


Abstract
The enormous sequence heterogeneity of telomerase RNA (TR) subunits has thus far complicated their characterization in a wider phylogenetic range. Our recent finding that land plant TRs are, similarly to known ciliate TRs, transcribed by RNA polymerase III and under the control of the type-3 promoter, allowed us to design a novel strategy to characterize TRs in early diverging Viridiplantae taxa, as well as in ciliates and other Diaphoretickes lineages. Starting with the characterization of the upstream sequence element of the type 3 promoter that is conserved in a number of small nuclear RNAs, and the expected minimum TR template region as search features, we identified candidate TRs in selected Diaphoretickes genomes. Homologous TRs were then used to build covariance models to identify TRs in more distant species. Transcripts of the identified TRs were confirmed by transcriptomic data, RT-PCR and Northern hybridization. A templating role for one of our candidates was validated in Physcomitrium patens. Analysis of secondary structure demonstrated a deep conservation of motifs (pseudoknot and template boundary element) observed in all published TRs. These results elucidate the evolution of the earliest eukaryotic TRs, linking the common origin of TRs across Diaphoretickes, and underlying evolutionary transitions in telomere repeats.
Graphical Abstract
Graphical Abstract.
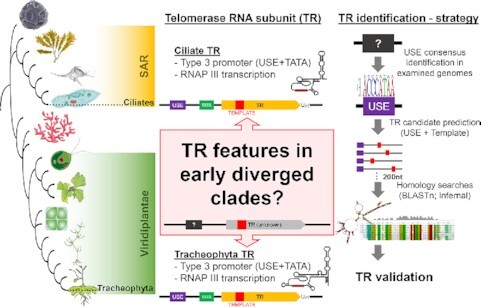
We present a smart strategy of telomerase RNA (TR) identification based on its conserved type-3 RNA Pol III promoter and TR template elements. We characterize TRs in early diverging Viridiplantae taxa, as well as in ciliates and other Diaphoretickes lineages. TRs are validated experimentally and show conservation of core TR structural domains. These results shed light on the evolution of a key eukaryotic non-coding RNA across more than a billion years.
INTRODUCTION
The origin of linear chromosomes, emergence of the end-replication problem and its solution through telomeres and their elongation by telomerase, have been associated with the earliest steps of eukaryotic evolution (1). Telomerase, a specific ribonucleoprotein enzyme complex, elongates telomeres by the catalytic activity of its Telomerase Reverse Transcriptase (TERT) subunit while using a short region of the associated Telomerase RNA (TR) as a template for synthesis. Although the ancient origin of telomerase corresponds with a number of conserved motifs shared between TERT and other reverse transcriptases (reviewed in (2)), its conservation is not entirely consistent with an apparent diversity of TRs even among relatively narrow taxonomic groups (3,4).
A new impetus in the research of how telomerase has evolved came with the recent identification of TRs across vascular plants, the first bona fide TRs in the plant kingdom (5–7). Despite the overall extensive TR variability in sequence, structure and biogenesis pathways, land plant TRs show a monophyletic origin and also a remarkable similarity to ciliate TRs in one particular aspect: both land plant and ciliate TRs are RNA Polymerase III (RNAPIII) transcripts (as the only known TRs so far) containing type 3 snRNA promoters (5,8–10) (Figure 1).
Figure 1.
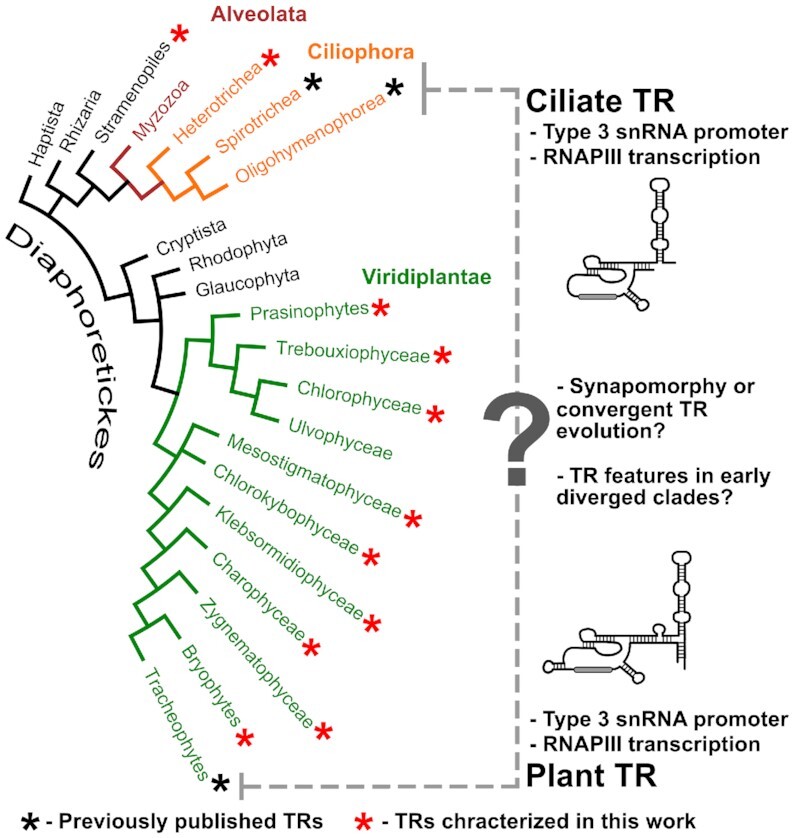
Cladogram of the eukaryote megagroup Diaphoretickes (according to (15)). Taxa with a TR gene identified in this study are highlited by a red asterisk, taxa with a known TR sequence by a black asterisk. These include OIigohymenophorea and Spirotrichea from Ciliates on the one side, and Tracheophytes on the other side. TRs in both, substantially distant, groups share the type 3 RNAPIII promoter which is characterized by its relatively conserved promoter sequence motif termed as Upstream Sequence Element (USE). In this study, we tested a hypothesis that TRs in other clades from Diaphoretickes also have the type 3 promoter whose USE sequence could be exploited for filtering/prediction of novel TR candidates.
Type 3 promoters are typical for a wide range of snRNAs (i.e. major spliceosomal RNAs (U1, U2, U4, U5, U6), U3 snoRNA, signal recognition particle RNA (SRP), and the RNA subunit of RNase for mitochondrial RNA processing (MRP). Type 3 promoters contain a conserved sequence motif upstream of the transcription start site (TSS) called the Upstream Sequence Element (USE). USE is specifically recognized by the snRNA activating protein complex (SNAPc), a core promoter factor, which activates snRNA transcription by its recruitment to USE and protein-protein interactions with other transcription factors (11), (Figure 2A). In contrast to the other RNAPIII promoters, type 3 promoters can selectively drive either RNAPII or RNAPIII transcription. In vascular plants and ciliates, this selective recruitment is determined by a specific distance between the USE and TATA box (12–14). For example, in Arabidopsis thaliana, USE-TATA spacing of 32–34 bp, and 23–24 bp is characteristic for RNAPII and III transcription, respectively (13). The enormous heterogeneity of TR sequences usually represents a major obstacle and complication when searching for TRs in evolutionary distant species/clades. However, for known TRs in plants and ciliates, the TR promoter type remains far more conserved during evolution than the TR sequence (5,9). Here, we investigate whether the type 3 promoter also plays a role in controlling TR transcription in early diverging plant and ciliate lineages or other lineages from the Diaphoretickes megagroup, as shown in Figure 1 according to (15). To do so, we predict TR candidates in genome assemblies, equipped only with the putative promoter motif (USE) and TR template region as searched features, as illustrated in Figure 2B. For the TR template, we consider all circular permutations of the telomeric C-rich strand motif + one nucleotide as a minimum TR-telomere annealing sequence.
Figure 2.

A comprehensive diagram showing key methodological aspects of this study. These include: (A) schematic view of a type 3 promoter including typical type 3 promoter-driven snRNA genes; (B) TR characterization strategy based on our assumption that TR promoter type can be far more conserved through evolution than TR sequence; (C) a detailed workflow of TR identification starting with USE characterization (in purple), followed by TR candidate prediction based on conserved USE and putative template sequence (in yellow), subsequent homology searches (in green) and finally experimental validation of novel TRs (in blue).
A multistep filtering scheme revealed putative TRs sharing sequence homology across Viridiplantae—linking Chlorophytes, streptophyte algae, bryophytes and, importantly, previously identified TRs from land plants (5). We validated our concept of using USE/Template to identify novel putative TRs by an independent prediction of TRs in Heterotrichea (Postciliodesmatophora), an early diverging clade to the other ciliates from Intramacronucleata. Similarly, we proposed a convincing TR candidate in the Stramenopile genus Blastocystis (Figure 1).
Selected TR candidates from Viridiplantae were supported using whole transcriptomic data, RT-PCR and Northern hybridization experiments, as well as the identification of key structural elements (e.g. pseudoknots) known to be ubiquitously present and required for function across known eukaryotic TRs. Finally, we confirmed the biological function for one of our in silico predicted TRs in the model bryophyte, Physcomitrium (formerly Physcomitrella) patens.
Thanks to the conserved type 3 promoter (= conserved USE) and the extensive availability of genomic/transcriptomic data in numerous species, we present here an unprecedented set of ∼120 putative telomerase RNAs. These results shed light on the evolution of a key eukaryotic non-coding RNA across more than a billion years, suggesting that TR evolution underlies evolutionary transitions in telomere DNA repeats including those described in algae (16).
MATERIALS AND METHODS
USE characterization
Typical type 3 promoters for snRNAs were sought in genome assemblies from Viridiplantae (without Tracheophytes), Rhodophyte, Haptista, Cryptophyta and SAR by using Infernal 1.1.2 tool (17) with covariance models (CMs) available at RFAM database (18). These included CMs for U1 (RFAM no.: RF00003); U2 (RF00004); U3 (RF01847—for Archaeplastida genomes and RF01848—for protist genomes), U4 (RF00015); U5 (RF00020); U6 (RF00026); MRP (RF00030) and SRP (RF01855—for Archaeplastida genomes and RF01856—for protist genomes) RNAs. Significant hits in Infernal corresponding to particular snRNAs were subjected to promoter region extraction—100nt upstream from the hit start (or more nucleotides in cases, where Infernal hit did not match at the beginning of CM). Extracted promoters from each genome were subjected to screen for shared sequence motifs among themselves using MEME tool (19). MEME results for each genome were manually checked for motif(s) presence, conservation and topology, to select genomes with highly conserved USE present in most snRNA species. For schematic workflow see Figure 2C, purple panel.
TR candidate prediction
Selected genomes that showed USE presence and conservation were subjected to TR-like loci prediction. All genomic regions starting with corresponding USE and extending 200nt downstream from USE were checked, if putative template region was present. Any circular permutation of C-strand of corresponding telomere motif + one nucleotide as a minimum alignment template portion was considered as a putative template (i.e., if telomere motif was CCCTAAA, the minimum putative template could be any of the following sequences—CCCTAAAC/CCTAAACC/…/ACCCTAAA). All TR-like loci were manually checked for our subjective rating of TR-like loci (e.g., overlapping annotated genes, ORF, sequence complexity, template topology or number of similar sequences in a genome). For illustration see Figure 2C, yellow panel.
Homology searches
The TR-like loci (USE+200nt with template-like sequence) were extended by 200nt further downstream or to the putative T-rich terminator (that is typical for land plant and ciliate TRs). These sequences were used as a query for homology searches (using BLASTn) in related genomes. If significant hit/s was/were observed, subject sequence/s were checked if corresponding motifs are present (template, USE, terminator and other homologous regions). In cases where homologs were identified in BLASTn, predicted transcribed regions were aligned and folded in LocaRNA (20). Alignments in Stockholm format (.stk) were used to build covariance models (CMs) in Infernal 1.1.2 (17). CMs were used to examine whether TR-like sequences can show homologs in genomes of evolutionarily more distant species. New hits produced by Infernal tool were checked for their significance (e-value) and whether they meet our TR search parameters (template, USE). If so, newly ‘verified’ Infernal hits were used for optimization of CMs, which were subsequently employed in iterative Infernal searches. The approach is depicted in Figure 2C – green panel.
TR structure prediction
Multiple sequence alignments (MSAs) were generated for putative TR sequences in each major lineage (Bryophyta, Chlamydomonadales, Heterotrichea, Marchantiophyta, Opalinata, and Trebouxiophycea; see Supplementary Figure S3 for specific taxa and sequences). MSAs were generated in Geneious (v11.0.4) (https://www.geneious.com) using MAFFT (21) with default parameters. To identify putative pseudoknot elements, long range interaction regions surrounding the template were identified within each MSA separately using conservation or covariation as a guide (demarcated as the core-delimiting P1C/P1B). This delimited region surrounding the template was examined for pseudoknot elements with a particular focus on anchor points (conserved elements within the region) within the MSA. Predicted pseudoknot elements (P2/P3) in Bryophyta and Marchantiophyta were further supported by consensus alignments with published TR sequences from Angiosperms (6,22). The secondary structure of the long stem loop comprised of elements P4, P5, P6/7/8 was inferred by examining delimited sequences (P1C/B to the 3′ end of the MSA) with RNAalifold (v2.4.16, (23)) and manual refinement of MSAs in Geneious. For TR lineages with overall poor sequence similarity, structural elements were assigned to more closely related TRs, with additional TRs progressively incorporated into the MSA.
RNA evidence
Plant material, RNA, DNA extraction, cDNA synthesis
Samples used in the experimental part of this study are summarized in Supplementary File S1A incl. specimen voucher, culture conditions or used tissue. Total RNA was isolated from samples (100 mg/sample) using the TRI-REAGENT (Sigma-Aldrich) according to the manufacturer's protocol. RNAs were subsequently purified from contaminating DNA using TURBO™ DNase (Invitrogen). DNAs used in this study were isolated according to (24). cDNA was prepared by reverse transcription of ∼1 μg of total RNA using the M-MuLV (NEB) reverse transcriptase and Random Nonamers (Sigma).
RNAseq data
The existence of transcripts associated with predicted TR loci showing homology across large taxonomic groups of green plants was demonstrated in species where total RNA-seq data (using rRNA depletion) were available in Sequence Read Archive (SRA) data at NCBI, or in data generated by us (for more details see ‘Data availability’ and Supplementary Table S2). RNA-seq libraries were prepared using TruSeq Stranded Total RNA with Ribo-Zero Plant kit (Illumina) from 1 μg of total RNA as input (concentration and quality checked on Agilent 2200 TapeStation system (Agilent Technologies). Strand-specific Paired-End libraries were sequenced on NovaSeq 6000 System (Illumina) using sequencing kit NovaSeq 6000 SP Reagent Kit v1.5 (200 cycles) (Illumina). In cases where corresponding genome assemblies were available, RNAseq reads were mapped to reference genome using RMTA (25), otherwise RNAseq data were assembled de novo using TRINITY v2.11.0 (26). The assembly was done with options for stranded RNA-seq with paired-end fastq data (Trinity –seqType fq –left inputfile_R1.fastq –right inputfile_R2.fastq –SS_lib_type RF –KMER_SIZE 25). TR transcript presence, length and orientation were checked in these data.
Northern hybridization
Total RNAs (5 μg or 10 μg) with RNA Loading Dye (NEB) were denatured for 5′ at 65°C in thermal cycler and separated by electrophoresis (Mini-Protean Tetracell BioRad apparatus) in 7% denaturing polyacrylamide gel with 8M urea in 1× Tris/borate/EDTA (TBE) solution at 100 V for 5′, then at 150 V for 50′, constantly heated in water bath at 45°C. Low Range ssRNA Ladder (NEB) was used as molecular weight standard. Gel was stained by SYBR™ Gold (Thermo Scientific) and transferred to Amersham Hybond-XL (GE Healthcare) membrane using Trans-Blot® Turbo™ Transfer System (BioRad). Membrane was washed in 1× TBE and stored for hybridization with radioactively labelled dsDNA TR probes (Supplementary File S1B). TR probes were labelled using the DecaLabel DNA labelling kit protocol (Thermo Scientific). The membrane was hybridized overnight at 55°C with the respective [32P-dATP]-labelled TR probe and signals were visualized using a PhosphoImager FLA-7000 (GE Healthcare). For more details about TR probes and used chemicals see Supplementary File S1C.
Prediction of telomere sequence using TRFi
The analysis of short tandem repeats in raw genomic data or genome assemblies was performed by Tandem Repeats Finder (TRFi) (27) with custom made scripts as described previously (28), using setup—repeat motif length: 5–15 nt long; minimum of such repeats in tandem: 5 or 3 units. Candidate repeats for telomere sequence usually occur among most abundant tandem motifs in whole genome data, as was demonstrated previously (22,28). Telomere candidate motifs from TRFi were compared with putative template regions of identified TRs. Its ‘terminal’ localization on genome contigs/scaffolds (if available) were visually checked in Geneious 8.1 (https://www.geneious.com).
TR mutants in Physcomitrium patens
The TR knock-out mutants were generated via gene targeting (GT) by a replacement of the TR locus with a 35S:HygR cassette via homology directed repair (HDR) using Cas9-induced DNA double-strand cleavage within the TR locus. The construct for GT (Supplementary File S1D) was assembled in GoldenBraid cloning system as 35S:HygR cassette flanked by 900 bp long 5′ targeting fragment and 842 bp long 3′ targeting fragment (which was synthesized in two parts due to elimination of BsaI restriction site). The Gateway destination vector containing Cas9 and nptII expression cassettes (pMK-Cas9-gate) and entry vector containing the PpU6 promoter and sgRNA (pENTR-PpU6sgRNA-L1L2) were kindly provided by prof. Bezanilla (29). A protospacer targeting TR locus was designed in the CRISPOR online software (30) using P. patens (Phytozome V11) and S. pyogenes (5′ NGG 3′) as the genome and PAM parameters, respectively. The protospacer with the highest specificity score was chosen and subsequently synthesized as two complementary oligonucleotides—TR_sgRNA_1 and TR_sgRNA_2 (Supplementary File S1B). Four nucleotides were added to the 5′ ends of the oligonucleotides such that, when annealed, they create sticky ends compatible with BsaI‐linearized pENTR-PpU6sgRNA-L1L2. 500 pmol of each oligonucleotide for sgRNA spacer were mixed together and incubated for 1 h in room temperature to anneal together. The final product was ligated into BsaI-linearized pENTR-pU6sgRNA-L1L2 using T4 DNA ligase (Thermo Scientific). Cas9/sgRNA expression vector was generated using Gateway LR reaction to recombine the entry vector pENTR-PpU6sgRNA-L1L2 with sgRNA spacer and destination vector pMK-Cas9-gate (Invitrogen). DNA constructs were delivered into protoplasts by PEG-mediated transformation as described in (31). To generate knock-out mutants of TR (pptr plants), protoplasts isolated from 7 days old protonema were co-transformed with ∼10 μg of circular Cas9/sgRNA expression vector and 30 μg of gene targeting construct – linear DNA fragment containing the 35S:HygrR flanked by targeting sequences amplified by PCR (Supplementary File S1D). After five days of regeneration the transformed protoplasts were transferred to a Petri dish with BCDAT medium supplemented with antibiotics—30 mg/l Hygromycin. After three rounds of selection the knock-out mutants were considered to be stable. Successful replacement of the TR locus with a targeting construct was detected by PCR (Supplementary File S1B) using ‘outward-pointing’ primers Hyg1023_F and Hyg1024_R specific to the 35S:Hygro cassette in combination with ‘inward-pointing’ 5′- and 3′-gene-specific primers KO1322_F and KO1323_R corresponding to sequences external to the targeting construct.
Telomere repeat amplification protocol (TRAP)
Protein extracts from 7-day-old protonema tissue of P. patens were prepared according to (5) and diluted to 250 ng μl−1. These extracts were subjected to the TRAP assay based on the elongation of substrate primer TS21 by the telomerase and subsequent PCR amplification of the extension products as described in (32,33).
Terminal restriction fragment analysis (TRF)
Telomere length analysis was performed as described in (34) by Southern hybridization of terminal restriction fragments (TRF) produced by digestion with Tru1I restriction endonuclease (Thermo Scientific). A membrane was hybridized overnight at 55°C with telomere probe (Supplementary File S1C). Telomere probe was synthesized by non-template PCR according to (35) and radioactively labelled with [32P-dATP] using DecaLabel DNA labelling kit (Thermo Scientific). Telomere signals were visualized using a PhosphoImager FLA-7000 (GE Healthcare). Telomere lengths were calculated by WALTER toolset v2.0 (36) using default setup with background correction.
RESULTS
TR candidate prediction
USE characterization
To predict TR candidates based on a putative template region (according to the known or predicted telomere repeat motifs, see below) and USE sequence within the conserved type 3 RNAPIII promoter, we first had to characterize the specific USE sequence itself. For this purpose, we used typical type 3 promoters of snRNA genes. These included U1, U2, U3, U4, U5, U6, SRP and MRP snRNAs. A subsequent screen for shared motifs among their promoter regions revealed USE presence or absence in respective snRNAs and a degree of USE sequence conservation (as illustrated in Figure 2C, marked in purple). Using this method, we screened available genome assemblies from the megagroup Diaphoretickes (Archeplastida + Hacrobia + SAR supergroup). Example results from the promoter motif screen are summarized in Supplementary Table S1 (shown in detail for green algae, bryophytes and Ciliates, in the other taxonomic groups only species with highly conserved type 3 promoters are reported).
TR template regions
The second search parameter for TR loci identification—the minimum putative TR template region—was predicted as described in Methods, based either on previously known telomere sequence motifs, or motifs newly identified by the analysis of short tandem repeats in genomic data of particular organisms (Table 1).
Table 1.
Genomes with well conserved USE selected for TR-like loci prediction
| Selected genomes (from STab.1) | USE consensus (counts in genome) | Telomere motif | TR-like sequences | Show TR homology |
|---|---|---|---|---|
| Galdieria sulphuraria (GCA_000341285.1) | TCCCAWCATC (1415) | CCCTAATAAA | 4 | 0 |
| Galdieria phlegrea(GCA_006232345.1) | TCCCAHCA (1510) | CCCTAATAAA | 2 | 0 |
| Ostreococcus lucimarinus(GCA_000092065.1) | CCCRTAA (716) | CCCTAAA | 6 | 0 |
| Micromonas pusilla (GCA_000090985.2) | ACCCAYAW (321) | CCCTAAA | 5 | 1 |
| Auxenochlorella protothecoides(GCA_000733215.1) | ACCCATAA (166) | CCCTAA | 7 | 1 |
| Blastocystis hominis(GCA_000151665.1) | AACCCRTAA (154) | CCCTAA | 3 | 1 |
| Stentor coerulens(GCA_001970955.1) | RTCCCWTA (9792) | CCCTAACA | 42 | 1 |
| Plasmodiophora brassicae(GCA_003833335.1) | GNCCCAYW (8997) | CCCTAAAA | 10 | 0 |
Genomes were selected based on conserved USE in most tested snRNAs (Supplementary Table S1) and known or predicted putative template region (corresponding to telomere motif) as searching parameters. The number of TR-like sequences showing homology (in BLASTn) in genomes of investigated relatives is shown in the right column.
TR-like loci identification
With increasing USE conservation and complexity we expect fewer ‘USE-like’ sequences per genome (i.e. fewer putative TR candidates). Similarly, we expect the type 3 promoter to be conserved in TRs with a higher probability compared to some of the tested snRNA species whose promoters lacked an USE. The effect of USE length, conservation, and TR template region length on the number of TR-like sequences was calculated (per random 1M long nucleotide sequence) (Supplementary Figure S1). Thus, we can estimate how many TR-like sequences can be expected in different genomes, i.e., how many sequences may need to be checked in subsequent homology searches. Based on these criteria and assumptions, only genomes showing a highly conserved USE present at the most of examined snRNA species were selected for TR-loci prediction (Table 1).
Selected genomes were subjected to extraction of TR-like loci, i.e., all genomic sequences starting with USE and harbouring a template-like region up to 200nt downstream of USE (as illustrated in Figure 2C, yellow panel). The number of predicted TR-like loci in selected genomes is shown in Table 1. Homology searches were then performed on all of these TR-like loci.
TR-homology searches
To test our hypothesis that the type 3 promoter identified in land plant and ciliate TR genes also controls the transcription of TR genes in early diverging plant and ciliate lineages, we tested for homology the identified TR-like loci across large taxonomic groups under the hypothesis that shared homology would significantly support their role as genuine TRs.
In Chlorophyta, we started homology searches with Auxenochlorella protothecoides. It possesses a small (22 Mb) and well assembled genome where only 7 TR-like loci could be identified for further analysis (Table 1). Moreover, there are genome assemblies available from its close relatives (P. cutis and P. wickerhamii) for TR homology searches to test whether a template and USE are conserved. Interestingly, from the initially identified 7 TR-like loci, only one recovered a sequence homolog based on BLASTn (e-value ≤ 1e–7) in both P. cutis and P. wickerhamii genomes. A closer look at these TR homologs revealed several conserved regions including USE, template or [U]n—rich terminator (Figure 3), and therefore, their putative transcribed regions were easily predictable. Considering the overall extensive TR sequence variability, BLASTn may not be sufficient to show a homology across evolutionary more distant species. However, as was recently performed for TRs from early diverging Animalia lineages (37), covariance models (CMs) are suitable for this problem (38) since TR secondary structures are more conserved than TR sequences (reviewed in (3)). Starting with a CM from the alignment of predicted TRs from A. protothecoides and two Protoheca, we recovered significant hits (e-value ≤ 1e–9) using Infernal (38) in other Trebouxiophyceae species (Figure 3). Importantly, knowledge of USE features in particular organisms (listed in Supplementary Table S1) was very useful in evaluating new hits produced by Infernal. The inclusion of USE provided a way to assess hits independently of Infernal searches, which were based on CMs built from putative transcribed regions, i.e., without promoters. Subsequent progressive optimization of CMs with newly identified TR candidates allowed us to identify putative TRs across the green algae clades (Chlorophyceae, Trebouxiophyceae, Chlorodendrophyceae, Pyramimonadophyceae and Mamiellaceae) as well as streptophyte algae and bryophytes. Importantly, hits identified using the Chlorophyta TR CM in bryophytes and streptophyte algae were in accordance with hits identified by the CM built from previously known Tracheophytes TRs (5,6). Finally, three phylogenetically discrete CMs were optimized (Supplementary File S2)—one from green algae TRs, the second from bryophytes and streptophyte algae TRs, and the third from known TRs from Tracheophytes. Although these CMs are prepared from TRs from different taxonomic groups (i.e. green algae), reciprocal Infernal searches identified putative TRs when comparing each major clade to another, including previously published TRs from Tracheophytes (5,6) (Figure 4, Supplementary Table S4). These results clearly demonstrate a homology between the previously characterized land plant TRs and our newly identified green algae and bryophyte TRs (Supplementary Table S2). Moreover, the shared TR homology in Viridiplantae was demonstrated in two independent ways—one starting from a predicted TR candidate in A. protothecoides (Chlorophytes) based on USE/Template knowledge, and secondly, starting from previously known Tracheophyta TRs. Both approaches produced the same TR candidates (Figure 4).
Figure 3.

Graphical alignment showing TR identification by homology searches in related genomes. De novo predicted TR candidate (based on USE and putative TR template) from A. protothecoides showed homologs in close relatives P. cutis and P. wickerhamii by using BLASTn. Their putative transcribed regions were used to generate a Covariance model (CM) for searching TR homologs in evolutionarily more distant species by the Infernal tool. New significant hits (e-value ≤ 1e–9) were manually checked for corresponding template, USE or other conserved regions, and used for optimization of CM for reiterative Infernal searches.
Figure 4.
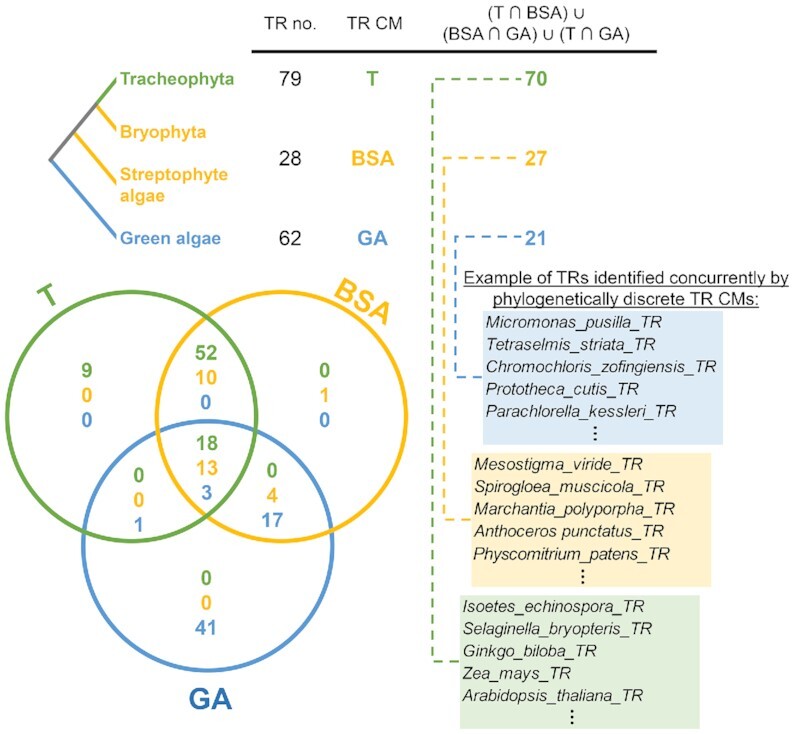
Summary of TR Infernal searches across Viridiplantae illustrated by Venn diagram confirms a homology of newly predicted TRs in this study with previously published TRs in Tracheophytes (5). Three phylogenetically discrete CMs (circles distinguished by colour) were optimized from previously published TRs from Tracheophytes (T, in green), bryophytes and streptophyte algae TRs (BSA, in yellow) and green algae TRs (GA, in blue), respectively (Supplementary File S2). Infernal search with these models (T; BSA; GA) against Viridiplantae TRs (Supplementary Table S4) identified concurrently corresponding TR sequences—visualized as numbers (for respective Venn diagram subsets) distinguished by colour (Tracheophyta TRs—green, bryophytes and streptophyte algae TRs—yellow, green algae TRs—blue).
In Heterotrichea, only three genome assemblies are publicly available at NCBI - Stentor coeruleus, Stentor roeselii and Condylostoma magnum. Due to a conserved USE and an unusual telomere motif CCCTAACA in Stentor coeruleus (39) and Stentor roeselii, and another unusual CCCTTACA motif predicted in this study in Condylostoma magnum (Supplementary Table S4), we were able to identify their TRs (Supplementary Table S2) in a similar manner as in Chlorophytes. Only one of 42 TR-like sequences in S. coeruleus showed homology in both S. roeselii and C. magnum genomes including USE and template region. To support this TR candidate as a genuine TR, other TRs from seven species across the Heterotrichea phylogeny were identified by BLASTn/Infernal in genomic/transcriptomic SRA data at NCBI (Supplementary Table S2). Heterotrichea (Postciliodesmatophora) represents a deeply branching group of Ciliates which diverged over ∼1200 MYA ago (40) from other ciliates from subphylum Intramacronucleata, in which the RNA and catalytic TERT protein subunits of telomerase were first characterized (41). Although predicted length and template topology of Heterotrichea TRs were similar to known TRs in Spriotrichea or Oligohymenophorea (Intramacronucleata), we could not support their homology by Infernal searches as we successfully did in Viridiplantae. No promising hits were observed between these taxonomic groups when using our CM from Heterotrichea TRs (Supplementary File S2) or CM built from Spriotrichea and Oligohymenophorea TRs that is available at RFAM (accession: RF00025).
As in early diverging plants and ciliates, we aimed to examine selected genomes from other eukaryotic lineages with conserved USEs (Table 1, Supplementary Table S1). These included genus Galdieria (red algae), plant parasites from the order Plasmodiophorida (Rhizaria) and gastrointestinal parasites from the genus Blastocystis (Stramenopiles).
Within the genus Galdieria, one genome assembly is available at NCBI for G. phlegrera and eight genome assemblies for G. sulphuraria strains. Only Galdieria species showed conserved USE in most tested snRNAs within red algae genomes. Moreover, in Galdieria an unusual telomere motif CCCTAATAAA was predicted (42), which could markedly limit the number of predicted TR-like loci. Although a few TR-like loci were predicted (Table 1), none of them showed TR homology across G. phleglera or G. sulphuraria accessions.
The order Plasmodiophorida (Rhizaria) includes sequenced genomes (available at NCBI) from Spongospora subterrena, Polymyxa betae and 50 assemblies for Plasmodiophora brassicae strains. Although TR-like loci were predicted based on conserved USE and putative template (Table 1), we could not support any of them in homology searches in available genomes from related species.
Genus Blastocystis includes a genetically heterogeneous group of gastrointestinal parasites. So far, 10 genomes are available for Blastocystis subtypes (STs). Unlike other analysed Stramenopiles, Blastocystis species and its closest sequenced relative Proteromonas lectrae showed USE conservation and presence in most snRNA promoters except MRP RNA (Table 1 and Supplementary Table S1). In the Blastocystis hominis genome (GCA_000151665.1—referred as subtype 7), only 154 USE-like sequences were present and 3 TR-like loci were predicted. One of these showed sequence homology in subtypes (ST6, ST9), but not in the others. However, a subsequent search using CMs identified homologs in all remaining subtypes (ST1, ST2, ST3, ST4-WR1, ST4-BT1, ST8, ASY1), but not in Proteromonas lectrae. Despite the high degree of genetic variation found within the genus, all homologs display a conserved template and USE and similar topology of conserved regions. Thus, while unable to confirm these loci in vivo, we believe that our approach has uncovered TRs in Blastocystis that share structural and regulatory features to plant and ciliate TRs.
Validation of predicted TRs
TR candidate presence in transcriptomes
Since predicted TR loci in this study are inferred from genomic data, we first verified their transcription by RNA-seq, Northern blot analysis, and RT-PCR (Figure 5). The vast majority of available RNA-seq datasets are poly(A) selected. As such, reads corresponding to the RNAPIII transcribed TRs are very rare or missing in such datasets. Thus, to verify TR expression we focused on publicly available rRNA depleted RNA-seq datasets available at NCBI’s Sequence Read Archive (SRA) for 7 species. We supplemented public transcriptomic data with our own for 8 species (Figure 3A, Supplementary Table S2). For one moss, four streptophyte algae and four green algae species where genome assemblies were available, RNA-seq reads were mapped to the genome. In all other cases (Dunaliella tertiolecta, Chlorella ohadii, Desmodesmus quadricauda and three Klebsormidium species), de novo assemblies were generated (see Supplementary Table S5 for RNA-seq information and assembly statistics). Importantly, we observed expression for putative TR loci in all 15 species for which total RNA-seq data were available, or which we sequenced in this work (Figure 5A).
Figure 5.

Evidence for the presence of transcripts of example predicted TRs from Viridiplantae employing: (A) TR presence in total RNA-seq data from rRNA depleted libraries.TR lengths were estimated based on the lengths of mapped RNA-seq data to reference genomes (if available) or de-novo assembled transcripts; (B) Northern hybridization using corresponding radioactively labelled TR probe (NB). Gels were stained with SYBR™ Gold and visualized in UV light (UV lanes). Low Range ssRNA Ladder (NEB) was used as a marker (M); (C) RT-PCR with TR specific primers (Supplementary File S1). GeneRuler 100 bp DNA Ladder (Thermo Scientific) was used as a marker (M).
In addition, these transcriptomic data allowed us to confirm the 5′ and 3′ transcript boundaries of putative TRs predicted based on the USE and RNAPIII terminator sequences (TR transcript sequences are listed in Supplementary Table S2, highlighted in yellow). These results were confirmed by Northern blot analyses in the moss Physcomitrium patens and two algae, Parachlorella kesslerii and Desmodesmus quadricauda (Figure 5B), or by RT-PCR in Parachlorella kesslerii, Scenedesmus quadricauda, Physcomitrium patens and Chlamydomonas reindhartii (Figure 5C).
In conclusion, we confirmed the expression of all 15 examined putative TR loci, which further supports the hypothesis that these loci are, in fact, TRs.
Pol II or III transcription?
Unlike the other known eukaryotic TRs, ciliate and land plant TRs are transcribed by RNAPIII (5,10,43). Since type 3 promoters have dual-polymerase activity recruiting either RNAPII or III transcription factors (TFs), the manner of TR transcription may not be conserved across these lineages. In plants and ciliates, RNAPIII specificity for type 3 promoters is defined primarily by the mutual positions of USE and TATA box (12,13,44). By comparing type 3 promoters of newly predicted TRs with promoters of either RNAPIII or RNAPII snRNAs, we assessed whether a particular TR is likely to be a RNAPIII or RNAPII transcript.
Sampling of our TRs from early diverging plants and ciliates (Figure 6), where USE and TATA can be identified, indicates that RNAPIII is involved in TR transcription in all sampled species. Interestingly, in the analysed green algae, the RNAP specificity of type 3 promoters appears to differ from land plants (determined by USE-TATA distance) (Figure 6). RNAPIII specificity in green algae more closely resembles human RNAPIII where TATA-rich type 3 promoters are primarily targeted by RNAPIII, while TATA-less type 3 promoters are RNAPII targets (reviewed in (45)). Contrary to the earlier view of Tetrahymena TR as an outlier in being transcribed by RNAPIII, we find that RNAPIII-dependent TR transcription may be deeply conserved across more than a billion years of evolution.
Figure 6.

A comparison of type 3 promoters of TRs with other snRNAs that are transcribed with either RNAPII or RNAPIII. A similarity with promoters of respective snRNAs is indicated by ‘∼‘. Mutual position of USE and TATA box (or TATA presence) is crucial for RNAP specificity (12,13,44). USE and TATA box (if present) are in capital letters in alignments. All analysed TR promoters correspond to snRNAs transcribed predominantly by RNAPIII (i.e. U6, SRP snRNA across eukaryotes, or U3 snRNA in Viridiplantae (46,47)).
TR templates coding for unusual telomere motifs
Knowledge of an organism's telomere sequence is essential, both for our prediction of TR-loci based on USE and template and for validation of newly identified TR-like sequences in homology searches. In Viridiplantae, the most common telomere DNA repeat is CCCTAAA. However, unusual or unknown telomere sequences were reported in several species/genera also within early diverging plants. These include Chlamydomonas reindhardtii (with CCCTAAAA) (48), genera Dunaliella and Stepanosphaera (with CCCTAA) and streptophyte algae genus Klebsormidium (with CCCTAAAA/CCTAAAA) (16), Chloropicon primus (with CCTAAAAA) (49) and species from genus Picochlorum, where raw genomic data lack any known telomere repeat motif (50). In addition, the amount of genome assemblies and other genomic data significantly increased since publication of the aforementioned works on unusual telomeres. By using modified Tandem Repeats Finder tool (TRFi) (22,28) and/or simple checking of scaffold termini, we re-visited these data to predict telomere repeats in some species (Supplementary Table S3). We identified a CCCTAA telomere motif in species from Prototheca genus, Haematococcus lacustris, and Chlorokybus atmophyticus where genomic data containing CCCTAA tandem repeats were highly abundant, while typical CCCTAAA telomere repeats were lacking or present at very low level. Alternate telomere motifs usually vary in the number of adenines or cytosines compared to the dominant CCCTAAA motif (reviewed in (51)). This variation can be caused either by a mutation in TR template or by a change in template usage, i.e., determination of which portion of the template anneals to telomere DNA and what is used for telomere extension by telomerase. In this respect, the origin of an exceptional telomere motif CCCTATA, predicted here for species from the genus Picochlorum (Supplementary Table S3), must be unambiguously associated with a corresponding mutation in its TR template region.
Similarly, in Heterotrichea, a CCCTAACA telomere motif was reported in Stentor coeruleus (39). We show here that the same motif is highly abundant in the available genomic data from related species Stentor roeselii and Blepharisma americanum, but not Condylostoma magnum, whose genome contigs were frequently capped by a similar CCCTTACA motif, while CCCTAACA tandem repeats were absent (Supplementary Table S3). Importantly, these exceptions with non-canonical telomere repeats are fundamentally important and helpful for validating predicted TR candidates. Conversely, species with possible unusual telomeres can be easily predicted based on TR knowledge. We applied this approach to demonstrate that template regions of predicted TRs from homology searches are congruent with predicted telomere motifs (its C rich strand) in the examples mentioned above (Figure 7).
Figure 7.
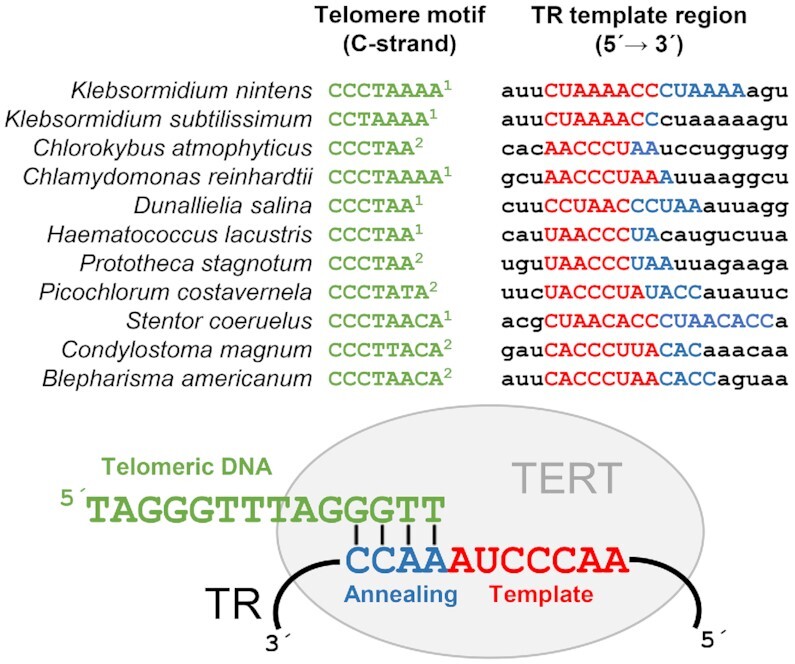
Species with diverse telomere DNA motifs (on the left, in green) and their putative TR template regions (on the right). 1 – previously published telomere motifs, 2 – newly identified telomere motifs in this work (Supplementary Table S3). Putative template regions are in capital letters, consisting of a template sequence (in red) and an annealing sequence (in blue). An example of telomerase RNA annealing to the 3′ end of telomere DNA is shown in a bottom part.
TR knock-out mutants
To directly demonstrate the function of a selected candidate TR in telomerase activity and telomere maintenance, we generated knock-out mutants in P. patens (pptr plants) by a replacement of the TR locus with a 35S:HygR cassette via homology directed repair (HDR) using Cas9-induced DNA double-strand cleavage within the TR locus (see Material and Methods section). Telomere lengths and telomerase activities were then analysed in pptr and WT plants in parallel. While the mean telomere lengths, obtained using terminal restriction fragment length analysis followed by evaluation using WALTER tool (36), showed values of 1.4 and 1.5 kb in two independent WT plants, mean telomere lengths in three pptr lines revealed values of ca. 300 and 480 bp, respectively (Figure 8A, B), thus corresponding to ca. 20–30% of the values in WT plants. Although the TRF lengths in mutants were substantially shorter, their signal intensities were several times higher than those of WT plants (under a comparable DNA input). Subsequent Southern hybridization experiments using partial digestion of genomic DNA with Tru1I (MseI), showed the presence of dimers and longer arrays of TRF unit produced by a complete Tru1I digestion. These results correspond to the tandem arrangement of units, which consist of (TTTAGGG)n repeats and the adjacent DNA regions harbouring the Tru1I site (Figure 8C). This arrangement is typical for telomerase-independent recombination process known as Alternative Lengthening of Telomeres (ALT). Therefore, we assume the surviving pptr plants may actually represent survivors in which ALT has been activated.
Figure 8.

Analysis of P. patens TR knock-out lines (pptr) shows telomere shortening and the loss of telomerase activity. (A) Terminal restriction fragment (TRF) analysis of regenerated P. patens pptr lines (samples 3, 4, 5) and wild-type plants (WT) (samples 1, 2) used as a background for TR knock-outs. For clarity, TRF signals under different exposition intensity are enclosed alongside. (B) Evaluation of telomere lengths from TRF signals using WALTER toolset v2.0. (C) Complete and partial DNA digestions of DNA of WT and pptr line (sample 3 in panel A) with decreasing activities of Tru1I (as indicated above lanes). While partial digestion of WT sample results in a shift of a smeared pattern of TRFs towards higher molecular weight, pptr sample shows a ladder of monomers, dimers and longer arrays of major and minor products (denoted by filled and empty arrowheads, respectively), Product lengths were evaluated using Clinx Image Analysis software (Clinx Science Instruments). (D) TRAP assay using cell extracts (250 ng of total protein) of WT (samples 1, 2) and pptr plants (samples 3, 4). Negative control (nc) indicates reaction without extract.
Telomerase activity in extracts of these WT and pptr plants were examined by Telomere Repeat Amplification Protocol (TRAP). These results showed the absence of telomere repeat products in pptr plants (showing only bands of primers) while the typical pattern of TRAP products was observed in WT plants (Figure 8D).
Conserved structural features can be identified within TR candidates
Aside from the template region, there are several key structural features known to be critical for TR function that we expect to infer in our TR candidates. Among the most characteristic structural features of TRs is the pseudoknot (PK) domain, which falls 3′ of the template region and forms long-range interactions that are necessary for telomerase activity (52–55). In addition to the PK domain, TRs typically contain a template boundary element (TBE) (56–58) and a stem-loop structure that closes the core template/PK region (referred to as P1c) (6,43,59). We undertook a phylogenetically informed comparative structural analysis to (i) determine if our putative TRs contained the critical structural features found in other eukaryotic TRs and (ii) compare these features between the long evolutionary distance sampled by our TR identification scheme. PK domains were readily identifiable within multiple sequence alignments of TRs from Bryophyta (mosses, species included in alignment = 11), Chlamydomonadales (green algae, n = 7), and Marchantiophyta (liverworts, n = 12) (Figure 9, Yellow box, and Supplementary Figures S2 and S3). Most sites within the P2/P3 stems of these PKs are >75% conserved within each lineage (red or purple nucleotides); non-conserved sites showed evidence of co-variation within lineages (blue nucleotides). In each lineage, the loop connecting P2 and the upstream region of P3 (J2/3u) contains invariant U residues, similar to those observed in vertebrates and Arabidopsis J2/3u loops and known to be critical for telomerase activity (6,54). Interestingly, consensus alignments of MSAs from each of these three lineages revealed sequence and positional conservation of the PK domains (Supplementary Figures S2 and S3), including a short ∼5 bp G/C rich P2 stem and a longer (7–12 bp) P3 stem. The J2/3u loops are short, ranging from 6 to 8 nts, whereas the downstream loop connecting P2 – P3 ranges from 6 to 29 nts, similar to those seen in Arabidopsis and vertebrate TRs. Furthermore, these alignments shared sequence and positional conservation with previously identified land plant TRs (5,6) (Figure 9D).
Figure 9.

Comparative phylogenetic structural predictions of Viridiplantae TRs reveal a deeply conserved core domain. (A) Predicted core Template/Pseudoknot (TPK) region of Bryophyta TRs based on the Physcomitrium patens TR. Invariant sites across the multiple sequence alignment (MSA) of 11 putative TRs are shown in red, with sites >90% conserved shown in purple and covariant sites where, for example, G = C binding switched to A = U binding, are shown in blue. (B) The predicted TPK region for Chlamydomonadales based off of a MSA of seven putative TR loci with Chlamydomonadales reinhardtii as reference. (C) Predicted TPK region for Marchantiophyta based off of a MSA of 12 putative TRs, with Marchantia polyporpha used as reference. (D) A proposed structural model of Viridiplantae TRs based off of the consensus sequences from Bryophyta, Chlamydomonadales, Marchantiophyta and Tracheophytes TPK regions. Full predicted structures or sequence alignments used to build structures are available in Supplementary Figures S2 and S3.
In addition to the PK domain, evidence for a conserved TBE (P1.1, Figure 9) similar to that previously predicted for Gymnosperms and Lycophytes was also uncovered immediately 5′ of the template region in TRs from the Bryophyta, Chlamydomonadales and Marchantiophyta lineages. Finally, in addition to the P1c domain, the putative TRs from each of these lineages contain an extensive stem similar to the P4-P6 domains observed in TRs from other plant, human, and ciliate lineages (6,43,59). Long range interactions between the extreme 5′ and 3′ ends, as well as a three-way junction composed of P1a/P1b/P6 interactions were observed in the Bryophyte and Marchantiophyta TRs (blue box, Supplementary Figure S2), whereas these interactions were absent in the predicted consensus structure for Chlamydomonadales TRs. In sum, the predicted structures for each of these three lineages closely matches expectations for a TBE, PK and P1c domain observed in all published TRs, strongly supporting the hypothesis that these RNAs are indeed deeply conserved TRs.
We also observed conserved PK domains in the Heterotrichea (n = 7), Opalinata (n = 8), and Trebouxiophyceae (n = 9) putative TRs. These PKs were supported by sequence conservation or covariation among the sampled taxa (Supplemental Figure S2 and S3). While the Heterotrichea TR PK was characterized by perfect base pairing along the P2 and P3 stems, both Opalinata and Trebouxiophyceae harboured mismatches in these domains (1 and 3 nts, respectively) that were not conserved in nature (i.e. some species did not have these mismatches, or the mismatched nucleotides themselves were highly variable; Supplemental Figure S3). Although there was less overall sequence conservation within structural domains of the TR representatives of these three lineages, we did observe intra-lineage evidence for the P1c domain in all three lineages and putative TBEs (P1.1) in Opalinata and Trebouxiophyceae, similar to the those presented here and reported elsewhere (6,43,59–61). This TBE element was noticeably lacking or with poor support in Heterotrichea (Supplemental Figure S2). In conclusion, our phylogenetically informed structural analysis revealed the presence of multiple conserved TR domains that are characteristic of other eukaryotic TRs, supporting our hypothesis that these RNAs are in fact TRs.
DISCUSSION
Our combination of complementary bioinformatic approaches (Figure 2C) proved to be successful in the identification of TR candidates across an unprecedented phylogenetic scale of early diverging eukaryotes. We were able to identify TRs across the Viridiplantae by taking advantage of newly characterized TRs in Streptophyta and Chlorophyta, as well as discover novel TRs in neighbouring branches of the Diaphoretickes megagroup—Ciliates (Alveolata), and Stramenopiles. Our basic assumption – conservation of the mode of TR transcription by RNAPIII across a wide range of early diverging eukaryotes, based on previous findings in land plants (5) and Ciliates (41)—made it possible to fill in the substantial knowledge gap concerning the early steps of TR evolution. Another critical prerequisite for this progress was the conservation of the type 3 RNAPIII promoter in TR genes. We also presumed that sequence conservation of the promoter is higher than that of TRs, where the conservation is limited only to specific motifs of TR structure. These presumptions allowed us to use the promoter sequence (in particular its USE) as a query, together with a presumed TR minimum template region, to identify an initial set of TR candidate genes.
Limitations of a strictly sequence homology based approach are apparent. Our approach relied on the availability of genomic and transcriptomic data in species from the examined clades. Since the first step of TR identification is based on a precise characterization of the type 3 promoter in a specific taxonomic group, the availability of genomic data was necessary to identify promoters of spliceosomal RNAs, U3, SRP and MRP snRNAs. The availability of genomic data in multiple closely related organisms allowed us to precisely define the degree to which the USE is conserved across each lineage. In turn, USE conservation delimited the species in which we could reliably search for candidate sequences with a suitable template domain. Candidate TRs were used to develop a CM for searching in additional species without the need to identify USEs. Covariation models exploit the conservable structural conservation within TRs, particularly the structural elements surrounding the template region (e.g. PK/TBE). The power of CMs is illustrated in Viridiplantae where three CMs were independently generated for TRs from (i) Tracheophyta, (ii) Bryophytes and streptophyte algae and (iii) green algae. These CMs, from distantly related lineages, enabled the reciprocal identification of a considerable number of TRs within the Viridiplantae (Figure 4 and Supplementary Table S4), reflecting the wide evolutionary conservation of TR secondary structural features.
The conservation of structural domains known to be important for TR/telomerase function, such as PK and TBE domains, were readily apparent in structural models generated from multiple sequence alignments. Our comparative phylogenetic analyses revealed key features within these domains that support the classification of these RNAs as TRs and furthermore, highlight the degree to which TRs are conserved across eukaryotes. We present a consensus PK model for all of Viridiplantae, with the functionally critical invariant U residues present in all J2/3u loops (Figure 9D). Viridiplantae PKs all contain a short P2 (∼5 bps) and a longer P3, similar to ciliates but in contrast to that seen in the vertebrate models. The P1.1/putative TBE is a conserved structural element across the Viridiplantae we sampled, however, it is not conserved at the sequence level. Indeed, despite its deep structural conservation, this element has been lost in many angiosperms, raising the possibility that an alternative feature serves as the template boundary. In TRs without an adjacent stem-loop structure, the single strand RNA-sequence itself is believed to serve as the TBE (62). Thus, the Viridiplantae P1.1 domain may instead be critical for TERT binding. In sum, our comparative phylogenetic approach of TRs across Viridiplantae has uncovered extensive conservation of core TR domains, reinforcing both the early evolutionary origins of this complex, as well as its critical role in eukaryotic biology.
Validation of results
Selected TR gene candidates were validated at several levels: The presence of TR transcripts was examined in RNA-seq data (either total RNA-seq data after rDNA depletion generated in this work, or similar data available in databases) (Figure 5A). In parallel, selected TR transcripts were demonstrated using Northern hybridization and RT-PCR (Figure 5B and C, respectively). After this confirmation of the TR transcript authenticity, we moved to TR functional testing. For this purpose, we chose TR from a model moss species, P. patens due to its amenability to genome editing. Knock-out pptr plants were generated, which indeed, did show a substantial telomere shortening and the loss of telomerase activity (Figure 8). Interestingly, the shorter terminal restriction fragments in pptr plants (panel A and B) showed considerably more intense signals than WT plants under the same DNA loading. Subsequent partial digestion experiment (panel C) indicated that telomeres in pptr plants were formed by tandemly arranged units composed of telomeric (TTTAGGG)n repeats and an adjacent DNA sequence harbouring a single MseI site. Thus, the structure of telomeres in pptr survivors is similar to that observed in ALT yeast survivors of the type I (63) or its subsequent unified form with type II survivors (as recently described by (64)). It is plausible that P. patens ALT survivors benefit (similarly to yeasts) from the high efficiency of homologous recombination in this organism.
An independent confirmation of the authenticity of identified TRs was obtained in organisms showing the presence of characteristic unusual telomere repeat motifs. Template regions of their candidate TRs corresponded to their telomere repeat motifs described in earlier studies or predicted here by TRFi tool (Figure 7, Supplementary Table S3).
In conclusion, we provide here an important advancement in the knowledge of telomerase RNAs in the early diverging organisms from Diaphoretickes megagroup. This megagroup, besides ecologically and biotechnologically important lower plants (Bryophytes and Streptophyte and Chlorophyte algae), also contains protozoan microorganisms from Stramenopiles, Alveolata and Ciliates including known pathogenic species (e.g. Blastocystis). We therefore assume this knowledge is not only important for a more comprehensive view of telomerase origin and evolution but also of a perspective practical importance in diagnostic and therapeutic targeting the telomerase of these pathogens as a critical factor for their survival.
DATA AVAILABILITY
RNA-seq data generated in this project are available at NCBI (BioProject ID: PRJNA700859).
All TR sequences from this work are available in Supplementary Table S2. TRs from genome assemblies available at NCBI were submitted as TPA (NCBI) under accessions: BK014486-BK014553
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Marek Eliáš, University of Ostrava, for insightful comments on Blastocystis phylogenesis, and Alena Lukešová (Biology Centre CAS) and Kateřina Bišová (Institute of Microbiology CAS), for providing us with Parachlorella, Desmodesmus and Klebsormidium species used in this work. We thank the computational resources supplied by the project ‘e-Infrastruktura CZ’ (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures. We acknowledge the CF Genomics supported by the NCMG research infrastructure (LM2018132 funded by MEYS CR) for their support with obtaining scientific data presented in this paper. CF Plant Sciences of CEITEC Masaryk University is gratefully acknowledged for their support with obtaining of the results presented in this paper.
Contributor Information
Petr Fajkus, Department of Cell Biology and Radiobiology, Institute of Biophysics of the Czech Academy of Sciences, Brno CZ-61265, Czech Republic; Mendel Centre for Plant Genomics and Proteomics, CEITEC Masaryk University, Brno CZ-62500, Czech Republic.
Agata Kilar, Mendel Centre for Plant Genomics and Proteomics, CEITEC Masaryk University, Brno CZ-62500, Czech Republic; Laboratory of Functional Genomics and Proteomics, NCBR, Faculty of Science, Masaryk University, Brno CZ-61137, Czech Republic.
Andrew D L Nelson, Boyce Thompson Institute, Cornell University, Ithaca, NY 14853, USA.
Marcela Holá, Institute of Experimental Botany of the Czech Academy of Sciences, Prague CZ-16000, Czech Republic.
Vratislav Peška, Department of Cell Biology and Radiobiology, Institute of Biophysics of the Czech Academy of Sciences, Brno CZ-61265, Czech Republic.
Ivana Goffová, Mendel Centre for Plant Genomics and Proteomics, CEITEC Masaryk University, Brno CZ-62500, Czech Republic; Laboratory of Functional Genomics and Proteomics, NCBR, Faculty of Science, Masaryk University, Brno CZ-61137, Czech Republic.
Miloslava Fojtová, Mendel Centre for Plant Genomics and Proteomics, CEITEC Masaryk University, Brno CZ-62500, Czech Republic; Laboratory of Functional Genomics and Proteomics, NCBR, Faculty of Science, Masaryk University, Brno CZ-61137, Czech Republic.
Dagmar Zachová, Mendel Centre for Plant Genomics and Proteomics, CEITEC Masaryk University, Brno CZ-62500, Czech Republic.
Jana Fulnečková, Department of Cell Biology and Radiobiology, Institute of Biophysics of the Czech Academy of Sciences, Brno CZ-61265, Czech Republic.
Jiří Fajkus, Department of Cell Biology and Radiobiology, Institute of Biophysics of the Czech Academy of Sciences, Brno CZ-61265, Czech Republic; Mendel Centre for Plant Genomics and Proteomics, CEITEC Masaryk University, Brno CZ-62500, Czech Republic; Laboratory of Functional Genomics and Proteomics, NCBR, Faculty of Science, Masaryk University, Brno CZ-61137, Czech Republic.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Czech Science Foundation [20-01331X]; ERDF [project SYMBIT, reg. no. CZ.02.1.01/0.0/0.0/15_003/0000477]; National Science Foundation [IOS-1758532, IOS-2023310 to A.D.L.N.]; Visegrad Scholarship Program [52010129 to A.K.]. Funding for open access charge: project SYMBIT financed by ERDF.
Conflict of interest statement. None declared.
REFERENCES
- 1. Koonin E.V. The origin of introns and their role in eukaryogenesis: a compromise solution to the introns-early versus introns-late debate?. Biol. Direct. 2006; 1:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sykorova E., Fajkus J.. Structure-function relationships in telomerase genes. Biol. Cell. 2009; 101:375–392. [DOI] [PubMed] [Google Scholar]
- 3. Podlevsky J.D., Chen J.J.L.. Evolutionary perspectives of telomerase RNA structure and function. RNA Biol. 2016; 13:720–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Waldl M., Thiel B.C., Ochsenreiter R., Holzenleiter A., Oliveira J.V.D., Walter M.E.M.T., Wolfinger M.T., Stadler P.F.. TERribly difficult: searching for telomerase RNAs in Saccharomycetes. Genes-Basel. 2018; 9:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fajkus P., Peska V., Zavodnik M., Fojtova M., Fulneckova J., Dobias S., Kilar A., Dvorackova M., Zachova D., Necasova I.et al.. Telomerase RNAs in land plants. Nucleic Acids Res. 2019; 47:9842–9856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song J.R., Logeswaran D., Castillo-Gonzalez C., Li Y., Bose S., Aklilu B.B., Ma Z.Y., Polkhovskiy A., Chen J.J.L., Shippen D.E.. The conserved structure of plant telomerase RNA provides the missing link for an evolutionary pathway from ciliates to humans. PNAS. 2019; 116:24542–24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dew-Budd K., Cheung J., Palos K., Forsythe E.S., Beilstein M.A.. Evolutionary and biochemical analyses reveal conservation of the Brassicaceae telomerase ribonucleoprotein complex. PLoS One. 2020; 15:e0222687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lingner J., Hendrick L.L., Cech T.R.. Telomerase RNAs of different ciliates have a common secondary structure and a permuted template. Genes Dev. 1994; 8:1984–1998. [DOI] [PubMed] [Google Scholar]
- 9. Hargrove B.W., Bhattacharyya A., Domitrovich A.M., Kapler G.M., Kirk K., Shippen D.E., Kunkel G.R.. Identification of an essential proximal sequence element in the promoter of the telomerase RNA gene of Tetrahymena thermophila. Nucleic Acids Res. 1999; 27:4269–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu J., Okada T., Fukushima T., Tsudzuki T., Sugiura M., Yukawa Y.. A novel hypoxic stress-responsive long non-coding RNA transcribed by RNA polymerase III in Arabidopsis. RNA Biol. 2012; 9:302–313. [DOI] [PubMed] [Google Scholar]
- 11. Mittal V., Ma B., Hernandez N.. SNAP(c): a core promoter factor with a built-in DNA-binding damper that is deactivated by the Oct-1 POU domain. Genes Dev. 1999; 13:1807–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Waibel F., Filipowicz W.. U6 Snrna genes of Arabidopsis are transcribed by RNA polymerase-III but contain the same 2 upstream promoter elements as RNA polymerase-II-transcribed U-Snrna genes. Nucleic Acids Res. 1990; 18:3451–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Waibel F., Filipowicz W.. RNA-polymerase specificity of transcription of Arabidopsis U-Snrna genes determined by promoter element spacing. Nature. 1990; 346:199–202. [DOI] [PubMed] [Google Scholar]
- 14. Dergai O., Cousin P., Gouge J., Satia K., Praz V., Kuhlman T., Lhote P., Vannini A., Hernandez N.. Mechanism of selective recruitment of RNA polymerases II and III to snRNA gene promoters. Genes Dev. 2018; 32:711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burki F., Shalchian-Tabrizi K., Pawlowski J.. Phylogenomics reveals a new ‘megagroup’ including most photosynthetic eukaryotes. Biol. Lett. 2008; 4:366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fulneckova J., Hasikova T., Fajkus J., Lukesova A., Elias M., Sykorova E.. Dynamic evolution of telomeric sequences in the green algal order Chlamydomonadales. Genome Biol. Evol. 2012; 4:248–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nawrocki E.P., Eddy S.R.. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R.. Rfam: an RNA family database. Nucleic Acids Res. 2003; 31:439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bailey T.L., Elkan C.. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:28–36. [PubMed] [Google Scholar]
- 20. Raden M., Ali S.M., Alkhnbashi O.S., Busch A., Costa F., Davis J.A., Eggenhofer F., Gelhausen R., Georg J., Heyne S.et al.. Freiburg RNA tools: a central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018; 46:W25–W29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Katoh K., Misawa K., Kuma K., Miyata T.. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002; 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fajkus P., Peska V., Sitova Z., Fulneckova J., Dvorackova M., Gogela R., Sykorova E., Hapala J., Fajkus J.. Allium telomeres unmasked: the unusual telomeric sequence (CTCGGTTATGGG)(n) is synthesized by telomerase. Plant J. 2016; 85:337–347. [DOI] [PubMed] [Google Scholar]
- 23. Lorenz R., Bernhart S.H., Siederdissen C.H.Z., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithm Mol Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dellaporta S.L., Wood J., Hicks J.B.. A plant DNA minipreparation: Version II. Plant Mol. Biol. Reporter. 1983; 1:19–21. [Google Scholar]
- 25. Peri S., Roberts S., Kreko I.R., McHan L.B., Naron A., Ram A., Murphy R.L., Lyons E., Gregory B.D., Devisetty U.K.et al.. Read mapping and transcript assembly: a scalable and high-throughput workflow for the processing and analysis of ribonucleic acid sequencing data. Front. Genet. 2020; 10:1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q.D.et al.. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011; 29:644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999; 27:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peska V., Sitova Z., Fajkus P., Fajkus J.. BAL31-NGS approach for identification of telomeres de novo in large genomes. Methods. 2017; 114:16–27. [DOI] [PubMed] [Google Scholar]
- 29. Mallett D.R., Chang M.Q., Cheng X.H., Bezanilla M.. Efficient and modular CRISPR-Cas9 vector system for Physcomitrella patens. Plant Direct. 2019; 3:e00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Concordet J.P., Haeussler M.. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018; 46:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Y.C., Vidali L.. Efficient polyethylene glycol (PEG) mediated transformation of the moss Physcomitrella patens. J. Visual. Exp. 2011; 2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fajkus J., Fulneckova J., Hulanova M., Berkova K., Riha K., Matyasek R.. Plant cells express telomerase activity upon transfer to callus culture, without extensively changing telomere lengths. Mol. Gen. Genet. 1998; 260:470–474. [DOI] [PubMed] [Google Scholar]
- 33. Goffova I., Vagnerova R., Peska V., Franek M., Havlova K., Hola M., Zachova D., Fojtova M., Cuming A., Kamisugi Y.et al.. Roles of RAD51 and RTEL1 in telomere and rDNA stability in Physcomitrella patens. Plant J. 2019; 98:1090–1105. [DOI] [PubMed] [Google Scholar]
- 34. Fojtová M., Fajkus P., Polanská P., Fajkus J.. Terminal restriction fragments (TRF) method to analyze telomere lengths. Bio-protocol. 2015; 5:e1671. [Google Scholar]
- 35. Ijdo J.W., Wells R.A., Baldini A., Reeders S.T.. Improved telomere detection using a telomere repeat probe (Ttaggg)N generated by PCR. Nucleic Acids Res. 1991; 19:4780–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lyčka M., Peska V., Demko M., Spyroglou I., Kilar A., Fajkus J., Fojtová M.. WALTER: an easy way to online evaluate telomere lengths from terminal restriction fragment analysis. BMC Bioinformatics. 2021; 22:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Logeswaran D., Li Y., Podlevsky J.D., Chen J.J.. Monophyletic origin and divergent evolution of animal telomerase RNA. Mol. Biol. Evol. 2021; 38:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eddy S.R., Durbin R.. Rna sequence-analysis using covariance-models. Nucleic Acids Res. 1994; 22:2079–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Slabodnick M.M., Ruby J.G., Reiff S.B., Swart E.C., Gosai S., Prabakaran S., Witkowska E., Larue G.E., Fisher S., Freeman R.M.et al.. The macronuclear genome of Stentor coeruleus reveals tiny introns in a giant cell. Curr. Biol. 2017; 27:569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fernandes N.M., Schrago C.G.. A multigene timescale and diversification dynamics of Ciliophora evolution. Mol. Phylogenet Evol. 2019; 139:106521. [DOI] [PubMed] [Google Scholar]
- 41. Greider C.W., Blackburn E.H.. A telomeric sequence in the rna of tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989; 337:331–337. [DOI] [PubMed] [Google Scholar]
- 42. Fulneckova J., Sevcikova T., Fajkus J., Lukesova A., Lukes M., Vlcek C., Lang B.F., Kim E., Elias M., Sykorova E.. A broad phylogenetic survey unveils the diversity and evolution of telomeres in eukaryotes. Genome Biol. Evol. 2013; 5:468–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romero D.P., Blackburn E.H.. A conserved secondary structure for telomerase RNA. Cell. 1991; 67:343–353. [DOI] [PubMed] [Google Scholar]
- 44. Orum H., Nielsen H., Engberg J.. Structural organization of the genes encoding the small nuclear RNAs U1 to U6 of Tetrahymena-Thermophila is very similar to that of plant small nuclear-RNA genes. J. Mol. Biol. 1992; 227:114–121. [DOI] [PubMed] [Google Scholar]
- 45. Schramm L., Hernandez N.. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002; 16:2593–2620. [DOI] [PubMed] [Google Scholar]
- 46. Kiss T., Marshallsay C., Filipowicz W.. Alteration of the RNA-polymerase specificity of U3 Snrna genes during evolution and in vitro. Cell. 1991; 65:517–526. [DOI] [PubMed] [Google Scholar]
- 47. Antal M., Mougin A., Kis M., Boros E., Steger G., Jakab G., Solymosy F., Branlant C.. Molecular characterization at the RNA and gene levels of U3 snoRNA from a unicellular green alga, Chlamydomonas reinhardtii. Nucleic Acids Res. 2000; 28:2959–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Petracek M.E., Lefebvre P.A., Silflow C.D., Berman J.. Chlamydomonas telomere sequences are A+T-rich but contain three consecutive G-C base pairs. PNAS. 1990; 87:8222–8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lemieux C., Turmel M., Otis C., Pombert J.F.. A streamlined and predominantly diploid genome in the tiny marine green alga Chloropicon primus. Nat. Commun. 2019; 10:4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krasovec M., Vancaester E., Rombauts S., Bucchini F., Yau S., Hemon C., Lebredonchel H., Grimsley N., Moreau H., Sanchez-Brosseau S.et al.. Genome analyses of the microalga Picochlorum provide insights into the evolution of thermotolerance in the green lineage. Genome Biol. Evol. 2018; 10:2347–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peska V., Garcia S.. Origin, diversity, and evolution of telomere sequences in plants. Front Plant Sci. 2020; 11:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Autexier C., Greider C.W.. Mutational analysis of the Tetrahymena telomerase RNA: identification of residues affecting telomerase activity in vitro. Nucleic Acids Res. 1998; 26:787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tzfati Y., Knight Z., Roy R., Blackburn E.H.. A novel pseudoknot element is essential for the action of a yeast telomerase. Genes Dev. 2003; 17:1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen J.L., Greider C.W.. Functional analysis of the pseudoknot structure in human telomerase RNA. PNAS. 2005; 102:8080–8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cash D.D., Cohen-Zontag O., Kim N.K., Shefer K., Brown Y., Ulyanov N.B., Tzfati Y., Feigon J.. Pyrimidine motif triple helix in the Kluyveromyces lactis telomerase RNA pseudoknot is essential for function in vivo. PNAS. 2013; 110:10970–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tzfati Y., Fulton T.B., Roy J., Blackburn E.H.. Template boundary in a yeast telomerase specified by RNA structure. Science. 2000; 288:863–867. [DOI] [PubMed] [Google Scholar]
- 57. Chen J.L., Greider C.W.. Template boundary definition in mammalian telomerase. Genes Dev. 2003; 17:2747–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jansson L.I., Akiyama B.M., Ooms A., Lu C., Rubin S.M., Stone M.D.. Structural basis of template-boundary definition in Tetrahymena telomerase. Nat. Struct. Mol. Biol. 2015; 22:883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen J.L., Blasco M.A., Greider C.W.. Secondary structure of vertebrate telomerase RNA. Cell. 2000; 100:503–514. [DOI] [PubMed] [Google Scholar]
- 60. Dandjinou A.T., Levesque N., Larose S., Lucier J.F., Elela S.A., Wellinger R.J., Grp R.. A phylogenetically based secondary structure for the yeast telomerase RNA. Curr. Biol. 2004; 14:1148–1158. [DOI] [PubMed] [Google Scholar]
- 61. Gunisova S., Elboher E., Nosek J., Gorkovoy V., Brown Y., Lucier J.F., Laterreur N., Wellinger R.J., Tzfati Y., Tomaska L.. Identification and comparative analysis of telomerase RNAs from Candida species reveal conservation of functional elements. RNA. 2009; 15:546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang Y.Q., Gallagher-Jones M., Susac L., Song H., Feigon J.. 2020) A structurally conserved human and Tetrahymena telomerase catalytic core. PNAS. 117:31078–31087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lundblad V., Blackburn E.H.. An alternative pathway for yeast telomere maintenance rescues Est1- senescence. Cell. 1993; 73:347–360. [DOI] [PubMed] [Google Scholar]
- 64. Kockler Z.W., Comeron J.M., Malkova A.. A unified alternative telomere-lengthening pathway in yeast survivor cells. Mol. Cell. 2021; 81:1816–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data generated in this project are available at NCBI (BioProject ID: PRJNA700859).
All TR sequences from this work are available in Supplementary Table S2. TRs from genome assemblies available at NCBI were submitted as TPA (NCBI) under accessions: BK014486-BK014553