

Abstract
Nine different antibody–drug conjugates (ADCs) are currently approved as cancer treatments, with dozens more in preclinical and clinical development. The primary goal of ADCs is to improve the therapeutic index of antineoplastic agents by restricting their systemic delivery to cells that express the target antigen of interest. Advances in synthetic biochemistry have ushered in a new generation of ADCs, which promise to improve upon the tissue specificity and cytotoxicity of their predecessors. Many of these drugs have impressive activity against treatment-refractory cancers, although hurdles impeding their broader use remain, including systemic toxicity, inadequate biomarkers for patient selection, acquired resistance and unknown benefit in combination with other cancer therapies. Emerging evidence indicates that the efficacy of a given ADC depends on the intricacies of how the antibody, linker and payload components interact with the tumour and its microenvironment, all of which have important clinical implications. In this Review, we discuss the current state of knowledge regarding the design, mechanism of action and clinical efficacy of ADCs as well as the apparent limitations of this treatment class. We then propose a path forward by highlighting several hypotheses and novel strategies to maximize the potential benefit that ADCs can provide to patients with cancer.
Antibody–drug conjugates (ADCs) are among the fastest growing drug classes in oncology. These therapeutic entities are composed of monoclonal antibodies (mAbs) linked to cytotoxic drugs and are designed, in principle, to widen the therapeutic window of those drugs by limiting their delivery specifically to cells that express the target antigen of the selected mAb1–4. Emerging evidence indicates that the efficacy of an ADC is dependent upon antibody-specific, linker-specific and payload-specific factors, each of which is a function of complex interactions between the ADC and various components of the tumour and the tumour microenvironment (TME)5. Many ADCs have demonstrated impressive activity against treatment-refractory cancers, resulting in approvals in numerous and diverse indications (TABLE 1); however, their broader use is limited by various challenges, including toxicities, suboptimal predictive biomarkers, unknown clinical value in combination with standard therapies and poorly understood pathways of drug resistance.
Table 1 |.
ADCs currently approved by the US FDA
| ADC | Target antigen | mAb isotype | Linker type | Payload | Payload class | Payload action | DAR | Disease indication (year of approval) |
|---|---|---|---|---|---|---|---|---|
| Gemtuzumab ozogamicin | CD33 | IgG4 | Cleavable | Ozogamicin | Calicheamicin | DNA cleavage | 2–3 | CD33+ R/R AML (2000)a |
| Brentuximab vedotin | CD30 | IgG1 | Cleavable | MMAE | Auristatin | Microtubule inhibitor | 4 | R/R sALCL or cHL (2011) R/R pcALCL or CD30+ MF (2017) cHL, sALCL or CD30+ PTCL (2018)b |
| Ado-trastuzumab emtansine (T-DM1) | HER2 | IgG1 | Non-cleavable | DM1 | Maytansinoid | Microtubule inhibitor | 3.5 (mean) | Advanced-stage HER2+ breast cancer previously treated with trastuzumab and a taxane (2013); early stage HER2+ breast cancer in patients with residual disease after neoadjuvant trastuzumab-taxane- based treatment (2019) |
| Inotuzumab ozogamicin | CD22 | IgG4 | Cleavable | Ozogamicin | Calicheamicin | DNA cleavage | 5–7 | R/R B-ALL (2017) |
| Fam-trastuzumab deruxtecan-nxki (T-DXd) | HER2 | IgG1 | Cleavable | DXd | Camptothecin | TOPO1 inhibitor | 8 | Advanced-stage HER2+ breast cancer after two or more anti-HER2-based regimens (2019) |
| Polatuzumab vedotin-piiq | CD79b | IgG1 | Cleavable | MMAE | Auristatin | Microtubule inhibitor | 3.5 (mean) | R/R DLBCL (2019)c |
| Sacituzumab govitecan-hziy | TROP2 | IgG1 | Cleavable | SN-38 (active metabolite of irinotecan) | Camptothecin | TOPO1 inhibitor | 8 | Advanced-stage, triple-negative breast cancer in the third-line setting or beyond (2020) |
| Enfortumab vedotin-ejfv | Nectin 4 | IgG1 | Cleavable | MMAE | Auristatin | Microtubule inhibitor | 4 | Advanced-stage urothelial carcinoma, following progression on a PD-l or PD-Ll inhibitor and platinum-containing chemotherapy (2020) |
| Belantamab mafodotin-blmf | BCMA | IgG1 | Non-cleavable | MMAF | Auristatin | Microtubule inhibitor | Unknown | R/R multiple myeloma in the fifth-line setting or beyond (2020) |
ADC, antibody–drug conjugate; AML, acute myeloid leukaemia; B-ALL, B cell acute lymphoblastic leukaemia; BCMA, B cell maturation antigen; cHL, classical Hodgkin lymphoma; DAR, drug-to-antibody ratio; DLBCL, diffuse large B cell lymphoma; mAb, monoclonal antibody; MF, mycosis fungoides; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; pcALCL, primary cutaneous anaplastic large cell lymphoma; PTCL, peripheral T cell lymphoma; R/R, relapsed and/or refractory; sALCL, systemic anaplastic large cell lymphoma; TOPO1, topoisomerase I; TROP2, tumour-associated calcium signal transducer 2.
As a single agent or in combination with daunorubicin and cytarabine. Gemtuzumab ozogamicin was withdrawn from the market in 2010 and re-approved in 2017 for newly diagnosed or R/R CD33-positive AML.
In combination with cyclophosphamide, doxorubicin and prednisone for newly diagnosed sALCL or CD30+ PTCL and in combination with doxorubicin, vinblastine and dacarbazine for newly diagnosed cHL.
In combination with bendamustine and rituximab.
Progress in synthetic biochemistry methods, including in mAb production, linker technology and novel payload discovery, has paved the way for a new generation of ADCs with the potential to improve upon the activity and toxicity profiles of earlier generations of ADCs6. At the time of publication, nine different ADCs have been approved for the treatment of patients with cancer (TABLE 1), with dozens more at various stages of preclinical and clinical development7. In this Review, we first discuss the historical development of ADCs, how they are constructed and insights from preclinical studies regarding their mechanism of action. We then discuss how these properties manifest clinically, using examples to explore the activity and toxicity profiles of ADCs. Finally, we conclude with potential strategies that could be tested to overcome these barriers and maximize the anticancer efficacy of ADCs in clinical practice.
A brief history of ADC development
The exponential growth in the development of ADC technology over the past decade is founded on over a century of research and vision, which can be traced back to the early 1900s when physician-scientist Paul Ehrlich first conceived of a ‘magic bullet’ that could deliver a toxic drug to certain cells while sparing others8,9. Advances in chemistry first enabled the linkage of cytotoxic agents with antibody species in the 1950s, when methotrexate was conjugated with polyclonal rodent immunoglobulins targeting leukaemia cells10. By the early 1970s, new techniques in hybridoma technology enabled the production of mAbs with greater homogeneity and targeting accuracy, spurring major leaps in the field, including the generation of ADCs that produced promising early results in both in vitro and in vivo models of cancer11–13.
Clinical trials of ADCs for the treatment of patients with cancer began in the 1980s, but problematic drug toxicities were observed without signs of clinical efficacy14–16. This pattern persisted for ~20 years before the CD33-targeted agent gemtuzumab ozogamicin became the first ADC to be approved by the US FDA in 2000. This ADC was initially approved for the treatment of relapsed and/or refractory (R/R) acute myeloid leukaemia, only to be voluntarily withdrawn from the market by the manufacturer in 2010 owing to concerns that its adverse event (AE) profile outweighed its efficacy17–19. However, in 2011, over a century after Ehrlich’s initial spark, the CD30-targeted ADC brentuximab vedotin was approved for the treatment of R/R classical Hodgkin lymphoma and systemic anaplastic large cell lymphoma (ALCL), followed shortly by the 2013 approval of the HER2-targeted ADC ado-trastuzumab emtansine (T-DM1) for the treatment of trastuzumab-resistant metastatic breast cancer20–22. The pace of ADC development seems to be increasing, with two ADCs approved in 2019 and an additional three approved in 2020 (TABLE 1).
ADC design and construction
Since the inception of ADCs, the basic approach to designing and building these agents has remained constant. All ADCs have three core components: an antibody that binds a tumour-associated antigen, a cytotoxic payload and a connecting linker. However, each of these components can vary widely between different ADCs in ways that strongly influence their pharmacological and clinical properties (FIG. 1).
Fig. 1 |. Modular components of ADCs.

a | Schematic representation of an antibody–drug conjugate (ADC), with the antibody in green, linker in blue and payload in yellow. This representative ADC has a drug-to-antibody ratio of 4. b | Illustration of the modular nature of ADCs, whereby an antibody with a given target can be attached to a payload via a cleavable or non-cleavable linker. Most approved ADCs utilize an immunoglobulin G1 (IgG1) backbone, although other antibody isotypes can be used to exploit different physiological attributes (such as serum half-life, complement component C1q-binding capacity and avidity for Fcγ receptors). Representative and commonly used examples of linkers and payloads are depicted, and their key properties are noted. The choice of linker and payload can determine the safety and efficacy of the ADC in different oncology indications. *Non-cleavable maleimidocaproyl (MC) and maleimidomethyl cyclohexane-1-carboxylate (MCC) linkers are often used with monomethyl auristatin F and emtansine payloads, respectively; MC and MCC linkers can be cleavable when conjugated to certain other payloads.
Antibody and target selection
The advent of antibody-based drugs has enabled substantial progress in the treatment of autoimmune, cardiovascular, benign haematological and bone diseases in addition to the treatment of cancer23. Although antibody fragments and bispecific antibodies present exciting opportunities for innovation, immunoglobulin G (IgG) remains the predominant antibody backbone used in this broad class of therapeutics as well as in ADCs specifically24. Human IgGs comprise four subclasses (IgG1, IgG2, IgG3 and IgG4), which differ in their constant domains and hinge regions. Subtle variations between these subclasses affect the solubility and half-life of mAbs as well as their affinity for different Fcγ receptors (FcγRs) expressed on immune effector cells25,26. The majority of ADCs are built upon the IgG1 architecture to optimize these factors, although some have IgG2 or IgG4 backbones (including gemtuzumab ozogamicin and inotuzumab ozogamicin, both of which use IgG4)7,27,28. In comparison with their IgG2 and IgG4 counterparts, IgG1 antibodies have similarly long serum half-lives but greater complement-fixation and FcγR-binding efficiencies. IgG3 might be the most immunogenic subclass, but such antibodies have generally been avoided in ADC design (for better or worse) owing to their relatively short circulating half-lives29 (FIG. 1). Owing to early problems encountered with the use of mouse antibodies, including acute hypersensitivity reactions and/or the generation of neutralizing anti-drug antibodies, modern ADCs standardly contain a chimeric or humanized antibody backbone, which minimizes but does not entirely preclude these issues28,30.
With regard to selecting the ideal mAb target, one guiding principle has been to identify cell-surface proteins that are highly expressed on tumour cells but not on non-malignant cells. ADCs are designed to deliver their toxic payload to any cell expressing the target antigen and, thus, targets that are preferentially expressed in tumours versus non-malignant tissues present a wider therapeutic window and decrease the chance of systemic toxicities2. Examples of successful targets among the ADCs currently approved for the treatment of solid tumours include HER2, TROP2 and nectin 4 (REFs22,31–34). Each of these proteins are expressed to some degree in non-malignant tissues, but they are often overexpressed by tumour cells, sometimes by several orders of magnitude35–37. In the context of haematological cancers, CD30, the target of brentuximab vedotin, is expressed by (and is characteristic of) the malignant lymphoid cells of Hodgkin lymphoma and ALCL but not by other cell types, with the exception of a small subset of lymphocytes38. Likewise, CD22, CD79b and B cell maturation antigen (BCMA), the respective targets of inotuzumab ozogamicin (approved for the treatment of R/R B cell acute lymphoblastic leukaemia), polatuzumab vedotin (approved for R/R diffuse large B cell lymphoma) and belantamab mafodotin (approved for R/R multiple myeloma) (TABLE 1) are highly specific for B cell lineages39,40. The threshold level of tumour-specific target expression required for ADC activity as well as the required degree of differential expression between tumour and non-malignant tissues are dependent on the particular ADC construct and therapeutic context.
Beyond tumour specificity, several additional target-related factors influence the efficacy of ADCs. For example, breast cancers with high levels of intratumour or intertumour heterogeneity in HER2 expression respond poorly to T-DM1 compared with those with homogeneous HER2 expression41,42. Furthermore, the rates of target turnover, internalization, lysosomal processing and degradation influence ADC activity, such that higher rates of turnover result in more efficient drug delivery and target replenishment and, thus, in greater antitumour activity43. Finally, targets that are functionally oncogenic, as opposed to simply being present on the surface of cancer cells, are less subject to downregulation of expression as a mechanism of drug resistance and can even be exploited for additional ADC activity via antibody-mediated suppression of downstream oncogenic signalling pathways44. These subtleties are explored in more detail later in this article.
Linker types and technologies
Linker technology has advanced substantially since the early days of ADC development, and this progress probably contributed to the clinical successes of this drug class achieved to date. The specific chemistry and synthetic techniques utilized in the manufacturing of ADCs are beyond the scope of this Review. Instead, we focus on some general principles and distinctions that are relevant to clinically observed ADC activity and toxicities.
The purpose of the linker is twofold. The first role is to ensure that the cytotoxic payload remains firmly attached to the antibody moiety while the drug circulates in plasma. Linkers that are unstable in plasma could release the payload prematurely, resulting in excess systemic toxicity and reduced payload delivery upon antigen engagement at the tumour site45. This issue is particularly relevant considering that many ADCs carry highly potent cytotoxic payloads with toxicity profiles that make them otherwise unsuitable for systemic delivery46. The second, often competing role of the linker is to enable efficient release of the payload within the tumour, particularly within cancer cells47. ADCs that do not properly deliver their payload forego the unique advantage that this drug class has over naked antibodies and traditional cytotoxic drugs.
Linkers broadly fall into two classes: cleavable and non-cleavable (FIG. 1). Cleavable linkers are designed to break down and release the cytotoxic payload of the ADC in response to tumour-associated factors such as acidic or reducing conditions or abundant proteolytic enzymes (for example, cathepsins). Examples of linkers cleaved by these mechanisms include pH-sensitive hydrazone linkers (such as the one used in gemtuzumab ozogamicin), reducible disulfide linkers (which are being used in several experimental agents, such as indatuximab ravtansine and mirvetuximab soravtansine) and various peptide-based, enzyme-cleavable linkers (including those used in brentuximab vedotin, polatuzumab vedotin, enfortumab vedotin, sacituzumab govitecan and trastuzumab deruxtecan (T-DXd))46 (TABLE 1). In real-world use, cleavable linkers exhibit varying degrees of stability in the circulation and can degrade in plasma over time45. For example, the hydrazone linker used in gemtuzumab ozogamicin is more labile than other cleavable linkers and undergoes some degree of hydrolysis at physiological pH, which might explain some of the off-target toxicities associated with this ADC45,48. By contrast, non-cleavable linkers tend to be more stable in plasma but rely on lysosomal degradation of the entire antibody–linker construct to release their payloads, often resulting in the retention of charged amino acids on the payload, which might affect its action or cell permeability45. Examples of non-cleavable linkers include thioether linkers (as used in T-DM1) and maleimide-based linkers (as used in belantamab mafodotin)49. Of note, data from several preclinical studies indicate that extracellular release of the cytotoxic payload might be an important component of ADC activity, thus framing linker stability as a complex optimization problem that is intrinsically linked to target and payload selection as well as to features of the TME46,50,51. Of the nine FDA-approved ADCs, two contain non-cleavable linkers: T-DM1 and belantamab mafodotin (TABLE 1).
Payloads
Early ADCs were designed to carry traditional chemotherapy drugs with known anticancer activity, such as methotrexate, doxorubicin or vinca alkaloids15,52,53. However, these ADCs were not more effective than their standard cytotoxic drug counterparts and sometimes required extremely high dosing for activity, which compromised hopes of limiting systemic toxicities54. Furthermore, data indicate that only a very small fraction of the administered dose of tumour-targeted mAbs reaches the tumour tissue (in the order of 0.1%), implying that payloads with greater cytotoxicity are needed to achieve therapeutic efficacy55–57. These observations led to experimentation with ADCs carrying highly potent chemotherapy drugs, such as auristatins, calicheamicins, maytansinoids and camptothecin analogues, which can be cytotoxic at sub-nanomolar concentrations3,58,59. Seven of the nine FDA-approved ADCs have payloads from one of these drug classes (TABLE 1). The auristatins include monomethyl auristatin E (MMAE) and monomethyl auristatin F (MMAF), which are synthetic derivatives of the peptide dolastatin 10 produced by Dolabella auricularia and are microtubule destabilizers60. Calicheamicins, such as ozogamicin, are DNA-binding compounds that cause double-stranded DNA breaks and are derived from actinomycetes bacteria61. Maytansinoids, such as DM1, are derived from maytansine (originally isolated from the Maytenus genus of plants) and bind to tubulin, thereby disrupting microtubule dynamic instability62. Finally, camptothecin analogues, originally derived from the bark of Camptotheca acuminata and including the exatecan derivative DXd and the irinotecan metabolite SN-38, inhibit topoisomerase I (TOPO1), leading to DNA breaks50,63. All of these mechanisms are commonly exploited in cancer therapy, although none of these payloads has been found to be suitable for systemic delivery as free drugs.
The drug-to-antibody ratio (DAR) is the average number of payload moieties attached to each mAb. This property, which varies between ADCs, has implications for drug pharmacology and activity7. DARs of currently approved ADCs range from 2 to 8 (TABLE 1). The synthesis techniques of some ADCs enable tight control over this parameter; however, others are manufactured using processes that rely on conjugating payloads to native cysteine or lysine residues of the mAb, resulting in products with substantial heterogeneity and DAR variation even within drug batches28. In general, ADCs with very high DARs are expectedly more potent in vitro, although some might be cleared faster from the plasma by the liver, which has been shown to reduce tumour ADC exposure and to result in comparable activity to ADCs with lower DARs in preclinical models64,65. This principle is illustrated by brentuximab vedotin, the in vitro activity of which directly correlates with the DAR; however, in mouse models, versions of the ADC with a DAR of 8 are cleared five times faster than those with a DAR of 2 and have a worse therapeutic index owing to increased toxicity without superior antitumour activity64. Data from preclinical studies suggest that this relationship between higher DARs and faster hepatic clearance is due to increased hydrophobicity of the antibody–linker complex, which can be avoided by using hydrophilic constructs66. With regard to ADCs for which drug conjugation — and thus the DAR — does not influence plasma clearance (such as sacituzumab govitecan), higher DARs are more directly associated with greater antitumour activity in vivo50,66.
The hydrophobicity of the detached payloads is also thought to be an important factor, in particular regarding the ‘bystander effect’. This phenomenon, which is discussed further in the following section, involves the diffusion of cell-permeable payloads from within cells expressing the target antigen into neighbouring cells, on which the drug can exert a cytotoxic effect regardless of target antigen expression67,68.
How ADCs work in vivo
ADCs are among the most complex biochemical platforms used in cancer medicine, integrating the effects of antibodies and cytotoxic drugs and thus exhibiting unique mechanisms of action and pharmacokinetic profiles. The subtleties that underlie ADC activity in the clinic are only just beginning to be understood69. The canonical model of ADC action posits the following: binding of the mAb to the target antigen, subsequent internalization and, finally, linker breakdown and intracellular payload release. While this model serves as a helpful overall framework, the reality is more complicated and differs appreciably between ADCs. To illustrate the mechanisms by which ADCs exert their therapeutic effects, we take a chronological approach from parenteral drug administration to cell death, highlighting some of the complexities along the way. We wish to emphasize that, for ADCs to act upon tumours, they themselves often need to be acted upon by tumour cells. In this way, ADCs can be conceived of as prodrugs, which in many cases require processing and metabolism by the target cells before their end activity can be fully realized (FIG. 2).
Fig. 2 |. Mechanisms of action of ADCs.

This figure depicts the current understanding of the chronology and complexity of antibody–drug conjugate (ADC) action. (1) Owing to incomplete conjugation during production and/or linker lability, ADCs circulate as three components: naked antibody, free payload and intact conjugate. The intact conjugate predominates with stable ADCs. (2) ADC penetration into tumours can be inefficient, and some payload might be released in the tumour microenvironment before antibody–antigen engagement. (3) The antibody component of many ADCs retains its activity profile and can therefore interfere with target function, dampen downstream signalling and/or engage with immune effector cells to elicit antitumour immunity before the payload is ever released. The extent to which such effects contribute to therapeutic activity or toxicities is often poorly characterized for a given ADC. (4) Following antigen engagement, most ADCs are internalized, predominantly through endocytosis along with their bound targets. Thus, the degree of target internalization and turnover might be an important contributor to ADC activity. (5) Once inside lysosomes or endosomes, acidic, proteolytic or redox conditions cause the ADC payloads to be released from their antibody carriers, following which the payloads can diffuse into the cytoplasm and throughout the cell to act on their target substrates, ultimately resulting in cell death. (6) Hydrophobic payloads can also diffuse through cell membranes, which can result in cytotoxic activity against neighbouring cells irrespective of their expression of the target antigen. This ‘bystander effect’ might be an important contributor to the efficacy of ADCs in tumours with heterogeneous expression of the antibody target. NK, natural killer.
Upon administration, the ADC formulation contains three major circulating components: the conjugate (which constitutes the overwhelming fraction), naked antibodies and free payload molecules. The relative proportions of these three components can vary between ADCs, depending in part on linker stability and product purity, and might change over time in the days following drug administration. For example, clinical pharmacokinetic studies tracking each component of T-DM1 revealed that peak serum concentrations of total trastuzumab (conjugated plus naked antibodies) exceeded those of the complete T-DM1 conjugate by approximately 20%, whereas concentrations of the DM1 payload were several orders of magnitude lower and were barely detectible by assay70. The half-life of total trastuzumab was 9–11 days, whereas the half-life of the T-DM1 conjugate was approximately 4 days, a discrepancy that might be explained by hepatic clearance of T-DM1, linker instability or antibody recycling70. These findings suggest that ADCs exist in vivo as a dynamic admixture of circulating components, which complicates pharmacological modelling and influences the clinical properties of these agents. Nevertheless, several attempts at pharmacokinetic and/or pharmacodynamic modelling of ADCs have been successful, highlighting the promise of such models and the need for further exploration in this area71–73.
In contrast to traditional cytotoxic therapies, mAbs are large molecules — a characteristic that limits their delivery to tumours. Furthermore, vascular anatomy, transcapillary pressure gradients and stromal tissue components can be highly aberrant in tumours, presenting an initial barrier to tumour penetration by ADCs74,75. After extravasation from capillaries, antibodies reach tumour cells via passive diffusion, often resulting in slow, inefficient and heterogeneous tissue penetration. Accurate measurements with ADCs are difficult to obtain in patients; however, evidence from labelled mAb-based studies as well as mathematical models suggests that only a fraction of a percent of the administered ADC dose actually reaches tumour cells, again highlighting the necessity for payload potency in ADC design76–78. Data from studies using labelled drug moieties in animal models indicate that, with existing ADCs, drug concentrations in tumour tissue often peak within 1–2 days following administration, reaching levels that can exceed their concentration in non-malignant tissue by 100-fold, although these parameters might vary substantially between different ADCs2,79,80. Adding further complexity to this picture, although the use of high-affinity antibodies targeting high-density and high-turnover proteins might seem ideal, these properties might result in reduced tumour penetration owing to a ‘binding-site barrier’, whereby the ADC is ‘spent’ on malignant cells located at the more-accessible surfaces of the tumour, thus protecting cells at less accessible, interior regions from drug exposure81,82.
Following tissue penetration, ADCs must engage with their target antigen for optimal cytotoxicity. Owing to the placement of linkers outside of the antigen-recognition domain of the mAb, ADCs typically bind to their target antigen with the same affinity as their unconjugated counterparts83. Upon antigen engagement, ADCs also seem to retain the functionality of their naked mAb counterparts and thus often begin to exert antitumour activity before the payload is released. Mechanistically, antigen-binding fragment (Fab)-mediated activity can disrupt target function by blocking ligand binding, interfering with dimerization and/or inducing endocytosis and degradation of the target protein84. The anti-HER2 mAb trastuzumab inhibits HER2 signalling predominantly by blocking ligand-independent HER2 dimerization, and data from preclinical studies suggest that this functionality remains intact with the HER2-targeted ADCs T-DM1 and T-DXd63,85. This finding supports the aforementioned hypothesis that, all else being equal, ADCs targeting an oncogenic and/or functional protein are likely to have greater antitumour activity than those with non-functional targets. Moreover, certain oncogenic drivers are more likely to be homogeneously and highly expressed in tumour tissue owing to evolutionary selection pressures86, which is another hypothetical advantage for ADCs targeting such functional targets. Beyond the Fab region, the Fc region of the mAb component of ADCs can orchestrate antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity and/or antibody-dependent cellular phagocytosis84,87. For example, T-DXd and T-DM1 are built upon the same ADCC-competent IgG1 backbone and can induce ADCC in vivo in preclinical models, suggesting that ADCs can function as a form of immunotherapy63,85.
After antigen binding, the internalization of the ADC–antigen complex is thought to be a crucial step in payload delivery for many ADCs7. ADC internalization can occur via the antigen-dependent processes of endocytosis or the antigen-independent process of pinocytosis, with clathrin-mediated endocytosis being the predominant mode of uptake88,89. Following internalization, ADC–antigen complexes are trafficked along the endosomal and/or lysosomal pathways in a manner that seems to depend on proper organelle acidification88,90,91. Payloads that are attached using acid-cleavable linkers are likely to be released in early endosomes, and those attached using linkers that are designed to be cleaved enzymatically or degraded via proteolysis are released in late endosomes or lysosomes88. Reducible linkers release the payload principally upon exposure to glutathione, which is found at higher concentrations intracellularly than in plasma45. The time from antigen engagement to terminal processing and payload release can be >24 hours91. While many ADCs are designed to release their payload inside tumour cells in this ‘Trojan horse’ fashion, accumulating evidence indicates that extracellular payload release in tumour tissue can occur with most linker types, owing to the redox environment, low pH and extracellular proteases found in the TME, and might have an important role in the anticancer activity of these agents92.
Regardless of the compartment in which the payload is released, certain ADCs are capable of exerting a ‘bystander effect’ on neighbouring cells, irrespective of target antigen expression68,93. For internalized ADCs, this property requires the diffusion of lipophilic payloads across cell membranes and is thought to be a major component of ADC activity against tumours with heterogeneous expression of the target antigen. In this setting, cleavable linkers that release uncharged payload molecules seem to be required for bystander killing to occur, whereas charged payloads released from ADCs with non-cleavable linkers are more likely to be retained intracellularly93,94. In preclinical studies, increasing the proportion of target antigen-positive versus antigen-negative cells in co-culture also increases the relative level of bystander killing of the latter population, suggesting that antigen-positive cells are needed to process the ADC and release the cytotoxic payload95. The speed at which the bystander effect manifests is also dependent on the percentage of antigen-positive cells, which itself decreases over time as the ADC exerts its cytotoxic effects95. ADCs that exhibit substantial extracellular payload release might depend less on these factors for the bystander effect92.
How ADC properties manifest clinically
The complexity of ADC activity raises fascinating questions about the relationship between ADC molecular structure and the macro-level activity and toxicity profiles observed in the clinic. The successes and failures of trials investigating ADCs for the treatment of cancer shed light on these relationships and provide lessons that can guide both drug and clinical trial design.
Activity in treatment-refractory cancers
All of the ADCs currently approved in solid tumour indications are labelled specifically for patients with treatment-refractory cancers based on demonstrated efficacy in such patient populations. Heavily pre-treated cancers tend to have a high degree of genomic instability, resulting in intertumour and intratumour heterogeneity, and often cultivate hypoxic and immunosuppressive TMEs that exclude the body’s natural defences and impede drug penetration96. Palliative chemotherapy is the standard approach to the treatment of such cancers; however, adequate dosing of chemotherapeutic agents is constrained by the toxicities caused by systemic exposure. This scenario sets the stage for ADCs, which enable the targeted delivery of highly potent and broadly cytotoxic agents selectively to tumour tissue and can have coincident immunostimulatory functions. Furthermore, hypoxic TMEs might facilitate linker cleavage and payload release, and the bystander effect provides indiscriminate cytotoxicity after tumour penetration, which can overcome tumour heterogeneity. Thus, ADCs seem suited to provide benefit even to patients with heavily pre-treated cancers.
Indeed, enfortumab vedotin, a nectin 4-targeted ADC carrying a MMAE microtubule inhibitor payload, produced an objective response rate (ORR) of 44% in patients with metastatic urothelial carcinoma previously treated with a median of three lines of therapy, including platinum-based chemotherapy and immune-checkpoint inhibitors34. In this setting, treatment with standard taxane-based microtubule inhibitor chemotherapy alone has an expected ORR of 10.5%97. Similarly, sacituzumab govitecan has been associated with an ORR of 33.3% in patients with heavily pre-treated metastatic triple-negative breast cancer (TNBC)33. T-DM1 resulted in an ORR of 43.6% in patients with metastatic HER2-positive breast cancer who had previously been treated with trastuzumab and a taxane22. In patients with early stage breast cancer who had residual invasive disease after neoadjuvant treatment with trastuzumab and a taxane, adjuvant T-DM1 also conferred a 50% reduction in the risk of disease relapse or death31. The next-generation HER2-targeted ADC, T-DXd, produced a striking ORR of 60.9% in patients with metastatic HER2-positive breast cancer who had previously received T-DM1 and median of five other prior therapies for advanced-stage disease32.
These clinical findings raise several important questions. First, why is ADC activity observed in cancers that are resistant to agents with the same target or primary mechanism of action as the ADC? T-DM1 links an anti-HER2 antibody (trastuzumab) with a microtubule-targeting payload (DM1) yet has considerable activity in patients with breast cancers resistant to concurrent administration of trastuzumab and microtubule-targeting chemotherapies, such as taxanes and/or vinca alkaloids. Similarly, T-DXd, which delivers a TOPO1 inhibitor payload, has clinical activity (ORR 51%) against gastrointestinal cancers that are only modestly responsive (ORR ~14%) to the closely related TOPO1 inhibitor irinotecan; in this trial population, the ORR of T-DXd was 41.7% among patients who were previously treated with irinotecan98. Indeed, with several ADCs, activity is observed in cancers that are considered ‘chemo-refractory’. This term is non-specific, although it likely encompasses a group of cancers for which pharmacological mechanisms of resistance preclude the achievement of therapeutic drug concentrations in cancer cells, rather than a categorical insensitivity of the cells to drugs directed at a given target or pathway. Thus, the basis of ADC activity in this clinical context might reflect a superior therapeutic index, whereby antibody-directed payload release enables sufficiently cytotoxic intratumoural drug levels to be achieved while minimizing systemic toxicity99.
Second, why does T-DXd demonstrate activity in cancers that are refractory to T-DM1 despite having the same anti-HER2 antibody backbone? In comparison with T-DM1, T-DXd has a cleavable linker and a higher DAR and, furthermore, carries a payload that is substantially different from that of T-DM1. TOPO1 inhibitors are not used in early lines of treatment for patients with breast cancer (whereas microtubule-targeting agents are used ubiquitously), leaving tumours naive to the mechanism of antitumour action exerted by the payload of T-DXd. Such a strategy of altering the mechanism of action with sequential therapies in the palliative setting is a well-established means of overcoming treatment resistance100. Of note, although both T-DXd and sacituzumab govitecan carry TOPO1 inhibitor payloads and have established activity in patients with metastatic breast cancer, irinotecan monotherapy has only modest efficacy in this patient population and is not currently listed in the National Comprehensive Cancer Network (NCCN) guidelines as a recommended treatment for the disease101,102. This distinction reinforces the likelihood that no singular aspect of ADCs accounts for their clinical activity but rather that the simultaneous changes in drug potency, mechanism of action and delivery to the tumour are all likely to be contributing factors. Furthermore, a modest efficacy observed with particular chemotherapeutic agents in a given cancer should not discourage investigation of ADCs with payloads sharing the same target and/or mechanisms in that disease setting. Indeed, clinically observed ‘insensitivity’ to such chemotherapies might be attributable to factors that can be overcome with antibody-dependent drug delivery.
Finally, as noted previously, intratumoural heterogeneity is a major basis for resistance to targeted therapy103. ADCs with cleavable linkers and membrane-permeable payloads seem to yield additional activity via the bystander effect, thus underscoring the indiscriminate cytotoxicity of chemotherapy against antigen-negative cells located in close proximity to antigen-positive cells67,104. However, the potential benefits of the bystander effect have to be weighed against any potential associated increase in the risk of toxicities. The consequences of the bystander effect on non-malignant tissue or immune mediators located in or near the TME are not yet known. Further research on this topic would enable investigators to better harness this property of ADCs for the clinical benefit of patients with cancer.
The issue of toxicity
ADCs were originally designed with the central motivation of limiting the toxicities caused by existing chemotherapeutic agents through improved tumour targeting; however, severe AEs were observed in many early trials of ADCs14–16,105. Traditional ‘unconjugated’ cytotoxic agents are distributed throughout the body and, thus, the toxicity profiles of these drugs are largely determined by their mechanism of action and how those mechanisms disrupt the function of non-malignant tissues such as the mucosa, nerves or skin106–108. With ADCs, the expression pattern of the target antigen influences the distribution of the cytotoxic drug and where it accumulates, which can occasionally lead to notable ‘on-target, off-tumour’ toxicities that are not necessarily payload dependent. For example, in the early 1990s, the ADC BR96-doxorubicin, which targets the Lewis Y antigen, was found to be highly active in mouse xenograft models of multiple tumour types54; however, unlike in mice, this antigen is expressed in non-malignant human tissues, particularly within the gastrointestinal tract109. When compared directly with unconjugated doxorubicin administered in a standard fashion, BR96-doxorubicin caused no discernible haematological or cardiac toxicities (which are characteristically associated with systemic exposure to doxorubicin) but caused grade ≥2 vomiting in nearly 90% of patients (compared to 22% with doxorubicin). Amylase and/or lipase elevations and haematemesis were also observed with the ADC but not with doxorubicin15,110. Similarly, the CD44v6-targeted ADC bivatuzumab mertansine caused skin toxicity in nearly 80% of patients, including toxic epidermal necrolysis that led to severe or fatal desquamation in some patients, which was thought to be related to expression of the target protein in the skin105,111.
Despite having the same payload and linker structure and comparable DARs, brentuximab vedotin, polatuzumab vedotin and enfortumab vedotin seem to have different toxicity profiles (although consideration of the limitations of cross-trial comparisons is warranted)112. For example, enfortumab vedotin has been associated with dysgeusia in 40% of patients, which might be related to nectin 4 expression in the salivary glands; however, this toxicity was not noted in the pivotal trials of brentuximab vedotin or polatuzumab vedotin34,37. In another example of target-dependent toxicity, the HER2-targeted ADCs T-DXd and trastuzumab duocarmycin, which have different payloads, both cause pulmonary toxicities via an unknown mechanism, and such toxicities have been observed, to a lesser extent, with T-DM1 and even trastuzumab22,32,113. Interestingly, cardiac toxicities seem to be less common with HER2-targeted ADCs than with unconjugated trastuzumab (although the risk of such AEs still warrants appropriate monitoring)114,115. The reason for this apparent discrepancy is unknown — one might expect worse cardiotoxicity with the ADC that delivers a cytotoxic payload directly to HER2-expressing cardiomyocytes, but this effect has not been observed clinically. Of note, interruption of the ERBB–neuregulin signalling axis has been implicated as a mediator of the cardiotoxicity associated with HER2-targeted therapies; thus, the unique effects ADCs might have on this pathway warrant further study116.
Despite these compelling examples of on-target toxicities, ‘off-target, off-tumour’ AEs seem to dominate the toxicity profiles of most existing ADCs112,117. Meta-analyses of the available data have shown that, independent of the target antigen, MMAE is associated with anaemia and/or neutropenia and peripheral neuropathy, DM1 is associated with thrombocytopenia and hepatotoxicity, and MMAF and DM4 are associated with ocular toxicity1,118,119. Such off-target toxicities might be attributable to payload release in the circulation, in non-tumour tissues or in the TME as well as to the subsequent effects of the payload on relevant non-malignant tissues112.
Importantly, such toxicity patterns might not correlate specifically with payload category or mechanisms of action, and notable within-class differences in toxicities between payloads can shed light on how subtle chemical changes in linker and payload structure manifest clinically. For example, regardless of the ADC target, ocular toxicities occur with MMAF but not with MMAE, despite the fact that both agents belong to the auristatin class118. When released from a non-cleavable linker, MMAF probably retains a charge and might therefore accumulate intracellularly in the corneal epithelia, whereas the more hydrophobic MMAE payload (which is often delivered via a cleavable linker) can diffuse out of the corneal epithelial cells119. However, ocular toxicities have been observed with nearly all ADCs that deliver DM4 and with some of those that deliver DM1, irrespective of linker type, which suggests that such toxicities are not unique to charged payloads119,120.
Notably, some ADCs with target antigens that are known to be expressed in the eye have no ocular toxicity120. With many ADCs, the target antigen might be present in non-malignant tissues but not at levels sufficient to induce toxicities. Some antigens, such as TROP2 (the target of sacituzumab govitecan), are indeed expressed in many non-malignant tissues but are spatially sequestered in a way that limits their accessibility unless they are aberrantly expressed, as they are on the surface of tumour cells35,121. In animal models with TROP2 expression patterns similar to that of humans, the toxicity profile of sacituzumab govitecan was comparable to that of the SN-38 payload administered alone, again supporting the predominance of off-target toxicities with ADCs122.
Further complicating the issue of toxicities, target-independent ADC uptake into non-malignant cells might also occur through mechanisms such as macropinocytosis and micropinocytosis or via binding to Fc receptors123. For example, T-DM1 might cause thrombocytopenia in part via HER2-independent, FcγR-related uptake by immature megakaryocytes, resulting in the disruption of megakaryocyte differentiation124. Data from preclinical studies suggest that macropinocytosis partially explains the ocular toxicities observed with ADCs targeting antigens that are not expressed in the eye125. Some researchers have even hypothesized that the FcγR-binding affinities of ADCs should be decreased to improve the therapeutic index of these agents69,126.
Interesting examples also exist of the same ADC causing different toxicity patterns depending on the tumour type in which they are investigated112. At the same dose, glembatumumab vedotin, a gpNMB-targeted ADC carrying MMAE, led to a severe rash in 30% of patients with melanoma (including one fatal case) but only in 4% of patients with breast cancer127,128. Explanations for such findings remain elusive, although we speculate that priming of the immune system against tumour-associated antigens might be involved.
These complex factors make the safety profiles of ADCs difficult to predict based on their composition alone, and these challenges underline the need for close monitoring, careful dose selection, and diligent AE reporting and attribution in clinical trials of these agents117,129,130. Further in-depth preclinical and translational studies of the mechanisms of ADC toxicities are also clearly warranted.
Resistance to ADCs
A detailed understanding of the mechanisms of resistance to a drug can provide insights into the fundamental mechanism of drug action and facilitate the development of predictive biomarkers to improve patient selection. Comprehensive information on the mechanisms of resistance to ADCs has not yet emerged; however, initial evidence suggests that cells can evade ADC activity at several mechanistic steps, including antibody–antigen engagement, ADC internalization and processing, or payload action (FIG. 3).
Fig. 3 |. Proposed mechanisms of resistance to ADCs.
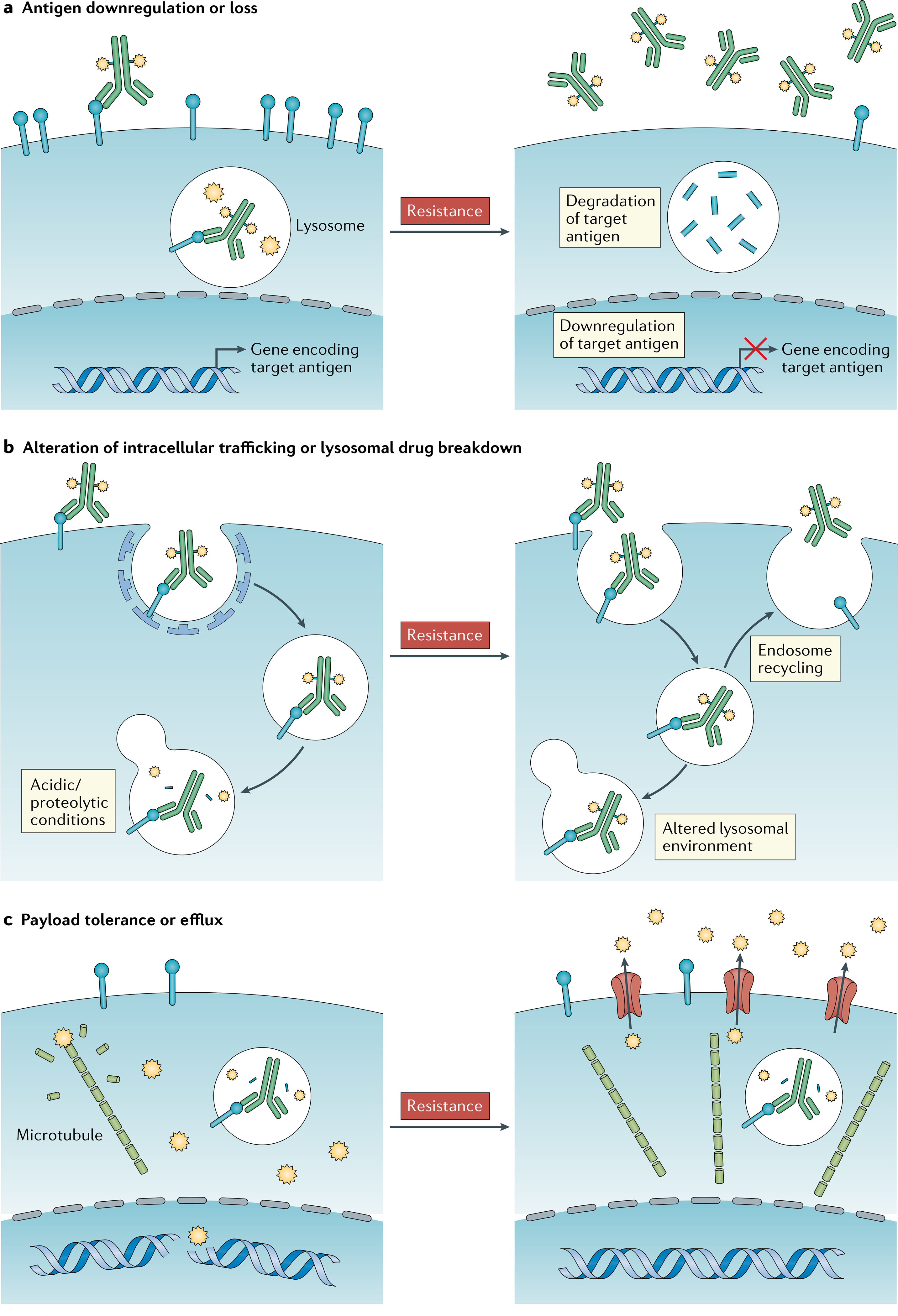
The following mechanisms of resistance to antibody–drug conjugates (ADCs) have been hypothesized and are supported largely by in vitro evidence but have not yet been confirmed in patients with cancer. a | Downregulation of the target antigen by tumour cells can prevent ADCs from docking on tumour cells, thus reducing the release of the payload therein. b | Recycling of endosomes to the cell surface might result in ejection of the ADC back to the exterior of tumour cells prior to payload release; the alteration of lysosomal acidification, redox environment or proteolytic processes might also prevent adequate payload release. c | The upregulation of ATP-binding cassette (ABC) transporter proteins in tumour cells can result in the active efflux of payload, thereby protecting cells from cytotoxic damage; however, not all payloads are ABC substrates.
De novo resistance and patient selection.
In contrast to leukaemias and lymphomas, in which the cell-surface markers exploited for drug targeting are often lineage defining and thus constitutively expressed, target antigen expression in many solid tumours is highly heterogeneous and can be dynamic. Therefore, selecting patients who are most likely to benefit from ADC treatment can involve measurement of target antigen expression in tumour tissue. Of the four ADCs approved by the FDA for the treatment of solid tumours (TABLE 1), T-DM1 and T-DXd are the only ones currently indicated exclusively for tumours expressing the respective target antigen (HER2 for both agents). In breast cancers, HER2 protein expression can vary by 3–4 logs — from virtually none to dramatic overexpression caused by high-level ERBB2 gene amplification131. The ASCO–College of American Pathology Guidelines for defining HER2 positivity, which encompass both immunohistochemistry (IHC) and fluorescent in situ hybridization, were developed solely to identify patients with marked ERBB2 gene amplification and/or HER2 overexpression as predictive biomarkers of therapeutic benefit from the unconjugated mAb therapy trastuzumab132. The value of these specific measurements in predicting the efficacy of HER2-targeted ADCs has never been defined. Notably, T-DM1 has activity in lung cancers with HER2 mutations, even in the absence of HER2 overexpression (ORR 44%)133, and both T-DXd and the experimental agent trastuzumab duocarmazine have produced early signs of activity in patients with breast cancers that express HER2 below the standard threshold for positivity104,113,134.
Sacituzumab govitecan is currently approved for patients with treatment-refractory metastatic TNBC regardless of TROP2 expression33. Nearly 90% of archival breast cancer samples from this patient population express moderate to high levels of TROP2 on IHC, which might obviate the need for biomarker-based patient selection; however, initial signals suggest a direct correlation between the level of TROP2 expression and therapy response121,135. No formal cut-off for ‘TROP2 positivity’ exists. In the trial that led to the approval of sacituzumab govitecan, 26% of patients had progressive disease and another 37% had stable disease as the best response, suggesting that de novo resistance to this agent is common33. Whether biomarker selection would improve the ORR in patients with treatment-refractory metastatic TNBC or whether these observations reflect the overall poor prognosis of such patients remains unclear. However, in a similar clinical setting, the gpNMB-targeted ADC glembatumumab vedotin had an ORR of 6% in patients with breast cancer and gpNMB expression in ≥5% of malignant epithelial cells on IHC but an ORR approaching 30% in those with gpNMB expression of ≥25%128. This finding suggests that patient selection is an important component of ADC development. According to IHC analysis, nectin 4, the target of enfortumab vedotin, is expressed by 83% of urothelial cancers34,37. Although the registrational trial of enfortumab vedotin did not require biomarker confirmation of target expression before enrolment34,37, all patients with available tissue samples had detectable tumoural expression of nectin 4, and data regarding the correlation between target expression and response to enfortumab vedotin are not yet mature.
Proposed mechanisms of acquired resistance.
As opposed to the demonstrated resistance mechanisms to tyrosine-kinase inhibitors, which often result from common drug-escape mutations involving the drug target136, acquired resistance to ADCs seems be more complicated and multifactorial, reflecting the general mechanistic complexity of this drug class137. The proposed mechanisms of acquired resistance to ADCs can be conceived in three major mechanistic categories: downregulation of antigen expression; alteration of intracellular trafficking pathways; and payload resistance, largely via upregulation of ATP-binding cassette (ABC) transporter proteins. Each of these three modes of resistance has been demonstrated preclinically and might occur concurrently in vivo, although limited clinical data currently exist to confirm these hypotheses.
As an example, breast cancer cell lines chronically exposed to T-DM1 downregulate the expression of HER2, decrease lysosomal acidification and slow proteolytic turnover, and upregulate the ABCB1 (MDR1) and/or ABCC1 (MRP1) drug efflux pumps138–140. These changes are respectively expected to result in decreased ADC binding, reduced linker cleavage and payload release, and ejection of payload moieties that enter the cytoplasm. Exposure to brentuximab vedotin similarly induces both CD30 downregulation and increased MDR1 expression in resistant lymphoma cells in vitro141. Clinically, low levels of CD33 expression and high protein efflux pump activity are central predictors of response to gemtuzumab ozogamicin in patients with acute myeloid leukaemia142.
ABC transporters such as MDR1, MRP1 and BCRP have long been known to play an active role in the cellular efflux of traditional antineoplastic agents, including anthracyclines, taxanes and camptothecins143. Notably, certain common ADC payloads, such as MMAE, DM1 and ozogamicin, are also known ABC transporter substrates and might be particularly susceptible to this mechanism of resistance144. However, other ADC payloads are poor ABC transporter substrates. For example, T-DXd is active in HER2-positive cancer cell lines with high levels of ABCC2 (MPR2) and ABCG2 (BCRP) expression following prolonged exposure and resistance to T-DM1 (REF.145), which might provide further explanation for the activity of this agent in T-DM1-resistant cancers32.
Activating mutations in PIK3CA are associated with resistance to trastuzumab in patients with advanced-stage breast cancer146,147, although the same finding has not been demonstrated with T-DM1 (REFs146,147). This finding reinforces the concept that ADC resistance mechanisms might have more in common with the paradigms of resistance to chemotherapies rather than to targeted therapies. Notwithstanding, more research on this topic is clearly needed.
Maximizing the potential of ADCs
On the basis of the evidence and principles outlined above, we offer thoughts and suggestions regarding how the full potential of this extremely promising and versatile drug class might be achieved in the treatment of patients with cancer. Decades of trial and error have led to a predominant focus on ADCs that target tumour-associated antigens, have cleavable linkers and deliver highly potent microtubule inhibitor or genotoxic payloads. The next decade promises to bring further innovations in the design and clinical application of these agents.
ADC-intrinsic strategies
ADCs are modular in nature (FIG. 1), which enables each component to be swapped or altered in a strategic fashion137. Typically, small-scale drug screens are conducted in vitro or in xenograft models to optimize each element of the ADC construct according to a given tumour subtype. Such approaches are often centred on comparisons of a single mAb modified with a limited selection of linker–payload combinations, which is a rational strategy but might miss opportunities to improve on antibody pharmacodynamics. This omission is potentially important given that different mAbs targeting the same antigen can have varying ligand-binding properties and differing effects on receptor dimerization and/or target internalization, which might in turn have marked effects on their in vivo activity148. Different mAbs might further differ in their Fc-dependent effector functions, as discussed above. Accordingly, mAbs that are optimized for other clinical applications might not actually be the best ADC backbones, especially given the growing evidence that ADC internalization and intracellular trafficking is central to the cytotoxic activity of ADCs. For example, in contrast with trastuzumab, the affinity of pertuzumab for HER2 is highly pH dependent, such that rapid dissociation of the antibody–antigen complex occurs in a low-pH environment. This finding led to the creation of an experimental recombinant pertuzumab-based ADC with enhanced cytotoxicity in preclinical models149.
Mutant proteins can be prone to higher rates of ubiquitylation, internalization and/or turnover than their wild-type counterparts, regardless of expression levels, which can in turn lead to marked clinical responses when mutant proteins are targeted by ADCs133,150. One could even envision ADCs built upon mAbs that specifically target proteins harbouring truncal oncogenic driver mutations (such as certain mutant forms of EGFR), which might maximize tumour specificity to degrees only achieved thus far with highly selective small-molecule tyrosine kinase inhibitors151,152.
The advent of bispecific antibodies presents additional opportunities for innovation. Such agents could potentially be leveraged to enhance antibody internalization and/or processing or to improve tumour specificity — possibilities that are now being actively explored153. For example, ADCs built on biparatopic antibodies, which target two separate epitopes of the same target antigen, can induce receptor clustering and rapid target internalization154. One bispecific ADC targeting HER2 and the lysosomal membrane protein CD63 seems to exhibit improvements in lysosomal accumulation and payload delivery relative to control constructs (monospecific HER2-targeted and CD63-targeted ADCs)155. A different experimental bispecific ADC targeting HER2 and the prolactin receptor (PRLR), which is rapidly recycled under normal physiological conditions, was more active than ADCs targeting HER2 alone in cells co-expressing HER2 and PRLR156. Fine-tuning the binding affinity properties of bispecific ADCs will be crucial to reducing on-target binding to systemically expressed epitopes outside of tumour cells, and the toxicity profiles of such agents remain to be determined153. Targeting two, carefully selected tumour antigens with a bispecific antibody could be a promising strategy if these obstacles are overcome.
Some ADC-like approaches abandon the traditional antibody backbone entirely in favour of ‘miniaturized’ small-molecule drug conjugates that utilize targeting moieties such as peptide fragments, single-chain variable fragments or diabodies (non-covalent dimers of single-chain variable fragments) to deliver their toxic payloads to antigen-expressing cells157,158. These efforts are largely motivated by hopes that smaller drug conjugates will have improved tumour tissue penetration and thus payload delivery. PEN-221, a 2 kDa peptide–DM1 conjugate targeting somatostatin receptor 2 (for reference, the size of a standard IgG is typically 150 kDa), is an example of such a construct that is being investigated in the treatment of neuroendocrine tumours and small-cell lung cancer (NCT02936323)159. The utility of small-molecule drug conjugates can be limited by rapid plasma clearance; however, if this issue is overcome, this drug class could have notable potential to reach tumour cells in otherwise difficult-to-reach environments such as poorly vascularized tumours or the central nervous system157,158. For example, ANG1005 (paclitaxel trevatide), which is composed of three paclitaxel moieties linked to a small peptide designed to cross the blood–brain barrier via low-density lipoprotein-related protein 1 (LRP1)-mediated transcytosis, received FDA orphan drug designation for the treatment of glioblastoma in 2014; the activity of this agent has also been evaluated in patients with brain metastases or leptomeningeal disease from solid tumours in phase II trials160.
Other experimental therapeutic strategies involve the use of non-internalizing ADCs to specifically target the tumour stroma, relying on extracellular payload release mediated by proteases or linker reduction in the TME161. Such approaches might prove effective against solid tumours, especially those with dense stromal components that can otherwise impede drug delivery75. Relatedly, the question of whether standard ADCs have activity against metastases in the central nervous system (and whether that activity is due to unconjugated payload transit across the blood–brain–tumour barrier) is an area of active investigation.
Substantial opportunities also exist to innovate on ADC payloads, moving beyond standard cytotoxic drugs to targeted or immunotherapeutic agents rationally selected for their antitumour activity. For example, mirzotamab clezutoclax is a B7-H3 (CD276)-targeted ADC carrying a pro-apoptotic BCL-XL inhibitor payload, which is being investigated in early phase clinical trials (NCT03595059)162. Other novel ADCs carry immunostimulatory agents, such as chemokines, Toll-like receptor agonists or STING agonists, and are designed to recruit and/or activate immune effector cells against tumour-associated antigens, building upon the existing immunogenicity of naked antitumour mAbs163–165. Several ADCs carrying cytotoxic radioisotopes have demonstrated clinical activity against lymphomas, including the CD20-targeted agents ibritumomab tiuxetan166, 131I-tositumomab167 and 131I-rituximab168. Similar agents are being studied for the treatment of prostate cancer (targeting prostate-specific membrane antigen), gastrointestinal tumours (targeting carcinoembryonic antigen) and glioblastoma (targeting EGFR), among other cancer types169. Oligonucleotides can be delivered using antibodies, raising the fascinating prospect of selectively modulating cellular signalling pathways at the level of translation in vivo170. Similar to the highly potent cytotoxic payloads used by approved ADCs, novel payloads need not be suitable for systemic delivery in their unconjugated form, which might broaden the choice of candidate payload agents.
ADC-extrinsic strategies
Beyond drug design and preclinical research, clinical and translational investigators have the practical responsibility to explore the true clinical potential of ADCs through the thoughtful design of clinical trials. This task is twofold: (1) to identify those patients who are most likely to benefit from ADC therapy (and spare others from unnecessary toxicities); and (2) to investigate rational therapeutic partners that might synergize with ADCs to augment their clinical efficacy.
With respect to the first task, the need for improved predictive biomarkers to direct ADC therapy is clear126,134. When biomarkers are applied to select patients for treatment with these agents in clinical trials, IHC is the primary modality used to measure the expression of target proteins. However, IHC is a semi-quantitative assay at best, and various cut-offs have been used to define target positivity without a clear rationale. The minimum threshold density of a given cell-surface antigen for ADC activity can range broadly, and once this threshold is met, cytotoxicity might or might not correlate with the degree of target antigen expression171. As discussed, properties such as target turnover, oncogenicity, heterogeneity and expression in non-malignant tissue, as well as features of the TME, are all likely to influence the therapeutic window and efficacy of ADCs. Thus, efforts to characterize tumour sensitivity to ADCs that go beyond qualitative measures of target expression would be of great benefit to this growing field of oncology172.
Awaiting such studies, several basic principles can inform the pairing of an ADC to a given cancer type in order to maximize the chances of clinical success. In general terms, ADCs are best suited to deliver cytotoxic drugs that have marked in vitro activity against a given tumour type but have an unacceptably narrow therapeutic window when administered systemically. On the one hand, we suspect that stable ADCs with non-cleavable linkers will be most appropriate for circumstances in which the target antigen is highly and homogeneously overexpressed in a tumour-specific manner42. This strategy will probably enable adequate cancer cell destruction while minimizing systemic toxicities. On the other hand, labile and/or cleavable ADCs can be expected to overcome tumour heterogeneity or low-level target expression via the bystander effect, sometimes at the expense of off-target toxicities67,95. With target antigens that are quickly turned over and processed or with cancers that are known to be particularly sensitive to a given payload, lower ADC doses or less-potent payloads might be sufficient133. ADCs are expected to have augmented activity against tumours in which the target antigen is functionally relevant but, in many cases, target antigens merely act as a docking station for ADCs on the tumour cells and oncologically inert cell-surface markers can still be leveraged for ADC internalization and payload delivery44.
In addition, several early phase clinical trials using rational therapy combinations to augment ADC activity are underway (FIG. 4; Supplementary Table 1). One promising strategy involves using partner drugs to therapeutically modulate target antigen dynamics and thus potentiate the susceptibility of tumour cells to ADCs either by stimulating target overexpression or by promoting target degradation. To this end, several novel approaches are being explored. For example, the concurrent use of irreversible kinase inhibitors against the ADC target (such as the pan-HER inhibitor neratinib with HER2-targeted ADCs) can stimulate antigen internalization and thus ADC endocytosis and activity150. Other approaches exploit feedback mechanisms; for example, MAPK pathway inhibitors can result in the upregulation of the receptor tyrosine kinase AXL and thereby augment the activity of the AXL-targeted ADC enapotamab vedotin against melanoma cell lines173. Similarly, HER3 upregulation is often observed in EGFR-mutant tumours that are resistant to EGFR inhibitors, which sensitizes such cells to the HER3-targeted ADC patritumab deruxtecan in vitro174. Approaches to actively modulate target expression and/or dynamics on tumour cells warrant consideration of the potential unintentional effects on target expression in non-malignant cells and of the toxicities that might occur as a result.
Fig. 4 |. Rational combination therapy strategies to augment ADC activity.
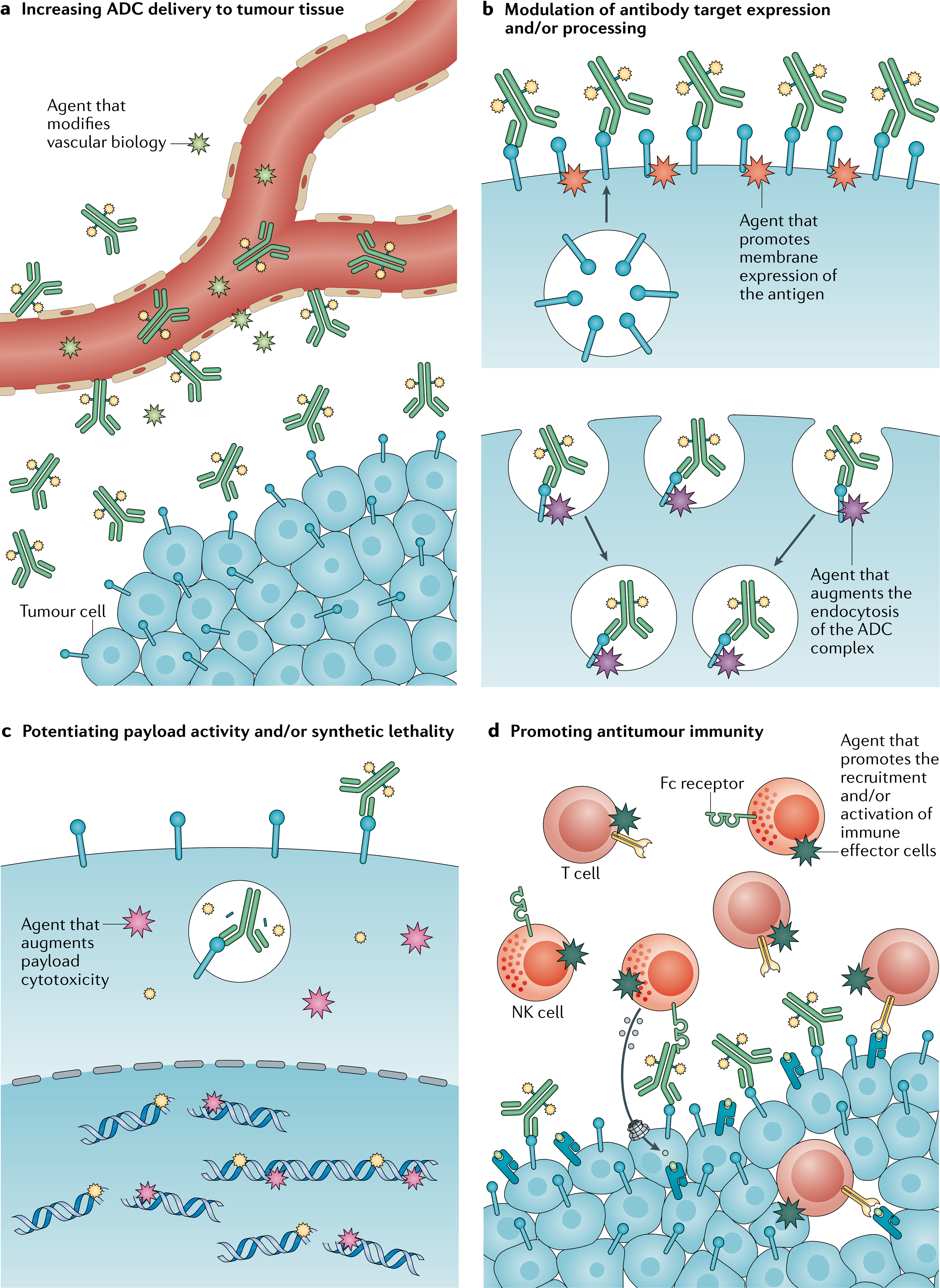
Trials of antibody–drug conjugates (ADCs) in combination with other anticancer therapies are ongoing. a | Antiangiogenic agents, such as those targeting the VEGF signalling pathway, might modify tumour vasculature in a way that improves ADC delivery to tumour tissues or enhances the cytotoxic effects of ADCs. b | Drugs that increase the cell-surface expression of the target antigen on tumour cells might promote antibody–antigen engagement. Alternatively, drugs that augment antigen turnover or degradation might promote ADC uptake and payload cleavage and release, thereby enhancing cytotoxicity. c | Payload activity can be potentiated with other agents that act synergistically through complementary mechanisms or synthetic lethality. d | Immunotherapies have the potential to build on the antitumour immunity induced by ADCs, either by enhancing antibody-dependent cellular cytotoxicity or by augmenting cell-mediated tumour recognition and immune effector function. NK, natural killer.
Beyond kinase inhibitors, strategies combining ADCs with other antibody-based therapies, such as the anti-VEFGA mAb bevacizumab, have demonstrated synergistic activity in preclinical models175–177 and early signs of clinical efficacy175–177, perhaps owing to enhanced drug delivery or other alterations in the TME that enhance tumour receptivity to ADCs. The elucidation of resistance mechanisms can also reveal potential therapeutic targets for combination treatments, such as the inhibition of polo-like kinase 1 in T-DM1 resistant cells178. Other promising combinatorial strategies include the addition of systemic cytotoxic agents with non-overlapping mechanisms of action in order to overcome tumour heterogeneity as well as synthetic lethality approaches based on payload-dependent cell damage, for example, using poly(ADP-ribose) polymerase inhibitors179,180. Indeed, three of the nine approved ADCs are labelled for use in combination with standard cytotoxic agents (TABLE 1).
More than 20 clinical studies investigating ADCs in combination with approved or experimental immunotherapies are currently under way (Supplementary Table 1). The proposed rationale supporting this strategy includes the ADC-mediated induction of immunogenic cell death and recruitment of tumour-infiltrating lymphocytes, which might promote the recognition of immunologically ‘cold’ tumours by immune effector cells and/or enhance ADC activity181. Early results from these trials are beginning to emerge. In the KATE2 trial, the addition of the anti-PD-L1 mAb atezolizumab did not seem to modulate the efficacy of T-DM1 in patients with previously treated, advanced-stage, HER2-positive breast cancer (REF.182). However, early signs of clinical activity with enfortumab vedotin plus the anti-PD-1 mAb pembrolizumab183 have prompted a randomized phase III study of this combination in patients with urothelial cancer (NCT04223856). Preclinical data suggest that certain ADC payloads have a greater capacity than others for dendritic cell priming or the recruitment of CD8+ effector T cells181, although whether this variation will manifest in meaningful differences in clinical activity remains unknown. Results from trials using various immunotherapeutic modalities to potentiate ADC activity will become available in the coming months and years.
Of note, all trials of combinatorial therapies risk subjecting patients to additional toxicities without concurrent therapeutic benefit184. Trials using such strategies should thus be rationally based on preclinical data to the greatest possible extent. Simply combining two independently effective drugs might not result in therapeutic synergy and, instead, could compromise the therapeutic index of both agents, especially when overlapping toxicities are expected.
Conclusions
ADCs are unique, powerful and capricious in ways that clinical and translational investigators are just beginning to comprehend. After decades of research and troubleshooting, technological advances and an improved mechanistic understanding of ADC activity have resulted in the development of several agents that provide demonstrable therapeutic benefit to patients with cancer. Many additional therapeutic candidates and innovative twists on the ADC approach are being actively investigated, some of which have the potential to change cancer care. Overall, the field of ADC development would benefit greatly from a more nuanced understanding of ADC processing and activity that occurs after antibody–antigen engagement both on a cell-specific and tumour-specific basis. Such knowledge would inform drug and trial design and facilitate optimal allocation of ADCs to those patients who are most likely to benefit from them. The roles of TME factors in ADC action also remain largely unexplored, as do the mechanisms of resistance to these agents in patients. As ADCs undergo broad clinical development, it is important to acknowledge that the rules that apply to standard chemotherapy or antibody-based therapies might not govern or predict the clinical properties of these agents. Models positing straightforward target-dependent drug delivery might be oversimplified and require revision to account for the mechanistic complexity of ADC action, both with respect to toxicity and efficacy. Lastly, biomarkers of response and resistance are essential for the safe and widespread use of this drug class. Such biomarkers could potentially be identified through tumour, plasma or radiological studies. If the subtleties of ADC–tumour interactions were to be better understood and harnessed, the true potential of this pharmacological platform could be broad-reaching and possibly transformative for the treatment of patients with cancer.
Supplementary Material
Key points.
Antibody–drug conjugates (ADCs) comprise three main components: an antibody, a linker and a payload. The clinical properties of ADCs depend on the characteristics of all three of these components.
The mechanism of action of ADCs is complex, often requiring drug internalization followed by intracellular processing and payload release. Unlike many standard therapies used in oncology, ADCs must be acted upon by cancer cells for optimal effectiveness.
The pharmacodynamic properties of ADCs make them uniquely suited for activity in treatment-refractory cancers, which is reflected in the current clinical indications for ADCs in oncology.
ADCs exhibit both on-target and off-target toxicities; while most toxicities seem to be related to the nature of the payload, notable examples of target-dependent toxicities exist.
Important and potentially practice-changing innovations in ADC design, biomarker development and combination therapies are ongoing in preclinical and clinical studies.
An improved understanding of the interactions between ADCs and tumours is essential for clinicians and scientists to realize the true potential of this drug class for the treatment of cancer.
Acknowledgements
All authors acknowledge support from the NCI Cancer Center Support Grant P30-CA008748. J.Z.D. acknowledges support from the Paul Calabresi Career Development Award for Clinical Oncology K12 CA184746 and a 2020 Conquer Cancer–Breast Cancer Research Foundation Young Investigator Award. S.C. acknowledges support from the Breast Cancer Research Foundation. The authors thank Linda Vahdat and Pedram Razavi of Memorial Sloan Kettering Cancer Center for editorial assistance and appreciate the helpful comments and suggestions provided by the journal editors and reviewers.
Competing interests
J.Z.D. has received Honoraria from OncLive. S.M. has received institutional research support from AstraZeneca, Daiichi Sankyo, Genentech, Novartis and Seattle Genetics; has participated in consulting/advisory boards for AstraZeneca, Daiichi Sankyo, Genentech, Macrogenics and Seattle Genetics; and has received speakers’ bureau from AstraZeneca, Daiichi Sankyo, Genentech and Seattle Genetics. S.C. has received consulting fees from Eli Lilly, Novartis and Paige.ai, and has received research support (via his institution) from Daiichi-Sankyo, Eli Lilly, Novartis and Sanofi.
Footnotes
Peer review information
Nature Reviews Clinical Oncology thanks Howard Burris III, Yasuhiro Matsumura, Dhaval K. Shah and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41571-021-00470-8.
References
- 1.Tolcher AW. Antibody drug conjugates: lessons from 20 years of clinical experience. Ann. Oncol 27, 2168–2172 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Alley SC, Okeley NM & Senter PD Antibody-drug conjugates: targeted drug delivery for cancer. Curr. Opin. Chem. Biol 14, 529–537 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Carter PJ & Senter PD Antibody-drug conjugates for cancer therapy. Cancer J. 14, 154–169 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Sievers EL & Senter PD Antibody-drug conjugates in cancer therapy. Annu. Rev. Med 64, 15–29 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Drake PM & Rabuka D Recent developments in ADC technology: preclinical studies signal future clinical trends. BioDrugs 31, 521–531 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deonarain MP, Yahioglu G, Stamati I & Marklew J Emerging formats for next-generation antibody drug conjugates. Expert Opin. Drug Dis 10, 463–481 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Beck A, Goetsch L, Dumontet C & Corvaia N Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov 16, 315–337 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Ehrlich P in The Collected Papers of Paul Ehrlich 596–618 (Pergamon, 1956). [Google Scholar]
- 9.Strebhardt K & Ullrich A Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 8, 473–480 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Mathe G, Lou TB & Bernard J Effet sur la leucemie 1210 de la souris dune combinaison par diazotation da-methopterine et de gamma-globulines de hamsters porteurs de cette leucemie par heterogreffe. Presse Med. 66, 571–571 (1958). [PubMed] [Google Scholar]
- 11.Rowland GF, Oneill GJ & Davies DAL Suppression of tumor-growth in mice by a drug-antibody conjugate using a novel approach to linkage. Nature 255, 487–488 (1975). [DOI] [PubMed] [Google Scholar]
- 12.Kohler G & Milstein C Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). [DOI] [PubMed] [Google Scholar]
- 13.Moolten FL & Cooperband SR Selective destruction of target cells by diphtheria toxin conjugated to antibody directed against antigens on the cells. Science 169, 68–70 (1970). [DOI] [PubMed] [Google Scholar]
- 14.Elias DJ et al. Phase I clinical comparative study of monoclonal antibody KS1/4 and KS1/4-methotrexate immunconjugate in patients with non-small cell lung carcinoma. Cancer Res. 50, 4154–4159 (1990). [PubMed] [Google Scholar]
- 15.Saleh MN et al. Phase I trial of the anti-Lewis Y drug immunoconjugate BR96-Doxorubicin in patients with Lewis Y-expressing epithelial tumors. J. Clin. Oncol 18, 2282–2292 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Schneck D et al. Disposition of a murine monoclonal antibody vinca conjugate (KS1/4-DAVLB) in patients with adenocarcinomas. Clin. Pharmacol. Ther 47, 36–41 (1990). [DOI] [PubMed] [Google Scholar]
- 17.Ford CH et al. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br. J. Cancer 47, 35–42 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sievers EL et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol 19, 3244–3254 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Bross PF et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res 7, 1490–1496 (2001). [PubMed] [Google Scholar]
- 20.Younes A et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol 30, 2183–2189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Younes A, Yasothan U & Kirkpatrick P Brentuximab vedotin. Nat. Rev. Drug Discov 11, 19–20 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Verma S et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med 367, 1783–1791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carter PJ & Lazar GA Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov 17, 197–223 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Schuurman J & Parren PW Editorial overview: special section: new concepts in antibody therapeutics: what’s in store for antibody therapy? Curr. Opin. Immunol 40, Vii–Xiii (2016). [DOI] [PubMed] [Google Scholar]
- 25.Vidarsson G, Dekkers G & Rispens T IgG subclasses and allotypes: from structure to effector functions. Front. Immunol 10.3389/fimmu.2014.00520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiller KE & Tessier PM Advances in antibody design. Annu. Rev. Biomed. Eng 17, 191–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu JF, Song YP & Tian W How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol 13, 45 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agarwal P & Bertozzi CR Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug Chem 26, 176–192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffmann RM et al. Antibody structure and engineering considerations for the design and function of antibody drug conjugates (ADCs). Oncoimmunology 7, e1395127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hock MB, Thudium KE, Carrasco-Triguero M & Schwabe NF Immunogenicity of Antibody drug conjugates: bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 17, 35–43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Minckwitz G et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med 380, 617–628 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Modi S et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med 382, 610–621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bardia A et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N. Engl. J. Med 380, 741–751 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg JE et al. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J. Clin. Oncol 37, 2592–2600 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stepan LP et al. Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: potential implications as a cancer therapeutic target. J. Histochem. Cytochem 59, 701–710 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pegram MD, Konecny G & Slamon DJ The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat. Res 103, 57–75 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Challita-Eid PM et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 76, 3003–3013 (2016). [DOI] [PubMed] [Google Scholar]
- 38.van der Weyden CA, Pileri SA, Feldman AL, Whisstock J & Prince HM Understanding CD30 biology and therapeutic targeting: a historical perspective providing insight into future directions. Blood Cancer J. 7, e603 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tedder TF, Tuscano J, Sato S & Kehrl JH CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu. Rev. Immunol 15, 481–504 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Pfeifer M et al. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 29, 1578–1586 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Gebhart G et al. Molecular imaging as a tool to investigate heterogeneity of advanced HER2-positive breast cancer and to predict patient outcome under trastuzumab emtansine (T-DM1): the ZEPHIR trial. Ann. Oncol 27, 619–624 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Metzger O et al. HER2 heterogeneity as a predictor of response to neoadjuvant T-DM1 plus pertuzumab: results from a prospective clinical trial. J. Clin. Oncol 37 (Suppl. 15), 502 (2019). [Google Scholar]
- 43.de Goeij BE et al. High turnover of tissue factor enables efficient intracellular delivery of antibody-drug conjugates. Mol. Cancer Ther 14, 1130–1140 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Damelin M, Zhong WY, Myers J & Sapra P Evolving strategies for target selection for antibody-drug conjugates. Pharm. Res 32, 3494–3507 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Jain N, Smith SW, Ghone S & Tomczuk B Current ADC linker chemistry. Pharm. Res 32, 3526–3540 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsuchikama K & An ZQ Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 9, 33–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drake PM & Rabuka D An emerging playbook for antibody-drug conjugates: lessons from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr. Opin. Chem. Biol 28, 174–180 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Hamann PR et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem 13, 47–58 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Erickson HK et al. The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol. Cancer Ther 11, 1133–1142 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Goldenberg DM, Cardillo TM, Govindan SV, Rossi EA & Sharkey RM Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 6, 22496–22512 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Polson AG et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: target and linker-drug selection. Cancer Res. 69, 2358–2364 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Kanellos J, Pietersz GA & Mckenzie IF Studies of methotrexate monoclonal-antibody conjugates for immunotherapy. J. Natl Cancer Inst 75, 319–332 (1985). [PubMed] [Google Scholar]
- 53.Starling JJ et al. In vivo antitumor activity of a monoclonal antibody-Vinca alkaloid immunoconjugate directed against a solid tumor membrane antigen characterized by heterogeneous expression and noninternalization of antibody-antigen complexes. Cancer Res. 51, 2965–2972 (1991). [PubMed] [Google Scholar]
- 54.Trail PA et al. Cure of xenografted human carcinomas by Br96-doxorubicin immunoconjugates. Science 261, 212–215 (1993). [DOI] [PubMed] [Google Scholar]
- 55.Teicher BA & Chari RVJ Antibody conjugate therapeutics: challenges and potential. Clin. Cancer Res 17, 6389–6397 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Sedlacek HH Antibodies as Carriers of Cytotoxicity (Karger, 1992). [Google Scholar]
- 57.Mach J-P et al. Tumor localization of radio-labeled antibodies against carcinoembryonic antigen in patients with carcinoma: a critical evaluation. N. Engl. J. Med 303, 5–10 (1980). [DOI] [PubMed] [Google Scholar]
- 58.Liu CN et al. Eradication of large colon tumor xenografts by targeted delivery of maytansinoids. Proc. Natl Acad. Sci. USA 93, 8618–8623 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Senter PD Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol 13, 235–244 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Waight AB et al. Structural basis of microtubule destabilization by potent auristatin anti-mitotics. PLoS ONE 11, e0160890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ricart AD Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res 17, 6417–6427 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Oroudjev E et al. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer Ther 9, 2700–2713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ogitani Y et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res 22, 5097–5108 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Hamblett KJ et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res 10, 7063–7070 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Sun XX et al. Effects of drug-antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjugate Chem. 28, 1371–1381 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Lyon RP et al. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol 33, 733–735 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Ogitani Y, Hagihara K, Oitate M, Naito H & Agatsuma T Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 107, 1039–1046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li F. et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. 76, 2710–2719 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Tolcher AW The evolution of antibody-drug conjugates: a positive inflexion point. Am. Soc. Clin. Oncol. Educ. Book 40, 1–8 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Girish S et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother. Pharmacol. 69, 1229–1240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bender BC et al. A population pharmacokinetic/pharmacodynamic model of thrombocytopenia characterizing the effect of trastuzumab emtansine (T-DM1) on platelet counts in patients with HER2-positive metastatic breast cancer. Cancer Chemother. Pharmacol 70, 591–601 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Singh AP & Shah DK Application of a PK-PD modeling and simulation-based strategy for clinical translation of antibody-drug conjugates: a case study with trastuzumab emtansine (T-DM1). AAPS J. 19, 1054–1070 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fujimori K, Covell DG, Fletcher JE & Weinstein JN A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J. Nucl. Med 31, 1191–1198 (1990). [PubMed] [Google Scholar]
- 74.Minchinton AI & Tannock IF Drug penetration in solid tumours. Nat. Rev. Cancer 6, 583–592 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Matsumura Y Cancer stromal targeting therapy to overcome the pitfall of EPR effect. Adv. Drug Deliv. Rev 154–155, 142–150 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Thurber GM & Wittrup KD A mechanistic compartmental model for total antibody uptake in tumors. J. Theor. Biol 314, 57–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chari RVJ Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc. Chem. Res 41, 98–107 (2008). [DOI] [PubMed] [Google Scholar]
- 78.Lu G et al. Co-administered antibody improves penetration of antibody-dye conjugate into human cancers with implications for antibody-drug conjugates. Nat. Commun 11, 5667 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alley SC et al. The pharmacologic basis for antibody-auristatin conjugate activity. J. Pharmacol. Exp. Ther 330, 932–938 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Giddabasappa A et al. Biodistribution and targeting of Anti-5T4 antibody-drug conjugate using fluorescence molecular tomography. Mol. Cancer Ther 15, 2530–2540 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Juweid M et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 52, 5144–5153 (1992). [PubMed] [Google Scholar]
- 82.Ritchie M, Tchistiakova L & Scott N Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 5, 13–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Acchione M, Kwon H, Jochheim CM & Atkins WM Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. mAbs 4, 362–372 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Redman JM, Hill EM, AlDeghaither D & Weiner LM Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol 67, 28–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Junttila TT, Li GM, Parsons K, Phillips GL & Sliwkowski MX Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat 128, 347–356 (2011). [DOI] [PubMed] [Google Scholar]
- 86.Moasser MM The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26, 6469–6487 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tai YT et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 123, 3128–3138 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kovtun YV & Goldmacher VS Cell killing by antibody-drug conjugates. Cancer Lett. 255, 232–240 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Jedema I et al. Internalization and cell cycle-dependent killing of leukemic cells by Gemtuzumab Ozogamicin: rationale for efficacy in CD33-negative malignancies with endocytic capacity. Leukemia 18, 316–325 (2004). [DOI] [PubMed] [Google Scholar]
- 90.Sutherland MSK et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J. Biol. Chem 281, 10540–10547 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Erickson HK et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 66, 4426–4433 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Staudacher AH & Brown MP Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Brit. J. Cancer 117, 1736–1742 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kovtun YV et al. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 66, 3214–3221 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Dokter W et al. The preclinical profile of the duocarmycin-based HER2-targeting ADC SYD985 predicts for clinical benefit in low HER2-expressing breast cancers. Mol. Cancer Ther 14, 692–7030 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Singh AP, Sharma S & Shah DK Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J. Pharmacokinet. Pharmacodyn 43, 567–582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vasan N, Baselga J & Hyman DM A view on drug resistance in cancer. Nature 575, 299–309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Drakaki A et al. Docetaxel with or without ramucirumab after immune checkpoint inhibition in platinum-refractory metastatic urothelial carcinoma (mUC): Prespecified subgroup analysis from the phase 3 RANGE trial. J. Clin. Oncol 36 (Suppl. 6), 434 (2018). [Google Scholar]
- 98.Shitara K et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med 382, 2419–2430 (2020). [DOI] [PubMed] [Google Scholar]
- 99.Barok M, Tanner M, Koninki K & Isola J Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 13, R46 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Coley HM Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev 34, 378–390 (2008). [DOI] [PubMed] [Google Scholar]
- 101.Perez EA et al. Randomized phase II study of two irinotecan schedules for patients with metastatic breast cancer refractory to an anthracycline, a taxane, or both. J. Clin. Oncol 22, 2849–2855 (2004). [DOI] [PubMed] [Google Scholar]
- 102.National Comprehensive Cancer Network. Breast cancer https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (2021).
- 103.Seol H et al. Intratumoral heterogeneity of HER2 gene amplification in breast cancer: its clinicopathological significance. Mod. Pathol 25, 938–948 (2012). [DOI] [PubMed] [Google Scholar]
- 104.Modi S et al. Antitumor activity and safety of trastuzumab deruxtecan in patients with HER2-low-expressing advanced breast cancer: results from a Phase Ib study. J. Clin. Oncol 38, 1887–1896 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tijink BM et al. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res 12, 6064–6072 (2006). [DOI] [PubMed] [Google Scholar]
- 106.Kerckhove N et al. Long-term effects, pathophysiological mechanisms, and risk factors of chemotherapy-induced peripheral neuropathies: a comprehensive literature review. Front. Pharmacol 8, 86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kamba T & McDonald DM Mechanisms of adverse effects of anti-VEGF therapy for cancer. Brit. J. Cancer 96, 1788–1795 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sorrentino MF, Kim J, Foderaro AE & Truesdell AG 5-fluorouracil induced cardiotoxicity: review of the literature. Cardiol. J 19, 453–458 (2012). [DOI] [PubMed] [Google Scholar]
- 109.Sakamoto S et al. Expression of Lewisa, Lewisb, Lewisx, Lewisy, siayl-Lewisa, and sialyl-Lewisx blood group antigens in human gastric carcinoma and in normal gastric tissue. Cancer Res. 49, 745–752 (1989). [PubMed] [Google Scholar]
- 110.Tolcher AW et al. Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J. Clin. Oncol 17, 478–484 (1999). [DOI] [PubMed] [Google Scholar]
- 111.Riechelmann H et al. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral. Oncol 44, 823–829 (2008). [DOI] [PubMed] [Google Scholar]
- 112.Donaghy H Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs 8, 659–671 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Banerji U et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2-expressing breast cancer: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 20, 1124–1135 (2019). [DOI] [PubMed] [Google Scholar]
- 114.Sendur MA, Aksoy S & Altundag K Cardiotoxicity of novel HER2-targeted therapies. Curr. Med. Res. Opin 29, 1015–1024 (2013). [DOI] [PubMed] [Google Scholar]
- 115.Ponde N et al. Trastuzumab emtansine (T-DM1)-associated cardiotoxicity: Pooled analysis in advanced HER2-positive breast cancer. Eur. J. Cancer 126, 65–73 (2020). [DOI] [PubMed] [Google Scholar]
- 116.Cote GM, Sawyer DB & Chabner BA ERBB2 inhibition and heart failure. N. Engl. J. Med 367, 2150–2153 (2012). [DOI] [PubMed] [Google Scholar]
- 117.Saber H & Leighton JK An FDA oncology analysis of antibody-drug conjugates. Regul. Toxicol. Pharm 71, 444–452 (2015). [DOI] [PubMed] [Google Scholar]
- 118.Masters JC, Nickens DJ, Xuan D, Shazer RL & Amantea M Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest. New Drugs 36, 121–135 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Eaton JS, Miller PE, Mannis MJ & Murphy CJ Ocular adverse events associated with antibody-drug conjugates in human clinical trials. J. Ocul. Pharmacol. Ther 31, 589–604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.de Goeij BE & Lambert JM New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol 40, 14–23 (2016). [DOI] [PubMed] [Google Scholar]
- 121.Bardia A et al. Efficacy and safety of anti-Trop-2 antibody drug conjugate sacituzumab govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 35, 2141–2148 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cardillo TM, Govindan SV, Sharkey RM, Trisal P & Goldenberg DM Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: preclinical studies in human cancer xenograft models and monkeys. Clin. Cancer Res 17, 3157–3169 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mahalingaiah PK et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther 200, 110–125 (2019). [DOI] [PubMed] [Google Scholar]
- 124.Uppal H et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin. Cancer Res 21, 123–133 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Zhao H et al. Modulation of micropinocytosis-mediated internalization decreases ocular toxicity of antibody-drug conjugates. Cancer Res. 78, 2115–2126 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Makawita S & Meric-Bernstam F Antibody-drug conjugates: patient and treatment selection. Am. Soc. Clin. Oncol. Educ. Book 40, 1–10 (2020). [DOI] [PubMed] [Google Scholar]
- 127.Ott PA et al. Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patients with advanced melanoma. J. Clin. Oncol 32, 3659–3666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yardley DA et al. EMERGE: a randomized phase II study of the antibody-drug conjugate glembatumumab vedotin in advanced glycoprotein NMB-expressing breast cancer. J. Clin. Oncol 33, 1609–1619 (2015). [DOI] [PubMed] [Google Scholar]
- 129.Saber H, Simpson N, Ricks TK & Leighton JK An FDA oncology analysis of toxicities associated with PBD-containing antibody-drug conjugates. Regul. Toxicol. Pharm 107, 104429 (2019). [DOI] [PubMed] [Google Scholar]
- 130.Drago JZ et al. Inferences about drug safety in phase 3 trials in oncology: Examples from advanced prostate cancer. J. Natl Cancer Inst. 10.1093/jnci/djaa134 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yardley DA et al. Quantitative measurement of HER2 expression in breast cancers: comparison with ‘real-world’ routine HER2 testing in a multicenter collaborative biomarker Study and correlation with overall survival. Breast Cancer Res. 17, 41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wolff AC et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. Arch. Pathol. Lab. Med 142, 1364–1382 (2018). [DOI] [PubMed] [Google Scholar]
- 133.Li BT et al. Ado-trastuzumab emtansine for patients With HER2-mutant lung cancers: results from a phase II basket trial. J. Clin. Oncol 36, 2532–2537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tarantino P et al. HER2-low breast cancer: pathological and clinical landscape. J. Clin. Oncol 38, 1951–1962 (2020). [DOI] [PubMed] [Google Scholar]
- 135.Hurvitz SA et al. Biomarker evaluation in the phase 3 ASCENT study of sacituzumab govitecan versus chemotherapy in patients with metastatic triple-negative breast cancer. https://www.abstractsonline.com/pp8/#!/9223/presentation/674 (2020).
- 136.Gainor JF & Shaw AT Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J. Clin. Oncol 31, 3987–3996 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Loganzo F., Sung M. & Gerber HP. Mechanisms of resistance to antibody-drug conjugates. Mol. Cancer Ther 15, 2825–2834 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Loganzo F et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther 14, 952–963 (2015). [DOI] [PubMed] [Google Scholar]
- 139.Li GM et al. Mechanisms of acquired resistance to trastuzumab emtansine in breast cancer cells. Mol. Cancer Ther 17, 1441–1453 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Rios-Luci C et al. Resistance to the antibody-drug conjugate T-DM1 is based in a reduction in lysosomal proteolytic activity. Cancer Res. 77, 4639–4651 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Chen R et al. CD30 downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to brentuximab vedotin. Mol. Cancer Ther 14, 1376–1384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Walter RB et al. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood 109, 4168–4170 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C & Gottesman MM Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov 5, 219–234 (2006). [DOI] [PubMed] [Google Scholar]
- 144.Jackson D & Stover D Using the lessons learned from the clinic to improve the preclinical development of antibody drug conjugates. Pharm. Res 32, 3458–3469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Takegawa N et al. DS-8201a, a new HER2-targeting antibody-drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2-positive gastric cancer T-DM1 resistance. Int. J. Cancer 141, 1682–1689 (2017). [DOI] [PubMed] [Google Scholar]
- 146.Chandarlapaty S et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin. Cancer Res. 18, 6784–6791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baselga J et al. Relationship between tumor biomarkers (BM) and efficacy in EMILIA, a phase III study of trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer (MBC). Cancer Res. 73, LB–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Scheuer W et al. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 69, 9330–9336 (2009). [DOI] [PubMed] [Google Scholar]
- 149.Kang JC et al. Engineering a HER2-specific antibody-drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat. Biotechnol 37, 523–526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li BT et al. HER2-mediated internalization of cytotoxic agents in ERBB2 amplified or mutant lung cancers. Cancer Discov. 10, 674–687 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Brevet M, Arcila M & Ladanyi M Assessment of EGFR mutation status in lung adenocarcinoma by immunohistochemistry using antibodies specific to the two major forms of mutant EGFR. J. Mol. Diagn 12, 169–176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hamblett KJ et al. AMG 595, an anti-EGFRvIII antibody–drug conjugate, induces potent antitumor activity against EGFRvIII-expressing glioblastoma. Mol. Cancer Ther 14, 1614–1624 (2015). [DOI] [PubMed] [Google Scholar]
- 153.Comer F, Gao C & Coats S in Innovations for Next-Generation Antibody-Drug Conjugates (ed. Damelin M) 267–280 (Springer International Publishing, 2018). [Google Scholar]
- 154.Li JY et al. A biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 35, 948–949 (2019). [DOI] [PubMed] [Google Scholar]
- 155.de Goeij BE et al. Efficient payload delivery by a bispecific antibody-drug conjugate targeting HER2 and CD63. Mol. Cancer Ther 15, 2688–2697 (2016). [DOI] [PubMed] [Google Scholar]
- 156.Andreev J et al. Bispecific antibodies and antibody-drug conjugates (ADCs) bridging HER2 and prolactin receptor improve efficacy of HER2 ADCs. Mol. Cancer Ther 16, 681–693 (2017). [DOI] [PubMed] [Google Scholar]
- 157.Zhuang C et al. Small molecule-drug conjugates: a novel strategy for cancer-targeted treatment. Eur. J. Med. Chem 163, 883–895 (2019). [DOI] [PubMed] [Google Scholar]
- 158.Casi G & Neri D Antibody-drug conjugates and small molecule-drug conjugates: opportunities and challenges for the development of selective anticancer cytotoxic agents. J. Med. Chem 58, 8751–8761 (2015). [DOI] [PubMed] [Google Scholar]
- 159.Whalen KA et al. Targeting the somatostatin receptor 2 with the miniaturized drug conjugate, PEN-221: a potent and novel therapeutic for the treatment of small cell lung cancer. Mol. Cancer Ther 18, 1926–1936 (2019). [DOI] [PubMed] [Google Scholar]
- 160.Kumthekar P et al. ANG1005, a brain-penetrating peptide-drug conjugate, shows activity in patients with breast cancer with leptomeningeal carcinomatosis and recurrent brain metastases. Clin. Cancer Res. 26, 2789–2799 (2020). [DOI] [PubMed] [Google Scholar]
- 161.Gebleux R, Stringhini M, Casanova R, Soltermann A & Neri D Non-internalizing antibody-drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. Int. J. Cancer 140, 1670–1679 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Carneiro BA & El-Deiry WS Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol 17, 395–417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mohit E & Rafati S Chemokine-based immunotherapy: delivery systems and combination therapies. Immunotherapy 4, 807–840 (2012). [DOI] [PubMed] [Google Scholar]
- 164.Cetinbas NM et al. Tumor cell-intrinsic STING pathway is activated in the presence of cues from immune cells and contributes to the anti-tumor activity of tumor cell-targeted STING agonist antibody-drug conjugates [abstract]. J. Immunother. Cancer 8, A373 (2020). [Google Scholar]
- 165.Moyes K et al. A systemically administered, conditionally active TLR8 agonist for the treatment of HER2-expressing tumors. Cancer Res. 79, 3271 (2019). [Google Scholar]
- 166.Witzig TE et al. Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol 20, 2453–2463 (2002). [DOI] [PubMed] [Google Scholar]
- 167.Kaminski MS et al. 131I-tositumomab therapy as initial treatment for follicular lymphoma. N. Engl. J. Med 352, 441–449 (2005). [DOI] [PubMed] [Google Scholar]
- 168.Leahy MF, Seymour JF, Hicks RJ & Turner JH Multicenter phase II clinical study of iodine-131-rituximab radioimmunotherapy in relapsed or refractory indolent non-Hodgkin’s lymphoma. J. Clin. Oncol 24, 4418–4425 (2006). [DOI] [PubMed] [Google Scholar]
- 169.Gill MR, Falzone N, Du Y & Vallis KA Targeted radionuclide therapy in combined-modality regimens. Lancet Oncol. 18, e414–e423 (2017). [DOI] [PubMed] [Google Scholar]
- 170.Dovgan I, Koniev O, Kolodych S & Wagner A Antibody-oligonucleotide conjugates as therapeutic, imaging, and detection agents. Bioconjug Chem. 30, 2483–2501 (2019). [DOI] [PubMed] [Google Scholar]
- 171.Perez HL et al. Antibody-drug conjugates: current status and future directions. Drug Discov. Today 19, 869–881 (2014). [DOI] [PubMed] [Google Scholar]
- 172.Dornan D & Settleman J in Antibody-Drug Conjugates and Immunotoxins: From Pre-Clinical Development to Therapeutic Applications (ed. Phillips GL) 77–90 (Springer, 2013). [Google Scholar]
- 173.Boshuizen J et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med 24, 203–212 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Yonesaka K et al. An HER3-targeting antibody-drug conjugate incorporating a DNA topoisomerase I inhibitor U3–1402 conquers EGFR tyrosine kinase inhibitor-resistant NSCLC. Oncogene 38, 1398–1409 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Ponte JF et al. Mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, potentiates the activity of standard of care therapeutics in ovarian cancer models. Neoplasia 18, 775–784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.O’Malley DM et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRalpha)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol. Oncol 157, 379–385 (2020). [DOI] [PubMed] [Google Scholar]
- 177.Moore KN et al. Phase 1b study of anti-NaPi2b antibody-drug conjugate lifastuzumab vedotin (DNIB0600A) in patients with platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol 158, 631–639 (2020). [DOI] [PubMed] [Google Scholar]
- 178.Saatci O et al. Targeting PLK1 overcomes T-DM1 resistance via CDK1-dependent phosphorylation and inactivation of Bcl-2/xL in HER2-positive breast cancer. Oncogene 37, 2251–2269 (2018). [DOI] [PubMed] [Google Scholar]
- 179.Zhong H et al. Improved therapeutic window in BRCA-mutant tumors with antibody-linked pyrrolobenzodiazepine dimers with and without PARP inhibition. Mol. Cancer Ther 18, 89–99 (2019). [DOI] [PubMed] [Google Scholar]
- 180.Cardillo TM et al. Synthetic lethality exploitation by an anti-trop-2-SN-38 antibody-drug conjugate, IMMU-132, Plus PARP inhibitors in BRCA1/2-wild-type triple-negative breast cancer. Clin. Cancer Res. 23, 3405–3415 (2017). [DOI] [PubMed] [Google Scholar]
- 181.Gerber HP, Sapra P, Loganzo F & May C Combining antibody-drug conjugates and immune-mediated cancer therapy: What to expect? Biochem. Pharmacol. 102, 1–6 (2016). [DOI] [PubMed] [Google Scholar]
- 182.Emens LA et al. Results from KATE2, a randomized phase 2 study of atezolizumab (atezo) plus trastuzumab emtansine (T-DM1) vs placebo (pbo)+T-DM1 in previously treated HER2+advanced breast cancer (BC). Cancer Res. 79, PD3–01 (2019). [Google Scholar]
- 183.Rosenberg JE et al. Study EV-103: Preliminary durability results of enfortumab vedotin plus pembrolizumab for locally advanced or metastatic urothelial carcinoma. J. Clin. Oncol 38, 441–441 (2020). [Google Scholar]
- 184.Macpherson IR & Cassidy J Challenges in combinational oncology studies. Pharm. Med 22, 85–97 (2008). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.