

Summary
Many embryonic organs undergo epithelial morphogenesis to form tree-like hierarchical structures. However, it remains unclear what drives the budding and branching of stratified epithelia, such as in embryonic salivary gland and pancreas. Here, we performed live-organ imaging of mouse embryonic salivary glands at single-cell resolution to reveal that budding morphogenesis is driven by expansion and folding of a distinct epithelial surface cell sheet characterized by strong cell-matrix adhesions and weak cell-cell adhesions. Profiling of single-cell transcriptomes of this epithelium revealed spatial patterns of transcription underlying these cell adhesion differences. We then synthetically reconstituted budding morphogenesis by experimentally suppressing E-cadherin expression and inducing basement membrane formation in 3D spheroid cultures of engineered cells, which required β1 integrin-mediated cell-matrix adhesion for successful budding. Thus, stratified epithelial budding, the key first step of branching morphogenesis, is driven by an overall combination of strong cell-matrix adhesion and weak cell-cell adhesion by peripheral epithelial cells.
In Brief
Using live organ imaging and transcriptomics, epithelial morphogenesis into tree-like hierarchical structures was profiled at single-cell resolution and the early steps were synthetically reconstituted in 3D spheroid cultures of engineered cells.
Graphical Abstract
Introduction
Branching morphogenesis is widely used by epithelial organs to maximize their functional surface area (Wang et al., 2017). All branching organs have a core epithelium encased by a layer of basement membrane (BM) surrounded by mesenchymal cells. The mesenchyme secretes growth factors critical for epithelial growth and morphogenesis (Affolter et al., 2009; Costantini and Kopan, 2010; Patel et al., 2006; Shih et al., 2013; Wang et al., 2017). However, when suitable growth factors and extracellular matrix are provided, the epithelium of many organs can branch without the mesenchyme (Ewald et al., 2008; Nogawa and Ito, 1995; Nogawa and Takahashi, 1991), indicating the core capacity for branching is intrinsic to the epithelium.
Branching epithelia can be single-layered with a lumen or stratified without a lumen. Branching of a single-layered epithelium involves buckling of the epithelial sheet (Nelson, 2016). The buckling of single-layered lung epithelium can be guided by external sculpting forces from airway smooth muscle cells (Goodwin et al., 2019; Kim et al., 2015) among other cell types (Young et al., 2020). In stratified epithelia, however, the concept of buckling cannot be easily applied due to the apparent lack of a sheet-like structure.
Embryonic salivary gland and pancreas are classical examples of stratified epithelia that undergo branching morphogenesis involving budding and ductal morphogenesis (Shih et al., 2013; Steinberg et al., 2005; Wang et al., 2017). In budding morphogenesis, numerous epithelial buds arise by repeated clefting of a single initial epithelial bud, and ductal morphogenesis generates tubular structures connecting terminal end buds together. Budding morphogenesis is characterized by extensive dynamics of epithelial cells and the BM matrix (Harunaga et al., 2014; Larsen et al., 2006; Shih et al., 2016), but it remains unclear how buds arise.
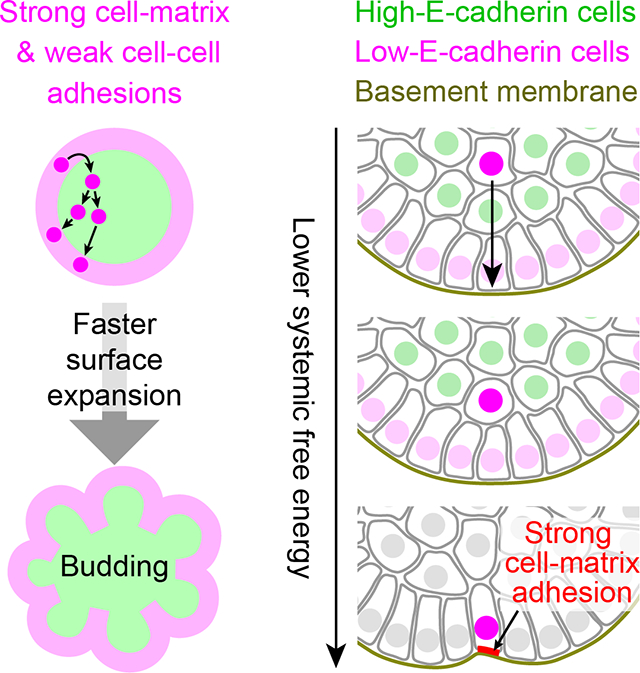
Here, we use volumetric live-organ imaging to follow individual cells within virtually the entire mouse embryonic salivary gland during branching morphogenesis. We find that surface-localized epithelial cells form an integral layer with the BM, which together expands and folds inward to drive budding morphogenesis. We use mathematical modeling and experimental perturbations to corroborate a model that a combination of weak cell-cell adhesion and strong cell-matrix adhesion of peripheral epithelial cells drives the expansion and folding of the surface epithelial sheet. Single-cell RNA sequencing and single-molecule RNA fluorescence in situ hybridization reveal distinct transcriptional features of these surface epithelial bud cells. Importantly, we demonstrate successful reconstitution of budding morphogenesis by experimentally reducing E-cadherin expression and inducing BM formation in 3D spheroid cultures of engineered epithelial cells that normally do not form buds. Our results reveal a fundamental self-organizing mechanism based on preferential cell-matrix adhesion vs. cell-cell adhesion that can explain how stratified epithelia undergo budding morphogenesis.
Results
Clefting in salivary glands is due to uniform expansion and inward folding of the surface cell sheet
To visualize cellular mechanisms of stratified epithelial branching, we developed live-organ imaging strategies using two-photon microscopy to image nearly the entire 3D volume of transgenic mouse embryonic salivary glands at high spatiotemporal resolution (Fig. S1A–C; Video S1). 3D cell tracking revealed extensive cell motility throughout the developing gland with cell migration rates increasing near the periphery of the branching epithelial buds as previously described (Hsu et al., 2013; Larsen et al., 2006) (Fig. S1D–E).
Next, we evaluated whether cells exchange freely between the outer epithelial layer and gland interior during morphogenesis, or whether branching salivary glands are composed of distinct interior and surface cell populations. We photoconverted patches of cells near the epithelial surface in transgenic salivary glands expressing KikGR, a photoconvertible fluorescent protein emitting green or red fluorescence before or after conversion (Hsu et al., 2013; Tsutsui et al., 2005). Most photoconverted peripheral epithelial cells moved rapidly along the tissue surface while maintaining intimate contact with the basement membrane (BM; Fig. 1A; Video S2), suggesting tight adherence of these cells to the BM. We then used an epithelial RFP reporter (Krt14p::RFP) with elevated expression in peripheral vs. interior epithelial cells (Fig. S1C) to enable automated rendering of the epithelial surface (Fig. 1B–C). We analyzed cell movements at the epithelial surface (located within 15 μm of the surface at any point within the tracked time window) to reveal that most cell movements remain confined to the surface (Fig. 1D–E; Video S3). During new bud formation by clefting, the peripherally enriched Krt14p::RFP reporter clearly delineated a distinct surface cell sheet, whose expansion and folding seemed to underlie clefting (Video S4).
Figure 1. Clefting in salivary glands is caused by uniform expansion and inward folding of the surface cell sheet.

(A) Left: schematic of KikGR photoconversion; Right: confocal images showing the middle slice of a branching epithelial bud in an E13 mouse salivary gland expressing KikGR. (B) Time-lapse two-photon microscopy images showing the maximum intensity projection of an E12.5 transgenic mouse salivary gland. (C) 3D rendering of epithelial surface using Krt14p::RFP at time points matching images in (B). (D) Surface-proximal epithelial cell tracks (tracking nuclear Histone-EGFP) color-coded by z-position at 20–22 h of the time-lapse sequence. Only tracks whose closest distance to the surface was ≤ 15 μm are shown. (E) Heatmap plot of the cell nucleus-to-surface distance vs. time for 500 randomly selected 3–10 hour-long surface-proximal tracks. (F) Time-lapse two-photon microscopy images showing the middle slice of an E13 mouse salivary gland. (G) Outlines of the epithelial surface at the middle two-photon image slice over 12.5 h at 5-min intervals. Blue to red, 0 to 12.5 h. (H) Plot of the bud perimeter and nuclear count along the surface cell layer at the middle slice over time. Dashed lines indicate fitted linear models. (I and J) Heatmaps of GFP intensity (I) and the curvature (J) along the surface epithelial layer at the middle slice over time. Arrowheads in (F, G, J) indicate clefts. Scale bars, 100 μm. See also Figure S1 and Videos S1–4.
We next determined whether new surface cells are added uniformly around the epithelial surface or locally at the cleft to distinguish between clefting as a systemic or local process. We traced nuclear histone-EGFP intensities of peripheral epithelial cells over time and computed local peripheral curvature to track surface deformation (Figs. 1F–J, S1F). Local expansion to form a cleft would predict an abrupt change in slope angles of temporal nuclear traces at cleft sites (Fig. S1G). Instead, the observed changes of slope angles were gradual, and surface expansion rates near clefts were indistinguishable from other locations, suggesting that clefting is a systemic activity (Figs. 1I–J, S1J–L). Moreover, increasing peripheral nuclear counts over time closely matched expansion of the bud perimeter, indicating constant peripheral cell density (Figs. 1H, S1H–I, M).
Taken together, we conclude that clefting in salivary glands is caused by uniform expansion and inward folding of the surface epithelial cell sheet.
Expansion of the surface cell sheet is driven by subsurface cell division and reinsertion as new surface cells
We next determined the origin of new epithelial surface cells. The distinct boundary of Krt14p::RFP expression levels between peripheral and interior epithelial cells hinted that new surface cells arise primarily from proliferation of preexisting surface cells (Fig. S1C, Video S4). However, no surface cells divided locally to directly produce two daughter cells remaining in the surface layer (n=289 surface-derived cell divisions; Fig. 2A). Instead, 92.4% of division-ready surface cells moved to a subsurface level to divide into two daughter cells in the gland interior (Type I; Figs. 2A, S2A), and the remaining 7.6% divided into one surface daughter cell and one interior daughter cell (Type II; Figs. 2A, S2B). Importantly, all surface-derived interior daughter cells eventually returned to the surface by reinserting between surface cells for delayed surface expansion (Figs. 2B, S2C–D; Video S5). Most cells returned within 4 hours, but a few took a little over 12 hours (Fig. 2B). Rapid-returning cells generally moved in proximity to the surface, while slow-returning cells stochastically wandered deep into the bud interior and were delayed or temporarily trapped before returning (Fig. S2E). Cell reinsertion sites were uniformly distributed around the surface (red dots in Fig. S2D), revealing the cellular basis of uniform surface expansion.
Figure 2. Expansion of the surface cell sheet is driven by subsurface cell division and reinsertion as new surface cells.

(A) Top and lower left: schematics and time-lapse two-photon images of 3 types of surface-derived cell division; Lower right: pie chart showing proportions of observed types. (B) Schematic and cumulative distribution plot of time intervals from anaphase onset of mother cell division to returning of 84 daughter cells to the epithelial surface. (C) Confocal image showing the middle slice of an epithelial bud from an E13.5 mouse salivary gland. (D) Schematic and plot of the surface-to-center line-scan profile of E-cadherin intensity. (E) Left: time-lapse two-photon images showing the middle slice of a branching epithelial bud in an E13 transgenic mouse salivary gland. Right: confocal images showing the middle slice of the same epithelial bud at 23 h after fixing and immunostaining. (F) Top: schematic of how E-cadherin intensity was measured. Bottom: scatter plot of the E-cadherin intensity of an edge vs. the mean RFP intensity of its two adjacent cells. Black line is the linear regression with 95% confidence interval (gray shading). (G) Plot of E-cadherin intensity at indicated categories of cell-cell boundaries. Error bars, 95% confidence intervals. ***, Tukey test p<0.001. n.s., not significant. (H) Schematic model of clefting in a stratified epithelium. Brighter color indicates dividing and newly divided cells. Note that the two steps happen concurrently but are drawn separately for clarity. Scale bars, 20 μm. See also Figures S2–3 and Videos S4–5.
What drives the robust surface return of surface-derived cells? Based on the lower E-cadherin expression level of peripheral epithelial cells compared to interior epithelial cells (Walker et al., 2008) (Fig. 2C–D), we hypothesized that differential cell-cell adhesion directed sorting out of low-E-cadherin surface-derived cells from high-E-cadherin interior cells (Steinberg, 1963).
To determine whether surface-derived cells maintained low E-cadherin expression when temporarily interior-located after cell division, we fixed transgenic glands immediately after live imaging and immunostained for E-cadherin (Fig. 2E) to identify surface-originating interior-located cells by cell tracking to compare their E-cadherin expression with surface cells. There was a clear negative correlation between E-cadherin intensity at cell-cell junctions and the average Krt14p::RFP intensity of the two adjacent cells (Fig. 2F). We then identified interior-located daughter cells after surface cell division by cell tracking and quantified E-cadherin intensities at cell-cell junctions between these cells and their neighbors. Importantly, E-cadherin intensity at these junctions were indistinguishable from randomly sampled junctions between high-RFP cells (mostly at the surface) and their neighbors (Fig. 2G). We conclude that surface-derived cells maintain low E-cadherin expression after moving into the gland interior, which probably underlies their robust return to the surface (Fig. 2B).
Accelerated branching of salivary glands upon basement membrane recovery from enzymatic disruption
Live imaging and the E-cadherin expression pattern led us to propose a model of salivary gland clefting based on the interplay between the basement membrane (BM) matrix and two cell types with distinct cell adhesion properties (Fig. 2H). In this testable model, provisional surface cells are first generated by proliferation of surface cells and temporarily stored in the interior domain to build up “branching potential,” i.e., the relative abundance of interior-located low E-cadherin cells. These cells then return to the subsurface layer by cell sorting driven by differential cell-cell adhesion, reinsert between surface cells adhering weakly to each other, and use strong cell-matrix adhesions to remain adherent to the BM. The expanded extra surface then folds to cause clefting and new bud formation (Fig. 2H).
This model has an interesting prediction: if the number of stored interior low-E-cadherin cells could be increased, it might be possible to accumulate “branching potential” separate from actual branching. We tested this by treating salivary glands with collagenase to disrupt the major BM component collagen IV. High-concentration collagenase treatment caused existing epithelial buds to fuse together to revert branching (Fig. 3A) (Grobstein and Cohen, 1965; Rebustini et al., 2007). Importantly, we discovered greatly accelerated catch-up branching after collagenase washout (Fig. 3A–B), likely resulting from attaching of accumulated surface-originated low-E-cadherin cells to the restored BM. The accumulation of surface-originated cells in the bud interior can be directly visualized by Krt14p::RFP after 24 h collagenase treatment (Fig. 3C), when both collagen IV and laminin were greatly reduced at the BM (Fig. 3D). We conclude that BM disruption can uncouple surface expansion from the buildup of an interior pool of low-E-cadherin cells. BM restoration enables rapid surface expansion and branching due to BM anchorage of these interior low E-cadherin cells.
Figure 3. Accelerated branching of salivary glands upon basement membrane recovery from enzymatic disruption.

(A) Phase contrast images of E12 + 1.5-day cultured salivary glands (0 h) treated for 24 h with solvent control or 20 μg/mL collagenase (24 h), followed by another 24 h after washout (washout + 24 h). (B) Plot of bud number per gland (left) or log2 bud ratio (right) over time. n=11 per group. (C) Two-photon images showing a single slice along the x-y plane (upper images) or the reconstructed x-z plane (lower images) before and after 24 h treatment with 20 μg/mL collagenase. Treatment began at E12.5 + 2-day culture, when Krt14::RFP expression becomes more distinct between the surface and interior epithelium. Yellow dashed line marks the surface-interior boundary. Arrows point to interior-located high-RFP cells. (D) Confocal images of control or collagenase treated glands at 24 h. (E) Left: time-lapse two-photon images of newly observed surface-derived cell division types in collagenase-treated glands. Right: stacked bar plot of surface-derived cell division types in control (same dataset as Fig. 2A; n=289) vs. collagenase-treated glands (n=130). Arrows, dividing cells. (F) Left: bar plot showing time intervals from anaphase onset to the return of interior daughter cells in control (n=84; same dataset as Fig. 2B) and collagenase-treated (n=11) glands. Right: combined bar and swarm plot showing the on-surface duration ratio of surface cells in control (n=30 tracks; 1957 frames; weighted average 0.930) and collagenase (n=39 tracks, 2408 frames; weighted average 0.701). Red and blue dots indicate newly returned and randomly selected pre-existing surface cells, respectively. Dashed line at 0.5 marks the ratio indicating no preference for surface or interior. (G) Confocal images and cellpose-segmented mesenchymal cell masks in control or collagenase-treated E13 salivary glands. (H) Confocal images showing maximum intensity projection of control or collagenase-treated E13 salivary glands immunostained with the mitosis marker phosto-Histone-H3 (pHH3). (I) Violin plots of cell size or shape metrics in control or collagenase-treated E13 salivary glands (left) and schematics of shape metrics definitions (right). Control: n=1715 cells from 7 glands; collagenase: n=1950 cells from 6 glands. (J) Violin plot of mesenchymal cell track speed in control (n=5878 tracks from 3 glands) or collagenase-treated (n=6139 tracks from 3 glands) glands. Dashed lines (I-J) represent data quartiles. (K) Bar plots showing the percentages of pHH3-positive area in the mesenchyme (left) or epithelium (right) of control (n=6) and collagenase (n=7) treated glands. Error bars, 95% confidence intervals. Statistics, Tukey test (B) or t-test (others). n.s., not significant. Scale bar in (E), 20 μm; others, 100 μm. See also Figures S2–3.
We next asked how BM disruption affects surface-originating cell divisions and cell dynamics. Interestingly, we observed a small proportion of in-plane divisions (Type III, 3.9%) and two new types of cell divisions where one or two daughter cells were temporarily extruded outward from the surface layer (Fig. 3E), indicating BM normally serves as a mechanical barrier to bias the extrusion towards the interior. Surface-derived daughter cells took much longer to initially return to the surface (Fig. 3F, left), and importantly failed to remain on the surface (Fig. 3F, right), suggesting adhesion to the BM normally keeps surface cells in place. In contrast, collagenase treatment only slightly affected mesenchymal cell shape and motility without changing cell proliferation rates in either the mesenchyme or epithelium (Fig. 3G–K), suggesting the effect of collagenase treatment was mainly due to BM disruption.
Mathematical modeling of budding morphogenesis
We derived a mathematical model of budding morphogenesis (see STAR Methods) incorporating the characteristic cell divisions (Fig. 2A), robust return of surface-originating cells (Fig. 2B), high affinity between surface cells and BM (Figs. 1D–E, 3F) and mainly constant cell division rates over time (Fig. S2F). The initial state is a 2-compartment sphere with a surface layer enveloping an interior core, where some division-ready cells in the surface layer are about to enter the subsurface to divide and with twice as many returning cells in the subsurface layer (Fig. S2G). A key parameter dictating the occupancy ratio of low-E-cadherin cells in surface vs. subsurface layers is the free energy difference ΔE of a unit event when a subsurface low-E-cadherin cell inserts into the surface layer (Fig. S2H–J). From experimentally measurable values including division-ready cell abundance (ω) and the geometric ratio (β), we estimated the interior-to-surface expansion ratio (α) (Fig. S2K). We found all data mapped to the parameter space permissive for surface folding (Fig. S3L), supporting our model.
The model predicts that increasing ΔE (i.e., making surface insertion harder) will inhibit budding (Fig. S2J). We tested this by weakening cell-matrix binding with integrin blocking antibodies or increasing BM thickness using inhibitors of matrix metalloproteinases, which all inhibited budding as predicted (Fig. S3A–C). We then evaluated the effects of low-concentration collagenase treatment to presumably soften the BM and observed dose-dependent inhibitory effects on both budding and collagen IV abundance in the BM (Fig. S3D–E). BM softening would simultaneously ease its stretching and weaken cell-matrix binding (Discher et al., 2005), which would have opposite effects on budding (Fig. S2I–J). Our results identify reduction of cell-matrix binding strength as the dominant factor (Fig. S3D).
Single-cell transcriptome profiling reveals spatial transcriptional patterns of the branching salivary gland epithelium
To explore regulatory mechanisms underlying differential cell adhesion properties among epithelial cells, we profiled single-cell transcriptomes of the E13 salivary gland epithelium by single-cell RNA sequencing (scRNA-seq). The 6,943 single-cell transcriptomes formed 7 main clusters with distinct marker genes (Fig. 4A–C). The cluster identities were assigned based on expression profiles of known marker genes, including the bud marker Sox10, duct marker Sox2, basal epithelial (outer bud and duct) marker Krt14 and the luminal (or inner) duct marker Krt19 (Lombaert and Hoffman, 2010; Szymaniak et al., 2017). We validated the bud enrichment of Sox10 expression and the duct enrichment of Sox2 expression by single-molecule RNA fluorescence in situ hybridization (smFISH) (Fig. S4A–B) (Raj et al., 2008; Wang, 2019). We also identified Cldn10 as a marker with strong inner bud enrichment and found that Claudin 10 protein was indeed highly expressed in the inner bud (Figs. 4B–C, S4C). Although perhaps counter-intuitive, calculations based on gland dimensions confirmed that there should be significantly more outer bud cells than inner bud cells (Fig. S4D).
Figure 4. Single-cell transcriptome profiling reveals spatial transcriptional patterns of the branching salivary gland epithelium.

(A, D, E) Scatter plots of 6,943 single-cell transcriptomes from the E13 mouse salivary gland epithelium shown in UMAP embedding and color coded by clusters (A and D) or cell cycle phase (E). Each dot represents one cell. Arrows in (D) indicate local RNA velocity estimated from unspliced and spliced transcripts of nearby cells. (B) Dot plot of selected cluster marker genes. (C) Scatter plots of single-cell transcriptomes of E13 salivary gland epithelium in UMAP embedding and color coded by the expression level of indicated genes. (F) Volcano plot comparing the expression levels of integrin and cadherin genes in the Inner bud vs. Outer bud (combining Outer bud 1–4). p-adjust, t-test with Benjamini-Hochberg correction. (G) Confocal images of Cdh1 mRNAs detected by single-molecule mRNA FISH in an E13 salivary gland. Each white dot is one Cdh1 mRNA molecule. (H) Plot of the Cdh1 mRNA density in outer or inner epithelial bud of E13 salivary glands. Measurements from the same image were connected by a line. p-value, paired two-sided t-test. (I) Table of the Pearson’s correlation coefficients and p-values between indicated genes. Blue and red shadings indicate negative and positive correlations, respectively. Scale bars, 20 μm. See also Figure S4.
To evaluate the dynamics of single-cell transcriptomes, we calculated RNA velocity (Bergen et al., 2020; La Manno et al., 2018), which predicts the future state of individual cells based on their unspliced and spliced mRNAs (Fig. 4D). This analysis revealed two prominent patterns: a cycling vector field covering all 4 outer bud clusters and a directional flow from the bud to duct clusters. The cycling vector field across outer bud clusters suggested outer bud cells were cell-cycling progenitors (Bergen et al., 2020), which was confirmed by the cell cycle phases of these cells (Fig. 4E). In fact, many marker genes of outer bud clusters were related to cell division or cell cycle regulation (Fig. 4B). On the other hand, the directional bud-to-duct flow suggested some bud cells would differentiate into duct cells (Fig. 4D).
We next compared expression patterns of major cell adhesion genes. For cell-matrix adhesion, all prominently expressed integrin genes (Itgb1, Itga6 and Itga9) had either comparable or slightly lower mRNA expression levels in the inner bud compared to outer bud cells (Figs. 4F, S4E). However, both ITGB1 and ITGA9 proteins had notable peripheral enrichment (Fig. S4F–J), which might enhance cell sorting by differential cell-cell adhesion. For cell-cell adhesion, the E-cadherin gene Cdh1 showed clear enrichment in the inner bud compared to outer bud cells (Fig. 4F, S4E), consistent with E-cadherin protein expression (Figs. 2C–E). We confirmed this pattern of Cdh1 mRNA expression by smFISH (Fig. 4G–H). This pattern was specific, since expression of the P-cadherin gene Cdh3 is similar between inner and outer bud cells (Figs. 4F, S4E). Among transcription factors involved in epithelial–mesenchymal transition (Stemmler et al., 2019), only Snai2 was prominently expressed, and its pattern was negatively correlated to Cdh1 and positively correlated to Krt14 (Fig. 4C, I), suggesting a regulatory role for Snai2 in shaping the expression pattern of Cdh1, consistent with prior in vitro findings (Bolós et al., 2003).
Reconstitution of epithelial branching morphogenesis using primary salivary gland epithelial cells
We next evaluated whether stratified epithelial branching could be reconstituted using primary salivary gland epithelial cells. Our lab had previously demonstrated self-assembly of dissociated salivary gland epithelial cells and partial primitive branching of self-assembled epithelial aggregates after embedding dissociated cells in solidified high-concentration Matrigel (basement membrane matrix extract) (Kleinman et al., 1986) and cultured on a polycarbonate filter (Wei et al., 2007). The results might have been limited by epithelial cell attachment to the filter. We optimized culture conditions to recapitulate prominent branching morphogenesis of either isolated single epithelial buds or completely dissociated single epithelial cells with rates comparable to intact salivary gland culture (Fig. 5A–G). Reconstituted branching from single buds or dissociated cells formed both end bud and duct structures (Fig. 5H), and live imaging confirmed that peripheral cells in both cultures undergo out-of-layer divisions characteristic of intact glands (Fig. 5I–J). Thus, we conclude that key aspects of stratified epithelial branching morphogenesis can be reconstituted from primary epithelial cells without the mesenchyme. Consequently, partial reconstitution of budding or branching morphogenesis might be possible using non-embryonic engineered cells.
Figure 5. Reconstitution of epithelial branching morphogenesis using primary salivary gland epithelial cells.

(A, D) Schematics of single-bud (A) and single-cell (D) isolation from E13 salivary glands. (B, E) Phase contrast images at indicated time points from single-bud (B) or single-cell (E) cultures. Arrows point to ductal structures. (C, F) Plots of bud number of single-bud (C) or single-cell (F) cultures. (G) Plot of bud ratios at 48 h to 24 h of intact, single-bud and single-cell cultures. (H) Merged confocal (KRT19) and two-photon (DAPI) microscopy images of intact, single-bud and single-cell cultures. KRT19 is a duct marker. (I-J) Time-lapse two-photon images (I) and stacked bar plot (J) showing two types of surface-derived cell divisions observed in single-cell and single-bud salivary gland cultures. Arrows in (I), dividing cells. Data for intact culture in (J) is the same as Fig. 2A. Error bars, 95% confidence intervals. Scale bar in (I), 20 μm; others, 100 μm.
Reconstitution of epithelial budding morphogenesis by engineering cell adhesion
Our model suggests that the key initial process of stratified epithelial budding is driven by cells with weak cell-cell adhesions plus strong cell-matrix adhesions. Reducing cell-cell adhesion can enhance branching morphogenesis in mammary gland and embryonic pancreas (Nguyen-Ngoc et al., 2012; Shih et al., 2016), but whether it alone is sufficient to drive branching remained unknown. We thus attempted to reconstitute epithelial branching by engineering cell adhesion. We chose the human adult colorectal adenocarcinoma cell line DLD-1 as a starting point, because DLD-1 expresses abundant E-cadherin and forms near-spherical spheroids in 3D cultures (Riedl et al., 2017). To modulate cell adhesion molecules, we established a clonal DLD-1 cell line with transgenes enabling CRISPR/dCas9-based inducible transcriptional repression and activation (Gao et al., 2016) (Fig. S5A–B). To stably express sgRNAs and monitor their expression, we constructed lentiviral vectors co-expressing sgRNAs with bright nuclear fluorescent reporters (Fig. S5C).
To reduce cell-cell adhesion strength, we identified two Cdh1 sgRNAs that efficiently reduced E-cadherin expression after cells were treated with abscisic acid (ABA), a dimerizer used to recruit the KRAB transcriptional repression domain (Figs. 6A, S5D–F). Without ABA, sg1-Cdh1 had minimal effects, whereas sg2-Cdh1 reduced E-cadherin to ~20% of controls (Fig. S5D–F), likely due to direct transcriptional blockade (Qi et al., 2013). The reduction of E-cadherin could be titrated by ABA concentrations, approaching maximum reduction at 3 days for sg1-Cdh1 and 2 days for sg2-Cdh1 (Fig. S5G–H). Inhibiting E-cadherin in DLD-1 resulted in only a moderate reduction of total β-catenin (Fig. S5I) and did not reduce cell proliferation or survival as reported for breast cancer cells (Padmanaban et al., 2019). Consistent with this, β-catenin in the cytoplasm and nucleus remain unchanged despite a severe loss from cell junctions upon E-cadherin downregulation (Fig. S5J). Importantly, we observed sorting out of low-E-cadherin cells in spheroid cultures of mixed sg-Control and sg-Cdh1 cells, suggesting E-cadherin reduction successfully lowered cell-cell adhesion strength (Fig. S6A).
Figure 6. Reconstitution of epithelial budding morphogenesis using engineered cells.

(A) Western blot of clonal Dia-C6 cells expressing Control (lacZ) or Cdh1 sgRNA treated with abscisic acid (ABA) or DMSO (vehicle). ABA is a dimerizer used to induce robust transcriptional repression in engineered cells. (B) Schematic of 3D spheroid cultures. (C) Merged phase contrast and epifluorescence images of spheroids from indicated experimental groups. (D-E) Bar plots of bud number or percentage of high-curvature perimeter length (|curvature|>20 mm−1). Error bars, 95% confidence intervals. ***, Tukey test p < 0.001. (F) Heatmap showing color-coded curvature along spheroid perimeters. Each column is one spheroid. Sample numbers in (C to F): n=11, 10, 16, 43 for groups 1–4 combining 2 independent experiments with similar results; only 21 randomly selected Group 4 samples were plotted in (F) to save space. (G) Maximum intensity projection of two-photon microscopy images of spheroids immunostained with laminin, a basement membrane (BM) marker. (H) Confocal images of a spheroid at the central slice. (I) Time-lapse confocal images of a branching spheroid. Atto-647N-labeled fibronectin was used to mark the BM (yellow); arrows and arrowheads indicate clefts. Scale bars, 100 μm. See also Figures S5–6 and Videos S6–7.
DLD-1 spheroids failed to spontaneously form a basement membrane (BM), a structure critical for salivary gland branching (Fig. 3). To induce BM formation, we supplemented culture media with a non-solidified, low-concentration suspension of the BM extract Matrigel. Strikingly, this led to robust budding morphogenesis in spheroids containing sg1-Cdh1 or sg2-Cdh1 cells after ABA-induced E-cadherin reduction (Figs. 6B–F, S6B–F; Video S6). Importantly, a condensed layer of BM formed around the spheroids with high levels of the BM components laminin and collagen IV (Fig. 6G–H). In spheroids with both sg-Cdh1 and sg-Control cells, cells contacting the BM were primarily sg-Cdh1 cells lacking E-cadherin expression (Figs. 6H, S6G). Furthermore, live-spheroid imaging revealed preferential outward expansion of contact surfaces between sg-Cdh1 cells (magenta) and the BM (yellow), whereas contact surfaces between sg-Control cells (green) and the BM were mainly found at the cleft bottom (Fig. 6H–I; Video S7). Bud formation could also occur in spheroids containing only low-E-cadherin cells, but these spheroids were often flatter, and their buds were less striking (Fig. S6H–I), suggesting high-E-cadherin cells may play a structural role by forming a more robust spheroid core. To summarize the process of reconstituted budding morphogenesis, cells with experimentally reduced E-cadherin expression sorted out to the surface by differential cell-cell adhesion and then interacted with the BM to promote budding as strong cell-matrix adhesions replaced weak cell-cell adhesions (Fig. S6J).
Reconstituted epithelial budding depends on integrin-mediated cell-matrix adhesion
Our model predicts that strong cell-matrix interactions are required for reconstituted budding morphogenesis. Inhibiting cell-matrix interactions by a function-blocking β1-integrin antibody inhibited bud formation (Fig. S7A–B). In addition, enzymatic disruption of the basement membrane (BM) reverted budding, which recovered upon BM reformation (Fig. S7C–D). We then identified an Itgb1 sgRNA to reduce β1-integrin expression in subsets of cells (Fig. S7E–G). Bud formation was completely blocked when β1-integrin was reduced in low-E-cadherin cells or in all cells, but not when only in high-E-cadherin cells (Figs. 7A–D, S7H), demonstrating that β1-integrin-dependent cell-matrix interactions were specifically required in low-E-cadherin cells for budding. Importantly, reducing E-cadherin or β1-integrin expression selectively inhibited cell attachment to E-cadherin extracellular domain or Matrigel-coated surfaces, respectively (Fig. S7I–L).
Figure 7. Reconstituted epithelial budding depends on integrin-mediated cell-matrix adhesion.

(A, E, H, K) Phase contrast images of spheroids from indicated experimental groups. 50 μM MnCl2 was used to enhance integrin-mediated cell-matrix adhesion (E). ABA was added in all cultures except in (E) to induce E-cadherin repression. Matrigel (A, E, K) or indicated ratios of Matrigel to laminin (H) were supplemented. (B, F, I, L) Bar plots of bud number per spheroid. (C, G, J, M) Bar plots of percent protruded bud area per spheroid. (D) Heatmap showing color-coded curvature along spheroid perimeters. Each column is one spheroid. (N) Swarm plot of apparent Young’s modulus measured by atomic force microscopy. Sample numbers: n=9, 15, 20, 10, 10, 10, 20 for groups a-g in (A-G) from one of two independent experiments with similar results. n=10 for all groups in (H-M). For groups in (N), n= 14, 15, 18, 20, 11 (left-to-right). Error bars, 95% confidence intervals. *, **, ***, Tukey test p < 0.05, 0.01 or 0.001. n.s., not significant. Statistics in (N) were performed on log-transformed data to meet the homogeneity of variance requirement of Tukey test. Scale bars, 100 μm. See also Figure S7.
We next tested whether enhancing cell-matrix adhesion strength could enhance budding. We capitalized on the low-level bud formation observed in mixed sg-Control/sg2-Cdh1 spheroid cultures without ABA, providing a sensitized assay (Fig. S6C–F). Using MnCl2 to enhance integrin-mediated cell-matrix adhesion strength (Bazzoni et al., 1995) produced a modest but definitive increase in bud formation (Fig. 7E–G). Thus, enhancing cell-matrix adhesion strength could enhance budding.
Finally, we tested whether changing the matrix composition or thickness of BM would affect the extent of budding. Matrigel contains ~60% laminin and ~30% collagen IV (Kleinman et al., 1986). When mixing Matrigel with an increasing ratio of laminin, we observed reduced budding morphogenesis (Fig. 7H–J), indicating the importance of optimal matrix composition. Budding was also inhibited when matrix metalloproteinases (MMPs) were inhibited (Fig. 7K–M), which significantly increased BM thickness (Fig. S7M). Spheroids with a BM were clearly stiffer than spheroids without matrix supplement, as measured by the apparent Young’s modulus using atomic force microscopy (Figs. 7N, S7N), supporting a putative mechanical role of the BM for the expanding surface cell layer. Interestingly, increasing BM thickness by MMP inhibition or changing its composition by laminin supplementation did not seem to alter the intrinsic stiffness of BM (Fig. 7N). The inhibitory effects on budding were probably due to reduced cell-matrix binding strength with altered composition and increased energy cost to stretch a thicker BM after MMP inhibition.
Discussion
Our work reveals the critical role of a specific combination of strong cell-matrix adhesions and weak cell-cell adhesions for driving budding morphogenesis of stratified epithelia, the key first step of branching morphogenesis. We discovered that budding morphogenesis of stratified salivary gland epithelium is driven by the comparatively faster expansion and inward folding of a cryptic surface epithelial sheet, which we could visualize by single-cell tracking. Mechanistically, the expansion of this epithelial sheet is driven by the subsurface cell division and back-insertion of surface-derived daughter cells that retain weak cell-cell adhesions and re-establish strong cell-matrix adhesions to the basement membrane (BM). Importantly, these two parameters were sufficient to successfully reconstitute budding morphogenesis of a stratified epithelium by experimentally reducing E-cadherin expression and inducing BM formation to provide cell-matrix adhesion.
Cell-matrix vs. cell-cell adhesion in epithelial morphogenesis
Cell-matrix and cell-cell adhesions play important roles in epithelial morphogenesis, e.g., in cell sorting by differential cell-cell adhesion (Steinberg, 1963) or alternative self-organization strategies (Cerchiari et al., 2015). In general, tissues minimize systemic interfacial energy by maximizing interfaces with stronger interactions. Following this principle, we find that strong cell-matrix adhesions combined with weak cell-cell adhesions are sufficient to drive clefting and bud formation of a stratified epithelium.
Weak cell-cell adhesions of peripheral epithelial cells (and their progeny) likely play two important roles in promoting expansion of the surface epithelial sheet. First, surface-derived epithelial cells that temporarily localize to the bud interior for cell division presumably rely on their inherited weak cell-cell adhesion to sort back out to the surface. Second, weak cell-cell adhesions between surface cells will allow returning cells to intercalate between them to engage with the BM.
Conversely, the strong cell-matrix adhesions of peripheral epithelial cells to the BM would both promote expansion of the surface epithelial sheet and maintain its integrity. When integrin-mediated cell-matrix adhesion is inhibited in salivary glands, surface cells frequently detach from the BM and migrate into the bud interior, which inhibits branching (Hsu et al., 2013) (Fig. S3A). After enzymatic BM disruption in branched salivary glands, pre-existing epithelial buds fuse (Grobstein and Cohen, 1965). In both normal branching and accelerated branching upon BM recovery, the surface cell sheet expands because cells with low E-cadherin prefer to engage with the BM rather than with other low E-cadherin cells (Fig. S2H). By increasing interfaces with stronger interactions, the epithelial cells and the BM comprise a system proceeding towards a state of lower overall interfacial energy – a prescribed end state.
Subsurface cell divisions in branching epithelia
Surface epithelial cells in the salivary gland epithelium mainly divide in the subsurface layer after delaminating from the surface (Fig. 2A). Similar out-of-layer cell divisions occur in the stratified embryonic pancreatic epithelium (Shih et al., 2016) and in single-layered embryonic lung and kidney epithelia (Packard et al., 2013; Schnatwinkel and Niswander, 2013).
We speculate division-ready cells delaminate due to mitotic cell rounding and cell crowding in the surface layer. Mitotic cells round up by outward osmotic pressure and inward contraction of the actomyosin cortex (Stewart et al., 2011) while losing adhesion to the matrix (Li and Burridge, 2019), resulting in extrusion. Because BM is stiffer than cells, cell extrusion is biased towards the interior mass of cells. In fact, we observed outward cell extrusion upon BM disruption (Fig. 3E).
A major advantage of out-of-layer divisions may be to facilitate re-distribution of rapidly dividing surface cells. If several adjacent surface cells were to divide locally to generate new surface daughter cells, they could build up considerable stress by over-stretching the associated BM. Instead, the out-of-layer division strategy is self-adjusting because extruded and dividing cells can insert between jostling soft cells compared to the stiff BM, and they can subsequently reinsert back into the surface layer at sites of easiest entry (lowest ΔE).
Branching morphogenesis of stratified vs. single-layered epithelia
With the insight that stratified epithelial budding can be conceptualized as folding of an expanding surface cell sheet, our findings reveal hidden similarities between the seemingly discrepant branching mechanisms used by single-layered and stratified epithelia. Both can now be seen as buckling of an epithelial sheet (Nelson, 2016), except that the surface cell sheet in a stratified epithelium is more cryptic until visualized by cell tracking.
Buckling of the two types of epithelial sheets is shaped by differing constraints imposed by surrounding tissues. Outside the BM of both is a mesenchyme consisting of cells and extracellular matrix. However, the epithelial sheet in a stratified epithelium is directly attached to an inner cell core, while a single-layered epithelium encloses a fluid-filled lumen. As a result, buckling of a single-layered epithelium is primarily constrained by the surrounding mesenchyme, as in mouse embryonic lung and intestine (Goodwin et al., 2019; Hughes et al., 2018; Kim et al., 2015). In contrast, buckling of the surface epithelial sheet in a stratified epithelium is constrained by both the surrounding mesenchyme and the interior epithelium. During branching of stratified terminal end buds of mammary gland epithelium, the bifurcation angle is constrained by localized matrix in the surrounding mesenchyme (Nerger et al., 2021). In the stratified salivary gland epithelium, however, the role of the interior epithelium appears more dominant than that of the surrounding mesenchyme. In our mesenchyme-free cultures of primary salivary gland epithelial cells, budding morphologies closely resembled intact salivary gland cultures with mesenchyme (Fig. 5H). In fact, as demonstrated by our model, preferential expansion of a surface layer attached to an inner cell core alone is sufficient to drive folding of the surface layer (Fig. S2G).
In conclusion, our study establishes the concept of the critical role of a specific combination of strong cell-matrix adhesion and weak overall cell-cell adhesion of peripheral epithelial cells for the expansion and buckling of a cryptic surface epithelial sheet, which in turn drives budding morphogenesis of a stratified epithelium. We anticipate that this unifying view of branching morphogenesis as buckling of an epithelial sheet will facilitate development of unifying physical models of branching morphogenesis that encompass single-layered and stratified epithelia. Importantly, understanding branching morphogenesis will pave the road to engineer functional branched organs from stem cells.
Limitations of the Study
Our mathematical model predicts the overall extent but not patterns of budding, which would require incorporating mechanical properties of cells and basement membrane. The proposed role of Snai2 in regulating Cdh1 expression is correlative and should be verified experimentally.
STAR METHODS
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kenneth M. Yamada (kenneth.yamada@nih.gov).
Materials Availability
Plasmids will be available from Addgene. Cell lines and mouse strains generated in this study will be made available upon request to the Lead Contact.
Data and Code Availability
Single-cell RNA sequencing data has been deposited in GEO (accession number GSE159780). All other source data and raw data that support the findings of this study are available in Figshare: https://doi.org/10.35092/yhjc.c.5404533. Customized scripts and usage instructions are available from Github: https://github.com/snownontrace/public-scripts-Wang2020-branching-morphogenesis.
Experimental Models and Subject Details
Mouse strains
All mouse experiments were performed under animal study protocols 14–745, 17–845, and 20–1040 approved by the NIDCR Animal Care and Use Committee (ACUC). Mouse housing, care and maintenance were provided by the NIDCR Veterinary Resources Core. All mouse embryos were used without sex identification (mixed sexes). All transgenic mice were in FVB/N background. The mT/mG;Histone-EGFP (Hadjantonakis and Papaioannou, 2004; Huebner et al., 2014; Muzumdar et al., 2007) mouse was a gift from A.J. Ewald (Johns Hopkins University). The Krt14p::RFP (Zhang et al., 2011) mouse was a gift from M.P. Hoffman (NIDCR, NIH) and was originally from E. Fuchs (Rockefeller University). Krt14p::RFP mice were crossed with mT/mG;Histone-EGFP mice to generate Krt14p::RFP;Histone-EGFP mice. Our KikGR mouse was generated as described (Hsu et al., 2013). Transgenic mice 8–16 weeks old were bred to obtain 12- or 13-day old embryos. For timing of embryonic stage, the day after a vaginal plug was found was considered to be embryonic day 1. For experiments using wildtype embryos, timed pregnant ICR (CD-1) outbred mice were obtained from Envigo.
Cell lines
The HEK293T cell line used for lentivirus packaging was obtained from Takara (632273). The DLD-1 cell line was obtained from ATCC (CCL-221). DLD-1 cells were co-transfected with a PiggyBac transposase vector (System Biosciences, PB210PA-1) and a PiggyBac-ABA-CRISPRi vector (pSLQ2818; Addgene, 84241) using Lipofectamine 3000 (Thermo Fisher Scientific, L3000015), selected by 5 μg/mL puromycin (MilliporeSigma, P8833) and sorted for the presence of tagBFP to generate “Di” cells. Di cells were co-transfected with the above transposase vector, PiggyBac-ABA-CRISPRi vector and a modified PiggyBac-GA-CRISPRa vector (pW210, see Plasmids) using Lipofectamine 3000, selected by 5 μg/mL puromycin and 250 μg/mL hygromycin (MilliporeSigma, H3274) and sorted for brighter tagBFP than Di cells to generate “Dia” cells. Single cell clones of Dia cells were isolated by limiting dilution, and selected clones were functionally validated. The clonal Dia-C6 cells were used for lenti-sgRNA transduction followed by 20 μg/mL blasticidin (InvivoGen, ant-bl-1) selection and fluorescence cell sorting for mNeonGreen or mScarlet (see Plasmids) to obtain final DLD-1 derived cell lines used for spheroid culture experiments.
Method Details
Plasmids
The modified PiggyBac-GA-CRISPRa vector (pW210) was generated by replacing the Zeocin resistance cassette of pSLQ2842 (Addgene, 84244) with a synthesized Hygromycin resistance cassette (IDT) by Gibson Assembly (Gibson et al., 2009). Lentiviral vectors for co-expressing sgRNAs and fluorescent nuclear reporters (pLenti-spsg-mNG/pW211, pLenti-spsg-mSL/pW212) were made by replacing the Cas9 expression cassette of lentiCRISPR v2 (Addgene, 52961) with an NLS-mNeonGreen-P2A-BlastR or NLS-mScarlet-I-BlastR cassette using Gibson Assembly. For lenti-sgRNA cloning, a pair of complementary oligos containing the desired sgRNA sequence (see sgRNA design) plus a 4-bp 5’-extension (“cacc” for the forward oligo and “aaac” for the reverse complementary oligo) was annealed to form an oligo duplex, which was ligated into Esp3I (NEB, R0734S) digested vectors by a 1:2 mixture of T4 ligase (NEB, M0202L) and T4 polynucleotide kinase (NEB, M0236L) in T4 ligase buffer. The ligation mix was transformed using NEB stable competent cells (NEB, C3040) for single colony isolation. The Miraprep (Pronobis et al., 2016) protocol was used to increase the yield of miniprep DNA, which was directly used for lentivirus packaging. Correct insertion of sgRNA sequence was confirmed by Sanger sequencing using primer 5’-gagggcctatttcccatgat-3’. Sanger sequencing was mostly performed by the NIDCR Combined Technical Research Core and occasionally by a local company (ACGT Inc., Germantown, MD).
Salivary gland isolation and culture
Mouse submandibular salivary glands were isolated at embryonic day 12 or 13 (E12 or E13) as previously described (Sequeira et al., 2013). Briefly, a scalpel (Fine Science Tools, 10011–00 and 10003–12) was used to decapitate the mouse embryo. While the detached head was held on its side with one prong of forceps (Fine Science Tools, 11251–20) pierced through the top, a scalpel was used to slice across the mouth opening to isolate the mandible and tongue, between which the submandibular glands were sandwiched. Under a dissecting microscope, the detached mandible tissue was placed on a glass plate with the tongue facing down. A pair of forceps was used to slice through the midline of the mandible tissue to expose the tongue and the two submandibular glands attached to the base of the tongue. After surrounding tissues were removed, glands were detached using forceps and collected into a 35-mm dish with 3 mL DMEM/F-12 (Thermo Fisher, 11039047) medium until all embryos were dissected. Isolated salivary glands were cultured on 13 mm diameter 0.1 μm pore polycarbonate filters (MilliporeSigma, WHA110405) floating on 200 μL Organ Culture Medium in the glass bottom area of a 50 mm MatTek dish (MatTek, P50G-1.5–14-F) at 37°C with 5% CO2. Organ Culture Medium was DMEM/F-12 supplemented with 150 μg/mL vitamin C (MilliporeSigma, A7506), 50 μg/mL transferrin (MilliporeSigma, T8158) and 1× PenStrep (100 units/mL penicillin, 100 μg/mL streptomycin; Thermo Fisher, 15140163).
Salivary gland collagenase treatment and washout
Paired salivary glands from the same embryo were separated into control and collagenase treatment groups. Purified collagenase (Elastin Products Company, CL103) was resuspended in water (Quality Biological, 351–029-131) for a 2 mg/mL stock (aliquoted and stored at −20°C). For collagenase washout, the polycarbonate filter with attached glands was transferred onto 2 mL fresh DMEM/F-12 in a 35 mm dish (Corning, 430165) and incubated for 15 min at 37°C for one wash. After 3× 15-min washes, the filter with glands was transferred onto 200 μL fresh Organ Culture Medium in a new 50 mm MatTek dish (see Salivary gland isolation and culture).
Isolation of salivary gland epithelial rudiments
Up to 6 intact salivary glands (see Salivary gland isolation and culture) were treated with 150 μL 2 units/mL dispase (Thermo Fisher, 17105041; diluted in DMEM/F-12) in a well of a Pyrex spot plate (Fisher Scientific 13–748B) for 15 min at 37°C. The glands were washed twice with 5% BSA (MilliporeSigma, A8577; diluted in DMEM/F-12) in the same well to quench the dispase activity. The mesenchyme of each gland was removed using a pair of forceps (Fine Science Tools, 11254–20) and a tungsten needle (Fine Science Tools, 10130–05 and 26016–12) under a dissecting microscope. The forceps were mostly used to hold the gland still, whereas the needle was gently inserted between the mesenchyme and epithelium to separate them. Isolated epithelial rudiments were transferred to a new well of the spot plate with 150 μL DMEM/F-12 medium using low-retention pipette tips (cut for larger opening; Rainin, 30389190). When needed, a pair of forceps was used to cut off single epithelial buds from the isolated epithelial rudiments.
Single-cell dissociation of salivary gland epithelium
Single-cell dissociation of the salivary gland epithelium was performed as previously described with modifications (Sekiguchi and Hauser, 2019). We thank R. Sekiguchi for sharing tissue dissociation protocols before publication. Depending on experiments, 8–12 isolated epithelial rudiments from E13 submandibular salivary glands (see Isolation of salivary gland epithelial rudiments) were rinsed in 1 mL HBSS (Thermo Fisher, 14170161) in a 2 mL protein LoBind tube (Eppendorf, 022431102). After the rudiments were pelleted by centrifugation at 100× g for 30 seconds, liquid was removed as much as possible using a 1 mL pipette followed by a 200 μL pipette under a dissecting microscope to avoid accidentally discarding the samples. 100 μL Accutase (Innovative Cell Technologies, AT104) was added to the tube, which was immediately incubated for 2 minutes in a 37°C water bath to disrupt cell-cell adhesion. While being monitored under a dissecting microscope, the epithelial rudiments were triturated using a 200 μL pipettor set at 50 μL using a low-retention tip (Rainin, 30389187) for 2 min, when most cells were clearly dissociated. 900 μL 10% fetal bovine serum (FBS; GE Healthcare/Cytiva, SH30070.03) diluted in PBS was added to quench the Accutase. The 1 mL cell suspension was passed through a 40 μm Flowmi (VWR, H13680–0040) cell strainer into a polyethylene terephthalate (PET) 15 mL tube (Corning, 430053). It is critical to use PET tubes instead of polypropylene tubes for efficient cell recovery. Cells were pelleted by centrifugation at 100× g for 2 minutes in a swinging-bucket rotor. After the supernatant was carefully removed, cells were resuspended in 1% FBS in PBS and pelleted again. About 50 μL liquid was retained to resuspend the cells, which was used for single cell capture (for scRNA-seq) or diluted in DMEM/F-12 medium for 3D culture.
Single-cell RNA sequencing (scRNA-seq) and data analysis
Single cell capture and library construction were performed using the 10x Genomics Chromium Single Cell 3’ Library & Gel Bead Kit (v2 Chemistry) following the manufacturer’s instructions. The libraries were sequenced on an Illumina NextSeq500 sequencer. The 10x Genomics Cell Ranger (v3.0.1) software suite was used for demultiplexing, read alignment and UMI (unique molecule identifier) counting. The above steps were performed by the NIDCR Genomics and Computational Biology Core.
To calculate RNA velocity, the spliced and unspliced RNAs for each gene and each cell were counted using the command line tool velocyto.py following instructions on its tutorial website (“velocyto run10x”; see the next paragraph for the link) (La Manno et al., 2018). The output loom file from the velocyto run contains the spliced and unspliced counts, which was merged into the AnnData object from the Scanpy (v1.6.0) (Wolf et al., 2018) analysis pipeline using the filtered features, barcodes and matrix output files from Cell Ranger. Genes with less than 20 total counts (spliced and unspliced) were filtered out, and the first and second order moments (i.e., mean and uncentered variance) of spliced and unspliced counts were computed. The moments were used to compute the RNA velocity vector (with the length of gene number after filtering) of each cell using the stochastic model of scVelo (v0.2.2) (Bergen et al., 2020). Finally, the velocity vector was used to compute a velocity graph that summarizes the transition probabilities between all cells. This velocity graph was used to project the velocities into a low-dimensional UMAP embedding.
Scripts for cell clustering, cell cycle phase assignment and RNA velocity calculation are available as a Jupyter Notebook on Github (see Data and Code Availability).
The velocyto.py tutorial website is: http://velocyto.org/velocyto.py/tutorial/cli.html#run10x-run-on-10x-chromium-samples
3D culture of primary salivary gland epithelial buds or dissociated cells
Ultra-low attachment 96-well V-bottom (S-bio, MS-9096VZ) plates were used for 3D culture of salivary gland epithelial buds (see Isolation of salivary gland epithelial rudiments) or dissociated cells (see Single-cell dissociation of salivary gland epithelium). One epithelial bud or ~3,000 dissociated epithelial cells were seeded per well in a 96-well plate with 50 μL DMEM/F-12 medium. Immediately after seeding, the plate was centrifuged at 100×g for 3 min to sediment the epithelial bud or dissociated cells. 50 μL 2× culture mix containing 400 ng/mL FGF7 (R&D Systems, 5028-KG-025), 2× ITS supplement (Thermo Fisher, 41400045) and 1 mg/mL growth factor-reduced Matrigel (Corning, 356231; stock 9–10 mg/mL) were added to each well. This culture media was modified from (Nakao et al., 2017). 20 ng/mL NRG1 (R&D Systems, 9875-NR-050) was also included in the 2× culture mix in earlier experiments, but it was omitted later as follow-up studies showed removing it did not affect epithelial growth or branching. Surrounding wells were filled with 100 μL HBSS to reduce culture media evaporation. The plate was cultured at 37°C with 5% CO2.
Single-molecule RNA fluorescence in situ hybridization (smFISH)
smFISH probe sets targeting Cdh1, Sox2 and Sox10 mRNAs were designed using the Stellaris probe designer and synthesized with either TAMRA-C9 or Quasar 670 dyes by LGC Biosearch Technologies. smFISH of wholemount E13 salivary glands was performed as previously described (Wang, 2019). Briefly, E13 salivary glands were fixed with 4% PFA in PBS at room temperature (RT) for 1 hour or overnight at 4°C, rinsed in PBSTx (PBS + 0.2% Triton-X-100), dehydrated sequentially in 30%, 50%, 70% and 100% methanol on ice, rehydrated sequentially in 70%, 50%, 30% methanol on ice, rinsed in PBSTx for 10 min at RT, permeabilized in 0.5% SDS in PBS at RT, equilibrated in smFISH Wash Solution (2× SSC and 10% formamide in DEPC-treated water) for 10 min at RT, hybridized in smFISH Hybridization Solution (2× SSC, 10% formamide, 10% dextran sulfate and 50 μg/mL yeast tRNAs in DEPC-treated water) containing 50 nM probes (1–2 nM each probe) at 37°C for 12 to 16 hours, washed in smFISH Wash Solution for 30 min at RT, stained with 0.5 μg/mL DAPI in smFISH Wash Solution for 2 hours at RT, washed 2 more times for 30 min at RT, rinsed in 2× SSC (30 mM sodium citrate and 300 mM sodium chloride; K D Medical, RGF-3240) and mounted in ProLong Diamond Anti-fade Mountant (Thermo Fisher, P36961) for imaging.
smFISH quantification
smFISH dot counting was performed using a suite of custom-written ImageJ macros as previously described (Wang, 2019). Briefly, smFISH images were smoothened by a Gaussian filter, contrast enhanced by a morphological top-hat filter (Legland et al., 2016), and local maxima points beyond a user-specified threshold level were identified and counted. An identical set of parameters was used to process all images from the same experiment.
2D cell culture
DMEM (Thermo Fisher, 11965118) and RPMI-1640 (ATCC, 30–2001) media were supplemented with 10% fetal bovine serum (FBS; GE Healthcare/Cytiva, SH30070.03), 2 mM L-glutamine (Thermo Fisher, 25030081) and 1× PenStrep to make DMEM Complete and RPMI-1640 Complete media. Phenol red-free DMEM (GE Healthcare/Cytiva, SH30284.01) or RPMI-1640 (Thermo Fisher, 11835030) were used when cells were used for imaging or cell sorting. HEK293T cells were cultured in DMEM Complete medium in 37°C incubators with 10% CO2. DLD-1 and DLD-1 derived cells were cultured in RPMI-1640 Complete medium in 37°C incubators with 5% CO2. For passage, cells were detached using trypsin-EDTA (Thermo Fisher, 25300120) after rinsing with HBSS (Thermo Fisher, 14170161). Cell density was determined using an automated cell counter (Nexcelom Cellometer Auto 2000).
3D spheroid culture
Ultra-low attachment 96-well U-bottom (Corning, 7007) or V-bottom (S-bio, MS-9096VZ) plates were used for 3D spheroid culture. DLD-1 cells expressing different sgRNAs were detached, pelleted by centrifugation at 1,000×g for 3 min, resuspended in RPMI-1640 Complete medium, counted and diluted to 60,000 cells/mL. For co-cultures of two cell types (e.g., sg-Control and sg-Cdh1 cells), appropriate volumes of the two cells were mixed in a separate tube to achieve desired mixing ratios. A multichannel pipette (Rainin, 17013810) was used to seed 50 μL cell suspensions in each well for 3,000 cells per spheroid. The 36 outer edge wells were filled with 100 μL HBSS to reduce medium evaporation over long culture periods (≥ 7 days). Immediately after seeding, the plate was centrifuged at 100×g for 3 min to sediment the cells. The next day, a 2× treatment mix of 44.8 μL RPMI-1640 Complete medium, 5 μL growth factor-reduced Matrigel (Corning, 356231; 9–10 mg/mL) and 0.2 μL DMSO (MilliporeSigma, D2650) or 50 mM abscisic acid (ABA; MilliporeSigma, A1049) was prepared for each well. A multichannel pipette was used to add 50 μL 2× treatment mix to each well for a final concentration of 5% Matrigel (450–500 μg/mL) and 100 μM ABA. Care was taken to minimize bubbles during pipetting. For integrin stimulation by MnCl2, 0.1 μL 50 mM MnCl2 was supplemented to every 50 μL 2× treatment mix. For integrin antibody blocking, two rat monoclonal antibodies (mAb13: anti-β1-integrin; mAb16: anti-α5-integrin) were diluted to 0.5 mg/mL in RPMI-1640 and passed through a desalting spin column (Thermo Fisher, 89883) pre-equilibrated for 4 times with RPMI-1640. Antibody concentrations were re-measured by absorbance at 280 nm on a nanodrop spectrophotometer (Denovix, DS-11). The per-well 2× treatment mix was adjusted to include 20 μL 0.5 mg/mL antibody solution, 19.4 μL RPMI-1640, 4.5 μL FBS, 0.45 μL 200 mM L-glutamine, 0.45 μL 100× PenStrep, 5 μL Matrigel and 0.2 μL 50 mM ABA. Unless specified otherwise, DLD-1 spheroids were all cultured for 6 days from the day of Matrigel and ABA supplementation.
sgRNA design
sgRNAs for target genes (Cdh1 or Itgb1) were designed on the CRISPOR (Haeussler et al., 2016) website using 500 bp sequences centered around the transcription start site (TSS ± 250 bp). sg1-Cdh1: 5’-gCCGAGAGGCTGCGGCTCCAA-3’. sg2-Cdh1: 5’-gTGGCCGGGGACGCCGAGCGA-3’. sg-Itgb1: 5’-GGACGCCGCGCGGAAAAGGT-3’. Control guide RNAs for both the S. pyogenes Cas9 (5’-gTGCGAATACGCCCACGCGAT-3’) and the S. aureus Cas9 (5’-gCCTTCCCAACAGTTGCGCAGC-3’) were designed from the bacterial lacZ gene against the human genome. An extra “g” was added to the 5’-end if the guide sequence did not begin with “g” to facilitate transcription by the U6 promoter.
Lentivirus packaging
All lentivirus work was performed using in a BSL2 room with a dedicated incubator. Lenti-sgRNA vectors (see Plasmids) were co-transfected with psPAX2 (Addgene, 12260) and pMD2.G (Addgene, 12259) into HEK293T cells by calcium co-precipitation to produce infectious lentiviral particles. Briefly, 4×106 HEK293T cells were seeded in a 10 cm dish one day before packaging. Next morning, culture media were changed and supplemented with 25 μM chloroquine (MilliporeSigma, C6628). Two 15 mL tubes (A and B) were used to prepare the transfection mix. 1 mL 2× HBS (50 mM HEPES, 280 mM NaCl, 1.5 mM Na2HPO4, pH 7.10) was added to tube A. 10 μg of each plasmid (the lenti-sgRNA vector, psPAX2 and pMD2.G) and 1 mL 0.3 M CaCl2 were sequentially added to tube B and mixed by pipetting. The DNA-CaCl2 mixture in tube B was then added dropwise into the 2× HBS in tube A and mixed by pipetting. The transfection mix was then added dropwise to the 10 cm dish. Culture media were changed twice at about 12- and 36-hours post transfection, and lentivirus-containing media were collected twice at about 36- and 60-hours post transfection into a 50 mL tube (stored at 4°C). Pooled lentivirus-containing media were passed through a 0.45 μm filter (MilliporeSigma, SE1M003M00) to remove cell debris. To concentrate the lentivirus, 4 mL 5× PEG reagent (System Biosciences, LV825A-1) was added and mixed by pipetting. After ≥ 12 hours incubation at 4°C (up to 4 days), lentivirus was pelleted by centrifugation at 1,500× g for 30 min at 4°C, and the pellet was resuspended in 400 μL DMEM/F-12 with 1× PenStrep and stored at −80°C.
Lentivirus titration
The titer of concentrated lentivirus was estimated using Lenti-X GoStix Plus (Takara, 631281) after 100× dilution. A GoStix Value (GV) of 50 was empirically considered to be equivalent to a lentivirus titer of 5×105 IFU/mL. The typical titer of concentrated sgRNA lentivirus was 1.5×108 IFU/mL.
Lentivirus transduction
One day before transduction, 1×105 cells were seeded in a well of a 12-well plate. Next day, 2 μL 4 mg/mL polybrene (MilliporeSigma, H9268) was added to 1 mL medium (final 8 μg/mL). An appropriate amount of lentivirus for an MOI (multiplicity of infection; ratio of infectious viral particles to cells) of 10–15 (typically 20 μL for concentrated sgRNA lentivirus) was then added. One day later, the virus-containing medium was replaced with regular medium after 4× HBSS washes, and cells were re-plated to a 75 cm2 flask in culture medium supplemented with 20 μg/mL blasticidin (InvivoGen, ant-bl-1) to begin the antibiotic selection.
Fluorescence activated cell sorting
For cell sorting, DLD-1 derived cells were trypsinized for 5 min longer than for passage (~15 min total) to increase the ratio of single cells, pelleted at 1,000× g for 3 min, and resuspended in phenol-red free, serum-free RPMI-1640 medium (Thermo Fisher, 11835030) for a cell density of 5–10×106/mL. The cell suspension was passed through a 40 μm Flowmi cell strainer (VWR, H13680–0040) and sorted on a BD FACSAria III or SONY SH800 cell sorter operated by the NIDCR Combined Technical Research Core.
Western blotting
DLD-1 derived cells were seeded in 12-well plates (Corning, 3512) at 20,000 cells/well on day 0, treated with DMSO vehicle or desired concentrations of ABA in DMSO on day 1 (or each day of days 1–5 per well for an ABA time course), and harvested on day 6. Culture media were changed once on day 4 or 5. For harvesting, 100 μL RIPA buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (MilliporeSigma, 11836170001) was added to each well after rinsing with PBS (Phosphate Buffered Saline; Lonza, 17–517Q). Cells were scraped into RIPA buffer on ice using 1 mL pipette tips. Cell suspensions were transferred to pre-cooled 1.5 mL tubes (Eppendorf, 022363212), incubated for 30 min on ice, and centrifuged at 13,000× rpm for 10 min at 4°C. Cleared cell lysates were transferred to a new set of pre-cooled 1.5 mL tubes and stored at −20°C. Protein concentrations of cell lysates were determined by Bradford assays (Bio-Rad, 5000201). Lysate aliquots with 16 μg protein were denatured in 1× Laemmli sample buffer (Bio-Rad, 1610747) for 5 min at 95°C. Samples of 8 μg protein or 10 μL protein ladder (Thermo Fisher, 26623) were loaded per lane onto a precast gel (Bio-Rad, 4561096) for electrophoresis. Proteins were transferred onto a nitrocellulose membrane (Bio-Rad, 1704159) using the Turbo Transfer system (Bio-Rad, 1704150). The membrane was stained with Ponceau S (MilliporeSigma, P7170) to assess transfer quality, washed for 5 min in TBST (Tris Buffered Saline with 0.1% Tween-20; Quality Biological, 351–086-101; MilliporeSigma, P2287) to remove Ponceau staining, blocked in Blocking Solution (5% nonfat dry milk in TBST) for 30 min at room temperature (RT), incubated in primary antibodies (see Antibody usage for Western blotting) diluted in Blocking Solution overnight at 4°C, washed 4× 5 min with TBST at RT, incubated in LI-COR secondary antibodies diluted in Blocking Solution for 1–2 hours at RT, washed 4× 5 min in TBST at RT, and imaged on a LI-COR Odyssey CLx imaging system controlled by LI-COR Image Studio software. Western blotting band intensities were quantified using LI-COR Image Studio Lite software.
Antibody usage for Western blotting
Primary antibodies used for Western blotting: anti-E-cadherin (Thermo Fisher, 13–1900), 0.5 μg/mL; anti-β-catenin (Abcam, ab32572), 1:5,000 (0.0126 μg/mL); anti-β1-integrin (MilliporeSigma, MABT821), 1 μg/mL; anti-GAPDH (Cell Signaling, 2118L), 1:2,000; anti-α-tubulin (MilliporeSigma, T6199), 0.5 μg/mL. All Western blotting secondary antibodies were from LI-COR and used at 1:5,000 for 800CW conjugates and 1:10,000 for 680RD conjugates.
Immunostaining of cells
Cells were seeded, immunostained and imaged in 8-well ibidi chambers (ibidi, 80826). All procedures were performed at room temperature with gentle rocking. Cells were fixed with 4% PFA in PBS for 15 min, permeabilized with PBSTx for 15 min, blocked in 5% donkey serum in PBS for 30 min, incubated in primary antibodies (see Antibody usage for immunostaining) diluted in PBS for 1 hour, washed 4× 5 min with PBS, incubated in 0.5 μg/mL DAPI (Thermo Fisher, D1306) and secondary antibodies diluted in PBS for 1 hour, washed 4× 5 min with PBS, stored at 4°C and imaged within 3 days.
Immunostaining of spheroids and salivary glands
For immunostaining not involving keratins or prelabeled hamster anti-β1 integrin antibodies, spheroids were rinsed in 2 mL PBS in a 35 mm dish and transferred into sample baskets (one basket per staining group; Intavis, 12.440) using low-retention pipette tips (cut for larger opening; Rainin, 30389187 or 30389190) under a dissecting microscope. For fixation, each basket was soaked in 1 mL fixative (4% PFA in PBS; Electron Microscopy Sciences, 15710) in a well of a 24-well plate (Corning, 3524) overnight at 4°C (all incubations were with gentle rocking). Cultured salivary glands were fixed on the filter by replacing Organ Culture Medium under the filter with 200 μL fixative for 1 hour at room temperature (RT) or overnight at 4°C. Fixed glands were detached from the filter and transferred into sample baskets in PBS in a 35 mm dish using a pair of forceps (Fine Science Tools, 11251–20) under a dissecting microscope. Fixed samples in baskets were permeabilized in PBSTx (PBS with 0.2% Triton-X-100; Thermo Fisher, 28314) for 30 min at RT, blocked in 5% donkey serum (Jackson ImmunoResearch, 017–000-121) in PBSTx for 2 hours at RT, incubated in primary antibodies (see Antibody usage for immunostaining) diluted in either PBSTx or 5% donkey serum for 2 days at 4°C, washed 4× 15 min in PBSTx at RT, incubated in secondary antibodies diluted in either PBSTx or 5% donkey serum for 2 days at 4°C, washed 4× 15 min in PBSTx at RT, rinsed in PBS and mounted under a dissecting microscope. To preserve fluorescence and to minimize compression, samples were mounted in 20 or 40 μL antifade mountant (Thermo Fisher, P36930) supported by one layer (for salivary glands) or two layers (for spheroids) of imaging spacers (Grace Bio-labs, 654004) attached to a glass slide (Thermo Fisher, 3011–002).
For immunostaining of mesenchyme-free salivary gland epithelial cultures with prelabeled hamster anti-β1 integrin antibodies (Atto-565-labeled Hamster anti-β1-integrin; see Protein labeling; clone Ha2/5, IgM, BD Biosciences 555002), the PFA fixation and permeabilization steps were identical to above, but a much higher antibody concentration (10 μg/mL) was used and the incubation time was extended to 6 days at 4°C, because of anticipated tissue penetration difficulties using IgM antibodies, which are 5 times as large as typical IgG antibodies.
For immunostaining of keratins (KRT19), spheroids and salivary glands were fixed in 1 mL ice-cold 1:1 mixture of acetone and methanol in a 1.5 mL Eppendorf tube and incubated for 1 hour to overnight at −20°C. The mixture of acetone and methanol was exchanged to 100% methanol for longer storage. Before staining, samples were re-hydrated in a sequential methanol gradient (70%, 50%, 30%, 0% methanol in PBSTx) on ice for 5 min incubation at each step. The remaining procedures were identical to those for PFA-fixed samples as described above (beginning from permeabilization by PBSTx).
Antibody usage for immunostaining
Primary antibodies used for immunostaining: anti-E-cadherin (Thermo Fisher, 13–1900), 1 μg/mL; anti-collagen type IV (MilliporeSigma, AB769), 2 μg/mL; anti-laminin (MilliporeSigma, L9393), 2.5 μg/mL; anti-histone H3 (phospho S10) (Abcam, ab47297), 1 μg/mL; anti-β1-integrin (MilliporeSigma, MABT821), 1 μg/mL; anti-Claudin 10 (Thermo Fisher, 38–8400), 1 μg/mL; anti-KRT19 (DSHB, TROMA-III-c), 2 μg/mL; anti-α6-integrin (clone GoH3, BD Biosciences, 555734), 2 μg/mL; anti-α9-integrin (R&D Systems, AF3827-SP), 1 μg/mL; Atto-565-labeled Hamster anti-β1-integrin (see Protein labeling; clone Ha2/5, BD Biosciences 555002), 10 μg/mL. All immunostaining secondary antibodies were from Jackson ImmunoResearch (an equal volume of glycerol was added for storage at −20°C after reconstitution as instructed) and used at 1:200 (1.5–3 μg/mL).
Integrin blocking for salivary gland culture
Hamster anti-β1-integrin (BD Biosciences,), isotype-matched control IgM (BD Biosciences,), rat anti-α6-integrin (BD Biosciences, 555734) and isotype-matched control IgG (BD Biosciences, 553992) were used at 100 μg/mL each for function blocking or control after overnight dialysis into 1 liter DMEM/F-12 medium supplemented with 1× PenStrep at 4°C with gentle agitation, using a dialysis cassette (Thermo Fisher, 66383).
Integrin blocking for 3D DLD-1 spheroid culture
Rat anti-β1-integrin (in-house clone mAb13; available from MilliporeSigma, MABT821) was used at 100 μg/mL for function blocking. Rat anti-α5-integrin (in-house clone mAb16; available from MilliporeSigma, MABT820) was used at 100 μg/mL. Stock antibodies (1.9–5 mg/mL) were diluted to 0.5 mg/mL in RPMI-1640 medium and further exchanged to RPMI-1640 medium using spin desalting columns (Thermo Fisher, 89882) following the manufacturer’s instructions.
Tissue clearing of spheroids
Tissue clearing was performed for the images shown in Fig. 6G to enable imaging over 200 μm thickness. For tissue clearing, regular immunostaining steps were carried out except for mounting. Spheroids were instead sequentially transferred to each well of a 3-well silicone chamber slide (ibidi, 80381), each containing 500 μL CytoVista clearing reagent (Thermo Fisher, V11315). After 5 min incubation in the last well, spheroids were transferred to 200 μL CytoVista clearing reagent per well of an 8-well glass-bottom ibidi chamber (ibidi, 80827) for imaging with two-photon microscopy (see Immunostaining light microscopy). Note that this clearing reagent preserved mScarlet but not mNeonGreen fluorescence.
Cell attachment assay
DLD-1 derived cells (sg-Control, sg1-Cdh1, sg2-Cdh1 and sg-Itgb1) were pre-treated with 100 μM ABA for 5–10 days before being used. The glass surface of MatTek 6-well plates (MatTek, P06G-0–14-F) was coated with 200 μL 91 μg/mL Matrigel in PBS or 8 μg/mL E-cadherin extracellular domain (E-cad-ECD; R&D Systems, 8505-EC-050) in PBS for 1–3 hours at 37°C. Coated wells were rinsed once with 3 mL PBS, blocked with 2 mL 5% bovine serum albumin (BSA; MilliporeSigma, 10735108001) in PBS for 30 min at 37°C, and washed twice with 3 mL PBS. During the blocking step, cells were detached, pelleted, resuspended, counted and diluted to 5×104 cells/mL. 2 mL cell suspension was seeded in each well. 3 or 4 wells were used per experimental group. After 15 min (for Matrigel coating) or 2 hours (for E-cad-ECD coating) incubation at 37°C with 5% CO2, unattached cells were removed from each well, which was then gently washed 3× with 3 mL PBS, fixed with 2 mL 4% PFA in PBS for 15 min at 37°C, washed 2× with 3 mL PBS, and imaged under a microscope (see Live-spheroid imaging and cell-attachment assay imaging) to quantify attached cell densities (see Image processing and analysis). For assay consistency, a 2 mL aspirating pipette was capped with a 200 μL pipette tip to attenuate vacuum strength, and the house vacuum valve was pre-adjusted using PBS to reach a liquid removal rate of ~1 mL/second. During liquid removal and addition, tips of aspirating or transferring pipettes were always placed to the side of the bottom of the MatTek well away from the coated glass surface, resulting in ~200 μL leftover liquid between washes. Care was taken throughout the assay to avoid agitating the plates. The incubation time for each coating was determined to be the time at which ~50% of control cells were attached in pilot assays.
Live-organ imaging of intact salivary glands by two-photon microscopy
All microscopy systems for live imaging were equipped with an environmental chamber to maintain samples at 37°C with 50% humidity and 5% CO2. The custom-built environmental chamber (Precision Plastics Inc., Beltsville, MD, USA) is comprised of black Lucite to block environmental light, and it encloses the entire upper body of the microscope with two front doors through which samples can be loaded onto the stage. Temperature and humidity was maintained by heated circulating air controlled by a temperature/humidity controller (Air-Therm ATX-H; Model # 300391; World Precision Instruments, Inc., Sarasota, FL, USA) connected to a humidifier (Model # V5100NS; Kaz USA Inc., Marlborough, MA, USA). CO2 level was maintained by CO2 infusion controlled by a controller unit (S/N: 827–1009; OKOLAB, Ambridge, PA, USA). Transgenic salivary glands expressing fluorescent markers were isolated and cultured on a floating 13 mm filter at 37°C with 5% CO2 (see Salivary gland isolation and culture) for at least 1 hour before being mounted for live imaging. Double-adhesive imaging spacers (Grace Bio-labs, 654008; cut into 8 separated wells, each 120 μm thick with 9 mm diameter opening) were sterilized by soaking in 70% ethanol for 3 min and attached to the glass bottoms of 50 mm MatTek dishes (MatTek, P50G-1.5–30-F). Under a dissecting microscope, 5 μL Organ Culture Medium was transferred to the center of the imaging spacer, and the filter with glands was flipped onto the imaging spacer so that glands were sandwiched between the filter and the glass bottom. Care was taken to ensure the filter was flat and center-aligned with the imaging spacer. The edge of the filter was pressed to ensure tight adherence to the imaging spacer. 2 mL Organ Culture Medium was then added to the MatTek dish, which was incubated at 37°C with 5% CO2 for at least 2 hours before imaging. A Nikon 40×, 1.15 NA, Apo LWD, water-immersion objective or a Nikon 25×, 1.05 NA, Plan Apo, silicone-immersion objective was used for live-organ imaging using two-photon microscopy on a Nikon A1R Confocal Microscope System equipped with a Ti:sapphire laser (Coherent, Chameleon Vision II). Image acquisition was controlled by Nikon NIS-Elements software. Images were acquired at 2 μm z intervals over 100 μm thickness and 5 min intervals for 20–36 hours. The tunable laser was used at 950 nm for simultaneous two-photon excitation of histone-EGFP and membrane-tdTomato (or heterozygous Krt14p::RFP), and 920 nm for histone-EGFP and homozygous Krt14p::RFP. The laser power was adjusted to compensate for z-depth changes using the “Z Intensity Correction” option with criteria that the bulk histograms of both channels spanned 500–1500 gray values (at typically 1–8% or 12–100 mW power).
Live imaging of single-bud and single-cell salivary gland epithelial cultures by two-photon microscopy
Single-bud and single-cell salivary gland epithelial cultures (referred to “spheroids” hereafter; see 3D culture of primary salivary gland epithelial buds or dissociated cells) from transgenic salivary glands were mounted in custom-assembled imaging chambers (see below) and imaged by two-photon microscopy using the same system and imaging setup steps as described in Live-organ imaging of intact salivary glands by two-photon microscopy. To assemble the imaging chamber, a 4-well silicone chamber was removed from an ibidi dish (ibidi, 80466) and attached to a glass-bottom Bioinert dish (ibidi, 81150). 2 mL 2% agarose (Millipore Sigma, A9539) in DMEM/F12 medium were dissolved by heating in a microwave and added outside of the silicone chamber to hold the chamber to prevent detaching from the Bioinert surface. 30 μL 2% agarose was added inside each chamber to form a thin agarose pad and incubated for 5 min at room temperature to allow the agarose to solidify. A 30 μL glass micropipette (Drummond Scientific Company, Cat # 2–000-030) was used to poke 1 or 2 micro wells in each chamber towards the center of the dish bottom. 100 μL DMEM/F12 medium was added to each well, and a pair of forceps was used to remove the agarose insert from each micro well to expose the glass surface. The medium in each well was then replaced by 80 μL DMEM/F12 medium supplemented with 200 ng/mL FGF7 (R&D Systems, 5028-KG-025), 1× ITS supplement (Thermo Fisher, 41400045) and 0.5 mg/mL growth factor-reduced Matrigel (Corning, 356231; stock 9–10 mg/mL). Spheroids were transferred from the 96-well plate into the imaging chamber using low-retention pipette tips (cut for larger opening; Rainin, 30389190) and each spheroid was gently pushed into one micro well for immobilization during live imaging. Immediately before imaging, 1 mL DMEM/F12 medium was added on top of the agarose pad outside the silicone chambers to help maintain humidity. Spheroid cultures from dissociated salivary gland epithelial cells need to be cultured for 1 day in 96-well plates to allow spheroid formation before being mounted for imaging.
KikGR photoconversion and live imaging by confocal microscopy
E13 KikGR transgenic glands were cultured on the filter with Organ Culture Medium supplemented with 5 μg/mL AF680-anti-collagen IV (see Protein labeling) for ~10 hours. Under a dissecting microscope, a 10 μL pipette tip was used to spot vacuum grease (MilliporeSigma, 18405) around glands on the filter, which was then flipped onto the glass area of a 35 mm dish (MatTek, P35G-1.5–20-C). The vacuum grease limited gland compression and also served as a bio-inert glue to adhere the filter to the glass. 2 mL Organ Culture Medium supplemented with 1 μg/mL AF680-anti-collagen IV (see Protein labeling) were added to the dish for imaging after 2 hours incubation at 37°C with 5% CO2. A Nikon 40×, 1.15 NA, Apo LWD, water-immersion objective was used for photoconversion and imaging on a Nikon A1R Confocal Microscope System equipped with 4 laser lines (405 nm, 488 nm, 561 nm, 640 nm; Nikon LU-N4). Photoconversion and image acquisition were controlled by Nikon NIS-Elements software. For photoconversion, a 405 nm laser was used at 1–5% power (0.15–0.75 mW) with the pinhole set at 1 AU (Airy Unit; 30.7 μm) to stimulate user-specified polygonal ROIs (Regions of Interest) in the “ND Stimulation” module of the software. Short 1–5 second pulses (depending on ROI sizes) were repeated until all green fluorescence inside ROIs was converted to red fluorescence. For image acquisition, 488 nm and 561 nm lasers were used at 2% power (0.3 mW), whereas the 640 nm laser was used at 5% (0.75 mW) with the pinhole set at 1.2 AU (58.7 μm). Images were acquired at 10 min time intervals and 2 μm z intervals.
Live-spheroid imaging and cell-attachment assay imaging
Live-spheroid imaging (time-lapse or single-time-point) and cell-attachment assay imaging were performed by phase contrast and epifluorescence microscopy using a Nikon 10×, 0.3 NA, Plan Fluor objective on a Nikon Ti-E brightfield microscope system with a Hamamatsu Orca Flash 4.0 V3 sCMOS camera. Spheroids were imaged in the same 96-well U-bottom or V-bottom plates for culture. Image acquisition was controlled by Nikon NIS-Elements software. The JOBS module of the software was used to automatically set up multiple positions in a 96-well plate (spheroids) or a 6-well plate (cell attachment assay). For time-lapse live-spheroid imaging by confocal microscopy, spheroids were incubated in 40 μg/mL Atto647N-fibronectin (see Protein labeling) overnight at 37°C with 5% CO2 and transferred into a 4-well 35 mm dish (ibidi, 80466), each well of which contained 2 or 3 spheroids in 100 μL spheroid culture medium with 10 μg/mL Atto647N-fibronectin. A Nikon 20×, 0.75 NA, Plan Apo objective was used on a Nikon A1R Confocal Microscope System equipped with 4 laser lines. A resonant scanner was used for high-speed laser scanning. Image acquisition was controlled by Nikon NIS-Elements software at 10 min time intervals and 2 μm z intervals.
Immunostaining light microscopy
Immunostained spheroids in Fig. 6G were imaged by two-photon microscopy using a Nikon 20×, 0.75 NA objective on a Nikon A1R Confocal Microscope System equipped with a Ti:sapphire laser (used at 760 nm, 3% or 90 mW) controlled by Nikon NIS-Elements software. All other immunostained spheroids and salivary glands were imaged by laser scanning confocal microscopy using Nikon 20×, 0.75 NA or 60×, 1.4 NA Plan Apo objectives on a Nikon A1R Confocal Microscope System controlled by Nikon NIS-Elements software or a Zeiss 20×, 0.75 NA or 63×, 1.4 NA Plan Apo objective on a Zeiss LSM 880 system controlled by Zeiss ZEN software. Immunostained tissue culture cells were imaged using a Nikon 40×, 1.25 NA, Plan Apo, silicone-immersion objective on a Nikon spinning disk confocal system equipped with a Yokogawa CSU-X1 unit and a Prime 95B sCMOS camera (Photometrics) controlled by Nikon NIS-Elements software.
Protein labeling
Human plasma fibronectin was purified as previously described (Akiyama, 1999). For fibronectin labeling, NHS-Atto647N (ATTO-TEC, AD 647N-31) was dissolved in DMSO to 10 mM and stored in a desiccated container at −20°C as 10 μL aliquots. Labeling buffer was 20 parts PBS with 1 part 0.2 M NaHCO3 (pH 9.0), which was adjusted to pH 8.3. For labeling, 1 mL fibronectin was exchanged to the labeling buffer using a spin desalting column (Thermo Fisher, 89891). The fibronectin concentration after buffer exchange was determined by absorbance at 280 nm (2.3 mg/mL), which was used to calculate the amount of NHS-Atto647N equivalent to 4× times molar excess of fibronectin (3.4 μL). The labeling mix was incubated for 45 min at room temperature, then unlabeled dye was removed by buffer exchange into PBS using a spin desalting column. Insoluble Atto647N-fibronectin was removed by centrifugation at 13,000× rpm for 30 min at 4°C. The concentration of cleared Atto647N-fibronectin and the degree of labeling (DOL) were calculated from absorbance at 280 nm and 646 nm with a dye-specific correction factor 0.03 (using the Atto calculation formula; 0.84 mg/mL, DOL 1.7). Atto647N-fibronectin was then aliquoted and stored at −80°C. A similar procedure was used to label the hamster anti-β1 antibody (Ha2/5) with NHS-Atto565 (ATTO-TEC, AD 565–31) and the goat collagen IV antibody (MilliporeSigma, AB769) with NHS-AF680 (Thermo Fisher, A20008). For collagen IV antibody labeling, a 50 mM borate buffer (pH 9.0) was used as the labeling buffer and labeled antibodies were stored in PBS at 4°C.
Image processing
All images acquired on the Nikon A1R Confocal System were denoised using the Denoise.ai function of the Nikon NIS-Elements software. All other image processing was performed in Fiji (Schindelin et al., 2012), an ImageJ distribution. Customized Python, Jython and ImageJ Macro scripts were used for automating or facilitating image analysis and data visualization in this study (see Data and Code Availability).
Atomic force microscopy (AFM)
DLD-1 spheroids containing only sg-Control cells cultured for 7 days were used for AFM measurements to minimize effects of complex tissue geometries. To immobilize spheroids, CellTak (Corning, 354240) was coated at ~3.5 μg/cm2 inside a 4-well silicone chamber (removed from ibidi, 80466; reusable for many times) attached to the center area of the glass bottom of a specialized AFM dish (World Precision Instruments, FluoroDish FD35–100). For coating of each dish, 5.4 μL 1 mg/mL CellTak was added to 300 μL 0.1 M sodium bicarbonate (pH 8.0; accurate pH is important). Immediately before dispensing, 2.7 μL 1N NaOH was added to the CellTak solution and vortexed. 75 μL was dispensed into each well and coating incubation was between 30 min and 4 hours. When the AFM system was ready, CellTak coating solution was removed and the coated wells were rinsed once with water. The borders of coated area were marked on the outside bottom of the AFM dish to facilitate sample locating during AFM measurements. The silicone chamber was removed and the AFM dish was filled with 3 mL warm RPMI-1640 Complete medium. 10–15 spheroids were rinsed 4–6 times in 2 mL RPMI-1640 Complete medium in a clean 24-well plate under a dissecting scope until milky carryover matrix condensates were removed. Spheroids were transferred using low-retention pipette tips (cut for larger opening; Rainin, 30389190). Cleaned spheroids were transferred into the AFM dish and arranged towards the center of the coated area. A pair of forceps with blunt tips were used to gently push each spheroid against the glass bottom to aid immobilization by CellTak. The AFM dish was then carefully transferred and mounted in the sample holder with a heating stage.
AFM measurements were performed using a Bruker JPK NanoWizard 4XP BioScience AFM system (Bruker, Santa Barbara, CA) mounted on an inverted Axio Observer.7 microscope system (Carl Zeiss, Gottingen, Germany) equipped with a Confocal Laser Scanning Microscope 900 with Airyscan 2 and Multiplex module (LSM 900, Carl Zeiss) and a 10× (0.6 NA, Plan-Apochromat) objective (Carl Zeiss). A petri dish heating stage (Bruker) was used to maintain a physiological temperature of 37°C for spheroids during measurements. Modified AFM microcantilevers with an attached 35 μm polystyrene microsphere were obtained from Novascan (Novascan, Ames, IA; Catalog # PT.PS.SN.35.CAL.). The AFM probe spring constant was obtained using the thermal tune methods built in the AFM system. Calibrated spring constant for cantilevers ranged from 0.173–0.577 N/m. After cantilever calibration, the AFM probe was moved on top of an immobilized spheroid to perform the quasi-static force spectroscopy measurements. The applied force was set to be 1–2 nN for spheroids cultured without the matrix and 3–5 nN for spheroids cultured with matrix supplement, yielding an indentation of 2–3 μm. The force curve ramp rate was set to 0.5 Hz and the speed ranged between 2–6 μm/s. For each spheroid, 5 consecutive force-indentation curves were collected to obtain the average value of measurements.
Quantification and Statistical Analysis
Statistical analysis
All statistical analysis was performed using customized Python scripts (see Data and Code Availability). Comparisons of 2 groups of paired or independent data were performed using paired or unpaired two-sided Student’s t-test, respectively. For comparisons of the expression of many genes between 2 subgroups of the scRNA-seq data, the Benjamini-Hochberg procedure was used to correct for multiple comparisons. Pairwise comparisons of 3 or more groups were performed using Tukey’s HSD test. When the raw data did not meet the homogeneity of variance requirement of Tukey’s test, statistical comparisons were performed on log-transformed data (e.g., comparisons of Young’s moduli derived from AFM). Error bars in all plots represent 95% confidence intervals. Details of sample number, data representation, and statistical comparison method of each plot can be found in the figures and figure legends.
Image analysis and quantification
Automatic 3D cell tracking and surface rendering were performed using Imaris 9.5.0 (Bitplane). Manual curation of semi-automatic cell tracking was performed in Fiji using the TrackMate (Tinevez et al., 2017) plugin. Manual surface reconstruction was performed by drawing polylines along the epithelial surface at sparse z planes (~6 μm intervals) on x-y view and resliced y-z view image stacks in Fiji, which were used for interpolation and plotting using customized Python scripts (see Data and Code Availability).
To determine the “on-surface ratio” of surface cells in control or collagenase-treated salivary glands, newly returned or randomly selected pre-existing surface cells were manually tracked using TrackMate (Fiji plugin) for an average of 5.4 hours (control) or 5.1 hours (collagenase). The “on-surface ratio” was calculated as the weighted average of the proportion of time frames that the tracked cell was at the epithelial surface (weighted by the total tracked time of each cell).
Automated segmentation of mesenchymal cells were performed using cellpose (cellpose.org) (Stringer et al., 2021). Segmented masks were used to compute shape metrics using customized Python scripts (see Data and Code Availability).
For quantification of phospho-Histone H3 (pHH3) staining, the pHH3 image was binarized by automatic thresholding followed by Gaussian filtering to measure the area covered by pHH3 fluorescence. To quantify the % pHH3 area in different regions of interest (ROIs), the epithelial ROI was segmented from E-cadherin staining, whereas the total cell-occupied tissue ROI was segmented from DAPI staining, both by automatic thresholding followed by Gaussian filtering. The mesenchymal ROI was computed by subtracting the epithelial ROI from the tissue ROI. When a whole epithelial bud was within the field of view, epithelial ROIs were shrunk by 15 μm to obtain the interior epithelium ROI, which was subtracted from the epithelial ROI to obtain a ring-shaped surface epithelium ROI. When only part of the epithelial bud was included in the image, the initial epithelial ROI and the 15 μm-shrunk interior epithelium ROI were both intersected with a user-drawn polygon ROI to obtain two pie-slice-shaped ROIs that only differed by a fan-shaped surface-layer ROI. The % pHH3 area of each ROI was calculated as the ratio of pHH3 area to the total ROI area. Parameters of Gaussian filtering and automatic thresholding were kept consistent for all images in the same dataset for comparison.
For automated cell counting in the attachment assay, nuclear fluorescence images were smoothed by Gaussian filter (sigma = 2 pixels), contrast enhanced by the MorphoLibJ (Legland et al., 2016) white top hat filter (disk, radius = 10), binarized and counted using the Analyze Particles function of ImageJ.
For manual bud counting, several efforts were made to minimize bias. First, file names of all images were scrambled before counting for observer blinding. Second, the same investigator performed the counting of all spheroids (K.M.) or all salivary glands (S.W.) to avoid between-person variance. Third, an explicit criterion was used such that a bud was counted only when the convex edge occupied at least one third of a circle. Finally, S.W. decoded and plotted the counting.
For quantification of the percentage of protruded bud area in DLD-1 spheroids, a similar observer-blinded procedure was used as above. First, file names of all images were scrambled. Second, K.M. manually drew a polygon along the spheroid perimeter excluding protruding buds delineated by a straight line drawn between the bottoms of bud clefts. This polygon encloses the spheroid interior and its area is measured as Area_in. Third, S.W. ran a script (available on Github; see Data and Code Availability) that automatically segmented the total occupied area of each spheroid from the bright fluorescence images by automatic thresholding after image smoothing with a Gaussian filter (Area_total; see Fig. S6B). Fourth, S.W. decoded the data from K.M. for the interior area of spheroids, calculated the percentage of protruded bud area (%Protruded bud area = 100 × (Area_total - Area_ in) / Area_total) and plotted them.
AFM data analysis and quantification
The DLD-1 spheroids apparent Young’s modulus (E, in Pa) was calculated by fitting each recorded force-indentation curve with the Hertz contact mechanics model for a rigid spherical probe indenting an infinite isotropic elastic half-space:
In the above equation, F is the applied force, E is the elastic Young’s modulus, υ is the Poisson’s ratio (assumed to be 0.5 for soft material), R is the radius of the AFM probe (17.5 μm), and δ is the spheroid indentation. AFM data analysis was performed using the JPK SPM Data Processing software. A 2 μm deformation range was selected from near the maximum indentation to fit the Hertz model.
Modeling of budding morphogenesis
Model assumptions and parameter constraints:
All epithelial cells are tightly packed with no empty spaces.
The average cell size Vcell is constant, so the net volume is proportional to cell number.
The initial epithelium contains an interior sphere with radius r enveloped by a surface layer with a constant thickness h = 15 μm. In branching salivary glands, h ≈ 15 μm when surface cells are very crowded and columnar, so this is an upper bound of possible surface layer thickness. The interior radius r is bounded by h ≤ r ≤ 6h, since bud diameters are approximately 60–200 μm. We note that the constant-thickness assumption disadvantages the system from increasing the surface area, because the surface area can be increased by reducing the surface-layer thickness without increasing the surface-layer volume.
The volume expansion or shrinkage of each compartment is contributed by a combination of local cell division and material (cell) transfer across the surface-interior interface. Specially, the interior cell layer at this interface is defined as the subsurface layer.
Without cell divisions, the material transfer at the surface-interior interface depends on the abundance of low-E-cadherin cells near the surface-interior interface. We assume low E-cadherin cells at this interface can swap between the surface (contacting the basement membrane; cell status S, cell number NS) and subsurface layer (contacting other cells; cell status B, cell number NB). Analogous to a chemical reaction B ↔ S, we assume their free energy difference ΔE = ES – EB determines which direction is thermodynamically favored (e.g., ΔE < 0 : surface is favored; ΔE > 0 : subsurface is favored). Due to high cell motility, we assume swapping is fast and equilibrium can be reached quickly. The cell number ratio at equilibrium , where c is a constant. Under normal conditions, the surface-residence ratio of both nascent and randomly sampled surface cells is about 0.930, corresponding to K = 13.285 and indicating a strong preference for low-E-cadherin cells to stay on surface.
When interior cells are ready to divide (division-ready cells), they all divide locally to produce two interior cells. Thus, the local cell division rate in the interior PIL is identical to the production rate of division-ready cells, which we denote as the apparent cell proliferation rate PIA.
In contrast, no surface cells divide locally to produce two surface cells. Thus, the local cell proliferation rate in the surface layer PSL = 0. As a result, the surface layer expansion is solely contributed by the material transfer between the interior and surface.
We assume division-ready cells at the surface all delaminate to divide in the interior to produce 2 interior cells. In practice, this is the case for the 92.4% of divisions that are Type I, but we note that the 7.2% Type II divisions can be viewed as a special case of Type I division in which one daughter cell has immediately returned to the surface. This simplification slightly disadvantages the expansion rate of the surface layer.
Each division-ready cell at the surface will cause one surface-to-interior transfer event and contributes two subsurface low-E-cadherin cells at steady state, because 100% of the daughter cells return, and most return within a short time. Denoting PSA as the apparent surface cell proliferation rate, i.e., the production rate of division-ready cells in the surface layer, we have a transient loss of NSPSA low-E-cadherin cells from the surface and a gain of 2NSPSA low-E-cadherin cells to the surface.
Another possible contribution to low-E-cadherin cells in the subsurface layer could theoretically come from putative events in which high-E-cadherin cells downregulate E-cadherin and then sort out to the subsurface layer. We think it is rare because all interior-to-surface transfer events have we observed could be traced back to be originating from the surface, whenever it was possible to trace them. For simplicity, we will ignore this term and note that this simplification will slightly disadvantage the expansion rate of the surface layer.
Parameter definitions:
r : radius of the interior sphere
h : thickness of the surface layer
Vcell : average volume of a single cell
VI : volume of the interior epithelium
Vepi : total volume of epithelium
VS : volume of the surface layer
NI : cell number of the interior epithelium
NS : cell number of the surface layer
RI : cell number expansion rate in the interior epithelium
RS : cell number expansion rate in the surface layer
α : ratio of RI to RS (expansion ratio)
β : ratio of interior radius to surface thickness (size ratio)
δ : ratio of VI to VS (volume ratio)
PIL : local cell proliferation rate in the interior epithelium, i.e., cells that complete division within the interior epithelium
PIA : apparent cell proliferation rate in the interior epithelium, i.e., the production rate of division-ready cells
PSL : local cell proliferation rate in the surface layer, i.e., cells that complete division within the surface layer
PSA : apparent cell proliferation rate in the surface layer, i.e., the production rate of division-ready cells
ω : ratio of PIA to PSA (apparent proliferation ratio)
τsi : material transfer rate from the surface layer to interior epithelium
τis : material transfer rate from the interior epithelium to surface layer
NB : number of low E-cadherin cells in the subsurface layer
NB0 : number of low E-cadherin cells in the subsurface layer at swapping equilibrium
NS0 : number of low E-cadherin cells in the surface layer at swapping equilibrium
K : ratio of low E-cadherin cell number in the subsurface layer to surface layer at swapping equilibrium (NB0/NS0)
ΔNS : change of low E-cadherin cells in the surface layer
ΔNB : change of low E-cadherin cells in the subsurface layer
ΔE : free energy difference when a subsurface low E-cadherin cell inserts into the surface layer
Below, we will first derive the boundary condition for the sphere to expand without changing shape (uniform spherical expansion).
The volumes of each compartment:
| (eq. 1) |
| (eq. 2) |
| (eq. 3) |
The cell number in each compartment:
| (eq. 4) |
| (eq. 5) |
The expansion rate of cell numbers in each compartment:
| (eq. 6) |
| (eq. 7) |
Substituting equations (1–5) into (6–7), we get:
| (eq. 8) |
| (eq. 9) |
From definitions 11–12:
| (eq. 10) |
| (eq. 11) |
Substituting equations (8, 9, 11) into (10), we get:
| (eq. 12) |
Equation 12 gives the boundary condition for uniform spherical expansion. When , the interior expands faster than the surface layer and will thus disrupt tissue integrity. Conversely, when , the surface layer expands faster and will promote budding.
We could experimentally quantify the apparent cell proliferation rates in each compartment (PIA and PSA) by immunostaining for a mitosis marker (phospho-Histone H3) and measuring the cross-sectional area of interior epithelium (S) using E-cadherin staining. In order to estimate α and β from experimentally measurable values, we derive the following parameter relationships.
From definitions 13–20:
| (eq. 13) |
| (eq. 14) |
| (eq. 15) |
| (eq. 16) |
From assumptions 4–8:
| (eq. 17) |
| (eq. 18) |
| (eq. 19) |
From assumptions 5, 9 and definitions 24–27:
| (eq. 20) |
| (eq. 21) |
| (eq. 22) |
Both the loss of surface cells and increase of new subsurface cells contribute to τis through re-distribution of low E-cadherin cells between surface and subsurface layers to achieve equilibrium, so:
| (eq. 23) |
Substituting equations (13–19, 23) into (10):
| (eq. 24) |
Substituting equations (1–3, 11) into equation 15:
| (eq. 25) |
We could use equation 24 to estimate α from experimentally measurable values as follows.
First, from experimental observation of the surface cell residence ratio under normal conditions (0.930), the estimated K = 13.285.
Second, the interior-to-surface volume ratio can be calculated from the size ratio using equation 25. To estimate the geometrical ratio , we measured the middle cross-sectional area of the interior epithelium S and estimated the horizontal interior radius as the radius of a circle with area S. The surface layer thickness is consistently ~15 μm. Thus, we have:
| (eq. 26) |
| (eq. 27) |
Third, to estimate the apparent cell proliferation ratio , we immunostained salivary glands with the mitosis marker phospho-Histone H3 (pHH3) and estimated as the interior-to-surface ratio of normalized pHH3-positive areas.
Substituting the above estimated values to equation 24 to calculate :
| (eq. 28) |
Parameter Changes in Experimental Perturbations
α is the interior-to-surface volume expansion ratio. Intuitively, a smaller value of α below the boundary condition of uniform spherical expansion (given by equation 12) promotes extra surface production and causes budding. From equation (24), we can see that α is positively correlated with ω, δ and ΔE, which are the interior-to-surface apparent proliferation ratio (ω), the interior-to-surface volume ratio (δ) and the free energy difference when a subsurface low E-cadherin cell inserts into the surface layer (ΔE).
When a subsurface cell inserts into the surface layer, the following changes will cost free energy:
-
1
To stretch and bend the basement membrane (E1).
-
2
To deform and move neighboring cells (E2).
-
3
To break contact between the new surface cell and previous neighbors (E3).
-
4
To break the contact opened up between surface cells (E4).
Second, it releases free energy from the following changes:
-
5
To establish contact between previous neighboring cells (E5).
-
6
To establish contact between the new surface cell and other surface cells (E6).
-
7
To establish contact between the new surface cell and basement membrane (E7).
Overall, the free energy difference is:
| (eq. 29) |
Comparing before and after states, most cells have retained the same types and sizes of contacting surface, except for the new surface cell, which has a net loss of cell-cell contact and a net gain of cell-matrix contact that is the size of its bottom (basal) area (SB). Denoting the binding energy between cell-cell or cell-matrix contact per unit area as ecc and ecm, we have:
| (eq. 30) |
| (eq. 31) |
Substituting equations (30–31) to (29), we get:
| (eq. 32) |
Equation 32 can help us to predict and understand the effects of various perturbations:
Down-regulating E-cadherin in peripheral cells reduces ecc and promotes budding.
Integrin blocking reduces ecm and inhibits budding.
Ablating the basement membrane by collagenase reduces ecm close to 0, which makes ΔE positive and thermodynamically not favorable.
Low-concentration collagenase treatment presumably softens the basement membrane, which makes it easier to stretch and thus reduces E1. At the same time, softened matrix has weaker interaction with cells and reduces ecm. In this case, whether ΔE increases or decreases depends on the magnitude of E1 and ecm changes. Our experimental results suggest the reduction of ecm dominates.
Inhibiting MMP activity by BB-94 or GM-6001 makes the basement membrane thicker and harder to stretch, which increases E1 and inhibits budding. It is important to note that although BB-94 treatment significantly increases the basement membrane thickness, the intrinsic stiffness as measured by its apparent Young’s modulus remains unchanged. Therefore, we do not expect ecm to change.
Supplementary Material
Figure S1 (Related to Figure 1). Live-imaging, cell tracking and surface expansion analysis of mouse embryonic salivary glands. (A) Schematic describing sample preparation for live imaging of wholemount mouse embryonic salivary glands. (B) Different views of a volumetric imaging data set at the first time point. (C) Two-photon microscopy slice showing the middle section of a branching salivary gland. (D) Salivary gland epithelial tracks at 20–22 hours of the time-lapse sequence color-coded by the mean track speed. (E) Plot of average track speed over distance to surface in 2 μm bins. (F) Two-photon microscopy images and epithelial surface outline color-coded by local curvature of an E13 transgenic mouse salivary gland. (G) Schematics of uniform vs. local expansion of the epithelial surface and their predicted peripheral GFP intensity profile over time. Blue and red dots indicate pre-existing and newly added surface cells, respectively. (H) Inverted histone-EGFP image from the same image in (F) at 0 h. The green curved line indicates the positions where peripheral GFP intensities were sampled. (I) Plot of GFP intensity along the green curve in (H). Green dots indicate automatically detected peaks used to quantify nuclear counts. (J-M) Outlines of the epithelial surface at the middle slice (J), heatmaps of the peripheral GFP intensity (K) and curvature (L), and plots of the bud perimeter (M; black) and peripheral nuclear count (M; green) over time. Dashed lines in (M) indicate fitted linear models. Scale bars, 100 μm.
Figure S2 (Related to Figures 2 and 3). Robust surface return of interior daughter cells after cell division of surface epithelial cells and mathematical modeling of budding morphogenesis. (A-B) Schematics and two-photon microscopy image sequences showing the two observed types of surface cell division. The 0 min images are the same as those in Fig. 2A. (C-D) 3D plots of cell tracks following 42 surface cell divisions and the surface return of their daughter cells. (E) 2D plots of cell tracks color-coded by post-division surface-returning time (left) or z-position (right). (F) Bar plots of cell division rates (% of phospho-Histone H3 area) in the surface epithelium (left), interior epithelium (middle) or their ratios (right) at 1, 7, 13, 20 hours of culture. n=7 glands for each time point. Quantifications were performed on maximum intensity projections of a 10 μm z-range. Error bars, 95% confidence intervals. n.s. (all), not significant for all pairwise comparisons. Statistics, Tukey test. (G) Schematic representation of the expansion of a 2-compartment sphere. (H) Schematic of the cell-sorting of an interior low E-cadherin cell to the subsurface level and then inserting into the surface layer. (I) Equation describing the contributions to the free energy difference of a surface-insertion event (ΔE). Note that ΔE<0 under normal conditions due to a preference for cell-matrix contact compared to cell-cell contact by low E-cad cells. (J) Equation describing the relationship between the interior-to-surface ratio of volume expansion rates (α) and the ratio of division-ready cell abundance (ω), the volume ratio (δ) and the free energy difference of a surface-insertion event (ΔE). See the Modeling section in STAR Methods for modeling assumptions and formula derivation steps. (K) Left: confocal immunofluorescence image of an E13 salivary gland. Anti-phospho-Histone H3 (pHH3) marks mitotic cells, and anti-collagen IV marks the basement membrane. Middle: a thresholded pHH3 image to illustrate how experimentally measurable parameters were quantified. Right: equations to calculate modeling parameters. (L) Plot of estimated interior-to-surface expansion ratio (α) over the interior-to-surface size ratio (β) for 12 epithelial buds from 3 submandibular salivary glands (SMG). Uniform spherical expansion requires the parameter pair to fall along the blue line marking the boundary condition. Note that all observed parameter pairs fall into the domain of faster surface expansion, which predicts budding. Scale bars in (A), 20 μm; (K), 100 μm.
Figure S3 (Related to Figures 2 and 3). Model testing by experimental perturbation of cell-matrix interaction strength or basement membrane properties. (A) Phase contrast images (left) and bud ratio plot (right) of E13 salivary glands treated with control IgG and IgM or a combination of α6-integrin and β1-integrin antibodies. n=5 glands each group. (B) Phase contrast images (left) and bud ratio plot (right) of E13 salivary glands treated with the matrix metalloproteinase inhibitors BB-94 (5 μM) or GM-6001 (25 μM). n=10, 5, 5 glands for control, BB-94 and GM-6001. (C) Confocal images showing the basement membrane components laminin and collagen IV in control, BB-94 or GM-6001 treated E13 salivary glands. Small insets in red boxes show that laminin staining in the control is almost invisible if image acquisition and scaling are optimized for the inhibitor-treated groups. (D) Phase contrast images (left) and bud ratio plot (right) of E13 salivary glands treated with indicated concentrations of collagenase. n=10, 10, 11, 8 glands for 0, 0.02, 0.2 and 2 μg/mL collagenase. (E) Confocal images showing the basement membrane component collagen IV in E13 salivary glands treated with increasing concentrations of collagenase. The lower boxes in (A, B, D) show how perturbations change the modeling parameters and cause observed inhibitory effects on budding and branching of salivary glands. ecm: binding energy per unit area between cell and the basement membrane matrix. ΔE: the free energy difference of a surface-insertion event. α: the interior-to-surface ratio of volume expansion rates. SB: new cell-matrix contact area in a surface-insertion event. E1: energy cost to stretch the basement membrane. Error bars, 95% confidence intervals. p-values in (A), two-tailed t-test; others, Tukey test. Scale bars, 100 μm.
Figure S4 (Related to Figure 4). Expression patterns of selected marker genes from the scRNA-seq data set. (A-B) Scatter plots in UMAP embedding (left) and confocal images of smFISH staining (right) of Sox10 (B) or Sox2 (C). (C) Confocal immunofluorescence images highlighting the inner bud marker Claudin10 (encoded by the Cldn10 gene). (D) The estimated ratio of numbers of outer bud cells vs. inner bud cells in E13 salivary gland epithelium is between 2.2 and 6.3. (E) Violin plots showing distributions of expression levels of indicated cadherin or integrin genes in the 7 clusters. (F-J) Two-photon (F) or confocal (G-J) images of salivary glands immunostained with indicated integrin antibodies. Note that β1-integrin staining was performed by 6-day incubation with mesenchyme-free salivary gland cultures to ensure antibody penetration due to high β1-integrin expression throughout the tissue. Scale bar in (D), 100 μm; others, 20 μm.
Figure S5 (Related to Figure 6). Establishing a clonal cell line for inducible modulation of cell adhesions. (A) Schematics illustrating engineering of a clonal Dia-C6 cell line from DLD-1. Dia cells stably express 4 transgenes that enable inducible transcriptional activation or repression. (B) Schematics of 2 PiggyBac vectors expressing 4 transgenes for inducible transcriptional modulation. (C) Schematics of lentivirus vectors used to deliver guide RNAs for transcriptional modulation. (D) Left: schematic of the ABA-inducible transcriptional repression system. Middle: maximum intensity projection of confocal images of cells expressing indicated sgRNAs. Green and magenta nuclei indicate cells expressing sgRNAs. Right: plot of mean E-cadherin intensities of indicated experimental conditions. n=12 fields of view for each group. (E, G, H, I) Western blots showing the expression levels of E-cadherin or β-catenin under the indicated experimental conditions. Percentage levels under each lane are normalized ratios of E-cadherin to GAPDH or β-catenin to α-tubulin intensities of that lane. Normalizations are versus the control group (sg-Control or Day 0, DMSO) from the same blot. (F) Plot of normalized ratios of E-cadherin to GAPDH intensities of Dia cells expressing sg-Control, sg1-Cdh1 or sg2-Cdh1 treated with DMSO or ABA for 5 days. Normalizations are versus the control group (sg-Control, DMSO) for each of the 3 biological replicates. (J) Confocal images of β-catenin immunofluorescence. Error bars in (D, F) are 95% confidence intervals. Scale bars, 100 μm.
Figure S6 (Related to Figure 6). Reducing cell-cell adhesion and inducing basement membrane formation is sufficient to induce budding morphogenesis of a stratified epithelium. (A) Top: schematics of the experimental design. Below: confocal fluorescence images of spheroids for indicated experimental conditions. (B) Left: phase contrast and epifluorescence images of a spheroid. Right: binary mask image segmented from merged green and red fluorescence, and its boundary outline color-coded by the local curvature. Each column of curvature heatmap represents a straightened outline color-coded by curvature. (C) Schematic and table showing experimental conditions of 6 groups. (D-E) Bar plots of bud number or percentage of high-curvature perimeter length (|curvature|>20 mm−1). (F) Heatmap showing color-coded curvature along spheroid perimeters. Each column is one spheroid. Only 21 of 43 Group 4 samples were included in the heatmap to save space. (G) Confocal images of spheroids from indicated experimental conditions immunostained with E-cadherin and laminin (yellow; a basement membrane marker). MIP, maximum intensity projection. (H) Merged phase contrast and epifluorescence images of spheroids using two different sgRNAs under the indicated experimental conditions with different mixing ratios of control and E-cadherin downregulated cells. (I) Plots of three different quantifications for the 8 experimental groups shown in (H). (J) Schematics illustrating the cell sorting and branching of a stratified epithelium with mixed populations of high vs. low E-cadherin-expressing cells. Error bars in all plots, 95% confidence intervals. ***, Tukey test p<0.001 compared to the first group of the plot. Scale bars, 100 μm.
Figure S7 (Related to Figure 7). Reconstituted epithelial budding requires β1-integrin-mediated cell-matrix adhesion. (A) Merged phase contrast and epifluorescence images of spheroids from indicated experimental conditions. The α5-integrin (clone mAb16) and β1-integrin (clone mAb13) blocking antibodies were used at 100 μg/mL. n=16 for each group from 3 independent experiments. (B) Plots of bud number or percent protruded bud area per spheroid for indicated experimental conditions. n=10 per group from the same experiment. (C) Phase contrast images of budded spheroids (sg2-Cdh1 + sg-Control, ABA + Matrigel for 7 days) treated with dispase and recovered for 6 days after washout. n=15 from 2 independent experiments. (D) Plots of bud number or percent protruded bud area per spheroid for groups shown in (C). (E) Maximum intensity projection of confocal immunofluorescence images of 2D-cultured cells expressing indicated sgRNAs. Green nuclei indicate cells expressing the sgRNAs. (F) Plot of mean β1-integrin intensities under the indicated experimental conditions. n=12 fields of view per group. (G) Western blot showing expression levels of β1-integrin under indicated experimental conditions. Percentage levels under each lane are normalized ratios of β1-integrin to GAPDH intensities of that lane. (H) Confocal images of spheroids under indicated experimental conditions. Note that green nuclei here indicate either sg-Control or sg-Itgb1 expression depending on the experimental group. White arrows point to cells that lost their β1-integrin expression. MIP, maximum intensity projection. (I) Schematics illustrating experimental procedures for the cell attachment assays. MG, Matrigel; E-cad-ECD, E-cadherin extracellular domain. (J) Phase contrast images of attached cells under indicated experimental conditions. (K-L) Plots of attached cells under indicated experimental conditions. n=52, 39, 39, 52 fields of view for groups in (I). n=39 fields of view per group in (J). Each experimental group had 3 or 4 biological replicates, and each had 13 fields of view. The full set of experiments was done twice with similar results. (M) Confocal images of laminin or collagen IV immunostaining in control, BB-94 (5 μM) or GM-6001 (25 μM) treated spheroids (7 days post treatment). (N) Left: brightfield image of the experimental setup to perform atomic force microscopy (AFM) on spheroids. The focus is on the AFM tip. Right: example force-indentation plot from a typical AFM measurement. Negative forces are from adhesion between the AFM probe and sample during withdrawal. Scale bars, 100 μm.
Video S1 (Related to Figure 1). Volumetric time-lapse video of a branching embryonic salivary gland. Salivary gland from a 13-day transgenic mouse embryo expressing membrane-tdTomato and histone-EGFP. Time-lapse volumetric two-photon microscopy images were acquired at 2-μm z steps over a 100 μm z range at 5-minute intervals for 20 hours. The first half of the video shows different views of the imaging volume at the first time point. The second half of the video shows time-lapse sequences of the middle x-y plane (left) or 3D rendering of the middle 40 μm-thick volume of a branching epithelial bud (right).
Video S2 (Related to Figure 1). Dynamics of surface-layer epithelial cells highlighted by KikGR photoconversion. Salivary gland from a 13-day transgenic mouse embryo expressing the photoconvertible fluorescent protein KikGR. Native KikGR has green fluorescence, which converts to red fluorescence after blue light excitation. Magenta color indicates photoconverted KikGR.
Video S3 (Related to Figure 1). Surface-proximal 3D cell migration tracks. Videos show only epithelial cell tracks for which the closest distance to the gland surface was ≤ 15 μm. White dots indicate cell centroids, and the tracks shown at each time point represent cell migration trajectories spanning the previous 2 hours. Tracks are color coded by mean track speed (left) or the z position (right).
Video S4 (Related to Figures 1 and 2). Different views of a branching epithelial bud from a developing salivary gland. Salivary gland from a 13-day transgenic mouse embryo expressing histone-EGFP and a heterozygous epithelial RFP reporter driven by the Krt14 promoter (Krt14p::RFP). Note that the surface-layer epithelial cells expressing high RFP comprise a cell sheet that folds inward during clefting.
Video S5 (Related to Figure 2). 3D tracking of daughter cells from surface-derived cell divisions. This video has 2 sequential parts. The first part shows “tracked cell highlighted” (left) and “tracked cell centered” (right) styles of an example cell tracking. The second part shows tracking of 16 pairs of daughter cells in the “tracked cell centered” style. Tracked cell is marked by a white dot. Distance indicated is from the coverslip. Time indicates minutes from the onset of anaphase (0 min).
Video S6 (Related to Figure 6). Time-lapse videos of 3D spheroid cultures of engineered DLD-1 cells under indicated experimental conditions. Images are merged phase contrast and epifluorescence channels. Green and magenta indicate cells expressing sg-Control or sg-Cdh1, respectively. Abscisic acid (ABA) is a dimerizer used to induce robust transcriptional repression in engineered DLD-1 cells. DMSO is the vehicle control. Live imaging started 5 days post Matrigel and ABA or DMSO treatment.
Video S7 (Related to Figure 6). Time-lapse videos of a budding spheroid culture of engineered DLD-1 cells. Images in the main window are maximum intensity projections of confocal fluorescence images, and images in the upper left inset are single-slice confocal fluorescence images. Green and magenta indicate cells expressing sg-Control or sg2-Cdh1, respectively. Yellow indicates the basement membrane marked by Atto-647N labeled fibronectin. Arrows and arrowheads indicate forming clefts. Live imaging started 4 days post Matrigel and ABA treatment.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-E-cadherin (clone ECCD-2) | Thermo Fisher | 13-1900 |
| Rat monoclonal anti-β1-integrin (clone mAb13) | MilliporeSigma | MABT821 |
| Rat monoclonal anti-α5-integrin (clone mAb16) | MilliporeSigma | MABT820 |
| Goat polyclonal anti-collagen type IV | MilliporeSigma | AB769 |
| Rabbit polyclonal anti-laminin | MilliporeSigma | L9393 |
| Rabbit polyclonal anti-histone H3 (phospho S10) | Abcam | ab47297 |
| Rabbit monoclonal anti-β-catenin (clone E247) | Abcam | ab32572 |
| Rabbit Claudin 10 Polyclonal Antibody | Thermo Fisher | 38-8400 |
| Rabbit monoclonal anti-GAPDH (clone 14C10) | Cell Signaling | 2118L |
| Mouse monoclonal anti-α-tubulin (clone DM1α) | MilliporeSigma | T6199 |
| Rat Anti-Human CD49f α6-integrin (clone GoH3) | BD Biosciences | 555734 |
| Rat IgG2a, κ Isotype Control (clone A110-2) | BD Biosciences | 553992 |
| Hamster Anti-Rat CD29 β1-integrin (clone Ha2/5) | BD Biosciences | 555002 |
| Hamster IgM, λ1 Isotype Control (clone G235-1) | BD Biosciences | 553957 |
| Rat monoclonal anti-KRT19 | DSHB | TROMA-III |
| Goat polyclonal anti-α9-integrin | R&D Systems | AF3827-SP |
| DyLight 405 donkey anti-rabbit | Jackson ImmunoResearch | 711-476-152 |
| Cy2 donkey anti-goat | Jackson ImmunoResearch | 705-225-147 |
| Alexa Fluor 488 donkey anti-rat | Jackson ImmunoResearch | 712-546-153 |
| Rhodamine Red-X donkey anti-rat | Jackson ImmunoResearch | 712-296-153 |
| Rhodamine Red-X donkey anti-rabbit | Jackson ImmunoResearch | 711-296-152 |
| Alexa Fluor 647 donkey anti-rat | Jackson ImmunoResearch | 712-606-153 |
| Alexa Fluor 647 donkey anti-rabbit | Jackson ImmunoResearch | 711-606-152 |
| 680RD goat anti-rabbit | LI-COR | 926-68071 |
| 800CW goat anti-rat | LI-COR | 926-32219 |
| 680RD goat anti-mouse | LI-COR | 926-68070 |
| 800CW goat anti-rabbit | LI-COR | 926-32211 |
| Chemicals, peptides, and recombinant proteins | ||
| NHS-Atto565 | ATTO-TEC | AD 565-31 |
| NHS-Atto647N | ATTO-TEC | AD 647N-31 |
| NHS-AF680 | Thermo Fisher | A20008 |
| L-Ascorbic Acid (Vitamin C) | MilliporeSigma | A7506 |
| Transferrin | MilliporeSigma | T8158 |
| Recombinant Mouse KGF/FGF-7 Protein | R&D Systems | 5028-KG-025 |
| Recombinant Mouse Neuregulin-1/NRG1 Protein | R&D Systems | 9875-NR-050 |
| Recombinant Human E-Cadherin Protein (E-cad-ECD; Asp155-Ile707, with a C-terminal 6-His tag) | R&D Systems | 8505-EC-050 |
| Critical commercial assays | ||
| Chromium Single Cell 3′ Library & Gel Bead Kit v2 | 10x Genomics | PN-120237 |
| Lenti-X GoStix Plus | Takara | 631281 |
| CytoVista Tissue Clearing Reagent | Thermo Fisher | V11315 |
| Dialysis Cassettes, 10K MWCO, 0.5 mL | Thermo Fisher | 66383 |
| Zeba Spin Desalting Columns, 7K MWCO, 5 mL | Thermo Fisher | 89891 |
| Zeba Spin Desalting Columns, 7K MWCO, 0.5 mL | Thermo Fisher | 89882 |
| Deposited data | ||
| Customized Python, Jython and ImageJ Macro Scripts for data analysis and plotting | This paper | https://github.com/snownontrace/public-scripts-Wang2020-branching-morphogenesis |
| Collection of raw data and source data tables (for all plots) of this paper | This paper | Figshare: https://doi.org/10.35092/yhjc.c.5404533 |
| Source data tables for all plots of this paper | This paper | Figshare: https://doi.org/10.35092/yhjc.12145626 |
| scRNA-seq dataset | This paper | GEO: GSE159780 |
| Experimental models: Cell lines | ||
| 293T | Takara | 632273 |
| DLD-1 (parental) | ATCC | CCL-221 |
| Dia-C6 (DLD-1 expressing 4 transgenes for inducible transcriptional modulation) | This paper | N/A |
| D188 (Dia-C6 with lenti-sg-Control and NLS-mScarlet-I) | This paper | N/A |
| D193 (Dia-C6 with lenti-sg-Control and NLS-mNeonGreen) | This paper | N/A |
| D266 (Dia-C6 with lenti-sg2-Cdh1 and NLS-mScarlet-I) | This paper | N/A |
| D267 (Dia-C6 with lenti-sg1-Cdh1 and NLS-mScarlet-I) | This paper | N/A |
| D301 (Dia-C6 with lenti-sg-Itgb1 and NLS-mNeonGreen) | This paper | N/A |
| D2 (Dia-C6 with lenti-sg1-Cdh1, NLS-mScarlet-I, lenti-sg-Itgb1 and NLS-mNeonGreen) | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: mTmG/HisG; FVB/N(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo Tg(HIST1H2BB-EGFP)1Pa/J | Huebner et al., 2014 | N/A |
| Mouse: K14RFP/HisG; FVB/N(Cg)-Tg(KRT14-RFP)#Efu Tg(HIST1H2BB-EGFP)1Pa/J | This paper | N/A |
| Mouse: KikGR; FVB/N(Cg)-Tg(KikGR)/N | Hsu et al., 2013 | N/A |
| Mouse: ICR (CD-1) outbred mice | Envigo | Hsd:ICR(CD-1) |
| Recombinant DNA | ||
| Plasmid: Super piggyBac Transposase expression vector | System Biosciences | PB210PA-1 |
| Plasmid: pSLQ2818 (PiggyBac-ABA-CRISPRi) | Gao et al., 2016 | Addgene: 84241 |
| Plasmid: pMD2.G | Didier Trono, unpublished | Addgene: 12259 |
| Plasmid: psPAX2 | Didier Trono, unpublished | Addgene: 12260 |
| Plasmid: pW210 (pPB-GA-inducible-CRISPRa-saCas9-VPR-hygR) | This paper | Addgene: 170808 |
| Plasmid: pW211 (lenti-spsg-pEF1s-NLS-mNeonGreen-P2A-BlastR) | This paper | Addgene: 170809 |
| Plasmid: pW212 (lenti-spsg-pEF1s-NLS-mScarlet-I-P2A-BlastR) | This paper | Addgene: 170810 |
| Plasmid: pW213 (lenti-sasg-pEF1s-NLS-mNeonGreen-P2A-BlastR) | This paper | Addgene: 170811 |
| Plasmid: pW214 (lenti-sasg-pEF1s-NLS-mScarlet-I-P2A-BlastR) | This paper | Addgene: 170812 |
| Plasmid: pW188 (lenti-spsgRNA-lacZ-pEF1s-NLS-mScarlet-I-P2A-BlastR) | This paper | Addgene: 170813 |
| Plasmid: pW193 (lenti-sasgRNA-lacZ-pEF1s-NLS-mNeonGreen-P2A-BlastR) | This paper | Addgene: 170814 |
| Plasmid: pW266 (lenti-spsgRNA-sg2-hsCDH1-pEF1s-NLS-mScarlet-I-P2A-BlastR) | This paper | Addgene: 170815 |
| Plasmid: pW267 (lenti-spsgRNA-sg1-hsCDH1-pEF1s-NLS-mScarlet-I-P2A-BlastR) | This paper | Addgene: 170816 |
| Plasmid: pW301 (lenti-spsgRNA-hsITGB1-sp-160rev-pEF1s-NLS-mNeonGreen-P2A-BlastR) | This paper | Addgene: 170817 |
| Software and algorithms | ||
| ImageJ (Fiji) | Schindelin et al., 2012 | https://fiji.sc |
| Imaris 9.5.0 | Bitplane | https://www.imaris.com |
| TrackMate | Tinevez et al., 2017 | https://imagej.net/TrackMate |
| MorphoLibJ | Legland et al., 2016 | https://imagej.net/MorphoLibJ |
| Cellpose | Stringer et al., 2021 | http://www.cellpose.org/ |
| Cell Ranger | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Scanpy | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/stable/ |
| Velocyto | La Manno et al., 2018 | http://velocyto.org/velocyto.py/index.html |
| scVelo | Bergen et al., 2020 | https://scvelo.readthedocs.io/ |
| CRISPOR | Haeussler et al., 2016 | http://crispor.org/ |
| smFISH quantification macros | Wang, 2019 | N/A |
| Other | ||
| Matrigel, Growth Factor Reduced Basement Membrane Matrix | Corning | 356231 |
| Laminin I, Cultrex 3D Culture Matrix (contains low amount of collagen IV) | Trevigen | 3446-005-01 |
| Accutase | Innovative Cell Technologies | AT104 |
| Dispase II | Thermo Fisher | 17105041 |
| Collagenase | Elastin Products Company | CL103 |
| 13 mm diameter 0.1 μm pore polycarbonate filters (for organ culture) | MilliporeSigma | WHA110405 |
| 50 mm MatTek dish with 14 mm opening (for organ culture) | MatTek | P50G-1.5-14-F |
| 50 mm MatTek dish with 30 mm opening (for live-organ imaging) | MatTek | P50G-1.5-30-F |
| MatTek glass-bottom 6-well plates | MatTek | P06G-0-14-F |
| ibidi μ-Dish 35 mm, with 4-well silicone culture-insert | ibidi | 80466 |
| ibidi μ-Dish 35 mm, with Bioinert surface | ibidi | 81150 |
| CellTak tissue adhesive | Corning | 354240 |
| FluoroDish for AFM measurements | World Precision Instruments | FD35-100 |
| Pyrex 9-well glass spot plate | Fisher Scientific | 13-748B |
| Dumont #5 - Forceps | Fine Science Tools | 11251-20 |
| Dumont #5 - Fine Forceps | Fine Science Tools | 11254-20 |
| Scalpel Handle - #3 | Fine Science Tools | 10003-12 |
| Scalpel Blades - #11 | Fine Science Tools | 10011-00 |
| Tungsten Needles | Fine Science Tools | 10130-05 |
| Moria Nickel Plated Pin Holder (use with Tungsten Needles) | Fine Science Tools | 26016-12 |
Highlights.
Single-cell tracking and RNA-seq identify mechanisms driving budding morphogenesis
In stratified epithelia, the surface cell sheet can expand and fold to form buds
A combination of strong cell-matrix and weak cell-cell adhesions is critical
Applying these principles permits synthetic reconstitution of budding morphogenesis
Acknowledgments
We thank all members of the Yamada Laboratory for helpful discussions, D. Wu (NIDDK), J. Lu (Shandong University), A. Desai (UCSD), R.A. Green (UCSD), and K. Oegema (UCSD) for critical reading of the manuscript, NIDCR Imaging Core, Genomics and Computational Biology Core, Combined Technical Research Core and Veterinary Resource Core for support, S. Qi (Stanford), F. Zhang (MIT), and D. Trono (EPFL) for plasmids, M.P. Hoffman (NIDCR), E. Fuchs (Rockefeller) and A.J. Ewald (JHU) for mice. Image analysis was performed in part using the NIH High Performance Computing system. This work was supported by the NIH Intramural Research Program (NIDCR, ZIA DE000525; NIBIB, ZIA EB000094). A.X.C.-R. and S.R.L were also supported by the NIH Distinguished Scholars Program. S.W. was supported in part by an NIDCR K99 Pathway to Independence Award (K99 DE27982).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Affolter M, Zeller R, and Caussinus E (2009). Tissue remodelling through branching morphogenesis. Nat. Rev. Mol. Cell Biol. 10, 831–842. [DOI] [PubMed] [Google Scholar]
- Akiyama SK (1999). Purification of fibronectin. Curr. Protoc. Cell Biol. 2, 1–13. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Shih DT, Buck CA, and Hemler ME (1995). Monoclonal antibody 9EG7 defines a novel β1 integrin epitope induced by soluble ligand and manganese, but inhibited by calcium. J. Biol. Chem. 270, 25570–25577. [DOI] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. [DOI] [PubMed] [Google Scholar]
- Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, and Cano A (2003). The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 116, 499–511. [DOI] [PubMed] [Google Scholar]
- Cerchiari AE, Garbe JC, Jee NY, Todhunter ME, Broaders KE, Peehl DM, Desai TA, LaBarge MA, Thomson M, and Gartner ZJ (2015). A strategy for tissue self-organization that is robust to cellular heterogeneity and plasticity. Proc. Natl. Acad. Sci. U. S. A. 112, 2287–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini F, and Kopan R (2010). Patterning a complex organ: Branching morphogenesis and nephron segmentation in kidney development. Dev. Cell 18, 698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Janmey P, and Wang Y-L (2005). Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139–1143. [DOI] [PubMed] [Google Scholar]
- Ewald AJ, Brenot A, Duong M, Chan BS, and Werb Z (2008). Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell 14, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Xiong X, Wong S, Charles EJ, Lim WA, and Qi LS (2016). Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods 13, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Goodwin K, Mao S, Guyomar T, Miller E, Radisky DC, Košmrlj A, and Nelson CM (2019). Smooth muscle differentiation shapes domain branches during mouse lung development. Development 146, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobstein C, and Cohen J (1965). Collagenase: effect on the morphogenesis of embryonic salivary epithelium in vitro. Science 150, 626–628. [DOI] [PubMed] [Google Scholar]
- Hadjantonakis AK, and Papaioannou VE (2004). Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Biotechnol. 4, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud J-B, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harunaga JS, Doyle AD, and Yamada KM (2014). Local and global dynamics of the basement membrane during branching morphogenesis require protease activity and actomyosin contractility. Dev. Biol. 394, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JC, Koo H, Harunaga JS, Matsumoto K, Doyle AD, and Yamada KM (2013). Region-specific epithelial cell dynamics during branching morphogenesis. Dev. Dyn. 242, 1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner RJ, Lechler T, and Ewald AJ (2014). Developmental stratification of the mammary epithelium occurs through symmetry-breaking vertical divisions of apically positioned luminal cells. Development 141, 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AJ, Miyazaki H, Coyle MC, Zhang J, Laurie MT, Chu D, Vavrušová Z, Schneider RA, Klein OD, and Gartner ZJ (2018). Engineered Tissue Folding by Mechanical Compaction of the Mesenchyme. Dev. Cell 44, 165–178.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Pang M-F, Varner VD, Kojima L, Miller E, Radisky DC, and Nelson CM (2015). Localized smooth muscle differentiation is essential for epithelial bifurcation during branching morphogenesis of the mammalian lung. Dev. Cell 34, 719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman HK, McGarvey ML, Hassell JR, Star VL, Cannon FB, Laurie GW, and Martin GR (1986). Basement membrane complexes with biological activity. Biochemistry 25, 312–318. [DOI] [PubMed] [Google Scholar]
- Larsen M, Wei C, and Yamada KM (2006). Cell and fibronectin dynamics during branching morphogenesis. J. Cell Sci. 119, 3376–3384. [DOI] [PubMed] [Google Scholar]
- Legland D, Arganda-Carreras I, and Andrey P (2016). MorphoLibJ: Integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32, 3532–3534. [DOI] [PubMed] [Google Scholar]
- Li Y, and Burridge K (2019). Cell-Cycle-Dependent Regulation of Cell Adhesions: Adhering to the Schedule. BioEssays 41, 1800165. [DOI] [PubMed] [Google Scholar]
- Lombaert IMA, and Hoffman MP (2010). Epithelial stem/progenitor cells in the embryonic mouse submandibular gland. Front. Oral Biol. 14, 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Nakao A, Inaba T, Murakami-Sekimata A, and Nogawa H (2017). Morphogenesis and Mucus Production of Epithelial Tissues of Three Major Salivary Glands of Embryonic Mouse in 3D Culture. Zoolog. Sci. 34, 475–483. [DOI] [PubMed] [Google Scholar]
- Nelson CM (2016). On buckling morphogenesis. J. Biomech. Eng. 138, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerger BA, Jaslove JM, Elashal HE, Mao S, Košmrlj A, Link AJ, and Nelson CM (2021). Local accumulation of extracellular matrix regulates global morphogenetic patterning in the developing mammary gland. Curr. Biol. 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, Yaswen P, Werb Z, and Ewald AJ (2012). ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. U. S. A. 109, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogawa H, and Ito T (1995). Branching morphogenesis of embryonic mouse lung epithelium in mesenchyme-free culture. Development 121, 1015–1022. [DOI] [PubMed] [Google Scholar]
- Nogawa H, and Takahashi Y (1991). Substitution for mesenchyme by basement-membrane-like substratum and epidermal growth factor in inducing branching morphogenesis of mouse salivary epithelium. Development 112, 855–861. [DOI] [PubMed] [Google Scholar]
- Packard A, Georgas K, Michos O, Riccio P, Cebrian C, Combes AN, Ju A, Ferrer-Vaquer A, Hadjantonakis AK, Zong H, et al. (2013). Luminal Mitosis Drives Epithelial Cell Dispersal within the Branching Ureteric Bud. Dev. Cell 27, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, and Ewald AJ (2019). E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VN, Rebustini IT, and Hoffman MP (2006). Salivary gland branching morphogenesis. Differentiation. 74, 349–364. [DOI] [PubMed] [Google Scholar]
- Pronobis MI, Deuitch N, and Peifer M (2016). The Miraprep: A protocol that uses a Miniprep kit and provides Maxiprep yields. PLoS One 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, and Lim WA (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin S. a, van Oudenaarden A, and Tyagi S (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebustini IT, Patel VN, Stewart JS, Layvey A, Georges-Labouesse E, Miner JH, and Hoffman MP (2007). Laminin α5 is necessary for submandibular gland epithelial morphogenesis and influences FGFR expression through β1 integrin signaling. Dev. Biol. 308, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl A, Schlederer M, Pudelko K, Stadler M, Walter S, Unterleuthner D, Unger C, Kramer N, Hengstschläger M, Kenner L, et al. (2017). Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT–mTOR–S6K signaling and drug responses. J. Cell Sci. 130, 203–218. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnatwinkel C, and Niswander L (2013). Multiparametric image analysis of lung-branching morphogenesis. Dev. Dyn. 242, 622–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi R, and Hauser B (2019). Preparation of Cells from Embryonic Organs for Single-Cell RNA Sequencing. Curr. Protoc. Cell Biol. 83, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira SJ, Gervais EM, Ray S, and Larsen M (2013). Genetic modification and recombination of salivary gland organ cultures. J. Vis. Exp. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Wang A, and Sander M (2013). Pancreas organogenesis: from lineage determination to morphogenesis. Annu. Rev. Cell Dev. Biol. 29, 81–105. [DOI] [PubMed] [Google Scholar]
- Shih HP, Panlasigui D, Cirulli V, and Sander M (2016). ECM signaling regulates collective cellular dynamics to control pancreas branching morphogenesis. Cell Rep. 14, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg MS (1963). Reconstruction of tissues by dissociated cells. Science 141, 401–408. [DOI] [PubMed] [Google Scholar]
- Steinberg Z, Myers C, Heim VM, Lathrop CA, Rebustini IT, Stewart JS, Larsen M, and Hoffman MP (2005). FGFR2b signaling regulates ex vivo submandibular gland epithelial cell proliferation and branching morphogenesis. Development 132, 1223–1234. [DOI] [PubMed] [Google Scholar]
- Stemmler MP, Eccles RL, Brabletz S, and Brabletz T (2019). Non-redundant functions of EMT-TFs. Nat. Cell Biol. 21, 102–112. [DOI] [PubMed] [Google Scholar]
- Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, and Hyman AA (2011). Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 469, 226–230. [DOI] [PubMed] [Google Scholar]
- Stringer C, Wang T, Michaelos M, and Pachitariu M (2021). Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106. [DOI] [PubMed] [Google Scholar]
- Szymaniak AD, Mi R, McCarthy SE, Gower AC, Reynolds TL, Mingueneau M, Kukuruzinska M, and Varelas X (2017). The hippo pathway effector YAP is an essential regulator of ductal progenitor patterning in the mouse submandibular gland. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Karasawa S, Shimizu H, Nukina N, and Miyawaki A (2005). Semi-rational engineering of a coral fluorescent protein into an efficient highlighter. EMBO Rep. 6, 233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JL, Menko AS, Khalil S, Rebustini I, Hoffman MP, Kreidberg JA, and Kukuruzinska MA (2008). Diverse roles of E-cadherin in the morphogenesis of the submandibular gland: insights into the formation of acinar and ductal structures. Dev. Dyn. 237, 3128–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S (2019). Single Molecule RNA FISH (smFISH) in Whole-Mount Mouse Embryonic Organs. Curr. Protoc. Cell Biol. 83, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sekiguchi R, Daley WP, and Yamada KM (2017). Patterned cell and matrix dynamics in branching morphogenesis. J. Cell Biol. 216, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Larsen M, Hoffman MP, and Yamada KM (2007). Self-Organization and Branching Morphogenesis of Primary Salivary Epithelial Cells. Tissue Eng. 13, 721–735. [DOI] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RE, Jones MK, Hines EA, Li R, Luo Y, Shi W, Verheyden JM, and Sun X (2020). Smooth Muscle Differentiation Is Essential for Airway Size, Tracheal Cartilage Segmentation, but Dispensable for Epithelial Branching. Dev. Cell 53, 73–85.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Stokes N, Polak L, and Fuchs E (2011). Specific microRNAs are preferentially expressed by skin stem cells to balance self-renewal and early lineage commitment. Cell Stem Cell 8, 294–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (Related to Figure 1). Live-imaging, cell tracking and surface expansion analysis of mouse embryonic salivary glands. (A) Schematic describing sample preparation for live imaging of wholemount mouse embryonic salivary glands. (B) Different views of a volumetric imaging data set at the first time point. (C) Two-photon microscopy slice showing the middle section of a branching salivary gland. (D) Salivary gland epithelial tracks at 20–22 hours of the time-lapse sequence color-coded by the mean track speed. (E) Plot of average track speed over distance to surface in 2 μm bins. (F) Two-photon microscopy images and epithelial surface outline color-coded by local curvature of an E13 transgenic mouse salivary gland. (G) Schematics of uniform vs. local expansion of the epithelial surface and their predicted peripheral GFP intensity profile over time. Blue and red dots indicate pre-existing and newly added surface cells, respectively. (H) Inverted histone-EGFP image from the same image in (F) at 0 h. The green curved line indicates the positions where peripheral GFP intensities were sampled. (I) Plot of GFP intensity along the green curve in (H). Green dots indicate automatically detected peaks used to quantify nuclear counts. (J-M) Outlines of the epithelial surface at the middle slice (J), heatmaps of the peripheral GFP intensity (K) and curvature (L), and plots of the bud perimeter (M; black) and peripheral nuclear count (M; green) over time. Dashed lines in (M) indicate fitted linear models. Scale bars, 100 μm.
Figure S2 (Related to Figures 2 and 3). Robust surface return of interior daughter cells after cell division of surface epithelial cells and mathematical modeling of budding morphogenesis. (A-B) Schematics and two-photon microscopy image sequences showing the two observed types of surface cell division. The 0 min images are the same as those in Fig. 2A. (C-D) 3D plots of cell tracks following 42 surface cell divisions and the surface return of their daughter cells. (E) 2D plots of cell tracks color-coded by post-division surface-returning time (left) or z-position (right). (F) Bar plots of cell division rates (% of phospho-Histone H3 area) in the surface epithelium (left), interior epithelium (middle) or their ratios (right) at 1, 7, 13, 20 hours of culture. n=7 glands for each time point. Quantifications were performed on maximum intensity projections of a 10 μm z-range. Error bars, 95% confidence intervals. n.s. (all), not significant for all pairwise comparisons. Statistics, Tukey test. (G) Schematic representation of the expansion of a 2-compartment sphere. (H) Schematic of the cell-sorting of an interior low E-cadherin cell to the subsurface level and then inserting into the surface layer. (I) Equation describing the contributions to the free energy difference of a surface-insertion event (ΔE). Note that ΔE<0 under normal conditions due to a preference for cell-matrix contact compared to cell-cell contact by low E-cad cells. (J) Equation describing the relationship between the interior-to-surface ratio of volume expansion rates (α) and the ratio of division-ready cell abundance (ω), the volume ratio (δ) and the free energy difference of a surface-insertion event (ΔE). See the Modeling section in STAR Methods for modeling assumptions and formula derivation steps. (K) Left: confocal immunofluorescence image of an E13 salivary gland. Anti-phospho-Histone H3 (pHH3) marks mitotic cells, and anti-collagen IV marks the basement membrane. Middle: a thresholded pHH3 image to illustrate how experimentally measurable parameters were quantified. Right: equations to calculate modeling parameters. (L) Plot of estimated interior-to-surface expansion ratio (α) over the interior-to-surface size ratio (β) for 12 epithelial buds from 3 submandibular salivary glands (SMG). Uniform spherical expansion requires the parameter pair to fall along the blue line marking the boundary condition. Note that all observed parameter pairs fall into the domain of faster surface expansion, which predicts budding. Scale bars in (A), 20 μm; (K), 100 μm.
Figure S3 (Related to Figures 2 and 3). Model testing by experimental perturbation of cell-matrix interaction strength or basement membrane properties. (A) Phase contrast images (left) and bud ratio plot (right) of E13 salivary glands treated with control IgG and IgM or a combination of α6-integrin and β1-integrin antibodies. n=5 glands each group. (B) Phase contrast images (left) and bud ratio plot (right) of E13 salivary glands treated with the matrix metalloproteinase inhibitors BB-94 (5 μM) or GM-6001 (25 μM). n=10, 5, 5 glands for control, BB-94 and GM-6001. (C) Confocal images showing the basement membrane components laminin and collagen IV in control, BB-94 or GM-6001 treated E13 salivary glands. Small insets in red boxes show that laminin staining in the control is almost invisible if image acquisition and scaling are optimized for the inhibitor-treated groups. (D) Phase contrast images (left) and bud ratio plot (right) of E13 salivary glands treated with indicated concentrations of collagenase. n=10, 10, 11, 8 glands for 0, 0.02, 0.2 and 2 μg/mL collagenase. (E) Confocal images showing the basement membrane component collagen IV in E13 salivary glands treated with increasing concentrations of collagenase. The lower boxes in (A, B, D) show how perturbations change the modeling parameters and cause observed inhibitory effects on budding and branching of salivary glands. ecm: binding energy per unit area between cell and the basement membrane matrix. ΔE: the free energy difference of a surface-insertion event. α: the interior-to-surface ratio of volume expansion rates. SB: new cell-matrix contact area in a surface-insertion event. E1: energy cost to stretch the basement membrane. Error bars, 95% confidence intervals. p-values in (A), two-tailed t-test; others, Tukey test. Scale bars, 100 μm.
Figure S4 (Related to Figure 4). Expression patterns of selected marker genes from the scRNA-seq data set. (A-B) Scatter plots in UMAP embedding (left) and confocal images of smFISH staining (right) of Sox10 (B) or Sox2 (C). (C) Confocal immunofluorescence images highlighting the inner bud marker Claudin10 (encoded by the Cldn10 gene). (D) The estimated ratio of numbers of outer bud cells vs. inner bud cells in E13 salivary gland epithelium is between 2.2 and 6.3. (E) Violin plots showing distributions of expression levels of indicated cadherin or integrin genes in the 7 clusters. (F-J) Two-photon (F) or confocal (G-J) images of salivary glands immunostained with indicated integrin antibodies. Note that β1-integrin staining was performed by 6-day incubation with mesenchyme-free salivary gland cultures to ensure antibody penetration due to high β1-integrin expression throughout the tissue. Scale bar in (D), 100 μm; others, 20 μm.
Figure S5 (Related to Figure 6). Establishing a clonal cell line for inducible modulation of cell adhesions. (A) Schematics illustrating engineering of a clonal Dia-C6 cell line from DLD-1. Dia cells stably express 4 transgenes that enable inducible transcriptional activation or repression. (B) Schematics of 2 PiggyBac vectors expressing 4 transgenes for inducible transcriptional modulation. (C) Schematics of lentivirus vectors used to deliver guide RNAs for transcriptional modulation. (D) Left: schematic of the ABA-inducible transcriptional repression system. Middle: maximum intensity projection of confocal images of cells expressing indicated sgRNAs. Green and magenta nuclei indicate cells expressing sgRNAs. Right: plot of mean E-cadherin intensities of indicated experimental conditions. n=12 fields of view for each group. (E, G, H, I) Western blots showing the expression levels of E-cadherin or β-catenin under the indicated experimental conditions. Percentage levels under each lane are normalized ratios of E-cadherin to GAPDH or β-catenin to α-tubulin intensities of that lane. Normalizations are versus the control group (sg-Control or Day 0, DMSO) from the same blot. (F) Plot of normalized ratios of E-cadherin to GAPDH intensities of Dia cells expressing sg-Control, sg1-Cdh1 or sg2-Cdh1 treated with DMSO or ABA for 5 days. Normalizations are versus the control group (sg-Control, DMSO) for each of the 3 biological replicates. (J) Confocal images of β-catenin immunofluorescence. Error bars in (D, F) are 95% confidence intervals. Scale bars, 100 μm.
Figure S6 (Related to Figure 6). Reducing cell-cell adhesion and inducing basement membrane formation is sufficient to induce budding morphogenesis of a stratified epithelium. (A) Top: schematics of the experimental design. Below: confocal fluorescence images of spheroids for indicated experimental conditions. (B) Left: phase contrast and epifluorescence images of a spheroid. Right: binary mask image segmented from merged green and red fluorescence, and its boundary outline color-coded by the local curvature. Each column of curvature heatmap represents a straightened outline color-coded by curvature. (C) Schematic and table showing experimental conditions of 6 groups. (D-E) Bar plots of bud number or percentage of high-curvature perimeter length (|curvature|>20 mm−1). (F) Heatmap showing color-coded curvature along spheroid perimeters. Each column is one spheroid. Only 21 of 43 Group 4 samples were included in the heatmap to save space. (G) Confocal images of spheroids from indicated experimental conditions immunostained with E-cadherin and laminin (yellow; a basement membrane marker). MIP, maximum intensity projection. (H) Merged phase contrast and epifluorescence images of spheroids using two different sgRNAs under the indicated experimental conditions with different mixing ratios of control and E-cadherin downregulated cells. (I) Plots of three different quantifications for the 8 experimental groups shown in (H). (J) Schematics illustrating the cell sorting and branching of a stratified epithelium with mixed populations of high vs. low E-cadherin-expressing cells. Error bars in all plots, 95% confidence intervals. ***, Tukey test p<0.001 compared to the first group of the plot. Scale bars, 100 μm.
Figure S7 (Related to Figure 7). Reconstituted epithelial budding requires β1-integrin-mediated cell-matrix adhesion. (A) Merged phase contrast and epifluorescence images of spheroids from indicated experimental conditions. The α5-integrin (clone mAb16) and β1-integrin (clone mAb13) blocking antibodies were used at 100 μg/mL. n=16 for each group from 3 independent experiments. (B) Plots of bud number or percent protruded bud area per spheroid for indicated experimental conditions. n=10 per group from the same experiment. (C) Phase contrast images of budded spheroids (sg2-Cdh1 + sg-Control, ABA + Matrigel for 7 days) treated with dispase and recovered for 6 days after washout. n=15 from 2 independent experiments. (D) Plots of bud number or percent protruded bud area per spheroid for groups shown in (C). (E) Maximum intensity projection of confocal immunofluorescence images of 2D-cultured cells expressing indicated sgRNAs. Green nuclei indicate cells expressing the sgRNAs. (F) Plot of mean β1-integrin intensities under the indicated experimental conditions. n=12 fields of view per group. (G) Western blot showing expression levels of β1-integrin under indicated experimental conditions. Percentage levels under each lane are normalized ratios of β1-integrin to GAPDH intensities of that lane. (H) Confocal images of spheroids under indicated experimental conditions. Note that green nuclei here indicate either sg-Control or sg-Itgb1 expression depending on the experimental group. White arrows point to cells that lost their β1-integrin expression. MIP, maximum intensity projection. (I) Schematics illustrating experimental procedures for the cell attachment assays. MG, Matrigel; E-cad-ECD, E-cadherin extracellular domain. (J) Phase contrast images of attached cells under indicated experimental conditions. (K-L) Plots of attached cells under indicated experimental conditions. n=52, 39, 39, 52 fields of view for groups in (I). n=39 fields of view per group in (J). Each experimental group had 3 or 4 biological replicates, and each had 13 fields of view. The full set of experiments was done twice with similar results. (M) Confocal images of laminin or collagen IV immunostaining in control, BB-94 (5 μM) or GM-6001 (25 μM) treated spheroids (7 days post treatment). (N) Left: brightfield image of the experimental setup to perform atomic force microscopy (AFM) on spheroids. The focus is on the AFM tip. Right: example force-indentation plot from a typical AFM measurement. Negative forces are from adhesion between the AFM probe and sample during withdrawal. Scale bars, 100 μm.
Video S1 (Related to Figure 1). Volumetric time-lapse video of a branching embryonic salivary gland. Salivary gland from a 13-day transgenic mouse embryo expressing membrane-tdTomato and histone-EGFP. Time-lapse volumetric two-photon microscopy images were acquired at 2-μm z steps over a 100 μm z range at 5-minute intervals for 20 hours. The first half of the video shows different views of the imaging volume at the first time point. The second half of the video shows time-lapse sequences of the middle x-y plane (left) or 3D rendering of the middle 40 μm-thick volume of a branching epithelial bud (right).
Video S2 (Related to Figure 1). Dynamics of surface-layer epithelial cells highlighted by KikGR photoconversion. Salivary gland from a 13-day transgenic mouse embryo expressing the photoconvertible fluorescent protein KikGR. Native KikGR has green fluorescence, which converts to red fluorescence after blue light excitation. Magenta color indicates photoconverted KikGR.
Video S3 (Related to Figure 1). Surface-proximal 3D cell migration tracks. Videos show only epithelial cell tracks for which the closest distance to the gland surface was ≤ 15 μm. White dots indicate cell centroids, and the tracks shown at each time point represent cell migration trajectories spanning the previous 2 hours. Tracks are color coded by mean track speed (left) or the z position (right).
Video S4 (Related to Figures 1 and 2). Different views of a branching epithelial bud from a developing salivary gland. Salivary gland from a 13-day transgenic mouse embryo expressing histone-EGFP and a heterozygous epithelial RFP reporter driven by the Krt14 promoter (Krt14p::RFP). Note that the surface-layer epithelial cells expressing high RFP comprise a cell sheet that folds inward during clefting.
Video S5 (Related to Figure 2). 3D tracking of daughter cells from surface-derived cell divisions. This video has 2 sequential parts. The first part shows “tracked cell highlighted” (left) and “tracked cell centered” (right) styles of an example cell tracking. The second part shows tracking of 16 pairs of daughter cells in the “tracked cell centered” style. Tracked cell is marked by a white dot. Distance indicated is from the coverslip. Time indicates minutes from the onset of anaphase (0 min).
Video S6 (Related to Figure 6). Time-lapse videos of 3D spheroid cultures of engineered DLD-1 cells under indicated experimental conditions. Images are merged phase contrast and epifluorescence channels. Green and magenta indicate cells expressing sg-Control or sg-Cdh1, respectively. Abscisic acid (ABA) is a dimerizer used to induce robust transcriptional repression in engineered DLD-1 cells. DMSO is the vehicle control. Live imaging started 5 days post Matrigel and ABA or DMSO treatment.
Video S7 (Related to Figure 6). Time-lapse videos of a budding spheroid culture of engineered DLD-1 cells. Images in the main window are maximum intensity projections of confocal fluorescence images, and images in the upper left inset are single-slice confocal fluorescence images. Green and magenta indicate cells expressing sg-Control or sg2-Cdh1, respectively. Yellow indicates the basement membrane marked by Atto-647N labeled fibronectin. Arrows and arrowheads indicate forming clefts. Live imaging started 4 days post Matrigel and ABA treatment.
Data Availability Statement
Single-cell RNA sequencing data has been deposited in GEO (accession number GSE159780). All other source data and raw data that support the findings of this study are available in Figshare: https://doi.org/10.35092/yhjc.c.5404533. Customized scripts and usage instructions are available from Github: https://github.com/snownontrace/public-scripts-Wang2020-branching-morphogenesis.