


Abstract
Impaired replication progression leads to de novo copy number variant (CNV) formation at common fragile sites (CFSs). We previously showed that these hotspots for genome instability reside in late-replicating domains associated with large transcribed genes and provided indirect evidence that transcription is a factor in their instability. Here, we compared aphidicolin (APH)-induced CNV and CFS frequency between wild-type and isogenic cells in which FHIT gene transcription was ablated by promoter deletion. Two promoter-deletion cell lines showed reduced or absent CNV formation and CFS expression at FHIT despite continued instability at the NLGN1 control locus. APH treatment led to critical replication delays that remained unresolved in G2/M in the body of many, but not all, large transcribed genes, an effect that was reversed at FHIT by the promoter deletion. Altering RNase H1 expression did not change CNV induction frequency and DRIP-seq showed a paucity of R-loop formation in the central regions of large genes, suggesting that R-loops are not the primary mediator of the transcription effect. These results demonstrate that large gene transcription is a determining factor in replication stress-induced genomic instability and support models that CNV hotspots mainly result from the transcription-dependent passage of unreplicated DNA into mitosis.
INTRODUCTION
Common fragile site (CFS) expression and copy number variant (CNV) formation are related forms of locus-specific genomic instability that manifest in cell systems in vitro following replication stress (1). Chromosome fragile sites are genomic loci that are prone to forming recurrent cytogenetic anomalies visible as gaps and breaks in metaphase cells. CFSs by their nature are studied in vitro, but correlative evidence accumulated over decades suggests that that these loci are also unstable in vivo, particularly in cancer (1).
Using genomic technologies, we have shown that specific loci are also more prone to forming CNVs, especially deletions, in response to stresses such as partial inhibition of replication by low doses of aphidicolin (APH) (2), hydroxyurea (HU) (3) or ionizing radiation (4). These CNV-prone loci, termed hotspots, overlap CFSs measured in the same cell line, suggesting that CFS expression and CNVs are different measurable outcomes of the same underlying genomic events (5). Whereas CFSs are valuable cytogenetic markers of replication stress-induced instability, CNVs are heritable genomic alterations with direct biological consequences.
As heritable mutations, it is easier to directly compare CNVs induced in vitro with those in normal and diseased human genomes. Such studies first revealed that in vitro CNVs share many properties with human CNVs observed in normal genomes and in constitutional genetic diseases, including a large median size measured in low hundreds of kbp, a high frequency of non-recurrent junctions mediated by nonhomologous mechanisms, and a subset of complex, multi-junction events (2,6). Moreover, cancer sequencing projects such as TCGA revealed that human cancers also accumulate CNVs at loci that correspond to CFSs and in vitro CNV hotspots (1,7–9). While some cancer CNV hotspots, such as those at tumor-suppressor loci, reflect phenotypic selection, others likely reflect a mechanistic predisposition to chromosome rearrangement.
An important feature of CFSs and CNV hotspots, whether in vitro or in cancer, is that they are cell type-specific (5,10–12). Thus, their instability must reflect dynamically changeable properties and not just primary locus sequences. CFSs and CNV hotspots are typically late replicating, disproportionately so under replication stress, as first demonstrated by fluorescence in situ hybridization (FISH)-based techniques (13). However, much more of the genome replicates in the last portions of S phase than comprises CFSs, and the replication timing of many chromosome domains is not strongly cell type-specific (14,15). It is more telling that the most unstable CNV hotspots correspond strongly to the largest human genes (>1 Mbp), including classic CFS genes such as FHIT and WWOX (1,5). We and others recently demonstrated that the specificity of CFS expression and CNV hotspots robustly correlates with the active transcription of the large isoforms of these genes (5,16). Similar conclusions were drawn from at least one other instability readout, an assay that traps cellular double-strand breaks (DSBs) using induced ‘bait’ DSBs (17).
Several mechanisms might account for the influence of large gene transcription on local instability. A first category invokes an interplay between transcription and replication timing during the cell cycle. While many smaller genes complete transcription during G1, large genes take many hours to transcribe, potentially resulting in transcription during S-phase in dividing cells (18–20). S-phase transcription can displace intragenic pre-replication complexes and prevent replication rescue by dormant origin firing (21–24). Consistently, a cell type-specific paucity of dormant origin firing has been reported for CFS loci (10,25,26). A second and distinct mechanistic category invokes a direct influence of transcription on the frequency of replication fork failure, a molecular genetic event likely to cause measurable downstream locus consequences such as metaphase anomalies, CNVs or DSBs. Direct replication-transcription collisions would be directional and biased toward the 3′ ends of genes, unlike the observed accumulation of CNVs in the centers of large hotspot genes (5,27). Alternatively, persistent transcription-dependent R-loops are a recognized mode of replication–transcription interactions resulting in fork failure and possibly CNVs as suggested by their reported role in CFS induction (18,28), with the RTEL1 protein recently implicated as a protective mechanism against R-loop formation at CFSs (29,30).
Blin et al. recently reported that, somewhat paradoxically, both increasing and decreasing large gene transcription can reduce CFS expression at the affected loci (16), with a similar effect noted for FHIT promoter ablation by Fernandes et al. (31). Limitations of prior work include the absence of a direct demonstration that preventing transcription of a large gene abrogates the induction of replication stress-dependent genomic mutations such as CNVs. Moreover, limited applications of such experimental tools have only allowed the relationships between transcription, replication and CNV formation to be established in a correlative manner. We used CRISPR-Cas9 technology to create mutant cell clones with deletions spanning the FHIT gene promoter and assessed the effects on both CNV formation and CFS expression. Results provide direct evidence for a cause-and-effect relationship between large gene transcription and heritable locus instability. We then used these cell tools to demonstrate that locus replication timing changes in response to APH based on transcription status in a manner that predicts unreplicated mitotic DNA as a primary substrate for CNV formation. In contrast, changing RNase H1 expression had minimal impact on APH-induced CNV formation, which we found to be consistent with low levels of R-loop formation in the central regions of large transcribed genes where CNVs form.
MATERIALS AND METHODS
Oligonucleotides, DNA extraction and PCR
Unless otherwise specified, oligonucleotides were purchased from Integrated DNA Technologies and designed using Primer3Plus and the GRCh38/hg38 and GRCm38/mm10 reference genomes. Primer and probe sequences are available in Supplementary Table S1. Unless otherwise specified, genomic DNA was extracted using Qiagen's Gentra PureGene Cell kit (#158388).
Phusion (NEB #M0530) was used as described by the manufacturer with 0.2 µM for each primer and 3% DMSO (v/v), and the following cycling conditions: initial denaturation at 98°C for 30 s followed by 35 cycles of 98°C for 5 s, 60°C for 10 s, and 72°C for 30 s, and a final extension at 72°C for 5 min. Native Taq polymerase (Thermo Fisher #18038042) was used as described by the manufacturer with 0.2 µM of each primer, 0.2 mM dNTPs, 1.5 mM MgCl2, and the following cycling conditions: initial denaturation at 94°C for 3 min followed by 40 cycles of 94°C for 45 s, 58–65°C for 30 s, and 72°C for 30 s to 1 min, final extension at 72°C for 5 min.
Tissue culture and aphidicolin treatment
GM11713, a mouse-human somatic cell hybrid line carrying hChr3 in the mouse A9 cell background, was obtained from the Coriell Institute. GM11713 and its derivatives were maintained in Eagle’s Minimum Essential Media with Earle's salts, non-essential amino acids and 10% fetal bovine serum, with continuous selection for the neomycin resistance gene on hChr3 with 250 μg/ml G418 added to the media at the time of use. The UMHF1 (HF1) human foreskin fibroblast immortalized cell line (32) was maintained in Dulbecco’s Modified Eagle Medium supplemented with 13% fetal bovine serum, 4 mM L-Glutamine and 1× penicillin-streptomycin. For APH treatment of cell lines, 200 μM APH (Sigma #A0781) dissolved in DMSO was diluted to 0.4, 0.6 or 0.8 μM in the appropriate cell culture media. Freshly prepared APH media were applied every 24 h during treatment. Unless otherwise noted, treated cells were allowed to recover for 24 h in non-APH media before further use. Cell clones were established by plating at low-density (100–200 cells per 100 mm dish), isolating single colonies with cloning rings, and expanding in multi-well dishes.
CRISPR-Cas9-mediated knockout of the FHIT promoter
Single guide RNAs (sgRNAs) surrounding the FHIT promoter region were designed using CRISPRdirect (33) and CHOPCHOP (34). sgRNAs oligonucleotides were integrated into pSpCas9(BB)-2A-GFP plasmids (Addgene #48138) as described (35). After confirmation by Sanger sequencing, plasmids were transfected into GM11713 using Lipofectamine 2000 (Thermo Fisher #11668027) per the manufacturer’s instructions. To identify cells that had undergone FHIT promoter deletion, we modified the protocol of Bauer et al. (36). Forty-eight hours post-transfection, the brightest 3% of GFP+ cells were sorted using iCyt’s Synergy Cell Sorting System at the Michigan Flow Cytometry Core and plated in a single well in a 96-well plate for recovery. When confluent, approximately 50 cells were plated per well in a 24-well plate. After expansion, PCR with primers flanking the two sgRNA target sites was used to determine which wells contained promoterless mutants. Cells from these wells were finally cloned and individual positive clones were identified using the same PCR reaction. The PCR products from two candidate mutants were confirmed by Sanger sequencing.
FHIT transcription analysis by qRT-PCR
Total RNA was isolated from cell lines using Qiagen's RNeasy mini kit (#74104) and reverse transcribed to complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription kit from Thermo Fisher (#4368814). qPCR was performed using Qiagen’s QuantiTect SYBR green PCR kit (#204143) and Applied Biosystems 7500 Real-Time PCR system. Three primer pairs specific to the FHIT mRNA are listed in Supplementary Table S1. The cycling conditions were 50°C for 10 min, then 95°C for 15 min, followed by 40 cycles of 15 s at 94°C, 30 s at 60°C, then 30 s at 72°C. Mouse ActB was used as a normalizing control gene and wild-type (WT) GM11713 as the reference sample to calculate ΔΔCt values.
GM11713 transcription analysis by Bru-seq nascent RNA sequencing
Bru-seq nascent RNA sequencing was performed as previously described (37). The resultant sequencing reads were aligned separately to both the GRCh38/hg38 and GRCm38/mm10 reference genomes to determine where RNA polymerase molecules were synthesizing DNA during a 30-min labeling period. The two reference genome alignments were carefully compared to understand the degree of cross-reactivity between the two species at all loci, which was found to be low.
CNV detection by droplet digital PCR
To establish a GM11713 reference region for droplet digital (ddPCR) analysis, genomic DNA was isolated and shotgun libraries were prepared by the Michigan Advanced Genomics Core and sequenced on Illumina HiSeq 4000 using a 2 × 150 read configuration. Reads were aligned to the GRCm38/mm10 reference genome and read depth determined in genomic bins. A region on mChrX was inferred and later confirmed by ddPCR to have a copy number of one allele per cell.
GM11713 and its derivatives were treated with 0.6 µM APH for 72 h as described above. Quantitative ddPCR was performed using the Bio-Rad system QX200, ddPCR Supermix for Probes (Bio-Rad #1863010 or #1863023) and 20× primer/probe sets (Supplementary Table S1) as described by the manufacturer. For multiplexing, the test assay probes were labeled with FAM and the reference assay probes were labeled with HEX. Final concentrations of primers and probes were 900 mM and 250 nM, respectively. Input genomic DNA was digested with HindIII prior to or during ddPCR reaction assembly and used at a final concentration of 5–10 ng/µl. Sample concentrations were optimized to produce 30–70% positive droplets per reaction. Data were analyzed using the Bio-Rad QuantaSoft software package where the ratio of test to reference molecules in a sample revealed allelic loss at the test probes, which was taken as evidence of deletion CNV formation. When possible, multiple tubes of the same reaction mixture were evaluated simultaneously and droplet count data was merged in QuantaSoft. Poisson 95% confidence intervals for ratio values were calculated in QuantaSoft and depicted as error bars. The sensitivity limit for detecting a true burden of deletion CNVs (i.e. reduced copy number) in a sample is determined by these Poisson measurement uncertainties, which are a function of the number of molecules counted for a given sample.
CNV detection by clonal PCR analysis
Following similar cell treatments as described above, individual GM11713 and promoterless cell clones were established, expanded and screened for DNA content using PCR assays directed at targets in and around the human FHIT gene (Supplementary Table S1). CNVs were detected by running the PCR amplicons on an agarose gel, where an absence of the product was considered as a deletion. All PCR reactions scored as deletions were confirmed by repeating those assays alongside appropriate positive control reactions applied to the same DNAs. Duplication CNVs could not be scored by this method but are much less common at large CNV hotspots (5).
Cytogenetic analysis
HF1 cells were fixed onto glass slides for standard Giemsa staining to score total gaps and breaks or for Giemsa-banding for fragile site analysis. For metaphase FISH, GM11713 and derivative cells were treated with or without 0.6 μM APH or 0.8 μM APH for 40 h and immediately prepared for metaphase spreads. Colcemid (Gibco #15210040) was added to cell cultures at 0.05 μg/ml for 45 min. Cells were harvested and treated with 75 mM KCl hypotonic solution at 37°C for 15 min followed by fixation with 3:1 methanol-acetic acid. Fixed cells were dropped onto slides to generate metaphase spreads and prepared for Giemsa banding by serial dehydration in 70%, 85% and 100% ethanol. The FISH reaction mixture of 2 μl FHIT probe (RP11-641C17, Green 5–Fluorescein, Empire Genomics), 2 μl NLGN1 probe (RP11-148D23, Red 5-ROX, Empire Genomics), 6 μl hybridization buffer (Empire Genomics), and 5 μl Vysis IntelliFISH (08N87-001, Abbott) was applied to each slide and denatured at 75°C for 2 min. Hybridization was carried out overnight at 37°C. Post hybridization, slides were washed in 0.3% Igepal (CA-630, Sigma), 0.4× SSC at 72°C for 2 min. Slides were then washed in 0.1% Igepal, 2× SSC at room temperature for 2 min. Slides were air dried and mounted with DAPI Prolong Gold (P36935, ThermoFisher Scientific) prior to image acquisition.
Fluorescent activated cell sorting based on DNA content
GM11713 and clonal derivative cell lines were seeded at 30% confluence into four T-75 flasks each. APH (0.6 μM) media was applied to two of the four flasks. After 24 h, cells were harvested, resuspended in PBS, counted on a hemocytometer, and fixed by the dropwise addition of 3 volumes of ice cold 100% ethanol. For DNA staining, aliquots of 2–4 × 106 fixed cells were transferred to new 15 ml conical tubes, centrifuged at 200 × g for 10 min at 4°C, rinsed with PBS, and resuspended to 3 × 106 cells/ml in PBS with 50 μg/ml propidium iodide (Sigma, #P4170) and 250 μg/ml DNase-free RNase A (Sigma, #10109142001). Cells were filtered through 37 μm mesh into 5 ml round bottom polypropylene tubes (Corning, #352235) and incubated in the dark at room temperature for 30 min.
Fluorescence activated cell sorting (FACS) to determine DNA content was performed in the Michigan Flow Cytometry Core on a BD FACSAria III flow cytometer (BD) configured with PBS sheath fluid, a 100 μm nozzle, a 561 nm excitation laser and a 610/20 nm filter. Using height versus area plots, intact single cells were gated to sort ∼2.5 × 105 G0/G1, S and G2/M cell populations from each sample into 1.5 ml low-bind microfuge tubes containing 100 μl PBS. The same gating was used for all samples. Cells were held on ice until all samples were sorted and then centrifuged in a swinging bucket rotor at 400 × g for 10 min at 4°C. All but approximately 180 μl of supernatant was removed from the cell pellet. Genomic DNA was purified from the remaining cell suspension using the Quick-DNA Microprep Plus Kit (Zymo Research #D4074).
Replication timing analysis
DNA from different cell cycle fractions was quantified with the HS DNA Qubit assay (Invitrogen Q32851) and used in ddPCR and next-generation sequencing. ddPCR reactions were the same as described above for CNV detection, except that cells for replication timing analysis were not allowed a recovery period to fix unreplicated DNA into CNVs. For genomic analysis of replication timing, Illumina Tru-seq whole genome libraries were prepared by the Michigan Advanced Genomics core with only as much PCR amplification as required to allow sequencing. All libraries were multiplexed into 7.5% of an Illumina NovaSeq 6000 S4 flow cell to target ∼5 × 107 read pairs per sample in the 2 × 150 read configuration. Reads were aligned using bwa mem with default parameters (38) to a specially constructed reference genome containing the entirety of the GRCm38/mm10 mouse reference build plus hChr3 from the GRCm37/hg19 reference, which mimicked the hybrid chromosome content of GM11713.
The next steps were executed using the read depth algorithm of svtools (39). Read pairs marked by bwa as proper (SAM flag 2) with a minimum mapping quality of 5 were used to construct a coverage map for each sample (svtools map). The G1 samples from WT GM11713 and promoterless clone 1 were taken as a reference set where no DNA replication was expected to have yet occurred. Thus, they nominally reflect a copy number of 1 throughout hChr3 while also accounting for underlying copy number variation in GM11713 chromosomes. Variable-width bins were defined throughout the genome such that the G1 bins had an average count of 350 read pairs and thus an equal and sufficient statistical weight (svtools bin). The average hChr3 bin size was 63.1 kbp, sufficient to provide multiple bins across a typical CFS gene. Analyzing individual chromosomes from G1 samples revealed a copy number difference of mouse chr18 when comparing GM11713 to promoterless clone 1; these regions were discounted in further analysis, as were rare bins with a low mappability (large bin size) or estimated copy number above 20 (e.g. rDNA).
In S phase, the DNA content of early replicating regions doubles first, leading to a skew in the measured read pair counts toward early replicating bins at the expense of late replicating bins, an effect expected to resolve in late G2/M when replication is completed (40). We expressed this relative replication timing as the log2 of the read pair count in a bin for a given sample divided by the weighted mean of the counts over all bins for that sample. The input bin values were weighted by the product of the bin length and estimated bin copy number to reflect the fraction of the nuclear DNA content replicated at different cell cycle stages. We applied a 3-bin moving average to provide a small degree of data smoothing for plot clarity. A final value of 0 represents a bin that matches the genome average. Values > and <0 indicate aggregate replication earlier and later than mid-S, respectively.
To explore the impact of APH treatment, we averaged the untreated replication timing value for each bin over the WT and clone 1 samples, independently for S and G2/M, as our best estimates of the baseline replication timing. We then calculated the deviation of the relative replication timing value for each bin in the APH-treated samples relative to that baseline. We call this difference the APH-induced replication delay, where a negative value indicates that an APH-treated sample took longer to complete replication as compared to the untreated samples.
For comparison to replication timing, Bru-seq nascent transcription data from WT GM11713 was parsed to find the strand with the maximum Bru-seq RPKM in each 1 kbp bin, which identified the transcribed strand, if any. Those RPKM values were averaged across the larger replication timing bins to determine the aggregate transcription level of the latter. To discover potentially unstable large genes, we filtered the hg19 GENCODE 27 and mm10 GENCODE 15 annotations (41) for genes >750 kbp that also had a Bru-seq RPKM of at least 0.1 sustained throughout the gene body, which was empirically determined to correspond to unambiguous transcription signal above background.
RNASEH1 knockdown and overexpression
For knockdown, HF1 was transduced by the Michigan Vector Core with two pTRIPZ lentiviral human RNASEH1 short hairpin RNA (shRNA) constructs and a single scrambled shRNA control construct (Dharmacon, #RHS4743). Knockdown clones KD1 and KD2 arose from Dharmacon V2THS-32362 and V3THS-365744 shRNA constructs, respectively. For overexpression, we amplified the full-length RNASEH1 cDNA from plasmid pCMV6-AC-RNaseH1 (Origene, SC319446) using PCR primers with AgeI and MluI overhangs (Supplementary Table S1) and cloned it into the pTRIPZ lentiviral vector at the corresponding restriction sites. The RNASEH1 pTRIPZ construct was verified by Sanger sequencing prior to transduction into HF1 by the Michigan Vector Core, alongside an empty pTRIPZ control construct. In all cases, transduced cells were selected with 0.5 μg/m puromycin for 10–14 days and cloned as described above. Either shRNA expression or RNASEH1 expression was induced in the derivative HF1 clones by treatment with 100 ng/ml of doxycycline (Sigma, D9891) for 48 h.
RNASEH1 expression analysis
RNA isolation and reverse transcription from the HF1 cell lines were performed as described above for GM11713, using primer pairs specific to RNASEH1 (Supplementary Table S1). The cycling conditions were: 50°C for 10 min, 95°C for 15 min, followed by 40 cycles of 15 s at 94°C, 30 s at 50°C and 30 s at 72°C. ACTB was used as a control gene and scrambled shRNA or empty pTRIPZ samples were used as reference samples to calculate ΔΔCt values.
Protein was isolated in RIPA buffer supplemented with 1× protease inhibitor cocktail (Sigma, #11836153001), sonicated for 30 s on ice, and centrifuged at 14 000 rcf for 15 min at 4°C. The Pierce BCA Protein Assay (ThermoFisher #23225) was used to determine protein concentration of the supernatants. Total protein (40 μg) was heated at 70°C for 10 min in NuPage LDS Buffer (ThermoFisher, NP0007) and 2.5% B-mercaptoethanol and resolved on 4–12% NuPage Bis-Tris polyacrylamide gels. Proteins were transferred to PVDF membranes and incubated with 1:500 anti-RnaseH1 mouse monoclonal antibody (Sigma, WH0246243M1) or 1:1000 anti-Alpha Actinin rabbit monoclonal antibody (Cell Signaling Technology, 6487T) in 5% milk and TBST overnight at 4°C. After three 5 min TBST washes, membranes were incubated with 1:4000 anti-mouse or 1:5000 anti-rabbit secondary antibody conjugated to HRP (GElifesciences). Proteins were detected using either SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher, 34577) or Pierce ECL Western Blotting Substrate (ThermoFisher, 32209).
Microarray detection of HF1 CNVs
HF1 derivative cell lines were treated with 100 ng/ml doxycycline for a first 48 h to initiate altered RNASEH1 expression. Drug treatment then continued with either 100 ng/ml doxycycline alone or 100 ng/ml doxycycline + 0.4 μM APH for another 72 h. Cells recovered for 24 h before plating for clones. Genomic DNA was extracted from individual clones using the Blood & Cell Culture DNA Mini Kit (Qiagen, 13323). Whole genomic SNP microarray analysis was performed using the Infinium Multi-Ethnic Global-8 v1.0 Kit Array (Illumina, WG-316–1002) by the Michigan Advanced Genomics Core. CNV detection was performed using our algorithm, msvtools, which detects de novo CNVs by comparing a sample’s log-R ratio and B allele frequency to the median consensus value from all untreated samples to avoid detecting CNVs present at baseline. CNVs unique to a single subclone were confirmed by inspection of its array data alongside other samples. The significance of any difference in CNV yields between sample sets was determined using the one-sided E-test for comparing two Poisson mean rates (42), where CNV rate is the number of CNVs observed divided by the number of clones examined. HF1 CNV hotspots were determined based on all available published (5) plus newly generated CNVs, where large genes with more than five CNVs were designated as a hotspot for the purpose of summary plots. HF1 CNVs were compared to replication timing data from BJ cells (GEO GSM1335322) (43) since both HF1 and BJ are immortalized foreskin fibroblast lines.
DRIP-seq
HF1 cell lines were handled as described for CNV studies and harvested immediately at the end of the treatment period. Biological replicates were processed through two variations of DNA:RNA hybrid immunoprecipitation (DRIP) and sequencing, qDRIP (44) and DRIP-seq (45), with the following modifications; only DRIP-seq data are reported as they provided the greatest specific signal and best comparison to published studies. The Blood and Cell Culture DNA Mini kit (Qiagen, 13323) was used to prepare nuclei prior to the cell lysis and genomic DNA isolation steps. For RNase H-treated control samples, 8 μg fragmented genomic DNA was digested in 1× buffer with 80 units of RNase H (NEB, M0297S). About 8 μg of each fragmented genomic DNA sample was immunoprecipitated with 20 μg S9.6 antibody (Kerafast, ENH001) and isolated with 72 μl Protein G magnetic beads (NEB, S1430S). DNA fragments released from the beads were captured on 1.8 volumes of AMPure beads (Beckman Coulter) and eluted in 100 µl of 10 mM Tris pH8. Enrichment was evaluated on 2 μl of each eluate by qPCR reactions with primer sets from three positive loci [ACTB (46), RPL13A (45) and MYADM (47)] and two negative loci [SNRPN (45) and ZNF554 (47)] (Supplementary Table S1) using the QuantiTect SYBR Green PCR kit (Qiagen, 204143) on an Applied Biosystems 7500 Real-Time PCR system with an initial denaturation at 95°C for 15 min, followed by 40 cycles of 94°C for 15 s and 60°C for 60 s. DRIP-seq eluates were sonicated to 150 bp average fragment size and converted into DNA libraries by the Michigan Advanced Genomics Core following by sequencing on an Illumina NovaSeq 6000 in the 2 × 150 read format to yield 41M to 68M read pairs per sample.
In parallel, we obtained FASTQ files from published DRIP-seq and associated data sets (29,30,48). They were chosen because we had available Bru-seq (37) or Gro-seq (49) nascent transcription data and because they included APH treated cells or RNAaseH1 overexpression, had previously suggested a role for R-loops in CFS expression, had a euploid karyotype (IMR90), or examined the U2OS cell line commonly used to study R-loops and CFSs.
DRIP-seq reads were subjected to adapter trimming, quality filtering and pair merging using fastp (50) and aligned using bwa mem (38) to the hg38 or mm10 reference genome with default parameters. Custom scripts purged duplicate molecules with identical paired alignment endpoints or single-ended reads with identical alignment positions and strands as relevant for the data set. Unique molecules (mapping quality >10) were queried for genomic R-loop enrichment by two parallel processes to support robust conclusions. First, we counted fractional mapped reads in 100 bp genomic bins based on the proportion of the source molecule that overlapped each bin. Subsequent analyses only utilized high quality bins that were not on the list of ENCODE problematic regions (51) and had a mappability of at least 0.9 (52) and a copy number >0, where reference copy numbers were derived from sex-specific chromosome numbers for euploid cell types and from the DepMap Project for U2OS cells (53). Counts were normalized to regional copy numbers to obtain mapped reads per allele. In our second approach, epic2 (54), a fast re-implementation of SICER (55), with default parameters (including a bin size of 200 and a false discovery rate of 0.05) was used to call diffuse enrichment peaks based on aligned reads in a test relative to a matched control sample (typically either an input or RNaseH pre-treated sample).
We used nascent transcription data to help analyze DRIP-seq signals based on the logic that only transcribed portions of the genome can show true R-loops. Bins, peaks or genes were considered untranscribed if their aggregate segmented Bru-seq RPKM (37) was <0.01 and transcribed if >0.1 (thresholds were 0.0025 and 0.025 for Gro-seq due to its distinct distribution of reads that includes pause sites). Regions between these thresholds were ambiguous with respect to transcription and not used for fitting or plotting. For our first approach, we used only untranscribed bins to fit a quadratic to the relationship between summed DRIP-seq read counts and bin GC content; such plots revealed the expected association of higher DRIP-seq signals with both transcription and higher GC content, which stabilizes R loops. Residual bin counts relative to this fit were normalized to the total molecule counts for each sample to yield adjusted reads per kb per million read (RPKM) units to support inter-sample comparisons (values can be negative due to the fit adjustment). For quality assessment, we plotted adjusted DRIP-seq RPKM for bins flanking transcription start sites (TSSs) of transcribed genes and over the first 100 kb of all relevant genes, similar to published studies (44).
Bins and enrichment peaks were compared to gene spans using bedtools intersect (56) to determine their overlap. For our first approach we calculated and plotted adjusted DRIP-seq RPKM, or the difference of this value between reference and control samples, as a function of either aggregate gene transcription RPKM (all genes) or gene length (transcribed genes only). For our second approach, analogous plots expressed each gene’s DRIP-seq enrichment as the fraction of its bases contained in epic2 peaks. The latter approach was independent of our bin normalization method but yielded similar conclusions for all samples. We finally calculated the aggregate DRIP-seq RPKM or fraction of bases within peaks for the untranscribed intergenic regions of the genome as a further reference for the R-loop signal expected at baseline, shown as red horizontal lines in figures.
RESULTS
Creation of mutant cell clones lacking the human FHIT promoter
We used the GM11713 mouse-human somatic hybrid cell line, which contains a single copy of human chromosome 3 (hChr3) tagged with a neomycin resistance gene (57). hChr3 contains the FHIT gene at 3p14.2, which encompasses the well-characterized FRA3B CFS, as well as the large NLGN1 gene at 3q26.3. GM11713 provided the first demonstration of replication stress-induced CNV formation at FHIT, NLGN1, and other loci (58). GM11713 only contains one copy of the human FHIT promoter to mutate and deletion CNVs on hChr3 are easy to validate as copy number zero events.
We used CRISPR-Cas9 technology to delete the FHIT promoter in GM11713 (Figure 1A). Two sites flanking the RefSeq-annotated FHIT transcription start site (TSS) were targeted with sgRNAs. The resulting deletion also removed CpG islands, sites with the H3K27ac mark, and transcription factor-binding sites, which characterize mammalian gene promoters (Figure 1A) (59,60). We reasoned that such a deletion would prevent FHIT transcription initiation while preserving the DNA corresponding to the FHIT deletion hotspot, which is located ∼500 kb away in the center of the gene body. Two promoterless deletion clones, identified as clone 1 and clone 2, were confirmed using PCR (Figure 1B). We further confirmed that the two clones had no gross internal deletions within FHIT by validating positive results with PCR assays targeting 7 exons and 1 intron throughout the gene body (Figure 1C). The clones had different breakpoint junctions and were thus independent derivatives of GM11713 (Supplementary Figure S1).
Figure 1.

FHIT promoter deletion in GM11713 cells. (A) UCSC Genome Browser screenshot showing the GRCh38/hg38 genomic region surrounding the targeted FHIT promoter deletion (shaded). Triangles mark the locations of two CRISPR-Cas9 guide RNAs. Tracks show CpG islands, layered ENCODE data histone marks, and a subset of the ORegAnno transcription factor binding sites (79). (B) Agarose gel showing PCR amplicons generated by the primers in (A), demonstrating the intended promoter deletion in two independently derived cell clones. See Supplementary Figure S1 for allelic details for each clone. (C) Triangles represent PCR assays used to validate the absence of unexpected internal deletions in the FHIT gene in clones 1 and 2. +/- symbols indicate whether the PCR assay yielded a product with WT GM11713 or the two promoterless clones.
Gene-specific knockdown of FHIT transcription
To confirm that FHIT transcription was selectively silenced in the two promoterless clones, we first performed qRT-PCR with three primer sets targeting different parts of the mature FHIT mRNA to rule out transcription from an alternate TSS. There was no significantly detectable FHIT transcription in either clone with any of the qRT-PCR assays as compared to clear evidence of transcription in WT GM11713 (Figure 2A). We also performed Bru-seq nascent RNA sequencing, which reveals the genomic location of RNA polymerase molecules incorporating ribonucleotides during a labeling window and thus all spans of active transcription (37). Neither promoter deletion mutant showed measurable transcription of any FHIT isoform (Figure 2B) as compared to contiguous transcription throughout the gene body in WT. Because GM11713 also carries mouse chromosomes that might confound transcription measurements, we sequenced our qRT-PCR products and separately aligned Bru-seq reads to the human (hg38) and mouse (mm10) reference genomes to rule out cross reactivity with murine Fhit. These steps confirmed the specificity of our assays for human FHIT and revealed that GM11713 does not express murine Fhit (Supplementary Figure S2). We conclude that clones 1 and 2 achieved complete and selective silencing of FHIT expression from hChr3 in GM11713 cells, despite an intact gene body.
Figure 2.
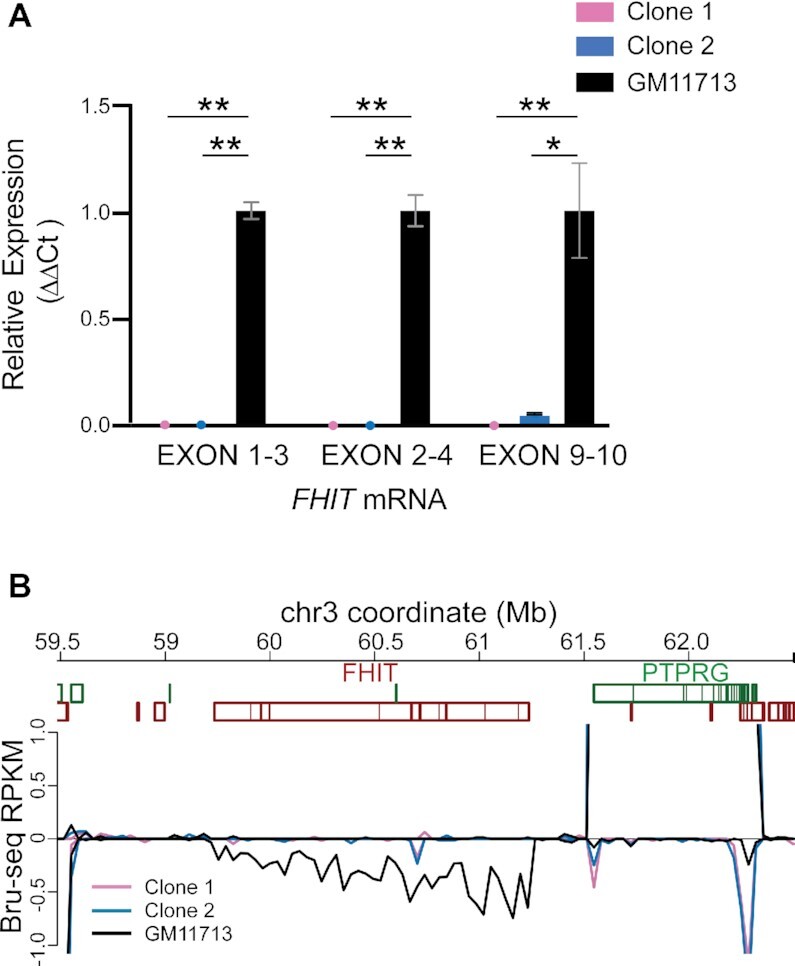
Promoterless mutants do not express FHIT. (A) Promoterless clones 1 and 2 showed almost no FHIT transcription relative to wild-type GM11713, as measured by qRT-PCR assays targeting three different exon–exon junctions of the mature FHIT mRNA. Statistical analyses were performed using a Student’s T-test. *, P ≤ 0.05; **, P ≤ 0.005. (B) Bru-seq nascent RNA transcription data are plotted for wild-type GM11713 and the two promoterless derivatives in and around FHIT, showing a selective loss of only FHIT expression only in the mutant clones. Negative RPKM values reflect transcription in the reverse direction, which matches the orientation of the FHIT gene (top line gene boxes, forward orientation; bottom line gene boxes, reverse orientation).
FHIT deletion CNVs in cell populations depend on transcription
We first used ddPCR to measure the frequency of replication stress-induced FHIT deletions in populations of GM11713 and derivative cells. We previously observed very high rates of FHIT deletion formation in APH-treated GM11713 that exceeded 20% (58), consistent with later observations that hotspot CNV formation is strongly biased to deletions with a peak occurrence in the centers of large transcription units (5). We therefore designed a test probe set within the CNV hotspot at FHIT exon 5, very near the center of the gene, and a reference probe set in a non-CNV hotspot region on mouse chromosome X (mChrX), which we determined from whole genome sequencing to be present at a single copy in GM11713 (Supplementary Figure S3). By comparing the ddPCR signal ratio at the test versus the reference loci, and assuming that deletions at the reference locus are rare, we could infer the frequency of deletions at FHIT exon 5 if it exceeded Poisson error limits.
Clones 1 and 2 and WT GM11713 were treated with 0.6 μM APH for 72 h followed by a 24-h period of cell outgrowth without APH in order to fix CNV mutations into the genome. We first noted that untreated GM11713 and its promoterless derivatives showed a FHIT exon 5 ddPCR test-to-reference ratio that was indistinguishable from 1, which demonstrated that hChr3 and the FHIT gene were stable in the absence of replication stress (Figure 3A). In contrast, APH-treated GM11713 showed a reduction in the test-to-reference ratio consistent with 19.5% of cells harboring a FHIT exon 5 deletion (Figure 3A), similar to the 23.3% of APH-treated clones that carried an exon 5 deletion in prior studies based on clonal PCR (58). Unlike WT GM11713, neither of the FHIT promoter-deletion lines showed a significant reduction in FHIT signal upon APH treatment (Figure 3A), consistent with the hypothesis that transcription is necessary for FHIT CNV formation during replication stress. Because loss of FHIT transcription might alter the physical distribution of CNVs away from the center of the gene, we used additional test probes that targeted the 5′ and 3′ ends of FHIT. Neither GM11713 nor the promoterless mutants showed a significant signal reduction with either of these probes, with or without APH treatment (Supplementary Figure S4).
Figure 3.
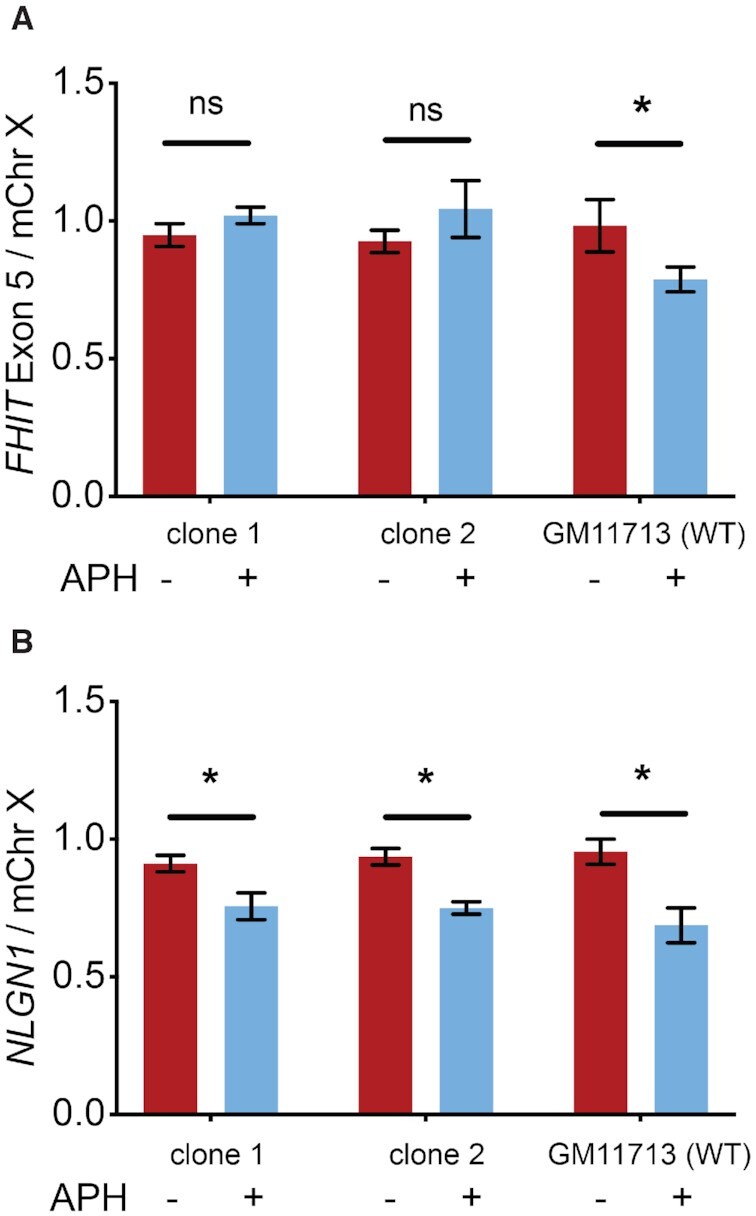
Transcription knockdown at FHIT reduces its instability at a population level. (A) ddPCR results show that, unlike APH-treated WT GM11713, APH-treated promoterless clones 1 and 2 did not have significantly more deletion CNVs near exon 5 of FHIT as compared to untreated controls. (B) As a control, promoterless clones 1 and 2 and WT GM11713 all showed significant APH-induced deletion CNVs at exon 4 of the unmanipulated NLGN1 gene, as compared to their untreated controls. Throughout, the plotted test to reference locus ratio represents the average of three biological replicates and error bars are the standard deviation. Statistical analyses were performed using a Student’s T-test. ns, P > 0.05; *, P ≤ 0.05.
To establish a positive control locus that would reveal the specificity of the observed CNV abrogation effect at FHIT, we designed an additional ddPCR test probe set targeted to exon 4 of NLGN1 at 3q26.21, which we previously showed is a CNV hotspot in GM11713 (2). The CRISPR-mediated ablation of the FHIT promoter did not prevent NLGN1 transcription, as expected (Supplementary Figure S5). Consistently, APH-induced deletion CNVs formed at NLGN1 at similar levels in both WT and FHIT-promoterless cells, demonstrating that the lack of induced CNVs in clones 1 and 2 was specific to FHIT (Figure 3B).
Cell clones confirm suppression of deletion CNVs in FHIT after promoter ablation
To further examine the frequency and physical distribution of FHIT CNVs, clones 1, 2 and WT GM11713 were again treated with 0.6 μM APH for 72 h to induce deletion CNVs. Individual cell subclones were isolated from each treatment group, expanded and screened for FHIT deletions using PCR assays distributed throughout FHIT and surrounding genomic regions with a higher probe density within the hotspot region surrounding exon 5 (Figure 4A). Since human FHIT is present at a single copy in GM11713 as validated by whole genome sequencing (see Methods), deletion CNVs were evident as absence of a PCR product. No FHIT deletions were detected in any untreated subclones (Figure 4B). After APH-treatment, 9/32 (28.1%) of WT GM11713 clones had a deletion within FHIT, consistent with the ddPCR results above and previous studies (58). Most were localized near the center of FHIT, consistent with the CNV pattern of many large genes, whereas two deletions spanned at least 1.5 Mbp distal to FHIT (Figure 4A). In contrast, no APH-induced FHIT deletions were detected in cells derived from FHIT promoter deletion clone 1 (0/18) or clone 2 (0/16) (Figure 4B), a significant reduction compared to WT (P < 0.0181 and P < 0.0204, respectively). Similar to the ddPCR results, a clonal PCR analysis analogous to that in Figure 4A confirmed the presence of APH-induced deletion formation at NLGN1 in both WT and FHIT promoterless clones (Figure 4B). Thus, distinct assays and multiple experiments confirm a selective suppression of APH-induced CNV formation at FHIT caused by ablation of that gene’s promoter.
Figure 4.

Transcription knockdown at FHIT reduces its instability at the clonal level. (A) Diagram showing the distribution of deletion CNVs detected in WT clones by PCR (no CNVs were detected in the mutant clones). Triangles show the location of PCR assays, vertical lines represent exons in FHIT, horizontal lines represent individual deletion CNVs, and the gray box shows the annotated FHIT gene. Deletion CNV ends on the diagram denote the locations of the last PCR assays that failed to yield a product. (B) Tables summarize the number of cell clones that either did or did not show a deletion CNV in FHIT (top) or NLGN1 (bottom), in APH-treated or untreated control samples. Statistical analyses between each mutant and WT were performed with Fisher's exact test. *, P< 0.05.
FRA3B expression depends on FHIT transcription
We further analyzed APH-induced CFS gaps and breaks at FHIT and NLGN1 in the WT and mutant GM11713 clones. FRA3B and FRA3O correspond to the FHIT and NLGN1 gene loci, respectively (61). Locus-specific FISH probes were used to facilitate the CFS analysis (Supplementary Figure S6). As previously shown and consistent with CNV data above, APH-induced CFS expression was readily observed at both FRA3B and FRA3O in GM11713 (Table 1). Transcriptional silencing of FHIT in the promoterless clones led to a near absence of CFS expression at FRA3B, confirming the parallel dependence of both CFS expression and deletion CNV formation on transcription (Table 1). In contrast, the positive control locus at FRA3O showed equivalent CFS expression regardless of transcription at the distant FHIT gene (Table 1).
Table 1.
Transcription is required for CFS expression at FHIT in a gene-specific manner
| # of gaps and breaks/cells scored (%) | ||
|---|---|---|
| Cells | FRA3B (FHIT) | FRA3O (NLGN1) |
| WT GM11713 + APH | 16/50 (32%) | 8/50 (16%) |
| KO Clone 1 + APH | 1/50 (2%) ** | 13/50 (26%) |
| KO Clone 2 + APH | 0/50 (0%) ** | 10/38 (26%) |
WT GM11713 and its promoterless derivatives were subjected to FISH-based CFS analysis at FRA3B and FRA3O. Statistical analysis was by Fisher’s exact test, comparing each clone to WT; **, P < 0.0001. Fifty cells from each cell type without APH treatment were also scored and none showed gaps or breaks at these loci.
Replication delay at the FRA3B CNV hotspot requires FHIT transcription
We next explored a potential mechanistic basis for the role of transcription in deletion formation at the FHIT CNV hotspot. We performed a replication timing analysis of untreated and APH-treated cells that were purified by FACS to be in the G1, mid-S or G2/M phases of the cell cycle (Figure 5A and B). We first used ddPCR to compare DNA content at a position in the center of the FHIT gene in intron 5 relative to a reference position just inside the 5′ portion of the gene in intron 1 because the centers of large genes typically replicate later (5). As expected, relative DNA content in the center of FHIT fell below 1 in S phase in both clones 1 and 2 because fewer cells had completed replication there relative to the 5′ end (Figure 5C). In untreated cells and APH-treated cells with a FHIT promoter deletion, the DNA content ratio returned to nearly 1 in the G2/M fraction as replication completed (Figure 5C). Only in APH-treated WT GM11713 with active FHIT transcription did we observe that DNA content remained low in G2/M, suggesting transcription-dependent replication failure (Figure 5C).
Figure 5.

APH-induced replication delay at selected large genes depends on transcription. (A and B) DNA content distribution of (A) untreated cells or (B) cells treated with APH, illustrating the gating that led to the G1, S and G2/M fractions. (C) Plot showing the relative replication timing of the center of the FHIT gene relative to a position near its 5′ end in both Clones, 1 and 2, as measured by ddPCR. The gene center replicates later, but fails to recover by M in WT cells treated with APH. (D) Correlation plot between genome-wide GM11713 replication timing in S (x-axis) and the replication delay induced by APH in S (y-axis), showing that the bins with the largest APH-induced replication delay (negative y-axis values) were later replicating without APH (negative x-axis values). (E) Similar to (D), showing residual replication delay still present in M phase (y-axis), where known hotspot genes FHIT and NLGN1 are the most prone to fail recovery by M. In (D) and (E), colored dots are bins in the central 50% of the indicated large genes (red, FHIT; blue, NLGN1; green, ZBTB20; magenta, ERC2), gray dots are all other hChr3 bins. (F) Detailed plot of bins surrounding NLGN1, showing replication timing for the indicated samples in S (top panel) and G2/M (bottom panel). The difference between APH and non-APH pairs is referred to as replication delay. (G) Similar to (F) for FHIT; note the different M-phase pattern for WT GM11713 and promoterless clone 1. Trace colors in (F) and (G) are: blue, WT; cyan, WT + APH; red, Clone 1; gold, Clone 1 + APH.
We next performed quantitative whole genome sequencing of similar cell cycle fractions for clone 1. The unreplicated DNA in G1 served as a reference for calculating the relative abundance of chromosome regions in S and G2/M, and thus of inferring the relative replication timing of different genomic bins (see Materials and Methods section and Supplementary Figures S7 and S8A) (40). We first noted a subtle but reproducible delay in replication in the genomic region near our FHIT promoter deletion, which is consistent with the well described association of replication origins with active gene promoters (Figure 5G) (14,62).
The genome-wide replication timing pattern for hChr3 reflected the well-described trends of mammalian chromosomes, including earlier replication of the most highly transcribed genome regions and later replication of the FHIT and NLGN1 hotspot genes as compared to other genes with similar transcription levels (Supplementary Figure S8B) (5,62). APH treatment accentuated this pattern, such that the replication delay induced by APH was directly correlated with late basal replication timing in S phase cells (Figure 5D, gray dots). The pattern was substantially but incompletely rectified by G2/M (Figure 5E, gray dots). We interpret these data to reflect that many late-replicating genome bins had not yet fully recovered from the effects of APH, given that our G2/M fraction contained some cells that were still finishing replication. The majority of such bins are within untranscribed genomic regions not known to be unstable (e.g. Supplementary Figure S9). However, CNV hotspots showed an especially strong APH-induced replication delay even when they had basal replication timing in mid-S (Figure 5D–E, red and blue dots). Detailed views of hotspot genes in S and G2/M confirmed that their replication delay occurred mainly in the centers of the genes and was very prolonged, including the control NLGN1 gene in all cell lines, and, even more strikingly, FHIT in WT cells (Figure 5F–G). In contrast, the FHIT gene in the promoterless clone showed a complete resolution of the APH-induced replication delay by M phase, even though it had a discernable effect in S (Figure 5G). Only bins in FHIT showed this altered replication delay in response to the FHIT promoter deletion (Supplementary Figure S8C). Thus, APH induces a disproportionate and prolonged replication delay in hotspot genes that correlates with position in the gene and is demonstrated at FHIT to be dependent on transcription.
Transcription is not sufficient to confer a prolonged replication delay at large genes
We further identified two additional genes on hChr3 that were transcribed as isoforms >750 kbp based on Bru-seq data: ZBTB20 and ERC2. Detailed examination revealed a pattern in which ZBTB20 mirrored the behavior of FHIT and NLGN1, nominating it as a candidate CNV hotspot in GM11713 (Figure 5D,E, green dots and Figure 6A), whereas ERC2 was an exception to this rule. ERC2 is large, transcribed, and showed an APH-induced replication delay in S-phase (Figure 6B, top panel). However, unlike the other genes, the ERC2 replication delay was resolved by G2/M (Figure 5D,E, purple dots and Figure 6B, bottom panel). Both the size and transcription level of ERC2 were between those of FHIT and NLGN1, so these properties cannot account for the distinct behavior of ERC2 (Table 2 and Supplementary Figure S10). ERC2 did have the earliest unstressed replication timing of the four genes, consistent with the idea that the recovery time available in S phase might be an important factor in determining instability (16). To add evidence on this point, we mined the GM11713 mouse chromosomes using the same gene size and transcription criteria and identified six genes with properties that might predict instability (Table 2). Detailed examination again revealed that some genes showed a prolonged M-phase replication delay while others did not (Table 2 and Figure 6C and D; Supplementary Figure S11). We observed a continuing trend toward later baseline replication timing in the genes that failed to recover from APH (correlation coefficient 0.89) a statistically significant result when comparing genes clustered by k-means into two groups based on replication delay (P = 0.026, Student’s t-test). A caveat is that all late replicating genomic regions showed the greatest tendency toward an APH replication delay (Figure 5D,E). Also, some mouse genes showed baseline copy number alterations, which might confound replication timing assessments and improve the stability of the deleted alleles but also provides evidence for their prior instability (Supplementary Figure S12).
Figure 6.

Large gene transcription is not sufficient to confer failed recovery of an APH-induced replication delay. Composite plots at four large genes show Bru-seq signal corresponding to nascent transcription (top panels) as well as relative replication timing in S and G2/M fractions (middle and bottom panels, respectively). (A) Plot illustrating delayed replication recovery from APH at transcribed gene ZBTB20, similar to NLGN1 and FHIT (Figure 5F,G). (B) In contrast, gene ERC2 showed a delay in S that was resolved by M, despite its active transcription. (C and D) are similar to (A) and (B) in showing mouse genes that do (Immp2l, C) and do not (Pard3b, D) manifest a prolonged APH-induced replication delay in M phase even though both are transcribed and show an APH delay in S. Trace colors in all panels are: blue, WT; cyan, WT + APH; red, Clone 1; gold, Clone 1 + APH.
Table 2.
Properties of candidate hotspot genes in GM11713
| Chrom | Gene | APH Rep. Delay in G2/M | Rep. Timing in S (no APH) | Size | Bru-Seq RPKM |
|---|---|---|---|---|---|
| hChr3 | FHIT | -0.356 | -0.063 | 1.5 Mbp | 0.332 |
| mChr12 | Immp2l | -0.285 | -0.156 | 931 kbp | 0.370 |
| hChr3 | NLGN1 | -0.261 | -0.261 | 888 kbp | 1.743 |
| hChr3 | ZBTB20 | -0.230 | -0.102 | 833 kbp | 4.902 |
| mChr14 | Gpc6 | -0.085 | -0.023 | 1.1 Mbp | 1.472 |
| mChr8 | Wwox | -0.073 | -0.040 | 913 kbp | 0.197 |
| mChr13 | Sugct | -0.060 | -0.019 | 837 kbp | 1.143 |
| hChr3 | ERC2 | -0.030 | 0.017 | 960 kbp | 0.744 |
| mChr14 | Lrmda | -0.025 | 0.085 | 1.0 Mbp | 0.264 |
| mChr1 | Pard3b | 0.006 | 0.054 | 1.0 Mbp | 0.873 |
| correlation coefficient | 0.89 | 0.04 | -0.14 | ||
The table includes all genes with an active GM11713 transcription unit of at least 750 kbp on human (h) and mouse (m) chromosomes. Correlation coefficients are for APH-induced replication delay in G2/M relative to basal replication timing in S, gene size in bp and Bru-seq transcription RPKM. The table is sorted by decreasing replication delay.
To correlate the replication timing patterns to CNV formation, we performed population-level CNV analysis by ddPCR at human ZBTB20 and ERC2 genes, similar to that described above for FHIT and NLGN1 (Figure 3). Test ddPCR assays targeted the middle of the large genes and reference assays targeted their 5′ or 3′ ends just beyond the coding sequences (Supplementary Table S1). To enhance precision, multiple parallel reactions were performed on each sample to assess up to 1.4 × 105 droplets on average. We did not detect significant deletion formation at either ZBTB20 or ERC2 upon APH treatment (Supplementary Figure S13A). We note that the ddPCR assay sensitivity is approximately 5 to 10% of cells with CNVs, as determined by Poisson sampling limits, and it is possible that one or both genes showed CNV formation below that level. Repeating this experiment with ddPCR assays for the six mouse genes with properties that might predict instability (Table 2) revealed that Immp2l incurred a high frequency of deletion CNV formation that paralleled its strong APH-dependent replication delay, whereas genes Lrmda, Sugct, Gpc6, Wwox and Pard3b did not (Supplementary Figure S13B). Collectively, these results indicate that large, transcribed genes show different levels of CNV susceptibility when exposed to replication stress.
Manipulating RNase H1 expression does not alter CNV formation rates
Another potential explanation for transcription-dependent instability of large genes invokes R-loop formation that conflicts with replication progression (18,63,64). We addressed this hypothesis by an experimental approach we previously used to establish many properties of CNV hotspots (2–5). We both knocked down and overexpressed RNase H1 in HF1 fibroblasts, an euploid TERT-immortalized male human fibroblast cell line, and monitored CNV formation by applying genomic microarray analysis to cell clones isolated after APH treatment. RNase H1 cleaves RNA in RNA:DNA hybrids to resolves R-loops; its manipulation is a common tool for changing cellular R-loop levels (18,28). Using lentiviral transduction, we established two independent HF1 clones expressing a doxycycline-inducible shRNA directed against RNASEH1 (KD1 and KD2), as well as two independent clones with doxycycline-inducible expression of RNASEH1 (OE1 and OE2). A scrambled shRNA and empty expression vector served as negative controls, respectively.
Altered RNase H1 levels were induced with doxycycline for 48 hours followed by treatment with 0.4 μM APH or DMSO for 72 h and a 24-h recovery period to allow CNV formation. We achieved doxycycline-inducible decreases and increases in RNase H1 expression in HF1 at both the RNA and protein levels throughout the experimental window (Figure 7A–C,E–G). However, neither manipulation led to a significant change in genome-wide CNV formation induced by treatment with APH (Figure 7D,H). CNVs were induced by APH as expected (Supplementary Table S2), but neither the rate nor the type of induced CNVs changed upon manipulation of RNase H1 levels (Supplementary Tables S3 and S4). There was a small but statistically significant decrease of baseline (i.e. untreated) CNV levels in the cells that overexpressed RNase H1 as compared to control (Figure 7F; P < 0.0187), suggesting that R-loops might contribute to CNV formation in the absence of added replication stress. We further monitored the effect of RNase H1 manipulation cytogenetically and did not observe significant differences between controls and either knockdown or overexpression cells in total gaps and breaks or between controls and knockdown cells in gaps and breaks at specific fragile sites (Table 3 and Supplementary Table S5).
Figure 7.

Altered RNase H1 expression does not change genome-wide APH-induced CNV frequencies in HF1 fibroblasts. (A) RNASEH1 RNA levels were determined by qRT-PCR for HF1 fibroblast cell clones carrying shRNA constructs that were either scrambled sequence controls or specific to RNASEH1. Expression decreased for each of two independent knockdown clones upon shRNA induction with 48 h of doxycycline. Results are the fold change relative to the untreated scrambled control and are the mean ± standard deviation of triplicate measurements. (B) RNASEH1 RNA knockdown persists through 48 h of doxycycline treatment followed by 72 h of APH treatment. (A) and (B) are from different experiments. (C) Western blots of the cell clones in (A) revealed parallel changes in RNASEH1 protein levels. Alpha-tubulin and actinin served as loading controls for experiments conducted at different times. (D) Genome-wide CNV formation was determined by microarray analysis of two cell clones expanded from RNASEH1 knockdown populations ± APH treatment, all treated with doxycycline. Results are expressed as the mean ± 95% confidence interval of the CNV rate (the number of de novo CNVs divided by the number of cell clones examined). (E–G) similar to (A), (B) and (C), showing increased RNase H1 RNA and protein levels for two independent clones that overexpressed RNase H1 in response to doxycycline, as compared to an empty expression vector. (H) similar to (D), showing CNV analysis for two clones with RNase H1 overexpression.
Table 3.
RNase H1 knockdown and overexpression do not alter chromosome breakage
| Sample | APH | Colchicine | Staining | # breaks / cells scored (rate) |
|---|---|---|---|---|
| Empty | 36 h | 45 min | solid | 74/50 (1.48) |
| OE2 | 36 h | 45 min | solid | 72/50 (1.44) |
| Scrambled | 36 h | 3 h | G-band | 38/50 (0.76) |
| KD2 | 36 h | 3 h | G-band | 36/50 (0.72) |
| KD2 | - | 3 h | G-band | 0/50 (0.00) |
Chromosome breaks were scored in HF1 cell lines with no observed difference between RNase H1 overexpression (OE) and empty vector or between knockdown (KD) and scrambled shRNA control. Method details are provided because they impact the sensitivity of break detection in different experiments.
HF1 data reinforced that large gene transcription was not sufficient to predict high CNV burden and hotspot formation, even though prior and current findings indicate that it is necessary (5). Three large genes, NEGR1, PRKG1 and MAGI2, had unusually high CNV burdens over all available data whereas many other genes, including ZBTB20, had at least 10-fold fewer CNVs despite similar gene size and transcription (Supplementary Table S6 and Figure S14A–C). As previously observed, HF1 deletion CNVs accumulated in the centers of large transcription units (Supplementary Figure S14D).
CNV hotspots and large genes have low internal R-loop burdens per unit length
To explore reasons for our RNase H1 results given prior literature (18,29,30,65–67), we performed DRIP-seq with the R-loop-specific S9.6 antibody to understand the genomic distribution of R-loops in HF1 cells and how it changed with RNaseH1 manipulation and APH treatment. We emphasized approaches where DRIP-seq signals were analyzed relative to nascent RNA sequencing data (see Materials and Methods), since R-loops should only form in transcribed DNA. Our primary approach based on 100 bp genomic bins validated that high HF1 DRIP-seq signals were associated with transcribed genes and higher GC content (Figure 8A and Supplementary Figure S15A–E), and a peak of R-loop signal at gene TSSs (Supplementary Figure S15F). Examining gene DRIP-seq signals as a function of gene size revealed that high inferred R-loop burdens were consistently associated with smaller genes; the largest genes, including each of 5 HF1 CNV hotspot genes (Supplementary Table S6), all showed a low R-loop burden when aggregated across the body of the gene that was only slightly above the signal level seen in the untranscribed genome even when zoomed in (Figure 8B and Supplementary Figure S16). The reason for this pattern was evident in the observation that R-loop signal was strongest at the 5′ ends of genes, decreasing to baseline at further distances from the TSS (Figure 8C) as seen by others (44). Larger genes have a large proportion of their span that is distant from the TSS and unlikely to have abundant R loops per unit length. Browser views of the PRKG1 hotspot illustrate promoter-associated R-loops with limited intragenic DRIP-seq signal (Supplementary Figure S17).
Figure 8.

Large transcribed genes are associated with low R-loop burden per unit length. (A) DRIP-seq signal expressed as adjusted RPKM (see Materials and Methods section) in HF1 cells bearing the empty vector (EV) as a function of Bru-seq transcription RPKM. One point is shown for each gene >10 kb in size. (B) DRIP-seq signal for HF1 EV cells as a function of gene length. Points are the same as (A) except now only including transcribed genes (Bru-seq RPKM > 0.1). (C) DRIP-seq signal expressed as the median value over bins from all transcribed genes as a function of distance from the TSS (fewer genes contribute to the median further from the TSS). (D) and (E) Similar to (B), now showing excess DRIP-seq signal as the difference between HF1 cells with RNase H1 knockdown (shRH1) relative to scrambled control (shCtrl) or RNase H1 overexpression (RH1 OE) relative to EV, respectively. (F) Excess DRIP-seq signal for APH-treated HF1 EV cells relative to untreated DMSO control. In all plots the red line denotes the aggregate y-axis value for untranscribed intergenic bins. The five most intense HF1 CNV hotspots are indicated with colored symbols and blue lines trace the median y-axis value. All plots use the same y-axis scale. (G) Replication associated phenomena at large transcribed genes. Gradient-colored bars depict where within large genes the indicated phenomena are most prominent based on data from this and other works. See text for discussion.
RNase H1 knockdown had the expected effect of increasing genomic R-loop signal as compared to either a scrambled shRNA control or an RNaseH pre-treated library (Figure 8D and Supplementary Figure S16B). The effect of RNase H1 overexpression was subtler and more variable, but most typically revealed a small decrease in R-loop burden in small transcribed genes (Figure 8E and Supplementary Figure S16C). These genetic effects were again only evident in smaller genes; even RNase H1 knockdown cells with increased R-loop levels showed minimal R-loop signal integrated across the body of large genes. Low-dose APH treatment did not change these conclusions such that large transcribed genes retained low baseline DRIP-seq signal in APH-treated HF1 cell while APH mostly tended to decrease R-loop levels at small genes in RNase H1 knockdown cells (Figure 8F and Supplementary Figure S18).
To validate these findings, we first re-analyzed our HF1 DRIP-seq data using an independent computational approach based on enrichment peak calling, which confirmed that only low baseline fractions of the largest transcribed genes corresponded to called peaks, even with RNase H1 knockdown (Supplementary Figure S19). We further re-analyzed DRIP-seq data from published studies (Supplementary Table S7), including two recent papers that reported RTEL1-dependent genomic R-loop suppression at fragile sites and other loci (29,30). The quality of these data sets varied but none showed a high R-loop burden in large transcribed genes as compared to either smaller genes or to the untranscribed genome (Supplementary Figure S20).
DISCUSSION
CFS expression and CNV formation are distinct manifestations of replication stress that have been linked to the transcription of large gene isoforms (5,16,18), but the relationship between these risk factors and outcomes is not established. Previous investigations focused on CFS expression in cell types with different transcription patterns (5,18). We addressed the limitations that arise when comparing different cell lines by ablating transcription of a large gene in isogenic clonal cell lines, similar to recent reports (16,31), and applied those tools to the direct study of CNV mutations. Small deletions of the FHIT promoter on hChr3 were highly effective at ablating just that gene’s transcription while preserving the gene body with its CFS and CNV hotspot. Two independent promoterless cell clones showed no detectable APH-induced deletion formation at the center of FHIT, in contrast to unedited WT cells. APH did induce CNVs and CFS expression in the mutant cell lines at a large CNV hotspot control locus in NLGN1, also on hChr3, demonstrating that the increased genomic stability was specific to FHIT. These results demonstrate that the FHIT promoter confers a mutagenic property on DNA ∼500 kbp distant from itself that nevertheless appears to be confined within the FHIT gene. This pattern is most consistent with transcription being the causal factor in the instability because a moving RNA polymerase provides an explicit mechanistic connection between the promoter and the gene body (Figure 8G).
Our data provide the most direct evidence to date that large gene transcription is required for APH-induced CNV hotspot formation at those loci and support studies showing its requirement for CFS expression (5,16,18,26,31), although that observation by itself does not explain the mechanistic link to replication. It was possible that the FHIT promoter acts by altering a local replication program, given that early replication timing is linked to gene transcription and that cis elements can control domain replication timing (62,68). However, replication timing analysis validated that large genes usually replicate in the latter half of S phase even when they are transcribed (5) and that the basal replication program around FHIT was not strongly altered by deletion of its promoter except for a possible slight replication delay in the region immediately surrounding the deleted promoter. In contrast, active transcription in WT cells correlated with an APH-induced replication delay at both FHIT and NLGN1. This effect was specifically reversed in the promoterless cells at FHIT but not NLGN1. The transcription-dependent replication delay peaked in the centers of the genes, mirroring the described spans of CNV hotspots (Figure 8G) (5), and was very prolonged, persisting into G2/M cell fractions. Thus, silencing FHIT transcription fundamentally altered its intragenic replication properties in the face of replication stress, suggesting that the main role of transcription in large gene instability is to impede replication recovery.
We proposed a model called Transcription-Dependent Double-Fork Failure (TrDoFF) to explain the relationship between transcription, replication, CFS expression and CNV formation (5). It proposes that pre-replication complexes deposited in G1 within large genes are rendered inactive by their ongoing transcription (21–24,69), leading to unusually large replication domains that are exponentially more prone to failure of both of the converging forks moving inward from the gene flanks (70). Ineffective dormant origin firing between those failed forks leads to unreplicated DNA that persists into G2/M. Here, we explicitly demonstrated the requisite transcription-dependent APH replication delay for FHIT instability, a finding supported by the observation of ‘significantly delayed regions’ under replication stress (26), a study that correlated APH replication delays to topologically associated domains (TADs) (71), and recent EdU-seq analyses (69,72).
Importantly, large gene transcription proved necessary but not sufficient for a prolonged APH-induced replication delay and associated CNV hotspot formation, at least within the recognized sensitivity limits of our ddPCR assay. Previously, we predicted CNV formation in the DAB1 gene in HF1 cells based only on nascent transcription data (5), but that was a single trial and the gene did not prove to be an intense CNV hotspot. Findings here and in other recent works expose the importance of basal replication timing in determining how much of an impact transcription has on large gene instability. Genes that normally replicate later in S appear to be most prone to failing replication recovery, leading to CFS and CNV formation under replication stress. Brison et al. also found that large genes showing replication delay beyond S-phase correspond to those with basal replication timing in the second half of S-phase (26), consistent with our comparison of basal replication timing and replication delay at large genes expressed in GM11713. Blin et al. (16) found that activating large gene transcription by a small degree induced instability, but intensive forced transcription advanced the replication timing to earlier in the cell cycle such that more transcription was paradoxically protective. Indeed, we previously found that the magnitude of a gene’s transcription did not significantly correlate with its instability (5). Sarni et al. (71) came to similar conclusions that CFS instability is dependent on multiple genomic factors including TADs.
Other mechanisms might influence how unstable a gene is. Most important here is the transcription-dependent phenomenon of DNA-RNA hybrids, or R-loops, which have been shown to promote replication fork stalling (66). Helmrich et al. provided early evidence that large CFS genes are characterized by enhanced R-loop formation (18), but that analysis was limited to a single probe in the FHIT gene. Our analysis of genome-wide DRIP-seq data generated by us and others showed that large transcribed genes consistently display low R-loop burdens only slightly above the background of the untranscribed genome and much less than many smaller genes. Others have shown that genetic alterations such as loss of RTEL1 and FANCD2 increase R-loops in genomic regions prone to impaired replication progression, including CFS genes (29,30,67), but even in those data sets the total DRIP-seq signal integrated over the bodies of large genes remained low. Caveats include that the few R-loop peaks observed in large genes might mediate the instability effect, but this is inconsistent with the fact that CNV endpoints arise diffusely in the body of large transcribed genes. The most important R-loops might also be context dependent, for example only occurring in a critical cell cycle stage, and thus missed by bulk DRIP-seq. Finally, replication forks moving long distances through large genes would integrate the potential for fork failure such that even limited R-loops per unit length might result in a substantial effect at these loci. Here, our observation that RNase H1 overexpression had a small effect in reducing the basal CNV rate is noteworthy as it potentially identifies R-loops as a source of low frequency endogenous replication failure in large genes, as distinct from the intense exogenous stress created by APH treatment, although much deeper data will be required to address this important issue.
Our R-loop analysis was consistent with the fact the we did not observe significant changes in APH-induced genome-wide CNV formation, chromosome breakage or specific fragile site induction after either knocking down or overexpressing RNase H1 in human fibroblasts. These results contrast with Helmrich et al. who did see effects of similar RNase H1 manipulations on APH-induced CFS expression in lymphoblasts (18). The reasons for these discrepant findings are not clear but could include the differences in the cell types studied. Nevertheless, our overall data suggest that the role for R-loop mediated fork failure in large gene instability is secondary to a dominant role of impaired recovery from double fork failures arising from all mechanisms (26,70). This interpretation is supported by the fact that replication delay and CNV formation are most pronounced at the centers of large genes and not at either end as would be expected of R-loop mediated fork stalling or transcription-replication collisions (Figure 8G) (27). It is further supported by our findings that the transcription level of large genes and CNV frequency do not strongly correlate (5) and those of by Brison et al. that inhibiting transcription in cells already engaged in S-phase did not prevent CFS instability (26).
The mechanism and timing of CNV formation, i.e. the conversion of unreplicated DNA into stable de novo CNV junctions, remains enigmatic (Figure 8G). Lesion resolution might occur in S or G2 by error-prone fork recovery mechanisms (73,74). Alternatively, CNV junction formation might be delayed until M-phase where it would likely be executed by mitotic DNA synthesis (MiDAS), a novel replication mechanism activated to resolve unreplicated DNA prior to chromosome separation in mitotic cells (75,76). MiDAS is especially intriguing as a CNV mechanism because it is required in order to observe cytogenetic lesions at CFSs (76) and shows an extremely strong correlation to CFS loci (69,72). However, CNV formation might also primarily occur in the following cell cycle when the above replication recovery mechanisms fail. Clarification of these critical issues will require high throughput monitoring of de novo CNV junctions since approaches used here cannot easily distinguish between unreplicated versus permanently deleted DNA.
This study underscores the importance of evolving cellular transcription profiles for dictating which sites will be the most prone to genome instability under replication stress. Cells go through numerous rounds of division during both normal development and cancer progression. If the long isoforms of large genes are transcribed in those cells, the corresponding loci can become susceptible to genome instability as the risk of double-fork failures increases (5). Several studies have reported focal deletions at large CFS genes in various tumors (1,7–9,77). While some of these events may impact the progression of cancers, it is likely that they often represent passenger mutations resulting from an inherent instability under replication stress. Most of the largest human genes are expressed primarily in the brain and have important roles in neurodevelopment (1,78). Several studies have reported deletions and DSB clusters at those genes (1,5,17), suggesting that they are at risk for genomic rearrangements in the somatic cells of the brain and necessitating a complete understanding of their mechanistic basis.
DATA AVAILABILITY
Our svtools and msvtools software suites are available for download via GitHub at https://github.com/wilsonte-umich?tab=repositories. GM11713 Bru-seq and whole genomic sequence and HF1 microarray and DRIP-seq data are available from the Database of Genotypes and Phenotypes (dbGaP) under accession phs002066.v1.p1.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Michelle Paulsen and Karan Bedi for assistance performing Bru-seq library preparation and initial data analysis, Lynn-Knudson-Horneber for assistance with G-banding and FISH imaging, JoAnn Sekiguchi and Shigeki Iwase for helpful discussions, and Bjoern Schwer for access to unpublished DRIP-seq data. We thank Olivia Koues, Michael Pihalja, Kamla Saiya-Cork, Tonya Kopas and additional staff members at the Michigan Advanced Genomics, Flow Cytometry and Vector Cores for their expert assistance.
Contributor Information
So Hae Park, Department of Human Genetics, University of Michigan, Ann Arbor, MI 48109, USA.
Pamela Bennett-Baker, Department of Human Genetics, University of Michigan, Ann Arbor, MI 48109, USA.
Samreen Ahmed, Department of Human Genetics, University of Michigan, Ann Arbor, MI 48109, USA; Department of Pathology, University of Michigan, Ann Arbor, MI 48109, USA.
Martin F Arlt, Department of Human Genetics, University of Michigan, Ann Arbor, MI 48109, USA.
Mats Ljungman, Department of Radiation Oncology, University of Michigan, Ann Arbor, MI 48109, USA.
Thomas W Glover, Department of Human Genetics, University of Michigan, Ann Arbor, MI 48109, USA; Department of Pathology, University of Michigan, Ann Arbor, MI 48109, USA.
Thomas E Wilson, Department of Human Genetics, University of Michigan, Ann Arbor, MI 48109, USA; Department of Pathology, University of Michigan, Ann Arbor, MI 48109, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the National Institutes of Health [CA200731 to T.E.W and T.W.G., GM007544 to S.H.P. and HG009382 to M.L.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Glover T.W., Wilson T.E., Arlt M.F.. Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer. 2017; 17:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arlt M.F., Mulle J.G., Schaibley V.M., Ragland R.L., Durkin S.G., Warren S.T., Glover T.W.. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009; 84:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arlt M.F., Ozdemir A.C., Birkeland S.R., Wilson T.E., Glover T.W.. Hydroxyurea induces de novo copy number variants in human cells. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:17360–17365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arlt M.F., Rajendran S., Birkeland S.R., Wilson T.E., Glover T.W.. Copy number variants are produced in response to low-dose ionizing radiation in cultured cells. Environ. Mol. Mutagen. 2014; 55:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson T.E., Arlt M.F., Park S.H., Rajendran S., Paulsen M., Ljungman M., Glover T.W.. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015; 25:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harel T., Lupski J.R.. Genomic disorders 20 years on-mechanisms for clinical manifestations. Clin. Genet. 2018; 93:439–449. [DOI] [PubMed] [Google Scholar]
- 7. Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhsng C.Z., Wala J., Mermel C.H.et al.. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013; 45:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M.et al.. The landscape of somatic copy-number alteration across human cancers. Nature. 2010; 463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bignell G.R., Greenman C.D., Davies H., Butler A.P., Edkins S., Andrews J.M., Buck G., Chen L., Beare D., Latimer C.et al.. Signatures of mutation and selection in the cancer genome. Nature. 2010; 463:893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Letessier A., Millot G.A., Koundrioukoff S., Lachages A.M., Vogt N., Hansen R.S., Malfoy B., Brison O., Debatisse M.. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011; 470:120–123. [DOI] [PubMed] [Google Scholar]
- 11. Le Tallec B., Dutrillaux B., Lachages A.M., Millot G.A., Brison O., Debatisse M.. Molecular profiling of common fragile sites in human fibroblasts. Nat. Struct. Mol. Biol. 2011; 18:1421–1423. [DOI] [PubMed] [Google Scholar]
- 12. Hosseini S.A., Horton S., Saldivar J.C., Miuma S., Stampfer M.R., Heerema N.A., Huebner K.. Common chromosome fragile sites in human and murine epithelial cells and FHIT/FRA3B loss-induced global genome instability. Genes Chromosomes Cancer. 2013; 52:1017–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Le Beau M.M., Rassool F.V., Neilly M.E., Espinosa R. 3rd, Glover T.W., Smith D.I., McKeithan T.W.. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998; 7:755–761. [DOI] [PubMed] [Google Scholar]
- 14. Marchal C., Sima J., Gilbert D.M.. Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 2019; 20:721–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hiratani I., Ryba T., Itoh M., Yokochi T., Schwaiger M., Chang C.W., Lyou Y., Townes T.M., Schubeler D., Gilbert D.M.. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008; 6:e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blin M., Le Tallec B., Nahse V., Schmidt M., Brossas C., Millot G.A., Prioleau M.N., Debatisse M.. Transcription-dependent regulation of replication dynamics modulates genome stability. Nat. Struct. Mol. Biol. 2019; 26:58–66. [DOI] [PubMed] [Google Scholar]
- 17. Wei P.C., Chang A.N., Kao J., Du Z., Meyers R.M., Alt F.W., Schwer B.. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell. 2016; 164:644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Helmrich A., Ballarino M., Tora L.. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell. 2011; 44:966–977. [DOI] [PubMed] [Google Scholar]
- 19. Bertoli C., Skotheim J.M., de Bruin R.A.. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013; 14:518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palozola K.C., Liu H., Nicetto D., Zaret K.S.. Low-level, global transcription during mitosis and dynamic gene reactivation during mitotic exit. Cold Spring Harb. Symp. Quant. Biol. 2017; 82:197–205. [DOI] [PubMed] [Google Scholar]
- 21. Snyder M., Sapolsky R.J., Davis R.W.. Transcription interferes with elements important for chromosome maintenance in Saccharomyces cerevisiae. Mol. Cell. Biol. 1988; 8:2184–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Looke M., Reimand J., Sedman T., Sedman J., Jarvinen L., Varv S., Peil K., Kristjuhan K., Vilo J., Kristjuhan A.. Relicensing of transcriptionally inactivated replication origins in budding yeast. J. Biol. Chem. 2010; 285:40004–40011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Powell S.K., MacAlpine H.K., Prinz J.A., Li Y., Belsky J.A., MacAlpine D.M.. Dynamic loading and redistribution of the Mcm2-7 helicase complex through the cell cycle. EMBO J. 2015; 34:531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Macheret M., Halazonetis T.D.. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature. 2018; 555:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ozeri-Galai E., Lebofsky R., Rahat A., Bester A.C., Bensimon A., Kerem B.. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell. 2011; 43:122–131. [DOI] [PubMed] [Google Scholar]
- 26. Brison O., El-Hilali S., Azar D., Koundrioukoff S., Schmidt M., Nahse V., Jaszczyszyn Y., Lachages A.M., Dutrillaux B., Thermes C.et al.. Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat. Commun. 2019; 10:5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hamperl S., Bocek M.J., Saldivar J.C., Swigut T., Cimprich K.A.. Transcription-replication conflict orientation modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell. 2017; 170:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Helmrich A., Ballarino M., Nudler E., Tora L.. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013; 20:412–418. [DOI] [PubMed] [Google Scholar]
- 29. Wu W., Bhowmick R., Vogel I., Özer Ö., Ghisays F., Thakur R.S., Sanchez de Leon E., Richter P.H., Ren L., Petrini J.H.et al.. RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat. Struct. Mol. Biol. 2020; 27:424–437. [DOI] [PubMed] [Google Scholar]
- 30. Kotsantis P., Segura-Bayona S., Margalef P., Marzec P., Ruis P., Hewitt G., Bellelli R., Patel H., Goldstone R., Poetsch A.R.et al.. RTEL1 regulates G4/R-loops to avert replication-transcription collisions. Cell Rep. 2020; 33:108546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fernandes P., Miotto B., Saint-Ruf C., Said M., Barra V., Nahse V., Ravera S., Cappelli E., Naim V.. FANCD2 modulates the mitochondrial stress response to prevent common fragile site instability. Commun. Biol. 2021; 4:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paulsen M.T., Veloso A., Prasad J., Bedi K., Ljungman E.A., Tsan Y.C., Chang C.W., Tarrier B., Washburn J.G., Lyons R.et al.. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:2240–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Naito Y., Hino K., Bono H., Ui-Tei K.. CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics. 2015; 31:1120–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Montague T.G., Cruz J.M., Gagnon J.A., Church G.M., Valen E.. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014; 42:W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bauer D.E., Canver M.C., Orkin S.H.. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. J. Vis. Exp. 2015; 95:e52118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paulsen M.T., Veloso A., Prasad J., Bedi K., Ljungman E.A., Magnuson B., Wilson T.E., Ljungman M.. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 2014; 67:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arlt M.F., Rajendran S., Holmes S.N., Wang K., Bergin I.L., Ahmed S., Wilson T.E., Glover T.W.. Effects of hydroxyurea on CNV induction in the mouse germline. Environ. Mol. Mutagen. 2018; 59:698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Farkash-Amar S., Simon I.. Genome-wide analysis of the replication program in mammals. Chromosome Res. 2010; 18:115–125. [DOI] [PubMed] [Google Scholar]
- 41. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arlt M.F., Rajendran S., Birkeland S.R., Wilson T.E., Glover T.W.. De novo CNV formation in mouse embryonic stem cells occurs in the absence of Xrcc4-dependent nonhomologous end joining. PLos Genet. 2012; 8:e1002981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu J., Li H., Hu M., Sasaki T., Baccei A., Gilbert D.M., Liu J.S., Collins J.J., Lerou P.H.. The distribution of genomic variations in human iPSCs is related to replication-timing reorganization during reprogramming. Cell Rep. 2014; 7:70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Crossley M.P., Bocek M.J., Hamperl S., Swigut T., Cimprich K.A.. qDRIP: a method to quantitatively assess RNA-DNA hybrid formation genome-wide. Nucleic Acids Res. 2020; 48:e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sanz L.A., Chedin F.. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2019; 14:1734–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chakraborty P., Huang J.T.J., Hiom K.. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018; 9:4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Loomis E.W., Sanz L.A., Chedin F., Hagerman P.J.. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLos Genet. 2014; 10:e1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gorthi A., Romero J.C., Loranc E., Cao L., Lawrence L.A., Goodale E., Iniguez A.B., Bernard X., Masamsetti V.P., Roston S.et al.. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature. 2018; 555:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen S., Zhou Y., Chen Y., Gu J.. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Amemiya H.M., Kundaje A., Boyle A.P.. The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep. 2019; 9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Karimzadeh M., Ernst C., Kundaje A., Hoffman M.M.. Umap and Bismap: quantifying genome and methylome mappability. Nucleic Acids Res. 2018; 46:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ghandi M., Huang F.W., Jane-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R. 3rd, Barretina J., Gelfand E.T., Bielski C.M., Li H.et al.. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019; 569:503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stovner E.B., Saetrom P.. epic2 efficiently finds diffuse domains in ChIP-seq data. Bioinformatics. 2019; 35:4392–4393. [DOI] [PubMed] [Google Scholar]
- 55. Zang C., Schones D.E., Zeng C., Cui K., Zhao K., Peng W.. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009; 25:1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ning Y., Lovell M., Taylor L., Pereira-Smith O.M.. Isolation of monochromosomal hybrids following fusion of human diploid fibroblast-derived microcells with mouse A9 cells. Cytogenet. Cell Genet. 1992; 60:79–80. [DOI] [PubMed] [Google Scholar]
- 58. Durkin S.G., Ragland R.L., Arlt M.F., Mulle J.G., Warren S.T., Glover T.W.. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Antequera F. Structure, function and evolution of CpG island promoters. Cell. Mol. Life Sci. 2003; 60:1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A.et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mrasek K., Schoder C., Teichmann A.C., Behr K., Franze B., Wilhelm K., Blaurock N., Claussen U., Liehr T., Weise A.. Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int. J. Oncol. 2010; 36:929–940. [DOI] [PubMed] [Google Scholar]
- 62. Rivera-Mulia J.C., Gilbert D.M.. Replication timing and transcriptional control: beyond cause and effect-part III. Curr. Opin. Cell Biol. 2016; 40:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prendergast L., McClurg U.L., Hristova R., Berlinguer-Palmini R., Greener S., Veitch K., Hernandez I., Pasero P., Rico D., Higgins J.M.G.et al.. Resolution of R-loops by INO80 promotes DNA replication and maintains cancer cell proliferation and viability. Nat. Commun. 2020; 11:4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Crossley M.P., Bocek M., Cimprich K.A.. R-loops as cellular regulators and genomic threats. Mol. Cell. 2019; 73:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Okamoto Y., Iwasaki W.M., Kugou K., Takahashi K.K., Oda A., Sato K., Kobayashi W., Kawai H., Sakasai R., Takaori-Kondo A.et al.. Replication stress induces accumulation of FANCD2 at central region of large fragile genes. Nucleic Acids Res. 2018; 46:2932–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Parajuli S., Teasley D.C., Murali B., Jackson J., Vindigni A., Stewart S.A.. Human ribonuclease H1 resolves R-loops and thereby enables progression of the DNA replication fork. J. Biol. Chem. 2017; 292:15216–15224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Madireddy A., Kosiyatrakul S.T., Boisvert R.A., Herrera-Moyano E., Garcia-Rubio M.L., Gerhardt J., Vuono E.A., Owen N., Yan Z., Olson S.et al.. FANCD2 facilitates replication through common fragile sites. Mol. Cell. 2016; 64:388–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sima J., Chakraborty A., Dileep V., Michalski M., Klein K.N., Holcomb N.P., Turner J.L., Paulsen M.T., Rivera-Mulia J.C., Trevilla-Garcia C.et al.. Identifying cis elements for spatiotemporal control of mammalian DNA replication. Cell. 2019; 176:816–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Macheret M., Bhowmick R., Sobkowiak K., Padayachy L., Mailler J., Hickson I.D., Halazonetis T.D.. High-resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell Res. 2020; 30:997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Blow J.J., Ge X.Q., Jackson D.A.. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011; 36:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sarni D., Sasaki T., Irony Tur-Sinai M., Miron K., Rivera-Mulia J.C., Magnuson B., Ljungman M., Gilbert D.M., Kerem B.. 3D genome organization contributes to genome instability at fragile sites. Nat. Commun. 2020; 11:3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ji F., Liao H., Pan S., Ouyang L., Jia F., Fu Z., Zhang F., Geng X., Wang X., Li T.et al.. Genome-wide high-resolution mapping of mitotic DNA synthesis sites and common fragile sites by direct sequencing. Cell Res. 2020; 30:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lee J.A., Carvalho C.M., Lupski J.R.. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007; 131:1235–1247. [DOI] [PubMed] [Google Scholar]
- 74. Hastings P.J., Ira G., Lupski J.R.. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLos Genet. 2009; 5:e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ozer O., Hickson I.D.. Pathways for maintenance of telomeres and common fragile sites during DNA replication stress. Open Biol. 2018; 8:180018–180028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Minocherhomji S., Ying S., Bjerregaard V.A., Bursomanno S., Aleliunaite A., Wu W., Mankouri H.W., Shen H., Liu Y., Hickson I.D.. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015; 528:286–290. [DOI] [PubMed] [Google Scholar]
- 77. Rajaram M., Zhang J., Wang T., Li J., Kuscu C., Qi H., Kato M., Grubor V., Weil R.J., Helland A.et al.. Two distinct categories of focal deletions in cancer genomes. PLoS One. 2013; 8:e66264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Smith D.I., Zhu Y., McAvoy S., Kuhn R.. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006; 232:48–57. [DOI] [PubMed] [Google Scholar]
- 79. Lesurf R., Cotto K.C., Wang G., Griffith M., Kasaian K., Jones S.J., Montgomery S.B., Griffith O.L.. ORegAnno 3.0: a community-driven resource for curated regulatory annotation. Nucleic Acids Res. 2016; 44:D126–D132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our svtools and msvtools software suites are available for download via GitHub at https://github.com/wilsonte-umich?tab=repositories. GM11713 Bru-seq and whole genomic sequence and HF1 microarray and DRIP-seq data are available from the Database of Genotypes and Phenotypes (dbGaP) under accession phs002066.v1.p1.