


Abstract
CRISPR Cas12a is an RNA-programmable endonuclease particularly suitable for gene regulation. This is due to its preference for T-rich PAMs that allows it to more easily target AT-rich promoter sequences, and built-in RNase activity which can process a single CRISPR RNA array encoding multiple spacers into individual guide RNAs (gRNAs), thereby simplifying multiplexed gene regulation. Here, we develop a flexible dCas12a-based CRISPRi system for Saccharomyces cerevisiae and systematically evaluate its design features. This includes the role of the NLS position, use of repression domains, and the position of the gRNA target. Our optimal system is comprised of dCas12a E925A with a single C-terminal NLS and a Mxi1 or a MIG1 repression domain, which enables up to 97% downregulation of a reporter gene. We also extend this system to allow for inducible regulation via an RNAP II-controlled promoter, demonstrate position-dependent effects in crRNA arrays, and use multiplexed regulation to stringently control a heterologous β-carotene pathway. Together these findings offer valuable insights into the design constraints of dCas12a-based CRISPRi and enable new avenues for flexible and efficient gene regulation in S. cerevisiae.
INTRODUCTION
Regulation of gene expression underpins numerous cellular processes, from the control of cell cycle progression to the dynamic adaptation of the proteome in response to environmental challenges (1). The ability to precisely and dynamically manipulate gene expression is also a crucial part of metabolic engineering because the proper control of a cell factory's function requires the regulation of selected genes, often in a timely manner, and the balancing of other native metabolic pathways to accommodate heterologous production of a compound of interest (2,3). This demand has resulted in the development of many molecular parts able to regulate gene expression (4–6).
Recently, the bacterial and archaeal adaptive immune system CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) has been adapted for activation (CRISPRa) and interference (CRISPRi) of gene expression (7–9). The easily programmable nature of the CRISPRa/i system, where a gene is targeted through the simple modification of a guide RNA (gRNA) to include a short ∼20 nucleotide (nt) complementary sequence, has resulted in CRISPRa/i becoming the predominant method for genome-wide studies of gene regulation (10–12). To date, the majority of CRISPRa/i systems have made use of dCas9 (7,8,10–13), which is sufficient to modulate transcription in prokaryotic cells without further modification. However, in eukaryotic cells performance can be enhanced by fusing the effector protein with a repression domain such as the Krüppel associated box (KRAB) domain of Kox1 involved in the formation of heterochromatin or the mammalian Mxi1 domain which interacts with the yeast histone deacetylase Sin3 homolog (8,14,15). It has been shown that fusing dCas9 with Mxi1 can greatly enhance repression, enabling a 53-fold decrease in constitutive GFP expression compared to just an 18-fold decrease when dCas9 alone was used to target the same fluorescent reporter (8). Analogously, CRISPR activation enhances gene expression through the fusion of transcriptional activator such as VPR or VP64 and the targeting of regions in close proximity to the promoter of a gene (13).
Beyond Cas9, CRISPRa/i systems have also been developed using class 2 type V Cas12a nucleases, formerly also known as Cpf1 (16–19). Cas12a nucleases are characterized by a different preference for the protospacer adjacent motif (PAM) sequence. Specifically, while the widely used SpCas9 recognizes NGG PAM sequences at the 3′ end of a genomic target, LbCas12a favours TTTV PAM sequences at the 5′ end of a genomic target (where V is either A, C or G). Furthermore, unlike Cas9, Cas12a does not require trans-activating CRISPR RNA (tracRNA) and DNA targeting can be achieved with CRISPR RNA (crRNA) as short as 42 bp (20). Ribonuclease activity of Cas12a also enables processing of multiple gRNAs encoded within a single crRNA array, enabling simultaneous modification of multiple genetic loci (21). Recognition of T-rich PAMs by Cas12a is especially useful for targeting promoter regions, which are generally AT-rich (22). A previous evaluation of dCas12a mediated downregulation of genes in Escherichia coli showed no strand bias within the promoter region, thus PAM sequences on either DNA strand can be used, further increasing the number of potential targets (18). In native yeast promoters, an enrichment of Ts within the core region has also been linked to the frequency of translation initiation events (23).
Saccharomyces cerevisiae is one of the most versatile and extensively used microorganism in industry and central to the production of pharmaceuticals, enzymes and food additives (24). Although CRISPR-Cas12a has been applied for genome editing of S. cerevisiae, its application for gene regulation has been limited to a selection of organisms, excluding baker's yeast (18,25–30). Furthermore, there has been a lack of systematic studies that characterise the many design choices when implementing dCas12a based CRISPRi systems in this host.
Here, we have addressed this issue by providing the first comprehensive assessment of dCas12a-based CRISPRi systems in S. cerevisiae. We vary and assess all key aspects of this system, including the position of nuclear localisation signals (NLS) fused to the dCas12a protein, the potential for enhanced repression through fusion of repression domains, the effectiveness of targeting different positions in promoters and ORFs, development of controllable gRNA expression regulation through the use of an RNA polymerase II (RNAP II) promoter and implementation of a dCas12a system for simultaneous downregulation of multiple genes. Finally, we demonstrate the potential application of this refined system for the stringent control of a heterologous β-carotene production pathway. This work provides valuable insight into the design constraints for effective CRISPRi when using dCas12a in S. cerevisiae and opens new avenues for stringent multiplexed control of gene expression and metabolic processes in this industrially important host.
MATERIALS AND METHODS
Strains and cultivation conditions
Strains constructed in this study were generated using S. cerevisiae CEN.PK113-7D as a parent strain, except for strains FR013 and FR014 which were constructed in the CEN.PK113-9D background giving a possibility for using auxotrophic markers (31). Cultures were grown in complex medium (YEPD) comprised of 2% Difco™ phytone peptone (Becton–Dickinson, Franklin Lakes, NJ, USA), 1% Bacto™ yeast extract (BD) and either d-glucose (2%, Sigma Aldrich, St Louis, MO, USA) or galactose (2%, Sigma Aldrich) when induction of the GAL10 promoter was required. Selection was achieved using nourseothricin (NTC, 200 μg/ml, Jena Bioscience, Germany) or geneticin (G418, 200 μg/ml, Sigma Aldrich), when appropriate. Solid medium was prepared by addition of Difco™ granulated agar (BD) to the medium to a final concentration of 2% (w/v). Propagation of plasmids was performed using Escherichia coli NEB 10-beta cells (New England BioLabs, Ipswich, MA, USA). Bacterial cultures were prepared in 2*PY medium comprised of tryptone peptone (1.6%, BD), Bacto™ yeast extract (1.0%, BD) and NaCl (0.5%, Sigma Aldrich) and containing ampicillin (100 μg/ml, Sigma Aldrich) or neomycin (50 μg/ml, Sigma Aldrich).
Strain construction
Yeast strains were constructed by genome editing of a single locus (INT4, Supplementary Table S1) using CRISPR–Cas9 or multiple loci (INT1,2,3, Supplementary Table S1) using CRISPR-LbCas12a in combination with a single crRNA array encoding three targets, as described previously (26,32). Briefly, strain CSN001 pre-expressing Cas9 from plasmid pCSN061 or strain CSN004 pre-expressing LbCas12a from plasmid pCSN067 (Supplementary Tables S2 and S3) were transformed with 1000 ng of the gRNA expression cassette and 100 ng of each DNA part and 100 ng of the gRNA recipient plasmid pRN1120 using the LiAc/ssDNA/PEG method (33). Reagents required for yeast transformation were obtained from Sigma Aldrich (lithium acetate dihydrate (LiAc) and deoxyribonucleic acid sodium salt from salmon testes (ssDNA)) and Merck (polyethylene glycol 4000 (PEG), Darmstadt, Germany). The recipient plasmid used in combination with the sgRNA of Cas9 was amplified from pRN1120 using primers pKC003-004 (Supplementary Table S4) whereas to generate the backbone for the Cas12a single crRNA array primers pKC007–008 were used. sgRNA for Cas9, crRNA and a single crRNA array for Cas12a (Supplementary Tables S5–S7) were obtained as synthetic DNA and amplified with different sets of primers (pKC001–002 and pKC005–006 for Cas9 and LbCas12a, respectively). Prior to the studies of gene expression regulation, plasmids used for the genome editing (pCSN060, pCSN067 and pRN1120) were removed by a sequential re-streaking and culturing of a strain and verification of the absence of resistance to markers used for selection of these plasmids.
A set of reporter strains with fluorescent protein genes integrated into genomic DNA (FR003, FR007–009, FR013; Supplementary Table S2) was created to study transcriptional silencing with plasmid borne dCas12a. Fluorescent protein genes were expressed from heterologous promoters to enable targeting of dCas12a without affecting the expression of any native gene. To create these reporter strains, expression cassettes for BFP, eGFP and mCherry were assembled via Golden Gate cloning. DNA fragments coding for the genes of interest (open-reading frames: ORFs) of the fluorescent protein genes were obtained as synthetic DNA (BaseClear, Leiden, the Netherlands) and BsaI recognition sites were added by a PCR reaction using primers pKC009–014. Each expression cassette was comprised of a heterologous promoter, an ORF and a terminator, flanked by 50 bp connector sequence on the 5′ and 3′ end to facilitate assembly via in vivo recombination in S. cerevisiae. Additionally, two flanking regions of ∼500 bp encoding the left and right region of an integration site were amplified from the genomic DNA of the host strain (primers pKC015–029,037). The same connector sequences as found in the expression cassettes were added to these flanking regions. Upon cleavage of double stranded DNA by a Cas protein and subsequent integration of the expression cassette, a sequence of ∼1000 bp between the flanking regions was removed. Strain FR003 contained expression cassettes of mCherry and eGFP integrated into the INT4 locus. In strains FR007–009 mCherry controlled by the FBA1 promoter from S. cerevisiae was integrated into INT3 whereas for eGFP three expression cassettes with different promoters were constructed and introduced into INT2. To prevent a lethal double strand break at INT1 when the single crRNA array was used in combination with Cas12a, a donor DNA encoding for a connector sequence and flanks of INT1 was supplied as the used array encodes three targets (INT1, INT2 and INT3). Strain FR013 was constructed to study multiplex silencing by integrating BFP, mCherry and eGFP controlled by three heterologous promoters into three independent loci (INT1,2,3). This strain was further engineered using Cas9 to integrate the dCas12a E925A NLS Mxi1 expression cassette in INT4 resulting in strain FR014. Genomic DNA was isolated from constructed strains using Zymolyase (Zymo Research, Irvine, CA, USA) (32) and the integrated construct was amplified to generate a sequencing template (primers pKC030-037). Integrated constructs were verified by Sanger sequencing using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (ThermoFisher Scientific, USA) and NucleoSEQ columns for dye terminator removal (Macherey-Nagel, Düren, Germany).
Strain DC001 expressing functional Cas12a and the set of strains DC002–013 (Supplementary Table S2) were created to compare the mutations D823A and E925A which confer nuclease deficiency to Cas12a and the functionality of five fused repression domains: Mxi1, KRAB, MIG1, TUP1 and UME6. To enhance stable expression, dCas12a was integrated into the genome at a single locus (INT4) together with mCherry and eGFP (32). Constructs encoding dCas12a with a C-terminal NLS sequence and repression domains were obtained as synthetic DNA (Invitrogen, Carlsbad, CA, USA). A cloning free strategy was used to generate expression cassettes of integrated genes (34). Promoters and terminators to be assembled in each transcriptional unit were first amplified to attach a 50 bp homology region to an integration site, connector, or a gene (primers pKC038–058). These homology regions facilitated in vivo recombination of nine parts to form expression cassettes of dCas12a, mCherry and eGFP separated by a 50 bp connector sequence. Constructs integrated in the edited locus of the created strains were sequence-verified using nanopore sequencing on a MinION device (Oxford Nanopore Technologies, UK). Genomic DNA was isolated as stated before and the sequencing template was amplified using a set of primers with unique 25 bp barcodes (pKC059–095). A combination of 5 reverse primers and 32 forward primers with unique barcodes allowed for 160 templates to be tagged which were subsequently pooled in equivalent amounts. Samples were prepped with a ligation kit (SQK-LSK 109, Oxford Nanopore Technologies, UK) and sequenced in a single run using a FLOW-MIN106 (9.4 SpotON) flow cell and the MinION MIN-101B sequencer.
The β-carotene producing strain CAR-034 was constructed by integrating the yeast codon optimized genes crtE, crtYB and crtI from Xanthophyllomyces dendrorhous (35) into three independent genomic loci using CRISPR Cas12a, as reported in (32). To test downregulation of β-carotene biosynthesis we integrated dCas12a E925A NLS Mxi1 into the INT4 locus of strain CAR-034, resulting in strains CAR-041 (dCas12a expressed from the TEF1 promoter) and CAR-042 (dCas12a expressed from the PGI1 promoter). Genome editing was conducted with CRISPR Cas9, as described in the previous section. Strain CAR-041 was used in single- and multiplex CRISPRi experiments with gRNA targeting either each of the crt ORFs or a single crRNA array encoding three spacers targeting heterologous promoters in front of all the crt genes. Expression of the crt genes was controlled by Sbay_TDH3 p, Smik_TEF1p and Kl_ENO1p promoters (as used in strain FR013 to control expression of fluorescent protein genes) (Supplementary Table S2). Strain CAR-042 was used in a spotting assay to assess the toxicity of dCas12a. Cultures grown overnight were diluted to OD600 of 1 and subsequently plated on YEPD agar plates in a series of 10-fold dilutions (i.e. 100, 10–1, 10–2, 10–3 fold dilutions).
Plasmid assembly
Plasmids pC-NLS and pC-NLS-Mxi1 (Figure 1A, Supplementary Table S3) used to express dCas12a were assembled by in vivo recombination in S. cerevisiae (32). The backbone was amplified from plasmid pCSN061 with primers pKC096 and pKC097 such that the Cas9 expression cassette was removed. Constructs of dCas12a E925A NLS without or with a Mxi1 domain (pC-NLS and pC-NLS-Mxi1, respectively) were obtained by PCR using genomic DNA from strains DC003 and DC010 as template with primers containing homology to the pCSN backbone (pKC098,099). To construct plasmids pC-Mxi1, pC-Mxi1-NLS, pC-NLS-Mxi1-NLS (Figure 1A, Supplementary Table S3) synthetic DNA encoding NLS and the repression domain Mxi1 was ordered from IDT (Leuven, Belgium) and assembled in the pCSN backbone with dCas12a E925A via Gibson assembly (36). The pCSN backbone with dCas12a E925A was amplified from plasmid pC-NLS with primers pKC100 and pKC101. To construct a functional dCas12a-eGFP fusion protein, a 60 bp linker was used (37). dCas12a-eGFP was assembled in a Gibson reaction using a PCR fragment encoding dCas12a in a pCSN backbone amplified from plasmid pC-NLS with primers KC043,101 and fragment encoding eGFP amplified with primers pKC103–104.
Figure 1.

Effect of NLS number and position in dCas12a E925A Mxi1 fusions. (A) Genetic diagram of plasmids used for expression of dCas12a E925A Mxi1 with NLS fusions (pC-Mxi1, pC-Mxi1-NLS, pC-NLS-Mxi1-NLS, pC-NLS, pC-NLS-Mxi1). (B) Repression pattern in strain FR003 with eGFP integrated into INT4 and (C) strain FR009 with eGFP integrated into INT2. CRISPRi was assessed using two gRNAs targeting the Kl_ENO1 promoter controlling eGFP expression. Bars represent fold repression between targeting and non-targeting gRNA ±1 standard error (n = 4). Dashed line indicates 1-fold change (i.e. no repression).
Molecular biology techniques
DNA parts subjected to cloning and sequencing were amplified using Q5 polymerase (NEB) and primers were obtained from IDT (Leuven, Belgium). PCR products were purified with the Wizard SV gel and PCR clean up kit (Promega, Madison, WI, USA). Golden Gate cloning was conducted using BsaI-HF v2 (NEB) and T4 DNA Ligase (Invitrogen). Plasmid isolation from bacterial cultures was performed using QIAprep Miniprep (QIAGEN, Venlo, the Netherlands) whereas for yeast cultures Zymoprep Yeast Plasmid Miniprep II (Zymo Research) was used.
gRNA design
Genomic targets were designed using the Benchling website. Specificity of the designed crRNAs was tested against the CEN.PK113-7D genome using Burrows–Wheeler Aligner (BWA) and visualized with SAMtools (38,39). Specific features of gRNA such as the length of the spacer, direct repeat and PAM sequence varied between nucleases and purpose (Supplementary Table S5). crRNAs for gene downregulation with dCas12a were designed to target either the template or non-template DNA strand for the promoter region, but only target the template strand of a gene sequence (Supplementary Table S6). The distance of a spacer from the transcription start site (TSS) was calculated according to Smith et al. (12) and using the YeasTSS database to elucidate the TSS of heterologous promoters used in this study (40). Spacers selected for single crRNA arrays are listed in Supplementary Tables S7 and S8. gRNAs were expressed using the SNR52 promoter (SNR52p) and SUP4 terminator (SUP4t) from a multicopy plasmid pRN1120 (NatMX marker) (26,41) with the exception of the experiment where gRNA expression from a promoter processed by RNAP II was tested (Supplementary Table S9). In this case, gRNA was expressed using the galactose-inducible GAL10 promoter and the GND2 terminator. A linear recipient plasmid fragment was obtained in a PCR reaction using primers pKC003,004 for Cas9 sgRNA and primers pKC007,008 for (d)Cas12a crRNA or single crRNA array. gRNAs were obtained from Twist Bioscience or IDT as expression cassettes flanked with 50 bp homology regions to the recipient plasmid pRN1120.
Micro-fermenter settings
Growth experiments were conducted in a BioLector (m2p-labs, Baesweiler, Germany). Cultures were prepared in YEPD medium with the appropriate antibiotic selection from individual colonies and grown overnight at 30°C, 250 rpm. The following day cultures were diluted in the medium to an OD600 of 0.05 and 1 ml was transferred to MTP-R48-B FlowerPlate (m2p-labs). For each strain three biological replicates were assayed. Cultures were grown in the BioLector under controlled conditions (30°C, 800 rpm, 80% rh, O2 20.95%) with biomass (em. 620 nm/ex. 620 nm and gain 20) and fluorescence measured at 15 min intervals for a total duration of 72 h. eGFP was detected with a filter gain of 100 and wavelengths em. 488 nm/ex. 520 nm while RFP at a gain of 50 and wavelengths em. 589 nm/ex. 610 nm.
Flow cytometry
For analysis by flow cytometry, cultures were prepared from four colonies picked from transformation plates and inoculated in YEPD media supplemented with NTC (and G418 for dCas12a expressed from a plasmid) followed by incubation at 30°C, 550 rpm and 80% rh for two days to reach full saturation. Subsequently, cultures were diluted 20x in physiological salt and analysed with a BD FACSAria Fusion (Becton–Dickinson). Detection of events was set such that 20,000 events were measured for single cells and double cells were excluded from the analysis. The signal of fluorescent proteins was detected with a bandpass filter set at 530/30 nm for eGFP, 450/50 nm for BFP and 610/20 nm for mCherry. The data was recorded using BD FACSDiva 8.0.2 software to retrieve the geometric mean of the fluorescence distribution which was averaged for quadruplicates. Fluorescence obtained for eGFP, mCherry and BFP in arbitrary units was converted to molecules of equivalent fluorophores using Rainbow calibration beads with 8-peaks (BioLegend, London, UK) and the FlowCal Python package (42). Specifically, the fluorescence of eGFP was expressed in Molecules of Equivalent FLuorescein (MEFL), mCherry in Molecules of Equivalent Phycoerythrin-TR (MEPTR) and BFP in Molecules of Equivalent BFP (MEBFP) using values of calibration beads detected with channels ECD, FITC and BFP for mCherry, eGFP and BFP, respectively.
Fluorescence microscopy
Nuclear DNA was stained with Vybrant® DyeCycle™ Violet (Invitrogen) and yeast cells were imagined with an Olympus BX53 microscope using CellSens software (Tokyo, Japan).
Carotenoid quantification
Isolation of carotenoids from cell pellets and subsequent quantification of phytoene, lycopene and β-carotene was conducted as described before (43). Briefly, cells in 0.15–0.45 ml of carotenogenic culture were pelletized and resuspended in 1 ml of tetrahydrofuran (Merck). Carotenoids were extracted using Precellys® homogeniser (Bertin, Montigny-le-Bretonneux, France) in two 20 s cycles at 6800 rpm with a 30 second pause in between. The resulting homogenised mixture was centrifuged at a speed of 13 000 rcf for 8 min at 4°C and the supernatant was collected and analysed using a UHPLC Ultimate 3000 (Thermo Fisher Scientific, Waltham, MA, USA), using a Waters XBridge® C18 column (3.5 μm, 2.1 mm × 50 mm, Milford, MA, USA) for the separation of products. Three mobile phases of ethyl acetate, water and acetonitrile were used for separation of carotenoids (Supplementary Table S10). Samples were stored at 12°C upon injection into the system (10 μl). Carotenoids were detected with Ultimate 3000 photodiode array detector (Thermo Fisher Scientific) at wavelengths 286, 475 and 450 nm to measure phytoene, lycopene and β-carotene, respectively. Data was recorded and analysed with Chromeleon software. Amounts of carotenoids were calculated based on standards: E/Z-Phytoene (Sigma Aldrich, #78903), lycopene (Sigma Aldrich, #75051) and β-carotene (U.S. Pharmacopeial, North Bethesda, MD, USA, #1065480).
Data analysis
Gene downregulation was expressed as fold repression by calculating the ratio between the fluorescence of a reporter gene upon expression of targeting gRNA (or a single crRNA array) and a non-targeting control gRNA (or single crRNA array). The standard error (SE) for these ratios was calculated as
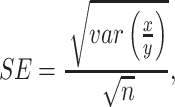 |
where x indicates fluorescence of a reporter gene when targeted with gRNA, y corresponds to fluorescence of a reporter upon treatment with non-targeting gRNA and n is the number of tested samples. To check the statistical validity of a difference between obtained results, an unpaired t-test was used. Plots were generated using custom Python scripts and the Matplotlib package and figures were prepared with BioRender.
RESULTS
Influence of NLS position on dCas12a-based CRISPRi efficiency
Fusion of a nuclear localization signal (NLS) to a Cas protein provides instructions for nuclear import and ultimately ensures the Cas protein is located within the same compartment as its target genomic DNA. In reported experiments using eukaryotes, Cas9 and Cas12a are most commonly fused to a single NLS at the C-terminus, which is often followed by a repression domain such as Mxi1 or KRAB to improve performance (8,17,41,44). The NLS composition (i.e. monopartite vs bipartite), their number, and position at either N- or C-terminus was reported to affect the genome editing efficiency of AsCas12a in mammalian cells as well as the linker used to fuse Cas9 to an NLS (30,45).
For our dCas12a system we selected the SV40 NLS and began by assessing the effect of its position on the performance of CRISPRi. We constructed five plasmids (pC-Mxi1, pC-Mxi1-NLS, pC-NLS-Mxi1-NLS, pC-NLS, pC-NLS-Mxi1) encoding dCas12a E925A and a Mxi1 repression domain with a single NLS or multiple NLSs at various positions (Figure 1A). These plasmids were evaluated using two strains (FR003, FR009) which contain an eGFP expression cassette integrated at a different locus in the genome (INT4 and INT2, respectively for FR003 and FR009). Two gRNAs (gENO2 and gENO6) targeting two different positions of the promoter in front of eGFP were selected to mediate downregulation. Fluorescence of the eGFP reporter protein was measured by flow cytometry and fluorescence was converted to calibrated units through the use of an external standard (Materials and Methods). Calibration of the fluorescence measurements improved the robustness of our results (ensuring technical errors were avoided) and has been demonstrated to enable better data reproducibility (46).
From these experiments (Figure 1B, C, Supplementary Figure S1), the strongest repression of eGFP integrated at locus INT4 was seen when dCas12a was fused to a single C-terminal NLS and Mxi1 repression domain (pC-NLS-Mxi1) and dCas12a-Mxi1 fusion missing an NLS (pC-Mxi1) (4.5 ± 0.3 and 4.1 ± 0.1 fold repression, respectively). In addition to constructs pC-NLS-Mxi1 and pC-Mxi1, which strongly downregulated eGFP at locus INT2 (7.1 ± 0.6 and 7.3 ± 1.6 fold repression, respectively), we observed an improved performance of pC-Mxi1-NLS (gENO2) harbouring a single NLS at the 3′ end of the Mxi1 domain (7.8 ± 0.5 fold repression). In contrast, a reduction in repression was seen for designs containing two C-terminal NLSs (pC-NLS-Mxi1-NLS), regardless of the target locus. Addition of the Mxi1 repression domain to dCas12a with a C-terminal NLS (pC-NLS, pC-NLS-Mxi1) in combination with gENO2 enhanced eGFP downregulation in both strains, matching findings in other organisms (8,17). Overall, fold repression of eGFP at locus INT2 was higher than for INT4, which is likely caused by the accessibility of the target locus. Based on these findings, we proceeded to use dCas12a-NLS-Mxi1 fusion for further experiments.
The ability for Cas9 lacking an NLS to edit yeast cells was reported previously (47), although editing efficiency was lower than when Cas9 was equipped with an NLS. To better understand our observation of gene downregulation using dCas12a without an NLS (pC-Mxi1), we constructed a fusion of dCas12a and eGFP to elucidate its cellular localization (Supplementary Figure S2). Fluorescence microscopy revealed that the dCas12a-eGFP fusion construct was located in both the cytoplasm and nucleus explaining the downregulation in gene expression observed. Functionality of the dCas12a-eGFP fusion construct was demonstrated in combination with the use of an effective gRNA to downregulate carotenoid production (Supplementary Figure S2C).
Evaluation of repression domains
CRISPR Type V effector proteins such as Cas12a contain a single nuclease RuvC domain to sequentially cleave both strands of targeted DNA (22), thus endonuclease deficiency can be conferred by a single mutation. Evaluation of the dCas12a CRISPRi system in E. coli revealed that a single mutation in the nuclease domain of Cas12a, either D832A or E925A, outperformed the combination of these mutations (18). Thus, we sought to compare dCas12a harbouring these single mutations by creating dCas12a mutants D832A and E925A. Gene downregulation can be further enhanced by equipping dCas12a with a repression domain, however, it has been shown that certain repressors can also abolish the activity of a dCas protein (27). To our knowledge, in previous studies repression domains used in combination with dCas12a were limited to KRAB and Mxi1 (3,27,48). We therefore extended this set by including the additional well-characterized MIG1, TUP1 and UME6 domains that have been shown to be effective in combination with dCas9 (17).
While dCas12a gene expression from a plasmid may provide variation in terms of copy number, integration of a single copy of the expression construct into the genome ensures stable expression during cell propagation. Expression of dCas12a solely or dCas12a-Mxi1 fusion from the same high strength promoter integrated into the genome resulted in at least 3-fold repression of eGFP in comparison with plasmid borne expression of the same construct, which is possibly a consequence of a selection pressure to maintain only the gRNA plasmid (Table 1). For this reason, we constructed 12 strains with genome-integrated dCas12, with and without fusion to the five different repression domains (strains DC002-013). This set of strains was constructed using CRISPR–Cas9 mediated genome edition in combination with assembly of the P-O-T cassettes of dCas12a, mCherry and eGFP via in vivo recombination guided by 50 bp homology flanks annealed to the individual parts. Subsequently, integration of dCas12a and the fluorescent reporter was verified by nanopore sequencing (Material and Methods). The combination of these two approaches for strain construction reduced the total construction time by omitting cloning steps and sequence verification of individual samples. A set of unique barcodes was used to generate a sequencing template allowing the resulting PCR amplicons to be pooled and analyzed in a single sequencing run (similar to the approach in (49)).
Table 1.
Comparison of dCas12a efficiency when expressed from plasmid (pC-NLS, pC-NLS-Mxi1) or integrated into the genome
| Fold repression | eGFP fluorescence (103 MEFL) | ||||
|---|---|---|---|---|---|
| gENO2 | gENO6 | gENO2 | gENO6 | gNone | |
| Integrated dCas12a-NLS | 8 ± 0.7 | 14.9 ± 1.1 | 21.6 ± 3.1 | 11.5 ± 1.8 | 171.7 ± 17.3 |
| Plasmid-based dCas12a-NLS | 2.2 ± 0.1 | 3.5 ± 0.2 | 37.7 ± 2.4 | 23.5 ± 3.3 | 81.8 ± 2.6 |
| Integrated dCas12a-NLS-Mxi1 | 12.6 ± 0.7 | 8.2 ± 0.5 | 13.2 ± 0.8 | 20.3 ± 1.3 | 166.5 ± 18.7 |
| Plasmid-based dCas12a-NLS-Mxi1 | 4.5 ± 0.3 | 2.3 ± 0.2 | 14.1 ± 1.1 | 27.3 ± 4.5 | 63.6 ± 8.1 |
Fold repression of eGFP expressed as an average of values normalized by a non-targeting gRNA (gNone) for four biological replicates ± 1 standard error.
dCas12a fusions with repression domains displayed varied efficiency in downregulating eGFP expression (Figure 2, Supplementary Figure S3). As observed for the previously tested plasmid pC_NLS, a reduction in eGFP fluorescence was noted for dCas12 without an additional repression domain. A similar repression level was observed for dCas12a D832A and dCas12a E925A targeting the promoter controlling eGFP expression (gENO2 and gENO4). The repression strength of dCas12a was further improved by a fusion to the MIG1 domain leading to almost a full blockage of eGFP repression (for dCas12a D832A 98% ± 4% and for dCas12a E925A 96% ± 1% decrease in eGFP fluorescence), in combination with the most efficient gRNA (gENO4). Fusion of the Mxi1 domain had a beneficial impact on the performance of dCas12a, particularly for gRNAs targeting eGFP ORF, which were ineffective for dCas12a alone. The strongest gRNA gENO4 and dCas12a D832A Mxi1 repressed eGFP fluorescence by 93 ± 27%, whereas for dCas12a D832A Mxi1 by 97 ± 5%. The combination of KRAB and Cas12a E925A abolished any repression and significantly reduced the efficiency of Cas12a D832A, which is in line with previous findings (27). Similarly, a general transcriptional repressor TUP1 diminished dCas12a mediated downregulation, although not to the extent observed for Cas12a E925A KRAB (2.6- and 1.1-fold repression for TUP1 and KRAB in combination with gRNA gENO4, respectively). No clear benefit was noted for fusion of the UME6 domain as an improved repression level was observed only for one gRNA (gENO4) and for the remaining gRNAs resembled repression levels of dCas12a lacking a repression domain. The presented results show boundaries of transferability of the design rules for S. cerevisiae elucidated for dCas9 to dCas12a. Although fusion of a KRAB domain to dCas9 improved downregulation, such effect was not observed for dCas12a used in Yarrowia lipolytica (27). In the study conducted by Lian et al. (17) the Mxi1 domain was outperformed by TUP1 in a fusion with dCas9, however, we observe an opposite behaviour for dCas12a. Our evaluation of repression domains using five gRNAs provides evidence for the advantage of using Mxi1 and MIG1 for dCas12a mediated CRISPRi.
Figure 2.

Comparison of repression domains and mutations defining nuclease-deficiency in dCas12a. (A). Downregulation efficiency of dCas12a D832A and repression domains genome integrated in strains DC002, DC004-008. (B). Downregulation efficiency of dCas12a E925A and repression domains genome integrated in strains DC003, DC009-013. Bars represent fold repression between targeting and non-targeting gRNA ±1 standard error (n = 4). Dashed line indicates 1-fold change (i.e. no repression). (C). dCas12a variants were assessed in downregulation of genome integrated eGFP (locus INT4) in strains DC002-013 using gRNAs targeting the Kl_ENO1 promoter or eGFP ORF.
eGFP downregulation targeting additional promoters
Understanding the requirements for effective gRNA design is complicated by a large number of different factors thought to affect efficiency. These include chromatin accessibility, target occupancy by the nucleosome and transcriptional factors, the target distance relative to the TSS and the DNA strand encoded by the spacer (12,18,27,50). We sought to test three different heterologous promoter targets integrated into the same locus to elucidate common features of functional gRNAs. For this purpose, we designed constructs where eGFP expression was controlled by either the TDH3 promoter from S. bayanus, the TEF1 promoter from S. mikatae or the ENO1 promoter from K. lactis, all integrated into the genome at the INT2 locus. A library of gRNAs was then designed for each strain with 4 to 6 gRNAs targeting the different heterologous promoters and four gRNAs targeting the eGFP ORF (Figure 3, Supplementary Table S6, Supplementary Figures S4 and S5). The efficiency of these gRNAs for gene repression was then tested with a plasmid carrying dCas12a-NLS-Mxi1 (pC-NLS-Mxi1), which we previously found achieved considerable repression when evaluating the effect of NLS configuration.
Figure 3.

Downregulation of eGFP expression by targeting dCas12a E925A Mxi1 to promoters controlling eGFP or eGFP ORF. (A) Repression pattern in strain FR007 with eGFP expressed from Sbay_TDH3p, (B) strain FR0008 with eGFP expressed from Smik_TEF1p, (C). strain FR009 with eGFP expressed from Kl_ENO1p. gRNAs targeting promoters in front of eGFP are depicted in grey and gRNAs targeting the eGFP ORF in green. NT indicates non-template strand and T – template strand. Bars represent fold repression between targeting and non-targeting gRNA ±1 standard error (n = 4). Dashed line indicates 1-fold change (i.e. no repression).
We found that targeting the promoter driving expression of eGFP rather than the ORF resulted in higher repression for functional gRNAs in all of the strains. The strongest repression was seen for gRNAs targeting promoters and resulted in 4.2-fold repression for Sbay_TDH3 (gTDH1), 4.0-fold for Smik_TEF1 (gTEF4) and 20-fold for Kl_ENO1 (gENO3). However, while all gRNAs were designed in the same way (Materials and Methods), when targeting the promoter or ORF at least one non-functioning gRNA was found per target (Figure 3). Notably, no transformants were obtained for gRNA gTEF2 targeting the Smik_TEF1 promoter. Due to the similarity between the TEF1 promoter from S. mikatae and S. cerevisiae, gTEF2 contained 18 complementary nucleotides to the promoters of both origins. The remaining two nucleotides mismatched the S. cerevisiae TEF1 promoter within the seed region and were therefore not expected to exhibit lethal activity towards the native promoter (51,52). The lack of transformants might be caused by a potential silencing effect of the native TEF1 gene. The target position of gRNAs exhibiting the strongest repression varied between promoters. The most efficient gRNAs targeting the Kl_ENO1 promoter were located 106–319 nt upstream the TSS, whereas for Sbay_TDH3 and Smik_TEF1 optimal targets were positioned 297 nt and 31 nt upstream TSS, respectively. Our observation leads to the conclusion that targets located at certain promoter regions are more effective, however, this region varies between promoters. Finally, no strand bias was found for gRNAs targeting Kl_ENO1 promoter nor other features such as PAM preference.
Controllable expression of gRNA from RNAP II promoter
In order to produce a functional gRNA the transcript must be precisely excised at specific sites, lack post-transcriptional modification of the 5′ and 3′ ends and should not be exported from the nucleus. These requirements are generally met by RNAP III promoters, exemplified by the snoRNA SNR52 promoter (41). An alternative approach is to combine the use of an RNAP II promoter with a gRNA flanked by self-cleaving ribozymes to cleave any signals for post-transcriptional modification or nuclear export (53). The intrinsic RNase activity of Cas12a also allows for an additional design of RNAP II expressed gRNAs, whereby a spacer is flanked by two direct repeats. In this case, the two direct repeats are recognized by Cas12a and the pre-crRNA is processed into its mature form of a spacer and a single direct repeat (28). Promoters recognized by RNAP II are generally well-characterized, exhibit a wide range of strengths and many are able to dynamically respond to changes in environmental conditions or the presence of an inducer molecule allowing for gRNA expression to be dynamically controlled (54). A recent study on Cas12a-mediated genome editing showed that driving gRNA expression from a RNAP II promoter increased gRNA availability and improved editing efficiency (28).
As inducible gRNA expression for dCas12a in S. cerevisiae has not been demonstrated before, we selected a crRNA encoding a gRNA that targeted the ENO1 promoter from K. lactis and compared repression when expressed from RNAP II and III promoters. For the RNAP II construct, the crRNA was expressed from an inducible GAL10 promoter which is activated by the presence of galactose. We tested three designs of galactose inducible gRNAs containing one direct repeat of Cas12a (gGAL1), a spacer flanked by two direct repeats (gGAL2) or self-cleaving ribozymes with a single direct repeat (gGAL3) (Figure 4A, Supplementary Figure S6). The RNAP III construct was expressed from the SNR52 promoter and contained a single direct repeat either with or without ribozymes (gRNA gSNR1 and gSNR2, respectively). Strong downregulation of eGFP was achieved with GAL10p expressed gRNAs with either two direct repeats (gGAL2) or ribozymes (gGAL3) which under inducing conditions displayed 35.7 ± 3.7 and 41.2 ± 3 fold repression, respectively. gGAL1 lead to much lower repression (1.8 ± 0.2) in comparison with gGAL2 and gGAL3 which illustrates that a single direct repeat is insufficient for pre-crRNA processing by Cas12a. We also observed a decrease in eGFP fluorescence for cells transformed with crRNA controlled by GAL10 promoter under non-inducive glucose conditions, which could be due to low levels of leaky expression (Figure 4A) (55).
Figure 4.

gRNA expression from RNAP III promoter and RNAP II promoter. (A) eGFP fluorescence upon expression of dCas12a NLS Mxi1 and gRNAs targeting the Kl_ENO1 promoter expressed from RNAP II promoter (GAL10p, gRNAs: gGAL1-3) and RNAP III promoter (SNR52p, gRNAs: gSNR1-2) under inducing (i.e. galactose, white bars) and repressing conditions (i.e. glucose, grey bars). Data normalized by a non-targeting control (gNone) tested under the same growth conditions as targeting gRNAs. Bars represent the mean from biological quadruplicates ±1 standard deviation. Dashed line indicates 1-fold change (i.e. no repression). (B) Biomass during a batch fermentation in microfermenter. pRN1120 indicates strain with empty gRNA recipient plasmid. Growth curves represent average of three biological replicates ±1 standard deviation.
Based on the comparison of repression levels achieved with GAL10p gRNAs with either two direct repeats or two ribozymes (gGAL2 versus gGAL3) and SNR52p gRNAs with one direct repeat or two ribozymes (gSNR1 versus gSNR2), there is no clear benefit to using self-cleaving ribozymes. The RNAP II based systems performed moderately better than the RNAP III-based systems (eGFP repression of 41-fold versus 26-fold for ribozyme flanked gRNAs gGAL3 and gSNR2 in galactose, respectively) (Figure 4A). This may be due to the known increased processivity of RNAP II over RNAP III (54). Numerous studies have shown that the availability of the gRNA is the limiting factor for CRISPR–Cas9 mediated genome editing in eukaryotic cells (28,56,57). Therefore, our results also suggest that despite using a multicopy plasmid in combination with a strong RNAP III promoter, the concentration of gRNA may still not be sufficient to fully saturate the available dCas12a pool.
To better understand the dynamics of dCas12a-based CRISPRi and the influence of the expression system on cell growth, a micro-fermentation experiment was performed (Material and Methods). Expression of a CRISPRi regulated eGFP and a constitutively expressed mCherry was found to be closely linked to the cell growth with expected decreases in the eGFP fluorescence for strains harbouring a targeting gRNAs and stable of mCherry fluorescence upon reaching the stationary phase (Supplementary Figure S7). Notably, antibiotic selection pressure used to maintain the empty pRN1120 plasmid caused a measurable decrease in growth rate (μ = 0.056 ± 0.001 h–1) when compared to the wild type strain (μ = 0.082 ± 0.006 h–1, Supplementary Figure S7A). In contrast, expression of the gRNA from the GAL10 promoter did not affect growth (μ = 0.059 ± 0.003 h–1 under inducing and μ = 0.055 ± 0.001 h–1 under non-inducing conditions). Both targeting and non-targeting gRNAs expressed by the SNR52 promoter negatively impacted cell growth (Figure 4B, μ = 0.044 ± 0.001 h–1 for gRNA gSNR1, μ = 0.043 ± 0.002 h–1 for gRNA gNone). We suspect that the expression of gRNAs from the RNAP III promoter leads to a competition for a shared pool of resources with native pathways leading to indirect impacts on normal cell growth (58).
Multiplexed downregulation
An advantage of dCas12a over dCas9 for CRISPRi is the automatic processing of CRISPR arrays by dCas12a, which removes the need for further accessory proteins (e.g. Csy4) (21,59,60). This allows for the expression of multiple targets to be easily regulated simultaneously or enables increased repression through the expression of multiple different gRNAs targeting the same gene. In addition, Cas12a has evolved to process long CRISPR arrays making this system robust to the production of multiple gRNAs, removing challenges faced when other approaches are used (e.g. expression of a gRNA array flanked by pre-tRNAs from an RNAP III promoter) (57).
To assess multiplexed regulation by dCas12a, we constructed a strain in which three fluorescence reporter proteins (BFP, mCherry and eGFP) were introduced into the genome at three integration sites and expressed by three different promoters (Sbay_TDH3, Smik_TEF1 and Kl_ENO1, respectively) (Figure 5A, Supplementary Figure S8). A single CRISPR array encoding three gRNAs targeting promoters driving expression of each reporter gene was then used to repress their expression simultaneously. The gRNA target sequences within each promoter were selected based on the capability to repress eGFP with similar strength (Figure 3): gRNA gTDH1 targeting Sbay_TDH3 (4.2-fold repression), gRNA gTEF4 targeting Smik_TEF1 (4.0-fold repression) and gRNA gENO6 targeting Kl_ENO1 (3.2-fold repression). To systematically assess the effect of gRNA order within an array, all possible permutations were tested with repression calculated by comparison to an array containing non-targeting gRNAs (Supplementary Table S8). A strong decrease in fluorescence of all reporters upon expression of the targeting arrays was observed (Figure 5B). Notably, gRNAs encoded in the first or the second position within the array caused stronger repression than the same gRNA encoded at the third position (unpaired t-test, P-value = 0.0001 for eGFP and mCherry and P-value = 0.01 for BFP, n = 8, Figure 5B). No significant difference was observed in repression efficiency for gRNAs targeting eGFP and BFP at position 1 and 2. Dependence of the gRNA context was recently reported for CRISPRi mediated by dFnCas12a in E. coli with improved repression for gRNAs encoded in position 2 compared to the same gRNA encoded in position 1 or 3 within an array (61).
Figure 5.

Simultaneous downregulation of fluorescent proteins with dCas12a E925A NLS Mxi1 and a single crRNA array. (A) Principle of multiplex downregulation. Strain FR014 harbours three fluorescent proteins expressed from three different heterologous promoters. dCas12a was genome integrated to ensure stable expression. Subsequently, the resulting strain was transformed with recipient plasmid pRN1120 and crRNA expression cassette provided as linear fragment. Single crRNA array is assembled into the recipient plasmid pRN1120 via in vivo recombination. The crRNA array is expressed from SNR52 promoter and SUP4 terminator and subsequently processed by dCas12a into three individual crRNA array comprised of a spacer (blue, red, green) and direct repeat (grey). dCas12a forms a complex with crRNA and is directed to the genomic target encoded in the spacer sequence. (B) Correlation between spacers order and repression of eGFP, BFP and mCherry. Bars represent fold change in BFP, mCherry and eGFP fluorescence normalized by a non-targeting array (array_10, not depicted) upon expression of dCas12a E925A NLS Mxi1 and single crRNA arrays. Six permutations in the order of spacers were tested and three control arrays with one spacer targeting the eGFP promoter and two non-targeting spacers. Bars represent fold repression between targeting and non-targeting gRNA ± 1 standard error (n = 4). Dashed line indicates 1-fold change (i.e. no repression).
Stringent regulation of heterologous β-carotene production
β-Carotene is a strongly coloured red-orange pigment abundant in plants, microalgae, fungi and bacteria and exhibits antioxidant properties (62,63). While there is increasing demand for β-carotene for food additives and nutraceuticals, supply is hampered by the low-productivity and harsh conditions needed for extraction from natural sources. Microbial production of β-carotene could alleviate this difficulty and has gained significant interest in recent years (35,64,65). To enable β-carotene production in industrially relevant microorganisms such as S. cerevisiae, the β-carotene biosynthesis pathway from Xanthophyllomyces dendrorhous can be heterologously expressed (Figure 6A) (35). This pathway comprises three genes: crtE, crtYB and crtI, encoding geranylgeranyl diphosphate synthase, bifunctional lycopene cyclase/phytoene synthase and phytoene desaturase, respectively. Only the simultaneous expression of all three crt genes results in the formation of β-carotene in S. cerevisiae and a clear phenotypical change where the cells turn from a white into an orange colour (35).
Figure 6.

Simultaneous downregulation of β-carotene production with dCas12a E925A NLS Mxi1 and single crRNA array. (A) Carotenoids production in S. cerevisiae is a multistep pathway catalysed by three enzymes: crtE, crtYB and crtI derived from Xanthophyllomyces dendrorhous. The pathway can be downregulated using dCas12a in sinlgeplex manner with a gRNA or multiplex using single crRNA arrays targeting promoters in front of crt genes resulting in decreased enzyme levels of crtE, crtYB and crtI and ultimately decreased amounts of carotenoids. Abbreviations: FPP, farnesyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate. (B) β-carotene and phytoene levels in strain CAR-041 upon singleplex downregulation with dCas12a and gRNAs harbouring a single spacer complementary to promoters in front crt genes. (C) Cell pellet obtained after growing transformants of strain CAR-041 with (left panel) gRNAs used in singleplex downregulation for targeting crtI, crtE and crtYB. For comparison transformants with non-targeting gRNA (gNone) and wild type strain; (right panel) single crRNA arrays targeting simultaneously crtE, crtYB and crtI, solely crtI and non-targeting control. Columns correspond to biological replicates. (D) β-carotene and phytoene levels in strain CAR-041 upon multiplex downregulation with dCas12a and single crRNA array. Arrays with three targeting spacers (array_1-6) targeting promoters Sbay_TDH3, Smik_TEF1 and Kl_ENO1 which control expression of crtE, crtYB and crtI, respectively are shown in dark grey whereas arrays with a single targeting spacer (array_7-9) targeting Kl_ENO1 promoter controlling crtI and two non-targeting spacer in light grey. Non-targeting control (array_10) is depicted in white. Bars represent mean ± 1 standard deviation (n = 2). (E) Effect of dCas12a expression in wild type and carotenogenic strains. DC001 Cas12a, DC002 dCas12a D832A, DC003 dCas12a E925A, CAR-034 carotenogenic strain, CAR-041 carotenogenic strain with genomically integrated dCas12a E925A Mxi1 controlled by the TEF1 promoter, CAR-042 carotenogenic strain with genome integrated dCas12a E925A Mxi1 controlled by the PGI1 promoter.
We sought to use our dCas12a CRISPRi system to stringently regulate β-carotene production in S. cerevisiae as a foundation for the dynamic control of this metabolic pathway. We made use of a previously constructed strain containing the heterologous genes required for β-carotene production (CAR-034) (32). This strain was further modified to have dCas12a E925A fused to Mxi1 and a single NLS integrated into its genome (strain CAR-041) and was transformed with a plasmid selected by dominant markers that expressed a single gRNA or crRNA array for regulation of expression of the crt genes. Crucially, the expression of the three genes (crtE, crtYB and crtI) was driven by the same heterologous promoters used for our assessment of multiplexed repression (Sbay_TDH3p, Smik_TEF1p and Kl_ENO1p, respectively), allowing the reuse of our previously tested gRNAs and crRNA arrays (Figures 3 and 5).
To begin, we used single gRNAs targeting either crtE (gTDH1–5), crtYB (gTEF3–4) or crtI (gENO1–6) in isolation to elucidate possible single points of control within the β-carotene pathway (Figure 6B, C). The most prominent impact was seen for repression of the crtI gene, which lead to no detectable β-carotene production. The corresponding gRNAs (gENO2,3,4,6) have previously been shown to repress eGFP by 70–95% (Figure 3C), suggesting strong expression of crtI is necessary for β-carotene production. The remaining gRNAs targeting crtI, gENO1,5 reduced β-carotene production by 2.8- and 5-fold, although repression of eGFP was not observed for these gRNAs (Figure 3C). For repression of crtE and crtYB, β-carotene was reduced but still detectable. As expected, repression of crtE which catalyses the first step in carotenogenesis, was not effective likely due to the presence of a native S. cerevisiae GGPP synthetase encoded by BTS1. Production of β-carotene despite repression of crtYB indicates that conversion of phytoene to lycopene by crtI limits the final lycopene cyclization to β-carotene by crtYB. The concentration of the intermediate metabolite phytoene increased upon expression of gRNAs targeting crtI, which likely relates to the accumulation of this compound due to limited conversion of the reaction catalysed by crtI into the subsequent compound lycopene. Furthermore, repression of crtYB decreased the phytoene production, however, only for gRNA gTEF4. Interestingly, the best performing gRNA for downregulation of the Sbay_TDH3 promoter when expressing eGFP (gTDH1) caused only a small reduction in phytoene concentration when used in the β-carotene pathway, whereas gRNA gTDH5 which repressed eGFP production only by 18%, halved phytoene (Figures 3C and 6B). It should be noted that eGFP and crtE were positioned on different chromosomes (11 and 10, respectively), which leads to the hypothesis that the functionality of these gRNAs might dependent on the target locus and/or genetic context. We also observed an over 8-fold drop in phytoene concentration when gRNA gTEF4 was used to repress crtYB, making it the limiting step in phytoene synthesis over crtE. Although we performed lycopene quantification using the UHPLC–DAD system, none was detected for any of the tested strains. Small amounts of lycopene could have accumulated in the tested strains, but this may not have been detected due to the sensitivity threshold of the quantification method. Strains with reduced production of β-carotene exhibited cell pellet with a faded colour, in comparison with the gNone control which had unaffected carotenoids production (Figure 6C). The colour intensity was not fully restored to the white appearance of the wild type pellet even when no β-carotene was detected. This can be explained by the presence of other colourful carotenoids and the detection limit of the UHPLC-DAD system utilized for quantification of β-carotene.
Finally, to see if a full shutdown of carotenoids production (i.e. all metabolites in the β-carotene pathway) could be achieved, we exploited the multiplexed downregulation of dCas12a capability by using crRNA arrays targeting a single or all promoters expressing the crt genes (Figure 6D, Supplementary Table S8). As expected, targeting solely crtI (arrays 7 and 9) disabled production of β-carotene but not phytoene (Figure 6C, D, Supplementary Table S8). However, expression of the crtI targeting gRNA from the second position (array_8) led to growth defects and only moderately reduced β-carotene production, suggesting expression of the gRNA was hampered and had broader off-target effects. In contrast, expression of arrays_1-6 simultaneously targeting crtE, crtYB and crtI were found to block the production of β-carotene and decreased phytoene by 5-fold (with the exception of one array), demonstrating the ability for multiplexed CRISPRi to stringently control the entire pathway.
The production of β-carotene is known to decrease cellular fitness (66–70). To evaluate the potential additional burden imposed by the CRISPRi system on the carotenogenic strain, we performed a spotting assay (Figure 6E). Firstly, we compared the growth of the wild type strain (CEN.PK113-7D) and strains with genomically-integrated Cas12a or dCas12a harboring a mutation in the nuclease domain (either D832A or E925A) expressed from the strong constitutive promoter TEF1 (strains DC001-003, respectively). Strains expressing (d)Cas12a formed smaller colonies compared to the wild type strain. Next, we tested the effect of carotenoids production in combination with weak and strong expression of dCas12a E925A to understand whether the toxicity of CRISPRi system is caused by a difference in protein expression (71). A carotenoid-producing strain with a strongly expressed dCas12a protein (CAR-041) exhibited smaller colonies sizes in comparison with a strain expressing solely dCas12a (DC002,003). However, cellular growth was restored in a carotenoids producing strain (CAR-042) when the strong TEF1 promoter was substituted with the weaker PGI1. These results demonstrate mild toxicity of CRISPRi dCas12a when highly expressed, which can be mitigated by using of a weaker promoter, in line with previous reports for FnCas12a (25).
DISCUSSION
The versatility and relevance of S. cerevisiae in industry motivates the significant demand for new molecular tools for gene regulation in this important host organism. Although dCas12a is a suitable system for expression control of metabolic pathways due to its T-rich PAM preference and a capability to process array for regulation of multiple targets, application of CRISPRi-dCas12a has never been reported for S. cerevisiae. Furthermore, studies on dCas12a in other organisms typically employ design rules previously established for dCas9, which may not be optimal for dCas12a. In this work, we have addressed these issues and demonstrated the use of dCas12a for multiplexed CRISPRi in S. cerevisiae. Our systematic study revealed some core considerations for effective regulation when using this system. First, we found efficient downregulation was present for dCas12a with either a C-terminal NLS and Mxi1 or when fused with a C-terminal Mxi1 domain. Using fluorescence microscopy, it was observed that dCas12a lacking an NLS can localize to the nucleus, which demonstrated that dCas12a can even be effective without NLS fusion. Although our results cannot explain the mechanism of the nuclear import and accumulation of dCas12a, a possible explanation for dCas12a localization in the nucleus is a cryptic location signal in dCas12a, as speculated before for Cas9 when genomic edits were detected for the nuclease fused to a nuclear export signal (NES) (47). Lysine is a particularly abundant amino acid in dCas12a (posing 12.5%), giving a possibility for a region with positively charged residuals to serve as a cryptic NLS undetected explicitly with available prediction tools (72–74). An alternative mechanism of dCas12a import to nucleus is passive diffusion (75,76). Considering recent publications (77,78) reporting slow dissociation rate of dCas9 once it is bound to a target, a low amount of dCas protein in the nucleus might be expected to mediate downregulation. To fully understand the mechanism of dCas12a import to the nucleus further research is necessary. Strong repression was observed when using dCas12a E925A with a single C-terminal NLS and Mxi1 or MIG1 repression domain, leading to a 97% reduction in an eGFP reporter protein. Fusion to KRAB and TUP1 repression domains diminished the repressing capability of dCas12a, which has not been observed previously when used with dCas9 (3). Second, we found that dCas12a-based CRISPRi is more effective when the promoter region is targeted rather than targeting the ORF. This feature is potentially useful to downregulate many different genes simultaneously using the same gRNA if its promoter target is used for expression. Third, analysis of crRNA arrays encoding three gRNAs for multiplexed downregulation revealed a position-dependent effect on the efficiency of each gRNA. Those located in the first two positions caused significantly better repression than those in the third position. We also demonstrated gRNA expression from an inducible RNAP II-controlled GAL10 promoter to enable inducible downregulation, opening avenues for dynamic control of gene expression using this system. Finally, as a proof-of-concept application, we showed that the inherent ability of dCas12a-based CRISPRi to preform multiplexed gene regulation can be used to stringently control a heterologous metabolic pathway producing β-carotene.
Our comparison of dCas12a-CRISPRi designs was evaluated for downregulation of eGFP and β-carotene biosynthesis, however, the presented results can serve multiple purposes. We observed a consistent performance of a dCas12a fusion with a Mxi1 domain and a C-terminal NLS regardless of a target and its chromosomal location, if the gRNA is functional. Therefore this construct is expected to be effective in regulating any metabolic pathway if working gRNAs are chosen. The modularity of the dCas12a system (NLS, repression domain, gRNA) allows to vary different elements. Among five repression domains we observed enhanced silencing activity of dCas12a for two of the domains and diminished activity of two others. Evaluation of a broader set of repression domains may lead to the discovery of even stronger repression systems. Although our work is limited to dCas12a from Lachnospiraceae bacterium ND2006 (LbCas12a), CRISPRi has been demonstrated for other orthologues: FnCas12a (18,27–29), AsCas12a (79) and EeCas12a (80). With the recent work of Toth et al. this set could be further extended by the implementation of novel Cas12a variants exhibiting a preference for alternative PAM sequences such as TNTN (81) developed from LbCas12a without affecting the nuclease domain.
As the complexity of synthetic genetic systems grows, there will be an increased need for high-performance molecular tools able to regulate gene expression in a flexible way. The CRISPRi system we developed in this study exploits the inherent benefits of dCas12a for gene regulation, such as its preference for a T-rich PAM and inherent capability to process crRNA arrays to enable multiplexed regulation with minimal effort for the design. It also demonstrates the value of comprehensive studies of design parameters for such systems, which open up avenues to both refine existing systems and ensure the best possible performance is achieved; aspects that will be essential for transitioning them into industrial use.
DATA AVAILABILITY
Sequences of plasmids, genome edited strains and examples of gRNA expression cassette are submitted to GenBank (MW584243-66, MW766349-52, MZ222243-46, Supplementary Table S11). Flow cytometry data is deposited at Flow Repository (FR-FCM-Z38Q). Reads from nanopore sequencing of strains DC001-013 are submitted at Sequence Read Archive (PRJNA688652). Plasmids pC-Mxi1, pC-Mxi1-NLS, pC-NLS-Mxi1-NLS, pC-NLS and pC-NLS-Mxi1 (ID 166728–166732) and dCas12a-eGFP (ID 171629) are deposited at Addgene.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Bianca Gielesen for the assistance with FACS, Reza Maleki Seifar for the assistance with UHPLC and Abel Folch-Fortuny for the support in statistical analysis. We express our gratitude to Liesbeth Veenhoff for valuable discussion on dCas12a localization.
Contributor Information
Klaudia Ciurkot, DSM Biotechnology Center, Delft 2613 AX, The Netherlands; Department of Chemistry, University of Hamburg, Hamburg 20146, Germany.
Thomas E Gorochowski, School of Biological Sciences, University of Bristol, Tyndall Avenue, Bristol BS8 1TQ, UK.
Johannes A Roubos, DSM Biotechnology Center, Delft 2613 AX, The Netherlands.
René Verwaal, DSM Biotechnology Center, Delft 2613 AX, The Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant [764591]; T.E.G. is supported by BrisSynBio, a BBSRC/EPSRC Synthetic Biology Research Centre [BB/L01386X/1]; Royal Society University Research Fellowship [UF160357]. Funding for open access charge: H2020 Marie Skłodowska-Curie Actions [764591].
Conflict of interest statement. None declared.
REFERENCES
- 1. De Nadal E., Ammerer G., Posas F.. Controlling gene expression in response to stress. Nat. Rev. Genet. 2011; 12:833–845. [DOI] [PubMed] [Google Scholar]
- 2. Nielsen J., Keasling J.D.. Engineering cellular metabolism. Cell. 2016; 164:1185–1197. [DOI] [PubMed] [Google Scholar]
- 3. Lian J., HamediRad M., Zhao H.. Advancing metabolic engineering of saccharomyces cerevisiae using the CRISPR/Cas system. Biotechnol. J. 2018; 13:1700601. [DOI] [PubMed] [Google Scholar]
- 4. Agrawal N., Dasaradhi P.V.N., Mohmmed A., Malhotra P., Bhatnagar R.K., Mukherjee S.K.. RNA Interference: biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 2003; 67:657–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beerli R.R., Barbas C.F.. Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002; 20:135–141. [DOI] [PubMed] [Google Scholar]
- 6. Khalil A.S., Lu T.K., Bashor C.J., Ramirez C.L., Pyenson N.C., Joung J.K., Collins J.J.. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 2012; 150:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A.. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013; 152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A.et al.. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013; 154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meaker G.A., Hair E.J., Gorochowski T.E.. Advances in engineering CRISPR–Cas9 as a molecular swiss army knife. Synth. Biol. 2020; 5:ysaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C.et al.. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014; 159:647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li S., Jendresen C.B., Landberg J., Pedersen L.E., Sonnenschein N., Jensen S.I., Nielsen A.T.. Genome-wide CRISPRi-based identification of targets for decoupling growth from production. ACS Synth. Biol. 2020; 9:1030–1040. [DOI] [PubMed] [Google Scholar]
- 12. Smith J.D., Suresh S., Schlecht U., Wu M., Wagih O., Peltz G., Davis R.W., Steinmetz L.M., Parts L., St.Onge R.P.. Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design. Genome Biol. 2016; 17:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farzadfard F., Perli S.D., Lu T.K.. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth. Biol. 2013; 2:604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schreiber-Agus N., Chin L., Chen K., Torres R., Rao G., Guida P., Skoultchi A.I., DePinho R.A.. An amino-terminal domain of Mxi1 mediates anti-myc oncogenic activity and interacts with a homolog of the yeast transcriptional repressor SIN3. Cell. 1995; 80:777–786. [DOI] [PubMed] [Google Scholar]
- 15. Witzgall R., O’Leary E., Leaf A., Onaldi D., Bonventre J. V.. The Kruppel-associated box-A (KRAB-A) domain of zinc finger proteins mediates transcriptional repression. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:4514–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tak Y.E., Kleinstiver B.P., Nuñez J.K., Hsu J.Y., Horng J.E., Gong J., Weissman J.S., Joung J.K.. Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors. Nat. Methods. 2017; 14:1163–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lian J., Hamedirad M., Hu S., Zhao H.. Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat. Commun. 2017; 8:1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miao C., Zhao H., Qian L., Lou C.. Systematically investigating the key features of the DNase deactivated Cpf1 for tunable transcription regulation in prokaryotic cells. Synth. Syst. Biotechnol. 2019; 4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shmakov S., Smargon A., Scott D., Cox D., Pyzocha N., Yan W., Abudayyeh O.O., Gootenberg J.S., Makarova K.S., Wolf Y.I.et al.. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017; 15:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., Van Der Oost J., Regev A.et al.. Cpf1 is a single RNA-Guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zetsche B., Heidenreich M., Mohanraju P., Fedorova I., Kneppers J., Degennaro E.M., Winblad N., Choudhury S.R., Abudayyeh O.O., Gootenberg J.S.et al.. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017; 35:31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swarts D.C., Jinek M.. Cas9 versus Cas12a/Cpf1: structure–function comparisons and implications for genome editing. Wiley Interdiscip. Rev. RNA. 2018; 9:e1481. [DOI] [PubMed] [Google Scholar]
- 23. Ottoz D.S.M., Rudolf F.. Constitutive and regulated promoters in yeast: how to design and make use of promoters in S. cerevisiae. Synthetic Biology: Parts, Devices and Applications. 2018; John Wiley & Sons; 106–130. [Google Scholar]
- 24. Borodina I., Nielsen J.. Advances in metabolic engineering of yeast Saccharomyces cerevisiae for production of chemicals. Biotechnol. J. 2014; 9:609–620. [DOI] [PubMed] [Google Scholar]
- 25. Swiat M.A., Dashko S., Ridder Den, Wijsman M., Van Der Oost M., Daran J., Daran-Lapujade P.. FnCpf1: a novel and efficient genome editing tool for Saccharomyces cerevisiae. Nucleic Acids Res. 2017; 45:12585–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Verwaal R., Buiting-Wiessenhaan N., Dalhuijsen S., Roubos J.A.. CRISPR/Cpf1 enables fast and simple genome editing of Saccharomyces cerevisiae. Yeast. 2018; 35:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang J., Peng Y.Z., Liu D., Liu H., Cao Y.X., Li B.Z., Li C., Yuan Y.J.. Gene repression via multiplex gRNA strategy in Y. lipolytica. Microb. Cell Fact. 2018; 17:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y., Boeke J.D.. CRISPR-Cas12a system in fission yeast for multiplex genomic editing and CRISPR interference. Nucleic Acids Res. 2020; 48:5788–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi S.Y., Woo H.M.. CRISPRi-dCas12a: a dCas12a-mediated CRISPR interference for repression of multiple genes and metabolic engineering in cyanobacteria. ACS Synth. Biol. 2020; 9:2351–2361. [DOI] [PubMed] [Google Scholar]
- 30. Liu P., Luk K., Shin M., Idrizi F., Kwok S., Roscoe B., Mintzer E., Suresh S., Morrison K., Frazão J.B.et al.. Enhanced Cas12a editing in mammalian cells and zebrafish. Nucleic Acids Res. 2019; 47:4169–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Dijken J.P., Bauer J., Brambilla L., Duboc P., Francois J.M., Gancedo C., Giuseppin M.L.F., Heijnen J.J., Hoare M., Lange H.C.et al.. An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microbial. Technol. 2000; 26:706–714. [DOI] [PubMed] [Google Scholar]
- 32. Ciurkot K., Vonk B., Gorochowski T.E., Roubos J.A., Verwaal R.. CRISPR/Cas12a multiplex genome editing of Saccharomyces cerevisiae and the creation of yeast pixel art. J. Vis. Exp. 2019; 147:e59350. [DOI] [PubMed] [Google Scholar]
- 33. Gietz R.D., Schiestl R.H., Willems A.R., Woods R.A.. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast. 1995; 11:355–360. [DOI] [PubMed] [Google Scholar]
- 34. Oldenburg K.R., Vo K.T., Michaelis S., Paddon C.. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997; 25:451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Verwaal R., Wang J., Meijnen J.P., Visser H., Sandmann G., Van Den Berg J.A., Van Ooyen A.J.J.. High-level production of beta-carotene in Saccharomyces cerevisiae by successive transformation with carotenogenic genes from Xanthophyllomyces dendrorhous. Appl. Environ. Microbiol. 2007; 73:4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 37. Mircetic J., Steinebrunner I., Ding L., Fei J.F., Bogdanova A., Drechsel D., Buchholz F.. Purified Cas9 fusion proteins for advanced genome manipulation. Small Methods. 2017; 1:1600052. [Google Scholar]
- 38. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMillan J., Lu Z., Rodriguez J.S., Ahn T.H., Lin Z.. YeasTSS: An integrative web database of yeast transcription start sites. Database. 2019; 2019:baz048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dicarlo J.E., Norville J.E., Mali P., Rios X., Aach J., Church G.M.. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013; 41:4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Castillo-Hair S.M., Sexton J.T., Landry B.P., Olson E.J., Igoshin O.A., Tabor J.J.. FlowCal: a user-friendly, open source software tool for automatically converting flow cytometry data from arbitrary to calibrated units. ACS Synth. Biol. 2016; 5:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bailey R., Madden K.T., Trueheart J.. Production of Carotenoids in Oleaginous Yeast and Fungi. 2006; U.S. Patent and Trademark Office; US8288149B2. [Google Scholar]
- 44. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M.. RNA-guided human genome engineering via Cas9. Science. 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao P., Zhang Z., Lv X., Zhao X., Suehiro Y., Jiang Y., Wang X., Mitani S., Gong H., Xue D.. One-step homozygosity in precise gene editing by an improved CRISPR/Cas9 system. Cell Res. 2016; 26:633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beal J., Haddock-Angelli T., Baldwin G., Gershater M., Dwijayanti A., Storch M., de Mora K., Lizarazo M., Rettberg R.. Quantification of bacterial fluorescence using independent calibrants. PLoS One. 2018; 13:e0199432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roggenkamp E., Giersch R.M., Schrock M.N., Turnquist E., Halloran M., Finnigan G.C.. Tuning CRISPR–Cas9 gene drives in Saccharomyces cerevisiae. G3 Genes, Genomes, Genet. 2018; 8:999–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu Y., Han J., Chen Z., Wu H., Dong H., Nie G.. Engineering cell signaling using tunable CRISPR-Cpf1-based transcription factors. Nat. Commun. 2017; 8:2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Currin A., Swainston N., Dunstan M.S., Jervis A.J., Mulherin P., Robinson C.J., Taylor S., Carbonell P., Hollywood K.A., Yan C.et al.. Highly multiplexed, fast and accurate nanopore sequencing for verification of synthetic DNA constructs and sequence libraries. Synth. Biol. 2019; 4:ysz025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jensen E.D., Ferreira R., Jakočiunas T., Arsovska D., Zhang J., Ding L., Smith J.D., David F., Nielsen J., Jensen M.K.et al.. Transcriptional reprogramming in yeast using dCas9 and combinatorial gRNA strategies. Microb. Cell Fact. 2017; 16:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kleinstiver B.P., Tsai S.Q., Prew M.S., Nguyen N.T., Welch M.M., Lopez J.M., McCaw Z.R., Aryee M.J., Joung J.K.. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016; 34:869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Swarts D.C., van der Oost J., Jinek M.. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell. 2017; 66:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gao Y., Zhao Y.. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J. Integr. Plant Biol. 2014; 56:343–349. [DOI] [PubMed] [Google Scholar]
- 54. McCarty N.S., Graham A.E., Studená L., Ledesma-Amaro R.. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020; 11:1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hofmann A., Falk J., Prangemeier T., Happel D., Köber A., Christmann A., Koeppl H., Kolmar H.. A tightly regulated and adjustable CRISPR-dCas9 based AND gate in yeast. Nucleic Acids Res. 2019; 47:509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O.et al.. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013; 31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ng H., Dean N.. Dramatic improvement of CRISPR/Cas9 editing in candida albicans by increased single guide RNA expression. mSphere. 2017; 2:e00385-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gorochowski T.E., Avcilar-Kucukgoze I., Bovenberg R.A.L., Roubos J.A., Ignatova Z.. A Minimal model of ribosome allocation dynamics captures trade-offs in expression between endogenous and synthetic genes. ACS Synth. Biol. 2016; 5:710–720. [DOI] [PubMed] [Google Scholar]
- 59. Fonfara I., Richter H., BratoviÄ M., Le Rhun A., Charpentier E.. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016; 532:517–521. [DOI] [PubMed] [Google Scholar]
- 60. McCarty N.S., Shaw W.M., Ellis T., Ledesma-Amaro R.. Rapid assembly of gRNA arrays via modular cloning in yeast. ACS Synth. Biol. 2019; 8:906–910. [DOI] [PubMed] [Google Scholar]
- 61. Liao C., Ttofali F., Slotkowski R.A., Denny S.R., Cecil T.D., Leenay R.T., Keung A.J., Beisel C.L.. Modular one-pot assembly of CRISPR arrays enables library generation and reveals factors influencing crRNA biogenesis. Nat. Commun. 2019; 10:2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yabuzaki J. Carotenoids database: structures, chemical fingerprints and distribution among organisms. Database. 2017; 2017:bax004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li C., Swofford C.A., Sinskey A.J.. Modular engineering for microbial production of carotenoids. Metab. Eng. Commun. 2020; 10:e00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jacobsen I.H., Ledesma-Amaro R., Martinez J.L.. Recombinant β-Carotene production by yarrowia lipolytica – assessing the potential of micro-scale fermentation analysis in cell factory design and bioreaction optimization. Front. Bioeng. Biotechnol. 2020; 8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu T., Ye L., Zhao D., Li S., Li Q., Zhang B., Bi C., Zhang X.. Membrane engineering - a novel strategy to enhance the production and accumulation of β-carotene in Escherichia coli. Metab. Eng. 2017; 43:85–91. [DOI] [PubMed] [Google Scholar]
- 66. Hong J., Park S.H., Kim S., Kim S.W., Hahn J.S.. Efficient production of lycopene in Saccharomyces cerevisiae by enzyme engineering and increasing membrane flexibility and NAPDH production. Appl. Microbiol. Biotechnol. 2019; 103:211–223. [DOI] [PubMed] [Google Scholar]
- 67. Zhou P., Ye L., Xie W., Lv X., Yu H.. Highly efficient biosynthesis of astaxanthin in Saccharomyces cerevisiae by integration and tuning of algal crtZ and bkt. Appl. Microbiol. Biotechnol. 2015; 99:8419–8428. [DOI] [PubMed] [Google Scholar]
- 68. Verwaal R., Jiang Y., Wang J., Daran J.M., Sandmann G., Van Den Berg J.A., Van Ooyen A.J.J.. Heterologous carotenoid production in Saccharomyces cerevisiae induces the pleiotropic drug resistance stress response. Yeast. 2010; 27:983–998. [DOI] [PubMed] [Google Scholar]
- 69. Chen Y., Xiao W., Wang Y., Liu H., Li X., Yuan Y.. Lycopene overproduction in Saccharomyces cerevisiae through combining pathway engineering with host engineering. Microb. Cell Fact. 2016; 15:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu P., Sun L., Sun Y., Shang F., Yan G.. Decreased fluidity of cell membranes causes a metal ion deficiency in recombinant Saccharomyces cerevisiae producing carotenoids. J. Ind. Microbiol. Biotechnol. 2016; 43:525–535. [DOI] [PubMed] [Google Scholar]
- 71. Eguchi Y., Makanae K., Hasunuma T., Ishibashi Y., Kito K., Moriya H.. Estimating the protein burden limit of yeast cells by measuring the expression limits of glycolytic proteins. Elife. 2018; 7:e34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brameier M., Krings A., MacCallum R.M.. NucPred - predicting nuclear localization of proteins. Bioinformatics. 2007; 23:1159–1160. [DOI] [PubMed] [Google Scholar]
- 73. Kosugi S., Hasebe M., Tomita M., Yanagawa H.. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lin J., Mondal A.M., Liu R., Hu J.. Minimalist ensemble algorithms for genome-wide protein localization prediction. BMC bioinformatics. 2012; 13:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Popken P., Ghavami A., Onck P.R., Poolman B., Veenhoff L.M.. Size-dependent leak of soluble and membrane proteins through the yeast nuclear pore complex. Mol. Biol. Cell. 2015; 26:1386–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Timney B.L., Raveh B., Mironska R., Trivedi J.M., Kim S.J., Russel D., Wente S.R., Sali A., Rout M.P.. Simple rules for passive diffusion through the nuclear pore complex. J. Cell Biol. 2016; 215:57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jones D.L., Leroy P., Unoson C., Fange D., Ćurić V., Lawson M.J., Elf J.. Kinetics of dCas9 target search in Escherichia coli. Science. 2017; 357:1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ma H., Tu L.C., Naseri A., Huisman M., Zhang S., Grunwald D., Pederson T.. CRISPR–Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 2016; 214:529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang X., Wang J., Wang J., Cheng Q., Zheng X., Zhao G.. Multiplex gene regulation by CRISPR-ddCpf1. Cell Discov. 2017; 3:17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kim S.K., Kim H., Ahn W.C., Park K.H., Woo E.J., Lee D.H., Lee S.G.. Efficient transcriptional gene repression by Type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth. Biol. 2017; 6:1273–1282. [DOI] [PubMed] [Google Scholar]
- 81. Toth E., Krausz L., Nyeste A., Welker Z., Husz K., Welker E.. Improved LbCas12a variants with altered PAM specificities further broaden the genome targeting range of Cas12a nucleases. Nucleic Acids Res. 2020; 48:3722–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequences of plasmids, genome edited strains and examples of gRNA expression cassette are submitted to GenBank (MW584243-66, MW766349-52, MZ222243-46, Supplementary Table S11). Flow cytometry data is deposited at Flow Repository (FR-FCM-Z38Q). Reads from nanopore sequencing of strains DC001-013 are submitted at Sequence Read Archive (PRJNA688652). Plasmids pC-Mxi1, pC-Mxi1-NLS, pC-NLS-Mxi1-NLS, pC-NLS and pC-NLS-Mxi1 (ID 166728–166732) and dCas12a-eGFP (ID 171629) are deposited at Addgene.