

Abstract
Interstitial lung disease is a relatively frequent manifestation of systemic sclerosis with approximately one-third of patients developing clinical restrictive lung disease. Fibrotic nonspecific interstitial pneumonia is the most common cause of diffuse parenchymal lung disease in patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD), followed by usual interstitial pneumonia (UIP). Radiographic pleuroparenchymal fibroelastosis-like changes may accompany other forms of interstitial lung disease, most commonly UIP. In an appropriate clinical setting with supportive high-resolution computed tomography findings, lung biopsy is not needed to confirm the presence of interstitial lung disease and surgical lung biopsies are often reserved for atypical presentations. In this review, we discuss the histological findings that define the most common patterns of SSc-ILD and outline other findings sometimes encountered in lung biopsies obtained from systemic sclerosis patients, including pulmonary vascular changes, aspiration, chronic pleuritis, and diffuse alveolar damage.
Keywords: interstitial lung disease, pathology, scleroderma, systemic sclerosis
Introduction
Interstitial lung disease is a common manifestation of systemic sclerosis and is included as a criteria for the classification of systemic sclerosis in the 2013 American College of Rheumatology/European League Against Rheumatism scoring system. 1 The largest autopsy studies to date have shown that moderate-to-severe pulmonary fibrosis occurs in approximately 75% of patients with systemic sclerosis,2,3 but only about one-third of patients will develop clinical restrictive lung disease. Risk factors for systemic sclerosis-associated interstitial lung disease (SSc-ILD) include diffuse cutaneous systemic sclerosis, male sex, African American race, and the presence of anti-Scl-70 antibodies.4,5 Interstitial lung disease ultimately accounts for about 20% of all causes of systemic sclerosis-related deaths. 6
Surgical lung biopsies are performed infrequently on systemic sclerosis patients, since multiple studies have shown fibrotic nonspecific interstitial pneumonia (NSIP) is the most common cause of diffuse parenchymal lung disease in patients with SSc-ILD.7,8 In the setting of supportive clinical findings and a restrictive ventilatory defect on pulmonary function tests, the presence of reticular and/or ground-glass opacities on high-resolution computed tomography (HRCT) is sufficient for the diagnosis of NSIP.9,10 Usual interstitial pneumonia (UIP) occurs less frequently than NSIP but, in the context of appropriate HRCT changes, confirmatory lung biopsy is often not needed for the diagnosis. Therefore, surgical lung biopsies tend to be reserved for atypical radiographic presentations or patients for whom there may be other clinical (e.g., smoking-related lung disease) or environmental (e.g., antigenic exposures to mold, thermophilic bacteria, or animal proteins) factors that raise concerns for diagnostic alternatives. Finally, pleuroparenchymal fibrosis (PPFE) is a rare cause of interstitial lung disease that has been radiographically described in systemic sclerosis patients with variable frequency, often accompanying other forms of lung disease.
In this review, we discuss the histological findings that define the most common patterns of SSc-ILD. Additionally, we outline other pulmonary findings sometimes encountered in lung biopsies obtained from systemic sclerosis patients.
Nonspecific interstitial pneumonia
NSIP is the most common interstitial pneumonia reported in patients with SSc-ILD, discovered in more than two-thirds of patients who undergo lung biopsy for evaluation of diffuse parenchymal lung disease.7,8 NSIP has been described as a manifestation of not only systemic sclerosis but also other forms of systemic connective tissue disease. It may also occur in the settings of drug-induced lung disease, certain environmental exposures, chronic lung disease following an episode of diffuse alveolar damage (DAD), or it may be idiopathic.11,12 Occasionally, patients who are initially diagnosed with idiopathic NSIP are later discovered to have underlying systemic connective tissue disease (CTD) and are re-categorized as CTD-associated NSIP.13,14 Pathologic assessment cannot reliably distinguish between those cases with an underlying etiology or association and idiopathic NSIP, emphasizing the importance of multidisciplinary discussion as part of the diagnostic pathway. While certain histologic features, such as degree of lymphoid hyperplasia, are reported more commonly in specific connective tissue diseases, the positive predictive value of these findings is low, and NSIP as a manifestation of SSc-ILD is morphologically indistinguishable from other causes on the basis of histology alone.11–15
Histologically, NSIP is characterized by expansion of the alveolar interstitium by a combination of inflammation and fibrosis. The interstitial abnormality may be diffuse or patchy with areas of intervening more normal-appearing lung parenchyma. Whether diffuse or patchy, the interstitial changes are qualitatively uniform, lacking the temporal and regional heterogeneity characteristic of UIP. NSIP also lacks the architecturally-distorting scarring or honeycomb change typical of UIP (Table 1). 11
Table 1.
Comparison of histologic features helpful in differentiating fibrotic NSIP, UIP, and PPFE.
| Histologic feature | Fibrotic NSIP | UIP | PPFE |
|---|---|---|---|
| Distribution of fibrosis | Uniform | Subpleural, lower lobe-predominant | Subpleural, upper lobe-predominant |
| Fibroblastic foci | Absent | Present | Absent |
| Confluent scarring | Absent | Present | Present |
| Honeycomb change | Absent | Present | Absent |
NSIP, nonspecific interstitial pneumonia; PPFE, pleuroparenchymal fibroelastosis; UIP, usual interstitial pneumonia.
The inflammatory infiltrate is a mixture of lymphocytes and plasma cells. The profusion of plasma cells and lymphoid aggregates with germinal centers is variable, but tends to be greater in patients with underlying connective tissue disease, including systemic sclerosis, compared with patients with idiopathic NSIP. 16 Fibroblast foci are rare and the fibrosis tends to consist of dense collagen deposition. Although most cases comprise a mixture of inflammation and fibrosis, one component may predominate, resulting in what is referred to as cellular or fibrotic NSIP (Figure 1a,b). In systemic sclerosis patients with NSIP, the fibrotic variant is more common than the cellular variant, representing more than three-quarters of cases.7,8 A distal airspace exudate in which macrophages predominate sometimes accompanies the interstitial abnormality, likely accounting for descriptions of desquamative interstitial pneumonia (DIP) in rare patients with systemic sclerosis. 17 With the current classification scheme, systemic sclerosis patients with “DIP-like” changes are best categorized as NSIP. 18
Figure 1.

NSIP is characterized by uniform expansion of the interstitium with preservation of the underlying alveolated lung architecture. In cellular NSIP (a), the interstitial abnormality is predominated by a chronic inflammatory infiltrate, comprised mainly of lymphocytes and plasma cells, whereas fibrotic NSIP (b) is relatively paucicellular and consists mainly of collagenous fibrosis.
NSIP, nonspecific interstitial pneumonia.
While studies have shown a survival benefit for patients with all causes of cellular NSIP compared with fibrotic NSIP,11,19,20 this has not been shown to be true in systemic sclerosis, where no significant survival difference has been found between the histologic subtypes. Functional severity of disease as measured by diffusing capacity of carbon monoxide at time of diagnosis appears to be a reproducible predictor of survival. 7 Regardless of subtype, systemic sclerosis patients with NSIP tend to experience longer survival than those with UIP. 8 In systemic sclerosis-associated NSIP, the presence of coexistent organizing pneumonia, a variant that has been referred to by some as NSIP with organizing pneumonia overlap, reportedly has a better outcome than NSIP alone.21,22
Usual interstitial pneumonia
Compared with NSIP, UIP is a relatively less common pulmonary manifestation of systemic sclerosis.7,8 Like NSIP, UIP is a histologic pattern observed in a variety of clinical settings. While UIP usually occurs in patients with the clinical syndrome of idiopathic pulmonary fibrosis (IPF), it may also be seen in the context of underlying systemic connective tissue disease, asbestosis, or drug-induced lung disease. 18 Although patients with systemic sclerosis-associated UIP (SSc-UIP) and other forms of CTD-associated UIP, such as rheumatoid arthritis, may have a greater degree of lymphoid hyperplasia encountered on lung biopsy than IPF patient samples, this finding is not specific. There are no unique histologic clues that are consistently helpful in differentiating SSc-UIP from other causes of CTD-associated UIP or IPF on the basis of morphology alone.23–25 Therefore, determining the possible underlying cause or association for a patient’s pathologically diagnosed UIP is entirely dependent upon the clinical context.
Although SSc-UIP may be morphologically indistinguishable from idiopathic UIP, the mechanisms underlying the pathogenesis of these interstitial pneumonias are thought to differ. While a comprehensive understanding of the initiating events and progression of IPF remain in evolution, there is compelling evidence to suggest that repetitive epithelial injury from a variety of toxic insults with aberrant wound repair process culminates in progressive pulmonary fibrosis. 26 Chronic epithelial injury remains a suspected underlying mechanism in systemic sclerosis; however, microvascular injury and endothelial cell dysfunction seem to play greater roles in the development of pulmonary fibrosis in this patient population.27,28
UIP is a fibrosing chronic interstitial pneumonia that results in interstitial expansion by fibrosis that has a predilection for the subpleural lung zones and interlobular septa, resulting in a “patchwork” distribution. Regional heterogeneity is a term often used to describe UIP, which refers not only to the peripheral, lower lobe predominance of this chronic interstitial pneumonia but also the variegated appearance between microscopic fields. Confluent areas of fibrous scarring are characteristic, but often variably conspicuous and result in architectural distortion and lung remodeling of the lung parenchyma. Cystically dilated terminal bronchioles embedded within areas of scarring, referred to as honeycomb change, result in traction of the more proximal airways. 29 The combination of lung contracture by scarring and honeycomb change may cause the visceral pleura to assume a cobblestoned appearance upon gross examination (Figure 2).
Figure 2.
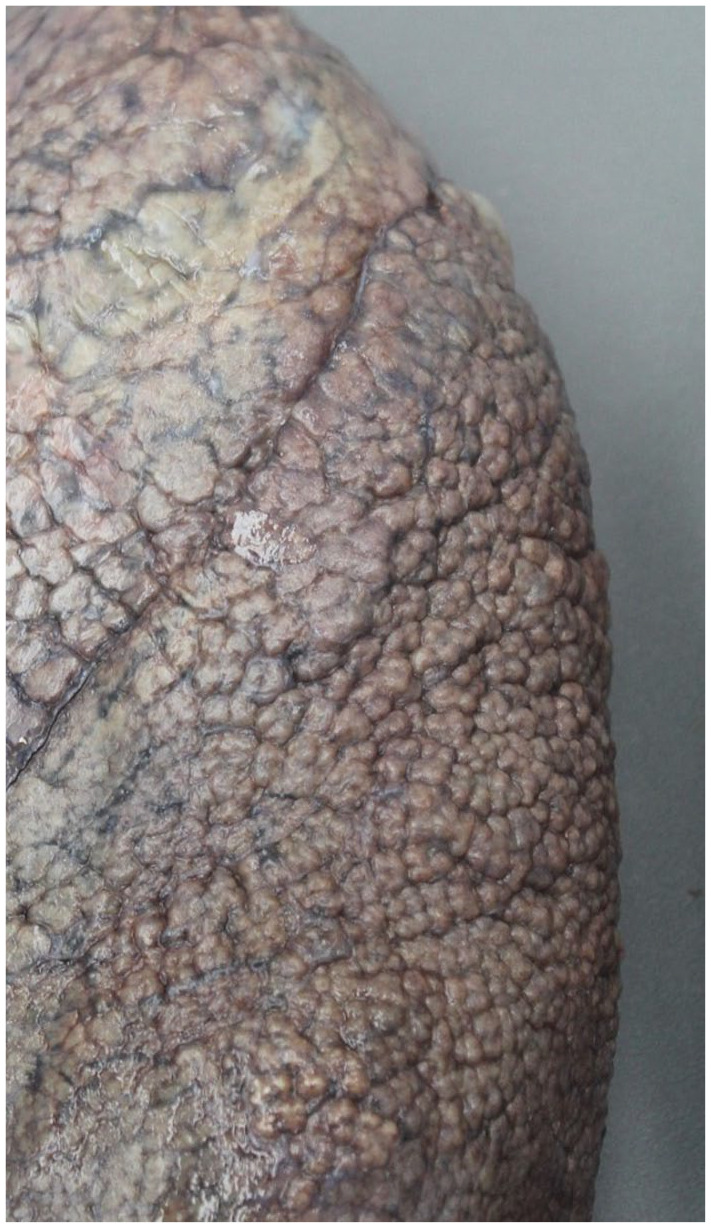
Gross image of the lung’s visceral pleural surface demonstrates a “cobblestone” appearance in UIP.
UIP, usual interstitial pneumonia.
The fibrosis usually consists of dense collagen deposition that may be accompanied by varying degrees of smooth muscle hyperplasia and focal osseous metaplasia. The junction between collagenous fibrosis and normal-appearing lung tissue is punctuated by variable numbers of fibroblast foci in which there are loose, subepithelial aggregates of fibroblasts and myofibroblasts, representing the advancing front of end-stage fibrosis and accounting for the application of the term temporal heterogeneity to describe UIP (Figure 3 and Table 1).
Figure 3.
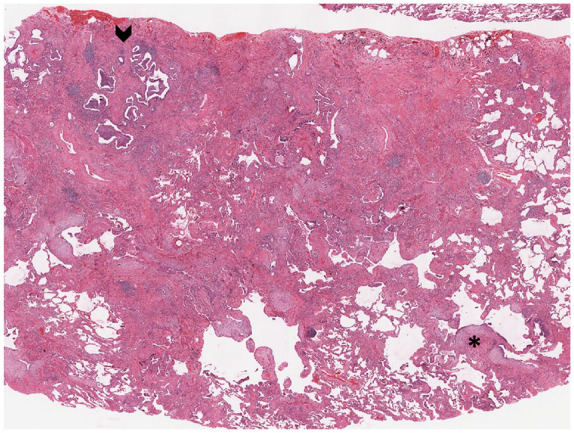
UIP results in a “patchwork” pattern of fibrosis that is accompanied by architecturally distorting scarring and microscopic honeycomb change (black arrow head). Scattered fibroblastic foci (asterisk) are seen as pale zones located at transition points between scarred lung and more normal-appearing lung parenchyma.
UIP, usual interstitial pneumonia.
Occasionally, patients initially thought to have NSIP on lung biopsy are later re-categorized as UIP either through continued radiographic follow up, re-biopsy, or lung transplant. Histologically, these cases are often discovered to have extensive “NSIP-like” areas in otherwise conventional UIP that is only appreciated with examination of the subsequent biopsy or lung explant specimen. Rather than reflecting an evolution from NSIP to UIP, the discrepancy between initial and final pathology is almost certainly sampling-related, based upon surgical selection of biopsy sites.30,31 Additionally, many patients with UIP may have radiologic features more typical of NSIP. 32 Given that, it seems almost certain that some patients presumed to have NSIP even after multidisciplinary discussion will prove to have UIP over time.
Patients with SSc-UIP tend to experience a progressive clinical course not unlike that seen in individuals with IPF. However, overall survival is better for patients with SSc-UIP than IPF, but worse than patients with NSIP.8,25,33–35 Regardless of severity of their histologic abnormality, age and functional capacity by pulmonary function testing are reported as the best predictors of survival in patients with CTD-associated UIP.25,33 Finally, a subset of systemic sclerosis who are commonly current or former smokers develop an interstitial pneumonia, most often UIP, in the setting of radiographic emphysema, an entity referred to as combined pulmonary fibrosis and emphysema. These patients have been reported to have a higher mortality risk than individuals with SSc-ILD alone.36,37
Pleuroparenchymal fibroelastosis
PPFE is a rare cause of restrictive lung disease that is most often idiopathic, but it has also been described in several clinical settings, including as a complication of bone marrow or lung transplant or arising in association with connective tissue disease.38–40 It frequently coexists with other pathologic patterns, such as UIP, so, in this scenario, the upper lobe abnormality might be regarded by some as “PPFE-like” rather than diagnostic of PPFE and UIP.41,42 Morphologically, PPFE is characterized by upper lobe-predominant, dense subpleural fibroelastosis and septal scarring, but without honeycomb change (Figure 4 and Table 1). The histologic findings overlap with apical cap and, on a single site lung biopsy, establishing a diagnosis of PPFE requires a supportive clinical and radiographic context.
Figure 4.
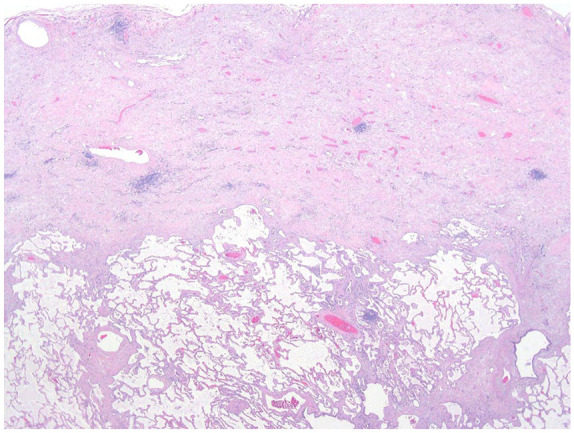
PPFE shows marked thickening of the visceral pleura and subpleural lung zone by paucicellular fibroelastosis. There is no honeycomb change, which is an important clue in differentiating PPFE from UIP.
PPFE, pleuroparenchymal fibroelastosis; UIP, usual interstitial pneumonia.
PPFE or PPFE-like lesions have been described in systemic sclerosis patients predominantly in the context of HRCT findings. Enomoto et al. found that PPFE-like changes occurred in approximately 40% of cases, but frequently accompanied other radiographic changes that most commonly fell into the category of UIP or possible UIP. 40 In the largest study to date, Bonifazi et al. reviewed HRCT findings in 359 systemic sclerosis patients and reported PPFE in 65 (18.1%) patients without noting other patterns of interstitial lung disease. 43 In the end, it is difficult to estimate the true prevalence of isolated PPFE as a manifestation of SSc-ILD. However, there is some evidence that supports that PPFE-like features in systemic sclerosis patients represent an independent predictor of poor prognosis, even in the setting of other histologic patterns of interstitial lung disease.40,43
Pulmonary vascular changes
Pulmonary arterial hypertension is well described in systemic sclerosis, particularly the limited variant, and some evidence seems to support that pulmonary vasculopathies occur in the setting of systemic sclerosis independent of the presence of diffuse parenchymal lung disease.44–46 While arterial fibrosis is the most common histologic finding in systemic sclerosis-related vasculopathies, venous changes may also occur and resemble pulmonary veno-occlusive disease.44,47 Notably, pulmonary vascular changes frequently accompany pulmonary fibrosis and separating this finding as a primary vasculopathy versus a secondary compensatory response is often not possible on the basis of histology alone.2,44 Additionally, histologic arterial changes do not predict for the presence of clinical pulmonary hypertension 44 ; therefore, right heart catheterization for assessment of mean pulmonary artery pressure remains the gold standard for clinical diagnosis.
Other pulmonary findings
Aspiration
Chronic aspiration of gastric contents due to impaired esophageal motility is common in patients with systemic sclerosis. 48 Aspirated foreign particulates may be encountered on lung biopsy and while sometimes an incidental finding, may be clinically unsuspected as a cause of a patient’s clinical presentation and/or radiographic abnormality. Microscopically, aspiration pneumonia most often results in intraluminal fibroblasts plugs of organizing pneumonia, affiliated with foreign aspirated material, and often accompanied by multinucleated giant cells (Figure 5). Less frequently, suppurative granulomas, peribronchiolar fibrosis, and acute bronchopneumonia may also occur. 49
Figure 5.

Aspirated particles of crospovidone, seen as deep blue material, are accompanied by organizing pneumonia in the example on the left, while on the right, macrophages attempt to engulf aspirated vegetable material.
Chronic pleuritis and pleural fibrosis
Pleural changes often accompany the underlying parenchymal abnormalities in systemic sclerosis with chronic pleuritis and fibrous pleural adhesions identified in more than three-quarters of patients. 2
Diffuse alveolar damage
DAD is a pattern of catastrophic acute lung injury most commonly seen in the clinical setting of acute respiratory distress syndrome. DAD results in a spectrum of histologic changes that include hyaline membranes in the early acute phase to edema, airspace collapse and organization/consolidation, and pneumocyte hyperplasia as DAD progresses into its later phases (Figure 6). In its resolving phases, fibrosis resembling NSIP may occur.
Figure 6.
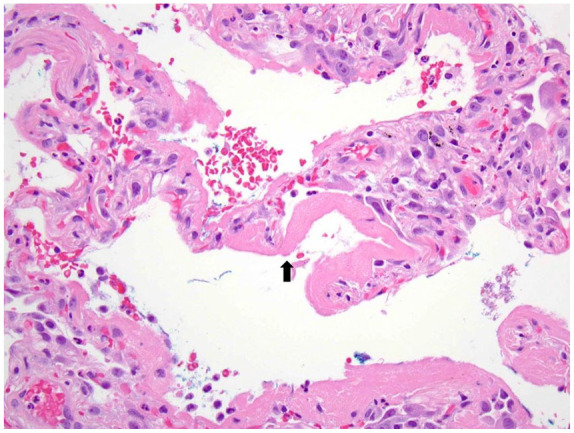
The histologic hallmark of DAD is the presence of bright pink hyaline membranes (black arrow) that outline distal airspaces.
DAD, diffuse alveolar damage.
DAD is the anticipated histologic finding in patients who suffer from acute exacerbation of an underlying chronic interstitial pneumonia. These changes have been described not only in patients with IPF, but also systemic sclerosis patients with NSIP who suffer from an acute decline in respiratory function.21,50 DAD in systemic sclerosis may also occur in the absence of underlying lung disease. 51 Regardless of the inciting etiology, the underlying cause of a patient’s DAD is not usually evident on the basis of histology alone. Therefore, determining if a patient’s DAD represents acute exacerbation or might be related to another cause, such as infection or drug reaction, is entirely dependent upon other clinical and laboratory data.
Conclusion
The patterns of interstitial lung disease observed in patients with systemic sclerosis overlap with those seen in patients with idiopathic interstitial pneumonias. While there may be subtle histologic clues when comparing cohorts of patients, none of these differences are pathognomonic in individual patients for whom the distinction is predicated on clinical and laboratory findings. Survival differences based on histologic pattern have been reported in some studies, while in others disease severity as reflected in measures of lung function and evidence of disease progression over time are more important predictors of outcome.
Footnotes
Conflict of interest statement: The authors declare that there is no conflict of interest.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs: Kristine E. Konopka  https://orcid.org/0000-0002-4582-0120
https://orcid.org/0000-0002-4582-0120
Jeffrey L. Myers
https://orcid.org/0000-0001-8247-3028
Contributor Information
Kristine E. Konopka, Department of Pathology, University of Michigan, Michigan Medicine, 2800 Plymouth Road, Building 35, Ann Arbor, MI 48109, USA.
Jeffrey L. Myers, Department of Pathology, University of Michigan, Ann Arbor, MI, USA
References
- 1. van den Hoogen F, Khanna D, Fransen J, et al. 2013. classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013; 65: 2737–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. D’Angleo WA, Fries JF, Masi AT, et al. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46: 428–440. [DOI] [PubMed] [Google Scholar]
- 3. Weaver AL, Divertie MB, Titus JL. Pulmonary scleroderma. Dis Chest 1968; 54: 490–498. [DOI] [PubMed] [Google Scholar]
- 4. Steen VD, Conte C, Owens GR, et al. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum 1994; 37: 1283–1289. [DOI] [PubMed] [Google Scholar]
- 5. McNearney TA, Reveille JD, Fischbach M, et al. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum 2007; 57: 318–326. [DOI] [PubMed] [Google Scholar]
- 6. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69: 1809–1815. [DOI] [PubMed] [Google Scholar]
- 7. Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med 2002; 165: 1581–1586. [DOI] [PubMed] [Google Scholar]
- 8. Fischer A, Swigris JJ, Groshong SD, et al. Clinically significant interstitial lung disease in limited scleroderma: histopathology, clinical features, and survival. Chest 2008; 134: 601–605. [DOI] [PubMed] [Google Scholar]
- 9. Daimon T, Johkoh T, Honda O, et al. Nonspecific interstitial pneumonia associated with collagen vascular disease: analysis of CT features to distinguish the various types. Intern Med 2009; 48: 753–761. [DOI] [PubMed] [Google Scholar]
- 10. Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004; 232: 560–567. [DOI] [PubMed] [Google Scholar]
- 11. Katzenstein AL, Fiorelli RF. Non-specific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994; 18: 136–147. [PubMed] [Google Scholar]
- 12. Cottin V, Donsbeck AV, Revel D, et al. Nonspecific interstitial pneumonia. Individualization of a clinicopathologic entity in a series of 12 patients. Am J Respir Crit Care Med 1998; 158: 1286–1293. [DOI] [PubMed] [Google Scholar]
- 13. Fujita J, Ohtsuki Y, Yoshinouchi T, et al. Idiopathic non-specific interstitial pneumonia: as an “autoimmune interstitial pneumonia.” Respir Med 2005; 99: 234–240. [DOI] [PubMed] [Google Scholar]
- 14. Kono M, Nakamura Y, Yoshimura K, et al. Nonspecific interstitial pneumonia preceding diagnosis of collagen vascular disease. Respir Med 2016; 117: 40–47. [DOI] [PubMed] [Google Scholar]
- 15. Fischer A, Antoniou K, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015; 46: 976–987. [DOI] [PubMed] [Google Scholar]
- 16. Ozasa M, Ichikawa H, Sato S, et al. Proposed method of histological separation between connective tissue disease-associated interstitial pneumonia and idiopathic interstitial pneumonias. PLoS One 2018; 13: e0206186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swartz JS, Chatterjee S, Parambil JG. Desquamative interstitial pneumonia as the initial manifestation of systemic sclerosis. J Clin Rheumatol 2010; 16: 284–286. [DOI] [PubMed] [Google Scholar]
- 18. American Thoracic Society and European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of Idiopathic Interstitial Pneumonias. This joint statement of the American thoracic society (ATS), and the European respiratory society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS executive committee, June 2001. Am J Respir Crit Care Med 2002; 165: 277–304. [DOI] [PubMed] [Google Scholar]
- 19. Travis WD, Matsui K, Moss J, et al. Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol 2000; 24: 19–33. [DOI] [PubMed] [Google Scholar]
- 20. Nicholson AG, Colby TV, du Bois RM, et al. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162: 2213–2217. [DOI] [PubMed] [Google Scholar]
- 21. Kambouchner M, Levy P, Nicholson AG, et al. Prognostic relevance of histologic variants in nonspecific interstitial pneumonia. Histopathology 2014; 65: 549–560. [DOI] [PubMed] [Google Scholar]
- 22. Ito Y, Arita M, Kumagai S, et al. Serologic and morphologic prognostic factors in patients with interstitial pneumonia with autoimmune features. BMC Pulm Med 2017; 17: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harrison NK, Myers AR, Corrin B, et al. Structural features of interstitial lung disease in systemic sclerosis. Am Rev Respir Dis 1991; 144: 706–713. [DOI] [PubMed] [Google Scholar]
- 24. Cipriani NA, Strek M, Noth I, et al. Pathologic quantification of connective tissue disease-associated versus idiopathic usual interstitial pneumonia. Arch Pathol Lab Med 2012; 136: 1253–1258. [DOI] [PubMed] [Google Scholar]
- 25. Moua T, Zamora Martinez AC, Baqir M, et al. Predictors of diagnosis and survival in idiopathic pulmonary fibrosis and connective tissue disease-related usual interstitial pneumonia. Respir Res 2014; 15: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sgalla G, Iovene B, Calvello M, et al. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res 2018; 19: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khanna D, Tashkin DP, Denton CP, et al. Etiology, risk factors, and biomarkers in systemic sclerosis with interstitial lung disease. Am J Respir Crit Care Med 2020; 201: 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beon M, Harley RA, Wessels A, et al. Myofibroblast induction and microvascular alteration in scleroderma lung fibrosis. Clin Exp Rheumatol 2004; 22: 733–742. [PubMed] [Google Scholar]
- 29. Tanabe N, McDonough JE, Vasilescu DM, et al. Pathology of idiopathic pulmonary fibrosis assessed by a combination of microcomputed tomography, histology, and immunohistochemistry. Am J Pathol 2020; 190: 2427–2435. [DOI] [PubMed] [Google Scholar]
- 30. Flaherty KR, Travis WD, Colby TV, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med 2001; 164: 1722–1727. [DOI] [PubMed] [Google Scholar]
- 31. Katzenstein A-LA, Zisman DA, Litzky LA, et al. Usual interstitial pneumonia: histologic study of biopsy and explant specimens. Am J Surg Pathol 2002; 26: 1567–1577. [DOI] [PubMed] [Google Scholar]
- 32. Flaherty KR, Thwaite EL, Kazerooni EA, et al. Radiologic versus histological diagnosis in UIP and NSIP: survival implications. Thorax 2003; 58: 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175: 705–711. [DOI] [PubMed] [Google Scholar]
- 34. Papiris SA, Vlachoyiannopoulos PG, Maniati MA, et al. Idiopathic pulmonary fibrosis and pulmonary fibrosis in diffuse systemic sclerosis: two fibroses with different prognoses. Respiration 1997: 64: 81–85. [DOI] [PubMed] [Google Scholar]
- 35. Wells AU, Cullinan P, Hansell DM, et al. Fibrosing alveolitis associated with systemic sclerosis has a better prognosis than long cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 1994; 149: 1583–1590. [DOI] [PubMed] [Google Scholar]
- 36. Champtiaux N, Cottin V, Chassagnon G, et al. Combined pulmonary fibrosis and emphysema in systemic sclerosis: a syndrome associated with heavy morbidity and mortality. Semin Arthritis Rheum 2019; 49: 98–104. [DOI] [PubMed] [Google Scholar]
- 37. Cottin V, Nunes H, Mouthon L, et al. Combined pulmonary fibrosis and emphysema syndrome in connective tissue disease. Arthritis Rheum 2011; 63: 295–304. [DOI] [PubMed] [Google Scholar]
- 38. Ofek E, Sato M, Saito T, et al. Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Mod Pathol 2013; 26: 350–356. [DOI] [PubMed] [Google Scholar]
- 39. Takeuchi Y, Miyagawa-Hayashino A, Chen F, et al. Pleuroparenchymal fibroelastosis and non-specific interstitial pneumonia: frequent pulmonary sequelae of hematopoietic stem cell transplantation. Histopathology 2015; 66: 536–544. [DOI] [PubMed] [Google Scholar]
- 40. Enomoto Y, Nakamura Y, Colby TV, et al. Radiographic pleuroparenchymal fibroelastosis-like lesion in connective tissue disease-related interstitial lung disease. PLoS One 2017; 12: e0180283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reddy TL, Tominaga M, Hansell DM, et al. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J 2012; 40: 377–385. [DOI] [PubMed] [Google Scholar]
- 42. Oda T, Ogura T, Kitamura H, et al. Distinct characteristics of pleuroparenchymal fibroelastosis with usual interstitial pneumonia compared to idiopathic pulmonary fibrosis. Chest 2014; 146: 1248–1255. [DOI] [PubMed] [Google Scholar]
- 43. Bonifazi M, Sverzellati N, Negri E, et al. Pleuroparenchymal fibroelastosis in systemic sclerosis: prevalence and prognostic impact. Eur Repir J 2020; 56: 1902135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yousem SA. The pulmonary pathologic manifestations of the CREST syndrome. Hum Pathol 1990; 21: 467–474. [DOI] [PubMed] [Google Scholar]
- 45. Stupi AM, Steen VD, Owens GR, et al. Pulmonary hypertension in the CREST syndrome variant of systemic sclerosis. Arthritis Rheum 1986; 29: 515–524. [DOI] [PubMed] [Google Scholar]
- 46. Salerni R, Rodnan GP, Leon DF, et al. Pulmonary hypertension in the CREST syndrome variant of progressive systemic sclerosis (scleroderma). Ann Intern Med 1977; 86: 394–399. [DOI] [PubMed] [Google Scholar]
- 47. Overbeek MJ, Vonk MC, Boonstra A, et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: a distinctive vasculopathy. Eur Respir J 2009; 34: 371–379. [DOI] [PubMed] [Google Scholar]
- 48. Johnson DA, Drane WE, Curran J, et al. Pulmonary disease in progressive systemic sclerosis. A complication of gastroesophageal reflux and occult aspiration? Arch Intern Med 1989; 149: 589–593. [PubMed] [Google Scholar]
- 49. Mukhopadhyay S, Katzenstein A-LA. Pulmonary disease due to aspiration of food and other particulate matter: a clinicopathologic study of 59 cases diagnosed on biopsy or resection specimens. Am J Surg Pathol 2007; 31: 752–759. [DOI] [PubMed] [Google Scholar]
- 50. Park I-N, Kim DS, Shim TS, et al. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest 2007; 132: 214–220. [DOI] [PubMed] [Google Scholar]
- 51. Muir TE, Tazelaar HD, Colby TV, et al. Organizing diffuse alveolar damage associated with progressive systemic sclerosis. Mayo Clin Proc 1997; 72: 639–642. [DOI] [PubMed] [Google Scholar]