


Abstract
Poly (ADP-ribose) polymerase inhibitor (PARPi)-based therapies initially reduce tumor burden but eventually lead to acquired resistance in cancer patients with BRCA1 or BRCA2 mutation. To understand the potential PARPi resistance mechanisms, we performed whole-genome CRISPR screens to discover genetic alterations that change the gene essentiality in cells with inducible depletion of BRCA2. We identified that several RNA Polymerase II transcription Mediator complex components, especially Cyclin C (CCNC) as synthetic survival targets upon BRCA2 loss. Total mRNA sequencing demonstrated that loss of CCNC could activate the transforming growth factor (TGF)-beta signaling pathway and extracellular matrix (ECM)-receptor interaction pathway, however the inhibition of these pathways could not reverse cell survival in BRCA2 depleted CCNC-knockout cells, indicating that the activation of these pathways is not required for the resistance. Moreover, we showed that the improved survival is not due to restoration of homologous recombination repair although decreased DNA damage signaling was observed. Interestingly, loss of CCNC could restore replication fork stability in BRCA2 deficient cells, which may contribute to PARPi resistance. Taken together, our data reveal CCNC as a critical genetic determinant upon BRCA2 loss of function, which may help the development of novel therapeutic strategies that overcome PARPi resistance.
INTRODUCTION
Integrity of human genome is continuously challenged by endogenous and exogenous lesions. In response to a variety of DNA insults, cells have evolved DNA damage response pathways to sense DNA lesions, arrest cell cycle, and recruit coordinated DNA repair factors to prevent the inheritance of unrepaired DNA. Among all DNA lesions, double-strand breaks (DSBs) are considered the most detrimental because they block all transactions on DNA. Failure to repair DSBs leads to cell lethality, whereas inappropriate repair of DSBs results in genome rearrangement and oncogenic transformation (1).
Typically, cells employ two major pathways to repair DSBs: the classical non-homologous end joining (C-NHEJ) pathway and homologous recombination (HR) pathway. In addition, at least two alternative repair pathways—alternative end joining (alt-EJ) and single-strand annealing (SSA), also have been shown to operate in various cellular contexts (2–4). All of these pathways involve specific repair factors and produce different repair outcomes. Whereas DSB repair by c-NHEJ, alt-EJ and SSA are considered error-prone, HR provides an error-free mechanism to precisely repair the breaks by using a sister or homologous chromatid (5,6).
DSB repair by HR is a complex process involving many gene products, and deficiencies in HR contribute to mutations associated with malignancy and reduced cell viability. BRCA1 and BRCA2 are tumor suppressors that play essential roles in promoting HR repair, which helps maintain genome integrity (7,8). Germline mutations of BRCA1 and BRCA2 are associated with about 40–80% of the hereditary breast and ovarian cancer cases and linked with increased risk of several other human malignancies, including prostate, pancreatic, stomach, and colorectal cancers (9–12).
During the HR repair process, BRCA1 acts as a versatile protein that links DNA damage sensing and repair effectors through its interaction with multiple protein complexes, whereas BRCA2 mediates the recruitment of the recombinase RAD51 to DSBs, which is an essential step for HR (12). In addition to their roles in HR, BRCA1 and BRCA2 have other functions in genome maintenance. For example, BRCA2 prevents MRE11-dependent degradation of nascent DNA strands at stalled replication forks through its C-terminal region, which is not required for HR (13). BRCA2 also associates with the TREX2 mRNA export factor PCID2 and RNA polymerase (Pol) II to prevent R-loop accumulation, which can lead to replication fork stalling and dysregulated transcriptional elongation (14,15).
BRCA1- and BRCA2-deficient cells are hypersensitive to treatment with inhibitors of poly ADP ribose polymerase (PARPi) through multiple mechanisms, including the synthetic lethality that results from unresolved DNA damage (16,17) and the replication arrest that results from physical obstruction of replication forks by PARP trapping (18). Several PARP inhibitors have been approved by the U.S. Food and Drug Administration for the treatment of cancer in patients with BRCA1 or BRCA2 mutations, such as olaparib (breast and ovarian cancer), rucaparib (ovarian cancer), and niraparib (ovarian cancer, regardless of BRCA mutation status) (19–22). However, about 60% of the patients did not respond to PARPi due to pre-existing or therapy-induced resistance, suggesting that a deeper understanding of BRCA1/BRCA2 biology and new anticancer strategies are needed to overcome PARPi resistance.
In this study, we employed CRISPR screening to identify genes that can rescue the lethality of BRCA2 loss in HEK293A cells. We first engineered a mini auxin-inducible degron (mAID) tag at the C-terminus of endogenous BRCA2 and utilized AID technology to inducibly deplete BRCA2 expression. We then performed unbiased whole-genome CRISPR guide RNA (gRNA) screens to discover genetic alterations that can alter the gene essentiality in BRCA2-deficient cells. We identified POLQ and APEX2 among the most synthetic lethal genes, which was consistent with previous studies (23–27). Moreover, we found that loss of several genes such as CCNC, MED12, CDK8 and MED24, which belong to the RNA Pol II transcription Mediator complex, led to improved survival of BRCA2-deficient cells even with the treatment of PARPi. Depletion of CCNC decreased DNA damage signaling in BRCA2 deficient cells upon PARPi treatment which may help cell survival. This enhanced survival was not due to restoration of HR. To explore the potential mechanisms underlying this synthetic viability relationship, we performed total mRNA sequencing (mRNA-Seq) with both control and BRCA2-depleted CCNC- and MED12-knockout (KO) cells. We found that genes involved in the regulation of cell proliferation, like those in the transforming growth factor (TGF)-beta signaling pathway and extracellular matrix (ECM)-receptor interaction pathway, were altered significantly. However these alterations may not be sufficient for rescuing cell lethality upon BRCA2 depletion. Interestingly, depletion of CCNC could restore replication fork stability in BRCA2 deficient cells. Taken together, our findings suggest that CCNC is one of the critical genetic determinants that dictate responses to treatment with PARPi in BRCA2-deficient cells with its role in replication fork protection. The information provided here may help the design of better treatment strategies for patients with BRCA2 mutated cancers.
MATERIALS AND METHODS
Cell lines
The HEK293A cell line was purchased from the American Type Culture Collection (ATCC) and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum. Capan-1 cells were a gift from Dr. Katharina Schlacher at MD Anderson Cancer Center and were grown in DMEM with 10% fetal bovine serum.
Chemicals
LY2157299 (S2230, Selleck Chemicals), CT251921 (HY-19984, MedChem Express), AZD-6738 (B1167, BioVision), Camptothecin (390238, Calbiochem), Olaparib (10188-, VMR international), beta-aminopropionitrile (A3134, Sigma-Aldrich), Mitomycin C (M4287, Sigma-Aldrich), Etoposide (E1383, Sigma-Aldrich), Hydroxyurea (H8627, Sigma-Aldrich), Talazoparib (204710, Medkoo).
Antibodies
CCNC (A301-989A, Bethyl Laboratories), CDK8 (4106S, Cell Signaling Technology), MED12 (4529S, Cell Signaling Technology), MED13 (A301-278A, Bethyl Laboratories), MED24 (A301-472A, Bethyl Laboratories), MED25 (sc-393759, Santa Cruz Biotechnology), BRCA2 (A303-434A, Bethyl Laboratories), Vinculin (#V9131, Sigma), HA (#H3663, Sigma), β-tubulin (#T5168, Sigma), GFP (sc-9996, Santa Cruz Biotechnology), RAD51 (ab63801, Abcam), γH2AX (05-636l, Millipore Sigma).
AID-Tagging of endogenous BRCA2
The CRISPR/Cas9 technology was used to knock in the mAID-EGFP tag to the C-terminus of endogenous BRCA2. HEK293A cells stably incorporating Tet-inducible expression of OsTIR1 were generated via infection with lentivirus expressing OsTIR1 (pInducer20 backbone; Addgene; #44012). After selection with G418, single clones were picked and confirmed using Western blotting. Next, HEK293A OsTIR1 pInducer20 cells were co-transfected with PX330 plasmid and a BRCA2–knock-in donor vector (PUC19 backbone) containing mAID, EGFP, P2A self-cleavage site, and blasticidin resistance selection gene flanked by approximately 1 kb of homology arms. Positive clones were first screened by genomic PCR and further validated via western blotting. BRCA2 CKI gRNA sequence is: 5′-ATATATCTAAGCATTTGCAA-3′.
Generation of KO cells
CCNC- and MED12-KO cells were generated by using the pLentiCRISPRv2 constructs in HEK293A OsTIR1 pInducer20 BRCA2 CKI mAID-EGFP cells. Cells were transiently transfected with the indicated pLentiV2 gRNA plasmids and selected with puromycin (2 μg/ml). Single clones were then plated into 96-well plates. After incubation for 2 weeks, the single clones were picked and validated by western blot. To generate pooled KO cells, HEK293A OsTIR1 pInducer20 BRCA2 CKI mAID-EGFP cells were infected with indicated pLentiCRISPRv2 lentivirus and selected with puromycin (2 μg/mL). CCNC gRNA-1 and MED12 gRNA-2 were used for the generation of KO clones. gRNA sequence used were listed below:
CCNC-KO gRNA-1: 5′-TAGGCAAAGATCCGTTCTGT-3′;
CCNC-KO gRNA-2: 5′-ATACCTAAAGCTATCATGAA-3′;
CDK8-KO gRNA-1: 5′-TGCAGCCCTCGTATTCAAAC-3′;
CDK8-KO gRNA-2: 5′-ACTATGCTGAACATGACCTC-3′;
CDK8-KO gRNA-3: 5′-TTCACTACCTGCATGCTAAC-3′;
MED12-KO gRNA-1: 5′-AGGATTGAAGCTGACGTTCT-3′;
MED12-KO gRNA-2: 5′-GGGAGCGTGACATTGTACGT-3′;
MED13-KO gRNA-1: 5′-AGCGTAACCGAAGACATAGC-3′;
MED13-KO gRNA-2: 5′-GATAGTACTAGCCACCATGG-3′;
MED24-KO gRNA-1: 5′-CCCTTCTTAGCGCCCTCCAC-3′;
MED24-KO gRNA-2: 5′-ATACCTTACTGATGGCTGTG-3′;
MED25-KO gRNA-1: 5′-GCGCCTATGAGTTTGTCACC-3′;
MED25-KO gRNA-2: 5′-TTACCTGGCAGTGGAGCCCG-3′
Whole-genome CRISPR gRNA screening
The TKOv3 gRNA library (total 71090 gRNAs) contains 70,948 gRNAs targeting 18 053 protein-coding genes (four gRNAs/gene) plus 142 control gRNAs targeting LacZ, EGFP, and luciferase was a gift from Dr Traver Hart (MD Anderson Cancer Center). CRISPR screening was conducted as described previously (28,29). Briefly, 120 million HEK293A OsTIR1-pInducer20 BRCA2 CKI mAID-EGFP cells were infected with the TKOv3 library lentiviruses at a low MOI (<0.3). Twenty-four hours after infection, the medium was replaced with fresh medium containing puromycin (2 μg/ml). After selection, cells were placed in two groups: one was cultured in the medium containing DMSO as control wild-type BRCA2 cells, and the other cultured in the medium containing Doxycycline (1 μg/ml; Sigma) and IAA (500 μM; Fisher Scientific) to induce BRCA2 degradation as BRCA2-depleted cells. Both groups contained two replicates, with each replicate containing about 20 million cells passaged every 3 days and maintained at about 200-fold coverage. At days 0 and 21, 30 million cells (>300-fold coverage) were collected for genomic DNA extraction using a QIAamp Blood Maxi Kit (QIAGEN). gRNA inserts were amplified via PCR using primers harboring Illumina TruSeq adapters with i5 and i7 barcodes as described previously (30), and the resulting PCR libraries were sequenced using an Illumina HiSeq 2500 system. Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) (31) and drug-Z (28) analysis were used to calculate the difference in gRNA enrichment between the control BRCA2-WT cells and cells with BRCA2 depletion.
Cell survival assay
For cell growth assays, cells were seeded in six-well plates (105 per well), passaged every 2 days, and cell numbers were determined. For clonogenic assays, equal numbers of cells were seeded onto six-well plates in triplicate, treated with various doses of radiation or different concentrations of various chemical drugs, and incubated for 10–14 days. Colonies were fixed and stained with 0.5% crystal violet. The colonies were counted using ImageJ software (NIH) or manually. All cell survival assays were performed at least in triplicate.
Immunofluorescence staining
The immunostaining experiment was performed similarly to that described previously (32). Briefly, cells grown on glass coverslips were fixed in 3% formaldehyde for 15 mins on ice and then permeabilized for 15 mins in 0.5% Triton X-100, which was followed by blocking with 5% bovine serum albumin for 1 h at room temperature. Cells were then incubated with primary antibodies diluted in 5% bovine serum albumin for 2 h at 37°C and indicated secondary antibody for 1 h at room temperature. Coverslips were mounted with the use of DAPI solution (Thermo Fisher Scientific).
DNA fiber analysis
DNA fiber analyses were carried out as reported with minor modification (33). Briefly, cells were first pulse-labeled with 25 μM IdU for 30 min, washed three times with PBS, then pulse-labeled with 250 μM CldU for 30 min followed by 4 hrs of treatment with 2mM HU. Cells were harvested and resuspended in PBS for a final concentration of 1500 cells/μl. Cells were then lysed in lysis buffer (200 mM Tris–HCl (pH 7.5), 50 mM EDTA, 0.5% SDS), DNA fibers were stretched onto positively charged glass slides and fixed in methanol:acetic acid (3:1). After rehydration in PBS twice for 5 min, DNA were denatured with 2.5 M HCl for 1 h, slides were then washed with PBS for three times and blocked with 5% BSA at 37°C for 1 h. The newly replicated ldU and CIdU tracks were immunostained using mouse-anti-BrdU (1/100, 347580, BD Biosciences) and rat-anti-BrdU (1/100, Ab6326, Abcam) primary antibodies, and Alexa Fluor 546 goat-anti-mouse IgG1 (1/500, A-21123, ThermoFisher Scientific) and Alexa Fluor 488 goat-anti-rat (1/500, A-11006, ThermoFisher Scientific) secondary antibodies respectively. Coverslips were mounted using ProLong Gold Antifade Mountant (ThermoFisher Scientific).
HR reporter assays
DSB repair efficiency by HR was measured in U2OS cells stably expressing HR reporter DR‐GFP reporter as previous described (34). Briefly, CCNC knocking down U2OS DR-GFP cells were generated by infection with CCNC g1 Lenti-virus. Then control and CCNC knocking-down cells were transfected with 50 pmol control siRNA or BRCA2 siRNA (SR300471, OriGene Technologies) with Lipofectamine RNAiMAX (ThermoFisher Scientific). Twenty-four hours later, 2 μg of I‐SceI expression vector were transfected into the cells with Lipofectamine™ 3000 (ThermoFisher Scientific). After culture for an additional 48 h, cells were collected and subjected to flow cytometry analysis to determine percentages of GFP‐positive cells.
RNA-Seq and data analysis
HEK293A BRCA2 CKI mAID-EGFP WT, CCNC-KO and MED12-KO with or without BRCA2 depletion cells (each with two biological replicates) were collected and total RNA was extracted using an RNeasy Mini Kit (QIAGEN; #74104) according to the manufacturer's instructions. The library was prepared using an Illumina TruSeq Stranded Total RNA Library Prep Kit, including rRNA depletion and sequencing at NextSeq 550 (Illumina) to generate 75-bp paired ends.
For RNA-seq data analysis, reads were adapter-trimmed and preprocessed with Cutadapt software (version 1.15) for quality control and data filtering. Genome mapping was conducted using STAR (version 2.5.3a) (35) and the human reference genome (GRCh38). Genes abundance was measured by HTseq-count uniquely-mapped reads number with default parameter and using the ENSEMBL v83 annotations. Only genes with >5 reads in at least one sample were retained. The raw read counts of retained genes were submitted for differential expression analysis by DESeq2 software (36). The resulting P-values were adjusted by the Benjamini and Hochberg approach (37) to control for false discovery rate (FDR). Genes with fold change (FC) >1.35 and FDR <0.05 were considered as differentially expressed genes (DEGs). Standard gene set enrichment analysis was performed with a hypergeometric test using RDAVID WebService (v1.19.0) (38).
Statistical analyses
Statistical analyses were performed with the use of Student t-tests (two-tailed) or one-way ANOVA in GraphPad Prism8 software. All results are presented as means (±S.D.) with at least three times repeats. (*P < 0.05, **P < 0.01, ***P < 0.001).
RESULTS
CRISPR screens to identify genes that can rescue cell lethality resulting from BRCA2 loss
BRCA2 loss of heterozygosity (LOH) is frequently observed in BRCA2-mutated tumors, but biallelic loss of BRCA2 causes embryonic lethality in mice and inhibits proliferation of normal somatic cells (39–41). In order to fully inactivate BRCA2 in normal cells, we introduced Auxin-inducible degron (AID) technology to inducibly deplete BRCA2 expression. This technology introduces a plant-specific degradation pathway controlled by a phytohormone, auxin, into non-plant cells (42). In cells expressing the specific auxin-perceptive F-box protein TIR1, which forms a functional Skp1-Cullin-F-box ubiquitin ligase, proteins fused with an AID tag derived from the indole-3-acetic acid 17 (IAA17) protein in Arabidopsis thaliana can be induced for rapid degradation by the addition of auxin to culture medium (42,43). We first generated HEK293A cells with doxycycline (Dox)-inducible expression of OsTIR1. We then used CRISPR/Cas9-mediated gene editing to knock in a mAID-EGFP tag at the C-terminus of BRCA2 at its endogenous genomic loci in these cells (Figure 1A and B). When we added Dox and auxin (using IAA) for 24 hrs, as shown in Figure 1C, we noted that endogenously EGFP-tagged BRCA2 protein was degraded. We also performed a colony survival assay and confirmed that degradation of BRCA2 led to a reduction in cell survival (Figure 1D).
Figure 1.

Genome-wide CRISPR/Cas9 screens identify genetic vulnerabilities in BRCA2-deficient cells. (A) Strategy for CRISPR-based mAID-EGFP tagging at the C-terminal BRCA2 genomic loci in HEK293A OsTIR1 pInducer20 cells. TAA: stop coden; BSD: blasticidin; HR: homologous recombination. (B) Genomic PCR was performed to validate the homozygous clone with correct insertion of mAID-EGFP tag. MW, molecular weight; WT: wild-type; KI: knock-in. (C) Western blot showing that after cells were treated with Dox and IAA (indole-3-aceticacid), BRCA2 was degraded within 24 h. (D) Colony survival assay was performed to determine cell viability upon BRCA2 depletion in HEK293A OsTIR1 pInducer20 BRCA2 mAID-EGFP KI cells. (E) Schematic of the workflow for the CRISPR screen performed in HEK293A OsTIR1 pInducer20 BRCA2 mAID EGFP KI cells with TKOv3 whole-genome gRNA library. MOI, multiplicity of infection; DMSO, dimethyl sulfoxide; NGS, next-generation sequencing. (F) Ranking of BRCA2–/– co-essential genes based on drugZ analysis of the results of CRISPR/Cas9 screen. The z-score was used to define possible vulnerability of cell lethality with BRCA2 ablation. The higher the score the more viable upon co-depletion of BRCA2. Likewise, the lower the score the greater vulnerability with BRCA2 co-depletion. All genes targeted in the TKOv3 library were scored according to the fold change in the abundance of their sgRNAs (IAA-based versus DMSO-based treatment). (G) The top 10 significantly enriched Gene Ontology (GO) Biological Process terms (P < 0.001) with the high-confidence candidate genes whose loss of function led to survival of BRCA2–/– cells.
We used the Toronto Knock Out Library version 3 (TKOv3), which contains 70 948 gRNAs targeting 18,053 protein-coding genes (44), to perform unbiased whole-genome gRNA screens for discovery of genetic alterations that can rescue the lethality of BRCA2 loss in the established BRCA2 mAID-EGFP-knock-in(KI) HEK293A cells (Figure 1E). Following infection of HEK293A OsTIR1 BRCA2 mAID-EGFP-KI cells with the TKOv3 gRNA library, we separated the cells into two groups- dimethyl sulfoxide (DMSO)-treated (i.e. BRCA2+/+) and Dox&IAA-treated (i.e. BRCA2–/–) groups. We allowed the cells to grow for approximately 20 doublings and then collected them for genomic sequencing. Deep sequencing confirmed total gRNA reads in the two groups. We then performed Drug Z analysis (44) and ranked the genes according to their drugZ scores which was used to define possible vulnerability with BRCA2 ablation (Figure 1F and Supplementary Table S1). As shown in Figure 1F, drugZ analysis identified APEX2, POLQ, and PARP1 rank on the top in the list that showed synthetic lethality with BRCA2 loss, which is consistent with previous studies (23–26), as well as a recent report by Dr. Stephen Elledge group using small DNA repair-focused short hairpin RNA and CRISPR gRNA libraries (27). These data suggested that our screening results are reliable and informative for further follow-up experiments. We also noticed that besides APEX2 and POLQ, single gRNAs (sgRNAs) targeting APEX1 or RNASEH2A also decreased significantly upon BRCA2 depletion (Supplementary Figure S1A, Tables S2 and S3), suggesting that these genes may be additional targets for synthetic lethality with BRCA2 loss.
We then examined the top 200 genes whose loss would lead to improved survival upon BRCA2 loss based on the drugZ score. By performing gene set enrichment analysis using Gene Ontology (GO), we found the top 10 most significantly enriched GO terms were related to transcription regulation, mRNA splicing, and others (Figure 1G). Surprisingly, we found that several RNA Pol II transcription Mediator complex components (e.g. CCNC, CDK8, MED24, MED25, MED16) were ranked on the top 10 among the synthetically survival genes. Deep sequencing showed that levels of sgRNA reads targeting several Mediator components were markedly higher in BRCA2-deficient cells than those in the control cells (Supplementary Figure S1B, Tables S2 and S3). These data suggested that loss of several RNA Pol II transcription Mediator complex components is probably required for cell survival upon BRCA2 depletion.
The RNA Pol II transcription Mediator complex is required for survival of BRCA2-deficient cells
Studies of genes resistant to lethality induced by BRCA2 loss should identify not only new components that may function in or together with BRCA2-dependent DNA damage repair but also potential biomarkers for PARPi-based therapy in BRCA-deficient cancer patients. Researchers initially discovered the RNA Pol II transcription Mediator complex in budding yeast through genetic and biochemical studies (45–48) and later in mammals, flies, and other eukaryotes (49). RNA Pol II transcription Mediator is an evolutionarily conserved multi-subunit protein complex (comprising 25 subunits in budding yeast and at least 31 subunits in humans) that is required for regulating RNA Pol II-dependent transcripts of protein-coding and noncoding RNA genes (50). It serves as a functional bridge to transduce signals from the transcription activators bound to enhancer regions to the transcription machinery, which is assembled at promoters as the preinitiation complex (PIC) to control transcription initiation (45,50,51). Mediator can be divided into four main modules: the head, middle, tail and transiently associated CDK8 kinase modules (50).
Based on the drugZ analysis (Figure 1F), we picked the top four synthetic survival candidates: CCNC, CDK8, MED24 and MED25, all of which belong to Mediator complex, for further validation of cell viability upon BRCA2 loss. We designed several sgRNAs that differed from the ones used in the TKOv3 library and showed that they are effective in knocking down the expression of their target genes without altering BRCA2 expression in pooled HEK293A BRCA2 mAID-EGFP-KI cells (Figure 2A). Moreover, when we added IAA and Dox to inducibly deplete BRCA2 in these pooled cells, BRCA2 was clearly degraded as in control cells (Figure 2A), ruling out the possibility that loss of these Mediator components somehow affects the removal of BRCA2 in our experimental system. We then performed long-term clonogenic survival assays and found that cells with co-depletion of CCNC, MED24, or CDK8 with BRCA2 exhibited more survival and PARPi resistance than did those with BRCA2 loss alone, whereas depletion of MED25 had only a mild effect (Figure 2B and C). CCNC and CDK8, together with the accessory subunits MED12 and MED13, constitute an independent and reversible/dissociable regulatory sub-complex called the CDK8-dependent kinase module, which play the core transcriptional function in the Mediator complex (52). Thus, we also studied the other two components, MED12 and MED13, in the CDK8-dependent kinase module. We found that co-depletion of MED12 or MED13 with BRCA2 showed respectively very significant or mild resistance to cell lethality due to BRCA2 loss and/or PARPi-based treatment (Figure 2D–F).
Figure 2.
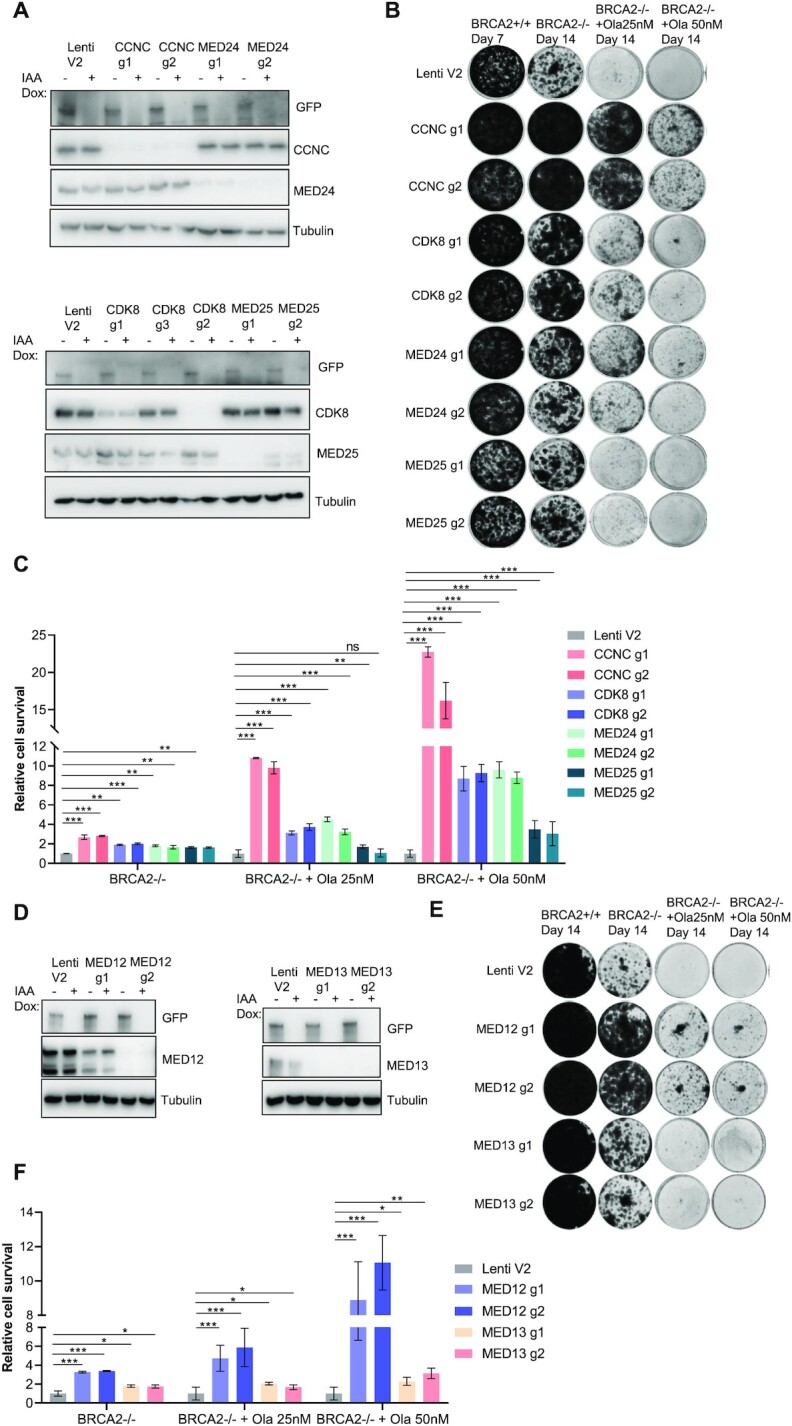
The RNA Pol II transcription Mediator complex is important for survival and PARPi sensitivity in BRCA2-depleted cells. (A) Western blots performed to determine the efficacy of sgRNAs targeting CCNC, CDK8, MED24 and MED25. HEK293A BRCA2 mAID-EGFP cells were infected with lentiviruses expressing the indicated sgRNAs and selected with puromycin. After treatment with Dox and IAA for 24 hrs, cells were collected and lysates were blotted with the indicated antibodies. (B) Results of long-term clonogenic assays performed using cells transduced with the indicated sgRNA constructs and given the indicated treatments. 5×103 cells were seeded in each well and incubated with the indicated treatments for 7 or 14 days. Colonies were fixed and stained with 0.5% crystal violet. Ola, olaparib. Hereafter BRCA2+/+ cells represent HEK293A BRCA2 mAID-EGFP cells without IAA and Dox treated cells; BRCA2-/- cells represent HEK293A BRCA2 mAID-EGFP cells with IAA and Dox treated cells. (C) Quantification of relative cell survival in the clonogenic assays in (B). All treated groups were normalized to the indicated untreated group. And the relative cell growth was compared with LentiV2 (V2) which was set as 1. The data are presented as means (±SD). **P < 0.01; ***P < 0.001; ns, not significant (Student t-test). (D) Western blots performed to determine the efficacy of sgRNAs targeting MED12 and MED13. (E) Results of long-term clonogenic assays performed to determine the viability of the cells in D. 5 × 103 cells were seeded in each well and incubated with the indicated agents for 14 days. Colonies were fixed and stained with 0.5% crystal violet. (F) Quantification of relative cell survival in the clonogenic assays in (E). The data are presented as means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 (Student's t-test).
Among all the genes examined in long-term clonogenic survival assays, CCNC depletion had the most dramatic effects on cell survival. This is probably due to the fact that CCNC is the only cyclin in the RNA Pol II transcription Mediator complex, whereas other genes in this complex have paralogs. For example, CDK8, MED12, and MED13 all have paralogs—CDK19, MED12L, and MED13L, respectively—that are assembled in a mutually exclusive manner (50,53). We performed a cell growth assay with pooled CCNC-KO HEK293A BRCA2 mAID-EGFP-KI cells and found that depletion of CCNC did not have much effect on cell growth (Supplementary Figure S2A). However, when we co-depleted CCNC and BRCA2 in these cells, the cell growth rate was considerably faster than that cells with only BRCA2 depletion. We then examined the cell-cycle distribution for cells with CCNC depletion and those with CCNC and BRCA2 co-depletion. As shown in Supplementary Figure S2B and S2C, neither depletion of CCNC nor co-depletion of CCNC and BRCA2 affected the cell-cycle profile, which agreed with previous studies demonstrating that CCNC is not a conventional cyclin, its expression is not altered in different cell-cycle phases, and it does not regulate cell-cycle progression (54,55).
We next generated fully CCNC-KO cells derived from HEK293A BRCA2 mAID-EGFP KI cells for further study (Figure 3A). By performing the clonogenic survival assay, we confirmed that CCNC-KO cells displayed markedly more survival and PARPi resistance than wild-type (WT) cells when BRCA2 was depleted (Figure 3B-E). Consistent results were obtained with two different PAPR inhibitors: Olaparib (Figure 3B and C) and Talazoparib (Figure 3D and E). We also knocked down CCNC in BRCA2 deficient Capan-1 cells (Figure 3F). In consistent with the results in HEK293A cells, we confirmed that depletion of CCNC showed both Olaparib (Figure 3G–H) and Talazoparib (Figure 3I–J) resistance in Capan-1 cells. We also found that overexpression of CCNC in both HEK293A BRCA2 mAID-EGFP and Capan-1 cells showed mild sensitivity to Olaparib treatment (Supplementary Figure S3), suggesting that CCNC expression is an important regulator of PARPi sensitivity in BRCA2 deficient cells.
Figure 3.

Loss of CCNC is important for survival and PARPi sensitivity in BRCA2-deficient Cells. (A) Validation of CCNC KO in HEK293A BRCA2 mAID-EGFP KI cells by western blot using the indicated antibodies. (B) Results of long-term clonogenic assays conducted using HEK293A BRCA2 mAID-EGFP CCNC-WT and -KO cells with the indicated treatments. Ola: Olaparib. (C) Quantification of relative cell survival in the clonogenic assays in (B). All treated groups were normalized to the indicated untreated group. The data are presented as means ± SD. ***P < 0.001 (Student's t-test). (D) Results of long-term clonogenic assays conducted using HEK293A BRCA2 mAID-EGFP CCNC-WT and -KO cells with the indicated treatments. Tal: Talazoparib. (E) Quantification of relative cell survival in the clonogenic assays in (D). All treated groups were normalized to the indicated untreated group. The data are presented as means ± SD. ***P < 0.001 (Student's t-test). (F) Capan-1 cells were infected with control pLentiCRISPRv2 (Lenti V2), or CCNC gRNA-1 (g1) virus and selected with puromycin. Western blot was conducted to validate CCNC knock down efficiency with the indicated antibodies. (G) Results of long-term clonogenic assays conducted using Capan-1 LentiV2 and CCNC g1 cells with different concentration of Olaparib treatments. Ola: Olaparib. (H) Quantification of relative cell survival in clonogenic assays in (G). All treated groups were normalized to the indicated untreated group. The data are presented as means ± SD. ***P < 0.001 (Student's t-test). (I) Results of long-term clonogenic assays conducted using Capan-1 LentiV2 and CCNC g1 cells with different concentration of Talazoparib treatments. Tal: Talazoparib. (J) Quantification of relative cell survival in the clonogenic assays in (I). All treated groups were normalized to the indicated untreated group. The data are presented as means ± SD. **P < 0.01, ***P < 0.001 (Student's t-test).
In summary, our data suggested that the RNA Pol II transcription Mediator complex especially CCNC may be a critical genetic determinant that dictates the responses to treatment with PARPi in BRCA2-deficient cancer cells.
CCNC-KO cells are resistant to treatment with several DNA-damaging agents
We next examined whether CCNC depletion has an effect on cellular sensitivity to various DNA-damaging agents even in the presence of BRCA2. Our group previously reported that loss of genes from the Mediator complex (e.g. CCNC, MED12, MED13, MED24) also showed resistance to treatment with ATR inhibitors through CRISPR screening (29). We therefore performed a colony formation assay with treatment with an ATR inhibitor (ATRi) or different DNA-damaging agents. As shown in Figure 4A, CCNC-KO cells displayed significant resistance to the ATRi (AZD6738), which agrees with our previous finding (29). We also found that CCNC-KO cells were resistant to treatment with mitomycin C (MMC), camptothecin (CPT) and PARP inhibitor (Olaparib) at different concentrations (Figure 4B–D) and mildly resistant to irradiation (IR) (Figure 4E). However, these cells did not exhibit much difference to treatment with hydroxyurea (HU) and exhibited slight sensitivity to etoposide treatment (Figure 4F and G). These findings hint that loss of CCNC may somehow protect replication fork, since MMC, CPT and Olaparib are all known to affect replication fork stability. However, CCNC-KO cells were not resistant to HU treatment, which also causes replication fork stalling and/or collapse. This may be explained by several different cell-killing mechanisms proposed for HU. The well-known function of HU is to inhibit ribonucleotide reductase (RNR), which decreases the deoxyribonucleotide triphosphate (dNTP) levels and therefore slows down the movement of DNA polymerases at replication forks (56,57). Recent studies also suggested that HU may have other targets such as other enzymes and the matrix proteases (58–60). Suppression of these targets may arrest cells in cytokinesis which also leads to cell lethality. HU treatment can also result in the accumulation of Reactive Oxygen Species (ROS) that also kill cells (61–63). Since HU have multiple inhibitory effects, the response of CCNC KO cells to HU in the long-term cell survival assay may rely on one or several of these mechanisms.
Figure 4.

Determination of the viability of CCNC-KO cells treated with different DNA-damaging agents. (A–G) Cell viability assays performed in HEK293A BRCA2 mAID-EGFP KI CCNC-WT and -KO cells. Cells were treated with the indicated doses of ATRi (ATR inhibitor AZD6738), CPT(Camptothecin), MMC (Mitomycin C), Ola (Olaparib), Irradiation (IR), HU (Hydroxyurea), ETO (Etoposide). The data are presented the mean ± SD. n = 2 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
The roles of CCNC and the Mediator complex in promoting cell survival in response to DNA damage appear to be complex because these cells were sensitive to treatment with some but not all DNA-damaging agents.
Regulation of gene expression by loss of CCNC or MED12
We then would like to evaluate how the Mediator complex affects cell survival upon BRCA2 loss. Considering that the major function of Mediator complex is in RNA Pol II-dependent transcriptional regulation and MED12 loss is reported to confer resistance to treatment with ALK, EGFR, BRAF, and MEK inhibitors and cytotoxic agents in various cancer cell types by activating TGF-beta signaling (64). We generated CCNC- and MED12-KO in HEK293A BRCA2 mAID-EGFP KI cells (Figure 3A and Supplementary Figure S5A) and performed total mRNA sequencing to determine how the gene expression is altered in both parental and BRCA2-depleted CCNC- and MED12-KO cells. As shown in Supplementary Figure S4A–S4D, we placed these RNA-seq data in four groups to perform the analysis: CCNC-KO versus CCNC-WT cells (referred to as CCNC KO) (Supplementary Figure S4A), BRCA2- and CCNC- double KO versus BRCA2-KO cells (referred to as B2 CCNC DKO) (Supplementary Figure S4B), MED12-KO versus MED12-WT cells (referred to as MED12 KO) (Supplementary Figure S4C), and BRCA2- and MED12- double KO versus BRCA2-KO cells (referred to as B2 MED12 DKO) (Supplementary Figure S4D). At a false discovery rate (FDR) <0.05 and fold change > 1.35, we identified 652 differentially expressed genes (DEGs) in CCNC KO, 486 DEGs in B2 CCNC DKO, 991 DEGs in MED12 KO, and 641 DEGs in B2 MED12 DKO (Supplementary Table S4). When we compared the DEGs in CCNC KO and B2 CCNC DKO, we found that 311 of them were the same, which represented 64% (311/486) of the DEGs in B2 CCNC DKO (Supplementary Figure S4E). MED12 KO and B2 MED12 DKO shared 471 DEGs, which corresponded to 73.5% (471/641) of the DEGs in B2 MED12 DKO (Supplementary Figure S4E). These data suggested that BRCA2 depletion did not dramatically affect gene expression in CCNC- or MED12-KO cells. We then performed pathway enrichment analysis of these DEGs using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotations (Supplementary Table S5). We found that all four groups were enriched in seven pathways: the TGF-beta signaling pathway, the ECM-receptor interaction pathway, Focal adhesion, Amoebiasis, the PI3K-Akt signaling pathway, small cell lung cancer, and pathways in cancer (Supplementary Figure S4F). These enriched pathways control cellular activities such as proliferation, adhesion, migration, differentiation, and apoptosis.
We further analyzed the DEGs involved in the TGF-beta signaling pathway in the four groups described above (Supplementary Figure S5B–S5E). We found that in the CCNC KO and B2 CCNC KO groups, genes that promote TGF-beta signaling, such as TGFB2, TGFBR1, and CDKN2B (65), were significantly upregulated, whereas genes that inhibit TGF-beta signaling, such as SMAD6, SMURF2, MYC, TGIF2 and ID4 (66–70), were dramatically downregulated (Supplementary Figure S5B and S5C), suggesting that TGF-beta signaling was activated by depletion of CCNC in both WT and BRCA2-depleted cells. We found similar patterns in MED12 KO and B2 MED12 DKO, with SMAD6, SMURF2, MYC, TGIF2, and ID1-4 downregulated and TGFB1, TGFB2, and CDKN2B upregulated (Supplementary Figure S5D and S5E). These analyses implied that activation of TGF-beta signaling by loss of CCNC or MED12 might be one of the mechanisms by which cell survival is enhanced as previously reported (64).
We therefore checked whether inhibition of TGF-beta signaling could restore the lethality and PARPi sensitivity in BRCA2 deficient cells. As shown in Supplementary Figure S6A and S6B, treatment with the TGF-βR inhibitor LY2157299 in CCNC-BRCA2 DKO or MED12-BRCA2 DKO cells could not restore the lethality and PARPi sensitivity in BRCA2 deficient cells, suggesting that the activation of TGF-beta signaling may not be sufficient for these functions. We also checked and found that inhibition of ECM-receptor interaction pathway with Beta-aminopropionitrile (BAPN) also failed to restore the lethality and PARPi sensitivity in BRCA2 deficient cells (Supplementary Figure S6C and D). Thus, although loss of CCNC or MED12 indeed influenced transcriptional regulation of several pathways which regulate a wide spectrum of cellular functions, at least two of these pathways could not account for or were not sufficient for the resistance phenotypes observed in these BRCA2 deficient cells.
Loss of CCNC leads to decreased DNA damage signal in BRCA2 deficient cells without restoration of HR
We then checked the DNA damage level in CCNC-BRCA2 DKO cells with or without PARPi treatment. As shown in Figure 5A and B, in non-treated HEK293A BRCA2 WT or depleted cells, CCNC KO did not result in much difference of γH2AX staining compared to cells with WT CCNC. However, when treated with Olaparib for 24 hrs, CCNC KO cells displayed significant decreased γH2AX foci number in both BRCA2 WT or depleted cells. We also repeated these experiments in Capan-1 cells and found that knocking down of CCNC in CAPAN-1 cells significantly decreased the γH2AX staining when treated with Olaparib (Figure 5C and D). We then checked whether the decreased DNA damage signal is due to the restoration of HR repair since it is one of the known PARPi resistance mechanisms (71). As shown in Figure 6A and B, depletion of CCNC did not affect RAD51 foci formation upon Olaparib treatment. Moreover, when BRCA2 was depleted, we could not detect any cells with RAD51 foci formation, supporting the known critical role of BRCA2 in promoting RAD51 loading and RAD51 foci formation. Co-depletion of CCNC with BRCA2 did not rescue RAD51 foci formation (Figure 6A and B), suggesting that the rescue of cell lethality due to CCNC loss does not result from restoration of HR. We also utilize U2OS HR reporter system to assess the impact of CCNC depletion on HR restoration. As shown in Figure 6C and D, knocking down of CCNC could not increase HR repair efficiency in either control siRNA (sictrl) or BRCA2 siRNA (siBRCA2) treated cells. All these data suggest that the resistance caused by CCNC depletion is not due to the restoration of HR repair.
Figure 5.

Depletion of CCNC decrease DNA damage signal in BRCA2 deficient cells. (A) CCNC KO and WT HEK293A BRCA2 mAID cells with or without depletion of BRCA2 were untreated (NT) or treated with Olaparib (2 μM) for 24 h and collected for immunostaining with γH2AX antibody. (B) Statistical quantification of γH2AX foci formation from (A). Mean number of γH2AX foci were analyzed in >100 cells. **P < 0.01. (C) Immunostaining of γH2AX in Capan-1 LentiV2 and CCNC g1 cells with or without Olaparib (2 μM) treatment for 24 h. (D) Statistical quantification of γH2AX foci formation from (C). Percentage of cells with > 10 γH2AX foci were counted. Data are represented as mean ± SD (n = 3), **P < 0.01, ns, not significant (Student's t-test).
Figure 6.

Depletion of CCNC does not restore HR repair. (A) Depletion of CCNC does not affect RAD51 foci formation upon DNA damage. HEK293A BRCA2 mAID CCNC-KO and WT cells with BRCA2 depletion (+IAA) or without BRCA2 depletion were treated with Olaparib (2 μM) for 24 h and collected for immunostaining with RAD51 antibody. (B) Statistical quantification of percentage of cells with >10 RAD51 foci from (A). (C) Validation of CCNC and BRCA2 knock down efficiency in U2OS DR-GFP cells by western blot with indicated antibodies. (D) Statistical quantification of HR repair efficiency in cells from (C). Data are represented as mean ± SD (n = 3). ns, not significant (Student t-test).
Loss of CCNC restores replication fork stability in BRCA2 deficient cells in response to replication stress
In addition of its role in HR repair, BRCA2 is also required for the protection of stalled replication forks, which could be degraded by nucleases such as MRE11 and MUS81 upon replication stress (13,72). The restoration of replication fork stability can also lead to PARPi resistance (71,73). We thus assessed the impact of CCNC depletion in replication fork protection in BRCA2 deficient cells upon replication stress using DNA fiber assay. As shown in Figure 7A and B, HEK293A BRCA2 depleted cells exhibited HU-induced nascent DNA degradation, which was presented by decreased CldU/ldU tract ratio, and this could be partially restored upon CCNC depletion. We also performed similar experiments in Capan-1 cells. We found that depletion of CCNC did not affect CldU/ldU tract ratio in normal condition. However, when cells were treated with HU to induce replication stress, CCNC depletion significantly increase the CldU/ldU tract ratio when compared with those in control cells (Figure 7C and D). These observations indicate that the restoration of replication fork stability by the loss of CCNC may explain PARPi resistance in BRCA2 deficient cells.
Figure 7.

Depletion of CCNC restore replication fork stability in BRCA2 deficient cells upon replication stress. (A) Schematic of ldU/CIdU pulse-labeling followed by a 4 hrs hydroxyurea (HU; 2 mM) treatment (top). Representative images of ldU and CIdU replication tracks in HU-treated HEK293A BRCA2 mAID CCNC-KO and WT cells with (+IAA) or without BRCA2 depletion are shown (bottom). (B) Dot plots of CIdU to ldU tract length ratios for individual replication forks in cells from (A). The median value of 100 or more IdU and CldU tracts per experimental condition is indicated. Statistical analysis was conducted using Mann-Whitney test (n.s. not significant, ***P < 0.001). (C) Schematic of ldU/CIdU pulse-labeling followed by a 4 h hydroxyurea (HU; 2 mM) treatment (top). Representative images of ldU and CIdU replication tracks in DMSO or HU-treated Capan-1 LentiV2 and CCNC g1 cells are shown (bottom). (D) Dot plots of CIdU to ldU tract length ratios for individual replication forks in cells from (C). The median value of 100 or more IdU and CldU tracts per experimental condition is indicated. Statistical analysis was conducted using Mann–Whitney test (n.s. not significant, ***P < 0.001).
DISCUSSION
PARPi-based therapy is becoming standard of care for breast and ovarian cancer patients with BRCA mutations. With PARPi resistance emerging in the clinic, a key objective is to enhance PARPi-based therapy and overcome therapy resistance. In this study, we used an AID system to inducibly degrade BRCA2 in HEK293A cells and carried out unbiased whole-genome CRISPR gRNA screens to identify genetic alterations that alter cell lethality caused by BRCA2 loss. We identified APEX2, APEX1, POLQ, and PARP1 as BRCA2 synthetic lethal targets, which is consistent with the results of several previous studies (23–27). Moreover, we discovered that loss of RNA Pol II transcription Mediator components like CCNC, MED12, CDK8 and MED24 led to improved survival of PARPi-treated and untreated BRCA2-depleted cells.
Because the Mediator is best known for its function with RNA Pol II in regulating gene transcription and MED12 loss is reported to confer resistance to treatment with ALK, EGFR, BRAF and MEK inhibitors and cytotoxic agents in various cancer cell types through activation of TGF-beta signaling, via promotion of TGF-βR2 maturation and induction of partial EMT (64), we performed total mRNA-Seq using control and BRCA2-depleted CCNC- and MED12-KO cells. We found that indeed many genes involved in the TGF-beta signaling pathway and ECM-receptor interaction pathway were changed at the transcriptional level. However, inhibition of TGF-beta signaling or ECM-receptor interaction pathway did not restore the lethality and PARPi sensitivity in BRCA2 deficient cells, suggesting that dysregulation of those signaling pathways could not fully account for the resistance mechanisms due to loss of CCNC or other Mediator components in BRCA2-deficient cells.
To explore the potential mechanisms underlying this synthetic viability relationship, we next checked that the role of CCNC in the regulation of HR and replication fork stability in BRCA2 deficient cells. We found that depletion of CCNC could not restore HR, but it decreased DNA damage signal in BRCA2 deficient cells upon PARPi treatment, suggesting that CCNC loss may help cell survival in a way other than the restoration of HR. Interestingly, we showed that depletion of CCNC was able to restore replication fork stability in BRCA2 deficient cells, which may account for decreased DNA damage signal. Taken together, our findings suggest that CCNC is one of the critical genetic determinants that dictate responses to treatment with PARPi in BRCA2-deficient cells with its potential role in regulating DNA replication fork stability upon replication stress. These data may help designing better treatment strategies for patients with BRCA2 mutated cancers.
The TGF-beta signaling pathway plays a complex role in tumorigenesis, including epithelial to mesenchymal transition (EMT) (74), angiogenesis (75), tumor-cell motility and metastasis (76), cancer-associated fibroblast (CAF) proliferation (77), and immunosuppression (78). MED12 loss is reported to confer resistance to treatment with ALK, EGFR, BRAF and MEK inhibitors and cytotoxic agents in various cancer cell types through activation of TGF-beta signaling via promotion of TGF-βR2 maturation and induction of partial EMT (64). In our study, we found that besides the loss of MED12, depletion of other Mediator components like CCNC, MED24, and CDK8 also significantly enhanced cell viability when BRCA2 ablation was induced, with or without PARPi treatment. Moreover, CCNC-KO cells exhibited resistance to treatment with multiple, although not all, DNA-damaging agents (Figure 4), raising the possibility that CCNC acts similarly to MED12 and confers resistance to a number of anticancer drugs. Thus, CCNC loss may be a general biomarker for drug resistance in multiple cancer types.
Several Mediator components have been linked with various diseases. For example, CCNC locus (6q21) was associated with loss or translocation in about 10% of childhood acute lymphoblastic leukemia (T-ALL) (79). Deletion in the proximal arm of 6q is observed in about one third of prostate cancers (80). Mutations of MED12 are found in X-linked dominant mental retardation as well as prostate cancer and uterine leiomyoma (81–84). A mutation in MED17 is associated with infantile cerebral atrophy (85), and a mutation in MED23 co-segregates with intellectual disability (86). It is possible that some BRCA2-mutant cancer patients may experience resistance to PARPi-based treatment or have pre-existing resistance to PARPi due to loss or functional inactivation of some of these mediator genes. We queried the TCGA PanCan 2018 database available at cBioPortal (https://www.cbioportal.org/) to determine the distribution of different alterations of CCNC and MED12 (gene amplification, deep deletions, mutations, fusions, and multiple alterations) at the DNA level in clinical tumor samples. Deep deletion of CCNC was most common in several types of cancer and some other genetic alterations were also found (Supplementary Figure S7A). Most cancer types had missense mutations in MED12, but few cancer types also had amplification and deep deletion of MED12 (Supplementary Figure S7B). We further checked whether the levels of CCNC and MED12 RNA expression in tumors correlate with patient overall survival (OS). By using RNA-Seq data in the pan-cancer dataset of Kaplan-Maier Plotter (kmplot.com), we found that the expression of both CCNC and MED12 was correlated with increased overall survival durations in kidney renal clear cell carcinoma, thymoma, and rectum adenocarcinoma patients (Supplementary Figure S7C and D), suggesting that altered expression of these genes might contribute to drug resistance and shorten survival of patients. However, this is not the case for all types of cancer. For example, the higher expression of CCNC and MED12 in breast cancer patients was associated with poor prognosis (Supplementary Figure S7C and D), indicating complex and likely tissue-specific functions of these Mediator complex components in tumor progression and therapy resistance that warrant further investigation.
Although detailed mechanisms of Mediator-loss induced resistance still need to be further investigated, our findings link it with replication fork protection in BRCA2 deficient cells upon replication stress. These observations may provide new insights into potential transcriptional-independent roles of Mediator in the cell. R-loops which are DNA–RNA hybrids formed during transcription can potentially interfere with DNA replication progression (87,88). BRCA2 has been reported to associate with the TREX2 mRNA export factor PCID2 and RNA polymerase (Pol) II to prevent R-loop accumulation, which can lead to replication fork stalling and dysregulated transcriptional elongation (14,15). Mediator may help dissolve R-loop formation or accumulation caused by BRCA2 loss. Of course, further studies are needed to test these hypotheses.
In summary, our unexpected results suggest that some of the RNA Pol II transcription Mediator components especially CCNC may be critical genetic determinants that dictate responses to PARPi treatment in BRCA2-deficient cells and act as general determinants of drug response in multiple cancer types, which may help in design of new strategies for the treatment of BRCA2 mutated and other types of cancers. This opens a new direction for studying cell survival following DNA damage repair defects and/or treatment with various anticancer agents, which may not directly influence DNA repair but promotes general survival following stress.
DATA AVAILABILITY
RNA-seq data generated in this study are available at NCBI GEO database with the accession number GSE150067.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Chen laboratory for their help and constructive discussion. We also thank Donald R Norwood in Scientific Publications, Research Medical Library, MD Anderson for help with the scientific editing of the manuscript.
Author contributions: M.T. and J.C. conceived the project. M.T., G.P., D.S., X.F., M.S., C.W. and Z.C. performed the experiments. G.P. and Z.Z. helped with RNA-Seq experiments. M.T. and J.C. wrote the manuscript with input from all authors.
Contributor Information
Mengfan Tang, Department of Experimental Radiation Oncology, Unit 1052, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Guangsheng Pei, Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Dan Su, Department of Experimental Radiation Oncology, Unit 1052, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Chao Wang, Department of Experimental Radiation Oncology, Unit 1052, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Xu Feng, Department of Experimental Radiation Oncology, Unit 1052, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Mrinal Srivastava, Department of Experimental Radiation Oncology, Unit 1052, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Zhen Chen, Department of Experimental Radiation Oncology, Unit 1052, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Zhongming Zhao, Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA; Human Genetics Center, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Junjie Chen, Department of Experimental Radiation Oncology, Unit 1052, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Cancer Prevention and Research Institute of Texas (CPRIT) [RP180813 to J.C., in part]; CPRIT [RP160667, in part]; National Institutes of Health (NIH) [R01CA210929, R01CA216911, R01CA216437 to J.C.]; J.C. also received support from the Pamela and Wayne Garrison Distinguished Chair in Cancer Research; MD Anderson's Flow Cytometry and Cellular Imaging Facility (supported by MD Anderson's NIH Cancer Center Support Grant) [P30CA016672]; UTHealth Cancer Genomics Core for RNA-seq data generation and analysis (supported by the CPRIT) [RP180734]. Funding for open access charge: CPRIT [RP180813].
Conflict of interest statement. None declared.
REFERENCES
- 1. Aparicio T., Baer R., Gautier J.. DNA double-strand break repair pathway choice and cancer. DNA Repair (Amst.). 2014; 19:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ceccaldi R., Rondinelli B., D’Andrea A.D.. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016; 26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kowalczykowski S.C. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2015; 7:a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pannunzio N.R., Watanabe G., Lieber M.R.. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018; 293:10512–10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prakash R., Zhang Y., Feng W., Jasin M.. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015; 7:a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodgers K., McVey M.. Error-prone repair of DNA double-strand breaks. J. Cell. Physiol. 2016; 231:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lord C.J., Ashworth A.. The DNA damage response and cancer therapy. Nature. 2012; 481:287–294. [DOI] [PubMed] [Google Scholar]
- 8. Jiang Q., Greenberg R.A.. Deciphering the BRCA1 tumor suppressor network. J. Biol. Chem. 2015; 290:17724–17732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gorodetska I., Kozeretska I., Dubrovska A.. BRCA genes: the role in genome stability, cancer stemness and therapy resistance. J. Cancer. 2019; 10:2109–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mersch J., Jackson M.A., Park M., Nebgen D., Peterson S.K., Singletary C., Arun B.K., Litton J.K.. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015; 121:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cavanagh H., Rogers K.M.. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin. Pract. 2015; 13:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roy R., Chun J., Powell S.N.. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer. 2011; 12:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M.. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011; 145:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shivji M.K.K., Renaudin X., Williams C.H., Venkitaraman A.R.. BRCA2 regulates transcription elongation by RNA polymerase II to prevent R-loop accumulation. Cell Rep. 2018; 22:1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhatia V., Barroso S.I., Garcia-Rubio M.L., Tumini E., Herrera-Moyano E., Aguilera A.. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014; 511:362–365. [DOI] [PubMed] [Google Scholar]
- 16. Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T.. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917. [DOI] [PubMed] [Google Scholar]
- 17. Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C.et al.. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921. [DOI] [PubMed] [Google Scholar]
- 18. Pommier Y., O’Connor M.J., de Bono J.. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016; 8:362ps317. [DOI] [PubMed] [Google Scholar]
- 19. Godet I., Gilkes D.M.. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr. Cancer Sci. Ther. 2017; 4: 10.15761/ICST.1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ison G., Howie L.J., Amiri-Kordestani L., Zhang L., Tang S., Sridhara R., Pierre V., Charlab R., Ramamoorthy A., Song P.et al.. FDA approval summary: Niraparib for the maintenance treatment of patients with recurrent ovarian cancer in response to platinum-based chemotherapy. Clin. Cancer Res. 2018; 24:4066–4071. [DOI] [PubMed] [Google Scholar]
- 21. Konecny G.E., Kristeleit R.S.. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions. Br. J. Cancer. 2016; 115:1157–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zimmer A.S., Gillard M., Lipkowitz S., Lee J.M.. Update on PARP inhibitors in breast cancer. Curr. Treat. Options Oncol. 2018; 19:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mateos-Gomez P.A., Gong F., Nair N., Miller K.M., Lazzerini-Denchi E., Sfeir A.. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015; 518:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ceccaldi R., Liu J.C., Amunugama R., Hajdu I., Primack B., Petalcorin M.I., O’Connor K.W., Konstantinopoulos P.A., Elledge S.J., Boulton S.J.et al.. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015; 518:258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dai C.H., Chen P., Li J., Lan T., Chen Y.C., Qian H., Chen K., Li M.Y.. Co-inhibition of pol theta and HR genes efficiently synergize with cisplatin to suppress cisplatin-resistant lung cancer cells survival. Oncotarget. 2016; 7:65157–65170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gourley C., Balmana J., Ledermann J.A., Serra V., Dent R., Loibl S., Pujade-Lauraine E., Boulton S.J.. Moving from poly (ADP-Ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J. Clin. Oncol. 2019; 37:2257–2269. [DOI] [PubMed] [Google Scholar]
- 27. Mengwasser K.E., Adeyemi R.O., Leng Y., Choi M.Y., Clairmont C., D’Andrea A.D., Elledge S.J.. Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Mol. Cell. 2019; 73:885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S.et al.. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015; 163:1515–1526. [DOI] [PubMed] [Google Scholar]
- 29. Wang C., Wang G., Feng X., Shepherd P., Zhang J., Tang M., Chen Z., Srivastava M., McLaughlin M.E., Navone N.M.et al.. Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene. 2019; 38:2451–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hart T., Tong A.H.Y., Chan K., Van Leeuwen J., Seetharaman A., Aregger M., Chandrashekhar M., Hustedt N., Seth S., Noonan A.et al.. Evaluation and design of Genome-Wide CRISPR/SpCas9 knockout screens. G3 (Bethesda). 2017; 7:2719–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S.. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014; 15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feng L., Fong K.W., Wang J., Wang W., Chen J.. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 2013; 288:11135–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quinet A., Tirman S., Jackson J., Svikovic S., Lemacon D., Carvajal-Maldonado D., Gonzalez-Acosta D., Vessoni A.T., Cybulla E., Wood M.et al.. PRIMPOL-Mediated adaptive response suppresses replication fork reversal in BRCA-deficient cells. Mol. Cell. 2020; 77:461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tang M., Feng X., Pei G., Srivastava M., Wang C., Chen Z., Li S., Zhang H., Zhao Z., Li X.et al.. FOXK1 participates in DNA damage response by controlling 53BP1 function. Cell Rep. 2020; 32:108018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anders S., Huber W.. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B (Methodological). 1995; 57:289–300. [Google Scholar]
- 38. Fresno C., Fernandez E.A.. RDAVIDWebService: a versatile R interface to DAVID. Bioinformatics. 2013; 29:2810–2811. [DOI] [PubMed] [Google Scholar]
- 39. Sharan S.K., Morimatsu M., Albrecht U., Lim D.S., Regel E., Dinh C., Sands A., Eichele G., Hasty P., Bradley A.. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997; 386:804–810. [DOI] [PubMed] [Google Scholar]
- 40. Kuznetsov S.G., Liu P., Sharan S.K.. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat. Med. 2008; 14:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ding X., Philip S., Martin B.K., Pang Y., Burkett S., Swing D.A., Pamala C., Ritt D.A., Zhou M., Morrison D.K.et al.. Survival of BRCA2-Deficient cells is promoted by GIPC3, a novel genetic interactor of BRCA2. Genetics. 2017; 207:1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Natsume T., Kiyomitsu T., Saga Y., Kanemaki M.T.. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 2016; 15:210–218. [DOI] [PubMed] [Google Scholar]
- 43. Holland A.J., Fachinetti D., Han J.S., Cleveland D.W.. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E3350–E3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hart T., Tong A.H.Y., Chan K., Van Leeuwen J., Seetharaman A., Aregger M., Chandrashekhar M., Hustedt N., Seth S., Noonan A.et al.. Evaluation and design of genome-wide CRISPR/SpCas9 knockout screens. G3-Genes Genom Genet. 2017; 7:2719–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thompson C.M., Young R.A.. General requirement for RNA polymerase II holoenzymes in vivo. Proc. Natl Acad. Sci. U.S.A. 1995; 92:4587–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim Y.J., Bjorklund S., Li Y., Sayre M.H., Kornberg R.D.. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell. 1994; 77:599–608. [DOI] [PubMed] [Google Scholar]
- 47. Thompson C.M., Koleske A.J., Chao D.M., Young R.A.. A multisubunit complex associated with the RNA polymerase II CTD and TATA-binding protein in yeast. Cell. 1993; 73:1361–1375. [DOI] [PubMed] [Google Scholar]
- 48. Flanagan P.M., Kelleher R.J. 3rd, Sayre M.H., Tschochner H., Kornberg R.D.. A mediator required for activation of RNA polymerase II transcription in vitro. Nature. 1991; 350:436–438. [DOI] [PubMed] [Google Scholar]
- 49. Bourbon H.M. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 2008; 36:3993–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soutourina J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018; 19:262–274. [DOI] [PubMed] [Google Scholar]
- 51. Soutourina J., Wydau S., Ambroise Y., Boschiero C., Werner M.. Direct interaction of RNA polymerase II and mediator required for transcription in vivo. Science. 2011; 331:1451–1454. [DOI] [PubMed] [Google Scholar]
- 52. Clark A.D., Oldenbroek M., Boyer T.G.. Mediator kinase module and human tumorigenesis. Crit. Rev. Biochem. Mol. Biol. 2015; 50:393–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Poss Z.C., Ebmeier C.C., Taatjes D.J.. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013; 48:575–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dynlacht B.D. Regulation of transcription by proteins that control the cell cycle. Nature. 1997; 389:149–152. [DOI] [PubMed] [Google Scholar]
- 55. Bregman D.B., Pestell R.G., Kidd V.J.. Cell cycle regulation and RNA polymerase II. Front. Biosci. 2000; 5:D244–D257. [DOI] [PubMed] [Google Scholar]
- 56. Krakoff I.H., Brown N.C., Reichard P.. Inhibition of ribonucleoside diphosphate reductase by hydroxyurea. Cancer Res. 1968; 28:1559–1565. [PubMed] [Google Scholar]
- 57. Nordlund P., Reichard P.. Ribonucleotide reductases. Annu. Rev. Biochem. 2006; 75:681–706. [DOI] [PubMed] [Google Scholar]
- 58. Scozzafava A., Supuran C.T.. Hydroxyurea is a carbonic anhydrase inhibitor. Bioorg. Med. Chem. 2003; 11:2241–2246. [DOI] [PubMed] [Google Scholar]
- 59. Campestre C., Agamennone M., Tortorella P., Preziuso S., Biasone A., Gavuzzo E., Pochetti G., Mazza F., Hiller O., Tschesche H.et al.. N-Hydroxyurea as zinc binding group in matrix metalloproteinase inhibition: mode of binding in a complex with MMP-8. Bioorg. Med. Chem. Lett. 2006; 16:20–24. [DOI] [PubMed] [Google Scholar]
- 60. Juul T., Malolepszy A., Dybkaer K., Kidmose R., Rasmussen J.T., Andersen G.R., Johnsen H.E., Jorgensen J.E., Andersen S.U.. The in vivo toxicity of hydroxyurea depends on its direct target catalase. J. Biol. Chem. 2010; 285:21411–21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yi D., Alvim Kamei C.L., Cools T., Vanderauwera S., Takahashi N., Okushima Y., Eekhout T., Yoshiyama K.O., Larkin J., Van den Daele H.et al.. The Arabidopsis SIAMESE-RELATED cyclin-dependent kinase inhibitors SMR5 and SMR7 regulate the DNA damage checkpoint in response to reactive oxygen species. Plant Cell. 2014; 26:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davies B.W., Kohanski M.A., Simmons L.A., Winkler J.A., Collins J.J., Walker G.C.. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol. Cell. 2009; 36:845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nakayashiki T., Mori H.. Genome-wide screening with hydroxyurea reveals a link between nonessential ribosomal proteins and reactive oxygen species production. J. Bacteriol. 2013; 195:1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Huang S., Holzel M., Knijnenburg T., Schlicker A., Roepman P., McDermott U., Garnett M., Grernrum W., Sun C., Prahallad A.et al.. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell. 2012; 151:937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang Y., Alexander P.B., Wang X.F.. TGF-beta family signaling in the control of cell proliferation and survival. Cold Spring Harb. Perspect. Biol. 2017; 9:a022145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jung S.M., Lee J.H., Park J., Oh Y.S., Lee S.K., Park J.S., Lee Y.S., Kim J.H., Lee J.Y., Bae Y.S.et al.. Smad6 inhibits non-canonical TGF-beta1 signalling by recruiting the deubiquitinase A20 to TRAF6. Nat. Commun. 2013; 4:2562. [DOI] [PubMed] [Google Scholar]
- 67. Nakashima H., Tsujimura K., Irie K., Ishizu M., Pan M., Kameda T., Nakashima K.. Canonical TGF-beta signaling negatively regulates neuronal morphogenesis through TGIF/Smad Complex-Mediated CRMP2 suppression. J. Neurosci. 2018; 38:4791–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Koganti P., Levy-Cohen G., Blank M.. Smurfs in protein homeostasis, signaling, and cancer. Front. Oncol. 2018; 8:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Frederick J.P., Liberati N.T., Waddell D.S., Shi Y., Wang X.F.. Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol. Cell. Biol. 2004; 24:2546–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. DiVito K.A., Simbulan-Rosenthal C.M., Chen Y.S., Trabosh V.A., Rosenthal D.S.. Id2, Id3 and Id4 overcome a Smad7-mediated block in tumorigenesis, generating TGF-beta-independent melanoma. Carcinogenesis. 2014; 35:951–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Noordermeer S.M., van Attikum H.. PARP inhibitor resistance: a tug-of-war in BRCA-mutated cells. Trends Cell Biol. 2019; 29:820–834. [DOI] [PubMed] [Google Scholar]
- 72. Schlacher K., Wu H., Jasin M.. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012; 22:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Taglialatela A., Alvarez S., Leuzzi G., Sannino V., Ranjha L., Huang J.W., Madubata C., Anand R., Levy B., Rabadan R.et al.. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell. 2017; 68:414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. David C.J., Huang Y.H., Chen M., Su J., Zou Y., Bardeesy N., Iacobuzio-Donahue C.A., Massague J.. TGF-beta tumor suppression through a lethal EMT. Cell. 2016; 164:1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sounni N.E., Dehne K., van Kempen L., Egeblad M., Affara N.I., Cuevas I., Wiesen J., Junankar S., Korets L., Lee J.et al.. Stromal regulation of vessel stability by MMP14 and TGFbeta. Dis Model Mech. 2010; 3:317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Friedl P., Alexander S.. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011; 147:992–1009. [DOI] [PubMed] [Google Scholar]
- 77. Calon A., Espinet E., Palomo-Ponce S., Tauriello D.V., Iglesias M., Cespedes M.V., Sevillano M., Nadal C., Jung P., Zhang X.H.et al.. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012; 22:571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Thomas D.A., Massague J.. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005; 8:369–380. [DOI] [PubMed] [Google Scholar]
- 79. Li H., Lahti J.M., Valentine M., Saito M., Reed S.I., Look A.T., Kidd V.J.. Molecular cloning and chromosomal localization of the human cyclin C (CCNC) and cyclin E (CCNE) genes: deletion of the CCNC gene in human tumors. Genomics. 1996; 32:253–259. [DOI] [PubMed] [Google Scholar]
- 80. Cooney K.A., Wetzel J.C., Consolino C.M., Wojno K.J.. Identification and characterization of proximal 6q deletions in prostate cancer. Cancer Res. 1996; 56:4150–4153. [PubMed] [Google Scholar]
- 81. Kampjarvi K., Park M.J., Mehine M., Kim N.H., Clark A.D., Butzow R., Bohling T., Bohm J., Mecklin J.P., Jarvinen H.et al.. Mutations in Exon 1 highlight the role of MED12 in uterine leiomyomas. Hum. Mutat. 2014; 35:1136–1141. [DOI] [PubMed] [Google Scholar]
- 82. Barbieri C.E., Baca S.C., Lawrence M.S., Demichelis F., Blattner M., Theurillat J.P., White T.A., Stojanov P., Van Allen E., Stransky N.et al.. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012; 44:685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nishikawa J.L., Boeszoermenyi A., Vale-Silva L.A., Torelli R., Posteraro B., Sohn Y.J., Ji F., Gelev V., Sanglard D., Sanguinetti M.et al.. Inhibiting fungal multidrug resistance by disrupting an activator-Mediator interaction. Nature. 2016; 530:485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Risheg H., Graham J.M. Jr, Clark R.D., Rogers R.C., Opitz J.M., Moeschler J.B., Peiffer A.P., May M., Joseph S.M., Jones J.R.et al.. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat. Genet. 2007; 39:451–453. [DOI] [PubMed] [Google Scholar]
- 85. Kaufmann R., Straussberg R., Mandel H., Fattal-Valevski A., Ben-Zeev B., Naamati A., Shaag A., Zenvirt S., Konen O., Mimouni-Bloch A.et al.. Infantile cerebral and cerebellar atrophy is associated with a mutation in the MED17 subunit of the transcription preinitiation mediator complex. Am. J. Hum. Genet. 2010; 87:667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hashimoto S., Boissel S., Zarhrate M., Rio M., Munnich A., Egly J.M., Colleaux L.. MED23 mutation links intellectual disability to dysregulation of immediate early gene expression. Science. 2011; 333:1161–1163. [DOI] [PubMed] [Google Scholar]
- 87. Al-Hadid Q., Yang Y.. R-loop: an emerging regulator of chromatin dynamics. Acta Biochim. Biophys. Sin. (Shanghai). 2016; 48:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hamperl S., Bocek M.J., Saldivar J.C., Swigut T., Cimprich K.A.. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 2017; 170:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data generated in this study are available at NCBI GEO database with the accession number GSE150067.