

Abstract
Smoking is a major risk factor for a variety of diseases, including cancer and immune-mediated inflammatory diseases. Tobacco smoke contains a mixture of chemicals, including a host of reactive oxygen- and nitrogen species (ROS and RNS), among others, that can damage cellular and sub-cellular targets, such as lipids, proteins, and nucleic acids. A growing body of evidence supports a key role for smoking-induced ROS and the resulting oxidative stress in inflammation and carcinogenesis. This comprehensive and up-to-date review covers four interrelated topics, including ‘smoking’, ‘oxidative stress’, ‘inflammation’, and ‘cancer’. The review discusses each of the four topics, while exploring the intersections among the topics by highlighting the macromolecular damage attributable to ROS. Specifically, oxidative damage to macromolecular targets, such as lipid peroxidation, post-translational modification of proteins, and DNA adduction, as well as enzymatic and non-enzymatic antioxidant defense mechanisms, and the multi-faceted repair pathways of oxidized lesions are described. Also discussed are the biological consequences of oxidative damage to macromolecules if they evade the defense mechanisms and/or are not repaired properly or in time. Emphasis is placed on the genetic- and epigenetic alterations that may lead to transcriptional deregulation of functionally-important genes and disruption of regulatory elements. Smoking-associated oxidative stress also activates the inflammatory response pathway, which triggers a cascade of events of which ROS production is an initial yet indispensable step. The release of ROS at the site of damage and inflammation helps combat foreign pathogens and restores the injured tissue, while simultaneously increasing the burden of oxidative stress. This creates a vicious cycle in which smoking-related oxidative stress causes inflammation, which in turn, results in further generation of ROS, and potentially increased oxidative damage to macromolecular targets that may lead to cancer initiation and/or progression.
Keywords: carcinogenesis, inflammatory disease, oxidative damage, reactive oxygen species (ROS), tar, tobacco
Graphic abstract
1. Introduction
The prevalence of smoking tobacco cigarettes in the United States has decreased from 42% to 14% since the Surgeon General’s original report on the adverse health consequences of smoking in 1964 [1, 2]. This decline in smoking prevalence has been credited to the compelling scientific evidence informing public health policies and practices, and regulation of the manufacture, distribution, and marketing of tobacco products [1–3]. The public health initiatives to combat smoking, among others, have been centered on harm reduction/elimination [1, 3–5]. The goal has been to deter nonsmokers from picking up smoking in the first place, and potentially becoming long-term smokers (i.e., prevent smoking initiation), as well as educate habitual smokers on the health risks associated with smoking (i.e., promote smoking cessation) [2, 3]. These efforts have largely been successful as evidenced by the substantial drop in the prevalence of smoking and the denormalization of smoking in the United States and many parts of the world [1–3]. Despite these successes, smoking-associated diseases and deaths still remain a significant global health problem [6, 7]. For example, smoking-related lung cancer is a leading cause of death, worldwide, accounting for an estimated 9.6 million deaths in 2018 [6, 7]. In the United States, nearly 30% of all cancer deaths and 80% of lung cancer deaths are attributed to smoking [7]. The discordant trends in smoking prevalence and disease incidence or mortality rates have been ascribed to a wide variety of factors [1]. Of these, the evolving landscape of tobacco products together with innovative advertising and aggressive marketing strategies employed by the manufacturers of these products deserves special attention [4]. Notwithstanding the tenacious smoking prevention and cessation efforts, the tobacco industry continues to entice new generation of smokers as well as users of novel tobacco products, such as electronic cigarettes and heat-not-burn devices (e.g., IQOS), to maintain an upward trajectory in profit [4, 5, 8].
Approximately 90% of all lung cancer cases are directly linked to smoking [7]. Lung cancer is a devastating disease with a 5-year survival rate of only 15% [7]. Other smoking-related cancers, such as liver and pancreatic cancers, also have low 5-year survival rates (18% and 9%, respectively) [7]. The existing data clearly show that smoking increases risk of cancer at multiple organ sites, often leading to premature deaths [7]. Approximately, one third of all cancer-related deaths in the United States are linked to chronic smoking [1, 7]. Worldwide, smoking is also the leading cause of otherwise preventable deaths [3, 7]. In the United States alone, nearly 500,000 deaths per year can be attributed to smoking-related diseases [2, 3]. There is ample evidence to support that habitual smoking is a primary risk factor for chronic obstructive pulmonary disease (COPD), cardiovascular disease (CVD), immune-mediated inflammatory diseases, and a variety of cancer types [1–3, 9, 10]. Specifically, smoking is the leading risk factor for head and neck cancers and lung cancer [1, 7]. There is also a strong link between smoking and bladder, pancreatic, and renal pelvis cancers. Smoking has also been implicated in the etiology of colon, liver, and stomach cancers [1, 7].
Tobacco smoke contains a complex mixture of chemicals, including a host of reactive oxygen- and nitrogen species (ROS and RNS), among others, that can damage macromolecular targets, such as lipids, proteins, and nucleic acids. Accumulating evidence shows an important role for smoking-induced ROS and the resulting oxidative stress in inflammation and cancer. This review article centers on elucidating the interconnections among smoking, oxidative stress, inflammation, and cancer by highlighting the macromolecular damage that occurs consequent to exposure to smoke-derived/induced ROS. A focal point of the article is the vicious cycle in which smoking-related oxidative stress causes inflammation, which in turn, results in further generation of ROS, and potentially increased oxidative damage to macromolecules that may trigger carcinogenesis. We note that there is a distinction between oxidation response elicited by ‘direct’ smoke-derived ROS and the response stimulated by ‘indirect’ smoke-induced ROS. It is important to distinguish the former from the latter as the direct oxidative damage by free radicals and ROS present in cigarette smoke may be less pronounced than the indirect damage/response triggered by other toxicants and carcinogens in the smoke, e.g., aldehydes or particulate matter, or their secondarily formed metabolites (see, Section 7). In addition, it is worth mentioning that cigarette smoke contains a wide variety of carcinogenic compounds, many of which exert their effects through mechanisms that do not involve oxidative damage [1, 11–14]. We acknowledge that there is a wealth of information on smoking-induced ROS and oxidative stress in humans, animal models, and in vitro cell culture systems [15–17]. We should, however, emphasize that the objective of this review is to showcase representative studies, but by no means, discuss the existing literature (in its entirety), to establish the interplays among smoking, oxidative stress, inflammation, and cancer.
This comprehensive and up-to-date review covers four important topics, including ‘smoking’, ‘oxidative stress’, ‘inflammation’, and ‘cancer’. Rather than capturing only one or two topics, the present review discusses each of the four topics, while exploring the intersections among the topics by highlighting the macromolecular damage attributable to ROS. There is an extensive body of literature related to the four topics discussed in this review (see, Supplementary Table S1). We have conducted a thorough PubMed search and literature review to select and discuss the most relevant and representative (I) cell culture; (II) animal; and (III) human studies related to each of the four topics covered by this review. Queries for PubMed search and the results are shown in Supplementary Table S1. We reiterate that the overarching goal of this review is to showcase representative studies selected from our PubMed search and literature review, but by no means, discuss the entire body of literature related to the four topics covered in this review. In the following sections, we will highlight the selected studies and discuss them in detail, as appropriate.
2. Cigarette filters and filtration vents
During the 1950s, many cigarette companies began incorporating a filter of some kind into the structure of their cigarettes [18, 19]. Introduction of the filter was aimed at reducing overall levels of tar and nicotine received by smokers in order to mitigate harm [19]. Tar contains many carcinogenic chemicals, including polycyclic aromatic hydrocarbons (PAHs), aromatic amines, tobacco-specific nitrosamines (TSNAs), and phenolic compounds [20]. In aqueous solution, tar also produces several oxidative agents through redox-cycling reactions, when its constituents come in contact with molecular oxygen in the human lungs [20, 21]. Thus, reduction of tar yield in cigarettes is desirable because not only does the tar comprise carcinogenic compounds but also the by-products of redox cycling in tar, such as free radicals and other oxidative agents, are linked to cancer development and increased inflammatory response [21–24]. Likewise, reduction of nicotine yield in cigarettes is warranted because nicotine reacts with nitrates from tobacco during combustion, thereby generating carcinogenic compounds, such as TSNAs [1]. Nicotine is also a highly addictive substance; smokers’ nicotine-dependence makes them continue to smoke, and experience withdrawal syndrome when refraining from smoking or attempting to quit [1, 25]. For decades, a long-held view on smoking addiction and mortality has been that ‘people smoke for the nicotine and die from the tar’ [1–4].
Today, cellulose acetate is the most commonly used filter in tobacco cigarettes [19]. This type of filter efficiently removes large quantities of tar from cigarette smoke [18]. However, even though filtered cigarettes deliver substantially reduced levels of tar and carcinogenic compounds, they do not completely eliminate exposure to toxicants and carcinogens present in cigarette smoke [18, 19]. Therefore, additional modifications in cigarette design were sought out to lower smokers’ exposure to harmful constituents of tobacco smoke. Accordingly, filtration vents were added to cigarette design to reduce smoke exposure by allowing air to enter the filter [19]. Theoretically, this would dilute the drawn smoke with air at the filter end [26]. Machine testing of the modified cigarettes with vented filters showed promising results, i.e., they had lower yields of tar and nicotine [26]. However, smoking machines do not recapitulate human behavior and adaptations in smoking patterns [27]. In real-life, reduction of the nicotine content in cigarettes forces smokers to smoke more in order to get the same amount of nicotine on which they are dependent [1, 27]. Furthermore, whilst filter ventilation creates more airflow through the rod of a cigarette and decreases resistance when taking a puff, it also makes smokers take longer puffs, which leads to higher smoke production [27]. Larger smoke volume together with a decrease in burning temperature from filter vents, which eases deeper inhalation, allows deposition of higher quantities of toxicants and carcinogens in the smokers’ lungs [19, 27]. The net result would be more lung tissues being exposed to larger amounts of smoke, which could translate to an overall increase in disease risk [1, 27].
3. Tobacco curing
In addition to cigarette filters and filtration vents, tobacco curing and processing are other important factors influencing the tar and nicotine content of cigarettes [28]. Curing tobacco is a process necessary to prepare the leaf for consumption because in its raw, freshly picked state, the green tobacco leaf is too wet to ignite and be smoked [28]. Curing and subsequent aging allow for the slow oxidation and degradation of carotenoids in the tobacco leaf. This process produces various compounds in the tobacco leaves that give cured and aged tobacco its unique flavor and aroma. Air-, fire-, flue-, or sun-curing, and fermentation/sweating are widely used methods for tobacco curing [28]. The distinct curing methods produce different tobacco blends with various chemical yields (e.g., tar and nicotine) when tobacco is smoked [18]. For example, air-cured (black) and fire-cured tobacco cigarettes have high nicotine content, whereas flue-cured (blond) tobacco cigarettes yield moderate to high levels of nicotine. Conversely, sun-cured tobacco cigarettes are low in nicotine. Moreover, blond tobacco smoke has lower content of TSNAs but higher amounts of PAHs than black tobacco smoke [28]. On the other hand, black tobacco smoke has larger yields of aromatic amines, arsenic, and cadmium compared to blond tobacco smoke [28]. Consistent with aromatic amines being a known bladder carcinogen, smokers of black tobacco cigarettes have greater risk of bladder cancer than smokers of blond tobacco cigarettes [1, 29].
4. Carcinogenic compounds in tobacco smoke
The International Agency for Research on Cancer (IARC) has classified carcinogens into 4 categories, including Classes 1, 2A, 2B, and 3 [11, 30]. Class 1 carcinogens are known to cause cancer in humans. Class 2A carcinogens are most likely to cause cancer in humans, whereas Class 2B agents could possibly be carcinogenic in humans. Class 3 comprises compounds for which carcinogenicity data are limited [11]. To decipher the role of smoking in cancer development, it is important to understand what carcinogenic compounds are produced when a cigarette is smoked [12–14, 31, 32]. Tobacco smoke contains more than 7,000 chemicals, of which nearly 70 have been identified as known or suspected carcinogens (see, Table 1) [11]. For example, benzo[a]pyrene (B[a]P), a PAH compound found in tobacco smoke, is causally linked to lung cancer development [33], whereas 4-aminobiphenyl, a primary smoke-derived aromatic amine, is a well-known bladder carcinogen [29]. Moreover, free radicals originating from tobacco smoke are implicated in the etiology of many subtypes of oral cancer, as they induce a state of chronic inflammation that is considered a common feature of oral carcinogenesis (see, next section) [1, 22, 23, 34].
Table 1.
Selected known or suspected carcinogens in mainstream cigarette smoke.
| Chemical family | Compound | Quantity per cigarette | IARC carcinogen class |
|---|---|---|---|
| Polycyclic aromatic hydrocarbons | Benzo[a]pyrene | 8.5–17.6 ng | 1 |
| Benz[a]anthracene | 20–70 ng | 2A | |
| Dibenz[a,h]anthracene | 4 ng | 2A | |
| Benzo[b]fluorathene | 4–22 ng | 2B | |
| Benzo[j]fluorathene | 6–21 ng | 2B | |
| Benzo[k]fluorathene | 6–12 ng | 2B | |
| Dibenzo[a,i]pyrene | 1.7–3.2 ng | 2B | |
| Dibenzo[a,e]pyrene | Present | 2B | |
| Indenol[1,2,3-cd]pyrene | 4–20 ng | 2B | |
| 5-methylchrysene | U-0.6 ng | 2B | |
| Heterocyclic compounds | Benzo [b]furan | Present | 2B |
| Dibenz[a,h]acridine | U-0.1 ng | 2B | |
| Dibenz[a,j]acridine | U-10 ng | 2B | |
| Dibenzo[c,g]carbazole | U-0.7 ng | 2B | |
| Furan | 20–40 μg | 2B | |
| Aromatic amines | 2-naphthylamine | 1–22 ng | 1 |
| 4-aminobiphenyl | 2–5 ng | 1 | |
| 2-toluidine | 30–200 ng | 2A | |
| 2,6-dimethylaniline | 4–50 ng | 2B | |
| Organic compounds | Vinyl chloride | 11–15 ng | 1 |
| Ethylene oxide | 7 μg | 1 | |
| Acrylamide | Present | 2A | |
| Acetamide | 38–56 μg | 2B | |
| Acrylonitrile | 3–15 μg | 2B | |
| 1,1-dimethylhydrazine | Present | 2B | |
| Propylene oxide | U-100 ng | 2B | |
| Urethane | 20–38 ng | 2B | |
| Phenolic compounds | Caffeic acid | < 3 μg | 2B |
| Catechol | 59–81 μg | 2B | |
| Inorganic compounds | Radioisotope polonium-210 | 0.03–1.0 pCi | 1 |
| Hydrazine | 24–43 ng | 2B | |
| N-nitrosamines | N-nitrosonornicotine | 154–196 ng | 1 |
| 4-(methy1nitrosamino)-1-(3-pyridyl)-1-butanoe | 110–133 ng | 1 | |
| N-nitrosodimethylamine | 0.1–180 ng | 2A | |
| N-nitrosodiethylamine | U-25 ng | 2A | |
| N-nitrosoethylmethylamine | U-13 ng | 2B | |
| N-nitrosopyrrolidine | 1.5–110 ng | 2B | |
| N-nitrosopiperidine | U-9 ng | 2B | |
| N-nitrosodiethanolamine | U-36 ng | 2B | |
| Heterocyclic aromatic amines | 3-amino-3-methylimidazo[4,5-f]quinoline | 0.3 ng | 2A |
| 2-amino-9H-pyrido[2,3-b]indole | 25–260 ng | 2B | |
| 2-amino-3-methyl-9H-pyrido[2,3-b]indole | 2–37 ng | 2B | |
| 3-amino-1,4-dimethyl-5H-pyrido[4,3-b]indole | 0.3–0.5 ng | 2B | |
| 3-amino-1-methyl-5H-pyrido[4,3-b]indole | 0.8–1.1 ng | 2B | |
| 2-amino-6-methylpyrido[1,2-a:3’,2’-d]imidazole | 0.37–0.89 ng | 2B | |
| 2-aminodipyrido[1,2-a:3’2’-d]imidazole | 0.25–0.88 ng | 2B | |
| 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine | 11–23 ng | 2B | |
| Metals | Arsenic | 40–120 ng | 1 |
| Beryllium | 0.5 ng | 1 | |
| Nickel | U-600 ng | 1 | |
| Chromium (hexavalent) | 4–70 ng | 1 | |
| Cadmium | 41–62 ng | 1 | |
| Lead (inorganic) | 34–85 ng | 2A | |
| Cobalt | 0.13–0.20 ng | 2B | |
| Nitro compounds | Nitromethane | 0.5–0.6 μg | 2B |
| 2-nitropropane | 0.7–1.2 ng | 2B | |
| Nitrobenzene | 25 μg | 2B | |
| Volatile compounds | Benzene | 12–50 μg | 1 |
| 1,3-butadiene | 20–40 μg | 2A | |
| Isoprene | 450–1,000 μg | 2B | |
| Aldehydes | Formaldehyde | 10.3–25 μg | 1 |
| Acetaldehyde | 770–864 μg | 2B |
Analytical chemistry studies have demonstrated that incomplete combustion of organic compounds in tobacco results in the formation of a wide range of carcinogens, such as PAHs and aromatic amines [1, 33]. Also, heat-associated degradation of certain tobacco constituents gives rise to various carcinogenic compounds [1]. For example, propylene glycol, which is used as a tobacco additive (serving as humectant), undergoes heat degradation to form propylene oxide, a Class 2B carcinogen [1, 11]. Another group of carcinogens found in tobacco smoke is N-nitrosamines [1]. Several tobacco-derived N-nitrosamines are among the most potent chemical carcinogens [11]. There are multiple varieties of N-nitrosamines, including volatile nitrosamines (VNAs) and TSNAs. These chemicals can be found naturally in tobacco leaves in limited quantities, but concentrations increase during the processing and curing of tobacco, as well as during its pyrolysis, i.e., when tobacco is smoked [11]. Yield of VNA and TSNA compounds also depends on the blend of tobacco [18]. Blends with higher nitrate levels produce more nitric oxide during smoking than blends with lower nitrate amounts [1]. Nitric oxide is quickly oxidized to form nitrogen dioxide which, along with other nitrogen oxides, reacts with amines and nicotine to produce VNAs and TSNAs, respectively [1, 11]. Many TSNAs generated through these reactions, such as N’-nitrosonornicotine (NNN) and 4-(methy1nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) are known human carcinogens (i.e., Class 1 carcinogens) [1, 11]. Oxidation can also lead to the production of ethylene oxide, a Class 1 carcinogen, through the reaction of oxygen and ethene in tobacco smoke [11].
5. Free radicals in tobacco smoke
Free radicals are atoms, molecules, or ions that are unstable, redox active, and highly reactive toward cellular and sub-cellular targets as they contain unpaired electrons [35, 36]. Free radicals are by-products of natural reactions occurring in the body, including metabolic processes and immune system response [22, 23, 35]. Exogenous sources of free radicals include substances present in the air we breathe, the food we eat, and the water we drink [23, 35, 37]. Environmental and lifestyle factors, such as cigarette smoking, represent major sources of exogenous free radical exposures to humans [20, 21, 37, 38]. Free radicals can damage cellular structure, e.g., cell membrane, or macromolecules, e.g., proteins, lipids, and nucleic acids, through a process involving abstraction of their electrons [35, 36, 39, 40]. This process is called “oxidation”, and the induced damage is termed “oxidative damage” [35, 36, 40].
Tobacco smoke is comprised of mainstream smoke (MS) and sidestream smoke (SS), both of which carry large quantities of free radicals [20, 21, 38, 41]. MS is generated when taking a puff from a cigarette, and is inhaled directly from the filter/cigarette end into the oral cavity and down to the respiratory tract [13, 14]. SS is formed by the burning of a cigarette from the lit end, and is produced in-between puffs [13, 14]. Both MS and SS can be partitioned into two phases according to the size of their constituents [13]. The two phases include tar (particulate) and gas phases [14]. The tar phase consists of compounds, which are 0.1–1 micrometers (μm) in diameter (average=0.2 μm), and the gas phase comprises chemicals with a diameter smaller than 0.1 μm [14, 20, 38]. Both the gas and tar phases of tobacco smoke contain huge amounts of free radicals; for example, the gas phase delivers upwards of 1015 free radicals with every puff inhaled, and tar, per gram, gives rise to nearly 1017 free radicals [20]. These free radicals are carbon-, nitrogen-, and oxygen-centered radical species, such as semiquinone, hydroxyl, and superoxide radicals [1, 20]. The small oxygen- and carbon-centered radicals in the gas phase are much more reactive than the tar-phase free radicals [1, 20].
6. Oxidative stress: smoke-induced ROS and cancer
ROS comprise both free radical and non-free radical oxygen intermediates, such as hydrogen peroxide (H2O2), superoxide (O2•−), singlet oxygen (1O2), and the hydroxyl radical (•OH) [22, 23, 36]. ROS are generated as a byproduct of the aerobic metabolism of oxygen and play key roles in homeostasis and cell signaling [36, 42]. ROS are also involved in other metabolic processes and immunity, e.g., via the nicotinamide adenine dinucleotide phosphate oxidase (NADPH) pathway [43, 44]. In addition, ROS are produced by phagocytic cells, such as neutrophils, eosinophils, and mononuclear phagocytes (e.g., macrophages) in response to stressors [36, 45]. The formation of ROS can also be stimulated by a variety of exogenous agents, including pollutants, dietary agents, drugs, lifestyle factors, or radiation [36]. Substantial quantities of ROS are produced in the mitochondria as a natural by-product of oxidative phosphorylation, which generates adenosine triphosphate (ATP). ATP is used as an energy source for most cellular functions, including active transport and cell signaling [46]. Production of ATP occurs mainly through aerobic respiration using the electron transport chain (ETC) mechanism [46]. ETC operates through the transfer of electrons from one complex to another via redox reactions, and ends with oxygen as the final electron acceptor [47]. Cells acquire large quantities of ATP through this process; however, due to electron leak, ETC can also result in the production of a wide range of ROS [47]. The generated ROS can directly or indirectly damage cellular and sub-cellular targets, thus resulting in adverse biological consequences [22, 23, 35, 36]. Cells have evolved elaborate antioxidant defense mechanisms to counteract the effects of ROS [48–51]. However, external factors, e.g., environment, can impose an additional burden of ROS on the cells, thus overloading the antioxidant defense system and disrupting the homeostasis between oxidants and antioxidants [37, 50]. This imbalance is known as “oxidative stress”, a condition in which the amount of ROS exceeds the capacity of the antioxidant system within an organism [40, 52]. In humans, environmental exposure and lifestyle factors, specifically cigarette smoking, are prominent sources of oxidative stress [35, 36, 40].
Oxidative stress can induce both apoptosis (programmed cell death) and cellular senescence (a state of permanent growth arrest without undergoing apoptosis) [53, 54]. Whether a cell undergoes apoptosis or senescence depends on the severity of damage and the tissue type; however, both events act as protective mechanisms to prevent damaged cells from proliferating [55]. This is to avoid genomic instability and propagation of the induced damage to progeny cells [55]. Upon evasion of apoptosis or senescence, however, oxidative stress and the excess ROS in the cell can further damage macromolecular targets, such as proteins, lipids, and nucleic acids [14, 35, 36, 39, 40, 56]. The induced damage to these macromolecules is significant because maintaining the integrity of DNA/RNA, proteins, and lipids is critical to determining the health vs. disease state [35, 40, 57]. Accumulating evidence supports a major role for oxidative stress in the development of a variety of human diseases, including cancer [39, 40, 57, 58]. Because genomic instability is a hallmark of cancer, the ROS that damage DNA, are of special importance in carcinogenesis [22, 23, 57]. Whilst the critical ROS comprise O2•−, H2O2, •OH, 1O2, the latter two are of most significance because they can directly attack and damage DNA [22, 23]. Although O2•− and H2O2 are not as reactive as •OH and 1O2, they are abundant by-products of aerobic metabolism, and can undergo Haber-Weiss reactions with iron to generate •OH [22, 59]. Thus, a buildup of O2•− and H2O2 is still a major contributor to the accumulation of oxidative DNA damage because both of these two ROS can be converted to •OH which, in turn, can inflict damage on DNA [22, 23].
7. Direct and indirect generation of ROS in cigarette smoke
Both the gas and tar phases of tobacco smoke yield large quantities of ROS [24, 41]. ROS in the gas phase are generated during the combustion of tobacco, and are inhaled by the smoker as part of the mainstream smoke [13, 60]. The tar phase contains several relatively stable free radicals, such as a quinone/hydroquinone (Q/QH2) complex held in the tarry matrix [20, 21]. This Q/QH2 polymer may function as an active redox system by reducing molecular oxygen in the smokers’ lungs to produce O2•−, which can eventually form other free radicals, such as H2O2 and •OH [20, 21]. It is important to recognize the distinction between ROS that are derived ‘directly’ from tobacco smoke and those that are formed ‘indirectly’ (e.g., from other toxicants or carcinogens or their secondarily formed metabolites) as they may impose distinct burden of oxidative stress and/or elicit different biological responses (see, below). Importantly, oxidative stress resulting from the gas- or tar-phase derived ROS can be augmented by a defect or saturated antioxidant defense system, or as a consequence of additional ROS or other reactive metabolites generated through biotransformation of tobacco smoke chemicals inhaled by smokers [49, 61].
As particulates from tobacco smoke are deposited into the lungs, a layer of tar begins to accumulate [20]. This forms an aqueous solution that undergoes redox cycling to produce various reactive species that can cause oxidative damage [21]. Major tobacco smoke constituents that cause oxidative damage through this process include phenolic compounds, quinones, heavy metals, and free radicals [1, 13, 20]. Phenolic compounds, such as hydroquinone and catechol, are known to undergo redox cycling in aqueous tar, thereby forming O2•− [20, 21]. Semiquinone radicals also give rise to O2•− production through the reduction of oxygen. O2•− can then be dismutated by superoxide dismutase (SOD) to form oxygen (O2) and hydrogen peroxide (H2O2); H2O2 plays a critical role in redox cycling with heavy metals (see, next section) [62–64]. Many free radicals, such as O2•−, react with other short-lived radicals (e.g., in the gas phase) to form other highly reactive species that impose additional burden of oxidative damage [65]. For instance, O2•− is known to readily react with nitric oxide (•NO) to form peroxynitrite (ONOO−), which is a very short-lived oxidant and nucleophile and extremely reactive with biomolecules, e.g., proteins and lipoproteins [65].
Furthermore, metals in tobacco smoke are also a main contributor to oxidative stress in smokers [1, 22, 23]. Tobacco leaves contain trace amounts of heavy metals absorbed from the soil during plant growth [66]. Many of these metals, such as chromium, cadmium, arsenic, beryllium, and nickel, are proven Class 1 carcinogens [1, 11]. Some metals like chromium, nickel, iron, and copper, which are active in redox reactions, produce ROS through Fenton-like reactions, thereby contributing to oxidative stress [1]. The latter group of metals undergo redox cycling, while in the presence of H2O2, to form ROS, such as •OH, which is highly reactive with DNA [22, 23]. •OH can induce oxidative DNA lesions, such as single strand DNA breaks, which are mutagenic if not repaired properly [23, 67]. Since most single strand DNA breaks possess a single nucleotide gap at the site of breakage [68], misinsertion of inappropriate nucleotide(s) during the gap filling step of DNA repair can lead to mutation (see, Section 12 for detailed information) [69, 70].
8. Antioxidant defense mechanisms
In humans, there are diverse antioxidant defense mechanisms, which regulate ROS levels [48–51]. These include the enzymatic and non-enzymatic antioxidants [48, 49]. The most prominent enzymatic antioxidants are SOD, glutathione peroxidases (GPx), and glutathione S-transferases (GST) [48, 62, 71]. The principal non-enzymatic antioxidant is intracellular glutathione (GSH) [48]. SOD enzymes catalyze conversion of O2•− to H2O2 [63, 64]. The generated H2O2 is then reduced by GPx enzymes using GSH as a cofactor, resulting in H2O and oxidized GSH (GSSG) [71, 72]. GST enzymes catalyze conjugation of GSH to lipophilic compounds, including free radicals and their byproducts, thus helping facilitate cellular detoxification [48]. Therefore, both GPx and GST enzymes rely on the GSH to perform their antioxidant functions [48, 71, 72]. Orhan et al. [73] reported significant decrease in SOD and GPx activities in erythrocytes of smokers as compared to nonsmokers, suggesting that smoking-associated ROS leads to saturation of these antioxidant enzymes, hence, reducing their bioavailability. GPx expression levels in smokers vary depending on tissue type. In patients with smoking-related COPD, GPx is up-regulated in the epithelial lung tissue, but downregulated in blood components, such as plasma and erythrocytes [74, 75]. SOD has been shown to offer protection against oxidative damage, such as oxidized DNA lesions and lipid peroxidation products [62, 63]. Transgenic mice expressing human CuZnSOD and exposed chronically to cigarette smoke (6 hours/day, 5 days/week, for 1 year) showed significant attenuation of oxidative DNA damage and lipid peroxidation products in the lungs as compared to wildtype littermates [76].
GSH is a non-enzymatic antioxidant, which detoxifies free radicals or the byproducts of their reactions either directly or indirectly through reactions catalyzed by GPx and GST enzymes [48, 71, 72]. GSH also enhances the activity of other antioxidants, such as vitamin C and E, thereby elevating the overall antioxidant defense capacity [48, 49]. However, the GSH antioxidant properties can be diminished or overwhelmed by excessive ROS generated during increased oxidative stress state, e.g., cigarette smoking [50, 61, 77]. Additionally, GSH activity and function can be impeded by trace metals present in tobacco smoke [48, 61]. For example, metals like arsenic, cadmium, mercury, and lead can interfere with GSH activity or function by binding to this tripeptide and reducing its availability for antioxidant reactions [1]. Other metals in tobacco smoke, such as chromium, nickel, iron, and copper can indirectly impact the antioxidant defense system by undergoing redox cycling, while in the presence of H2O2, thereby producing an additional burden of ROS [35, 36, 40]. The impairment of antioxidant defense mechanisms leaves ROS levels unregulated, thus favoring a condition in which macromolecular targets can be readily attacked by the excess ROS [35, 36, 39, 40]. The induced damage to critical targets, such as proteins, lipids, and nucleic acids may then lead to disruption of key cellular functions, resulting in a disease state [36, 39, 40]. Accumulating evidence supports a critical role for oxidative damage to macromolecules in the development of a variety of smoking-associated diseases, including cancer [39, 40, 57].
9. Oxidative damage to lipids: connection with smoking and cancer
Oxidation of lipids is termed lipid peroxidation and the products of this reaction can serve as biomarkers for assessing overall levels of oxidative stress [78–80]. The increase in lipid peroxidation products as a result of smoking is noteworthy because these products can form lesions on DNA and proteins that may have deleterious biological consequences [78, 81]. ROS can induce degradation of polyunsaturated fatty acids (PUFAs), resulting in the formation of a variety of products, including malondialdehyde (MDA), 4-hydroxy-nonenal (HNE) and the F2-isoprostane 15(S)-8-iso-prostaglandin F2α (15(S)-8-iso-PGF2α) [79, 82]. MDA is a reactive aldehyde and a highly electrophilic species that can covalently bind to DNA and proteins, forming complexes, called DNA- and protein-“adducts”, respectively [1, 79, 81–83]. For instance, MDA reacts with deoxyadenosine and deoxyguanosine in DNA, forming multiple DNA adducts, of which pyrimido[1,2-a]-purin-10(3H)-one (M1dG) is a predominant adduct with mutagenic properties [79, 84, 85]. Also, MDA can interact with the guanidine group of arginine residues and form 2-aminopyrimidines [82, 86]. While there is some debate over which lipid peroxidation product is the most accurate and reliable biomarker of oxidative damage to lipids, measurement of MDA levels in blood plasma has been widely used to estimate the overall load of lipid oxidation in humans [87, 88].
Many analytical methods are available for measuring MDA concentrations in biological samples [88]. Most methods exploit one of the two chemical properties of MDA for quantification purposes; these features include: (1) the C—H acidity of its methylene H atoms in aqueous solution; and (2) the reactivity of its two aldehyde groups towards nucleophiles, [89]. The most widely used methods employ chemical conversion of MDA to derivatives with improved physicochemical properties for chromatographic separation and detection [88]. The most common derivatization method utilizes thiobarbituric acid (TBA), which is directed towards the aldehyde groups of MDA. The method targets MDA and other as yet unidentified TBA-reactive substances present in biological samples [88]. The selectivity of this method is enhanced by extracting the MDA-(TBA)2 derivative with n-butanol, and separating the derivative by high performance liquid chromatography (HPLC) prior to its visible absorbance or fluorescent detection. The spectrophotometric and spectrofluorometric TBA-based methods are the most widely used techniques for quantification of MDA in biospecimens [88, 90].
Generally, reliable quantification of MDA in biological samples, particularly in lipid-rich matrices, such as plasma or serum, is highly challenging due to (1) pre-analytical issues, such as variations in specimen collection and storage (conditions and time/duration), (2) analytical issues, such as artifactual formation of MDA during sample processing, work up and analysis, as well as comparability of the applied methods in terms of sensitivity and/or specificity [91, 92]. In addition, many reactive carbonyl group-containing compounds, originating from endogenous or exogenous sources (e.g., diet or lifestyle), may interfere with MDA measurement in biospecimens [87]. This may complicate the interpretation of the results, especially in human biomonitoring studies, wherein the origin of the quantified MDA is rarely attributable to a single source [88]. Analytical methods for measurement of MDA in biological samples have been thoroughly reviewed and discussed in recent reviews [88, 92]. Whilst TBA-based measurements of MDA have largely shown increased levels of this prototype lipid peroxidation product in blood plasma of smokers as compared to nonsmokers [93–100], divergent results have also been reported [101, 102]. For the most part, the discrepancies in results have been ascribed to the methodological differences (per-analytical and analytical issues), and/or biological variables (e.g., characteristics of the study population), as described above.
Elevated MDA levels have been observed in smoking-associated cancers [79, 103–105]. A recent study has demonstrated significantly higher levels of MDA in blood plasma of colorectal cancer patients as compared to age- and sex-matched healthy controls [104]. The authors have reported significant predictive values for MDA determination in colorectal cancer patients by showing positive correlations between MDA levels and depth of tumor invasion as well as carcinoembryonic antigen (CEA)/C-reactive protein(CRP) levels. Of note, CEA/CRP are the most commonly evaluated laboratory markers of colorectal cancer [104]. Similarly, significantly higher levels of serum MDA have been reported in breast carcinoma patients as compared to patients with benign tumors as well as normal controls [106].
Lipid peroxidation also leads to the formation of other aldehydes, such as acrolein and 4-HNE [78, 79, 86]. Acrolein is endogenously generated as a result of oxidative degradation of low-density lipoproteins (LDLs) [78, 86]. Acrolein can also be formed exogenously consequent to combustion of organic materials. Tobacco smoke is a major source of exogenous acrolein [1, 86, 107]. As a highly reactive compound, acrolein can readily engage in Michael type reactions with cellular nucleophiles to form alkylated adducts with GSH, protein sulfhydryls, thiol-containing enzymes, and DNA [108]. Hydroxypropanodeoxyguanosine (HOPdG) is the predominant promutagenic acrolein-DNA adduct [79, 107, 108]. Binding of acrolein to intracellular GSH results in depletion of the supply of this antioxidant [109]. 4-HNE is another highly reactive aldehyde, capable of binding to GSH via GST activity, resulting in decreased antioxidant capacity [110, 111]. 4-HNE has been implicated in cancer and inflammatory diseases owing to its ability to form 4-HNE-protein adducts that can impair cell signaling and alter gene expression [83, 103, 112].
10. Oxidative damage to proteins: connection with smoking and cancer
Oxidative damage to proteins is divided in two categories, including the reversible and irreversible protein modifications [52, 113]. Carbonylation is a primary form of irreversible oxidative protein modification as the modified protein is not functional due to aggregation and degradation [114]. Carbonylation occurs mainly in side chains of native amino acids in proteins, e.g., histidine, cysteine, and lysine, thereby giving rise to carbonyl derivatives, such as aldehyde and ketones [82, 114]. The specificity of multiple amino acids to undergo carbonylation has made this modification a widely used biomarker for assessing oxidative damage to proteins [114, 115]. Oxidative stress, often metal catalyzed, is a main source of protein carbonylation [116]. There is growing evidence to support a link between smoking-induced oxidative stress and protein carbonylation [114, 116, 117]. Colombo et al. [118] have shown that treatment of human bronchial epithelial cells with increasing concentrations of cigarette-smoke concentrate induces oxidative stress and increases protein carbonylation in a concentration-dependent manner. Also, Sumanasekera et al. [61] have reported that cardiac stem cells treated with cigarette-smoke concentrate exhibit increased oxidative modification of proteins via carbonylation. Of significance, the induced protein carbonylation was attenuated by pre-treatment of the cells with antioxidant, ascorbic acid (vitamin C) [61].
Another form of irreversible protein modification due to oxidative stress is nitration [119]. Accumulating data show protein nitration in smoking-induced oxidative stress [117]. It is known that free radicals in tobacco smoke, such as O2•− and •NO, can give rise to ONOO−, which, in turn, nitrate tyrosine residues in proteins, in addition to oxidizing lipoproteins [119]. The resulting nitrotyrosine is considered a broad spectrum biomarker of oxidative stress, oxidative protein damage, cell damage, inflammation, as well as •NO production [115, 117, 120]. Female mice exposed to cigarette smoke for a period of six months have shown significant increase in 3-nitrotyrosine levels in the lungs as compared to control mice exposed to clean air [121].
Oxidation of cysteine residues in proteins is a prime example of reversible protein modification [58, 117]. Reversible oxidation of cysteine amino acid in proteins can serve as an “on and off” switch to regulate protein function and redox signaling pathways in response to stress conditions [58]. Thus, reversible cysteine modifications are often involved in regulating redox signaling pathways and protein function during oxidative stress [117]. Reversible cysteine redox modifications include S-sulfonation, S-nitrosylation, S-glutathionylation, and disulfide bond formation [58]. Chen et al. [122] have reported significantly higher extents of glutathionylation at α-Cys-104 and β-Cys-93 in hemoglobin of smokers as compared to nonsmokers. Moreover, the authors observed statistically significant correlations between glutathionylation extents at α-Cys-104 and β-Cys-93 and the number of cigarettes smoked per day as well as smoking index (number of cigarette per day × years smoked) [122].
Altogether, protein modifications induced by oxidative stress can have adverse biological consequences; however, they may also protect against subsequent injury to cellular and sub-cellular targets [117]. For example, while both carbonylation and nitration cause detrimental effects on the target proteins, evidence is also emerging that such modifications can play protective roles in cellular function under stress conditions [116]. Furthermore, reversible cysteine oxidation may also protect the target proteins and other proteins from further damage [58, 117]. The reversibly oxidized cysteines are involved in redox signaling cascades that can elicit positive stress responses to prevent further oxidation and eventual degradation of the target and other functionally-important proteins [116, 117]. The protective aspects of post-translational modifications of proteins, however, may be severely diminished or overshadowed by excessive oxidative stress posed by external sources, such as chronic smoking [116]. The excess ROS generated by long-term smoking can significantly outweigh the potentially beneficial consequences of protein oxidative modifications, thus rendering crucial proteins involved in, e.g., cell cycle and DNA repair, ineffective or non-functional (see, next section) [116, 123]. Aberrations in the activity or function of many of these critically important proteins are a common feature of human cancer.
11. Oxidative damage to DNA: connection with smoking and cancer
Oxidative damage to DNA can produce a wide variety of lesions with varying mutagenic potentials [22, 23, 51, 86]. Oxidative DNA lesions can be formed through two distinct pathways, including: (1) direct oxidation of a base (purine/pyrimidine) in DNA; and (2) misincorporation of oxidized deoxynucleoside triphosphates (dNTPs) into DNA by DNA polymerase(s) [124–126]. All four bases of the DNA can undergo direct oxidation, forming various oxidized purines, such as 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxodG) and 8-oxo-7,8-dihydro-2’-deoxyadenosine (8-oxodA) and oxidized pyrimidines, such as thymine glycol and 5-hydroxyuracil [22, 23]. However, guanine has the lowest redox potential of all four DNA bases, and is, therefore, the most vulnerable residue to oxidation (2′-deoxyguanosine (dG): 1.29 V; 2’-deoxyadenosine (dA): 1.42 V; 2’-deoxycytidine (dC): 1.6V; and thymidine (T): 1.7 V) [127]. 8-oxodG has an even lower redox potential than guanine (0.74 V), thus being susceptible to further oxidation [22, 128–130]. The oxidation products are hydantoin-type DNA adducts, such as spiroiminodihydantoin and guanidinohydantoin, both of which being more mutagenic than 8-oxodG by multiple orders of magnitude [128–130]. 8-oxodG can also be reduced to form other oxidized/ring-opened purines, such as 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG), although to a much lesser extent [131, 132]. Oxidation of dNTPs and their subsequent misincorporation into the DNA can be followed by erroneous replication of the oxidized nucleotides during translesion DNA synthesis, thereby giving rise to mutation [124, 133].
8-oxodG is the most thoroughly investigated oxidized base lesion whose increased level over the steady-state level in cellular DNA (i.e., baseline/background vs. induced 8-oxodG levels) is often considered an indicator (biomarker) of oxidative stress [22, 130]. 8-oxodG has garnered much attention owing to its propensity for formation, persistence, and accumulation in vivo, as well as its mutagenic potential (see, below). In normal human cells, an estimated 300 – 1,000 oxidized guanine bases per cell per day are formed [134]. The background levels of 8-oxodG in blood lymphocytes of healthy young male subjects were estimated to be between 0.3 and 4.2 per 106 dG, determined as the median of the values obtained from a consortium of mainly European laboratories [135]. Quantification of 8-oxodG in blood leukocytes or measurement of the excreted modified base or nucleoside in urine are commonly used for biomonitoring purposes [23, 136–138]. Elevated levels of 8-oxodG in DNA isolated from peripheral blood and urine of smokers as compared to nonsmokers have been reported in a large number of investigations [137, 139–143]. Similarly, increased levels of 8-oxodG have been found in precancerous and cancerous tissues or cancer cell lines as compared to normal cells, as well as in experimental animals or cells exposed in vivo/in vitro to tobacco smoke or its constituents as compared to controls [143–146]. Nonetheless, contradictory results have also been reported in other studies [147–149] (and reviewed in refs. [141–143, 146]). The disparate results in different studies may have arisen from analytical challenges in measuring 8-oxodG without artifactual oxidation of normal dG during sample processing and analysis, high background levels of this lesion in cellular DNA, multi-source nature of 8-oxodG in biospecimens (endogenous and exogenous), different methodologies used, conditions and time of sample collection, inter- and intra-individual variations in subjects’ metabolic capacity, DNA repair activity, or antioxidant defenses, and various confounding factors [23, 138, 150, 151].
8-oxodG is a miscoding lesion during DNA replication [152, 153]. As such, an adenine is preferentially incorporated opposite 8-oxodG in the template DNA during translesion synthesis, which upon the next round of replication, produces G:C→T:A transversion mutation [152–154]. Although 8-oxodG predominantly induces G:C→T:A transversion [152–154], bypass of this lesion in mammalian cells can also lead to G:C→A:T transition and G:C→C:G transversion [124, 155]. The latter types of mutation presumably arise from hydantoin-type DNA lesions that are photooxidation products of 8-oxodG [128]. Elevated levels of 8-oxodG and/or significant induction of G:C→T:A transversion as well as G:C→A:T transition and G:C→C:G transversion mutations have been reported in numerous studies investigating the effects of oxidative stress associated with tobacco smoke exposure in vitro or in vivo [12, 23, 77, 142, 156–159]. Oxidized dNTPs are often poor substrates for DNA polymerases although exceptions exist for certain polymerases [124]. For example, in humans, polymerase η (Polη) incorporates oxidized guanine nucleosides (8-OH-dGTPs) opposite a template adenine with almost the same efficiency as that of normal 2’-deoxythymidine triphosphate (dTTP) [160]. Such misincorporation leads to A:T→C:G transversion because the oxidized dGTP pairs with an incoming dCMP in the next round of DNA replication [161, 162]. The frequency of spontaneous A:T→C:G transversions has been shown to increase more than 1000-fold over the background in MutT mutant E. coli, which are deficient in hydrolyzing 8-OH-dGTP to the mono-phosphate form and sanitizing the nucleotide pool [163–165]. The extremely high levels of A:T→C:G transversions in the MutT mutant strain are nearly completely suppressed when the cells are cultured in anaerobic conditions, indicating the important role of oxidative stress in the induced mutagenesis [166]. Moreover, when the cDNA for MutT homolog 1 (MTH1), a human counterpart of E. coli MutT, is expressed in MutT mutant cells, the markedly high rate of spontaneous A:T→C:G mutations reverts to normal [167]. Russo et al. [168] have demonstrated that over-expression of hMTH1 significantly attenuates the mutation rates in DNA mismatch repair (MMR) defective mouse- and human cells, suggesting that the high spontaneous mutation rates in MMR-defective cells are mostly due to incorporation of oxidized dNTPs into DNA, and less likely, caused by spontaneous replication errors. The authors concluded that incorporation of oxidized purines from the dNTP pool can significantly contribute to the extreme genetic instability of MMR-defective human tumors [168].
In humans, DNA damage induced by smoking-associated oxidative stress can be efficiently repaired by specialized DNA repair enzymes (see, next section) [51, 169]. However, when not repaired properly, oxidative DNA damage can undergo erroneous replication and lead to mutation [169]. Functionally-important mutations in key genes involved in, e.g., cell growth, differentiation, and survival, are of most relevance to cancer [51, 77, 170]. Specifically, DNA damage-driven mutagenesis in distinct genomic loci may activate normally inactive protooncogenes or inactivate otherwise active tumor suppressor genes, thereby giving rise to cancer development [14, 51, 77, 171]. Mutations in the TP53 gene are the most frequent genetic alteration in human cancer [171, 172]. Over half of all human cancers harbor mutations in the TP53 tumor suppressor gene. Inactivating mutations in the TP53 gene have been observed in nearly all types of smoking-associated cancer [13, 171, 172]. Barta et al. [173] have reported inactivating mutations in the TP53 tumor suppressor gene in lung tumors of current and former smokers, with the majority of mutations being G:C→T:A transversions at known lung cancer mutational ‘hotspots’, including codons 157, 158, 245, 248, and 273 [13, 32, 172]. This mutational fingerprint is considered a ‘smoking signature’, which has been ascribed to tobacco smoke constituents, such as PAHs and specific ROS [31, 51, 172–174]. We note that many chemicals in a mixture, such as tobacco smoke, induce similar type(s) of mutation, e.g., both ROS and B[a]P (a prototype PAH), give rise predominantly to G:C→T:A transversion [13, 31, 32, 146, 170, 172, 174]. Thus, it is a formidable challenge to tease out the contribution of each chemical to the induced mutation spectrum in cells exposed in vitro/in vivo to complex mixture of chemicals, such as tobacco smoke [12, 14, 31, 32, 171, 175].
Furthermore, recent work in our laboratory has shown down regulation of the tumor suppressor genes notch receptor 1 (NOTCH1) and HECT and RLD domain containing E3 ubiquitin protein ligase 2 (HERC2) in healthy smokers as compared no nonsmokers [176]. Lack of NOTCH1 is associated with the development of head and neck cancer, comprising malignancies in the oral cavity, nasal cavity, paranasal sinuses, pharynx, and larynx [177, 178], and a dysfunctional HERC2 gene has been observed in colorectal and gastric cancers [179]. Recently, Ogawa et al. [180] have demonstrated that treatment of the human airway basal stem cells with cigarette-smoke extract resulted in increased KRAS and RAS protein family activation in vitro. Consistent with this finding, they also observed that airway epithelial cells brushed from healthy smokers had elevated RAS activation compared to nonsmokers [180]. The protooncogene K-RAS is one of the three human RAS genes (other members: H-RAS and N-RAS) that are among the most frequently mutated genes in human cancer [181]. Activating mutations in the RAS protooncogenes have been found in 20–25% of all human cancers, including lung, colon, and breast cancers, and up to 90% of certain types of tumors, e.g., pancreatic cancer [181, 182].
12. DNA repair of oxidized lesions: modulation of mutagenesis and carcinogenesis
Mammalian cells possess an elaborate DNA repair machinery to correct the plethora of endogenous and exogenously derived DNA damage [51, 52, 169]. In humans, DNA repair is a highly complex process involving cooperation of many proteins some of which have specialized and/or overlapping functions [23, 52]. There are three excision repair pathways that repair single stranded DNA damage; these include: nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR) [22, 169, 183]. Whereas NER is involved in recognition and removal of bulky lesions from the genome overall [169], BER recognizes non-bulky lesions and repairs specific damaged bases by distinct DNA glycosylases [130]. MMR exclusively repairs DNA damage with mismatched Watson-Crick base pairs [183]. As a versatile DNA repair pathway, NER is divided in two sub-pathways: (1) global genome repair (GGR), which is involved in the repair of lesions from any location in the genome; and (2) transcription-coupled repair (TCR) that repairs lesions exclusively on the active template strand of DNA [184, 185]. GGR is initiated when lesions alter the helical structure of DNA, whereas TCR is triggered when RNA polymerase is stalled during transcription due to blockage from a damaged template strand [169, 184, 185]. While the two sub-pathways differ in how they recognize DNA damage, they share the same process for lesion incision, repair, and ligation [22, 23]. In both cases, recognition of the damage leads to removal of a short single-stranded DNA segment that contains the lesion. The undamaged single-stranded DNA remains intact and is used as a template by DNA polymerases for synthesis of a short complementary sequence. The synthesized sequence is then ligated by DNA ligase to form double stranded DNA [169, 184, 185].
BER is primarily involved in the repair of small, non-helix-distorting base lesions in the genome, particularly those induced by oxidative stress [22, 130, 186]. BER is initiated by DNA glycosylases, which recognize and remove specific damaged- or inappropriate bases (i.e., due to nucleotide misincorporation), thus forming apurinic/apyrimidinic (AP) sites [23]. The AP sites are subsequently cleaved by an AP endonuclease. The resulting single-strand breaks are then processed by either short-patch (where a single nucleotide is replaced) or long-patch BER (where 2–10 new nucleotides are synthesized) [22].
All proteins involved in DNA repair are essential for restoring the genome integrity consequent to assault by stressors, such as ROS [169]. Therefore, if any gene responsible for transcription of these proteins becomes dys- or non-functional due to, e.g., DNA damage and mutation, the entire DNA repair pathway may turn ineffective [22, 51]. For example, 8-oxoguanine DNA glycosylase (OGG1) recognizes and excises oxidized guanine, such as 8-oxodG, from the DNA sequence, whereas MutY DNA glycosylase (MUTYH) removes adenine when it is mispaired with 8-oxo-dG during DNA replication [130, 186, 187]. Both enzymes, which are part of the BER pathway, leave the DNA with an abasic site, which is marked by an apurinic/apyrimidinic endodeoxyribonuclease (APEX1) that creates a single-strand break in the DNA. The poly (ADP-ribose) polymerase 1 (PARP1) enzyme then recognizes the single-strand break, and recruits DNA polymerase beta (POLB) to fill in the correct nucleotide. Finally, ligation is performed by a DNA ligase to restore the original DNA sequence (see, Figure 1) [22, 52]. There are many other enzymes that participate in the BER pathway for repair of oxidative damage, including various DNA glycosylases, e.g., formamidopyrimidine DNA glycosylase (Fpg in E. coli and hOGG1 in human) and multiple Nei-like (NEIL) glycosylases [126, 170, 186]. Mutations resulting from oxidative lesions in genes encoding any of these proteins may adversely impact the corresponding enzyme activity/function and cause the entire BER pathway to dys- or malfunction [22, 23, 77].
Figure 1. BER-mediated repair of 8-oxodG.
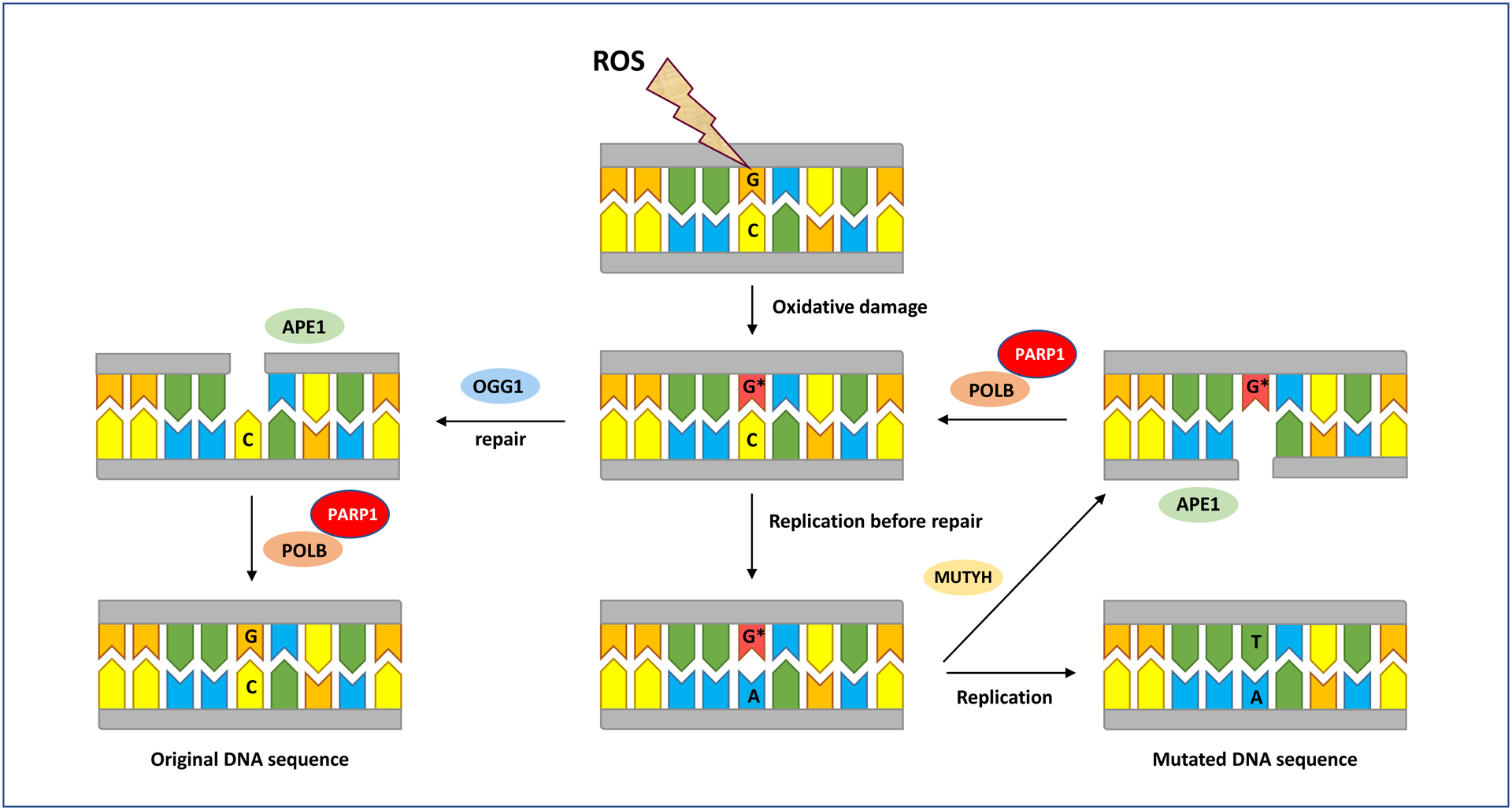
ROS oxidize guanine residues in DNA leading to formation of 8-oxodG (G*). This damage is removed by OGG1 leaving an apurinic (AP) site, which is recognized by APE1 (encoded by the APEX1 gene in humans) that nicks the DNA at the site of damage. PARP1 then recruits POLB to fill the gap with a correct guanine. The gap is sealed by DNA ligase and the original DNA sequence is conserved. If replication occurs before OGG1 has excised the lesion, 8-oxodG pairs with adenine, which upon replication, results in G:C→T:A mutation. Alternatively, MUTYH excises the mispaired adenine with 8-oxodG, POLB preferentially inserts a cytosine in its place, and the above cycle continues to repeat itself. As discussed in the text, the DNA repair and mutagenic pathways for oxidative damage are highly complex, involving numerous determinants, including various proteins, enzymes, substrates, cofactors, etc. Of these, we have focused on the most prominent determinants that are specifically covered in this review. We note that this figure is a simplified visualization of the highly complex and multi-component reactions occurring during oxidative DNA damage, repair, and mutagenesis, as described in the text. Interested readers are referred to elegant papers, including references [22, 23, 101, 103, 119, 128, 141, 144, and 145], which have discussed other specialized aspects of DNA damage/repair & mutagenesis that are outside the scope of this review. APE1: apurinic/apyrimidinic endodeoxyribonuclease; MUTYH: mutY DNA glycosylase; OGG1: 8-oxoguanine DNA glycosylase; PARP1: poly(ADP-ribose) polymerase 1; POLB: DNA polymerase beta.
Studies involving knockout of OGG1 in various model systems have shown that in the absence of this gene product, oxidative DNA lesions, such as 8-oxodG, significantly accumulate, resulting in induction of G:C→T:A transversion mutations [188]. Not being excised by OGG1 enzyme, 8-oxodG adopts a similar Watson-Crick conformation to that of thymine, thus, mispairing with adenine during DNA replication and giving rise to G:C→T:A transversion mutation [187]. Furthermore, Ogg1 knockout mice have been shown to be predisposed to develop lung adenoma/carcinoma [189]. The Ogg1 knockout mice had a mean number of 0.71 tumors per mouse, which was five times higher than that observed in wildtype counterparts (0.14 tumors/mouse) [189]. Arai et al. [190] chronically treated Ogg1 knockout mice and wildtype counterparts with potassium bromate (KBrO3), an oxygen radical forming agent, added to drinking water, for 29 weeks, and examined the formation of 8-oxodG in kidney, and tumor occurrence in multiple organs after an additional 23 weeks. In the treated Ogg1 knockout mice (both males and females), 8-oxodG levels were approximately 250-fold higher than those in similarly treated wildtype mice. The untreated knockout mice (Ogg1−/−) had also 15-fold higher levels of 8-oxodG than untreated wildtype counterparts (Ogg1+/+). However, no tumor formation was observed in any of the examined organs (i.e., kidney, lung, liver, spleen, thymus, stomach, and intestine) in Ogg1−/− or Ogg1+/+ mice, with or without KBrO3 treatment and of either sex. The authors ascribed the absence of tumors in the treated- and untreated Ogg1 knockout mice, the latter being in contrast to the findings of Sakumi et al. [189], to the younger age of mice at time of sacrifice (60 weeks vs. 78 weeks in ref. [189]) [190]. Moreover, double knockout of Myh/Ogg1 in mice has been shown to cause age-associated accumulation of 8-oxodG in lung and small intestine [191], consistent with the increased incidence of tumors in the same organs in doubly defective Myh−/−/Ogg1−/− mice, as reported by Xie et al. [192]. Myh is the mouse homolog of human MUTYH, which removes adenine mispaired with 8-oxodG from the DNA [130, 186, 187]. Double and triple knockouts for Myh, Ogg1, and Msh2 in mice have revealed that deficiencies in Myh and Ogg1 predisposed nearly 66% of mice to tumors, predominantly lung and ovarian tumors, and lymphomas. Remarkably, G:C→T:A mutations were found in 75% of the lung tumors at an activating mutational hotspot in the K-ras oncogene (i.e., codon 12), but none in their adjacent normal tissues. In addition, Msh2 heterozygosity increased malignant lung tumor incidence in Myh−/−Ogg1−/− mice [192]. Msh2 is a MMR gene involved in oxidative DNA damage repair [183].
The nudix hydrolase 1 (NUDT1 or MTH1), the human homologue of E. coli MutT, is another critical protein that protects against mutagenesis induced by oxidative stress [161, 162]. NUDT1 hydrolyzes oxidized guanine in the nucleotide pool, thus preventing the misincorportation of 8-OH-dGTP opposite adenine during DNA replication, which produces A:T→C:G transversion mutation (see, Figure 2) [161, 162]. Mice deficient in MTH1 displayed enhanced tumor formation in the lung, liver and stomach, suggesting a contributory role of oxidized dNTP pool in carcinogenesis [193]. Oxidative damage to the nucleotide pool is increasingly considered a key factor in the genesis and progression of cancer [133, 168].
Figure 2. Direct and indirect accumulation of 8-oxodG in DNA and prevention of mutagenesis by NUDT1.
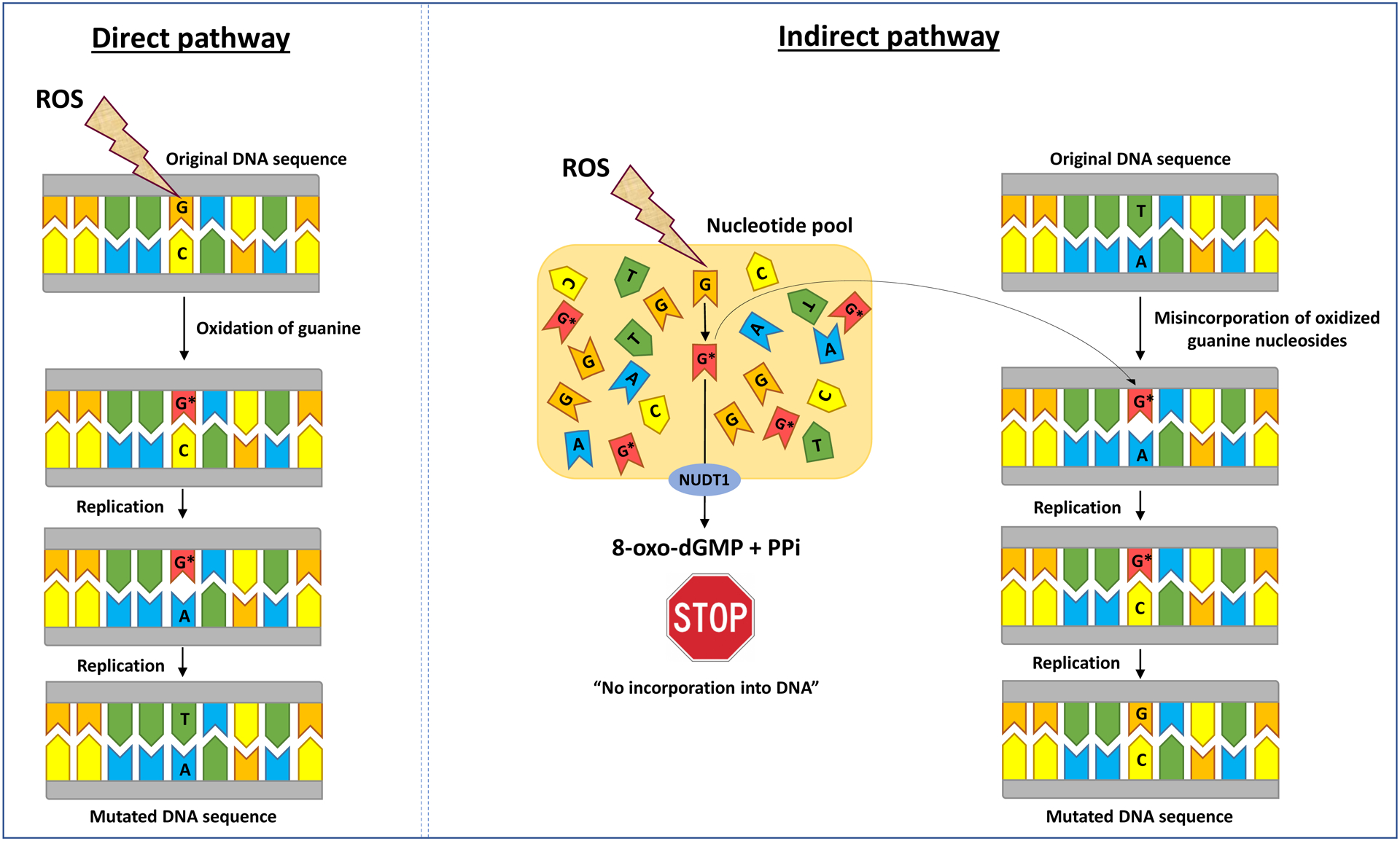
8-oxodG (G*) accumulates in DNA through two distinct pathways, including: (1) direct oxidation of guanine residues; and (2) misincorporation of oxidized guanine nucleosides (8-oxo-7,8-dihydro-2′- deoxyguanosine 5′-triphosphate (8-OHdGTP)), which upon two rounds of replication, result in G:C→T:A and A:T→C:G transversion mutations, respectively. With a properly functioning NUDT1 (also known as MutT homolog 1 (MTH1)) enzyme, 8-OHdGTP is hydrolyzed to a monophosphate form (8-oxo-dGMP) plus pyrophosphate (PPi), which can no longer be incorporated into the DNA, thus preventing mutagenesis. We note that this figure is a simplified visualization of the highly complex and multi-component reactions occurring during oxidative DNA damage, repair, and mutagenesis, as described in the text. Interested readers are referred to elegant papers, including references [22, 23, 101, 103, 119, 128, 141, 144, and 145], which have discussed other specialized aspects of DNA damage/repair & mutagenesis that are outside the scope of this review. NUDT1: nudix hydrolase 1 also known as MTH1.
13. Feedback loop between oxidative stress and inflammation: impact of smoking and implications for carcinogenesis
The innate immune system plays a central role in the inflammatory response [9]. In response to inflammatory stimuli, white blood cells (WBC, also known as leukocytes), such as neutrophils, enter areas of tissue injury as a result of signals released by small proteins, such as cytokines [9, 170]. Of note, when monocytes (the largest of all white blood cells) enter the tissue, they enlarge and mature into macrophages, which are part of the mononuclear phagocyte system [194]. In addition to phagocytosis, macrophages play an important role in innate immunity and also help initiate adaptive immunity by recruiting other immune cells, such as lymphocytes. For example, macrophages serve as antigen presenting cells to T lymphocytes (T cells) [194]. A key component of the defense mechanism of neutrophils and macrophages is ROS production, which is leveraged to combat pathogens and help with tissue repair [170]. However, the ROS generated in response to tissue injury may cause damage to macromolecules as they are not specific to combating pathogens only [86, 195].
Upon oxidative damage to cells, arachidonic acid is released from the cell membrane [196, 197]. This is critical to the induction of inflammatory response because certain enzymes, such as cyclooxygenases (COX1 and COX2) and lipoxygenase (LPO), convert arachidonic acid to inflammatory mediators, e.g., prostaglandins and leukotrienes, respectively [197, 198]. The inflammatory mediators act as signaling molecules to recruit neutrophils and subsequently macrophages to the damaged site. The recruited macrophages release cytokines, such as tumor necrotic factor-alpha (TNF-α), interleukin-1 (IL-1), and interleukin-8 (IL-8). The release of these inflammatory cytokines triggers further recruitment of neutrophils and macrophages. This creates a cyclic process leading to the elevation of various cytokine and chemokine levels [196]. These signaling proteins are linked to the activation of inflammatory response primarily through the nuclear factor-κB (NF-κB) in which TNF-α causes a signaling cascade and eventual activation of the NF-κB protein complex. This allows NF-κB to enter the nucleus of the cell and increase transcription of pro-inflammatory and anti-apoptotic genes, as well as increase production of cytokines and chemokines [196, 199]. NF-κB activation can occur as a result of exposure to cigarette smoke [200]. Even secondhand smoke has been shown to affect inflammatory response and activate the NF-κB pathway in mice sub-chronically exposed to this carcinogen [201, 202]. Experimental studies in animal models and observational studies in humans have shown that exposure to tobacco smoke or its constituents triggers inflammatory cell flux and accumulation in the lung parenchyma and bronchoalveolar lavage (BAL) fluid, followed by significant increases in inflammatory cytokine/chemokine levels, which may be reversed, at least partially, upon cessation of exposure (reviewed in refs. [17, 203–208]). Furthermore, disruption of various cell signaling pathways by smoking-associated ROS, mostly mediated through the transcription factors NF-κB and signal transducer and activator of transcription 3 (STAT3), hypoxia-inducible factor-1α, kinases, growth factors, cytokines and other proteins, and enzymes, have been linked to cellular transformation, inflammation, proliferation, invasion, angiogenesis, and metastasis of cancer (reviewed in refs. [209–213]).
Figure 3 is a schematic representation of the feedback loop between smoking-induced oxidative stress and the inflammatory response relevant to carcinogenesis. It depicts how tobacco smoke induces oxidative stress, which may damage cellular and sub-cellular targets [14, 22, 23, 52, 56, 170]. The inflicted damage leads to activation of transcription factors, such as NF-κB and other signaling molecules involved in the inflammatory response [170, 195, 214]. Inflammation gives rise to production of ROS, reactive nitrogen species (RNS), and other reactive moieties, which promote oxidative stress and increased oxidative damage to critical macromolecules, with a potential to initiate/promote carcinogenesis [52, 86, 170]. This creates a vicious cycle in which increased oxidative damage exacerbates inflammation, which, in turn, further elevates damage to macromolecular targets that may potentially lead to cancer initiation and/or progression (see, Fig. 3) [170, 195].
Figure 3. The cycle of smoking-induced oxidative damage and inflammation.
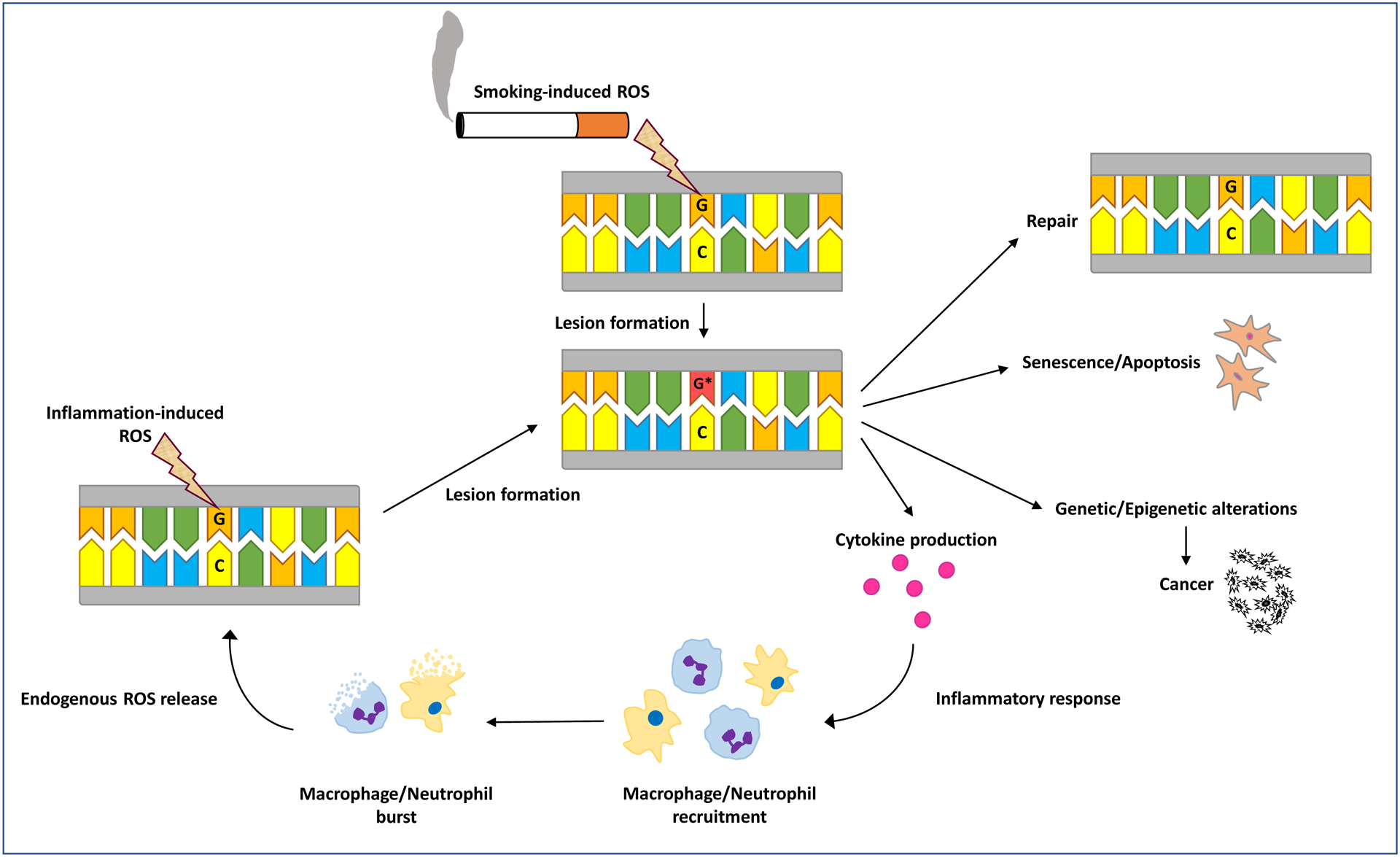
A simplified schematic diagram of the feedback loop between smoking-induced oxidative stress and the inflammatory response is shown (see, text for detailed description).
Altogether, it is estimated that chronic inflammation contributes to 15–25% of human cancers [215, 216]. The inflammatory mediators known to contribute to cancer include prostaglandins, cytokines, such as TNF-α, IL-1β, IL-6, IL-15, and chemokines, such as IL-8 and the chemokine (C-X-C motif) ligand 1 (CXCL1), formerly known as the GROα [217, 218]. These inflammatory mediators, and others, responding to various stressors, such as tobacco smoke, create an environment that fosters proliferation and survival, while also promoting oxidative stress and damage to macromolecular targets that may cause genetic and epigenetic alterations, which may trigger carcinogenesis [193, 217–219].
14. Interplay of epigenetics, smoking, and cancer
Epigenetics is a rapidly evolving field in cell biology [220–223]. Epigenetics refers to heritable, yet, reversible changes in gene expression that do not involve alterations in the underlying DNA sequence [220–222]. Epigenetic changes include aberrant DNA methylation, histone modifications and variants, microRNA (miRNA) deregulation, chromatin remodeling, and nucleosome positioning [221, 224, 225]. Of these, aberrant DNA methylation is the best-studied epigenetic alteration in human cancer [220, 222, 223, 226]. More recently, DNA hydroxymethylation has come to the forefront of epigenetic research as this chemical modification of DNA seems to play an important role in gene deregulation in many diseases, including cancer [227, 228]. 5-hydroxymethycytosine (5-hmC), the oxidation product of 5-methylcytosine (5-mC), is being increasingly viewed as an informative epigenetic mark for the study of human cancer and other diseases [227, 228].
In mammals, DNA methylation occurs almost exclusively at CpG dinucleotides whereby a methyl group is added to the C-5 position of cytosine, forming 5-mC [220, 222, 226]. This reaction is catalyzed by the maintenance DNA methyltransferase (DNMT1) and de novo DNA methyltransferase enzymes (DNMT3A and DNMT3B), with S-adenosylmethionine (SAM) as the methyl donor [220, 222, 226, 229]. Locus-specific gain of methylation (hypermethylation) at CpG dinucleotides, within CpG islands, clustered at the promoter, untranslated 5’- region, and exon 1 of known genes (promoter CpG islands) or localized within gene bodies is a common event in a wide variety of diseases, including cancer [225, 226, 229–231] and immune-related (inflammatory) diseases [232–235]. Also, global loss of methylation (hypomethylation) at repetitive DNA elements, such as long- and short interspersed nuclear elements (LINE and SINE, respectively), and long terminal repeat retrotransposons (LTR), as well as at single copy genes is a frequent occurrence in human cancer and other chronic diseases [220, 222, 226, 229]. DNA hypermethylation is believed to contribute to carcinogenesis primarily through deregulation of gene expression, e.g., through transcriptional silencing of tumor suppressor genes [220, 222, 229], whereas DNA hypomethylation is thought to play an integral role in cancer development by reactivating latent retrotransposons leading to genomic instability, and activating protooncogenes [226, 236, 237].
In the mammalian genome, the ten-eleven translocation (TET) family of methylcytosine dioxygenases (TET1, TET2, and TET3) is responsible for catalyzing 5-mC oxidation to 5-hmC [222, 227, 228]. In contrast to 5-mC, which is able to bind transcriptional repressors, 5-hmC can inhibit this binding, and therefore counteract the repressive effect of 5-mC [222, 228]. While 5-mC is often, but not always, associated with gene repression, particularly at gene promoters, 5-hmC facilitates transcription by contributing to an open chromatin state [222, 227]. Depending on the genomic loci marked by these two epigenetic modifications, both 5-mC and 5-hmC can positively or negatively influence gene transcription [222, 227, 228]. For the most part, there is a ‘complex’, and not necessarily ‘direct’, relationship between the genomic distribution and levels of 5-mC and 5-hmC and gene expression in individual cells and across different cell types [222, 227, 228]. For example, there is evidence to suggest that DNA hydroxymethylation may serve as a proxy for DNA methylation or vice versa (e.g., the more 5-mC, the greater potential for conversion to 5-hmC). Thus, the genomic distribution and level of 5-mC may influence those of 5-hmC as well as affect gene transcription [222, 228]. However, demethylation of DNA also appears to be regulated by oxidative state, where oxidative stress sequentially hydroxylates 5-mC to 5-hmC, followed by successive oxidation to 5-formylcytosine and 5-carboxylcytosine, which would eventually lead to reduction of both 5-mC and 5-hmC levels [222, 227]. It is important to note that global 5-mC or 5-hmC levels may not necessarily reflect locus/gene-specific patterns of DNA methylation or hydroxymethylation, respectively [222, 227, 228].
A growing body of literature has investigated the modulation of DNA methylation and hydroxymethylation patterns consequent to exposure to tobacco smoke [238–247]. Decreased global 5-mC levels concomitant with increased levels of global 5-hmC have been observed in cells directly exposed to tobacco smoke or its constituents [248–250]. Coulter et al. [248] have reported reduced global 5-mC levels together with elevated levels of global 5-hmC in kidney cells exposed in vitro to hydroquinone, a benzene metabolite and a major component of cigarette smoke. Ringh et al. [250] have performed genome-wide DNA methylation and hydroxymethylation analyses in BAL cells from smokers as compared to nonsmokers. The majority of differentially methylated CpGs in smokers were found hypomethylated, whereas approximately all of the differentially hydroxymethylated regions were hyperhydroxymethylated [250].
Conversely, Tellez-Plaza et al. [251] have shown a positive correlation between global DNA methylation and global DNA hydroxymethylation in blood samples collected from the same individuals at two time points (7–10 years apart). The authors confirmed their findings in an independent population of healthy men, supporting consistency in the direction of the observed association in two distinct human populations [251]. Recently, we have quantified DNA methylation levels in LINE-1 repeats, as an indicator of global 5-mC content of DNA [236, 237], and measured global levels of 5-hmC in the blood cells of healthy smokers as compared to nonsmokers, matched for age, gender, and race [252]. We observed significant loss of methylation in LINE-1 repeat elements as well as reduction of global 5-hmC levels in smokers as compared to nonsmokers. A direct and statistically-significant correlation was found between methylation levels in the LINE-1 elements and global 5-hmC levels in the study subjects. Also, inverse and statistically-significant correlations were observed between both the LINE-1 methylation levels and global 5-hmC levels and the intensity and duration of smoking, expressed as pack year [252]. We note that the discrepancies in the results of studies investigating DNA methylation and/or hydroxymethylation in various human populations have mainly been ascribed to differences in characteristics of the study populations, tissues analyzed (e.g., target vs. surrogate tissues or directly vs. indirectly exposed tissues to carcinogens, such as tobacco smoke), cell-type specificity of epigenetic marks, methodologies used, and varying sample sizes leading to different statistical power [240–247, 250–252].
Tobacco smoke constituents induce a wide variety of DNA lesions [14, 22, 23, 51, 56, 86], many of which can interfere with the binding of DNMTs to DNA, leading to genomic DNA hypomethylation [253]. In addition, certain components of tobacco smoke, such as heavy metals, e.g., cadmium and nickel, are known inhibitors of DNMTs activity [240, 254, 255]. Mammalian DNMTs contain several zinc binding sites that appear to play a critical role in regulating their function, and zinc can often be replaced by cadmium in biomolecules [256]. It is plausible that the inhaled cadmium by smokers may replace zinc binding sites, thereby inhibiting the activity and function of DNMTs, and ultimately leading to global loss of DNA methylation [252]. Takiguchi et al. [254] have reported that cadmium exposure of rat liver cells inhibits DNMTs activity in a concentration-dependent manner, and at higher doses induces DNA hypomethylation. Of note, the methyl-group donor, SAM is required both for DNA methylation and metabolism of chemicals, e.g., arsenic, present in tobacco smoke [257]. Thus, competitive demand for SAM between metabolism of specific metals (e.g., arsenic) and methylation of DNA in smokers may also lead to global DNA hypomethylation [252].
Furthermore, active loss of DNA methylation may arise from dys- or malfunction of the DNA repair machinery consequent to exposure to tobacco smoke [51, 52, 169, 240, 258]. The ROS-inducing agents in tobacco smoke [24, 41] may promote DNA hypomethylation through different mechanisms [253, 258]. The oxidized DNA lesions, such as 8-oxodG, formed at guanine within CpG dinucleotides [22, 23], have been shown to strongly inhibit methylation of the preceding cytosine [259]. Also, an unrepaired and miscoding 8-oxodG, which undergoes erroneous replication to induce G→T mutation, can cause a net loss of CpG dinucleotides [260]. Recently, Furlan et al. [261] have shown that accumulation of oxidative DNA damage in a compromised BER model of colorectal cancer is linked to significant demethylation of LINE-1 elements. In addition, under oxidative stress and elevated ROS levels and reduced availability of SAM, depletion of the methyl pool in a folate-deficient rat model has led to DNA hypomethylation [262].
Because both DNA methylation and hydroxymethylation are regulated by redox reactions [222, 228], it is possible that the elevated burden of oxidative stress imposed by smoking would uniformly impair the one-carbon transfer and the citric acid metabolic pathways that are involved in the formation of 5-mC and 5-hmC, respectively [227]. This would affect the global levels of 5-mC and 5-hmC alike. Previous studies by others have reported significant decrease in 5-hmC levels concomitant with loss of function mutations of TETs in various smoking-associated cancers [222, 227, 228, 246, 263]. For instance, Zhang et al. [246] have reported that global 5-hmC content in laryngeal squamous cell carcinoma patients was inversely related to smoking.
While the focus of this section is on DNA methylation and hydroxymethylation in relation to oxidative stress, smoking, and cancer, we refer the readers to elegant and comprehensive reviews on other epigenetic mechanisms, such as histone modifications and variants, miRNA deregulation, chromatin remodeling, and nucleosome positioning [221, 223, 224, 264, 265], which fall outside the scope and space limit of this review article.
15. Interconnections among smoking, oxidative stress, inflammation, and cancer: convergence on macromolecular damage
Tobacco smoke contains large quantities of ROS and RNS [24, 41], which are fundamental contributors to oxidative stress [22, 23]. Accumulating evidence shows an important role for oxidative stress in the pathogenesis of a variety of diseases, including cancer and immune-mediated inflammatory diseases [39, 40, 57, 58]. Chronic smoking is an established risk factor for the development of various types of malignancy as well as inflammatory diseases. ROS are oxygen-containing intermediates that can indiscriminately damage macromolecules, such as proteins, lipids, and nucleic acids [14, 35, 36, 39, 40, 56]. In humans, a highly versatile defense system, comprised of enzymatic and non-enzymatic antioxidants, prevents oxidative damage to these macromolecular targets [48–51]. However, continued smoking can overwhelm the defense mechanisms, providing an opportunity for the produced ROS to exert their damaging effects [40, 77, 86]. The human genome also contains highly complex and specialized repair mechanisms to correct the ROS-induced damage to critical macromolecules [23, 51, 52, 86, 126, 183]. If not repaired properly and in time, the induced damage may lead to functionally-important genetic and epigenetic alterations, with potentially severe biological consequences [23, 51, 52, 86, 126, 183]. Alterations of the genome and epigenome consequent to smoking-induced ROS and oxidative stress are being increasingly recognized as key drivers of carcinogenesis as well as biomarkers of inflammatory diseases [176, 225, 232, 234, 252, 253, 258, 266–272].
Oxidative damage to cellular and sub-cellular targets can also elicit an inflammatory response, which involves, among others, ROS production that initiates an intricate and inter-related cascade of events [170, 195, 214]. Triggering of the inflammatory response results in migration of neutrophils to the site of tissue damage [9, 170]. These and other inflammatory cells release ROS to combat foreign pathogens at the site of injury, and help restore the damaged tissue [9]. However, the released ROS are non-specific and may have unintentional consequences, in addition to having beneficial effects [199, 218]. More specifically, while the released ROS assist with tissue restoration, they may also elevate the burden of oxidative stress and potentially damage crucial macromolecules [199]. Thus, in an effort to counteract oxidative damage, cells involved in immune response may actually cause more harm [170]. This creates a vicious cycle in which oxidative stress causes inflammation, which in turn, results in further generation of ROS and potentially increased oxidative damage to macromolecular targets that may lead to disease development, including various types of malignancy [170, 214].
16. How lifestyle factors may shape the human genome and susceptibility to cancer: a new evolutionary study underscoring the potential role of smoking
The Amish are a conservative religious minority group who live in farm settlements, use horses for work and travel, exercise vigorously, and avoid many aspects of modern life, including lifestyle factors, such as tobacco smoking and alcohol use. They are reproductively isolated and highly inbred [273, 274]. Recently, Kessler et al. [275] have used a high-coverage whole-genome sequencing dataset from diverse populations of European, African, and Native American (Latino) ancestries, including Amish individuals from a founder population with European ancestry, to directly analyze de novo mutation rates and patterns. While demonstrating that single-nucleotide mutation rate is similar across various human ancestries and populations, they discovered a significantly reduced mutation rate (about 7% decrease) in the Amish founder population, which seemed to be driven by reductions in C→A and T→C mutations. Together with the estimation that mutation rate has zero narrow-sense heritability (h2), their findings suggest that the environment, including lifestyle factors, may play a bigger role in modulating the mutation rate/pattern than previously appreciated [275]. The Amish lifestyle comprises preindustrial era habits and activities, and while current Amish communities are diverse and have adopted some aspects of modern life, they continue to limit the influence of technology in their daily lives [273, 276]. According to Kessler et al. [275], it is plausible that the Amish are exposed to fewer and/or lower levels of environmental mutagens and carcinogens, and that their “clean living” may account for the observed reduced mutation rate and distinct mutation spectrum. It is known that rural areas, similar to those inhibited by the Amish, have fewer and/or lower concentrations of carcinogens and mutagens than industrialized areas [277–279]. Recent analysis of mutation spectra has also called into question the classic view that de novo mutations arise predominantly from replicative errors; instead, exogenous mutagens are suggested to play a larger role in mutation accumulation than previously recognized [280]. If the Amish bear a lower burden of environmentally driven mutagenesis, they would then be expected to have a decreased incidence of cancer than the general population. In fact, significant reductions in cancer rate have been found in multiple Old Order Amish populations, particularly among males [274, 281]. Importantly, the incidence rate for tobacco-related cancers in the Amish adults was 37% of the rate for non-Amish counterparts (P < 0.0001) [274]. Similarly, decreased overall mortality rate has been reported in Amish men compared to counterpart males of European American ancestry, which has been ascribed to lifestyle factors, such as reduced tobacco use and elevated physical activities (59). While Kessler et al. [275] acknowledge that the underpinning of reduction in mutation rate in the Amish remains to be fully determined [275], the potential role played by the environment, particularly lifestyle factors, in shaping the human genome and susceptibility to diseases, such as cancer, deserves special attention.
17. Concluding remarks: current challenges and future directions
It is estimated that up to a quarter of all human cancers are attributable to chronic inflammation [215, 216]. Whilst the involvement of ROS in the inflammatory response and disruption of key cell signaling pathways linked to cancer initiation and progression is greatly recognized (reviewed in refs. [209–213]), the exact role played by smoking-induced ROS and the resulting oxidative stress in carcinogenesis continues to be investigated [9, 86, 195, 216]. Current challenges facing the research field and outstanding questions remaining to be answered are identified below:
Reliable and reproducible measurement of ROS-induced macromolecular damage relevant to carcinogenesis remains, at least partly, an analytical challenge, especially when different methods and non-harmonized bench protocols in various laboratories are employed.
Adventitious oxidation of macromolecular targets during sample collection, storage, processing, and analysis is variably handled by different research groups.
The origin of the quantified ROS-induced macromolecular damage in humans is hardly attributable to a single source, such as smoking. This underscores the importance of being cognizant of and controlling for confounders (e.g., age, gender, diet, and other lifestyle factors) when designing a study, analyzing the data, and interpreting the results.
There are considerable inter- and intraindividual variations in the formation and/or repair of ROS-induced macromolecular damage, dependent on a number of factors, including subject’s metabolic capacity, repair activity, or antioxidant defense system. This underlines the need for well-designed studies with large sample sizes and sufficient statistical power to allow for meaningful and conclusive results.
In a complex mixture of chemicals, such as tobacco smoke, both ROS and ROS-inducing agents as well as other toxicants and carcinogens can exert similar effects on cellular and sub-cellular targets, resulting in comparable, if not, identical, macromolecular damage. Assigning the detected damage in a biospecimen to a specific chemical or a class of chemicals remains a challenging task. Future technological advances and development of new methods for identification and quantification of macromolecular damage with highest sensitivity and specificity should help mitigate this problem, to the utmost extent possible.
We conclude by proposing the utility of detection of macromolecular damage discussed in this review for assessing the biological consequences of novel tobacco product use. Today, new and emerging nicotine delivery systems, such as electronic cigarettes and heat-not-burn devices, are becoming increasingly popular, especially among adolescents and young adults, worldwide [4, 5]. The uncontrolled and non-regulated marketing and advertising of these novel tobacco products, especially during the early stages of their introduction into the market, have led to a perception that these alternative tobacco products are “safe” or “less-unhealthy” than conventional tobacco cigarettes [4, 5]. Currently, around 35 million people worldwide use these new nicotine delivery systems [2, 282]. Investigating the safety of use of these alternative tobacco products and assessing their health risks or potential benefits compared to smoking remain a high priority for research [4, 5, 282]. Recent studies have shown that in vitro or in vivo exposure to these novel tobacco products are associated with elevation of biomarkers of oxidative stress and increased molecular changes linked to carcinogenesis [4, 176, 252]. Future studies should compare and contrast the biological consequences of smoking vs. alternative tobacco product use by investigating the damaging effects of the respective products on crucial macromolecular targets. These investigations will provide urgently needed empirical evidence on which future regulations for manufacturing, marketing, and distribution of new and emerging tobacco products can be based. Ultimately, this research will help accomplish the universal goal of preventing or reducing the burden of tobacco-related diseases.
Supplementary Material
Highlights.
Tobacco smoke contains a host of ROS that are main determinants of oxidative stress.
Oxidative stress is a key contributor to both cancer and inflammatory diseases.
Smoking-induced ROS can damage macromolecular targets.
Smoking-elicited inflammatory response may generate further ROS/macromolecular damage.
The induced damage may lead to functionally important genetic/epigenetic alterations.
Genome/epigenome alterations are key drivers of carcinogenesis.
Acknowledgements
This work was supported by grants from the National Institute of Dental and Craniofacial Research of the National Institutes of Health (1R01DE026043) and the University of California Tobacco-Related Disease Research Program (28IR-0058 and T31IR-1839) to A.B.
Abbreviations
- 4-HNE
4-hydroxynonenal
- 5-caC
5-carboxylcytosine
- 5-fC
5-formylcytosine
- 5-hmC
5-hydroxymethylcytosine
- 5-mC
5-methylcytosine
- 8-oxodA
8-oxo-7,8-dihydro-2’-deoxyadenosine
- 8-oxodG
8-oxo-7,8-dihydro-2’-deoxyguanosine
- 15(S)-8-iso-PGF2α
F2-isoprostane 15(S)-8-iso-prostaglandin F2α
- AA
aromatic amines
- AP
apurinic/apyrimidinic
- APE1
apurinic/apyrimidinic endodeoxyribonuclease 1
- ATP
adenosine triphosphate
- BAL
bronchoalveolar lavage
- B[a]P
benzo[a]pyrene
- BER
base excision repair
- CEA
carcinoembryonic antigen
- COPD
chronic obstructive pulmonary disease
- COX
cyclooxygenases
- CRP
C-reactive protein
- CVD
cardiovascular disease
- CXCL1
chemokine (C-X-C motif) ligand 1
- dA
2’-deoxyadenosine
- dC
2’-deoxycytidine
- dG
2′-deoxyguanosine
- DNMT
DNA methyltransferase enzymes
- dTTP
2’-deoxythymidine triphosphate
- ETC
electron transport chain
- FapyG
2,6-diamino-4-hydroxy-5-formamidopyrimidine
- Fpg
formamidopyrimidine DNA glycosylase
- GGR
global genome repair
- GPx
glutathione peroxidases
- GSH
glutathione
- GSSG
oxidized GSH
- GST
glutathione S-transferases
- HERC2
HECT and RLD domain containing E3 ubiquitin protein ligase 2
- HOPdG
hydroxypropanodeoxyguanosine
- H2O2
hydrogen peroxide
- HPLC
high performance liquid chromatography
- hs-CRP
high-sensitivity C-reactive protein
- HTPs
heated tobacco products
- IARC
International Agency for Research on Cancer
- IL-1
interleukin-1
- IL-8
interleukin-8
- LDLs
low-density lipoproteins
- LINE
long- and short interspersed nuclear elements
- LPO
lipoxygenase
- LTR
long terminal repeat retrotransposons
- MDA
malondialdehyde
- M1dG
pyrimido[1,2-a]-purin-10(3H)-one
- MMR
mismatch repair
- MS
mainstream smoke
- MTH1
MutT homolog 1
- MUTYH
mutY DNA glycosylase
- NADPH
nicotinamide adenine dinucleotide phosphate
- NEIL
Nei-like glycosylases
- NER
nucleotide excision repair
- NF-κB
nuclear factor-κB
- NNK
4-(methy1nitrosamino)-1-(3-pyridyl)-1-butanone
- NNN
N’-nitrosonornicotine
- •NO
nitric oxide
- NOTCH1
notch receptor 1
- NUDT1
nudix hydrolase 1
- 1O2
singlet oxygen
- O2
oxygen
- O2•−
superoxide
- OGG1
8-oxoguanine DNA glycosylase
- •OH
hydroxyl radical
- ONOO−
peroxynitrite
- PAHs
polycyclic aromatic hydrocarbons
- PARP1
poly(ADP-ribose) polymerase 1
- POLB
DNA polymerase beta
- Q/QH2
quinone/hydroquinone
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SAM
S-adenosylmethionine
- SINE
short interspersed nuclear elements
- SNCG
synuclein gamma
- SOD
superoxide dismutase
- SPP1
secreted phosphoprotein 1
- SS
sidestream smoke
- SSB
single strand breaks
- STAT3
signal transducer and activator of transcription 3
- TBA
thiobarbituric acid
- TCR
transcription-coupled repair
- TET
ten-eleven translocation
- TLS
translesion synthesis
- T
thymidine
- TNF- α
tumor necrotic factor-alpha
- TSNAs
tobacco-specific nitrosamines
- VNAs
volatile nitrosamines
- WBC
white blood cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare no conflict of interest. The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, writing of the report, or in the decision to submit for publication.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].The US Surgeon General, The health consequences of smoking - 50 years of progress: A report of the Surgeon General, US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2014. http://www.surgeongeneral.gov/library/reports/50-years-of-progress/ [Google Scholar]
- [2].World Health Organization (WHO), WHO report on the global tobacco epidemic, 2017: Monitoring tobacco use and prevention policies, Geneva, Switzerland, 2017. Licence: CC BY-NC-SA. [Google Scholar]
- [3].World Health Organization (WHO), WHO report on the global tobacco epidemic 2019: Offer help to quit tobacco use, Geneva, Switzerland, 2019. [Google Scholar]
- [4].National Academies of Sciences, Engineering, and Medicine, Public health consequences of e-cigarettes, Washington, DC, 2018. 10.17226/24952 [DOI] [Google Scholar]
- [5].Besaratinia A, Tommasi S, Vaping epidemic: challenges and opportunities, Cancer Causes Control, 31 (2020) 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jemal A, Torre L, Soerjomataram I, Bray F, The Cancer Atlas, Third edition ed., American Cancer Society, Atlanta, GA, 2019. [Google Scholar]
- [7].American Cancer Society (ACS), Cancer Facts & Figures 2020, Atlanta, GA, 2020. [Google Scholar]
- [8].Liber A, Cahn Z, Larsen A, Drope J, Flavored E-Cigarette Sales in the United States Under Self-Regulation From January 2015 Through October 2019, American journal of public health, 110 (2020) 785–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huynh K, Inflammation: Targeting inflammatory pathways to treat atherosclerosis and cancer, Nat Rev Cardiol, 14 (2017) 629. [DOI] [PubMed] [Google Scholar]
- [10].Boukhenouna S, Wilson MA, Bahmed K, Kosmider B, Reactive Oxygen Species in Chronic Obstructive Pulmonary Disease, Oxid Med Cell Longev, 2018 (2018) 5730395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].International Agency for Research on Cancer (IARC), IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Tobacco Smoke and Involuntary Smoking, Lyon, France, 2004. [PMC free article] [PubMed] [Google Scholar]
- [12].DeMarini DM, Genotoxicity of tobacco smoke and tobacco smoke condensate: a review, Mutat Res, 567 (2004) 447–474. [DOI] [PubMed] [Google Scholar]
- [13].Besaratinia A, Pfeifer GP, Second-hand smoke and human lung cancer, The lancet oncology, 9 (2008) 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hecht SS, Lung carcinogenesis by tobacco smoke, Int J Cancer, 131 (2012) 2724–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Filaire E, Dupuis C, Galvaing G, Aubreton S, Laurent H, Richard R, Filaire M, Lung cancer: what are the links with oxidative stress, physical activity and nutrition, Lung Cancer, 82 (2013) 383–389. [DOI] [PubMed] [Google Scholar]
- [16].Goldkorn T, Filosto S, Chung S, Lung injury and lung cancer caused by cigarette smoke-induced oxidative stress: Molecular mechanisms and therapeutic opportunities involving the ceramide-generating machinery and epidermal growth factor receptor, Antioxidants & redox signaling, 21 (2014) 2149–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Münzel T, Hahad O, Kuntic M, Keaney JF, Deanfield JE, Daiber A, Effects of tobacco cigarettes, e-cigarettes, and waterpipe smoking on endothelial function and clinical outcomes, Eur Heart J, 41 (2020) 4057–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].O’Connor RJ, Hurley PJ, Existing technologies to reduce specific toxicant emissions in cigarette smoke, Tobacco control, 17 Suppl 1 (2008) i39–48. [DOI] [PubMed] [Google Scholar]
- [19].Song MA, Benowitz NL, Berman M, Brasky TM, Cummings KM, Hatsukami DK, Marian C, O’Connor R, Rees VW, Woroszylo C, Shields PG, Cigarette Filter Ventilation and its Relationship to Increasing Rates of Lung Adenocarcinoma, J Natl Cancer Inst, 109 (2017) djx075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pryor WA, Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity, Environ Health Perspect, 105 Suppl 4 (1997) 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stone KK, Bermúdez E, Pryor WA, Aqueous extracts of cigarette tar containing the tar free radical cause DNA nicks in mammalian cells, Environ Health Perspect, 102 Suppl 10 (1994) 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dizdaroglu M, Oxidatively induced DNA damage and its repair in cancer, Mutation research. Reviews in mutation research, 763 (2015) 212–245. [DOI] [PubMed] [Google Scholar]
- [23].Cadet J, Davies KJA, Medeiros MH, Di Mascio P, Wagner JR, Formation and repair of oxidatively generated damage in cellular DNA, Free Radic Biol Med, 107 (2017) 13–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Goel R, Bitzer Z, Reilly SM, Trushin N, Foulds J, Muscat J, Liao J, Elias RJ, Richie JP Jr., Variation in Free Radical Yields from U.S. Marketed Cigarettes, Chem Res Toxicol, 30 (2017) 1038–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Benowitz NL, Hukkanen J, Jacob P 3rd, Nicotine chemistry, metabolism, kinetics and biomarkers, Handbook of experimental pharmacology, (2009) 29–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stephens WE, Dependence of tar, nicotine and carbon monoxide yields on physical parameters: implications for exposure, emissions control and monitoring, Tobacco control, 16 (2007) 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kozlowski LT, O’Connor RJ, Cigarette filter ventilation is a defective design because of misleading taste, bigger puffs, and blocked vents, Tobacco control, 11 Suppl 1 (2002) I40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].World Health Organization (WHO), Tobacco and its environmental impact: an overview, Geneva, Switzerland, 2017. Licence: CC BY-NC-SA 3.0 IGO. [Google Scholar]
- [29].Besaratinia A, Tommasi S, Genotoxicity of tobacco smoke-derived aromatic amines and bladder cancer: current state of knowledge and future research directions, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 27 (2013) 2090–2100. [DOI] [PubMed] [Google Scholar]
- [30].Pearce N, Blair A, Vineis P, Ahrens W, Andersen A, Anto JM, Armstrong BK, Baccarelli AA, Beland FA, Berrington A, Bertazzi PA, Birnbaum LS, Brownson RC, Bucher JR, Cantor KP, Cardis E, Cherrie JW, Christiani DC, Cocco P, Coggon D, Comba P, Demers PA, Dement JM, Douwes J, Eisen EA, Engel LS, Fenske RA, Fleming LE, Fletcher T, Fontham E, Forastiere F, Frentzel-Beyme R, Fritschi L, Gerin M, Goldberg M, Grandjean P, Grimsrud TK, Gustavsson P, Haines A, Hartge P, Hansen J, Hauptmann M, Heederik D, Hemminki K, Hemon D, Hertz-Picciotto I, Hoppin JA, Huff J, Jarvholm B, Kang D, Karagas MR, Kjaerheim K, Kjuus H, Kogevinas M, Kriebel D, Kristensen P, Kromhout H, Laden F, Lebailly P, LeMasters G, Lubin JH, Lynch CF, Lynge E, t Mannetje A, McMichael AJ, McLaughlin JR, Marrett L, Martuzzi M, Merchant JA, Merler E, Merletti F, Miller A, Mirer FE, Monson R, Nordby KC, Olshan AF, Parent ME, Perera FP, Perry MJ, Pesatori AC, Pirastu R, Porta M, Pukkala E, Rice C, Richardson DB, Ritter L, Ritz B, Ronckers CM, Rushton L, Rusiecki JA, Rusyn I, Samet JM, Sandler DP, de Sanjose S, Schernhammer E, Costantini AS, Seixas N, Shy C, Siemiatycki J, Silverman DT, Simonato L, Smith AH, Smith MT, Spinelli JJ, Spitz MR, Stallones L, Stayner LT, Steenland K, Stenzel M, Stewart BW, Stewart PA, Symanski E, Terracini B, Tolbert PE, Vainio H, Vena J, Vermeulen R, Victora CG, Ward EM, Weinberg CR, Weisenburger D, Wesseling C, Weiderpass E, Zahm SH, IARC monographs: 40 years of evaluating carcinogenic hazards to humans, Environ Health Perspect, 123 (2015) 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR, Mutational signatures associated with tobacco smoking in human cancer, Science (New York, N.Y.), 354 (2016) 618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, Getz G, Rozen SG, Stratton MR, The repertoire of mutational signatures in human cancer, Nature, 578 (2020) 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Luch A, Nature and nurture - lessons from chemical carcinogenesis, Nat Rev Cancer, 5 (2005) 113–125. [DOI] [PubMed] [Google Scholar]
- [34].India Project Team of the International Cancer Genome Consortium, Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups, Nature communications, 4 (2013) 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yan M, Lo JC, Edwards JT, Baran PS, Radicals: Reactive Intermediates with Translational Potential, Journal of the American Chemical Society, 138 (2016) 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dickinson BC, Chang CJ, Chemistry and biology of reactive oxygen species in signaling or stress responses, Nature chemical biology, 7 (2011) 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA, Environmental and chemical carcinogenesis, Seminars in cancer biology, 14 (2004) 473–486. [DOI] [PubMed] [Google Scholar]
- [38].Pryor WA, Stone K, Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite, Ann N Y Acad Sci, 686 (1993) 12–27; discussion 27–18. [DOI] [PubMed] [Google Scholar]
- [39].Frijhoff J, Winyard PG, Zarkovic N, Davies SS, Stocker R, Cheng D, Knight AR, Taylor EL, Oettrich J, Ruskovska T, Gasparovic AC, Cuadrado A, Weber D, Poulsen HE, Grune T, Schmidt HH, Ghezzi P, Clinical Relevance of Biomarkers of Oxidative Stress, Antioxidants & redox signaling, 23 (2015) 1144–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Helfinger V, Schröder K, Redox control in cancer development and progression, Molecular aspects of medicine, 63 (2018) 88–98. [DOI] [PubMed] [Google Scholar]
- [41].Horinouchi T, Higashi T, Mazaki Y, Miwa S, Carbonyl Compounds in the Gas Phase of Cigarette Mainstream Smoke and Their Pharmacological Properties, Biological & pharmaceutical bulletin, 39 (2016) 909–914. [DOI] [PubMed] [Google Scholar]
- [42].Turrens JF, Mitochondrial formation of reactive oxygen species, J Physiol, 552 (2003) 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rada B, Leto TL, Oxidative innate immune defenses by Nox/Duox family NADPH oxidases, Contrib Microbiol, 15 (2008) 164–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Belikov AV, Schraven B, Simeoni L, T cells and reactive oxygen species, J Biomed Sci, 22 (2015) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen X, Song M, Zhang B, Zhang Y, Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment, Oxid Med Cell Longev, 2016 (2016) 1580967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dosch M, Gerber J, Jebbawi F, Beldi G, Mechanisms of ATP Release by Inflammatory Cells, Int J Mol Sci, 19 (2018) 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhao RZ, Jiang S, Zhang L, Yu ZB, Mitochondrial electron transport chain, ROS generation and uncoupling (Review), Int J Mol Med, 44 (2019) 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hayes JD, Flanagan JU, Jowsey IR, Glutathione transferases, Annual review of pharmacology and toxicology, 45 (2005) 51–88. [DOI] [PubMed] [Google Scholar]
- [49].Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O, Oxidative stress and antioxidant defense, World Allergy Organ J, 5 (2012) 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M, Targeting Free Radicals in Oxidative Stress-Related Human Diseases, Trends in pharmacological sciences, 38 (2017) 592–607. [DOI] [PubMed] [Google Scholar]
- [51].Poetsch AR, The genomics of oxidative DNA damage, repair, and resulting mutagenesis, Computational and structural biotechnology journal, 18 (2020) 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Alnajjar KS, Sweasy JB, A new perspective on oxidation of DNA repair proteins and cancer, DNA Repair (Amst), 76 (2019) 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M, Cellular Senescence: Defining a Path Forward, Cell, 179 (2019) 813–827. [DOI] [PubMed] [Google Scholar]
- [54].Munoz-Espin D, Serrano M, Cellular senescence: from physiology to pathology, Nature reviews. Molecular cell biology, 15 (2014) 482–496. [DOI] [PubMed] [Google Scholar]
- [55].Galadari S, Rahman A, Pallichankandy S, Thayyullathil F, Reactive oxygen species and cancer paradox: To promote or to suppress?, Free Radic Biol Med, 104 (2017) 144–164. [DOI] [PubMed] [Google Scholar]
- [56].Phillips DH, Venitt S, DNA and protein adducts in human tissues resulting from exposure to tobacco smoke, Int J Cancer, 131 (2012) 2733–2753. [DOI] [PubMed] [Google Scholar]
- [57].Saikolappan S, Kumar B, Shishodia G, Koul S, Koul HK, Reactive oxygen species and cancer: A complex interaction, Cancer letters, 452 (2019) 132–143. [DOI] [PubMed] [Google Scholar]
- [58].Yan LJ, Protein redox modification as a cellular defense mechanism against tissue ischemic injury, Oxid Med Cell Longev, 2014 (2014) 343154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Huang Z, Chen Y, Zhang Y, Mitochondrial reactive oxygen species cause major oxidative mitochondrial DNA damages and repair pathways, Journal of biosciences, 45 (2020) 84. [PubMed] [Google Scholar]
- [60].Goel R, Bitzer ZT, Reilly SM, Foulds J, Muscat J, Elias RJ, Richie JP, Influence of Smoking Puff Parameters and Tobacco Varieties on Free Radicals Yields in Cigarette Mainstream Smoke, Chem Res Toxicol, 31 (2018) 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sumanasekera WK, Dao HT, Shekhovtsova V, Schultz K, Jani M, Gyamfi F, Tran DM, Le N, The mechanistic role of oxidative stress in cigarette smoke-induced cardiac stem cell dysfunction and prevention by ascorbic acid, Cell Biol Toxicol, 35 (2019) 111–127. [DOI] [PubMed] [Google Scholar]
- [62].Che M, Wang R, Li X, Wang HY, Zheng XFS, Expanding roles of superoxide dismutases in cell regulation and cancer, Drug discovery today, 21 (2016) 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Griess B, Tom E, Domann F, Teoh-Fitzgerald M, Extracellular superoxide dismutase and its role in cancer, Free Radic Biol Med, 112 (2017) 464–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Deepa SS, Van Remmen H, Brooks SV, Faulkner JA, Larkin L, McArdle A, Jackson MJ, Vasilaki A, Richardson A, Accelerated sarcopenia in Cu/Zn superoxide dismutase knockout mice, Free Radic Biol Med, 132 (2019) 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Samuel EL, Marcano DC, Berka V, Bitner BR, Wu G, Potter A, Fabian RH, Pautler RG, Kent TA, Tsai AL, Tour JM, Highly efficient conversion of superoxide to oxygen using hydrophilic carbon clusters, Proc Natl Acad Sci U S A, 112 (2015) 2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bandeira CM, de Almeida A, Carta CFL, Almeida AA, de Figueiredo FAT, Sandrim VC, Gonçalves AJ, Almeida JD, Tobacco influence in heavy metals levels in head and neck cancer cases, Environ Sci Pollut Res Int, 25 (2018) 27650–27656. [DOI] [PubMed] [Google Scholar]
- [67].Caldecott KW, Single-strand break repair and genetic disease, Nature reviews. Genetics, 9 (2008) 619–631. [DOI] [PubMed] [Google Scholar]
- [68].Caldecott KW, DNA single-strand break repair and spinocerebellar ataxia, Cell, 112 (2003) 7–10. [DOI] [PubMed] [Google Scholar]
- [69].Ray Chaudhuri A, Nussenzweig A, The multifaceted roles of PARP1 in DNA repair and chromatin remodelling, Nature reviews. Molecular cell biology, 18 (2017) 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Abbotts R, Wilson DM 3rd, Coordination of DNA single strand break repair, Free Radic Biol Med, 107 (2017) 228–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Benhar M, Roles of mammalian glutathione peroxidase and thioredoxin reductase enzymes in the cellular response to nitrosative stress, Free Radic Biol Med, 127 (2018) 160–164. [DOI] [PubMed] [Google Scholar]
- [72].Jiao Y, Wang Y, Guo S, Wang G, Glutathione peroxidases as oncotargets, Oncotarget, 8 (2017) 80093–80102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Orhan H, Evelo CT, Sahin G, Erythrocyte antioxidant defense response against cigarette smoking in humans--the glutathione S-transferase vulnerability, J Biochem Mol Toxicol, 19 (2005) 226–233. [DOI] [PubMed] [Google Scholar]
- [74].Vlahos R, Bozinovski S, Glutathione peroxidase-1 as a novel therapeutic target for COPD, Redox Rep, 18 (2013) 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bentley AR, Emrani P, Cassano PA, Genetic variation and gene expression in antioxidant related enzymes and risk of COPD: a systematic review, Thorax, 63 (2008) 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Foronjy RF, Mirochnitchenko O, Propokenko O, Lemaitre V, Jia Y, Inouye M, Okada Y, D’Armiento JM, Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in mice, Am J Respir Crit Care Med, 173 (2006) 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Klaunig JE, Oxidative Stress and Cancer, Current pharmaceutical design, 24 (2018) 4771–4778. [DOI] [PubMed] [Google Scholar]
- [78].Łuczaj W, Gęgotek A, Skrzydlewska E, Antioxidants and HNE in redox homeostasis, Free Radic Biol Med, 111 (2017) 87–101. [DOI] [PubMed] [Google Scholar]
- [79].Eckl PM, Bresgen N, Genotoxicity of lipid oxidation compounds, Free Radic Biol Med, 111 (2017) 244–252. [DOI] [PubMed] [Google Scholar]
- [80].Umeno A, Biju V, Yoshida Y, In vivo ROS production and use of oxidative stress-derived biomarkers to detect the onset of diseases such as Alzheimer’s disease, Parkinson’s disease, and diabetes, Free Radic Res, 51 (2017) 413–427. [DOI] [PubMed] [Google Scholar]
- [81].Winczura A, Zdżalik D, Tudek B, Damage of DNA and proteins by major lipid peroxidation products in genome stability, Free Radic Res, 46 (2012) 442–459. [DOI] [PubMed] [Google Scholar]
- [82].Hauck AK, Bernlohr DA, Oxidative stress and lipotoxicity, Journal of lipid research, 57 (2016) 1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Barrera G, Pizzimenti S, Daga M, Dianzani C, Arcaro A, Cetrangolo GP, Giordano G, Cucci MA, Graf M, Gentile F, Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders, Antioxidants (Basel), 7 (2018) 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Knutson CG, Akingbade D, Crews BC, Voehler M, Stec DF, Marnett LJ, Metabolism in vitro and in vivo of the DNA base adduct, M1G, Chem Res Toxicol, 20 (2007) 550–557. [DOI] [PubMed] [Google Scholar]
- [85].Wauchope OR, Mitchener MM, Beavers WN, Galligan JJ, Camarillo JM, Sanders WD, Kingsley PJ, Shim HN, Blackwell T, Luong T, deCaestecker M, Fessel JP, Marnett LJ, Oxidative stress increases M1dG, a major peroxidation-derived DNA adduct, in mitochondrial DNA, Nucleic Acids Res, 46 (2018) 3458–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yu Y, Cui Y, Niedernhofer LJ, Wang Y, Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage, Chem Res Toxicol, 29 (2016) 2008–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ayala A, Muñoz MF, Argüelles S, Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal, Oxid Med Cell Longev, 2014 (2014) 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tsikas D, Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges, Anal Biochem, 524 (2017) 13–30. [DOI] [PubMed] [Google Scholar]
- [89].Esterbauer H, Schaur RJ, Zollner H, Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes, Free Radic Biol Med, 11 (1991) 81–128. [DOI] [PubMed] [Google Scholar]
- [90].Domijan AM, Ralić J, Radić Brkanac S, Rumora L, Žanić-Grubišić T, Quantification of malondialdehyde by HPLC-FL - application to various biological samples, Biomed Chromatogr, 29 (2015) 41–46. [DOI] [PubMed] [Google Scholar]
- [91].Tsikas D, Rothmann S, Schneider JY, Gutzki FM, Beckmann B, Frölich JC, Simultaneous GC-MS/MS measurement of malondialdehyde and 4-hydroxy-2-nonenal in human plasma: Effects of long-term L-arginine administration, Anal Biochem, 524 (2017) 31–44. [DOI] [PubMed] [Google Scholar]
- [92].Khoubnasabjafari M, Ansarin K, Jouyban A, Critical Review of Malondialdehyde Analysis in Biological Samples, Curr. Pharmac. Anal, 12 4–17. [Google Scholar]
- [93].Bridges AB, Scott NA, Parry GJ, Belch JJ, Age, sex, cigarette smoking and indices of free radical activity in healthy humans, Eur J Med, 2 (1993) 205–208. [PubMed] [Google Scholar]
- [94].Lapenna D, Mezzetti A, de Gioia S, Pierdomenico SD, Daniele F, Cuccurullo F, Plasma copper and lipid peroxidation in cigarette smokers, Free Radic Biol Med, 19 (1995) 849–852. [DOI] [PubMed] [Google Scholar]
- [95].Nielsen F, Mikkelsen BB, Nielsen JB, Andersen HR, Grandjean P, Plasma malondialdehyde as biomarker for oxidative stress: reference interval and effects of life-style factors, Clin Chem, 43 (1997) 1209–1214. [PubMed] [Google Scholar]
- [96].Kim HS, Lee BM, Protective effects of antioxidant supplementation on plasma lipid peroxidation in smokers, J Toxicol Environ Health A, 63 (2001) 583–598. [DOI] [PubMed] [Google Scholar]
- [97].Vassalle C, Lubrano V, L’Abbate A, Clerico A, Determination of nitrite plus nitrate and malondialdehyde in human plasma: analytical performance and the effect of smoking and exercise, Clin Chem Lab Med, 40 (2002) 802–809. [DOI] [PubMed] [Google Scholar]
- [98].Li N, Jia X, Chen CY, Blumberg JB, Song Y, Zhang W, Zhang X, Ma G, Chen J, Almond consumption reduces oxidative DNA damage and lipid peroxidation in male smokers, J Nutr, 137 (2007) 2717–2722. [DOI] [PubMed] [Google Scholar]
- [99].Bloomer RJ, Decreased blood antioxidant capacity and increased lipid peroxidation in young cigarette smokers compared to nonsmokers: Impact of dietary intake, Nutr J, 6 (2007) 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lykkesfeldt J, Malondialdehyde as biomarker of oxidative damage to lipids caused by smoking, Clin Chim Acta, 380 (2007) 50–58. [DOI] [PubMed] [Google Scholar]
- [101].Pré J, Le Floch A, Lipid-peroxidation products and antioxidants in plasma of cigarette smokers, Clin Chem, 36 (1990) 1849–1850. [PubMed] [Google Scholar]
- [102].Kamal AA, el Khafif M, Koraah S, Massoud A, Caillard JF, Blood superoxide dismutase and plasma malondialdehyde among workers exposed to asbestos, Am J Ind Med, 21 (1992) 353–361. [DOI] [PubMed] [Google Scholar]
- [103].Wang Y, Wang W, Yang H, Shao D, Zhao X, Zhang G, Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of Toll-like receptor 4 signaling, Free Radic Biol Med, 131 (2019) 237–242. [DOI] [PubMed] [Google Scholar]
- [104].Zińczuk J, Maciejczyk M, Zaręba K, Romaniuk W, Markowski A, Kędra B, Zalewska A, Pryczynicz A, Matowicka-Karna J, Guzińska-Ustymowicz K, Antioxidant Barrier, Redox Status, and Oxidative Damage to Biomolecules in Patients with Colorectal Cancer. Can Malondialdehyde and Catalase Be Markers of Colorectal Cancer Advancement?, Biomolecules, 9 (2019) 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Brancato B, Munnia A, Cellai F, Ceni E, Mello T, Bianchi S, Catarzi S, Risso GG, Galli A, Peluso ME, 8-Oxo-7,8-dihydro-2’-deoxyguanosine and other lesions along the coding strand of the exon 5 of the tumour suppressor gene P53 in a breast cancer case-control study, DNA Res, 23 (2016) 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Karki K, Pande D, Negi R, Khanna S, Khanna RS, Khanna HD, Expression of serum toll-like receptor 9 and oxidative damage markers in benign and malignant breast diseases, DNA Cell Biol, 33 (2014) 630–636. [DOI] [PubMed] [Google Scholar]
- [107].Kim SI, Pfeifer GP, Besaratinia A, Lack of mutagenicity of acrolein-induced DNA adducts in mouse and human cells, Cancer Res, 67 (2007) 11640–11647. [DOI] [PubMed] [Google Scholar]
- [108].Zhang S, Villalta PW, Wang M, Hecht SS, Detection and quantitation of acrolein-derived 1,N2-propanodeoxyguanosine adducts in human lung by liquid chromatography-electrospray ionization-tandem mass spectrometry, Chem Res Toxicol, 20 (2007) 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Horiyama S, Kunitomo M, Yoshikawa N, Nakamura K, Mass Spectrometric Approaches to the Identification of Potential Ingredients in Cigarette Smoke Causing Cytotoxicity, Biological & pharmaceutical bulletin, 39 (2016) 903–908. [DOI] [PubMed] [Google Scholar]
- [110].Dalleau S, Baradat M, Guéraud F, Huc L, Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance, Cell Death Differ, 20 (2013) 1615–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Singhal SS, Singh SP, Singhal P, Horne D, Singhal J, Awasthi S, Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling, Toxicology and applied pharmacology, 289 (2015) 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Jaganjac M, Milkovic L, Gegotek A, Cindric M, Zarkovic K, Skrzydlewska E, Zarkovic N, The relevance of pathophysiological alterations in redox signaling of 4-hydroxynonenal for pharmacological therapies of major stress-associated diseases, Free Radic Biol Med, 157 (2020) 128–153. [DOI] [PubMed] [Google Scholar]
- [113].Hawkins CL, Davies MJ, Detection, identification, and quantification of oxidative protein modifications, J Biol Chem, 294 (2019) 19683–19708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Hauck AK, Huang Y, Hertzel AV, Bernlohr DA, Adipose oxidative stress and protein carbonylation, J Biol Chem, 294 (2019) 1083–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Campolo N, Issoglio FM, Estrin DA, Bartesaghi S, Radi R, 3-Nitrotyrosine and related derivatives in proteins: precursors, radical intermediates and impact in function, Essays Biochem, 64 (2020) 111–133. [DOI] [PubMed] [Google Scholar]
- [116].Dalle-Donne I, Colombo G, Gornati R, Garavaglia ML, Portinaro N, Giustarini D, Bernardini G, Rossi R, Milzani A, Protein Carbonylation in Human Smokers and Mammalian Models of Exposure to Cigarette Smoke: Focus on Redox Proteomic Studies, Antioxidants & redox signaling, 26 (2017) 406–426. [DOI] [PubMed] [Google Scholar]
- [117].Moldogazieva NT, Lutsenko SV, Terentiev AA, Reactive Oxygen and Nitrogen Species-Induced Protein Modifications: Implication in Carcinogenesis and Anticancer Therapy, Cancer Res, 78 (2018) 6040–6047. [DOI] [PubMed] [Google Scholar]
- [118].Colombo G, Garavaglia ML, Astori E, Giustarini D, Rossi R, Milzani A, Dalle-Donne I, Protein carbonylation in human bronchial epithelial cells exposed to cigarette smoke extract, Cell Biol Toxicol, 35 (2019) 345–360. [DOI] [PubMed] [Google Scholar]
- [119].Bartesaghi S, Radi R, Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration, Redox Biol, 14 (2018) 618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Cai Z, Yan LJ, Protein Oxidative Modifications: Beneficial Roles in Disease and Health, J Biochem Pharmacol Res, 1 (2013) 15–26. [PMC free article] [PubMed] [Google Scholar]
- [121].Tam A, Churg A, Wright JL, Zhou S, Kirby M, Coxson HO, Lam S, Man SF, Sin DD, Sex Differences in Airway Remodeling in a Mouse Model of Chronic Obstructive Pulmonary Disease, Am J Respir Crit Care Med, 193 (2016) 825–834. [DOI] [PubMed] [Google Scholar]
- [122].Chen HJ, Lin WP, Chiu SD, Fan CH, Multistage mass spectrometric analysis of human hemoglobin glutathionylation: correlation with cigarette smoking, Chem Res Toxicol, 27 (2014) 864–872. [DOI] [PubMed] [Google Scholar]
- [123].Farmer EE, Davoine C, Reactive electrophile species, Current opinion in plant biology, 10 (2007) 380–386. [DOI] [PubMed] [Google Scholar]
- [124].Nohmi T, Kim SR, Yamada M, Modulation of oxidative mutagenesis and carcinogenesis by polymorphic forms of human DNA repair enzymes, Mutat Res, 591 (2005) 60–73. [DOI] [PubMed] [Google Scholar]
- [125].Hidaka K, Yamada M, Kamiya H, Masutani C, Harashima H, Hanaoka F, Nohmi T, Specificity of mutations induced by incorporation of oxidized dNTPs into DNA by human DNA polymerase eta, DNA Repair (Amst), 7 (2008) 497–506. [DOI] [PubMed] [Google Scholar]
- [126].Dutta A, Yang C, Sengupta S, Mitra S, Hegde ML, New paradigms in the repair of oxidative damage in human genome: mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins, Cellular and molecular life sciences : CMLS, 72 (2015) 1679–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Steenken S, Jovanovic SV, How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution, J. Am. Chem. Soc, 119 (1997) 617–618. [Google Scholar]
- [128].Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM, The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo, Biochemistry, 42 (2003) 9257–9262. [DOI] [PubMed] [Google Scholar]
- [129].Neeley WL, Essigmann JM, Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products, Chem Res Toxicol, 19 (2006) 491–505. [DOI] [PubMed] [Google Scholar]
- [130].David SS, O’Shea VL, Kundu S, Base-excision repair of oxidative DNA damage, Nature, 447 (2007) 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Boiteux S, Gajewski E, Laval J, Dizdaroglu M, Substrate specificity of the Escherichia coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of purine lesions in DNA produced by ionizing radiation or photosensitization, Biochemistry, 31 (1992) 106–110. [DOI] [PubMed] [Google Scholar]
- [132].Cadet J, Ravanat JL, Martinez GR, Medeiros MH, Di Mascio P, Singlet oxygen oxidation of isolated and cellular DNA: product formation and mechanistic insights, Photochemistry and photobiology, 82 (2006) 1219–1225. [DOI] [PubMed] [Google Scholar]
- [133].Caliri AW, Tommasi S, Bates SE, Besaratinia A, Spontaneous and photosensitization-induced mutations in primary mouse cells transitioning through senescence and immortalization, J Biol Chem, 295 (2020) 9974–9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Loft S, Poulsen HE, Cancer risk and oxidative DNA damage in man, J Mol Med (Berl), 74 (1996) 297–312. [DOI] [PubMed] [Google Scholar]
- [135].Gedik CM, Collins A, Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 19 (2005) 82–84. [DOI] [PubMed] [Google Scholar]
- [136].Valavanidis A, Vlachogianni T, Fiotakis C, 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis, J Environ Sci Health C Environ Carcinog Ecotoxicol Rev, 27 (2009) 120–139. [DOI] [PubMed] [Google Scholar]
- [137].Mesaros C, Arora JS, Wholer A, Vachani A, Blair IA, 8-Oxo-2’-deoxyguanosine as a biomarker of tobacco-smoking-induced oxidative stress, Free Radic Biol Med, 53 (2012) 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Møller P, Cooke MS, Collins A, Olinski R, Rozalski R, Loft S, Harmonising measurements of 8-oxo-7,8-dihydro-2’-deoxyguanosine in cellular DNA and urine, Free Radic Res, 46 (2012) 541–553. [DOI] [PubMed] [Google Scholar]
- [139].Lodovici M, Caldini S, Luceri C, Bambi F, Boddi V, Dolara P, Active and passive smoking and lifestyle determinants of 8-oxo-7,8-dihydro-2’-deoxyguanosine levels in human leukocyte DNA, Cancer Epidemiol Biomarkers Prev, 14 (2005) 2975–2977. [DOI] [PubMed] [Google Scholar]
- [140].Seet RC, Lee CY, Loke WM, Huang SH, Huang H, Looi WF, Chew ES, Quek AM, Lim EC, Halliwell B, Biomarkers of oxidative damage in cigarette smokers: which biomarkers might reflect acute versus chronic oxidative stress?, Free Radic Biol Med, 50 (2011) 1787–1793. [DOI] [PubMed] [Google Scholar]
- [141].Halliwell B, Oxidative stress and cancer: have we moved forward?, The Biochemical journal, 401 (2007) 1–11. [DOI] [PubMed] [Google Scholar]
- [142].Poulsen HE, Oxidative DNA modifications, Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie, 57 Suppl 1 (2005) 161–169. [DOI] [PubMed] [Google Scholar]
- [143].Lee JD, Cai Q, Shu XO, Nechuta SJ, The Role of Biomarkers of Oxidative Stress in Breast Cancer Risk and Prognosis: A Systematic Review of the Epidemiologic Literature, J Womens Health (Larchmt), 26 (2017) 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Gackowski D, Kowalewski J, Siomek A, Olinski R, Oxidative DNA damage and antioxidant vitamin level: comparison among lung cancer patients, healthy smokers and nonsmokers, Int J Cancer, 114 (2005) 153–156. [DOI] [PubMed] [Google Scholar]
- [145].Yano T, Shoji F, Baba H, Koga T, Shiraishi T, Orita H, Kohno H, Significance of the urinary 8-OHdG level as an oxidative stress marker in lung cancer patients, Lung Cancer, 63 (2009) 111–114. [DOI] [PubMed] [Google Scholar]
- [146].Olinski R, Gackowski D, Cooke MS, Endogenously generated DNA nucleobase modifications source, and significance as possible biomarkers of malignant transformation risk, and role in anticancer therapy, Biochim Biophys Acta Rev Cancer, 1869 (2018) 29–41. [DOI] [PubMed] [Google Scholar]
- [147].Harman SM, Liang L, Tsitouras PD, Gucciardo F, Heward CB, Reaven PD, Ping W, Ahmed A, Cutler RG, Urinary excretion of three nucleic acid oxidation adducts and isoprostane F(2)alpha measured by liquid chromatography-mass spectrometry in smokers, ex-smokers, and nonsmokers, Free Radic Biol Med, 35 (2003) 1301–1309. [DOI] [PubMed] [Google Scholar]
- [148].Gackowski D, Speina E, Zielinska M, Kowalewski J, Rozalski R, Siomek A, Paciorek T, Tudek B, Olinski R, Products of oxidative DNA damage and repair as possible biomarkers of susceptibility to lung cancer, Cancer Res, 63 (2003) 4899–4902. [PubMed] [Google Scholar]
- [149].Feng S, Roethig HJ, Liang Q, Kinser R, Jin Y, Scherer G, Urban M, Engl J, Riedel K, Evaluation of urinary 1-hydroxypyrene, S-phenylmercapturic acid, trans,trans-muconic acid, 3-methyladenine, 3-ethyladenine, 8-hydroxy-2’-deoxyguanosine and thioethers as biomarkers of exposure to cigarette smoke, Biomarkers, 11 (2006) 28–52. [DOI] [PubMed] [Google Scholar]
- [150].Pilger A, Rüdiger HW, 8-Hydroxy-2’-deoxyguanosine as a marker of oxidative DNA damage related to occupational and environmental exposures, Int Arch Occup Environ Health, 80 (2006) 1–15. [DOI] [PubMed] [Google Scholar]
- [151].Poulsen HE, Nadal LL, Broedbaek K, Nielsen PE, Weimann A, Detection and interpretation of 8-oxodG and 8-oxoGua in urine, plasma and cerebrospinal fluid, Biochim Biophys Acta, 1840 (2014) 801–808. [DOI] [PubMed] [Google Scholar]
- [152].Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA, 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions, J Biol Chem,267 (1992) 166–172. [PubMed] [Google Scholar]
- [153].Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM, Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome, Biochemistry, 29 (1990) 7024–7032. [DOI] [PubMed] [Google Scholar]
- [154].Tudek B, Laval J, Boiteux S, SOS-independent mutagenesis in lacZ induced by methylene blue plus visible light, Molecular & general genetics : MGG, 236 (1993) 433–439. [DOI] [PubMed] [Google Scholar]
- [155].Pavlov YI, Minnick DT, Izuta S, Kunkel TA, DNA replication fidelity with 8-oxodeoxyguanosine triphosphate, Biochemistry, 33 (1994) 4695–4701. [DOI] [PubMed] [Google Scholar]
- [156].De Flora S, Izzotti A, D’Agostini F, Bennicelli C, You M, Lubet RA, Balansky RM, Induction and modulation of lung tumors: genomic and transcriptional alterations in cigarette smoke-exposed mice, Experimental lung research, 31 (2005) 19–35. [DOI] [PubMed] [Google Scholar]
- [157].Besaratinia A, Li H, Yoon JI, Zheng A, Gao H, Tommasi S, A high-throughput next-generation sequencing-based method for detecting the mutational fingerprint of carcinogens, Nucleic Acids Res, 40 (2012) e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Kim SI, Yoon JI, Tommasi S, Besaratinia A, New experimental data linking secondhand smoke exposure to lung cancer in nonsmokers, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 26 (2012) 1845–1854. [DOI] [PubMed] [Google Scholar]
- [159].Micale RT, La Maestra S, Di Pietro A, Visalli G, Baluce B, Balansky R, Steele VE, De Flora S, Oxidative stress in the lung of mice exposed to cigarette smoke either early in life or in adulthood, Archives of toxicology, 87 (2013) 915–918. [DOI] [PubMed] [Google Scholar]
- [160].Shimizu M, Gruz P, Kamiya H, Masutani C, Xu Y, Usui Y, Sugiyama H, Harashima H, Hanaoka F, Nohmi T, Efficient and erroneous incorporation of oxidized DNA precursors by human DNA polymerase eta, Biochemistry, 46 (2007) 5515–5522. [DOI] [PubMed] [Google Scholar]
- [161].Kasai H, Chemistry-based studies on oxidative DNA damage: formation, repair, and mutagenesis, Free Radic Biol Med, 33 (2002) 450–456. [PubMed] [Google Scholar]
- [162].Michaels ML, Miller JH, The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine), Journal of bacteriology, 174 (1992) 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Maki H, Sekiguchi M, MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis, Nature, 355 (1992) 273–275. [DOI] [PubMed] [Google Scholar]
- [164].Michaels ML, Cruz C, Grollman AP, Miller JH, Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA, Proc Natl Acad Sci U S A, 89 (1992) 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H, Sekiguchi M, Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis, J Biol Chem, 268 (1993) 23524–23530. [PubMed] [Google Scholar]
- [166].Sakai A, Nakanishi M, Yoshiyama K, Maki H, Impact of reactive oxygen species on spontaneous mutagenesis in Escherichia coli, Genes to cells : devoted to molecular & cellular mechanisms, 11 (2006) 767–778. [DOI] [PubMed] [Google Scholar]
- [167].Furuichi M, Yoshida MC, Oda H, Tajiri T, Nakabeppu Y, Tsuzuki T, Sekiguchi M, Genomic structure and chromosome location of the human mutT homologue gene MTH1 encoding 8-oxo-dGTPase for prevention of A:T to C:G transversion, Genomics, 24 (1994) 485–490. [DOI] [PubMed] [Google Scholar]
- [168].Russo MT, Blasi MF, Chiera F, Fortini P, Degan P, Macpherson P, Furuichi M, Nakabeppu Y, Karran P, Aquilina G, Bignami M, The oxidized deoxynucleoside triphosphate pool is a significant contributor to genetic instability in mismatch repair-deficient cells, Mol Cell Biol, 24 (2004) 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Paz-Elizur T, Sevilya Z, Leitner-Dagan Y, Elinger D, Roisman LC, Livneh Z, DNA repair of oxidative DNA damage in human carcinogenesis: potential application for cancer risk assessment and prevention, Cancer letters, 266 (2008) 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Kay J, Thadhani E, Samson L, Engelward B, Inflammation-induced DNA damage, mutations and cancer, DNA Repair (Amst), 83 (2019) 102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Phillips DH, Mutational spectra and mutational signatures: Insights into cancer aetiology and mechanisms of DNA damage and repair, DNA Repair (Amst), 71 (2018) 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Hollstein M, Alexandrov LB, Wild CP, Ardin M, Zavadil J, Base changes in tumour DNA have the power to reveal the causes and evolution of cancer, Oncogene, 36 (2017) 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Barta JA, Pauley K, Kossenkov AV, McMahon SB, The lung-enriched p53 mutants V157F and R158L/P regulate a gain of function transcriptome in lung cancer, Carcinogenesis, 41 (2020) 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, Gomez C, Degasperi A, Harris R, Jackson SP, Arlt VM, Phillips DH, Nik-Zainal S, A Compendium of Mutational Signatures of Environmental Agents, Cell, 177 (2019) 821–836.e816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Korenjak M, Zavadil J, Experimental identification of cancer driver alterations in the era of pan-cancer genomics, Cancer science, 110 (2019) 3622–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Tommasi S, Caliri AW, Caceres A, Moreno DE, Li M, Chen Y, Siegmund KD, Besaratinia A, Deregulation of Biologically Significant Genes and Associated Molecular Pathways in the Oral Epithelium of Electronic Cigarette Users, Int J Mol Sci, 20 (2019) 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Sakamoto K, Notch signaling in oral squamous neoplasia, Pathology international, 66 (2016) 609–617. [DOI] [PubMed] [Google Scholar]
- [178].Leemans CR, Snijders PJF, Brakenhoff RH, The molecular landscape of head and neck cancer, Nat Rev Cancer, 18 (2018) 269–282. [DOI] [PubMed] [Google Scholar]
- [179].Sanchez-Tena S, Cubillos-Rojas M, Schneider T, Rosa JL, Functional and pathological relevance of HERC family proteins: a decade later, Cellular and molecular life sciences : CMLS, 73 (2016) 1955–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Ogawa F, Walters MS, Shafquat A, O’Beirne SL, Kaner RJ, Mezey JG, Zhang H, Leopold PL, Crystal RG, Role of KRAS in regulating normal human airway basal cell differentiation, Respir Res, 20 (2019) 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Malumbres M, Barbacid M, RAS oncogenes: the first 30 years, Nat Rev Cancer, 3 (2003) 459–465. [DOI] [PubMed] [Google Scholar]
- [182].Prior IA, Lewis PD, Mattos C, A comprehensive survey of Ras mutations in cancer, Cancer Res, 72 (2012) 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Brierley DJ, Martin SA, Oxidative stress and the DNA mismatch repair pathway, Antioxidants & redox signaling, 18 (2013) 2420–2428. [DOI] [PubMed] [Google Scholar]
- [184].Morita R, Nakane S, Shimada A, Inoue M, Iino H, Wakamatsu T, Fukui K, Nakagawa N, Masui R, Kuramitsu S, Molecular mechanisms of the whole DNA repair system: a comparison of bacterial and eukaryotic systems, Journal of nucleic acids, 2010 (2010) 179594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Georgakopoulos-Soares I, Koh G, Momen SE, Jiricny J, Hemberg M, Nik-Zainal S, Transcription-coupled repair and mismatch repair contribute towards preserving genome integrity at mononucleotide repeat tracts, Nature communications, 11 (2020) 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Boiteux S, Coste F, Castaing B, Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: Properties and biological roles of the Fpg and OGG1 DNA N-glycosylases, Free Radic Biol Med, 107 (2017) 179–201. [DOI] [PubMed] [Google Scholar]
- [187].Banda DM, Nuñez NN, Burnside MA, Bradshaw KM, David SS, Repair of 8-oxoG:A mismatches by the MUTYH glycosylase: Mechanism, metals and medicine, Free Radic Biol Med, 107 (2017) 202–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Duan S, Han X, Akbari M, Croteau DL, Rasmussen LJ, Bohr VA, Interaction between RECQL4 and OGG1 promotes repair of oxidative base lesion 8-oxoG and is regulated by SIRT1 deacetylase, Nucleic Acids Res, 48 (2020) 6530–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Sakumi K, Tominaga Y, Furuichi M, Xu P, Tsuzuki T, Sekiguchi M, Nakabeppu Y, Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption, Cancer Res, 63 (2003) 902–905. [PubMed] [Google Scholar]
- [190].Arai T, Kelly VP, Minowa O, Noda T, Nishimura S, The study using wild-type and Ogg1 knockout mice exposed to potassium bromate shows no tumor induction despite an extensive accumulation of 8-hydroxyguanine in kidney DNA, Toxicology, 221 (2006) 179–186. [DOI] [PubMed] [Google Scholar]
- [191].Russo MT, De Luca G, Degan P, Parlanti E, Dogliotti E, Barnes DE, Lindahl T, Yang H, Miller JH, Bignami M, Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases, Cancer Res, 64 (2004) 4411–4414. [DOI] [PubMed] [Google Scholar]
- [192].Xie Y, Yang H, Cunanan C, Okamoto K, Shibata D, Pan J, Barnes DE, Lindahl T, McIlhatton M, Fishel R, Miller JH, Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors, Cancer Res, 64 (2004) 3096–3102. [DOI] [PubMed] [Google Scholar]
- [193].Tsuzuki T, Egashira A, Igarashi H, Iwakuma T, Nakatsuru Y, Tominaga Y, Kawate H, Nakao K, Nakamura K, Ide F, Kura S, Nakabeppu Y, Katsuki M, Ishikawa T, Sekiguchi M, Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase, Proc Natl Acad Sci U S A, 98 (2001) 11456–11461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Cohen BJ, Hull KL, The Blood, Memmler’s The Human Body in Health and Disease Wolters Kluwer, Philadelphia, PA, 2015, pp. 289–311. [Google Scholar]
- [195].Kawanishi S, Ohnishi S, Ma N, Hiraku Y, Murata M, Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis, Int J Mol Sci, 18 (2017) 1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Leuti A, Maccarrone M, Chiurchiù V, Proresolving Lipid Mediators: Endogenous Modulators of Oxidative Stress, Oxid Med Cell Longev, 2019 (2019) 8107265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Tallima H, El Ridi R, Arachidonic acid: Physiological roles and potential health benefits -A review, Journal of advanced research, 11 (2018) 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Hanna VS, Hafez EAA, Synopsis of arachidonic acid metabolism: A review, Journal of advanced research, 11 (2018) 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Deeb RS, Hajjar DP, Repair Mechanisms in Oxidant-Driven Chronic Inflammatory Disease, The American journal of pathology, 186 (2016) 1736–1749. [DOI] [PubMed] [Google Scholar]
- [200].Ma L, Jiang M, Zhao X, Sun J, Pan Q, Chu S, Cigarette and IL-17A synergistically induce bronchial epithelial-mesenchymal transition via activating IL-17R/NF-κB signaling, BMC Pulm Med, 20 (2020) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Tommasi S, Zheng A, Besaratinia A, Exposure of mice to secondhand smoke elicits both transient and long-lasting transcriptional changes in cancer-related functional networks, Int J Cancer, 136 (2015) 2253–2263. [DOI] [PubMed] [Google Scholar]
- [202].Tommasi S, Yoon JI, Besaratinia A, Secondhand Smoke Induces Liver Steatosis through Deregulation of Genes Involved in Hepatic Lipid Metabolism, Int J Mol Sci, 21 (2020) 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].van der Vaart H, Postma DS, Timens W, ten Hacken NH, Acute effects of cigarette smoke on inflammation and oxidative stress: a review, Thorax, 59 (2004) 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Gonçalves RB, Coletta RD, Silvério KG, Benevides L, Casati MZ, da Silva JS, Nociti FH Jr., Impact of smoking on inflammation: overview of molecular mechanisms, Inflamm Res, 60 (2011) 409–424. [DOI] [PubMed] [Google Scholar]
- [205].Milnerowicz H, Ściskalska M, Dul M, Pro-inflammatory effects of metals in persons and animals exposed to tobacco smoke, J Trace Elem Med Biol, 29 (2015) 1–10. [DOI] [PubMed] [Google Scholar]
- [206].de Carvalho FO, Felipe FA, de Melo Costa AC, Teixeira LG, Silva É R, Nunes PS, Shanmugam S, de Lucca Junior W, Quintans JS, de Souza Araújo AA, Inflammatory Mediators and Oxidative Stress in Animals Subjected to Smoke Inhalation: A Systematic Review, Lung, 194 (2016) 487–499. [DOI] [PubMed] [Google Scholar]
- [207].Strzelak A, Ratajczak A, Adamiec A, Feleszko W, Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review, Int J Environ Res Public Health, 15 (2018) 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Garth J, Barnes JW, Krick S, Targeting Cytokines as Evolving Treatment Strategies in Chronic Inflammatory Airway Diseases, Int J Mol Sci, 19 (2018) 3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Peebles KA, Lee JM, Mao JT, Hazra S, Reckamp KL, Krysan K, Dohadwala M, Heinrich EL, Walser TC, Cui X, Baratelli FE, Garon E, Sharma S, Dubinett SM, Inflammation and lung carcinogenesis: applying findings in prevention and treatment, Expert Rev Anticancer Ther, 7 (2007) 1405–1421. [DOI] [PubMed] [Google Scholar]
- [210].Janakiram NB, Rao CV, The role of inflammation in colon cancer, Advances in experimental medicine and biology, 816 (2014) 25–52. [DOI] [PubMed] [Google Scholar]
- [211].Prasad S, Gupta SC, Tyagi AK, Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals, Cancer letters, 387 (2017) 95–105. [DOI] [PubMed] [Google Scholar]
- [212].Todoric J, Antonucci L, Karin M, Targeting Inflammation in Cancer Prevention and Therapy, Cancer Prev Res (Phila), 9 (2016) 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Mandal P, Molecular signature of nitric oxide on major cancer hallmarks of colorectal carcinoma, Inflammopharmacology, 26 (2018) 331–336. [DOI] [PubMed] [Google Scholar]
- [214].Srivastava S, Singh D, Patel S, Singh MR, Role of enzymatic free radical scavengers in management of oxidative stress in autoimmune disorders, International journal of biological macromolecules, 101 (2017) 502–517. [DOI] [PubMed] [Google Scholar]
- [215].Mantovani A, Allavena P, Sica A, Balkwill F, Cancer-related inflammation, Nature, 454 (2008) 436–444. [DOI] [PubMed] [Google Scholar]
- [216].Chiba T, Marusawa H, Ushijima T, Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation, Gastroenterology, 143 (2012) 550–563. [DOI] [PubMed] [Google Scholar]
- [217].Malerba M, Montuschi P, Non-invasive biomarkers of lung inflammation in smoking subjects, Curr Med Chem, 19 (2012) 187–196. [DOI] [PubMed] [Google Scholar]
- [218].Headland SE, Norling LV, The resolution of inflammation: Principles and challenges, Seminars in immunology, 27 (2015) 149–160. [DOI] [PubMed] [Google Scholar]
- [219].Liu J, Liang Q, Frost-Pineda K, Muhammad-Kah R, Rimmer L, Roethig H, Mendes P, Sarkar M, Relationship between biomarkers of cigarette smoke exposure and biomarkers of inflammation, oxidative stress, and platelet activation in adult cigarette smokers, Cancer Epidemiol Biomarkers Prev, 20 (2011) 1760–1769. [DOI] [PubMed] [Google Scholar]
- [220].Baylin SB, Jones PA, Epigenetic Determinants of Cancer, Cold Spring Harbor perspectives in biology, 8 (2016) a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Esteller M, Pandolfi PP, The Epitranscriptome of Noncoding RNAs in Cancer, Cancer discovery, 7 (2017) 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Feinberg AP, Koldobskiy MA, Gondor A, Epigenetic modulators, modifiers and mediators in cancer aetiology and progression, Nature reviews. Genetics, 17 (2016) 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Zhang W, Qu J, Liu GH, Belmonte JCI, The ageing epigenome and its rejuvenation, Nature reviews. Molecular cell biology, 21 (2020) 137–150. [DOI] [PubMed] [Google Scholar]
- [224].Blanco E, González-Ramírez M, Alcaine-Colet A, Aranda S, Di Croce L, The Bivalent Genome: Characterization, Structure, and Regulation, Trends in genetics : TIG, 36 (2020) 118–131. [DOI] [PubMed] [Google Scholar]
- [225].Michalak EM, Burr ML, Bannister AJ, Dawson MA, The roles of DNA, RNA and histone methylation in ageing and cancer, Nature reviews. Molecular cell biology, 20 (2019) 573–589. [DOI] [PubMed] [Google Scholar]
- [226].Jones PA, Issa JP, Baylin S, Targeting the cancer epigenome for therapy, Nature reviews. Genetics, 17 (2016) 630–641. [DOI] [PubMed] [Google Scholar]
- [227].Shi DQ, Ali I, Tang J, Yang WC, New Insights into 5hmC DNA Modification: Generation, Distribution and Function, Front Genet, 8 (2017) 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Wu X, Zhang Y, TET-mediated active DNA demethylation: mechanism, function and beyond, Nature reviews. Genetics, 18 (2017) 517–534. [DOI] [PubMed] [Google Scholar]
- [229].Kelly AD, Issa JJ, The promise of epigenetic therapy: reprogramming the cancer epigenome, Current opinion in genetics & development, 42 (2017) 68–77. [DOI] [PubMed] [Google Scholar]
- [230].Klutstein M, Nejman D, Greenfield R, Cedar H, DNA Methylation in Cancer and Aging, Cancer Res, 76 (2016) 3446–3450. [DOI] [PubMed] [Google Scholar]
- [231].Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, Smits KM, Veeck J, Herman JG, Van Neste L, Van Criekinge W, De Meyer T, van Engeland M, Analysis of DNA methylation in cancer: location revisited, Nature reviews. Clinical oncology, 15 (2018) 459–466. [DOI] [PubMed] [Google Scholar]
- [232].Placek K, Schultze JL, Aschenbrenner AC, Epigenetic reprogramming of immune cells in injury, repair, and resolution, The Journal of clinical investigation, 129 (2019) 2994–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Dor Y, Cedar H, Principles of DNA methylation and their implications for biology and medicine, Lancet (London, England), 392 (2018) 777–786. [DOI] [PubMed] [Google Scholar]
- [234].Xu S, Pelisek J, Jin ZG, Atherosclerosis Is an Epigenetic Disease, Trends Endocrinol Metab, 29 (2018) 739–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Greenberg MVC, Bourc’his D, The diverse roles of DNA methylation in mammalian development and disease, Nature reviews. Molecular cell biology, 20 (2019) 590–607. [DOI] [PubMed] [Google Scholar]
- [236].Piskareva O, Lackington W, Lemass D, Hendrick C, Doolan P, Barron N, The human L1 element: a potential biomarker in cancer prognosis, current status and future directions, Current molecular medicine, 11 (2011) 286–303. [DOI] [PubMed] [Google Scholar]
- [237].Xiao-Jie L, Hui-Ying X, Qi X, Jiang X, Shi-Jie M, LINE-1 in cancer: multifaceted functions and potential clinical implications, Genetics in medicine : official journal of the American College of Medical Genetics, 18 (2016) 431–439. [DOI] [PubMed] [Google Scholar]
- [238].Hutt JA, Vuillemenot BR, Barr EB, Grimes MJ, Hahn FF, Hobbs CH, March TH, Gigliotti AP, Seilkop SK, Finch GL, Mauderly JL, Belinsky SA, Life-span inhalation exposure to mainstream cigarette smoke induces lung cancer in B6C3F1 mice through genetic and epigenetic pathways, Carcinogenesis, 26 (2005) 1999–2009. [DOI] [PubMed] [Google Scholar]
- [239].Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, Davis S, Zhang Y, Hussain M, Xi S, Rao M, Meltzer PA, Schrump DS, Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate, Oncogene, 29 (2010) 3650–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Lee KW, Pausova Z, Cigarette smoking and DNA methylation, Front Genet, 4 (2013) 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Besaratinia A, Cockburn M, Tommasi S, Alterations of DNA methylome in human bladder cancer, Epigenetics, 8 (2013) 1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Fasanelli F, Baglietto L, Ponzi E, Guida F, Campanella G, Johansson M, Grankvist K, Johansson M, Assumma MB, Naccarati A, Chadeau-Hyam M, Ala U, Faltus C, Kaaks R, Risch A, De Stavola B, Hodge A, Giles GG, Southey MC, Relton CL, Haycock PC, Lund E, Polidoro S, Sandanger TM, Severi G, Vineis P, Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts, Nature communications, 6 (2015) 10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Ivorra C, Fraga MF, Bayon GF, Fernandez AF, Garcia-Vicent C, Chaves FJ, Redon J, Lurbe E, DNA methylation patterns in newborns exposed to tobacco in utero, Journal of translational medicine, 13 (2015) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Teschendorff AE, Yang Z, Wong A, Pipinikas CP, Jiao Y, Jones A, Anjum S, Hardy R, Salvesen HB, Thirlwell C, Janes SM, Kuh D, Widschwendter M, Correlation of Smoking-Associated DNA Methylation Changes in Buccal Cells With DNA Methylation Changes in Epithelial Cancer, JAMA oncology, 1 (2015) 476–485. [DOI] [PubMed] [Google Scholar]
- [245].Li X, Liu Y, Salz T, Hansen KD, Feinberg A, Whole-genome analysis of the methylome and hydroxymethylome in normal and malignant lung and liver, Genome research, 26 (2016) 1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Zhang Y, Wu K, Shao Y, Sui F, Yang Q, Shi B, Hou P, Ji M, Decreased 5-Hydroxymethylcytosine (5-hmC) predicts poor prognosis in early-stage laryngeal squamous cell carcinoma, American journal of cancer research, 6 (2016) 1089–1098. [PMC free article] [PubMed] [Google Scholar]
- [247].Jenkins TG, James ER, Alonso DF, Hoidal JR, Murphy PJ, Hotaling JM, Cairns BR, Carrell DT, Aston KI, Cigarette smoking significantly alters sperm DNA methylation patterns, Andrology, 5 (2017) 1089–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Coulter JB, O’Driscoll CM, Bressler JP, Hydroquinone increases 5-hydroxymethylcytosine formation through ten eleven translocation 1 (TET1) 5-methylcytosine dioxygenase, J Biol Chem, 288 (2013) 28792–28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Li Z, Li N, Guo C, Li X, Qian Y, Yang Y, Wei Y, The global DNA and RNA methylation and their reversal in lung under different concentration exposure of ambient air particulate matter in mice, Ecotoxicology and environmental safety, 172 (2019) 396–402. [DOI] [PubMed] [Google Scholar]
- [250].Ringh MV, Hagemann-Jensen M, Needhamsen M, Kular L, Breeze CE, Sjoholm LK, Slavec L, Kullberg S, Wahlstrom J, Grunewald J, Brynedal B, Liu Y, Almgren M, Jagodic M, Ockinger J, Ekstrom TJ, Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells, EBioMedicine, 46 (2019) 290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Tellez-Plaza M, Tang WY, Shang Y, Umans JG, Francesconi KA, Goessler W, Ledesma M, Leon M, Laclaustra M, Pollak J, Guallar E, Cole SA, Fallin MD, Navas-Acien A, Association of global DNA methylation and global DNA hydroxymethylation with metals and other exposures in human blood DNA samples, Environ Health Perspect, 122 (2014) 946–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Caliri AW, Caceres A, Tommasi S, Besaratinia A, Hypomethylation of LINE-1 repeat elements and global loss of DNA hydroxymethylation in vapers and smokers, Epigenetics, 15 (2020) 816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Ziech D, Franco R, Pappa A, Panayiotidis MI, Reactive oxygen species (ROS)--induced genetic and epigenetic alterations in human carcinogenesis, Mutat Res, 711 (2011) 167–173. [DOI] [PubMed] [Google Scholar]
- [254].Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP, Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation, Experimental cell research, 286 (2003) 355–365. [DOI] [PubMed] [Google Scholar]
- [255].Lee YW, Broday L, Costa M, Effects of nickel on DNA methyltransferase activity and genomic DNA methylation levels, Mutat Res, 415 (1998) 213–218. [DOI] [PubMed] [Google Scholar]
- [256].Chuang LS, Ng HH, Chia JN, Li BF, Characterisation of independent DNA and multiple Zn-binding domains at the N terminus of human DNA-(cytosine-5) methyltransferase: modulating the property of a DNA-binding domain by contiguous Zn-binding motifs, J Mol Biol, 257 (1996) 935–948. [DOI] [PubMed] [Google Scholar]
- [257].Olmedo P, Goessler W, Tanda S, Grau-Perez M, Jarmul S, Aherrera A, Chen R, Hilpert M, Cohen JE, Navas-Acien A, Rule AM, Metal Concentrations in e-Cigarette Liquid and Aerosol Samples: The Contribution of Metallic Coils, Environ Health Perspect, 126 (2018) 027010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Fleming AM, Burrows CJ, 8-Oxo-7,8-dihydroguanine, friend and foe: Epigenetic-like regulator versus initiator of mutagenesis, DNA Repair (Amst), 56 (2017) 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Weitzman SA, Turk PW, Milkowski DH, Kozlowski K, Free radical adducts induce alterations in DNA cytosine methylation, Proc Natl Acad Sci U S A, 91 (1994) 1261–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Kuchino Y, Mori F, Kasai H, Inoue H, Iwai S, Miura K, Ohtsuka E, Nishimura S, Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues, Nature, 327 (1987) 77–79. [DOI] [PubMed] [Google Scholar]
- [261].Furlan D, Trapani D, Berrino E, Debernardi C, Panero M, Libera L, Sahnane N, Riva C, Tibiletti MG, Sessa F, Sapino A, Venesio T, Oxidative DNA damage induces hypomethylation in a compromised base excision repair colorectal tumourigenesis, British journal of cancer, 116 (2017) 793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Miller JW, Nadeau MR, Smith J, Smith D, Selhub J, Folate-deficiency-induced homocysteinaemia in rats: disruption of S-adenosylmethionine’s co-ordinate regulation of homocysteine metabolism, The Biochemical journal, 298 (Pt 2) (1994) 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Yang Q, Wu K, Ji M, Jin W, He N, Shi B, Hou P, Decreased 5-hydroxymethylcytosine (5-hmC) is an independent poor prognostic factor in gastric cancer patients, Journal of biomedical nanotechnology, 9 (2013) 1607–1616. [DOI] [PubMed] [Google Scholar]
- [264].Berdasco M, Esteller M, Clinical epigenetics: seizing opportunities for translation, Nature reviews. Genetics, 20 (2019) 109–127. [DOI] [PubMed] [Google Scholar]
- [265].Martire S, Banaszynski LA, The roles of histone variants in fine-tuning chromatin organization and function, Nature reviews. Molecular cell biology, 21 (2020) 522–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Zhong J, Agha G, Baccarelli AA, The Role of DNA Methylation in Cardiovascular Risk and Disease: Methodological Aspects, Study Design, and Data Analysis for Epidemiological Studies, Circulation research, 118 (2016) 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Yang IV, Lozupone CA, Schwartz DA, The environment, epigenome, and asthma, The Journal of allergy and clinical immunology, 140 (2017) 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Hama N, Totoki Y, Miura F, Tatsuno K, Saito-Adachi M, Nakamura H, Arai Y, Hosoda F, Urushidate T, Ohashi S, Mukai W, Hiraoka N, Aburatani H, Ito T, Shibata T, Epigenetic landscape influences the liver cancer genome architecture, Nature communications, 9 (2018) 1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Zhang W, Song M, Qu J, Liu GH, Epigenetic Modifications in Cardiovascular Aging and Diseases, Circulation research, 123 (2018) 773–786. [DOI] [PubMed] [Google Scholar]
- [270].Zeng Z, Mukherjee A, Zhang H, From Genetics to Epigenetics, Roles of Epigenetics in Inflammatory Bowel Disease, Front Genet, 10 (2019) 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Zarzour A, Kim HW, Weintraub NL, Epigenetic Regulation of Vascular Diseases, Arteriosclerosis, thrombosis, and vascular biology, 39 (2019) 984–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Stratton MS, Farina FM, Elia L, Epigenetics and vascular diseases, Journal of molecular and cellular cardiology, 133 (2019) 148–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Nolt SM, The Amish: A Concise Introduction, Johns Hopkins University Press (JHUP), Baltimore, MD, 2016. [Google Scholar]
- [274].Westman JA, Ferketich AK, Kauffman RM, MacEachern SN, Wilkins JR 3rd, Wilcox PP, Pilarski RT, Nagy R, Lemeshow S, de la Chapelle A, Bloomfield CD, Low cancer incidence rates in Ohio Amish, Cancer Causes Control, 21 (2010) 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Kessler MD, Loesch DP, Perry JA, Heard-Costa NL, Taliun D, Cade BE, Wang H, Daya M, Ziniti J, Datta S, Celedón JC, Soto-Quiros ME, Avila L, Weiss ST, Barnes K, Redline SS, Vasan RS, Johnson AD, Mathias RA, Hernandez R, Wilson JG, Nickerson DA, Abecasis G, Browning SR, Zöllner S, O’Connell JR, Mitchell BD, O’Connor TD, De novo mutations across 1,465 diverse genomes reveal mutational insights and reductions in the Amish founder population, Proc Natl Acad Sci U S A, 117 (2020) 2560–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Mitchell BD, Lee WJ, Tolea MI, Shields K, Ashktorab Z, Magder LS, Ryan KA, Pollin TI, McArdle PF, Shuldiner AR, Schäffer AA, Living the good life? Mortality and hospital utilization patterns in the Old Order Amish, PLoS One, 7 (2012) e51560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Menzie CA, Potocki BB, Santodonato J, Exposure to carcinogenic PAHs in the environment, Environ. Sci. Technol, 26 (1992) 1278–1284. [Google Scholar]
- [278].Nielsen PS, Okkels H, Sigsgaard T, Kyrtopoulos S, Autrup H, Exposure to urban and rural air pollution: DNA and protein adducts and effect of glutathione-S-transferase genotype on adduct levels, Int Arch Occup Environ Health, 68 (1996) 170–176. [DOI] [PubMed] [Google Scholar]
- [279].Tsihrintzis VA, Hamid R, Modeling and Management of Urban Stormwater Runoff Quality: A Review, Water Resources Management, 11 (1997) 136–164. [Google Scholar]
- [280].Gao Z, Moorjani P, Sasani TA, Pedersen BS, Quinlan AR, Jorde LB, Amster G, Przeworski M, Overlooked roles of DNA damage and maternal age in generating human germline mutations, Proc Natl Acad Sci U S A, 116 (2019) 9491–9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Hamman RF, Barancik JI, Lilienfeld AM, Patterns of mortality in the the Old Order Amish. I. Background and major causes of death, Am J Epidemiol, 114 (1981) 845–861. [DOI] [PubMed] [Google Scholar]
- [282].Besaratinia A, Tommasi S, Vaping: A growing global health concern, EClinicalMedicine, 17 (2019) 100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.