


Abstract
Although ribosome-profiling and translation initiation sequencing (TI-seq) analyses have identified many noncanonical initiation codons, the precise detection of translation initiation sites (TISs) remains a challenge, mainly because of experimental artifacts of such analyses. Here, we describe a new method, TISCA (TIS detection by translation Complex Analysis), for the accurate identification of TISs. TISCA proved to be more reliable for TIS detection compared with existing tools, and it identified a substantial number of near-cognate codons in Kozak-like sequence contexts. Analysis of proteomics data revealed the presence of methionine at the NH2-terminus of most proteins derived from near-cognate initiation codons. Although eukaryotic initiation factor 2 (eIF2), eIF2A and eIF2D have previously been shown to contribute to translation initiation at near-cognate codons, we found that most noncanonical initiation events are most probably dependent on eIF2, consistent with the initial amino acid being methionine. Comprehensive identification of TISs by TISCA should facilitate characterization of the mechanism of noncanonical initiation.
Graphical Abstract
Graphical Abstract.
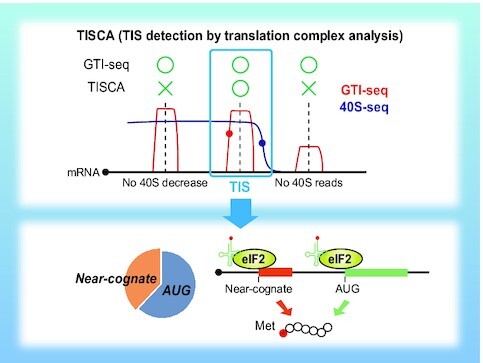
TISCA reliably identifies translation initiation sites (TISs) and provides insight into the mechanism of translation initiation at near-cognate codons. TISCA efficiently reduces the background noise in GTI-seq analysis. The absence and nonrelease of eIF3-seq footprints are mostly eliminated as noise. Red and blue circles show the 5′ end of a GTI-seq peak and an eIF3 release point, respectively. TIS candidates are located at +11 to +13 nt from the GTI-seq peak and at −22 to −26 nt from the eIF3 release point (top). TISCA identifies a substantial number of near-cognate initiation codons, most of which are translated in a manner dependent on eIF2–Met-tRNAi (bottom).
INTRODUCTION
The translation reaction of protein synthesis in eukaryotes consists of three stages: initiation, elongation and termination (1–5). The initiation stage includes binding of the initiator methionyl-tRNA (Met-tRNAi) to the 40S ribosome small subunit [formation of the 43S preinitiation complex (PIC)], binding of the 43S PIC to the 5′ end of the mRNA (formation of the 48S PIC), scanning and recognition of the initiation codon, and binding of the 60S ribosome large subunit (formation of the 80S ribosome). Eukaryotic initiation factor 2 (eIF2) consists of a heterotrimer of α, β and γ subunits and binds to Met-tRNAi in a GTP-dependent manner to form a ternary complex, which then combines with the 40S subunit, eIF1, eIF1A, eIF5 and the eIF3 complex (which comprises 12 subunits: eIF3A to eIF3I and eIF3K to eIF3M), resulting in formation of the 43S PIC. Binding of the eIF4F complex (which consists of the cap binding protein eIF4E, the DEAD-box RNA helicase eIF4A, and the scaffolding protein eIF4G) to the cap structure at the 5′ end of the mRNA is followed by recruitment of the 43S PIC to the mRNA to form the 48S PIC. The 48S PIC then scans the start codon in an ATP-dependent manner, and the formation of base pairs between Met-tRNAi and the start codon results in the hydrolysis of GTP bound to eIF2. Subsequently, eIF1, eIF1A and eIF2-GDP dissociate from the 48S PIC, whereas eIF5B promotes recruitment of the 60S ribosome subunit to form the 80S ribosome. Even after binding of the 60S subunit, eIF3 remains associated with the complex and moves with the ribosome during the early stage of elongation (6–8).
Ribosome profiling (also known as Ribo-seq) is a technique that allows measurement of translation efficiency and the identification of novel open reading frames (ORFs) (9–11). For this approach, treatment of cells with cycloheximide (CHX) halts translation at all ribosomes, whereas that with harringtonine or lactimidomycin (LTM) induces selective stalling of initiating ribosomes and thereby enriches ribosomes at translation initiation sites (TISs). Ribo-seq combined with treatment of cells with either harringtonine or LTM is referred to as translation initiation sequencing (TI-seq), with the combination of Ribo-seq with LTM in particular also being known as global TI-seq (GTI-seq) (11,12). Application of these methods has resulted in the identification of a larger number of near-cognate initiation codons than anticipated. However, the results are likely to include a substantial number of experimental artifacts (13), and it has remained a challenge to estimate how many of the identified codons represent actual translation initiation events.
Translation from near-cognate initiation codons, which have a sequence that differs from the AUG codon by one nucleotide (for example, CUG, GUG, ACG and AUU), has also been detected by analysis of individual genes including those that encode eIF4G2 (GUG codon), TEF-1 (AUU codon) and c-Myc (CUG codon) (14–16). Phylogenetic analysis has identified potential NH2-terminal extensions with near-cognate initiation codons in 42 human genes (17). Proteomics approaches have revealed that methionine is still incorporated at the NH2-terminus of proteins translated from near-cognate codons, suggesting that the eIF2–Met–tRNAi complex engages in wobble base-pairing with the near-cognate codons (18,19).
Whereas eIF2 binds specifically to Met-tRNAi, eIF2A and eIF2D, which bind to tRNAs in a GTP-independent manner and also contribute to translation initiation, were shown to bind to and deliver other tRNAs such as Leu-tRNACUG and Val-tRNAGUG in addition to Met-tRNAi for recognition of the initiation codon (20–23). Of note, loss of eIF2A reduces the efficiency of CUG-dependent translation but does not affect AUG-dependent translation (22). Furthermore, eIF2A-dependent translation of upstream ORFs (uORFs) promotes development of squamous cell carcinoma, and loss of eIF2A suppresses tumor formation (24). However, in addition to its role in translation initiation, eIF2D has been shown to facilitate recycling of the 40S subunit at the stop codon (25), and the function of eIF2A and eIF2D in near-cognate translation initiation has remained largely unknown.
Whereas Ribo-seq reveals the dynamics of 80S ribosomes, translation complex profile sequencing (TCP-seq) allows the monitoring of both 40S and 80S ribosomes (26). (Hereafter, all variants of the small ribosomal subunit including 40S, 43S and 48S will be referred to simply as 40S.) In TCP-seq, all translation complexes are fixed by treatment with formaldehyde instead of CHX, which is followed by partial digestion of RNA with RNase I and separation of 40S and 80S ribosomes by density gradient ultracentrifugation for sequencing analysis. Selective TCP-seq (Sel-TCP-seq), a combination of TCP-seq and immunopurification of ribosomes associated with a factor of interest, has recently been developed to reveal the dynamics of each translational factor (27,28).
Here, we combined aspects of Sel-TCP-seq, GTI-seq and Ribo-seq to establish a more accurate method for TIS identification termed TISCA (TIS detection by translation Complex Analysis), and we verified its superiority over existing methods by analysis of proteomics data. TISCA also revealed that translation initiation at near-cognate codons of most ORFs is likely to be dependent on the eIF2–Met-tRNAi complex. The comprehensive identification of near-cognate initiation codons should provide further insight into the molecular mechanism of noncanonical translation initiation.
MATERIALS AND METHODS
Cell lines and antibodies
HEK293T cells were obtained from American Type Culture Collection and were checked for mycoplasma contamination with the use of MycoAlert (Lonza). The cells were cultured under an atmosphere of 5% CO2 at 37°C in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Life Technologies) and antibiotics. Primary antibodies included the following: eIF2A (Bethyl Laboratories, A301-949A), eIF2D (Proteintech, 12840-1-AP), eIF2α (Cell Signaling Technology, 5324S), Ser51-phosphorylated eIF2α (Cell Signaling Technology, 3398S), eIF3B (Thermo Fisher Scientific, PA5-23278), eIF3D (Bethyl Laboratories, A301-758A), RPS3 (Cell Signaling Technology, 9538), RPL5 (Cell Signaling Technology, 14568), ATF4 (Proteintech, 10835-1-AP), V5 tag (Thermo Fisher Scientific, R960CUS), FLAG tag (Merck, F1804), HSP90 (BD Biosciences, #610419), and normal rabbit immunoglobulin G (IgG) (SouthernBiotech, 0111-01).
Cloning of eIF3 subunits
Complementary DNAs for human eIF3 subunits containing the V5 tag were amplified by the polymerase chain reaction (PCR) and subcloned into the EcoRV site of pcDNA3 (Invitrogen) with the use of a NEBuilder HiFi DNA Assembly Master Mix kit (New England Biolabs).
RNA interference
HEK293T cells were transfected with 10 nM small interfering RNAs (siRNAs) with the use of Lipofectamine RNAiMAX (Invitrogen). Silencer Select Pre-Designed siRNAs (Thermo Fisher Scientific)—including negative control no. 1 (4390843), siEIF2α no. 1 (s4555) and siEIF2α no. 2 (s4556)—were used.
Generation of eIF2A- and eIF2D-deficient cell lines
Sense and antisense oligonucleotides (Supplementary Table S1) encoding single guide RNAs (sgRNAs) for human eIF2A or eIF2D were cloned into pX330 (Addgene). HEK293T cells were transfected for 24 h with the resulting plasmids as well as pPUR (Clontech) with the use of the X-tremeGENE 9 DNA Transfection Reagent (Sigma-Aldrich). The cells were then subjected to selection in puromycin-containing medium for 2 days before culture for an additional week in puromycin-free medium. Clonal cell lines were established by limiting dilution.
Generation of eIF3D tag-KI cell lines
Sense and antisense oligonucleotides (Supplementary Table S1) encoding sgRNAs were cloned into pX330. Targeting constructs containing selection markers for homologous recombination were cloned in pBluescript II SK (Stratagene) with the use of a NEBuilder HiFi DNA Assembly Master Mix kit. HEK293T cells were transfected for 24 h with targeting vectors and pX330 encoding sgRNAs and were then subjected to selection in medium containing puromycin or blasticidin S for 2 weeks before establishment of clonal cell lines by limiting dilution. The cells also had an insertion of the 3× FLAG tag sequence at the NH2-terminus of eIF4E in addition to the V5 tag at the COOH-terminus of eIF3D. For generation of eIF3D-V5/3×FLAG-eIF4E double–knock-in (KI) cells, the eIF3D-V5 KI clonal cell line was established before introduction of the 3×FLAG sequence at the eIF4E gene locus.
Immunoblot analysis
Protein samples were subjected to SDS-polyacrylamide gel electrophoresis on 5–20% ExtraPAGE One Precast Gels (Nacalai Tesque). Membranes were incubated consecutively with primary antibodies and horseradish peroxidase–conjugated secondary antibodies (Promega), and signals were visualized with SuperSignal West Pico PLUS or Dura (Thermo Fisher Scientific) reagents and with a ChemiDoc imaging system (Bio-Rad).
Immunoprecipitation of overexpressed eIF3 subunits
HEK293T cells were transfected for 48 h with plasmids encoding V5-tagged subunits of the eIF3 complex with the use of the X-tremeGENE 9 DNA Transfection Reagent. Cells were lysed in ice-cold lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol (DTT), 1% Triton X-100, and EDTA-free protease inhibitor (Roche)], and the RNA concentration of the lysates was determined with the use of a Qubit RNA BR assay kit (Thermo Fisher Scientific). Dynabeads Protein G (Thermo Fisher Scientific) were conjugated with antibodies to V5 by incubation overnight at 4°C in lysis buffer supplemented with 0.5% bovine serum albumin, and they were washed with lysis buffer three times before use. The cell lysates were incubated with the antibody-coated beads for 90 min at 4°C, and the resulting immunoprecipitates were washed three times with wash buffer [50 mM Tris–HCl (pH 7.5), 300 mM NaCl, 1 mM DTT, 0.1% Triton X-100]. RNA was purified from the immunoprecipitates with the use of a Direct-zol RNA Kit (Zymo Research) and the addition of ISOGEN (Nippon Gene) directly to the beads. The amount of purified RNA was measured with the use of a Qubit RNA HS assay kit (Thermo Fisher Scientific).
Sucrose density gradient analysis
Cells were seeded at a density of 1.2 × 107 per 15-cm dish, with two dishes for fixed samples and four dishes for unfixed samples per assay, and they were cultured overnight to ensure subconfluence. For fixed samples, the cells were treated for 10 min at room temperature with phosphate-buffered saline (PBS) containing 0.25% formalin, after which glycine was added to a final concentration of 125 mM and the cells were collected. Fixed or unfixed cells were lysed in lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1% Triton X-100, EDTA-free protease inhibitor] supplemented with CHX (100 μg/ml) and SUPERase•In RNase inhibitor (10 U/ml, Invitrogen), and the lysates were incubated for 10 min on ice and then centrifuged at 20 380 × g for 10 min at 4°C. The resulting supernatants were loaded on a 10–50% sucrose gradient buffer [20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, CHX (100 μg/ml), SUPERase•In (10 U/ml)] and centrifuged at 36 000 rpm (160 000 × g) for 3 h at 4°C in a Beckman SW41 rotor. The gradient was fractionated with the use of a TRIAX gradient-profiling system (BioComp) and AC-5700P microcollector (ATTO). Polysome profiles were determined by measurement of absorbance at 260 nm.
Ribo-seq and GTI-seq
Libraries were prepared according to a method described previously (12,29), with slight modifications. For eIF2α knockdown experiments, cells were seeded at a density of 2.0 × 106 per 10-cm dish and the following analyses were performed 48 h after siRNA transfection. For other experiments, cells were seeded at a density of 2.5 × 106 per 10-cm dish and cultured overnight to ensure subconfluence. For arsenite treatment and GTI-seq, 40 μM sodium arsenite or 50 μM LTM was added to the medium either 60 or 30 min, respectively, before cell lysis. The medium was aspirated, and the cells were immediately placed on ice, washed once with ice-cold PBS, and lysed with lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1% Triton X-100 and EDTA-free protease inhibitor] supplemented with either CHX (100 μg/ml) for Ribo-seq or 50 μM LTM for GTI-seq. The lysates were then incubated with TURBO DNase (Thermo Fisher Scientific) on ice for 10 min and centrifuged at 20 380 × g for 10 min at 4°C, and the resulting supernatants were collected carefully, assayed for RNA concentration with a Qubit RNA BR assay kit, incubated with RNase I (20 U per 10 μg of RNA, Epicentre) for 45 min at 25°C, and placed on ice before the addition of 200 U SUPERase•In RNase inhibitor. The samples were then loaded on a 1 M sucrose cushion buffer [20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, SUPERase•In (10 U/ml), 1 mM DTT and either CHX (100 μg/ml) for Ribo-seq or 50 μM LTM for GTI-seq], and the gradients were centrifuged at 100 000 rpm (417 200 × g) for 1 h at 4°C in a Beckman TLA110 rotor. The resulting pellets were suspended in ribosome splitting buffer [20 mM Tris–HCl (pH 7.5), 300 mM NaCl, 5 mM EDTA, SUPERase•In (20 U/ml), 1 mM DTT, 1% Triton X-100] and subjected to purification with the use of an Amicon Ultra filtration device (100-kDa cutoff, Millipore) to deplete rRNAs. The purified RNA was subjected to selection on the basis of a size range of 17–34 nucleotides (nt) by electrophoresis through a 15% polyacrylamide and Tris-borate-EDTA (TBE)–urea gel (SuperSep RNA, Fujifilm). The footprint fragments were treated with T4 PNK (New England Biolabs) to repair the 2′-3′ cyclic phosphates, a DNA linker including barcode sequences (NI-810 to NI-817) was ligated with the use of T4 RNA Ligase 2, truncated K227Q (New England Biolabs), and the resulting products were purified on a 15% polyacrylamide and TBE–urea gel. Ribosomal RNAs were further depleted with the use of RiboZero Gold (Illumina). Reverse transcription was performed with the NI-802 primer, and the resulting products were purified on a 15% polyacrylamide and TBE–urea gel. The purified cDNAs were circularized with circLigase II (Lucigen), and index sequences were then added by amplification in a PCR reaction with the common primer (NI-798) and primers including index sequences (NI-799 and NI-822 to NI-826). Products of the desired size were purified on a 15% polyacrylamide nondenaturing gel (SuperSep DNA, Fujifilm), and the libraries were sequenced with a NovaSeq 6000 system (Illumina).
Sel-TCP-seq
Cells were seeded at a density of 1.2 × 107 per 15-cm dish, with 8 to 12 dishes per assay, and they were cultured overnight to ensure subconfluence before fixation for 10 min at room temperature with PBS containing 0.25% formalin followed by quenching by the addition of glycine (final concentration of 125 mM) and collection. The cells were washed three times with ice-cold PBS, lysed in lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1% Triton X-100, EDTA-free protease inhibitor] supplemented with CHX (100 μg/ml), and incubated for 10 min on ice. The lysates were centrifuged at 20 380 × g for 10 min at 4°C, and the resulting supernatants were collected carefully, assayed for RNA concentration with a Qubit RNA BR assay kit, incubated with RNase I (20 U per 10 μg of RNA) at 25°C for 45 min, and then placed on ice before the addition of SUPERase•In RNase inhibitor. The samples were loaded on a 5% to 30% sucrose gradient buffer [20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, CHX (100 μg/ml), SUPERase•In (10 U/ml)] and centrifuged at 38 000 rpm (180 000 × g) for 4 h at 4°C in a Beckman SW41 rotor. The gradient was fractionated with the use of a TRIAX gradient-profiling system and AC-5700P microcollector. Polysome profiles were determined by measurement of absorbance at 260 nm. Dynabeads Protein G were conjugated with antibodies to V5 or to eIF3B, or with control rabbit IgG, by incubation overnight at 4°C in lysis buffer supplemented with 0.5% bovine serum albumin, and they were washed three times with lysis buffer before use. The 40S or 80S fractions were incubated with the beads for 90 min at 4°C, and the resulting immunoprecipitates were washed four times with wash buffer [50 mM Tris–HCl (pH 7.5), 300 mM NaCl, 1 mM DTT, 0.1% Triton X-100] before the addition of 150 μl of decrosslinking buffer [1% SDS, 10 mM EDTA, 10 mM Tris–HCl (pH 7.5), 10 mM glycine], 750 μl of ISOGEN LS, and 200 μl of chloroform followed by incubation for 45 min at 65°C. RNA was then purified by precipitation with isopropanol, selected according to size in the range of 17 to 100 nt by electrophoresis in a 15% polyacrylamide and TBE-–urea gel, and subjected to dephosphorylation, DNA linker ligation, and rRNA depletion with RiboZero Gold as described for the Ribo-seq protocol. Reverse transcription was performed with the use of the SI-019 primer, and the resulting products were purified on a 15% polyacrylamide and TBE–urea gel. The second linker (SI-018) was ligated by incubation overnight at 25°C with T4 RNA Ligase 1 (ssRNA Ligase), High Concentration (New England Biolabs). The products were then amplified in a PCR reaction with the same primers as used for the Ribo-seq protocol, and those of the desired size were purified by electrophoresis through a 15% polyacrylamide nondenaturing gel. The libraries were sequenced with a NovaSeq 6000 system.
RNA-seq
Total RNA was extracted from cell lysates with the use of a PureLink RNA Mini kit (Thermo Fisher Scientific). The quality of the purified RNA was assessed with a 2100 Bioanalyzer (Agilent). After mRNA selection with the use of a NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs), libraries were prepared with the use of a NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs). The cDNAs were sequenced with a NovaSeq 6000 system.
Read processing and analysis
Adaptor sequences were trimmed from raw reads with the use of cutadapt. Reads of low quality were discarded with the use of fastq_quality_trimmer and fastq_quality_filter of the FASTX-Toolkit. Ribosomal RNA reads were removed by alignment with human rRNA sequences with the use of STAR, and the remaining reads were aligned with the human transcriptome (GRCh38.p13) and human genome (hg38) also with the use of STAR. Multiple mappings were allowed. Metagene analyses were performed with custom Python script and with the use of Numpy (v1.17.3) and Pysam (v0.15.3). The coverage of Sel-TCP-seq reads was calculated across the 5′ untranslated region (5′-UTR), coding sequence (CDS), and 3′-UTR of transcripts. Counts for the 5′ end or 3′ end of Sel-TCP-seq reads per fragment length were calculated around annotated start codon sites. The heat maps of counts per fragment length were colored according to the sum of counts from all transcripts at a given position for a given fragment length. For tRNA content analysis, reads mapped to hg38 tRNA sequences downloaded from GtRNAdb were counted with the use of ShortRead (R package). Read counts were summed per tRNA anticodon type. Reads of Sel-TCP-seq and RNA-seq mapped to the 5′-UTR of human coding transcripts were counted with the use of featureCounts and transcripts per kilobase million (TPM) values were calculated. Scanning efficiency (SE) was calculated for each transcript according to the formula: SE = TPM of the 5′-UTR in Sel-TCP-seq/TPM of the 5′-UTR in RNA-seq. Sequence logo analysis was performed with WebLogo (30). TI-seq data sets were retrieved from GEO and SRA [SRA056377 (12), GSE94460 (31), SRA160745 (32), GSE41605 (33), GSE139391 (28), GSE131650 (34)].
Identification of TIS positions
Pickup of ORFs and transcripts
Ribo-seq data were analyzed with RibORF in order to obtain a list of all provable ORF sequences and to calculate their pred.pvalues, which reflect the homogeneity of reads on the ORF and the 3-nt periodicity (35). In RibORF analysis, AUG and near-cognate codons (CUG, GUG, UUG, ACG, AGG, AAG, AUC, AUU and AUA) were used as start codons. Among all ORF candidates, those with a pred.pvalue of >0.7 were selected, and only transcripts containing these ORFs were subjected to the subsequent analysis.
Detection of GTI-seq peaks
With the use of GTI-seq results, the mapped read data were converted into read aggregation plots by Numpy (v1.17.3) and Pysam (v0.15.3). After normalization by the total number of reads, the points at which the derivative of GTI-seq normalized footprint density exceeded the threshold (μ + 4σ, where μ and σ represented the mean and standard deviation of the derivative of GTI-seq normalized footprint density on the transcript, respectively) were defined as GTI-seq peaks for each transcript.
40S decreasing point identification
With the use of the Sel-TCP-seq data for eIF3 subunits, the mapped read data were converted into two types of read aggregation plots by Numpy (v1.17.3) and Pysam (v0.15.3): full length of reads (eIF3_full) and 3′ end of reads (eIF3_3′). The local maximum points of eIF3_3′ were extracted with the use of Scipy (v1.4.0rc1). The ratio of the area of eIF3_full on the 5′ and 3′ sides of these local maximum points was defined as the 40S decreasing score ( = area of 5′ side/area of 3′ side, see also schematic representation in Supplementary Figure S3D). When calculating the area, we left a gap of 10 bases before and after the local maximum points and calculated the area from that position to 15 bases farther on (±10 to 25 nt from each point). Only points with a 40S decreasing score of >1 were retained for the following analysis.
TIS detection and frame-fitting
TIS candidates were identified at positions +11 to 13 nt from GTI-seq peaks, and they were adopted only if 40S decreasing points existed at positions +22 to 26 nt from the candidate sites. For frame-fitting, we predicted all ORFs that could arise from the TIS candidates. The ORFs that perfectly matched those identified by RibORF were then identified as true ORFs, and the TISs derived from these ORFs were defined as true TISs in our analysis.
Immunoaffinity purification–MS analysis for eIF3 subunit detection
The 40S fraction of cell lysates was subjected to immunoprecipitation with antibodies to V5 as described for Sel-TCP-seq analysis, and precipitated proteins were eluted by incubation of the beads for 10 min at room temperature with 20 μl of V5 peptide (Sigma-Aldrich) at 0.5 mg/ml. The purified proteins were subjected to removal of cross-links by incubation for 45 min at 95°C in SDS sample buffer and were then fractionated by SDS-polyacrylamide gel electrophoresis on a 10% gel and stained with silver. Protein bands were excised from the gel and subjected to in-gel digestion, and the resulting peptides were dissolved in a solution comprising 0.1% trifluoroacetic acid and 2% acetonitrile for analysis with an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific) coupled with a nanoLC instrument (Advance, Michrom BioResources) and HTC-PAL autosampler (CTC Analytics). Acquired mass spectrometry (MS) data were analyzed with MaxQuant and the human UniProt KB database.
MS data analysis for identification of NH2-termini
The three proteomics data sets for protein NH2-terminal analysis with HEK293T cells were retrieved from ProteomeXchange Consortium via PRIDE and JPOST. These data sets consisted of 54 raw files obtained with the Nrich method with artificial acetylation (PXD0055831) (36), 48 raw files obtained with the TAILS method (PXD006633) (19), and 9 raw files obtained with Lysarginase and strong cation exchange chromatography (PXD010551) (37). The raw files were processed with MaxQuant v.1.6.14.0 and searched against the database concatenated with the protein sequence generated by TISCA and the human Swiss-Prot database including all isoforms (version 2020_5, 42 363 sequences). Peptide tolerance for the first search and main search allowed 20 and 4.5 ppm, respectively. The MS/MS match tolerance allowed 20 ppm. For the Nrich data set, the COOH-terminal side of arginine was selected as the cleavage site. Oxidation of methionine as well as light and heavy acetylation of peptide NH2-termini were set as variable modifications. Methylthiolation of cysteine and heavy acetylation of lysine were set as fixed modifications. For the TAILS data set, the COOH-terminal side of arginine was selected as the cleavage site. Dimethylation of peptide NH2-termini, acetylation of protein NH2-termini, and oxidation of methionine were set as variable modifications. Carbamidomethylation of cysteine and dimethylation of lysine were set as fixed modifications. For the data set obtained by the method of Chang et al., the NH2-terminal sides of lysine and arginine were selected as the cleavage sites. Oxidation of methionine and acetylation of protein NH2-termini were set as variable modifications. Carbamidomethylation of cysteine was set as a fixed modification. A maximum of two missed cleavages was allowed, and the minimum peptide length was set to seven amino acids for all the data sets. The cutoff for the false discovery rate (FDR) was set to 0.01 at the peptide spectrum match (PSM) and protein level for analysis of TISs. Three experts manually inspected hundreds of MS/MS spectra, and the acceptance criterion was set at a posterior error probability (PEP) of <0.005, which corresponds to an FDR of <0.13%. For comparison of TISCA with previous TIS prediction methods, the three data sets were searched against databases consisting of the human Swiss-Prot database and the protein sequences predicted by TISCA or by the other methods. A cutoff for FDR was not set so as to obtain all peptides matched to target and decoy sequences.
Quantification and statistical analysis
We used DESeq2 to analyze differential expression from RNA-seq reads (38), and the generalized linear model in RiboDiff to analyze differential translation efficiency (39). Gene set enrichment analysis (GSEA) was performed with the use of GSEApy (v0.9.9) (40). Pearson's correlation was calculated in Python 3.6.8 with the use of Pandas (v0.25.3). Student's t test was performed in Python 3.6.8 with the use of Scipy (v1.2.1). For extraction of differentially expressed genes, we used an adjusted P value (q value, FDR of <0.05). A P value of <0.05 was considered statistically significant.
RESULTS
eIF3 footprint analysis
The 12 subunits of the eIF3 complex are categorized structurally into PCI (proteasome, COP9, eIF3)–MPN (Mpr1–Pad1 N-terminal) core subunits and peripheral subunits (41–43). The PCI-MPN core consists of an octamer of eIF3A, -C, -E, -F, -H, -K, -L and -M in mammals, whereas the peripheral subunits include eIF3D and the yeast-like core (YLC) module consisting of eIF3B, eIF3G, eIF3I and the COOH-terminus of eIF3A. In particular, eIF3D is located at the periphery of eIF3 and is attached to the PCI-MPN core via eIF3E (44), and it promotes eIF4E-independent but cap-dependent translation of several specific mRNAs through direct interaction with the 5′ cap in chronic glucose-deprived conditions (45,46).
Sel-TCP-seq analysis of the eIF3 complex has been performed with antibodies to eIF3B in human cells (27,28). To observe the dynamics of the other eIF3 subunits, we attempted to insert into each subunit a tag sequence that does not interfere with the structure and function of the eIF3 complex and which is insensitive to the formalin fixation adopted in Sel-TCP-seq. To this end, we transfected HEK293T cells with cDNAs encoding each eIF3 subunit fused to a V5 epitope tag at its NH2- or COOH-terminus and then subjected lysates of the transfected cells to immunoprecipitation with antibodies to V5. Determination of the amount of RNA co-immunoprecipitated with each eIF3 subunit revealed that eIF3D tagged with the V5 epitope at its COOH-terminus showed the greatest efficiency for RNA recovery (Supplementary Figure S1A). We therefore manipulated HEK293T cells with the use of the CRISPR–Cas9 system to insert the V5 tag sequence at the COOH-terminus of endogenous eIF3D (Supplementary Figure S1B), and we confirmed that the V5 tag sequence was inserted into at least one allele of the eIF3D gene, with the other allele remaining intact (Supplementary Figure S1C). Sucrose density gradient analysis followed by immunoblot analysis revealed that the distribution of eIF3D-V5 in eIF3D-V5 knock-in (KI) cells was similar to that of eIF3D in wild-type (WT) cells (Supplementary Figure S1D and S1E). Furthermore, liquid chromatography (LC) and MS/MS analysis of immunoprecipitates prepared from the 40S fraction of eIF3D-V5 KI cells with antibodies to V5 detected all subunits of the eIF3 complex (Supplementary Figure S1F). These results thus suggested that insertion of the V5 tag sequence at the COOH-terminus of endogenous eIF3D does not interfere with the structure and function of the eIF3 complex.
We next performed Sel-TCP-seq analysis of eIF3D-V5 and eIF3B. The cells were fixed with 0.25% formalin, lysed, treated with RNase I, and separated into 40S and 80S fractions by sucrose gradient centrifugation. The 40S fraction was subjected to immunoprecipitation with antibodies to V5 or to eIF3B, and the footprints were sequenced (Figure 1A, Supplementary Figure S2A and S2B). Metagene analysis of the footprints revealed that eIF3D-V5 and eIF3B localized predominantly to the 5′ untranslated region (5′-UTR) of mRNAs, whereas the 40S input for eIF3D-V5 showed some degree of 80S contamination (Figure 1B, Supplementary Figure S2C). The scanning efficiency ratio (footprint reads normalized by RNA-seq reads) for eIF3D-V5 was substantially correlated with that for eIF3B (Figure 1C), suggesting that most eIF3D-V5 and eIF3B molecules function in the same complex. The tRNAs co-immunoprecipitated with eIF3D-V5 or eIF3B were comprised mostly of Met-tRNAi (∼70% of the total), validating the high purity of scanning ribosomes (Figure 1D). Of note, eIF3D-V5 showed a lower percentage of reads that mapped to coding sequence (CDS), indicating that the RNA footprints of eIF3D-V5 were more enriched for the 5′-UTR compared with those of eIF3B (Figure 1E). There was no significant difference in 3′-UTR read occupancy between eIF3D-V5 and eIF3B, likely as a result of the smaller numbers of reads for the 3′-UTR (Supplementary Figure S2D).
Figure 1.

Sel-TCP-seq analysis of eIF3 subunits in HEK293T cells. (A) Schematic representation of the Sel-TCP-seq protocol for analysis of eIF3D-V5 and eIF3B. IP, immunoprecipitation. (B) Metagene plots for eIF3D-V5 and eIF3B footprints. All reads were mapped to all human protein-coding transcripts with a 5′-UTR and 3′-UTR of >100 nt (n = 37 916). (C) Correlation of scanning efficiency (SE) for eIF3D-V5 with that for eIF3B. All human protein-coding transcripts with a coefficient of variation of >0.5 in two technical replicates were plotted (n = 34 574). Pearson's correlation coefficient (r) is indicated. (D) Percentage of Met-tRNAi reads for tRNAs bound to eIF3D-V5 or eIF3B complexes. NS, not significant (Student's t test). (E) Percentage of reads mapped to CDS for eIF3D-V5 and eIF3B footprints. The P value was determined with Student's t test.
This tag KI approach allowed us to perform Sel-TCP-seq analysis not only for eIF3B but also for eIF3D with a reduced level of background noise. As mentioned above, the eIF3D subunit of the eIF3 complex is unique in that it binds to the 5′ cap of mRNA and controls eIF4E-independent translation in chronic glucose-deprived conditions (45,46). Application of our KI strategy to in vivo studies by generation of mice harboring the epitope tag sequence inserted at the eIF3D gene locus should provide insight into the translational dynamics of the eIF3 complex.
Identification of TISs by combined analyses of translational dynamics
The length of footprints and the positions of their 5′ and 3′ ends reflect the properties of translation. Most footprints obtained by Ribo-seq are 21 and 29 nt, reflecting the size of the 80S complex at the different stages of translation (47,48). The 5′ end of the footprints accumulates at –12 nt relative to the start codon, and their 3-nt periodicity indicates codon sliding. In contrast, the size of 40S-derived footprints in TCP-seq shows a wide distribution, ranging from ∼24 to ∼70 nt, and their 3′ end accumulates at around +24 nt relative to the start codon (27,28,49).
We performed GTI-seq with HEK293T cells treated with 50 μM LTM for 30 min as well as Ribo-seq with HEK293T cells. Consistent with the previous GTI-seq results, analysis of our GTI-seq data showed that the 5′ end of the footprints accumulated predominantly at –12 nt relative to the start codon but was also detected to a lesser extent at positions –13 and –11 (Figure 2A). Furthermore, a robust background of GTI-seq peaks in coding regions was observed (Supplementary Figure S3A). We therefore analyzed previously reported TI-seq data obtained with LTM or harringtonine, and found that most data showed a substantial proportion of reads in the coding region, with the enrichment ratio at the initiation codon varying among experiments (Supplementary Figure 3A). This noisy characteristic of TI-seq might result in false positives and a low reproducibility of TIS identification by GTI-seq alone. On the other hand, consistent with the previous results, we found that the 3′ end of the footprints derived from eIF3D-V5 and eIF3B accumulated as peaks located +23 or +24 nt downstream of the start codon (Figure 2B, Supplementary Figure S3B and S3C), indicating that the 40S footprints decrease at +23 or +24 nt. However, the decrease in the 40S footprints was not as sharp as the GTI-seq peak (Figure 2A, B), making it difficult to predict TISs precisely on the basis of Sel-TCP-seq data alone. We therefore surmised that combinatorial analysis by GTI-seq and Sel-TCP-seq might greatly reduce the noise of GTI-seq analysis. Identified GTI-seq peaks that were not associated with a decrease in the 40S footprints, or without a sufficient amount of 40S footprints, would be considered false positives. In addition, frame-fitting with the use of Ribo-seq data might further improve identification of TISs (Figure 2C).
Figure 2.

Identification of ORFs and TISs in HEK293T cells by TISCA. (A) Histogram for the 5′ end position of GTI-seq footprints relative to the start codon for all human protein-coding transcripts. (B) Histograms for the 3′ end position of eIF3D-V5 or eIF3B footprints relative to the start codon for all human protein-coding transcripts as determined by Sel-TCP-seq. (C) Schematic overview of TISCA. TISs were identified from the combination of GTI-seq and 40S dynamics, followed by frame-fitting on the basis of Ribo-seq results. (D) Read aggregation plots for GTI-seq and Sel-TCP-seq analysis of eIF3D-V5 on ATF4 mRNA. Red and blue circles show GTI-seq peaks and 40S decreasing points, respectively; red and dashed black vertical lines indicate TIS positions of uORFs and of the main ORF (mORF), respectively; and black inverted triangles denote the TIS positions identified by TISCA. (E, F) Pie charts indicating the composition of initiation codons (E) and the types of translated ORFs (F) identified by TISCA. (G) Number of TISs for each transcript identified by TISCA.
Figure 3.

Detailed analysis of initiation codons identified by TISCA. (A–C) Read aggregation plots for GTI-seq as well as for Sel-TCP-seq of eIF3D-V5 for EIF2AK1 (A), LMNB2 (B), and SAFB (C) mRNAs. Red and blue circles show GTI-seq peaks and 40S decreasing points, respectively; black inverted triangles indicate TIS positions identified by TISCA; red and black vertical lines denote novel TIS positions identified by TISCA and canonical TIS positions, respectively; and white inverted triangles with blue vertical lines indicate TIS positions identified only by GTI-seq. (D) Motif analysis for surrounding sequences of AUG, CUG, GUG, UUG and ACG initiation codons identified by TISCA.
We first listed all ORFs and their corresponding transcripts with the use of RibORF, an algorithm for ORF prediction on the basis of Ribo-seq data taking into account frame preference and signal uniformity (50). We adopted all AUG codons and near-cognate codons (CUG, GUG, UUG, ACG, AGG, AAG, AUC, AUU and AUA) as candidates for TISs, resulting in the generation of several ORF candidates with different TISs from a single frame. We next identified the positions of the 5′ end of GTI-seq peaks as well as the eIF3-bound 40S read decreasing point (40S decreasing point), where the 3′ end of the 40S footprints accumulates and the number of sequencing reads decreases (Supplementary Figure S3D). We then defined a TIS candidate as a site that met both of the following location criteria: (i) +11 to +13 nt from the 5′ end of the GTI-seq peak and (ii) –22 to –26 nt from the 40S decreasing point (Figure 2C). Furthermore, to apply frame-fitting to the ORFs corresponding to the TIS candidates, we listed all ORFs starting at the TIS candidates and selected only those that were a perfect match with the ORFs predicted by RibORF analysis (Figure 2C, Supplementary Table S2). We termed this method TISCA (TIS detection by translation Complex Analysis), and its application indeed allowed us to identify the TISs of three uORFs (uORF0, uORF1 and uORF2) of ATF4, consistent with previous results (51) (Figure 2D).
The most common initiation codon identified by TISCA was AUG (62.1%), as expected (Figure 2E). Of the various near-cognate codons also identified, CUG initiation codons (12.0%) were the most common, again consistent with previous studies (12,34). ORFs corresponding to noncoding RNAs were found to constitute 17.0% of the total ORFs (Figure 2F). Furthermore, a substantial number of transcripts contained more than one TIS (Figure 2G), likely reflecting atypical ORFs such as those derived from extension or truncation of known ORFs as well as uORFs (Figure 2F).
Elimination of noise peaks in GTI-seq by TISCA
The canonical ORF for eIF2 alpha kinase 1 (EIF2AK1) with an AUG initiation codon was detected by TISCA, and we further identified a novel extended ORF with a GUG initiation codon at +12 nt relative to the GTI-seq peak and –23 nt relative to the 40S decreasing point (Figure 3A). The novel GUG initiation codon is located 54 nt (18 amino acids) upstream of the canonical AUG initiation codon. Furthermore, in addition to the canonical ORF for lamin B2 (LMNB2) with an AUG initiation codon, we identified a noncanonical truncated ORF with an AUG initiation codon, which was also identified previously (52), located at +12 nt from the GTI-seq peak and –22 nt from the 40S decreasing point (Figure 3B). The noncanonical AUG initiation codon is located 60 nt (20 amino acids) downstream of the canonical AUG initiation codon. These results thus revealed that TISCA is able to reliably identify TISs located either upstream or downstream of the canonical TIS.
TISCA was also able to remove artifacts of GTI-seq analysis through application of the data for 40S subunit dynamics. For example, in the case of the mRNA for scaffold attachment factor B (SAFB), GTI-seq peaks that were either not associated with a 40S decreasing point or without reads were apparent in addition to the TIS identified by TISCA (Figure 3C), suggesting that these peaks were artifacts of GTI-seq analysis. TISCA thus eliminated two GTI-seq peaks for SAFB mRNA (Figure 3C). These results indicated that TISCA is able to reduce misidentification of TISs by GTI-seq alone through additional consideration of another key feature of translation—the presence and decline of the 40S complex.
Identification of AUG-initiated truncated ORFs that result from leaky scanning
A consensus sequence flanking the AUG initiation codon, known as the Kozak motif, is required for efficient initiation of translation (53,54). In the case of genes with an initial AUG codon in a weak Kozak context, the 40S complex continues to scan beyond this point in a process known as leaky scanning, and translation is initiated at the next downstream AUG codon (55). Reporter construct analysis has recently validated that 10 human genes are translated from an AUG initiation codon downstream of the canonical AUG initiation codon (56), and TISCA identified 4 of the 10 truncated ORFs (Supplementary Figure S4A). Both canonical and truncation AUG initiation codons were identified for the AASDHPPT gene, whereas only the truncation AUG initiation codon was identified for the other three genes (CMPK1, ISL2 and LIMK1), suggesting that the translation initiation efficiency of the canonical AUG initiation codon for these genes is low (Supplementary Figure S4A).
In the case of the six truncated ORFs not identified by TISCA, five of these (for PABPC4L, ASPHD1, RELB, FRMD3 and C1orf94) were not identified as a result of their low expression levels (TPM of <1) in HEK293T cells (Supplementary Figure S4B). Despite the high expression level of ZBTB8OS mRNA, however, it was not identified by TISCA. The TIS of the truncated ORF of ZBTB8OS is located at +12 nt from a GTI-seq peak and –22 nt from a 40S decreasing point, but it did not meet the criteria for frame-fitting based on Ribo-seq results (pred.P-value of >0.7) (Supplementary Figure S4C and D), whereas the canonical ORF did not satisfy all the criteria, suggesting that the truncated ORF of ZBTB8OS may be translated.
Kozak-like sequence contexts of near-cognate codons
Kozak-like motifs also have been found to be pivotal for efficient translation initiation at near-cognate codons (57). We therefore next performed motif analysis for the near-cognate codons identified by TISCA. The sequences surrounding AUG initiation codons identified by TISCA showed the same pattern as the typical Kozak motif, and the CUG, GUG, UUG and ACG initiation codons also manifested Kozak-like motifs (Figure 3D). Guanine was most frequently found at position +4, a purine base (adenine or guanine) at position –3, and guanine at position –6. In addition, cytosine was observed at positions –1, –2, –4 and –5 with relatively high frequency. These results suggested that near-cognate codons also require Kozak-like motifs for efficient translation initiation.
Proteomics analysis of NH2-termini identified by TISCA
We next attempted to validate the accuracy of TISCA by shotgun proteomics targeting of the NH2-termini of proteins encoded by the identified ORFs. We referred to published data sets for shotgun NH2-terminal proteomics, in which protein NH2-terminal peptides were enriched by three different methods as applied to HEK293T cells (19,36,37). For the actual NH2-terminal amino acid residue of proteins encoded by ORFs with near-cognate codons, there are two possible scenarios: (i) the amino acid corresponding to the initiation codon is directly incorporated (it is therefore not methionine), or (ii) methionine is incorporated regardless of the initiation codon, probably as a result of wobble base-pairing of Met-tRNAi with the initiation codon. We therefore prepared two lists of amino acid sequences on the basis of our TISCA data, one in which the first amino acid residue is the amino acid encoded by the corresponding codon, and the other in which the encoded amino acid is replaced with methionine (Figure 4A). A peptide sequence database was generated from these TISCA-based lists (TISCAdb) and from the Swiss-Prot human protein sequence database (SPdb), and the MS/MS data obtained from HEK293T cells by the three different NH2-terminal proteomics analyses were compared with this database to identify protein NH2-terminal peptides. Among the identified peptides, only acetylated NH2-terminal peptides with a posterior error probability (PEP) of <0.005 were accepted to ensure identification confidence (58). Given that ∼90% of human proteins are acetylated at the NH2-terminus (59), we only accepted peptides with NH2-acetylation as a signature modification indicative of a TIS. With this approach, we identified a total of 4285 TISs (Figure 4B, Supplementary Table S3), of which 1027 TISs were identified specifically by the SPdb search, 2919 by both the SPdb and TISCAdb searches, and 339 specifically by the TISCAdb search (Figure 4C). The percentage of translated near-cognate codons was 3.0% for all TISs identified by the TISCAdb search and 28.0% for those identified by the TISCAdb search alone (Figure 4D). We also identified three translated near-cognate codons (GUG for eIF4G2 and CUG for R3HCC1 and RNF187) among TISs identified commonly by the SPdb and TISCAdb searches.
Figure 4.

Proteomics analysis of TISs identified by TISCA. (A) Outline for the generation of amino acid sequence lists for comparison with proteomics data. For the ORFs with near-cognate initiation codons identified by TISCA, two types of sequences were generated: one in which the first amino acid was the residue actually encoded by the corresponding codon, and the other in which the first amino acid was methionine regardless of the encoded residue. (B) Venn diagram showing numbers of TISs identified in the three data sets generated by the proteomics analyses of Chang et al. (37), Na et al. (19) and Yeom et al. (36). (C) Classification of TISs identified by proteomics analyses. The TISs were classified as those identified only in Swiss-Prot, both in Swiss-Prot and by TISCA, or by TISCA alone. (D) Pie charts indicating initiation codon usage of TISs identified from all TISCA data or from TISCA-specific data. (E) Number of novel TISs sorted by the type of initial amino acid and initiation codon. (F) Number of novel TISs in the Met or cleaved categories that were identified as possible substrates for MetAP. ORFs containing alanine, cysteine, glycine, proline, serine, threonine, or valine as the second amino acid were considered to encode potential MetAP substrates. (G) Density plots of translation efficiency (TE) as determined by Ribo-seq analysis for ORFs initiated at AUG or near-cognate codons. Multiple overlapping ORFs were excluded from the analysis.
The novel ORFs identified only by the TISCAdb search were assigned to three categories on the basis of the type of NH2-terminal amino acid: non-methionine, methionine, and cleaved (Figure 4E). Most peptides were included in the methionine or cleaved categories, with some derived from the same ORF being included in both of these categories. Methionine aminopeptidase (MetAP) cleaves the NH2-terminal methionine and has a preference for substrates that contain alanine, cysteine, glycine, proline, serine, threonine, or valine at the position adjacent to the NH2-terminal methionine (60). Almost all proteins with near-cognate initiation codons in the cleaved category contained such amino acids recognizable by MetAP at the NH2-terminal penultimate position, suggesting that the first amino acid is methionine (Figure 4F). In contrast, most such proteins in the methionine category had penultimate amino acids that were unlikely to be recognized by MetAP. Both the EIF2AK1 extended ORF with a GUG initiation codon and the LMNB2 truncated ORF with an AUG initiation codon shown in Figure 3A and B, respectively, were identified and classified in the cleaved category.
We detected only three ORFs in which amino acids (leucine, valine, and threonine) other than methionine were incorporated at the corresponding first codon (CUG, GUG and ACG, respectively). Two types of NH2-terminal peptide containing methionine or threonine at the first codon (ACG) detected by TISCA were identified by proteomics analysis for the CDC-like kinase 2 (CLK2) gene (Supplementary Figure S5A and B), consistent with a previous study (19). Only one peptide with threonine, as opposed to many peptides with methionine, was identified by proteomics analysis, suggesting that the protein with an initial methionine is the dominant form. Of interest, the TIS corresponding to the AUG codon for the canonical ORF of CLK2 deposited in the Swiss-Prot database was not identified by either TISCA or proteomics analysis (Supplementary Figure S5A). For the seryl-tRNA synthetase (SARS) gene, a peptide with leucine as the first amino acid was detected by TISCA as well as by proteomics analysis (Supplementary Figure S5A and C). In addition, a canonical AUG initiation codon that was also identified by TISCA was present two amino acids upstream of the CUG initiation codon, and the peptide initiating at this AUG codon was indeed identified by two of the independent proteomics analyses (Supplementary Figure S5A and C). Furthermore, one peptide with valine (GUG) as the initial amino acid for the exocyst complex component 7 (EXOC7) gene was identified by both TISCA and proteomics analysis (Supplementary Figure S5A and D). Given that the PEP was only slightly below the threshold (= 0.0040) and that the signals for GTI-seq and Sel-TCP-seq were noisy (Supplementary Figure S5D), however, this peptide was likely the result of a false identification.
Near-cognate codons accounted for 37.9% of TISs in the TISCA-based prediction (Figure 2E) but only 3.0% in the proteomics analyses (Figure 4D). Consistent with previous findings that the translation initiation activity of ORFs initiated at non-AUG codons is lower than that of ORFs initiated at AUG (61–63), our Ribo-seq results revealed that the translation efficiency of ORFs with near-cognate initiation codons was lower than that of those with the AUG codon (Figure 4G), suggesting that peptides corresponding to the former ORFs may not have been detected by proteomics analysis as a result of their low abundance. It is also possible that TISCA detected ‘noisy or abortive initiation’ that may occur infrequently as misinitiation, given that it is more sensitive than proteomics analysis.
Translation initiation with AAG as an initiation codon
Proteomics analysis identified several ORFs apparently translated from AAG or AGG codons, but, as far as we are aware, translation initiation at such codons has not previously been described other than in high-throughput studies such as those based on GTI-seq (64). An ORF with an AGG initiation codon was identified by TISCA in the putative WAS protein family homolog 3 (WASH3P) gene. A novel truncation AUG initiation codon was located immediately after the novel AGG initiation codon (Supplementary Figure S6A), and the sequence of the identified peptide was identical to the product predicted to be derived from the truncation AUG initiation codon (Supplementary Figure S6B). This peptide sequence would be classified in the Met category if translated from the truncation AUG initiation codon, and in the cleaved category if translated from the AGG initiation codon. Given that the putative product translated from the AGG initiation codon does not contain an amino acid sequence recognizable by MetAP (Figure 4F), the identified peptide is likely a product of the truncation AUG initiation codon.
ORFs with an AAG initiation codon were identified for three genes (SNRPG, ERI2 and HIST1H2BH) by proteomics analysis. In the case of SNRPG and ERI2, the signals of GTI-seq and Sel-TCP-seq were noisy, and the identified peptides appeared to be misidentified given that their spectra showed many unannotated peaks and the region covered by the b- and y-ions was limited to two or three residues at the COOH-terminus, although their PEP values met the criterion (Supplementary Figure S6C and D). On the other hand, in the case of HIST1H2BH, the canonical AUG initiation codon was located at the 5′ end of the mRNA, and the signals of GTI-seq and Sel-TCP-seq showed reliable patterns only for the AAG initiation codon, suggesting that this gene is translated only from the AAG initiation codon (Supplementary Figure S6E). Although the spectrum of the peptide showed many unannotated peaks, the y-ions covered almost the entire peptide. Furthermore, an additional two spectra with different charge states were found. Although these spectra did not meet the PEP criterion as a result of the large number of unannotated peaks, the observed y-ion profiles were similar in all the three spectra, suggesting that they were not randomly matched (Supplementary Figure S6E). These results thus suggest that at least the HIST1H2BH gene is indeed translated from an AAG initiation codon.
Improved accuracy of TIS prediction by TISCA
To evaluate the performance of TISCA in prediction of TISs, we compared it with that of existing methods. Ribo-TISH, RiboCode and RiboTaper all adopt an unsupervised approach to predict ORFs de novo from Ribo-seq data, whereas ORF-RATER and riboHMM both rely on a supervised approach that requires training with annotated ORFs (13). Among these existing methods, Ribo-TISH and ORF-RATER use not only Ribo-seq data but also TI-seq data, which allows more accurate TIS identification (13,31).
Ribo-TISH analysis identified 12 059 TISs, including 35.6% with near-cognate codons, using the same data set as that used for TISCA (Figure 5A). The number of TISs commonly predicted by both Ribo-TISH and TISCA was 6747, and the numbers of those predicted by only Ribo-TISH or only TISCA were 5312 and 10 170, respectively (Figure 5B). The three NH2-terminal proteome data sets were then searched against the Ribo-TISH–based database (Ribo-TISHdb) and TISCAdb sequences to identify acetylated NH2-terminal peptides. Again, only acetylated NH2-terminal peptides with a PEP of <0.005 were accepted to ensure identification confidence. A total of 2063 TISs was commonly identified by both TISCAdb and Ribo-TISHdb searches, with an additional 1198 and 289 TISs being identified specifically by the TISCAdb search and the Ribo-TISHdb search, respectively (Figure 5C, Supplementary Table S4). The number of peptides identified only by the Ribo-TISHdb search was thus much smaller than that identified only by the TISCAdb search (Figure 5D).
Figure 5.

Performance evaluation for TIS prediction by TISCA compared with that by previous methods. (A) Pie chart showing initiation codon composition for TISs identified by Ribo-TISH. (B, C) Venn diagrams showing numbers of TISs predicted by TISCA and Ribo-TISH (B) as well as the numbers of these TISs detected by proteomics analyses (C). (D) Histogram of Andromeda scores for acetylated peptides identified from the databases containing Ribo-TISH and TISCA sequences. Common refers to peptides identified by both Ribo-TISH and TISCA. (E) Pie chart showing initiation codon composition for TISs identified by ORF-RATER. (F, G) Venn diagrams showing numbers of TISs predicted by TISCA and ORF-RATER (F) as well as the numbers of these TISs detected by proteomics analyses (G). (H) Histogram of Andromeda scores for acetylated peptides identified from the databases containing ORF-RATER and TISCA sequences. Common refers to peptides identified by both ORF-RATER and TISCA.
ORF-RATER analysis identified 11 141 TISs, including 19.6% with near-cognate codons, again with the same data set as that used for TISCA (Figure 5E and F). ORF-RATER thus identified more AUG codons, probably as a result of the use of ORFs with AUG codons as training data. The three NH2-terminal proteome data sets were then searched against the ORF-RATER–based database (ORF-RATERdb) and TISCAdb sequences to identify acetylated NH2-terminal peptides. We found that the number of TISs and peptides identified specifically by TISCAdb was greater than that identified by ORF-RATERdb alone (Figure 5G and H, Supplementary Table S5). Collectively, these results suggested that TISCA is able to identify more TISs with higher reliability compared with the existing methods.
Translation at near-cognate initiation codons is largely dependent on eIF2
Both eIF2A and eIF2D have been shown to facilitate incorporation of amino acids other than methionine at TISs by recruiting tRNAs other than Met-tRNAi, such as Leu-tRNACUG (22–24). However, our analysis of proteomics data revealed that methionine was incorporated at the NH2-terminal position of most proteins translated from near-cognate initiation codons. To examine the possible contribution of eIF2A and eIF2D to noncanonical translation initiation, we generated HEK293T cells lacking eIF2A, eIF2D, or both of these proteins with the use of the CRISPR-Cas9 system (Figure 6A, Supplementary Figure S7A) and then subjected these cells to Ribo-seq analysis. We also performed Ribo-seq analysis with HEK293T cells transfected with siRNAs targeting eIF2α. Such transfection for 48 h reduced the abundance of eIF2α mRNA by >90%, whereas it reduced that of the eIF2α protein by only ∼50% (Figure 6B and C). Given that longer siRNA transfection times (≥72 h) elicited apoptosis, it was technically difficult to efficiently deplete eIF2α at the protein level. We therefore also performed Ribo-seq analysis with HEK293T cells treated with arsenite for 60 min in order to suppress eIF2-dependent translation by promoting the phosphorylation of eIF2α (65). We confirmed that arsenite treatment of HEK293T cells induced eIF2α phosphorylation and the expression of ATF4, which reflects suppression of eIF2 function (Figure 6D).
Figure 6.

eIF2 dependency of translation initiation at near-cognate codons. (A) Immunoblot analysis of eIF2A and eIF2D in HEK293T cells deficient in eIF2A (eIF2A KO), eIF2D (eIF2D KO), or both eIF2A and eIF2D (DKO) as well as in WT cells. Two independent clones of each type were examined, and HSP90 was analyzed as a loading control. (B) Fold change in RNA abundance for eIF2α mRNA in HEK293T cells transfected with siRNAs specific for eIF2α (eIF2α KD #1 or #2) relative to HEK293T cells transfected with a control siRNA. padj, adjusted P value. (C) Immunoblot analysis of eIF2α in HEK293T cells transfected with siRNAs as in (B). Lanes 1 and 2 correspond to biological replicates. (D) Immunoblot analysis of ATF4 as well as of phosphorylated (p-) and total forms of eIF2α in WT HEK293T cells treated with 40 μM arsenite for the indicated times. (E) Numbers of ORFs showing a significant change (adjusted P value of <0.05) in translation efficiency (TE) in eIF2A KO, eIF2D KO, and DKO cells, in eIF2α KD cells, as well as in arsenite-treated WT HEK293T cells compared with control cells. For eIF2A KO, eIF2D KO, DKO and eIF2α KD cells, ORFs not commonly up-regulated or down-regulated in two clones or with two different siRNAs were removed. (F) GSEA plots for ORFs initiated at near-cognate codons whose translation efficiency was down-regulated by arsenite treatment. Translation of these ORFs is compared between eIF2α KD cells and control HEK293T cells. NES, normalized enrichment score. (G, J) Read aggregation plots for GTI-seq and Sel-TCP-seq of eIF3D-V5 for NPM1 (G) and HNRNPA2B1 (J) mRNAs. Red and blue circles show GTI-seq peaks and 40S decreasing points, respectively; black inverted triangles indicate TISs identified by TISCA; and red and black vertical lines denote novel TISs identified by TISCA and canonical TISs, respectively. (H, K) Read aggregation plots of Ribo-seq for NPM1 (H) and HNRNPA2B1 (K) mRNAs in HEK293T cells transfected with control or eIF2α siRNAs. The average of two replicates is shown. (I, L) Fold change in translation efficiency for mORF and the extended ORF of NPM1 (I) and HNRNPA2B1 (L) as analyzed by RiboDiff in eIF2A KO, eIF2D KO, DKO and eIF2α KD cells as well as in arsenite-treated WT cells compared with control cells.
For ORFs initiated from near-cognate codons that overlap with AUG-dependent ORFs, we quantified the specific changes in translation efficiency by excluding the overlapping region (Supplementary Figure S7B). We therefore omitted evaluation of translation efficiency for ORFs that completely overlap with canonical AUG-dependent ORFs, such as those corresponding to truncated versions of the latter. ORFs that do not overlap with AUG-dependent ORFs (for example, uORFs) were quantified without any processing. The knockout (KO) of eIF2A, eIF2D or both proteins as well as knockdown (KD) of eIF2α had only a small effect on translation efficiency, whereas arsenite treatment affected the translation efficiency of ORFs to a much greater extent (Figure 6E, Supplementary Figure S7B, Supplementary Table S6).
The marked effect of arsenite treatment on translation efficiency may have been due to induction of cellular stress rather than to inhibition of eIF2. Although siRNA transfection did not efficiently deplete eIF2α protein, if the effect of arsenite treatment on non-AUG initiation is indeed dependent of eIF2, the effect should also be reproduced, at least to a lesser extent, by eIF2α depletion. To test this notion, we performed GSEA with a gene set consisting of non-AUG–initiated ORFs whose translational efficiency was significantly reduced by arsenite treatment. The translational efficiency of the arsenite-sensitive ORFs was markedly down-regulated by eIF2α KD (Figure 6F), whereas it was not significantly affected by KO of eIF2A, eIF2D, or both proteins, with the exception of a weakly significant inhibition apparent in eIF2D KO #1 cells (Supplementary Figure S8). These results suggested that the attenuation of non-AUG translation initiation by arsenite treatment is dependent, at least in large part, on eIF2.
This trend can be exemplified by representative genes. The main ORF of the nucleophosmin 1 (NPM1) gene is translated from an AUG initiation codon, but we also identified a novel extended ORF with a CUG initiation codon by TISCA (Figure 6G). The translational efficiency of the extended ORF with the CUG initiation codon was significantly reduced by arsenite treatment and by transfection with two different eIF2α siRNAs (Figure 6H and I). The translation efficiency of the main ORF with the AUG initiation codon was significantly reduced by arsenite treatment and by transfection only with siRNA #2 (Figure 6I). We also identified a novel extended ORF with an ACG initiation codon for the heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) gene by TISCA (Figure 6J). The translation efficiency for the extended ORF was significantly reduced both by arsenite treatment and by eIF2α KD, whereas that for the AUG-dependent main ORF was not affected (Figure 6K and L). These results thus suggested that translation from these near-cognate codons is mediated by authentic eIF2 rather than by eIF2A or eIF2D.
Enhancement of eIF2A and eIF2D dependence by eIF2 suppression at a limited number of ORFs with near-cognate initiation codons
Suppression of eIF2 function by arsenite treatment was previously shown to increase the dependence of translation on eIF2A (24). We therefore examined the changes in translation efficiency for ORFs initiated at near-cognate codons in cells deficient in eIF2A, eIF2D, or both proteins in the presence of arsenite (Figure 7A, Supplementary Figure S9A, Supplementary Table S7). However, the loss of eIF2A, eIF2D or both factors had a limited effect on such translation efficiency even in arsenite-treated cells, although the fold changes in translation efficiency were greater than those apparent under the arsenite-free condition (Figures 6E, and 7A), indicative of a weak but increased dependence on eIF2A and eIF2D in the setting of eIF2 suppression.
Figure 7.

Contribution of eIF2A and eIF2D to translation initiation at near-cognate codons in the setting of eIF2 inhibition. (A) Numbers of ORFs showing a significant change (adjusted P value of <0.05) in translation efficiency in eIF2A KO, eIF2D KO and DKO cells compared with control cells under the arsenite-treated condition. ORFs not commonly up-regulated or down-regulated in two clones were removed. (B) Read aggregation plots for GTI-seq and Sel-TCP-seq of eIF3D-V5 for RPL38 mRNA. Red and blue circles show GTI-seq peaks and 40S decreasing points, respectively; black inverted triangles indicate TIS positions identified by TISCA; and red and black vertical lines denote novel TIS positions identified by TISCA and canonical TIS positions, respectively. euORF, extended upstream ORF. (C) Read aggregation plots of Ribo-seq for RPL38 mRNA in WT and DKO cells treated with arsenite. The average of two replicates is shown. (D) Fold change in translation efficiency for ORFs of RPL38 as analyzed by RiboDiff in eIF2A KO, eIF2D KO, and DKO cells with or without arsenite treatment compared with control cells not treated with arsenite.
As a representative example, a novel uORF and its extended uORF were identified in the 60S ribosomal protein L38 (RPL38) mRNA by TISCA (Figure 7B). In both eIF2A- and eIF2D-deficient cells, the translation efficiency of the uORF and its extended form was reduced only with arsenite treatment, remaining unaltered in the absence of arsenite (Figure 7C and D). Although changes in the translation of uORFs have previously been shown to affect the translation efficiency of downstream ORFs in the opposite direction (66), our results showed that a decrease in translation efficiency of a uORF did not result in up-regulation of the translation efficiency of the main ORF for RPL38 (Figure 7D).
We also analyzed the HNRNPA2B1 gene in WT and eIF2A/eIF2D KO cells under the arsenite-treated condition. The translation efficiency of the extended ORF with an ACG initiation codon, as well as that of the main ORF, did not differ significantly between WT and the double-knockout (DKO) cells even under the arsenite-treated condition (Supplementary Figure S9B and S9C). These results further support the conclusion that translation of the extended ORF of HNRNPA2B1 is dependent on eIF2. The lack of an effect of suppression of eIF2, eIF2A and eIF2D on translation efficiency of the main ORF might reflect a contribution of other tRNA binding proteins such as DENR/MCTS1 (21), although this issue requires further investigation.
Collectively, these findings suggest that, although translation initiation at most near-cognate codons is dependent largely on eIF2 and Met-tRNAi, its dependence on eIF2A and eIF2D in a limited number of cases is increased in the setting of eIF2 suppression.
DISCUSSION
We have here described the development of a novel analytic framework for identification of TISs based on the combination of data sets for translational dynamics such as Sel-TCP-seq, GTI-seq and Ribo-seq. This novel approach, designated TISCA, allows a comprehensive characterization of AUG- and non-AUG-dependent translation initiation. With this new method, we have shown that translation initiation at near-cognate codons is most likely to be dependent on eIF2 and Met-tRNAi, although this issue warrants further investigation to verify the contribution of eIF2 to the translation initiation at near-cognate codons.
It has remained unclear whether all the footprints revealed by Ribo-seq represent natural translation. To address this issue, various computational approaches that take certain features such as fragment size, signal uniformity, and 3-nt periodicity into account have been developed (13). Methods that rely only on Ribo-seq data such as RibORF and RiboCode identify multiple candidate ORFs with the same stop codon and different start codons (35,67). In this case, the ORFs with the most upstream in-frame ATG are retained, and the possibility of ORFs starting from near-cognate codons is ignored, rendering accurate identification of TISs difficult. Ribo-TISH and ORF-RATER are also able to exploit TI-seq data, which allows more accurate TIS identification compared with the methods that rely on Ribo-seq data alone. However, although TI-seq allows enrichment for the 80S ribosome at the start codon, it does not completely eliminate ribosomes engaged in translation elongation, which hinders precise TIS identification. Integration of (G)TI-seq data with data for Sel-TCP-seq in TISCA supports the efficient removal of noise associated with (G)TI-seq and thereby allows a more accurate identification of TISs and consequent correct identification of ORFs.
Comprehensive analysis by FACS-seq has shown that a Kozak-like sequence is required for efficient translation initiation at near-cognate codons (57). Although previous studies with Ribo-seq found weak enrichment for a Kozak-like motif at near-cognate codons (68,69), our analysis revealed a Kozak-like sequence context around the near-cognate codons identified by TISCA. A purine base at position –3 and a guanine at position +4 were previously shown to greatly increase translational activity of near-cognate codons (57), and these same sequence contexts were apparent for the near-cognate codons identified by TISCA. Translation efficiency was previously found to be more sensitive to the surrounding sequence context for near-cognate codons than for AUG (57). The mismatch between near-cognate initiation codons and the anticodon reduces the binding energy, with the result that the nucleotide context around these initiation codons affects the translation efficiency of ORFs with such mismatched sequences to a greater extent.
In a preliminary study, we applied TISCA to several fully noncognate codons as candidate initiation codons, and identified some fully noncognate TISs that met the TISCA identification criteria (data not shown). However, none of these TISs was confirmed by proteomics analysis, suggesting that the corresponding expression levels may be very low or that they are false positives. This issue warrants examination in more detail by future studies. In addition, inclusion of all noncognate codons is impractical, given that it requires a huge amount of computational time. We therefore focused only on near-cognate initiation codons in this study.
Despite previous studies (20–23), the role of eIF2A and eIF2D in initiating translation from near-cognate codons remains unclear. Mice deficient in eIF2A are viable and do not manifest any obvious phenotypes, suggesting that this factor may not play a major role in translation (70). The International Mouse Phenotyping Consortium (IMPC) analysis revealed that mice deficient in eIF2D are also viable and have no apparent developmental defects. In addition, we generated HEK293T cells lacking eIF2A, eIF2D, or both proteins, and these mutant cells were viable. Ribo-seq analysis indicated that translation initiation at near-cognate codons was dependent largely on eIF2, with inhibition of eIF2 affecting the dependence on eIF2A and eIF2D only slightly. These results suggest that eIF2A and eIF2D might contribute to the regulation of specific physiological functions by fine-tuning translation initiation at specific ORFs.
Although TISCA substantially reduced the noise of GTI-seq analysis as a result of application of the 40S decreasing point as well as identified more TISs with higher reliability compared with existing tools, it still gives rise to a certain number of misidentifications, such as the apparent ORF with the GUG initiation codon identified for EXOC7 (Supplementary Figure S5D). It will therefore be important in the future to further improve the accuracy of this method with the use of different computational approaches and other experimental data sets. In addition, the LTM treatment adopted in GTI-seq analysis may induce some experimental artifacts, with the adoption of a more physiological alternative method being preferable.
The comprehensive identification of near-cognate codons is important to provide insight into the molecular mechanism of noncanonical translation initiation. In the present study, we have identified a large number of TISs with high reliability by combining analyses of translation dynamics and we have clarified their pivotal features. However, TIS usage varies in a manner dependent on cellular stress induction and among tissue types (32), with the result that development of improved methods to identify TISs in vivo will be needed for a more comprehensive understanding of such variation. The development of such methods will also allow characterization of changes in TIS usage associated with diseases such as cancer and diabetes, which should lead to a better understanding of disease pathogenesis and may inform the development of new treatments.
DATA AVAILABILITY
All sequence data have been deposited in GEO under the accession number GSE174329. The MS data have been deposited with the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the JPOST partner repository under the data set identifier PXD025794. TISCA pipelines and example data are available at https://github.com/KazuIchihara/TISCA.
Supplementary Material
ACKNOWLEDGEMENTS
We thank L. Cui and T. Akinaga for technical assistance as well as A. Ohta for help with preparation of the manuscript. Computations were performed in part on the NIG supercomputer at ROIS National Institute of Genetics.
Author contributions. K. Ichihara performed computational analysis, developed algorithms, and wrote the manuscript. A.M. conceived and designed the project, performed experiments and wrote the manuscript. H.N. performed proteomics data analysis. Y.K. prepared libraries for sequencing. H.S., Y.S., S.I., K. Imami and Y.I. supervised experimental design. K.I.N. coordinated the study and wrote the manuscript.
Contributor Information
Kazuya Ichihara, Department of Molecular and Cellular Biology, Medical Institute of Bioregulation, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka, Fukuoka 812-8582, Japan.
Akinobu Matsumoto, Department of Molecular and Cellular Biology, Medical Institute of Bioregulation, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka, Fukuoka 812-8582, Japan.
Hiroshi Nishida, Department of Molecular and Cellular Bioanalysis, Graduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan.
Yuki Kito, Department of Molecular and Cellular Biology, Medical Institute of Bioregulation, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka, Fukuoka 812-8582, Japan.
Hideyuki Shimizu, Department of Molecular and Cellular Biology, Medical Institute of Bioregulation, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka, Fukuoka 812-8582, Japan.
Yuichi Shichino, RNA Systems Biochemistry Laboratory, RIKEN Cluster for Pioneering Research, Wako, Saitama 351-0198, Japan.
Shintaro Iwasaki, RNA Systems Biochemistry Laboratory, RIKEN Cluster for Pioneering Research, Wako, Saitama 351-0198, Japan; Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwa, Chiba 277-8561, Japan; AMED-CREST, Japan Agency for Medical Research and Development, Wako, Saitama 351-0198, Japan.
Koshi Imami, Department of Molecular and Cellular Bioanalysis, Graduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan.
Yasushi Ishihama, Department of Molecular and Cellular Bioanalysis, Graduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan.
Keiichi I Nakayama, Department of Molecular and Cellular Biology, Medical Institute of Bioregulation, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka, Fukuoka 812-8582, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
KAKENHI grants from Japan Society for the Promotion of Science (JSPS); Ministry of Education, Culture, Sports, Science and Technology of Japan [20H05928 to A.M., 18H05215 to to K.I.N.] (in part). Funding for open access charge: KAKENHI grant from Japan Society for the Promotion of Science (JSPS); Ministry of Education, Culture, Sports, Science and Technology of Japan [18H05215 to K.I.N.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hinnebusch A.G. Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem. Sci. 2017; 42:589–611. [DOI] [PubMed] [Google Scholar]
- 2. Jackson R.J., Hellen C.U., Pestova T.V.. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010; 11:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pelletier J., Sonenberg N.. The organizing principles of eukaryotic ribosome recruitment. Annu. Rev. Biochem. 2019; 88:307–335. [DOI] [PubMed] [Google Scholar]
- 4. Shirokikh N.E., Preiss T.. Translation initiation by cap-dependent ribosome recruitment: recent insights and open questions. Wiley Interdiscip. Rev.-RNA. 2018; 9:e1473. [DOI] [PubMed] [Google Scholar]
- 5. Sonenberg N., Hinnebusch A.G.. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009; 136:731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hronova V., Mohammad M.P., Wagner S., Panek J., Gunisova S., Zeman J., Poncova K., Valasek L.S.. Does eIF3 promote reinitiation after translation of short upstream ORFs also in mammalian cells. RNA Biol. 2017; 14:1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mohammad M.P., Pondelickova V.M., Zeman J., Gunisova S., Valasek L.S.. In vivo evidence that eIF3 stays bound to ribosomes elongating and terminating on short upstream ORFs to promote reinitiation. Nucleic Acids Res. 2017; 45:2658–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin Y., Li F., Huang L., Polte C., Duan H., Fang J., Sun L., Xing X., Tian G., Cheng Y.et al.. eIF3 associates with 80S ribosomes to promote translation elongation, mitochondrial homeostasis, and muscle health. Mol. Cell. 2020; 79:575–587. [DOI] [PubMed] [Google Scholar]
- 9. Ingolia N.T. Ribosome profiling: new views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014; 15:205–213. [DOI] [PubMed] [Google Scholar]
- 10. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ingolia N.T., Lareau L.F., Weissman J.S.. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011; 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee S., Liu B., Lee S., Huang S.X., Shen B., Qian S.B.. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calviello L., Ohler U.. Beyond read-counts: Ribo-seq data analysis to understand the functions of the transcriptome. Trends Genet. 2017; 33:728–744. [DOI] [PubMed] [Google Scholar]
- 14. Hann S.R., King M.W., Bentley D.L., Anderson C.W., Eisenman R.N.. A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt's lymphomas. Cell. 1988; 52:185–195. [DOI] [PubMed] [Google Scholar]
- 15. Takahashi K., Maruyama M., Tokuzawa Y., Murakami M., Oda Y., Yoshikane N., Makabe K.W., Ichisaka T., Yamanaka S.. Evolutionarily conserved non-AUG translation initiation in NAT1/p97/DAP5 (EIF4G2). Genomics. 2005; 85:360–371. [DOI] [PubMed] [Google Scholar]
- 16. Xiao J.H., Davidson I., Matthes H., Garnier J.M., Chambon P.. Cloning, expression, and transcriptional properties of the human enhancer factor TEF-1. Cell. 1991; 65:551–568. [DOI] [PubMed] [Google Scholar]
- 17. Ivanov I.P., Firth A.E., Michel A.M., Atkins J.F., Baranov P.V.. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res. 2011; 39:4220–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liang H., Chen X., Yin Q., Ruan D., Zhao X., Zhang C., McNutt M.A., Yin Y.. PTENbeta is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun. 2017; 8:14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Na C.H., Barbhuiya M.A., Kim M.S., Verbruggen S., Eacker S.M., Pletnikova O., Troncoso J.C., Halushka M.K., Menschaert G., Overall C.M.et al.. Discovery of noncanonical translation initiation sites through mass spectrometric analysis of protein N termini. Genome Res. 2018; 28:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dmitriev S.E., Terenin I.M., Andreev D.E., Ivanov P.A., Dunaevsky J.E., Merrick W.C., Shatsky I.N.. GTP-independent tRNA delivery to the ribosomal P-site by a novel eukaryotic translation factor. J. Biol. Chem. 2010; 285:26779–26787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skabkin M.A., Skabkina O.V., Dhote V., Komar A.A., Hellen C.U.T., Pestova T.V.. Activities of ligatin and MCT-1/DENR in eukaryotic translation initiation and ribosomal recycling. Genes Dev. 2010; 24:1787–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Starck S.R., Jiang V., Pavon-Eternod M., Prasad S., McCarthy B., Pan T., Shastri N.. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science. 2012; 336:1719–1723. [DOI] [PubMed] [Google Scholar]
- 23. Kearse M.G., Wilusz J.E.. Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 2017; 31:1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sendoel A., Dunn J.G., Rodriguez E.H., Naik S., Gomez N.C., Hurwitz B., Levorse J., Dill B.D., Schramek D., Molina H.et al.. Translation from unconventional 5′ start sites drives tumour initiation. Nature. 2017; 541:494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Young D.J., Makeeva D.S., Zhang F., Anisimova A.S., Stolboushkina E.A., Ghobakhlou F., Shatsky I.N., Dmitriev S.E., Hinnebusch A.G., Guydosh N.R.. Tma64/eIF2D, Tma20/MCT-1, and Tma22/DENR recycle post-termination 40S subunits in vivo. Mol. Cell. 2018; 71:761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Archer S.K., Shirokikh N.E., Beilharz T.H., Preiss T.. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature. 2016; 535:570–574. [DOI] [PubMed] [Google Scholar]
- 27. Wagner S., Herrmannova A., Hronova V., Gunisova S., Sen N.D., Hannan R.D., Hinnebusch A.G., Shirokikh N.E., Preiss T., Valasek L.S.. Selective translation complex profiling reveals staged initiation and co-translational assembly of initiation factor complexes. Mol. Cell. 2020; 79:546–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bohlen J., Fenzl K., Kramer G., Bukau B., Teleman A.A.. Selective 40S footprinting reveals cap-tethered ribosome scanning in human cells. Mol. Cell. 2020; 79:561–574. [DOI] [PubMed] [Google Scholar]
- 29. McGlincy N.J., Ingolia N.T.. Transcriptome-wide measurement of translation by ribosome profiling. Methods. 2017; 126:112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang P., He D.D., Xu Y., Hou J.K., Pan B.F., Wang Y.F., Liu T., Davis C.M., Ehli E.A., Tan L.et al.. Genome-wide identification and differential analysis of translational initiation. Nat. Commun. 2017; 8:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao X.W., Wan J., Liu B., Ma M., Shen B., Qian S.B.. Quantitative profiling of initiating ribosomes in vivo. Nat. Methods. 2015; 12:147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stern-Ginossar N., Weisburd B., Michalski A., Vu T.K.L., Hein M.Y., Huang S.X., Ma M., Shen B., Qian S.B., Hengel H.et al.. Decoding human cytomegalovirus. Science. 2012; 338:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J., Brunner A.D., Cogan J.Z., Nunez J.K., Fields A.P., Adamson B., Itzhak D.N., Li J.Y., Mann M., Leonetti M.D.et al.. Pervasive functional translation of noncanonical human open reading frames. Science. 2020; 367:1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ji Z. RibORF: Identifying genome-wide translated open reading frames using ribosome profiling. Curr. Protoc. Mol. Biol. 2018; 124:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yeom J., Ju S., Choi Y., Paek E., Lee C.. Comprehensive analysis of human protein N-termini enables assessment of various protein forms. Sci. Rep. 2017; 7:6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang C.H., Chang H.Y., Rappsilber J., Ishihama Y.. Isolation of acetylated and unmodified protein N-terminal peptides by strong cation exchange chromatographic separation of TrypN-digested peptides. Mol. Cell Proteomics. 2020; 20:100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhong Y., Karaletsos T., Drewe P., Sreedharan V.T., Kuo D., Singh K., Wendel H.G., Ratsch G.. RiboDiff: detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics. 2017; 33:139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valasek L.S., Zeman J., Wagner S., Beznoskova P., Pavlikova Z., Mohammad M.P., Hronova V., Herrmannova A., Hashem Y., Gunisova S.. Embraced by eIF3: structural and functional insights into the roles of eIF3 across the translation cycle. Nucleic Acids Res. 2017; 45:10948–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bochler A., Querido J.B., Prilepskaja T., Soufari H., Simonetti A., Del Cistia M.L., Kuhn L., Ribeiro A.R., Valasek L.S., Hashem Y.. Structural differences in translation initiation between pathogenic trypanosomatids and their mammalian hosts. Cell Rep. 2020; 33:108534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Querido J.B., Sokabe M., Kraatz S., Gordiyenko Y., Skehel J.M., Fraser C.S., Ramakrishnan V.. Structure of a human 48S translational initiation complex. Science. 2020; 369:1220–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith M.D., Arake-Tacca L., Nitido A., Montabana E., Park A., Cate J.H.. Assembly of eIF3 mediated by mutually dependent subunit insertion. Structure. 2016; 24:886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee A.S., Kranzusch P.J., Doudna J.A., Cate J.H.. eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature. 2016; 536:96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lamper A.M., Fleming R.H., Ladd K.M., Lee A.S.Y.. A phosphorylation-regulated eIF3d translation switch mediates cellular adaptation to metabolic stress. Science. 2020; 370:853–856. [DOI] [PubMed] [Google Scholar]
- 47. Lareau L.F., Hite D.H., Hogan G.J., Brown P.O.. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. Elife. 2014; 3:e01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu C.C.C., Zinshteyn B., Wehner K.A., Green R.. High-resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress. Mol. Cell. 2019; 73:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Giess A., Torres Cleuren Y.N., Tjeldnes H., Krause M., Bizuayehu T.T., Hiensch S., Okon A., Wagner C.R., Valen E.. Profiling of small ribosomal subunits reveals modes and regulation of translation initiation. Cell Rep. 2020; 31:107534. [DOI] [PubMed] [Google Scholar]
- 50. Ji Z., Song R.S., Regev A., Struhl K.. Many lncRNAs, 5 ' UTRs, and pseudogenes are translated and some are likely to express functional proteins. Elife. 2015; 4:e08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu P.D., Harding H.P., Ron D.. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004; 167:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schumacher J., Relchenzeller M., Kempf T., Schnolzer M., Herrmann H.. Identification of a novel, highly variable amino-terminal amino acid sequence element in the nuclear intermediate filament protein lamin B-2 from higher vertebrates. FEBS Lett. 2006; 580:6211–6216. [DOI] [PubMed] [Google Scholar]
- 53. Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986; 44:283–292. [DOI] [PubMed] [Google Scholar]
- 54. Simonetti A., Guca E., Bochler A., Kuhn L., Hashem Y.. Structural insights into the mammalian late-stage initiation complexes. Cell Rep. 2020; 31:107497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999; 234:187–208. [DOI] [PubMed] [Google Scholar]
- 56. Benitez-Cantos M.S., Yordanova M.M., O’Connor P.B.F., Zhdanov A.V., Kovalchuk S.I., Papkovsky D.B., Andreev D.E., Baranov P.V.. Translation initiation downstream from annotated start codons in human mRNAs coevolves with the Kozak context. Genome Res. 2020; 30:974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Noderer W.L., Wang C.L.. Complete motif analysis of sequence requirements for translation initiation at non-AUG start codons. Nucleic Acids Res. 2018; 46:985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kall L., Storey J.D., MacCoss M.J., Noble W.S.. Posterior error probabilities and false discovery rates: Two sides of the same coin. J. Proteome Res. 2008; 7:40–44. [DOI] [PubMed] [Google Scholar]
- 59. Lange P.F., Huesgen P.F., Nguyen K., Overall C.M.. Annotating N termini for the human proteome project: N termini and N alpha-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome. J. Proteome Res. 2014; 13:2028–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Varland S., Osberg C., Arnesen T.. N-terminal modifications of cellular proteins: the enzymes involved, their substrate specificities and biological effects. Proteomics. 2015; 15:2385–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ivanov I.P., Loughran G., Sachs M.S., Atkins J.F.. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1). Proc. Natl. Acad. Sci. USA. 2010; 107:18056–18060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loughran G., Sachs M.S., Atkins J.F., Ivanov I.P.. Stringency of start codon selection modulates autoregulation of translation initiation factor eIF5. Nucleic Acids Res. 2012; 40:2898–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wei J.J., Zhang Y., Ivanov I.P., Sachs M.S.. The stringency of start codon selection in the filamentous fungus neurospora crassa. J. Biol. Chem. 2013; 288:9549–9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Michel A.M., Andreev D.E., Baranov P.V.. Computational approach for calculating the probability of eukaryotic translation initiation from ribo-seq data that takes into account leaky scanning. BMC Bioinformatics. 2014; 15:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McEwen E., Kedersha N., Song B.B., Scheuner D., Gilks N., Han A.P., Chen J.J., Anderson P., Kaufman R.J.. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 2005; 280:16925–16933. [DOI] [PubMed] [Google Scholar]
- 66. Brar G.A., Yassour M., Friedman N., Regev A., Ingolia N.T., Weissman J.S.. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012; 335:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xiao Z., Huang R., Xing X., Chen Y., Deng H., Yang X.. De novo annotation and characterization of the translatome with ribosome profiling data. Nucleic Acids Res. 2018; 46:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eisenberg A.R., Higdon A.L., Hollerer I., Fields A.P., Jungreis I., Diamond P.D., Kellis M., Jovanovic M., Brar G.A.. Translation initiation site profiling reveals widespread synthesis of non-AUG-initiated protein isoforms in yeast. Cell Syst. 2020; 11:145–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang S., Hu H., Jiang T., Zhang L., Zeng J.. TITER: predicting translation initiation sites by deep learning. Bioinformatics. 2017; 33:i234–i242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Golovko A., Kojukhov A., Guan B.J., Morpurgo B., Merrick W.C., Mazumder B., Hatzoglou M., Komar A.A.. The eIF2A knockout mouse. Cell Cycle. 2016; 15:3115–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data have been deposited in GEO under the accession number GSE174329. The MS data have been deposited with the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the JPOST partner repository under the data set identifier PXD025794. TISCA pipelines and example data are available at https://github.com/KazuIchihara/TISCA.