

Abstract
Decellularized extracellular matrices (dECMs) have shown enormous potential for the biofabrication of tissues due to their biomimetic properties that promote enhanced cellular interaction and tissue regeneration. However, biofabrication schemes requiring electrostimulation pose an additional constraint due to the insulating properties of natural materials. Here, we propose a methacryloyl-modified decellularized small intestine submucosa (SISMA) hydrogel, embedded with graphene oxide (GO) nanosheets, for extrusion-based 3D bioprinting applications that require electrostimulation. Methacryloyl biochemical modification is performed to enhance the mechanical stability of dECM constructs by mediating photo-crosslinking reactions, and a multistep fabrication scheme is proposed to harness the bioactive and hydrophilic properties of GO and electroconductive properties of reduced GO. For this, GO was initially dispersed in SISMA hydrogels by exploiting its hydrophilicity and protein adsorption capabilities, and in situ reduction was subsequently performed to confer electroconductive abilities. SISMA-GO composite hydrogels were successfully prepared with enhanced structural characteristics, as shown by the higher crosslinking degree and increased elastic response upon blue-light exposure. Moreover, GO was homogeneously dispersed without affecting photocrosslinking reactions and hydrogel shear-thinning properties. Human adipose-derived mesenchymal stem cells were successfully bioprinted in SISMA-GO with high cell viability after 1 week and in situ reduction of GO during this period enhanced the electrical conductivity of these nanostructures. This work demonstrates the potential of SISMA-GO bioinks as bioactive and electroconductive scaffolds for electrostimulation applications in tissue engineering and regenerative medicine.
Keywords: Extracellular matrix bioink, Graphene oxide, Reduced graphene oxide, Electroconductive hydrogel, Photocrosslinking
1. Introduction
Decellularized extracellular matrices (dECMs) have gained significant attention over the past few decades as biomaterials for tissue engineering due to their promising potential as biomimetic scaffolds[1,2]. Composition-wise, dECMs hold a complex of structural proteins such as collagen, laminin, elastin, and fibronectin, which in native tissues grant adequate mechanical rigidity and structural stability for cellular growth, migration, and proliferation, and provide attachment sites for cell adhesion[1]. Additional proteins present in dECMs such as glycoproteins, proteoglycans, and bound growth factors mediate morphological organization and physiological function[3] while glycosaminoglycans (GAGs) provide an extremely hydrophilic environment essential for withstanding high compressive forces[4]. Although ECM composition varies depending on tissue function, its overall structure is similar for all tissues and, therefore, dECM hydrogels closely mimic the native cellular environment. Moreover, several studies have revealed that the presence and cumulative action of the bioactive molecules described above can promote regeneration processes and restore tissue homeostasis[5-7], underscoring the potential of dECM hydrogels for cellular scaffolding and advanced biomanufacturing applications above other natural materials.
Despite their great composition-wise resemblance to native tissues, solubilization procedures during dECM hydrogel preparation have shown to detrimentally alter the native structure of its fibrous proteins, mainly because of the use of proteolytic enzymes to aid matrix digestion[8]. This results in diminished biomechanical stiffness and higher biodegradability rate, which limits their prolonged functionality[9,10]. Specifically, temperature-dependent gelation, the principal crosslinking mechanism in dECM- and collagen-based hydrogels, is often not enough for maintaining structural stability within three-dimensional structures due to the weak forces (e.g., hydrogen bonds) holding together the digested components in the solubilized matrix[10]. As a result, dECM hydrogels usually need to be combined with other natural materials, such as hyaluronic acid, alginate, gelatin, chitosan, and silk fibroin, to facilitate alternative crosslinking mechanisms and improve their mechanical rigidity[11-13]. In this regard, the addition of crosslinking agents, such as glutaraldehyde and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC)/N-hydroxysulfosuccinimide (NHS), allows the formation of dense hydrogel networks[10,14]. However, unreacted reagents and reaction by-products from these approaches usually reduce the viability of the obtained hydrogel-based cellular constructs[15,16]. The biochemical modification of natural materials with methacryloyl groups has also emerged as a suitable alternative for photocrosslinking schemes[17], a method where material excipients are largely avoided for formulating the hydrogels and that favors crosslinking agents of low cytotoxicity[18-20]. In particular, improved mechanical stability has been reported in kidney- and bone-derived dECM bioinks upon methacryloyl modification, allowing tunable degrees of crosslinking with moderate ultraviolet or blue-light irradiation dosages[13,21]. Moreover, numerous biocompatible photoinitiators are commercially available to mediate this reaction (e.g., LAP, eosin Y, and riboflavin [RF])[13,20,22,23]. Therefore, methacryloyl modification of dECMs represents one of the most valuable avenues for the development of biomimetic hydrogels that closely recapitulate the biological and structural environment of native tissues and this, in turn, makes them suitable candidates for 3D bioprinting applications. In particular, these hydrogels can be precisely tuned for extrusion-based bioprinting (EBB) since their pseudoplastic rheological behavior facilitates their extrusion at high viscosity values, which are needed to achieve adequate shape fidelity and stability upon deposition[24]. EBB is one of the most adopted bioprinting technologies for the biofabrication of tissues and its advantages over other 3D bioprinting techniques have been extensively described elsewhere[25,26].
However, for the particular case of biomanufacturing tissues that require electrical stimulation to acquire their final biological function, dECM hydrogels face relevant limitations as building blocks, mainly because of their insulating properties[27]. Moreover, electrostimulation approaches have been reported to be beneficial in wound healing[28,29], bone regeneration[30,31], cell differentiation[32-34], and tissue maturation[35,36]. The incorporation of electroconductive nanostructured materials into hydrogels, such as graphene derivatives, gold nanoparticles, and carbon nanotubes, has shown great promise to alleviate these shortcomings[37]. Graphene, in particular, is a flexible nanomaterial made of sp2 hybridized carbon atoms organized in a single two-dimensional layer, which generate a π-electron cloud responsible for electroconductivity[38]. However, due to its hydrophobic surface, the colloidal stability of graphene in hydrophilic media is highly limited. Consequently, its oxidized derivative, graphene oxide (GO), has attracted significant attention due to its superior hydrophilicity. This can be attributed to the presence of functional groups, such as hydroxyl and carboxyl moieties, which facilitate its dispersion in aqueous media. Moreover, they can promote protein adsorption from either culture media or cellular secretions, which improves overall cell-hydrogel interactions by increasing available cell anchoring sites[39,40]. However, the introduction of functional groups on GO disrupts the highly ordered carbon structures and results in reduced electroconductivity when compared to pristine graphene or reduced GO (rGO)[41]. Leveraging the bioactive and electroconductive properties of GO nanostructures is, therefore, a major challenge for the development of electroconductive hydrogels for tissue engineering applications.
Accordingly, the aim of this study is to develop an extrudable bioink based on methacryloyl-modified dECM and fully exfoliated GO nanosheets to render the potential for the electrostimulation of 3D bioprinted tissue constructs. We propose a fabrication scheme for the incorporation of GO that exploits both its bioactive and hydrophilic properties during the initial maturation stage of extruded constructs and simultaneously improves GO’s electroconductivity upon in situ reduction. In particular, we harness the protein adsorption capacity of GO to induce homogeneous nanosheet dispersion within the dECM hydrogel, thus facilitating the formation of interconnected electrical networks and aiding initial cellular anchoring. The electro- and photo-addressable hydrogel obtained here, and the corresponding bioprinting scheme, hold much promise in the biofabrication of electrosensitive tissue constructs. Moreover, it may enable cell differentiation and tissue maturation processes by highly controllable electrostimulation strategies.
2. Materials and methods
2.1. Materials
Methacrylic acid (MA), EDC, NHS, dimethylformamide (DMF), picrylsulfonic acid, RF, formalin solution, TOX8 resazurin-based assay, rhodamine B, sodium bicarbonate, and low glucose Dulbecco’s Modified Eagle Medium (DMEM) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium dodecyl sulfate was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Fetal bovine serum (FBS) and penicillin/streptomycin (P/S) were purchased from BioWest and hydrogen peroxide (H2O2), sodium chloride (NaCl), sodium hydroxide (NaOH), glacial acetic acid, sulfuric acid, phosphoric acid, potassium permanganate (K2MnO4), Tris-HCl, and hydrochloric acid (HCl) were purchased from PanReac AppliChem (Chicago, IL, USA), and graphite flakes from Graphene Supermarket (Ronkonkoma, NY, USA). Propidium iodide, AlexaFluor™ 488 Phalloidin, and Hoechst 3342 were purchased from Invitrogen (Waltham, MA, USA).
2.2. Isolation and decellularization of small intestinal submucosa (SIS)
Porcine small intestines were obtained from a local butcher shop and SIS was isolated and decellularized by following the protocol previously reported by Sánchez-Palencia and colleagues[42]. Briefly, the jejunum was thoroughly washed with tap water and the tunica mucosa and serosa muscularis layers were mechanically removed to isolate the SIS. For decellularization, isolated tissue samples were chemically treated with a proprietary solution consisting of H2O2, sodium hypochlorite, and autoclaved type II water for 15 min under constant agitation at room temperature (~23°C). Next, three successive washes with autoclaved type II water and one with sterile 1× phosphate-buffered saline (PBS) were performed to remove cellular and chemical remnants and for restoring ionic balance, respectively. Finally, the obtained membranes were dried at room temperature under laminar flow overnight and subsequently pulverized with the aid of a cryogenic mill (6875 Freezer/Mill, SPEX SamplePrep, Metuchen, NJ, USA). Dry SIS powder samples were then stored at −20°C until further use.
2.3. GO synthesis and characterization
GO synthesis was performed by following the Tour’s methods with slight modifications[43]. Briefly, a solution prepared by mixing sulfuric acid and phosphoric acid at a 9:1 ratio was carefully poured into a flask containing graphite flakes and potassium permanganate. The resulting blend was then heated at 60°C and left for 12 h under constant vigorous magnetic stirring. Next, ice cold water and hydrogen peroxide (H2O2) were added to the blend and the obtained GO was washed successively by filtration with polyester fiber, centrifugation, and resuspension in a solution containing HCl, ethanol and type I water. The final pellet was resuspended in type II water and lyophilized for 24 h.
Adequate GO synthesis was confirmed through Raman spectroscopy by assessing the difference between excitation and emission intensity within a range of 0 and 3000 Raman shift (cm−1). A point-wise laser excitation of 532 nm was shone at different locations along the synthesized GO sheets to evaluate homogeneity of the sample.
2.4. Pre-gel preparation and biochemical modification
Dry SIS powder was solubilized at 4 mg/mL in a solution containing 0.5 M acetic acid and porcine pepsin at a 1 mg/mL concentration. Solubilization was performed under constant vigorous magnetic stirring and at room temperature (~23°C) for 48 h. The free-amine groups present in the obtained SIS pre-gels were quantified by performing the 2,4,6-trinitrobenzene sulfonic acid (TNBSA) assay (see section 2.6).
Next, the obtained pre-gel was biochemically modified by covalently linking the C-terminal of MA to primary-amine residues present in fibrous structural protein backbones of SIS. This was accomplished by adapting the protocol reported by Gaudet et al., which was developed for the biochemical modification of collagen[44]. Briefly, a working solution of MA (in a 1:20 molar excess with respect to free amines in SIS), EDC (1:1 molar ratio with MA) and NHS (1:1 molar ratio with MA) was prepared in DMF and heated for 15 min at 40°C under constant magnetic stirring for activating the terminal carboxyl groups of MA. The activated solution was then poured into the SIS pre-gel and the biochemical modification reaction was left for 24 h under constant magnetic stirring at 4°C. The resulting modified SIS (SISMA) pre-gel was dialyzed against 0.5 M acetic acid for 48 h and subsequently lyophilized for 24 h. The biochemically modified SIS samples were stored at −20°C and, if used for bioprinting experiments, sterilized by treatment with ethylene oxide (EtO).
2.5. Quantification of GO protein adsorption capacity
Dispersion of GO in acid solutions and biological buffers is very limited due to the presence of protonated carboxyl groups and surface charging that induces sheet agglomeration[45]. Consequently, its homogeneous incorporation into ECM and collagen-based hydrogels is a challenging task. To address this issue, we hypothesized that the dispersion of GO would be improved by promoting protein adsorption before its incorporation into SIS and SISMA hydrogels. To test this hypothesis, GO’s protein adsorption capacity was evaluated by quantifying the difference in protein concentration of cell culture media in the presence and absence of dispersed GO. Briefly, GO was dispersed at a concentration of 0.5 mg/mL in a DMEM cell culture media solution supplemented with 10% (v/v) FBS and 1% (v/v) P/S for 2 h at 4°C. The resulting solution was then centrifuged at 12,000 rpm for 5 min to isolate the GO from the medium, and protein concentration in the supernatant was finally quantified with the Quanti-Pro bicinchoninic acid assay Assay Kit (Sigma-Aldrich) and a bovine serum albumin standard curve (Figure S1 (651.7KB, pdf) ). The same procedure was conducted, as a control, for the same supplemented DMEM but in the absence of GO. Absorbance was measured at 562 nm with the aid of a microplate photometer (Multiskan™ FC, Thermo Scientific, Waltham, MA, USA) and protein adsorption on the GO sheets was calculated by implementing Equation 2.5.1.
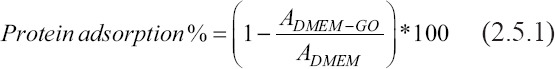
where ADMEM-GO is the absorbance measured from the supplemented DMEM in the presence of GO and ADMEM is the absorbance from the supplemented DMEM alone.
2.6. Hydrogel preparation
A working solution (pH 8.5) consisting of DMEM supplemented with 10% (v/v) FBS, 1% (v/v) P/S, 0.1% (w/v) RF, and Tris-HCl 0.1 M was prepared. Next, GO was dispersed at a 0.5 mg/mL concentration in this working solution and left for 12 h at 4°C to promote protein binding. This was followed by resuspension of lyophilized SISMA at 40 mg/mL in a 0.02 M acetic acid solution aided by a spatula. This mixture was subsequently mixed at a 1:1 volume ratio with the working solution containing GO (SISMA-GO). For the hydrogels produced in the absence of GO, the same procedure was performed but with a working solution without the dispersed nanomaterial.
2.7. Quantification of functionalization efficiency
Since MA is conjugated to free-amine residues, these were quantified before and after biochemical modification as an indicator of reaction efficiency. This was achieved through a TNBSA assay according to the protocol described by Capella-Monsonís et al.[46]. Briefly, SIS and SISMA hydrogels were diluted in 0.1 M sodium bicarbonate (pH 8.5) to a final concentration of 0.2% (w/v), and 250 μL of a 0.01% (w/v) picrylsulfonic acid solution were then added to 500 μL of the diluted hydrogel sample. The blends were then mixed thoroughly with a benchtop vortex and incubated for 2 h at 37°C under constant agitation. Finally, 100 μL of each sample after incubation were transferred to a 96-well microplate and the absorbance was measured at 405 nm with the aid of a microplate photometer (Multiskan™ FC, Thermo Scientific, Waltham, MA, USA). The reaction product is highly chromogenic, meaning that a higher color intensity is associated with a higher concentration of primary amines. Finally, the reaction efficiency was calculated with Equation 2.7.1.
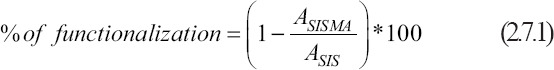
where ASISMA and ASIS are the absorbance’s of SISMA and SIS samples, respectively.
2.8. GO dispersion in SISMA
To evaluate if protein adsorption allowed proper GO dispersion within the hydrogel, serum proteins were fluorescently labeled with rhodamine B and exposed to GO such that, after dispersion, they can be imaged through confocal microscopy. Briefly, the carboxy terminal of rhodamine B was activated with EDC/NHS at 37°C for 15 min and subsequently added to a 10% (v/v) solution of FBS. The mixture was left to react for 24 h at room temperature (~23°C) under constant magnetic stirring. GO was then added at a concentration of 0.5 mg/mL and, after 2 h of exposure to the labeled proteins, it was thoroughly washed by several cycles of centrifugation and resuspension in type II water until the supernatant showed no traces of rhodamine B. Finally, protein-adsorbed GO was resuspended in type II water and mixed at a 1:1 volume ratio with a 40 mg/mL SISMA solution prepared in 0.02 M acetic acid. The samples were imaged at 559 nm with an Olympus FV1000 Confocal Microscope (Tokyo, Japan, Objective 10X) and particle count and area were later analyzed with the ImageJ® software. Z-stack reconstruction was also performed by capturing images at different positions throughout the hydrogel depth to assess the spatial distribution of GO sheets within the hydrogel.
2.9. Rheological evaluation
To assess the rheological response of the developed hydrogels, as well as the influence of photocrosslinking on their mechanical properties, flow sweep, temperature sweep, and time sweep experiments were performed on a Discovery Series Hybrid Rheometer-1 (TA Instruments, New Castle, DE, USA) using a parallel plate geometry with a 20 mm gap. Flow sweep experiments of SISMA and SISMA-GO hydrogels were conducted from 0.01 to 200 1/s at 1% strain and room temperature. The thermal response of these hydrogels was evaluated between 15°C and 37°C with a temperature ramp of 20°C/min, at 1% strain and 10 rad/s frequency. Finally, to compare the change in storage modulus upon photocrosslinking, time sweep assays were carried out before and after blue light irradiation (5 min at 62 mW/cm2 constant intensity) under oscillatory mode, and constant strain of 1% and 10 rad/s.
Shear-thinning properties of the hydrogels were estimated by fitting the viscosity (η) versus shear rate (γ) plot to the power law regression model described by Equation 2.9.1.

K and n were recovered for all samples to assess shear thinning behavior[24].
2.10. Scanning electron microscopy (SEM) imaging of crosslinked structures
To evaluate the resulting hydrogel microstructure upon thermal- or photo-crosslinking, SISMA-GO samples were imaged through SEM. Gelation temperature was achieved after incubation of the samples for 15 min at 37°C. Photocrosslinking was achieved upon blue-light irradiation for 5 min at a constant intensity of 62 mW/cm2. Samples were freeze dried and observed under vacuum conditions at 200× magnification with a 20 kV accelerating voltage (JSM 6490-LV, JEOL, Tokyo, Japan).
2.11. Isolation and in vitro culture of human adipose tissue-derived mesenchymal stem cells (hAD-MSCs)
For bioprinting experiments, hAD-MSCs were isolated following the protocol reported by Linero et al.[47] according to which residual adipose tissue (obtained from abdominoplasty procedures with the previous approval from the research ethics committee at the Universidad de Los Andes Act No. 942, 2018 and subsequent signing of patient informed consent) was subjected to mechanical distortion combined with enzymatic digestion. The isolated cells were subcultured until passage four using low glucose DMEM, supplemented with 10% (v/v) FBS and 1% (v/v) P/S. Their stem cell status was then characterized through flow cytometry (BD FACSCanto™ Flow Cytometer, BD Biosciences, San Jose, CA, USA) with the aid of a human mesenchymal stem cell validation kit (Human Mesenchymal Stem Cell Multi-Color Flow Kit, R&D Systems, Minneapolis, MN, USA) and data acquisition and analysis was done with the BD FACSDiva™ software version 6.1.3. Cells demonstrated positive expression of CD73, CD105, and CD90 as well as negative expression of all markers included in the negative marker cocktail. Quadrants have been set based on isotype controls (Figure S2 (651.7KB, pdf) ).
2.12. Biofabrication of cell-laden SISMA-GO constructs
After sterile SISMA and SISMA-GO hydrogels were successfully prepared, hAD-MSC in low glucose DMEM supplemented with 10% (v/v) FBS and 1% (v/v) P/S was homogeneously mixed at a 1:10 volume ratio with each hydrogel formulation to a final cell density of 4.5 × 106 cells/mL. The prepared bioink (i.e., hydrogel with embedded cells) was then loaded into a sterile printing cartridge. Bioprinting experiments were performed on an INKREDIBLE+ bioprinter (CELLINK AB, Gothenburg, Sweden). Printing parameters were manually optimized to achieve appropriate printing velocity and extrusion pressure according to the bioink’s characteristics (i.e., viscosity, printability, and shape fidelity), as well as nozzle diameter for the intended construct geometry. In this regard, SISMA and SISMA-GO hydrogels achieved proper filament formation through a 25 G blunt needle nozzle at a constant pneumatic pressure of 10 kPa and 5 mm/s printing velocity. A 100 mm2 squared construct with a 0.4 mm height was bioprinted with 10% infill density in a 6-well culture plate and was immediately crosslinked with 405 nm blue-light irradiation (62 mW/cm2) for 1 min (Figure S3 (651.7KB, pdf) ). After photocrosslinking, 4 mL of cell medium were added to each well and the constructs were incubated for up to 7 days. The cell medium was refreshed every other day.
2.13. Cell viability, morphology and proliferation assessment
To assess the viability of hAD-MSCs embedded in SISMA and SISMA-GO constructs, nuclei and dead cell staining was performed with Hoechst 3342 and Propidium Iodide stains, respectively, at 2 h, 3 days, and 7 days after bioprinting. Accordingly, a working solution of the fluorophores was prepared at a 1:1000 ratio with 1× PBS each, and subsequently added to the culture medium at a 1:4 ratio. After 45 min, the medium was removed; the constructs were fixed with 4% (v/v) paraformaldehyde for 5 min and stored in PBS until microscopic observation. Confocal microscopy (Olympus FV1000, Tokyo, Japan) was used to visualize nuclei and dead cell locations with a 358 nm and 559 nm laser excitation, respectively. Emission channels were overlapped to identify live and dead cells, where live cells comprised locations with only nuclei being stained and dead cell locations with both nuclei and propidium iodide stained. Cell viability was assessed by stacking 25 images, captured from adjacent Z-planes separated by 2 µm each, and counting the live cells from the total number of cells. Four different areas of each hydrogel were evaluated for statistical analysis.
Cell morphology changes were assessed in SISMA and SISMA-GO bioprinted constructs by staining F-actin filaments with AlexaFluor™ 488 Phalloidin and cell nuclei with Hoechst 3342 at day 0 and day 6. Briefly, tissues were fixed with 10% (v/v) formalin for 30 min and washed with 1× PBS 3 times to remove remnants of formalin. Cell membranes were then permeabilized with a 0.25% (w/v) solution of Triton-X in 1× PBS for 5 min and subsequently washed 3 times with 1× PBS. Finally, a working solution of AlexaFluor™ 488 Phalloidin and Hoechst 3342 was prepared at a 1:400 and 1:1000 ratios in 1× PBS, respectively, and then added to the constructs and incubated for 1 h. The stained constructs were washed 3 times with 1× PBS and immediately visualized with confocal microscopy using 488 nm and 358 nm laser excitations, respectively. Five images at a 20× magnification were captured and stacked from adjacent Z-planes, separated by 2 µm each, and emission channels were overlapped. Similarly, single-cell morphology was observed by capturing and stacking 45 images collected at 60× magnification.
Cell proliferation in SISMA and SISMA-GO constructs was also assayed by quantifying cell metabolic activity at days 0, 4, and 7 aided by a resazurin-based assay (TOX8). Reduced resazurin by viable cells emits red fluorescence and, therefore, the amount of dye conversion was fluorometrically measured. Accordingly, resazurin dye solution was added to the cell culture medium of each construct at a 10% (v/v) concentration and then incubated for 3 h. The medium was then removed and fluorescence intensity was quantified with a FluoroMax® fluorometer (Horiba, Kyoto, Japan) at 560 nm excitation. Fresh culture medium was added to the assayed constructs to continue incubation until the next measurement.
2.14. Characterization of GO reduced in situ
GO’s in situ reduction was confirmed with Fourier Transform Infrared (FT-IR) spectroscopy and thermogravimetric analysis (TGA). Infrared spectra of graphite, GO, and rGO were recorded using an A250/D FT-IR (Bruker, Germany) within the range of 4000 – 400 cm−1 and spectral resolution of 2 cm−1. The presence or absence of characteristic peaks of each material was analyzed to assess structural changes within these. TGA (TA Instruments) was also performed on these three materials from 30 to 800°C, with a temperature ramp of 10°C/min, to compare thermal stability differences that may arise from structural defects within these. Sample masses of 5 mg were used for this analysis. Reduction was further confirmed with Raman spectroscopy (0 – 3000 Raman shift [cm−1]) of the bioprinted constructs after 3 days of incubation in DMEM culture medium supplemented with 0.5 mM ascorbic acid at 37°C. The treated constructs were lyophilized to avoid noise by water presence during data collection and point wise laser excitation of 532 nm was performed at different locations within the samples.
2.15. Electroconductivity of GO reduced in situ
Since GO was incorporated at a low concentration of 0.25 mg/mL, contacts between all GO sheets are limited by their spatial distribution within the hydrogel. Therefore, considering that the hydrogel alone is not electroconductive, when a current is applied, it will only flow through interconnected networks at the microscale. However, such networks might not necessarily be all well connected. Since electric currents would be difficult to measure under the conditions of this scenario and present high variability, electroconductivity of the GO reduced in situ was measured by creating a densely packed rGO’s squared sheet of 1 cm2. For this, GO dispersed in DMEM culture medium supplemented with 0.5 mM ascorbic acid was incubated at 37°C for 3 days and subsequently centrifuged at 5000 rpm. The resulting pellet was lyophilized for 24 h and the obtained dry powder was resuspended in 96% (v/v) ethanol at a high concentration of 20 mg/mL to finally deposit it over a squared 1 cm2 glass slide. To favor sheet formation by ethanol evaporation, the preparation was left overnight at room temperature. Then, variable currents (Keithley 2450 SourceMeter, SMU Instruments) were applied to the resulting sheet using the four-point probe method to measure the resulting voltage values. Resistance of the rGO sheet was calculated according to Ohm’s law by fitting a linear regression of the measured values (Figure S4 (651.7KB, pdf) ). Conductivity was posteriorly calculated according to Equation 2.14.1.

where L is the thickness of the rGO sheet, A is the contact area of the sheet with the probes, and R is the previously calculated resistance of the sheet. This procedure was also performed for a GO sheet to estimate the possible effects of ascorbic acid and temperature treatment on the electroconductivity.
2.16. Statistical analysis
All experiments were performed in triplicate, unless otherwise stated and quantitative data are expressed as the mean ± standard deviation. Statistical analysis was performed with Student t-tests and P < 0.05 was considered as statistically significant.
3. Results and discussion
3.1. Preparation and functionalization of decellularized small intestine submucosa hydrogels
The SISMA-GO composite bioink was developed following the scheme shown in Figure 1. SIS was isolated from porcine intestines and subsequently decellularized with a sodium hypochlorite-based treatment (see section 2). Dry SIS powder was then solubilized with the aid of pepsin in an acidic solution to obtain a SIS pre-gel. Conjugation of carboxyl groups of MA to free amines present in the backbone of the ECM’s proteins was later performed to confer photosensitive properties to the material in the presence of photoinitiator molecules. Considering that collagen is the main structural component of dECMs, the proposed mechanism for EDC/NHS mediated conjugation of MA to collagen backbones is shown in Figure 2, where EDC and NHS are zero-length crosslinkers that activate the nucleophilic reaction but are not included in the conjugation[48]. This is convenient since this biochemical modification involves no bulky elements that significantly modify the native dECM composition and, before cell embedding, the reaction by-products are eliminated with the aid of dialysis. ECM free amines were previously quantified with the TNBSA assay to determine the necessary reagent concentrations. An efficient biochemical conjugation reaction was demonstrated by an 81.5 ± 0.8% reduction in free-amine content after methacryloyl conjugation to SIS pre-gels (i.e., SISMA) (Figure S5 (651.7KB, pdf) ). This strongly suggests a high degree of functionalization when compared with previously reported values of other functionalization methods with MA[43,49,50], which ranged between 20% and 30%. Furthermore, it was comparable to other methods based on methacrylic anhydride, which reported values between 71% and 87%[13,51,52]. Dialyzed SISMA samples were then lyophilized and resolubilized to incorporate the GO nanosheets and the photoinitiator (i.e., RF). Moreover, a pH 8.5 DMEM solution supplemented with serum proteins and Tris-HCl buffer was added during the resuspension procedure to raise the pH of the hydrogel to 7 and to adjust the ionic balance for posterior cell incorporation.
Figure 1.
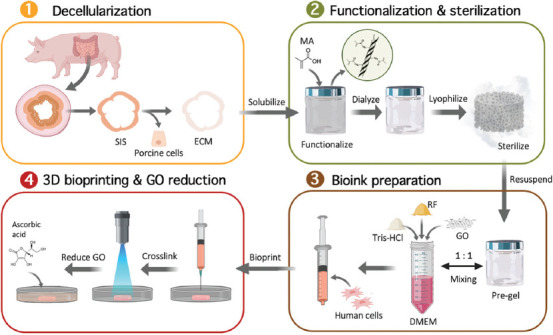
SISMA-graphene oxide (GO) composite bioink preparation and bioprinting schematic. The different steps in the preparation process are shown from small intestine submucosa dissection to bioprinting and tissue maturation with in situ reduction of GO (Created with BioRender.com).
Figure 2.
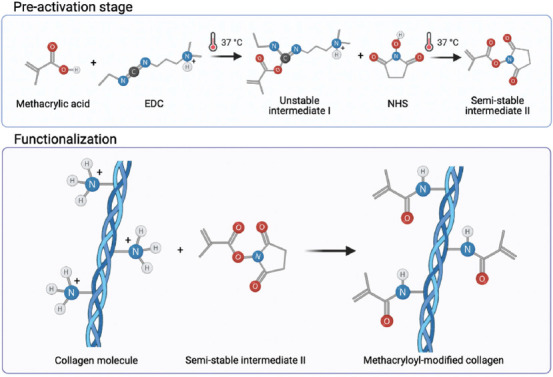
Proposed mechanism for the methacryloyl-modification of small intestinal submucosa collagen, as a general example of this modification on extracellular matrix proteins (Created with BioRender.com).
3.2. GO synthesis and dispersion in SISMA hydrogels
GO nanosheets were successfully synthesized upon graphite exfoliation and oxidation procedures. However, the colloidal stability of GO in SISMA hydrogels was a special concern since previous studies have reported that GO forms aggregates in acidic media and biological buffers[53], two of the essential media for SISMA hydrogel preparation. The electrostatic repulsion between GO nanosheets has shown to be the main driving force for their colloidal stability[45], and this phenomenon is impaired at low pH due to the protonation of GO’s carboxylic groups. Similarly, the sp2-hybridized carbon backbone of GO has a high degree of surface planarity that facilitates electrostatic surface charging in the presence of electrolyte solutions, resulting in irreversible sheet aggregation[54]. Accordingly, we hypothesized that by coating GO surface with hydrophilic serum proteins from culture media, GO aggregation could be largely avoided. This would lead to a superior exfoliation level within the hydrogel (Figure 3A). Protein adsorption on GO after exposure to culture media was confirmed by a 23.57 ± 3.51% decrease in medium protein concentration, as observed in Figure 3B. This corresponds to approximately 2.29 µg of serum proteins adsorbed per µg of GO. Nanocomposite exfoliation in SISMA hydrogels was then assessed by fluorescently labeling the serum proteins before its dispersion in SISMA. Confocal microscopy image reconstruction through z-stacking of the imaged GO-embedded hydrogels showed low aggregate formation and an overall homogeneous dispersion of GO nanosheets within the hydrogel (Figure 3C). Moreover, the particle area distribution within the hydrogel (Figure 3D) shows a right-tailed distribution centered at 0.386 μm2 with a few out-of-distribution cases, indicating that low GO aggregation occurred. The high frequency at this specific area value may have been a result of the image resolution limits, which means that smaller GO sheets may exist within the hydrogel. This suggests that protein coating before dispersion may contribute to the adequate GO exfoliation levels observed in SISMA hydrogels. Our exfoliation strategy is advantageous with respect to previous methods as it does not require any alterations to GO’s chemical structure with complex surface modification strategies[55-57]. We aimed at avoiding this functionalization approach because altering the surface groups of GO could hinder the reduction efficiency after bioprinting. Moreover, it avoids rough exfoliation methods, such as long periods of sonication[58,59], which can alter protein structure within bioactive hydrogels[60].
Figure 3.
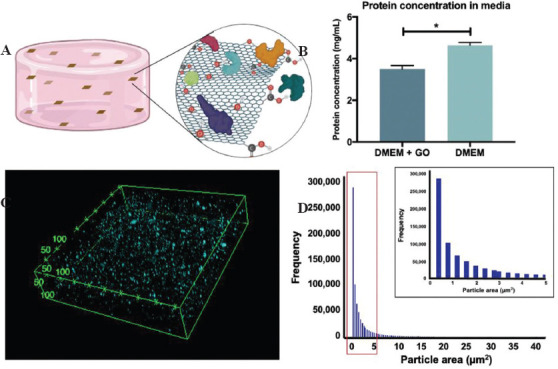
Graphene oxide (GO) protein adsorption and dispersion in composite hydrogels. (A) Schematic of hypothesized GO exfoliation in SISMA hydrogels as a result of exposure to serum proteins before dispersion (Created with BioRender.com). (B) Protein concentration in culture medium before and after the addition of GO. A statistically significant decrease is observed (P < 0.05, n = 3), which is attributed to serum protein adsorption on GO surface. (C) Confocal imaging of fluorescently labeled serum proteins adsorbed on GO shows a homogeneous dispersion within the hydrogel. Reference dimensions are in µm. (D) GO particle area distribution within the hydrogel seen as a right-tailed distribution centered at 0.386 μm2.
3.3. Characterization of SISMA-GO hydrogel crosslinking
The microscopic structural morphology of SISMA-GO composite hydrogels after thermal gelation and photo-crosslinking was evaluated through SEM imaging (Figure 4A). Fibrillar patterns resembling the hierarchical organization of collagen fibrils in native tissues were observed in thermally crosslinked hydrogels (yellow arrows). Self-assembling of collagen into fibers has been previously reported upon an increase in temperature, a process that is mediated by hydrogen bonding between collagen fibrils[61]. However, as shown in the magnified view (Figure 4A, right panel), the hydrogel is highly porous, a characteristic of weak crosslinking mechanisms[62]. On the contrary, fiber arrangement on photo-crosslinked hydrogels exhibits a much less porous structure (i.e., higher crosslinking degree), but is much more disorganized. This could be a result of the differences in crosslinking dynamics between both methods. In the presence of blue-light irradiation (405 nm), RF degrades and generates free radicals that destabilize the double bonds in methacryloyl moieties of the functionalized ECM and, as a consequence, induce covalent crosslinking between adjacent proteins[63]. Since at the moment of covalent bond formation collagen fibers were not aligned, SISMA hydrogels exhibited no observable hierarchical structure even after incubation at 37°C. This is probably because upon this thermal stimulus the formed covalent bonds have limited rotation for their 3D rearrangement.
Figure 4.
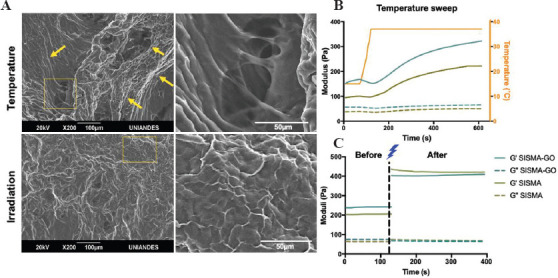
Thermal and photo-induced crosslinking of SISMA-graphene oxide (GO). (A) SEM images of composite hydrogels crosslinked with temperature and with blue-light irradiation. Thermal gelation showed alignment of collagen fibers but a highly porous structure, while photo-crosslinking showed a more disordered but less porous structure. (B) Temperature sweep experiments in SISMA-GO and SISMA hydrogels showed slow temperature-induced gelation profiles, as demonstrated by a similar increase in their storage moduli (G’). (C) Time sweeps before and after irradiation demonstrated a higher increase in G’ than with the temperature stimulus, and negligible effect of GO on photocrosslinking.
Thermal- and photo-induced crosslinking of SISMA-GO hydrogels was further compared by evaluating their rheological behavior in the presence of both stimuli (Figure 4B and C). SISMA hydrogels were also assessed as a control to ensure that the presence of GO led to no detrimental impact on the crosslinking dynamics. Both crosslinking mechanisms effectively increased the storage moduli (G’) in both hydrogels while the loss moduli (G’’) remained unchanged. This confirms that both mechanisms increase the elastic response of the hydrogels and, consequently, their structural stability. Moreover, there is no crossover between G’ and G” curves during crosslinking, which indicates that the hydrogels exhibit a predominantly elastic behavior over time. This is particularly important in hydrogels intended for EBB, since it is crucial for assuring filament formation during extrusion, and for maintaining shape fidelity upon deposition[24]. However, while the temperature stimulus increased G’ values of SISMA-GO to 300 Pa after 5 min, blue-light irradiation led to a value of about 400 Pa after 1 min, confirming that photo-stimulation yields faster and stronger crosslinking dynamics.
Moreover, since the formed covalent bonds during photo-stimulation are expected to produce more stable structures than the weak hydrogen bonds formed during thermal-stimulation, we evaluated both the soluble and gel fractions of thermally and photo-crosslinked SISMA-GO constructs as a function of mass loss over time, such that their stability can be estimated. After a 24-h incubation period, their gel fractions approached 69.32 ± 3.41% and 90.39 ± 1.92%, respectively (Figure S6 (651.7KB, pdf) ). These results suggest that photo-crosslinked SISMA-GO constructs exhibit a much slower degradation rate and are, therefore, more structurally stable over time. Likewise, swelling tests confirmed that, upon incubation, irradiated SISMA-GO constructs only swell up to 27.90 ± 4.88% of their initial weight, and this value remains unchanged after the first 8 h of incubation (Figure S7 (651.7KB, pdf) ). This confirms that the formed covalent bonds limit excessive hydrogel swelling and maintain stable hydrogel structures over time.
3.4. 3D bioprinting of cell-laden constructs
Before incorporating hAD-MSCs into the composite hydrogel, we conducted several tests to ensure it presented the required bioink behavior. Manual extrusion tests were carried out to select the adequate SISMA concentration that allowed a stable filament formation during the process (Figure 5A). Based on these results, a grid-like pattern was deposited with a 3D bioprinter to evaluate the shape fidelity of the concentration that granted the best filament formation (20 mg/mL). A defined construct outline with sharp edges was observed, which adequately maintained its shape during the bioprinting process and before photocrosslinking (Figure 5B, left panel). Similarly, an 8-layered cubic construct was successfully bioprinted (Figure 5B, right panel), which further demonstrates the suitability of our bioink for maintaining deposited patterns during the additive manufacturing of multilayered 3D constructs. Flow sweep experiments depicted in Figure 5C show that SISMA and SISMA-GO hydrogels presented the characteristic shear-thinning behavior of bioinks with superior printability, in which a decrease in viscosity occurs upon the application of a shearing force[64]. After fitting the data to a power law model (Figure S8 (651.7KB, pdf) ), the obtained n coefficients from Equation 2.9.1 were 0.208 and 0.223 for SISMA and SISMA-GO hydrogels, respectively, indicating a pseudoplastic behavior with strong shear-thinning (close to n = 2)[24]. This is important since high shear stress values predominate during the extrusion process and shear thinning fluids attenuate the perceived stress by cells by stress-relaxation mechanisms that ultimately help maintaining high cell viability[65]. Again, there was no observable difference between the shear-thinning behavior of SISMA-GO and SISMA hydrogels, meaning that GO shows no significant impact on the rheological response of the produced bioinks.
Figure 5.
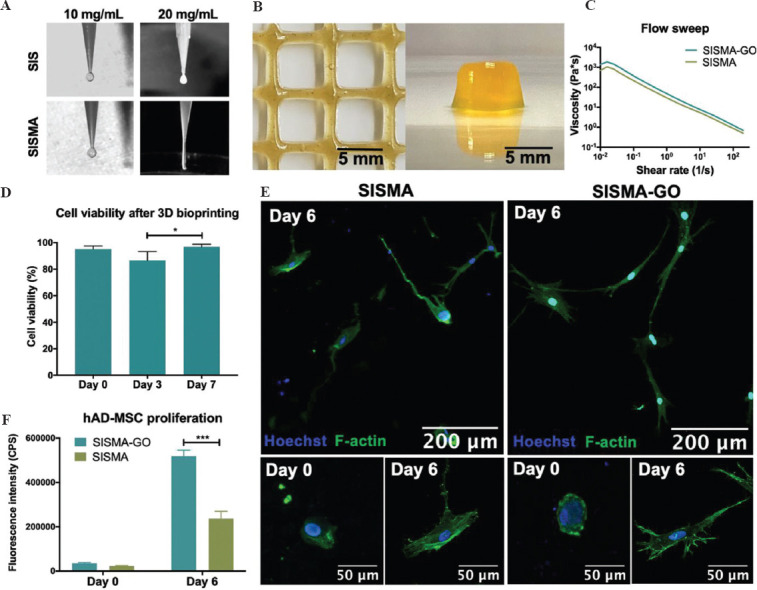
SISMA-graphene oxide (GO) bioprinting. Adequate filament formation during extrusion aided by a 3D bioprinter (A) and shape fidelity upon deposition (B) was observed for SISMA-GO composite hydrogels, highlighting their suitability for bioprinting applications. (C) Flow sweep experiments of SISMA-GO and SISMA hydrogels depict an appropriate shear-thinning behavior, with no observable difference in the presence of GO. (D) Cell viability of human adipose tissue-derived mesenchymal stem cells (hAD-MSC) 2 h, 3 days, and 7 days after bioprinting indicates high biocompatibility with SISMA-GO hydrogels (n = 4). (E) Confocal microscopy images of hAD-MSCs embedded in bioprinted SISMA and SISMA-GO constructs with stained nuclei in blue and stained F-actin filaments in green. Elongated morphologies are observed after 6 days of incubation, indicating the presence of cell-matrix interactions that mediate cell adhesion and spreading. (F) hAD-MSC proliferation in SISMA and SISMA-GO constructs, as measured by resazurin reduction as a consequence of metabolic activity.
hAD-MSCs were embedded and bioprinted in SISMA-GO hydrogels to evaluate the biocompatibility of the materials and the suitability of the proposed bioprinting scheme. In addition, the proposed ascorbic acid-based scheme for in situ reduction was also performed on day 3 to assess whether any change was observable on construct viability. Cell viability was assessed with nuclei (blue) and dead cell (red) staining, where nuclei signal alone marked live cells and dual staining (seen as magenta) marked dead cells (Figure S9 (651.7KB, pdf) ). Since propidium iodide has been reported to interact with GO sheets[66], spots with only red staining were disregarded as they correspond to GO-PI complexes. The bioprinted constructs exhibit high cell viability at all measured time points, with a slight decrease on day 3 but a prompt recovery by day 7 (Figure 6D). This small drop may be attributed to unreacted by-products from the photocrosslinking reaction, since excess free radicals may induce the formation of reactive oxygen species, which have shown to induce DNA damage and compromise cell viability[67]. The ascorbic acid supplementation on day 3 may have also contributed to the observed drop. Despite this, a timely recovery was observed and high cell viability (96.9 ± 0.9%) was achieved 1 week after bioprinting, confirming that neither GO incorporation nor the proposed reduction scheme compromises cellular survival. In addition, embedded hAD-MSCs in both SISMA and SISMA-GO constructs transitioned from rounded to elongated morphology after 6 days of incubation (Figure 5E), which suggests the formation of cell-matrix interactions that mediate cell adhesion (i.e., cell-matrix adhesion complexes)[68]. Moreover, as observed in the magnified images of day 6 (Figure 5, bottom panel), hAD-MSCs in SISMA-GO constructs exhibit a greater amount of filopodia than in SISMA constructs, indicating that the presence of GO likely aids in cell adhesion. Notably, GO nanosheets also favored hAD-MSCs proliferation, as evidenced by the increased proliferation rate when compared to SISMA constructs (Figure 5F). This is consistent with the previous reports where proteins adsorbed on GO surfaces have been shown to favor integrin-based cell adhesion by providing anchoring sites that facilitate cell migration, proliferation and function[69].
Figure 6.
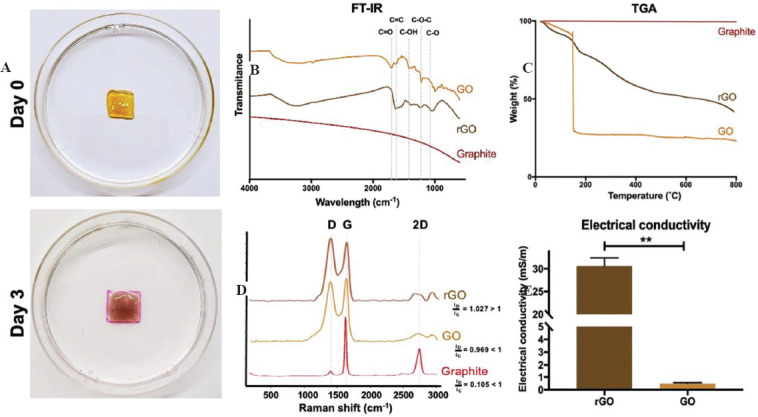
In situ reduction of graphene oxide (GO). (A) SISMA-GO constructs before and after 3-days incubation with ascorbic acid show hydrogel darkening attributed to GO reduction. (B) FTIR analysis confirms the elimination of carboxyl and hydroxyl peaks upon ascorbic acid incubation, and a (C) decreased thermal degradation rate is observed with thermogravimetric analysis. (D) Raman spectra of graphite, GO and GO after incubation in ascorbic acid (reduced GO [rGO]) present an increase in D peak intensity (ID) with surface modifications and it surpasses G peak intensity (IG) after ascorbic acid-based reduction. ID/IG ratios are indicative of structural disorder. (E) Improved electrical conductivity of rGO sheets with respect to GO, as measured with the four-point probe method.
3.5. Improved electroconductivity of GO reduced in situ
Several GO reduction methods have been reported that yield high reduction efficiencies, usually by employing high temperatures and by adding strong acids or bases at high concentrations[70]. However, these methods are not compatible with our biofabrication scheme due to their high cytotoxicity, especially considering that we need an in situ reduction of GO to harness its bioactive effects during the initial maturation stages of the bioprinted constructs. As a result, we propose an alternative reduction scheme based on ascorbic acid exposure and incubation at 37°C, to maintain high levels of electroconductivity without detrimentally affecting cell viability. Ascorbic acid concentration was selected according to previously reported values that are non-cytotoxic for mesenchymal stem cells[71]. GO reduction was evaluated after 3 days of exposure to ascorbic acid to subsequently assess the electroconductive potential of the bioinks. As observed in Figure 6A, after 3 days of incubation, the construct displays a much darker color (characteristic of rGO) in comparison to the color immediately after printing. The reduced weight loss profile of rGO when compared to GO (~20% difference) observed with TGA experiments (Figure 6B) suggests that the improved thermal stability may be a result of a decrease in GO’s surface functional groups with our method. Similarly, FT-IR spectroscopy analysis of GO and rGO sheets (Figure 6C) showed that carboxyl (1721 cm-1) and hydroxyl (1364 cm-1) peaks in rGO were strongly diminished, but epoxy peaks (1215 and 1051 cm-1) were conserved[72], suggesting a partial reduction was achieved. Moreover, Raman spectra of graphite, GO and rGO (Figure 6B) display the characteristic G and D peaks, where G represents the degree of sp2 hybridization and D represents structural disorder (increases with surface modification)[73]. When comparing GO to graphite spectra, a clear increase in the D peak intensity is observed, which indicates disruptions in the sp2-hybridized structure as a result of the presence of superficial carboxyl and hydroxyl groups. This, in turn, is an indicator of correct oxidation of the carbon structure, especially because the structural disorder ratio (ID/IG) is <1, which is typical of oxidized graphene[73]. The observed D peak intensity in the rGO spectrum surpasses that of the G peak, which indicates that incubation with ascorbic acid increased structural disorder. This structural disorder ratio above 1 has been reported for rGO because previously oxidized structures fail to recover the original carbon backbone of graphene[74]. Moreover, the process permanently destroys some functional groups, thereby affecting the carbon backbone even further[74].
To further characterize GO’s reduction, the conductivity of GO before and after incubation with ascorbic acid was assessed with the four-point probe method. As shown in Figure 6C, ascorbic acid-based reduction significantly increased the electroconductivity of GO sheets by nearly two orders of magnitude (i.e., from 0.49±0.04 mS/m to 30.59±1.27 mS/m). We decided to measure electroconductivity on the rGO sheets directly, instead of the composite hydrogel, to isolate the effect of the reduction from that of electrolytes in the culture medium and avoid measurement variabilities due to the localized electrical percolation networks[75]. This is a promising result for the intended electrostimulation applications since increased electroconductivity within the sheets may help direct electrical currents through the bioink. However, despite the large increase in conductivity with respect to GO, rGO sheets still exhibit a lower conductivity than those reported for rGO by other reduction methods[76-78]. This was an expected result due to the mild temperature and acid concentration used during the process to maintain viability unaffected. Nonetheless, considering electrostimulation studies have been performed in hydrogels without conductive materials due to the conductivity of cell culture medium (1,700 mS/m[79]) and that of cell surfaces (10-2 – 10-5 mS/m[80,81]), the presence of rGO sheets could enhance previous efficiencies by aiding the electrical connectivity and directing currents within the hydrogel. This is specifically advantageous if currents are applied during early maturation stages, since cells are yet to form interconnected networks themselves.
4. Conclusions
Tailoring the mechanical and biological properties of biomaterials for the biofabrication of tissues with specific maturation profiles has been an ongoing research focus for their translation to medical scenarios. The herein developed photocrosslinkable nanocomposite bioink showed great promise for 3D bioprinting applications, considering that it can be directly printed with little composition modifications and without the need of other excipient materials. The limited modification to the overall ECM composition enabled by methacryloyl modification maximizes its bioactive and biomimetic advantages, while also enhancing its mechanical properties upon the formation of stable interconnected crosslinked networks. This integrates two of the most important aspects for recapitulating the environment of native tissues and, in turn, for bioink formulation. In addition, GO incorporation followed by its in situ reduction harnessed the bioactivity and hydrophilicity of GO to improve its colloidal stability within the bioink, and once it was stably dispersed, improved its electroconductive properties by two orders of magnitude. As such, its bioactive, mechanically stable, and electroconductive properties are a clear advantage above previous bioink formulations.
The enhanced electrical conductivity of rGO sheets increases the potential of SISMA-GO for the biofabrication of tissues that require electrostimulation while avoiding current limitations of electroconductive hydrogels, such as cytotoxicity and poor dispersibility[82]. As such, it could contribute significantly to the development of electrosensitive tissues (e.g., skeletal muscle, cardiac muscle, and neural tissue), directing differentiation profiles (e.g., chondrogenesis[83], and osteogenesis[84]), and tissue regeneration[85]. Future work will be focused on optimizing the in situ reduction procedure to further improve the observed electroconductivity, and assessing its direct contribution to different electro stimulation applications. This work paves the way for exploiting the potential of SISMA-GO within the field of electrosensitive tissue engineering and regenerative medicine.
Author contributions
L.R. and J.A.S. performed most of the experiments and J.C performed experiments related to nanomaterial characterization. L.R. analyzed the data and wrote the manuscript with contributions from J.A.S., review and editing, L.R., J.C.C., and C.M.-C.; supervision, J.C.C. and C.M.-C.; project administration, J.C.C. and C.M.-C.; funding acquisition, J:C:C ans C.M.-C.
Funding sources
This work was funded by the Department of Biomedical Engineering and by the Vice-Chancellor for Research and Creation at Universidad de los Andes. Publishing expenses was funded by Convocatoria CI-001 Publica tus conocimientos o expone tus nuevas creaciones by the Vice-Chancellor for Research and Creation at Universidad de los Andes.
Conflict of interest
The authors declare no conflicts of interest.
References
- 1.Kim B, Das S, Jang J, et al. Decellularized Extracellular Matrix-based Bioinks for Engineering Tissue and Organ-specific Microenvironments. Chem Rev. 2020;120:10608–61. doi: 10.1021/acs.chemrev.9b00808. [DOI] [PubMed] [Google Scholar]
- 2.Ng W, Chua C, Shen Y. Print Me An Organ!Why We Are Not There Yet. Prog Polym Sci. 2019;97:101145. [Google Scholar]
- 3.Frantz C, Stewart KM, Weaver VM. The Extracellular Matrix at a Glance. J Cell Sci. 2010;123:4195–200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaefer L, Schaefer RM. Proteoglycans:From Structural Compounds to Signaling Molecules. Cell Tissue Res. 2009;339:237–46. doi: 10.1007/s00441-009-0821-y. [DOI] [PubMed] [Google Scholar]
- 5.Kim BS, Kim H, Gao G, et al. Decellularized Extracellular Matrix:A Step Towards the Next Generation Source for Bioink Manufacturing. Biofabrication. 2017;9:034104. doi: 10.1088/1758-5090/aa7e98. [DOI] [PubMed] [Google Scholar]
- 6.Kohane DS, Langer R. Polymeric Biomaterials in Tissue Engineering. Pediatr Res. 2008;63:487–91. doi: 10.1203/01.pdr.0000305937.26105.e7. [DOI] [PubMed] [Google Scholar]
- 7.Badylak SF. The Extracellular Matrix as a Scaffold for Tissue Reconstruction. Semin Cell Dev Biol. 2002;13:377–83. doi: 10.1016/s1084952102000940. [DOI] [PubMed] [Google Scholar]
- 8.Saldin LT, Cramer MC, Velankar SS, et al. Extracellular Matrix Hydrogels from Decellularized Tissues:Structure and Function. Acta Biomater. 2017;49:1–15. doi: 10.1016/j.actbio.2016.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pouliot RA, Young BM, Link PA, et al. Porcine Lung-Derived Extracellular Matrix Hydrogel Properties Are Dependent on Pepsin Digestion Time. Tissue Eng C Methods. 26:332–46. doi: 10.1089/ten.tec.2020.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang C. Enhanced physicochemical properties of collagen by using EDC/NHS-crosslinking. Bull Mater Sci. 2012;35:913–8. [Google Scholar]
- 11.Curley CJ, Dolan EB, Otten M, et al. An Injectable Alginate/Extra Cellular Matrix (ECM) Hydrogel Towards Acellular Treatment of Heart Failure. Drug Deliv Transl Res. 2018;9:1–13. doi: 10.1007/s13346-018-00601-2. [DOI] [PubMed] [Google Scholar]
- 12.Choi B, Kim S, Lin B, et al. Cartilaginous Extracellular Matrix-Modified Chitosan Hydrogels for Cartilage Tissue Engineering. ACS Appl Mater Interfaces. 2014;6:20110–21. doi: 10.1021/am505723k. [DOI] [PubMed] [Google Scholar]
- 13.Ali M, Kumar A Pr, Yoo JJ, et al. A Photo-Crosslinkable Kidney ECM-Derived Bioink Accelerates Renal Tissue Formation. Adv Healthc Mater. 2019;8:1800992. doi: 10.1002/adhm.201800992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sionkowska A, Skopinska-Wisniewska J, Gawron M, et al. Chemical and Thermal Cross-linking of Collagen and Elastin Hydrolysates. Int J Biol Macromol. 2010;47:570–7. doi: 10.1016/j.ijbiomac.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Davidenko N, Schuster CF, Bax DV, et al. Control of Crosslinking for Tailoring Collagen-based Scaffolds Stability and Mechanics. Acta Biomater. 2015;25:131–42. doi: 10.1016/j.actbio.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy N, Reddy R, Jiang Q. Crosslinking Biopolymers for Biomedical Applications. Trends Biotechnol. 2015;33:362–9. doi: 10.1016/j.tibtech.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 17.O'Connell CD, Zhang B, Onofrillo C, et al. Tailoring the mechanical properties of gelatin methacryloyl hydrogels through manipulation of the photocrosslinking conditions. Soft Matter. 2018;14:2142–51. doi: 10.1039/c7sm02187a. [DOI] [PubMed] [Google Scholar]
- 18.Ashwin S, Reed G, Michael M. Myelinated 3D Neural Culture Platform as a Physiological Model of Diabetic Peripheral Neuropathy. Front Bioeng Biotechnol. 2016;4:02188. [Google Scholar]
- 19.Williams CG, Malik AN, Kim TK, et al. Variable Cytocompatibility of Six Cell Lines with Photoinitiators Used for Polymerizing Hydrogels and Cell Encapsulation. Biomaterials. 2005;26:1211–8. doi: 10.1016/j.biomaterials.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 20.Diamantides N, Wang L, Pruiksma T, et al. Correlating Rheological Properties and Printability of Collagen Bioinks:The Effects of Riboflavin Photocrosslinking and pH. Biofabrication. 2017;9:034102. doi: 10.1088/1758-5090/aa780f. [DOI] [PubMed] [Google Scholar]
- 21.Parthiban SP, Athirasala A, Tahayeri A, et al. BoneMA Synthesis and Characterization of a Methacrylated Bone-derived Hydrogel for Bioprinting of Vascularized Tissues. BioRxiv. 20202020:974063. doi: 10.1088/1758-5090/abb11f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SH, Yeon YK, Lee JM, et al. Precisely Printable and Biocompatible Silk Fibroin Bioink for Digital Light Processing 3D Printing. Nat Commun. 2018;9:1620. doi: 10.1038/s41467-018-03759-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poldervaart MT, Goversen B, de Ruijter M, et al. 3D Bioprinting of Methacrylated Hyaluronic Acid (MeHA) Hydrogel with Intrinsic Osteogenicity. PLoS One. 2017;12:e0177628. doi: 10.1371/journal.pone.0177628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paxton N, Smolan W, Böck T, et al. Proposal to Assess Printability of Bioinks for Extrusion-based Bioprinting and Evaluation of Rheological Properties Governing Bioprintability. Biofabrication. 2017;9:044107. doi: 10.1088/1758-5090/aa8dd8. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Chen M, Fan X, et al. Recent Advances in Bioprinting Techniques:Approaches, Applications and Future Prospects. J Transl Med. 2016;14:271. doi: 10.1186/s12967-016-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derakhshanfar S, Mbeleck R, Xu K, et al. 3D Bioprinting for Biomedical Devices and Tissue Engineering:A Review of Recent Trends and Advances. Bioact Mater. 2018;3:144–56. doi: 10.1016/j.bioactmat.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwala S. Electrically Conducting Hydrogels for Health care:Concept Fabrication Methods, and Applications. Int J Bioprint. 2020;6:273. doi: 10.18063/ijb.v6i2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thakral G, LaFontaine J, Najafi B, et al. Electrical Stimulation to Accelerate Wound Healing. Diabetic Foot Ankle. 2013;4:22081. doi: 10.3402/dfa.v4i0.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashrafi M, Alonso-Rasgado T, Baguneid M, et al. The Efficacy of Electrical Stimulation in Lower Extremity Cutaneous Wound Healing:A Systematic Review. Exp Dermatol. 2017;26:171–8. doi: 10.1111/exd.13179. [DOI] [PubMed] [Google Scholar]
- 30.Meng S, Rouabhia M, Zhang Z. Electrical Stimulation Modulates Osteoblast Proliferation and Bone Protein Production through Heparin-Bioactivated Conductive Scaffolds. Bioelectromagnetics. 2012;34:189–99. doi: 10.1002/bem.21766. [DOI] [PubMed] [Google Scholar]
- 31.Leppik L, Zhihua H, Mobini S, et al. Combining Electrical Stimulation and Tissue Engineering to Treat Large Bone Defects in a Rat Model. Sci Rep. 2018;8:6307. doi: 10.1038/s41598-018-24892-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C, Bai X, Ding Y, et al. Electrical Stimulation as a Novel Tool for Regulating Cell Behavior in Tissue Engineering. Biomater Res. 2019;23:25. doi: 10.1186/s40824-019-0176-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu C, Pan S, Ma Y, et al. Effect of Electrical Stimulation Combined with Graphene-oxide-based Membranes on Neural Stem Cell Proliferation and Differentiation. Artif Cells Nanomed Biotechnol. 2019;47:1867–76. doi: 10.1080/21691401.2019.1613422. [DOI] [PubMed] [Google Scholar]
- 34.Kwon HJ, Lee GS, Chun H. Electrical Stimulation Drives Chondrogenesis of Mesenchymal Stem Cells in the Absence of Exogenous Growth Factors. Sci Rep. 2016;6:39302. doi: 10.1038/srep39302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sadeghian RB, Ebrahimi M, Salehi S. Electrical Stimulation of Microengineered Skeletal Muscle Tissue:Effect of Stimulus Parameters on Myotube Contractility and Maturation. J Tissue Eng Regen Med. 2017;12:912–22. doi: 10.1002/term.2502. [DOI] [PubMed] [Google Scholar]
- 36.Hirt MN, Boeddinghaus J, Mitchell A, et al. Functional Improvement and Maturation of Rat and Human Engineered Heart Tissue by Chronic Electrical Stimulation. J Mol Cell Cardiol. 2014;74:151–61. doi: 10.1016/j.yjmcc.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Ashtari K, Nazari H, Ko H, et al. Electrically Conductive Nanomaterials for Cardiac Tissue Engineering. Adv Drug Deliv Rev. 2019;144:162–79. doi: 10.1016/j.addr.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Javadi M, Gu Q, Naficy S, et al. Conductive Tough Hydrogel for Bioapplications. Macromol Biosci. 2017;18:1700270. doi: 10.1002/mabi.201700270. [DOI] [PubMed] [Google Scholar]
- 39.Zhou X, Cui H, Zhang LG, et al. 3D Bioprinted Graphene Oxide-incorporated Matrix for Promoting Chondrogenic Differentiation of Human Bone Marrow Mesenchymal Stem Cells. Carbon. 2017;116:615–24. [Google Scholar]
- 40.Ku SH, Park CB. Myoblast Differentiation on Graphene Oxide. Biomaterials. 2013;34:2017–23. doi: 10.1016/j.biomaterials.2012.11.052. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Zhang B, Xu Q, et al. Development of Graphene Oxide/Polyaniline Inks for High Performance Flexible Microsupercapacitors via Extrusion Printing. Adv Funct Mater. 2018;28:1706592. [Google Scholar]
- 42.Sánchez-Palencia DM, D'Amore A, González-Mancera A, et al. Effects of Fabrication on the Mechanics Microstructure and Micromechanical Environment of Small Intestinal Submucosa Scaffolds for Vascular Tissue Engineering. J Biomech. 2014;47:2766–73. doi: 10.1016/j.jbiomech.2014.04.048. [DOI] [PubMed] [Google Scholar]
- 43.Marcano DC, Kosynkin DV, Berlin JM, et al. Improved Synthesis of Graphene Oxide. ACS Nano. 2010;4:4806–14. doi: 10.1021/nn1006368. [DOI] [PubMed] [Google Scholar]
- 44.Gaudet ID, Shreiber DI. Characterization of Methacrylated Type-I Collagen as a Dynamic Photoactive Hydrogel. Biointerphases. 2012;7:25. doi: 10.1007/s13758-012-0025-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shih CJ, Lin S, Sharma R, et al. Understanding the pH-Dependent Behavior of Graphene Oxide Aqueous Solutions:A Comparative Experimental and Molecular Dynamics Simulation Study. Langmuir. 2011;28:235–41. doi: 10.1021/la203607w. [DOI] [PubMed] [Google Scholar]
- 46.Capella-Monsonís H, Coentro JQ, Graceffa V, et al. An Experimental Toolbox for Characterization of Mammalian Collagen Type I in Biological Specimens. Nat Protoc. 2018;13:507–29. doi: 10.1038/nprot.2017.117. [DOI] [PubMed] [Google Scholar]
- 47.Linero I, Chaparro O. Paracrine Effect of Mesenchymal Stem Cells Derived from Human Adipose Tissue in Bone Regeneration. PLoS One. 2014;9:e107001. doi: 10.1371/journal.pone.0107001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hermanson GT. Bioconjugate Techniques. Elsevier; Amsterdam, Netherlands: 2013. Zero-Length Crosslinkers; pp. 259–73. [Google Scholar]
- 49.Lim KS, Schon BS, Mekhileri NV, et al. New Visible-Light Photoinitiating System for Improved Print Fidelity in Gelatin-Based Bioinks. ACS Biomater Sci Eng. 2016;2:1752–62. doi: 10.1021/acsbiomaterials.6b00149. [DOI] [PubMed] [Google Scholar]
- 50.Yoon HJ, Shin SR, Cha JM, et al. Cold Water Fish Gelatin Methacryloyl Hydrogel for Tissue Engineering Application. PLoS One. 2016;11:e0163902. doi: 10.1371/journal.pone.0163902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drzewiecki KE, Parmar AS, Gaudet ID, et al. Methacrylation Induces Rapid Temperature-Dependent, Reversible Self-Assembly of Type-I Collagen. Langmuir. 2014;30:11204–11. doi: 10.1021/la502418s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ravichandran R, Islam MM, Alarcon EI, et al. Functionalised Type-I Collagen as a Hydrogel Building Block for Bio-orthogonal Tissue Engineering Applications. J Mater Chem B. 2016;4:318–26. doi: 10.1039/c5tb02035b. [DOI] [PubMed] [Google Scholar]
- 53.Tang H, Zhang S, Huang T, et al. Effects of pH and Electrolytes on the Sheet-to-Sheet Aggregation Mode of Graphene Oxide in Aqueous Solutions. Environ Sci Nano. 2020;7:984–95. [Google Scholar]
- 54.Hong BJ, Compton OC, An Z, et al. Successful Stabilization of Graphene Oxide in Electrolyte Solutions:Enhancement of Biofunctionalization and Cellular Uptake. ACS Nano. 2011;6:63–73. doi: 10.1021/nn202355p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cha C, Shin SR, Gao X, et al. Controlling Mechanical Properties of Cell-Laden Hydrogels by Covalent Incorporation of Graphene Oxide. Small. 2013;10:514–23. doi: 10.1002/smll.201302182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang CT, Shrestha LK, Ariga K, et al. A Graphenepolyurethane Composite Hydrogel as a Potential Bioink for 3D Bioprinting and Differentiation of Neural Stem Cells. J Mater Chem B. 2017;5:8854–64. doi: 10.1039/c7tb01594a. [DOI] [PubMed] [Google Scholar]
- 57.Li Z, Shen J, Ma H, et al. Preparation and Characterization of pH and Temperature-Responsive Hydrogels with Surface-functionalized Graphene Oxide as the Crosslinker. Soft Matter. 2012;8:3139. [Google Scholar]
- 58.Sayyar S, Murray E, Thompson BC, et al. Processable conducting graphene/chitosan hydrogels for tissue engineering. J Mater Chem B. 2015;3:481–90. doi: 10.1039/c4tb01636j. [DOI] [PubMed] [Google Scholar]
- 59.Liu S, Mou S, Zhou C, et al. Off-the-Shelf Biomimetic Graphene OxideCollagen Hybrid Scaffolds Wrapped with Osteoinductive Extracellular Matrix for the Repair of Cranial Defects in Rats. ACS Appl Mater Interfaces. 2018;10:42948–58. doi: 10.1021/acsami.8b11071. [DOI] [PubMed] [Google Scholar]
- 60.Rahman MM, Byanju B, Grewell D, et al. High-power Sonication of Soy Proteins:Hydroxyl Radicals and their Effects on Protein Structure. Ultrason Sonochem. 2020;64:105019. doi: 10.1016/j.ultsonch.2020.105019. [DOI] [PubMed] [Google Scholar]
- 61.Fratzl P. Collagen. Springer; United States: 2008. Collagen:Structure and mechanics an introduction; pp. 1–13. [Google Scholar]
- 62.Mane NC, Ponrathnam S. Effect of Chemical Crosslinking on Properties of Polymer Microbeads:A Review. Can Chem Trans. 2016;3:473–85. [Google Scholar]
- 63.Huang R, Choe E, Min DB. Kinetics for Singlet Oxygen Formation by Riboflavin Photosensitization and the Reaction between Riboflavin and Singlet Oxygen. J Food Sci. 2006;69:C726–32. [Google Scholar]
- 64.Jiang T, Munguia-Lopez JG, Flores-Torres S, et al. Extrusion Bioprinting of Soft Materials:An Emerging Technique for Biological Model Fabrication. Appl Phys Rev. 2019;6:011310. [Google Scholar]
- 65.Wilson SA, Cross LM, Peak CW, et al. Shear-Thinning and Thermo-Reversible Nanoengineered Inks for 3D Bioprinting. ACS Appl Mater Interfaces. 2017;9:43449–58. doi: 10.1021/acsami.7b13602. [DOI] [PubMed] [Google Scholar]
- 66.Liu F, Gao Y, Li H, et al. Interaction of Propidium Iodide with Graphene Oxide and its Application for Live Cell Staining. Carbon. 2014;71:190–5. [Google Scholar]
- 67.Mironi-Harpaz I, Wang DY, Venkatraman S, et al. Photopolymerization of Cell-encapsulating Hydrogels:Crosslinking Efficiency Versus Cytotoxicity. Acta Biomater. 2012;8:1838–48. doi: 10.1016/j.actbio.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 68.Lock JG, Wehrle-Haller B, Strömblad S. Cell-Matrix Adhesion Complexes:Master Control Machinery of Cell Migration. Semin Cancer Biol. 2008;18:65–76. doi: 10.1016/j.semcancer.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 69.Kumar S, Parekh SH. Linking Graphene-based Material Physicochemical Properties with Molecular Adsorption Structure and Cell Fate. Commun Chem. 2020;3:8. doi: 10.1038/s42004-019-0254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh RK, Kumar R, Singh DP. Graphene Oxide:Strategies for Synthesis Reduction and Frontier Applications. RSC Adv. 2016;6:64993–5011. [Google Scholar]
- 71.Choi KM, Seo YK, Yoon HH, et al. Effect of Ascorbic Acid on Bone Marrow-derived Mesenchymal Stem Cell Proliferation and Differentiation. J Biosci Bioeng. 2008;105:586–94. doi: 10.1263/jbb.105.586. [DOI] [PubMed] [Google Scholar]
- 72.Khalili D. Graphene Oxide:A Promising Carbocatalyst for the Regioselective Thiocyanation of Aromatic Amines Phenols, Anisols and Enolizable Ketones by Hydrogen Peroxide/KSCN in Water. New J Chem. 2016;40:2547–53. [Google Scholar]
- 73.Wu JB, Lin ML, Cong X, et al. Raman Spectroscopy of Graphene-Based Materials and its Applications in Related Devices. Chem Soc Rev. 2018;47:1822–73. doi: 10.1039/c6cs00915h. [DOI] [PubMed] [Google Scholar]
- 74.Muzyka R, Drewniak S, Pustelny T, et al. Characterization of Graphite Oxide and Reduced Graphene Oxide Obtained from Different Graphite Precursors and Oxidized by Different Methods Using Raman Spectroscopy. Materials. 2018;11:1050. doi: 10.3390/ma11071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marsden AJ, Papageorgiou DG, Vallés C, et al. Electrical Percolation in Graphene-polymer Composites. 2D Materials. 2018;5:032003. [Google Scholar]
- 76.Rao S, Upadhyay J, Polychronopoulou K, et al. Reduced Graphene Oxide:Effect of Reduction on Electrical Conductivity. J Compos Sci. 2018;2:25. [Google Scholar]
- 77.Mohan VB, Brown R, Jayaraman K, et al. Characterisation of Reduced Graphene Oxide:Effects of Reduction Variables on Electrical Conductivity. Mater Sci Eng B. 2015;193:49–60. [Google Scholar]
- 78.Kim M, Lee C, Jang J. Fabrication of Highly Flexible Scalable, and High-Performance Supercapacitors Using Polyaniline/Reduced Graphene Oxide Film with Enhanced Electrical Conductivity and Crystallinity. Adv Funct Mater. 2013;24:2489–99. [Google Scholar]
- 79.Lang Q, Wu Y, Ren Y, et al. AC Electrothermal Circulatory Pumping Chip for Cell Culture. ACS Appl Mater Interfaces. 2015;7:26792–801. doi: 10.1021/acsami.5b08863. [DOI] [PubMed] [Google Scholar]
- 80.Adams TN, Turner PA, Janorkar AV, et al. Characterizing the Dielectric Properties of Human Mesenchymal Stem Cells and the Effects of Charged Elastin-Like Polypeptide Copolymer Treatment. Biomicrofluidics. 2014;8:054109. doi: 10.1063/1.4895756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cho S, Thielecke H. Electrical Characterization of Human Mesenchymal Stem Cell Growth on Microelectrode. Microelectron Eng. 2008;85:1272–4. [Google Scholar]
- 82.Walker BW, Lara RP, Mogadam E, et al. Rational Design of Microfabricated Electroconductive Hydrogels for Biomedical Applications. Prog Polym Sci. 2019;92:135–57. doi: 10.1016/j.progpolymsci.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barlian A, Judawisastra H, Alfarafisa NM, et al. Chondrogenic Differentiation of Adipose-derived Mesenchymal Stem Cells Induced by L-Ascorbic Acid and Platelet Rich Plasma on Silk Fibroin Scaffold. PeerJ. 2018;6:e5809. doi: 10.7717/peerj.5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kumar A, Nune KC, Misra RD. Understanding the Response of Pulsed Electric Field on Osteoblast Functions in Three-dimensional Mesh Structures. J Biomater Appl. 2016;31:594–605. doi: 10.1177/0885328216658376. [DOI] [PubMed] [Google Scholar]
- 85.Ross CL. The Use of Electric Magnetic, and Electromagnetic Field for Directed Cell Migration and Adhesion in Regenerative Medicine. Biotechnol Prog. 2016;33:5–16. doi: 10.1002/btpr.2371. [DOI] [PubMed] [Google Scholar]