocking anti-CD11b or isotype control (IgG1) (d) or ASC was pretreated with Cp40 (e). Symbols represent individual samples (n) and bars represent ± SEM. Statistical comparisons were by ANOVA with Tukey post hoc test (*p<0.05). (f,g) Percent neutrophils adhered to T cells was unaffected by ASC exposure. ASC induced elongation of neutrophils at early time points. Percent neutrophils adhered to T cells and neutrophil morphology following ASC exposure were quantified over time by live cell imaging. Neutrophil morphology was categorized as rounded or elongated, reflecting activation. Data are representative of two independent experiments unless otherwise noted.
Neutrophil suppressors inhibit nuclear NFAT translocation and IL-2 production in T cells
We next examined activation of downstream NF-κB and NFAT by quantifying nuclear translocation on Imagestream. Although nuclear translocation of NF-κB was not altered in neutrophil + ASC cocultures, the translocation of NFAT was reduced (Fig. 6a–b, Supplementary Fig. S6a–b). To assess downstream functional effects of neutrophil-suppressed T cells, we evaluated intracellular IL2 production. T cells cultured with ASC or neutrophils alone showed decreased production of IL2, however, the combination further decreased IL2 production to levels seen in unstimulated cells (Fig. 6c, Supplementary Fig. S6c).
Figure 6: Neutrophil suppressors inhibit nuclear NFAT translocation, IL2 generation, glucose uptake, mitochondrial function and mTOR activation in stimulated T cells.
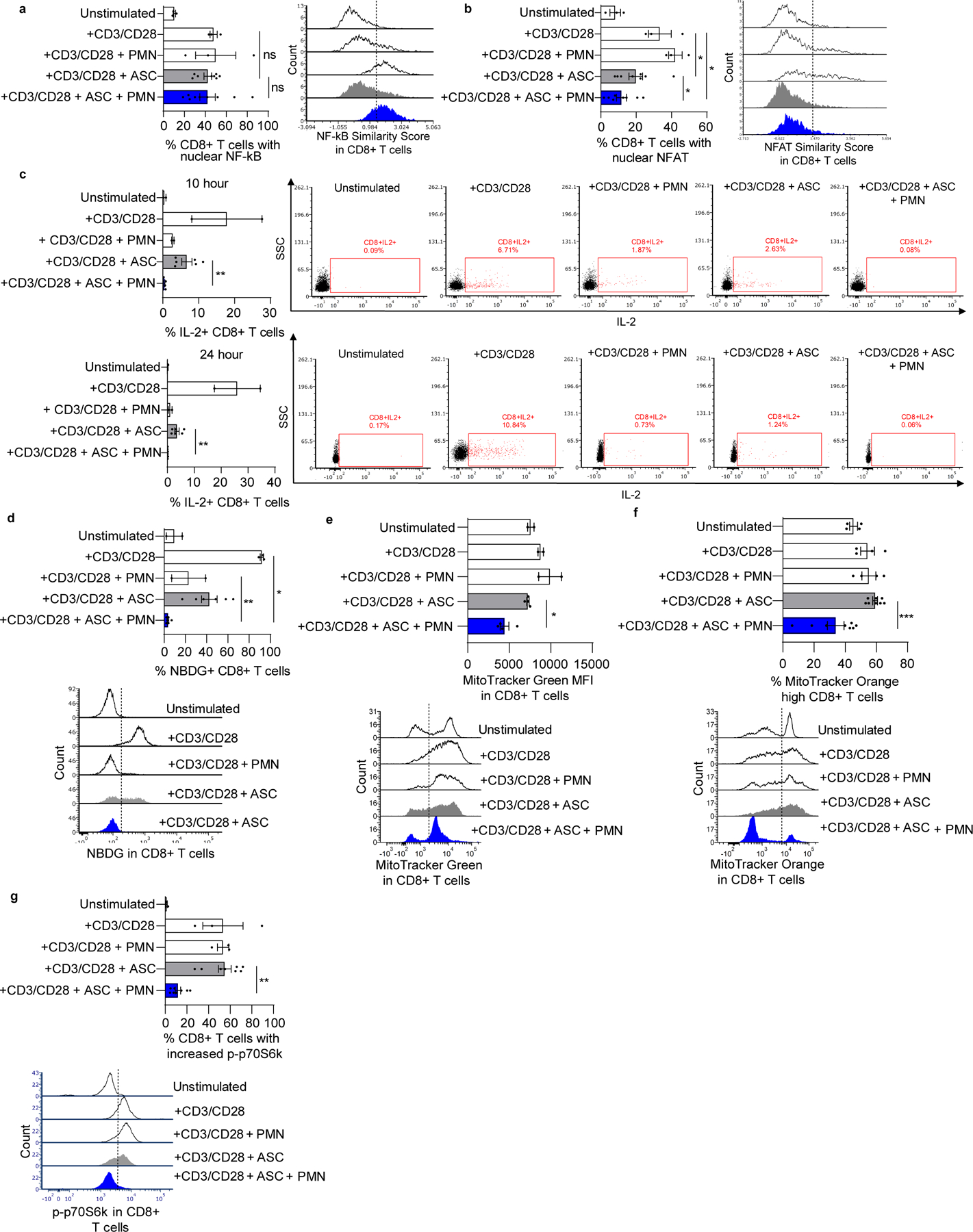
(a,b) Neutrophil suppressors inhibit activation of NFAT but not NF-κB in anti-CD3/CD28 stimulated T cells. Nuclear translocation of NF-κB and NFAT was evaluated 2hrs after stimulation using ImageStream. Representative flow overlays are shown on the right of each graph. (c) Sustained suppression of intracellular IL2 in CD8+ T cells by neutrophils and ASC. T cells were incubated with neutrophils and ASC (n=6). Brefeldin A was added at 4 and 18hrs after coculture and IL2 was evaluated 6hrs later by flow cytometry. Representative flow plots are shown on the right of each graph. (d) Combination of neutrophils and ASC decreased glucose uptake by T cells. T cells were cocultured with neutrophils and ASC (n=6) for 48hrs, followed by addition of NBDG. NBDG uptake by T cells was evaluated at 30 minutes by flow cytometry. Data pooled from two independent experiments. Representative overlay is shown below. (e,f) T cells were cocultured with neutrophils and ASC (n=8) for 48hrs then stained with MitoTracker Green FM or MitoTracker Orange CMTMRos to measure mitochondrial mass and mitochondrial membrane potential, respectively. Left panels show aggregate data, right panels show representative flow plot. Data in g is representative of two independent experiments. Data in h are pooled from two independent experiments. Representative overlays are shown below each graph. (g) T cells were cocultured with neutrophils and ASC (n=8) for 48hrs. mTOR activation was assayed by quantifying the percentage of cells showing phosphorylated p70S6 kinase (p-p70S6k) by flow cytometry. Left panel shows aggregate data; right panel shows representative flow plot. CD4+ T cells analyzed in the same experiments showed similar results (Supplementary Fig. S6). Symbols represent individual samples (n) and bars represent ± SEM. Statistical comparisons were by two-tailed Mann-Whitney (*p<0.05, **p<0.01, ***p<0.001).
Suppressor neutrophils cause severe metabolic disruptions in stimulated T cells
T-cell activation, proliferation, and effector functions require increased protein synthesis and metabolic changes. We evaluated whether ASC and neutrophils cause metabolic changes in T cells by measuring glucose uptake and mitochondrial mass and membrane potential, which increase upon proliferation (48). Glucose uptake by T cells was suppressed by ASC alone and to a greater degree by neutrophils alone, whereas neutrophils + ASC reduced glucose uptake to unstimulated levels (Fig. 6d, Supplementary Fig. S6d). At 48hrs, neutrophils + ASC resulted in a decrease in mitochondrial mass (Fig. 6e, Supplementary Fig. S6e) and mitochondrial membrane potential below unstimulated levels (Fig. 6f, Supplementary Fig. S6f).
Upon TCR stimulation, PI3K–AKT–mTOR plays a critical role in upregulation of metabolism and protein synthesis required for growth and proliferation. Based on neutrophil suppressors having broad effects on T-cell activation and metabolic pathways, we explored whether T-cell mTOR activation was impaired. Anti-CD3/CD28 stimulation led to a significant increase in the percentage of T cells with phosphorylated p70 ribosomal protein S6 kinase (p70S6k), a marker of mTOR activation. Neutrophils + ASC reduced p70S6k phosphorylation close to unstimulated levels (Fig. 6g, Supplementary Fig. S6g), whereas no effect was seen with neutrophils alone or ASC alone. These results indicate that neutrophil suppressors inhibit key metabolic and protein synthesis pathways required for T-cell activation.
Discussion
We show that OC ascites fluid induces a neutrophil suppressor phenotype accompanied by functional and anatomic changes, including alterations in nuclear morphology and surface protein expression. Following ASC exposure, normal donor neutrophils developed hypersegmented nuclei, a feature associated with neutrophils activated by Helicobacter pylori (13). A substantial proportion of ASC-activated neutrophils became low-density, a characteristic of PMN-MDSC and N2 neutrophils. Transformation of normal high-density circulating neutrophils into low-density suppressor neutrophils occurred following adoptive transfer in a murine tumor model (26) and in human neutrophils stimulated with formylated peptide (12). Low-density neutrophils may be a general response to activation by microbial products or DAMPs, increased extracellular fluid uptake, and/or neutrophil degranulation. Consistent with this latter idea, we found that ASC augmented neutrophil secretory vesicle and granule mobilization to the plasma membrane, an effect dependent on C5aR signaling. In addition to modulation of neutrophil suppressor function, neutrophil granule mobilization can facilitate metastasis (49). CyTOF analysis of neutrophils exposed to ASC showed that neutrophils could be divided into 15 phenotypically distinct clusters, whose relative populations change over 3hrs. Linking these heterogeneous neutrophil populations to specific suppressor functions will be pursued in future studies.
Our results show that ASC can induce features of PMN-MDSC in mature neutrophils: low density, increased LOX-1 expression, and T-cell suppressor function. These results are consistent with the growing understanding of granulocyte diversity in the TME (50,51), and the notion of granulocyte subsets acquiring suppressor function through distinct pathways that include abnormal granulopoiesis (bona fide PMN-MDSC) and signaling pathways induced in the TME that render normal mature neutrophils suppressive.
Injury and signaling cues from neutrophils resulted in T-cell immunoparalysis, characterized by suppression of NFAT nuclear translocation, impaired IL2 production, and inhibition of glucose uptake, mitochondrial mass and depolarization, and mTOR activation. Although suppression of anti-CD3/CD28-stimulated T-cell proliferation measured at 96hrs required the combination of neutrophils and ASC, we observed variable suppressive effects of neutrophils alone and ASC alone for a number of T cell functions at earlier time points (Fig. 6). Song et al. (52) found that OC ascites fluid triggered stress response pathways in T cells characterized by IRE1α-XBP1 activation that reduced the abundance of glutamine carriers and suppressed mitochondrial activity required for T-cell activation. Our results raise the notion for cumulative effects of neutrophils and ASC on signaling and metabolic responses in stimulated T cells determining whether proliferation will occur. This T-cell immunoparalysis wasn’t associated with increased T-cell death when assessed at 72hrs (8), and was distinct from established checkpoint pathways. Of particular clinical relevance, ASC-activated neutrophils also caused complement-dependent suppression of TALs. Our overall model for neutrophil-driven T-cell suppression is summarized in Fig. 7.
Figure 7: Model of neutrophil-mediated suppression of T cells in the TME.
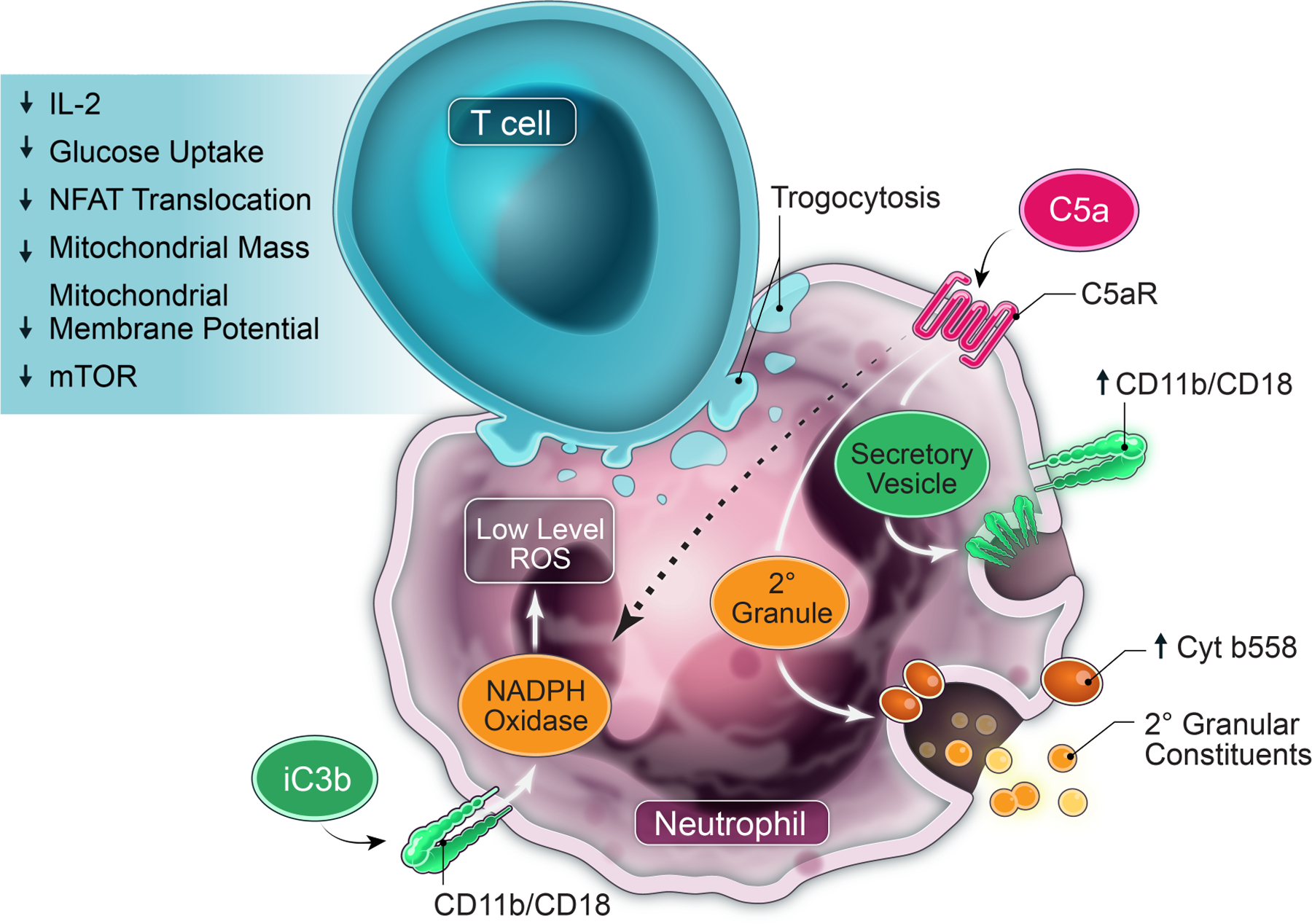
Ovarian cancer ascites fluid and other malignant effusions have multiple products, including DAMPs (25), cytokines, and chemokines (10), that chemoattract and activate neutrophils. Our previously published (8) and current results show that multiple neutrophil effector functions including signaling via CD11b/CD18 (CR3; receptor for iC3b) and C5aR, NADPH oxidase, SNARE-mediated intracellular transport and exocytosis of granular and/or vesicular constituents, protein synthesis, and PS drive this suppressor function. Following exposure of neutrophils to ASC, a positive feedback loop through C5aR activation increases surface expression of CD11b/CD18, and secondary and tertiary granule products, including flavocytochrome b558. ASC-activated neutrophils also increase surface expression of properdin, which stabilizes the alternative pathway C3 convertase to further activate complement. Activated neutrophils cause trogocytosis of T-cell membranes that is CD11b-dependent, likely by enhancing neutrophil adhesion to T cells, though an alternative mechanism could be iC3b binding and signaling. In response to these cumulative signaling and injurious cues, T cells acquire a profound immunoparalysis, characterized by suppression of critical signaling and metabolic pathways necessary for activation and proliferation. These signaling pathways driving neutrophil suppressor function and T-cell non-responsiveness are potential targets for therapeutic modulation.
The relevant components in ASC that induce suppressor neutrophils include C3 activation products and C5a, which appear to be generated by both the classical and alternative complement pathways. Potential antitumor effects of complement activation include complement-dependent cytotoxicity of tumor cells and enhanced trafficking and efficacy of adoptive cell therapy in mice (53). Targeting complement to enhance antitumor activity is garnering significant interest (9). We reported that neutrophil suppressor function was fully dependent on C3, but only partially abrogated by inhibition of C5 (8). We now show evidence for a positive feedback loop driving the generation of suppressor neutrophils in which both C5aR and C3 convertases participate. In addition, C5a is a potent neutrophil chemoattractant that may increase neutrophil trafficking to the TME. In addition to driving neutrophil suppressor function, activated neutrophils and complement can stimulate local thrombosis and metastasis and stimulate proliferative signaling in tumor cells. NETs, complement, and coagulation pathways can amplify each other (54), and create niches for metastatic seeding in the TME (55,56). Complement proteins generated by tumor cells, including OC, can have autocrine signaling that augments tumor growth (57). Inflammatory cells, including neutrophils, can also produce complement proteins (34), and may contribute to tumor growth. In addition, multiple studies in tumor-bearing mice point to complement signaling promoting tumor growth and obstructing antitumor immunity. For example, in tumor-bearing mice, PMN-MDSC infiltration is reduced by inhibition of C5a or C5aR (58). Of specific relevance to OC, genetic and pharmacologic inhibition of complement inhibits tumor growth through pathways dependent on VEGF and neovascularization in murine OC (59). Our work adds to this body of literature by showing a critical role for C3 activation in the human TME driving neutrophil suppressor function and provides additional premise for targeting complement to enhance antitumor immunity.
The neutrophil suppressor phenotype requires NADPH oxidase, but it appears to be independent of extracellular ROS or nitrosylation of the TCR. Although one mechanism for NADPH oxidase–mediated suppression of T-cell responsiveness is a direct effect of released ROS on the TCR (40), another possibility (not mutually exclusive) is NADPH oxidase modulating intracellular signaling in neutrophils required for suppressor function. SOD and catalase had no effect on neutrophil suppressor function, and we did not observe increased peroxynitrite formation on T cells in coculture experiments. These results lead us to posit that NADPH oxidase–regulated intracellular signaling rather than ROS release is driving neutrophil suppressor function. Neutrophil NADPH oxidase can enhance externalization of PS and lyso-PS, and the delayed neutrophil clearance (efferocytosis) by macrophages in CGD is likely related to impaired externalization of these lipids (43). Anti-PS abrogated neutrophil suppressor function. PS has suppressive functions mediated by a number of pathways. Binding of PS or lyso-PS to GPR174 increases levels of cyclic-adenosine monophosphate and inhibits T-cell production of IL2 (60,61). Further studies will delineate the relationship between NADPH oxidase and PS expression in malignant fluid and mechanisms by which PS on neutrophil surfaces drive suppressor function. Finally, induction of suppressor function in neutrophils is not a universal feature of the TME, and different inflammatory or tumor-derived cues can prime neutrophil responses that enhance T-cell immunity (62,63).
Negorev et al. (64) have reported that neutrophils adhere to anti-CD3/CD28 coated microbeads and block TCR engagement and downstream signaling in T cells. In their study, they perform neutrophil and T-cell cocultures in RPMI with 10% heat-inactivated FBS, the same conditions as our media controls. We observed modest or no inhibition of anti-CD3/CD28 stimulated T-cell proliferation when neutrophils were cocultured with media. Potentially, this discrepancy is due to their use of Ficoll gradient sedimentation versus our use of negative selection for neutrophil purification. In live cell imaging, neutrophils bound to microbeads to a similar degree in Cp40-ascites and SCR-ascites, indicating that binding by itself was insufficient to suppress T-cell proliferation. In addition, neutrophils + ASC inhibited proliferation of T cells stimulated with soluble anti-CD3/CD28 (8).
Similar to ASC and malignant effusions from other cancers, POF after OC surgical debulking or diagnostic lung surgery induced a complement-dependent suppressor phenotype in normal donor neutrophils. We consider acquisition of suppressor function in the TME as a general response to injury cues rather than to tumor-specific factors. Consistent with the notion of activated neutrophils inhibiting T-cell responses, a subset of circulating neutrophils with T-cell suppressive activity were expanded in patients with acute systemic inflammation (65). In murine influenza, neutrophils in the lungs limited T-cell infiltrates and lung pathology (66). Mechanisms that drive neutrophil suppression in response to these acute insults likely differ from chronic inflammatory and injurious cues in the TME. Although the role of neutrophils in amplifying tissue injury is well-recognized, neutrophils can also facilitate injury repair (67). CGD is characterized by excessive inflammatory responses to both infections and non-infectious injury (e.g., inflammatory bowel disease, granulomatous cystitis, and wound dehiscence). Seen in this light, the role of NADPH oxidase in the neutrophil suppressor phenotype in OC ascites fluid may be consistent with its general function to limit inflammation and to promote wound repair. Aarts et al. (24) show that neutrophils activated by formylated peptide and other stimuli acquire a suppressor phenotype that requires neutrophil–T-cell contact, CD11b/CD18, neutrophil degranulation, and NADPH oxidase, and is associated with trogocytosis of T-cell membranes. Together, these results support the notion of various DAMPs and microbial products inducing responses in neutrophils that limit excessive T-cell responses. Although suppressor neutrophils may be beneficial to wound healing and potentially avert autoimmunity, they are expected to impair antitumor immunity.
The mechanism by which suppressor neutrophils impair T-cell activation is complex, and likely involves trogocytosis and activation of an injury response within the T cells themselves, resulting in suppression of mTOR and NFAT signaling. Although neutrophil suppressors did not significantly affect T-cell viability (8), they inhibited multiple signaling nodes required for T-cell activation. Our results point to an early (within 30–60 minute) commitment by T cells stimulated with anti-CD3/CD28 to either a responsive or immunoparalysis phenotype following exposure to ASC-activated neutrophils. These results raise the possibility of suppressor neutrophils acting downstream of TCR ligation. Suppression of early signaling events would be expected to terminate NFAT translocation and IL2 generation. In addition, direct neutrophil-mediated injury to T cells (e.g., through trogocytosis) may induce injury repair responses at the expense of proliferation and cytokine generation.
In summary, our results demonstrate complement-dependent priming of neutrophil effector functions in the TME inducing T-cell non-responsiveness. Suppressor neutrophils cross-signal to T cells and deliver injury and signaling cues resulting in T-cell immunoparalysis through mechanisms distinct from established checkpoint pathways. Our results open new areas of research related to mechanisms driving immunosuppression in the TME and for targeting C3 and potentially downstream complement components to reverse the neutrophil suppressor phenotype.
Supplementary Material
Synopsis:
The ovarian cancer microenvironment induces suppressor neutrophils that inhibit T-cell signaling and metabolic functions. Acquisition of the suppressor phenotype is dependent on several pathways including complement signaling, which can be targeted therapeutically to enhance antitumor immunity.
Acknowledgements:
This work was supported by Roswell Park Cancer Center Support Grant P30CA016056, 5R01CA188900 (BHS and KBM), NIAID R01 AI119965 (LAHA), NSF predoctoral fellowship (SLSDT), DoD W81XWH-16-1-0464 (JAL), NIH NIAID U01AI138318-03 (JAL), and a grant from the Charles and Marjorie Holloway Foundation (MBY). Cytometry services were provided by the Flow and Image Cytometry Shared Resource at the Roswell Park Comprehensive Cancer Center which is supported in part by the NCI Cancer Center Support Grant 5P30 CA016056. The ImagestreamX-MKII instrument was funded through NIH Shared Instrument Grant 1S10OD018048. This work used resources and services provided by the Translational Imaging Shared Resource at Roswell Park that are supported by NCI P30CA16056. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This study used the Clinical Research Services, the University at Buffalo Confocal Microscope and Flow Cytometry Facility, the Harvard Medical Area CyTOF core, and the University of Iowa Central Microscopy Research Facility. We thank Dr. Jesus G. Valenzuela (NIH NIAID) for the donation of SALO.
Financial support:
This work was supported by Roswell Park Cancer Center Support Grant P30CA016056, 5R01CA188900 (BHS and KBM), NIAID R01 AI119965 (LAHA), a NSF predoctoral fellowship (SLSDT), DoD W81XWH-16-1-0464 (JAL), NIH NIAID U01AI138318-03 (JAL), and a grant from the Charles and Marjorie Holloway Foundation (MBY).
Footnotes
Conflict of interest statement: Anna Schubart, Holger Sellner, and Jörg Eder are employees of Novartis. The other authors declare no conflicts of interest.
References
- 1.Eng KH, Morrell K, Starbuck K, Spring-Robinson C, Khan A, Cleason D, et al. Prognostic value of miliary versus non-miliary sub-staging in advanced ovarian cancer. Gynecol Oncol 2017;146(1):52–7 doi 10.1016/j.ygyno.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szender JB, Emmons T, Belliotti S, Dickson D, Khan A, Morrell K, et al. Impact of ascites volume on clinical outcomes in ovarian cancer: A cohort study. Gynecol Oncol 2017. doi 10.1016/j.ygyno.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Worzfeld T, Pogge von Strandmann E, Huber M, Adhikary T, Wagner U, Reinartz S, et al. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front Oncol 2017;7:24 doi 10.3389/fonc.2017.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Idorn M, Olsen M, Halldorsdottir HR, Skadborg SK, Pedersen M, Hogdall C, et al. Improved migration of tumor ascites lymphocytes to ovarian cancer microenvironment by CXCR2 transduction. Oncoimmunology 2018;7(4):e1412029 doi 10.1080/2162402X.2017.1412029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan AN, Kolomeyevskaya N, Singel KL, Grimm MJ, Moysich KB, Daudi S, et al. Targeting myeloid cells in the tumor microenvironment enhances vaccine efficacy in murine epithelial ovarian cancer. Oncotarget 2015;6(13):11310–26 doi 10.18632/oncotarget.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mastio J, Condamine T, Dominguez G, Kossenkov AV, Donthireddy L, Veglia F, et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J Exp Med 2019;216(9):2150–69 doi 10.1084/jem.20181952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaul ME, Levy L, Sun J, Mishalian I, Singhal S, Kapoor V, et al. Tumor-associated neutrophils display a distinct N1 profile following TGFbeta modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology 2016;5(11):e1232221 doi 10.1080/2162402X.2016.1232221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singel KL, Emmons TR, Khan ANH, Mayor PC, Shen S, Wong JT, et al. Mature neutrophils suppress T cell immunity in ovarian cancer microenvironment. JCI Insight 2019;4(5):e122311 doi 10.1172/jci.insight.122311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol 2017. doi 10.1038/nri.2017.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolomeyevskaya N, Eng KH, Khan AN, Grzankowski KS, Singel KL, Moysich K, et al. Cytokine profiling of ascites at primary surgery identifies an interaction of tumor necrosis factor-alpha and interleukin-6 in predicting reduced progression-free survival in epithelial ovarian cancer. Gynecol Oncol 2015;138(2):352–7 doi 10.1016/j.ygyno.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 2010;363(27):2600–10 doi 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res 2001;61(12):4756–60. [PubMed] [Google Scholar]
- 13.Whitmore LC, Weems MN, Allen LH. Cutting Edge: Helicobacter pylori Induces Nuclear Hypersegmentation and Subtype Differentiation of Human Neutrophils In Vitro. J Immunol 2017;198(5):1793–7 doi 10.4049/jimmunol.1601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Shao D, Ricklin D, Hilkin BM, Nester CM, Lambris JD, et al. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology 2015;220(8):993–8 doi 10.1016/j.imbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schubart A, Anderson K, Mainolfi N, Sellner H, Ehara T, Adams CM, et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc Natl Acad Sci U S A 2019;116(16):7926–31 doi 10.1073/pnas.1820892116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolbink GJ, Bollen J, Baars JW, ten Berge RJ, Swaak AJ, Paardekooper J, et al. Application of a monoclonal antibody against a neoepitope on activated C4 in an ELISA for the quantification of complement activation via the classical pathway. J Immunol Methods 1993;163(1):67–76 doi 10.1016/0022-1759(93)90240-8. [DOI] [PubMed] [Google Scholar]
- 17.Hack CE, Paardekooper J, Smeenk RJ, Abbink J, Eerenberg AJ, Nuijens JH. Disruption of the internal thioester bond in the third component of complement (C3) results in the exposure of neodeterminants also present on activation products of C3. An analysis with monoclonal antibodies. J Immunol 1988;141(5):1602–9. [PubMed] [Google Scholar]
- 18.Ferreira VP, Fazito Vale V, Pangburn MK, Abdeladhim M, Mendes-Sousa AF, Coutinho-Abreu IV, et al. SALO, a novel classical pathway complement inhibitor from saliva of the sand fly Lutzomyia longipalpis. Sci Rep 2016;6:19300 doi 10.1038/srep19300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen JY, Galwankar NS, Emch HN, Menon SS, Cortes C, Thurman JM, et al. Properdin Is a Key Player in Lysis of Red Blood Cells and Complement Activation on Endothelial Cells in Hemolytic Anemias Caused by Complement Dysregulation. Front Immunol 2020;11:1460 doi 10.3389/fimmu.2020.01460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saggu G, Cortes C, Emch HN, Ramirez G, Worth RG, Ferreira VP. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol 2013;190(12):6457–67 doi 10.4049/jimmunol.1300610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maibaum J, Liao SM, Vulpetti A, Ostermann N, Randl S, Rudisser S, et al. Small-molecule factor D inhibitors targeting the alternative complement pathway. Nat Chem Biol 2016;12(12):1105–10 doi 10.1038/nchembio.2208. [DOI] [PubMed] [Google Scholar]
- 22.Zunder ER, Finck R, Behbehani GK, Amir el AD, Krishnaswamy S, Gonzalez VD, et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat Protoc 2015;10(2):316–33 doi 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chevrier S, Crowell HL, Zanotelli VRT, Engler S, Robinson MD, Bodenmiller B. Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry. Cell Syst 2018;6(5):612–20 e5 doi 10.1016/j.cels.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aarts CEM, Hiemstra IH, Beguin EP, Hoogendijk AJ, Bouchmal S, van Houdt M, et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv 2019;3(22):3562–74 doi 10.1182/bloodadvances.2019031609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singel KL, Grzankowski KS, Khan A, Grimm MJ, D’Auria AC, Morrell K, et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br J Cancer 2019;120(2):207–17 doi 10.1038/s41416-018-0339-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep 2015;10(4):562–73 doi 10.1016/j.celrep.2014.12.039. [DOI] [PubMed] [Google Scholar]
- 27.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150 doi 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol 2016;1(2) doi 10.1126/sciimmunol.aaf8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 2019;30(7):1080–7 doi 10.1093/annonc/mdz135. [DOI] [PubMed] [Google Scholar]
- 30.Bjorge L, Hakulinen J, Vintermyr OK, Jarva H, Jensen TS, Iversen OE, et al. Ascitic complement system in ovarian cancer. Br J Cancer 2005;92(5):895–905 doi 10.1038/sj.bjc.6602334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alcorlo M, Tortajada A, Rodriguez de Cordoba S, Llorca O. Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc Natl Acad Sci U S A 2013;110(33):13504–9 doi 10.1073/pnas.1309618110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blatt AZ, Saggu G, Kulkarni KV, Cortes C, Thurman JM, Ricklin D, et al. Properdin-Mediated C5a Production Enhances Stable Binding of Platelets to Granulocytes in Human Whole Blood. J Immunol 2016;196(11):4671–80 doi 10.4049/jimmunol.1600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camous L, Roumenina L, Bigot S, Brachemi S, Fremeaux-Bacchi V, Lesavre P, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood 2011;117(4):1340–9 doi 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 34.Wirthmueller U, Dewald B, Thelen M, Schafer MK, Stover C, Whaley K, et al. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J Immunol 1997;158(9):4444–51. [PubMed] [Google Scholar]
- 35.Barrett CD, Hsu AT, Ellson CD, B YM, Kong YW, Greenwood JD, et al. Blood clotting and traumatic injury with shock mediates complement-dependent neutrophil priming for extracellular ROS, ROS-dependent organ injury and coagulopathy. Clin Exp Immunol 2018;194(1):103–17 doi 10.1111/cei.13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witthaut R, Farhood A, Smith CW, Jaeschke H. Complement and tumor necrosis factor-alpha contribute to Mac-1 (CD11b/CD18) up-regulation and systemic neutrophil activation during endotoxemia in vivo. J Leukoc Biol 1994;55(1):105–11 doi 10.1002/jlb.55.1.105. [DOI] [PubMed] [Google Scholar]
- 37.McLeish KR, Uriarte SM, Tandon S, Creed TM, Le J, Ward RA. Exocytosis of neutrophil granule subsets and activation of prolyl isomerase 1 are required for respiratory burst priming. J Innate Immun 2013;5(3):277–89 doi 10.1159/000345992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Detmers PA, Lo SK, Olsen-Egbert E, Walz A, Baggiolini M, Cohn ZA. Neutrophil-activating protein 1/interleukin 8 stimulates the binding activity of the leukocyte adhesion receptor CD11b/CD18 on human neutrophils. J Exp Med 1990;171(4):1155–62 doi 10.1084/jem.171.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borregaard N, Heiple JM, Simons ER, Clark RA. Subcellular localization of the b-cytochrome component of the human neutrophil microbicidal oxidase: translocation during activation. J Cell Biol 1983;97(1):52–61 doi 10.1083/jcb.97.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 2007;13(7):828–35 doi 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002;416(6878):291–7 doi 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 42.Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ 2016;23(6):962–78 doi 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frasch SC, Berry KZ, Fernandez-Boyanapalli R, Jin HS, Leslie C, Henson PM, et al. NADPH oxidase-dependent generation of lysophosphatidylserine enhances clearance of activated and dying neutrophils via G2A. J Biol Chem 2008;283(48):33736–49 doi 10.1074/jbc.M807047200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10(+) and immature CD10(−) neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood 2017;129(10):1343–56 doi 10.1182/blood-2016-04-713206. [DOI] [PubMed] [Google Scholar]
- 45.Werfel T, Sonntag G, Weber MH, Götze O. Rapid increases in the membrane expression of neutral endopeptidase (CD10), aminopeptidase N (CD13), tyrosine phosphatase (CD45), and Fc gamma-RIII (CD16) upon stimulation of human peripheral leukocytes with human C5a. The Journal of Immunology 1991;147(11):3909–14. [PubMed] [Google Scholar]
- 46.Mercer F, Ng SH, Brown TM, Boatman G, Johnson PJ. Neutrophils kill the parasite Trichomonas vaginalis using trogocytosis. PLoS Biol 2018;16(2):e2003885 doi 10.1371/journal.pbio.2003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ, et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep 2018;23(13):3946–59 e6 doi 10.1016/j.celrep.2018.05.082. [DOI] [PubMed] [Google Scholar]
- 48.Baixauli F, Acin-Perez R, Villarroya-Beltri C, Mazzeo C, Nunez-Andrade N, Gabande-Rodriguez E, et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab 2015;22(3):485–98 doi 10.1016/j.cmet.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mollinedo F Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol 2019;40(3):228–42 doi 10.1016/j.it.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 50.Veglia F, Hashimoto A, Dweep H, Sanseviero E, De Leo A, Tcyganov E, et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J Exp Med 2021;218(4) doi 10.1084/jem.20201803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 2021. doi 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 2018;562(7727):423–8 doi 10.1038/s41586-018-0597-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Facciabene A, De Sanctis F, Pierini S, Reis ES, Balint K, Facciponte J, et al. Local endothelial complement activation reverses endothelial quiescence, enabling t-cell homing, and tumor control during t-cell immunotherapy. Oncoimmunology 2017;6(9):e1326442 doi 10.1080/2162402X.2017.1326442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol 2019;16(1):19–27 doi 10.1038/s41423-018-0024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci U S A 2014;111(30):E3053–61 doi 10.1073/pnas.1411082111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020;583(7814):133–8 doi 10.1038/s41586-020-2394-6. [DOI] [PubMed] [Google Scholar]
- 57.Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine effects of tumor-derived complement. Cell Rep 2014;6(6):1085–95 doi 10.1016/j.celrep.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares MJ, Agorreta J, et al. A Combined PD-1/C5a Blockade Synergistically Protects against Lung Cancer Growth and Metastasis. Cancer Discov 2017;7(7):694–703 doi 10.1158/2159-8290.CD-16-1184. [DOI] [PubMed] [Google Scholar]
- 59.Nunez-Cruz S, Gimotty PA, Guerra MW, Connolly DC, Wu YQ, DeAngelis RA, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia 2012;14(11):994–1004 doi 10.1593/neo.121262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shinjo Y, Makide K, Satoh K, Fukami F, Inoue A, Kano K, et al. Lysophosphatidylserine suppresses IL-2 production in CD4 T cells through LPS3/GPR174. Biochem Biophys Res Commun 2017;494(1–2):332–8 doi 10.1016/j.bbrc.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 61.Gavin MA, Gragerov A, Espling E, Rohde A, Sexton T, Doulami C, et al. Phosphatidylserine suppresses T cells through GPR174, and co-inhibition of adenosine receptors and GPR174 synergistically enhances T cell responses. Cancer Immunol Res 2020;8(3 Suppl): Abstract nr B45 2020. [Google Scholar]
- 62.Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest 2014;124(12):5466–80 doi 10.1172/JCI77053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell 2016;30(1):120–35 doi 10.1016/j.ccell.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Negorev D, Beier UH, Zhang T, Quatromoni JG, Bhojnagarwala P, Albelda SM, et al. Human neutrophils can mimic myeloid-derived suppressor cells (PMN-MDSC) and suppress microbead or lectin-induced T cell proliferation through artefactual mechanisms. Sci Rep 2018;8(1):3135 doi 10.1038/s41598-018-21450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest 2012;122(1):327–36 doi 10.1172/JCI57990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tak T, Rygiel TP, Karnam G, Bastian OW, Boon L, Viveen M, et al. Neutrophil-mediated Suppression of Influenza-induced Pathology Requires CD11b/CD18 (MAC-1). Am J Respir Cell Mol Biol 2018;58(4):492–9 doi 10.1165/rcmb.2017-0021OC. [DOI] [PubMed] [Google Scholar]
- 67.Wang J Neutrophils in tissue injury and repair. Cell Tissue Res 2018;371(3):531–9 doi 10.1007/s00441-017-2785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.