


Abstract
HIV-1 integration favors recurrent integration gene (RIG) targets and genic proviruses can confer cell survival in vivo. However, the relationship between initial RIG integrants and how these evolve in patients over time are unknown. To address these shortcomings, we built phenomenological models of random integration in silico, which were used to identify 3718 RIGs as well as 2150 recurrent avoided genes from 1.7 million integration sites across 10 in vitro datasets. Despite RIGs comprising only 13% of human genes, they harbored 70% of genic HIV-1 integrations across in vitro and patient-derived datasets. Although previously reported to associate with super-enhancers, RIGs tracked more strongly with speckle-associated domains. While depletion of the integrase cofactor LEDGF/p75 significantly reduced recurrent HIV-1 integration in vitro, LEDGF/p75 primarily occupied non-speckle-associated regions of chromatin, suggesting a previously unappreciated dynamic aspect of LEDGF/p75 functionality in HIV-1 integration targeting. Finally, we identified only six genes from patient samples—BACH2, STAT5B, MKL1, MKL2, IL2RB and MDC1—that displayed enriched integration targeting frequencies and harbored proviruses that likely contributed to cell survival. Thus, despite the known preference of HIV-1 to target cancer-related genes for integration, we conclude that genic proviruses play a limited role to directly affect cell proliferation in vivo.
INTRODUCTION
Retroviral replication proceeds through an obligate integrated DNA or proviral intermediate. Integration can have far-reaching consequences on the host organism. Dysregulation of cellular protooncogene expression can significantly stimulate cell growth, leading to clonal expansion and tumor formation in animals infected with murine leukemia virus (MLV; see Supplementary Table S1 for a list of non-standard abbreviations used in this manuscript) or avian sarcoma-leukosis virus (1–3). Clonal expansion of cells infected with human T cell lymphotropic virus 1 similarly underlies adult T cell leukemia (4,5). Although most cells infected with human immunodeficiency virus 1 (HIV-1) die as a consequence of active virus production, a small fraction become latently infected and persist under antiretroviral therapy (ART) (6–11). It has been estimated that at least 40% of cells latently infected with HIV-1 undergo clonal expansion (12–15) and that clonal expansion initiates soon after HIV-1 infection (16). The persistence of the latent cell reservoir is the principle barrier to curing HIV-1 infection (6,7,17).
Retroviral integration is non-random, with different virus types favoring particular aspects of host chromatin. For example, gammaretroviruses such as MLV favor gene promoters and active enhancers while lentiviruses, which include HIV-1, favor interior regions of actively transcribed genes [reviewed in (18) and (19)]. Viral integrase and capsid protein interactions with host proteins primarily determines HIV-1 integration targeting preferences. HIV-1 integration favors transcriptionally active speckle-associated domains (SPADs) and disfavors heterochromatin such as lamina-associated domains (LADs) (20–23). Cleavage and polyadenylation specificity factor 6 (CPSF6), which functions in mRNA 3′ end cleavage and polyadenylation, is a direct binding partner of HIV-1 capsid (24,25). In the absence of this interaction, viral preintegration complexes (PICs) mislocalize from interior regions of cell nuclei to the nuclear periphery, with concomitant changes from SPAD-proximal to LAD-proximal integration (20,21,26,27). Genic integration also relies on the interaction of integrase with lens epithelium-derived growth factor (LEDGF)/p75 (28–30). In LEDGF/p75 knockout (LKO) cells, residual intragenic integration occurs toward gene 5′ end regions, indicating that LEDGF/p75 directs HIV-1 integration to the interior regions of gene bodies (27,31). LEDGF/p75 interacts with numerous splicing factors (31) and can facilitate transcription elongation in vitro (32), indicating potential roles for mRNA splicing and/or transcriptional elongation in LEDGF/p75-dependent integration targeting.
Characterization of recurrently targeted genes (also known as recurrent integration genes or RIGs) has also informed HIV-1 integration targeting preferences (20–22,33). RIGs were first defined via cross-sample occurrence as genes targeted for integration in at least two independent studies (22,33). A potential drawback of this approach was susceptibility to type I or false positive identification errors. Comparatively long genes, for example, might artificially score due to their size. Alternative approaches for RIG identification took random targeting frequencies into consideration, effectively minimizing type I errors (20,21). However, given the common approach of utilizing only limited subsets of available data (e.g. the top 100 RIGs compared across 3–4 samples), such approaches were susceptible to type II or omission of true positive errors.
Here, we devised a novel approach to comprehensively classify genes targeted for integration in a way that minimizes both type I and type II errors. To decrease the likelihood of type I errors, RIGs were distinguished from non-RIGs by comparing observed versus expected integration frequencies from effectively all possible matched random datasets simultaneously. By applying this approach to 10 in vitro integration datasets, we determined that gene targeting patterns are similar across cell types and uncovered repeatedly avoided genes, which we termed recurrent avoided genes (RAGs). The likelihood of type II errors was minimized by combining identified RIGs and RAGs from each dataset cumulatively. From this, we estimate we have identified 84% and 45% of all RIGs and RAGs, respectively, in the human genome. Comparisons with integration sites derived from patients prior to ART treatment and during ART suppression revealed that ∼70% of genic integration events in vitro and in vivo occurred in identified RIGs, despite the fact that RIGs account for only ∼13% of human genes. We also show that RIGs in vitro are highly likely to be recurrently targeted in vivo. These data highlight general similarities in genic targeting preferences during the initial phase of HIV-1 infection, modeled here in vitro, and HIV-1 infection in patients. To glean additional insight across these datasets, we mapped integration sites with respect to 10 compartmentalized regions of cell nuclei (34) to produce one of the highest resolution assessments of the genic landscape of HIV-1 integration to-date.
Our methodology additionally allowed us to comprehensively analyze patient-derived genes for characteristic features of selection bias based on integration site overrepresentation, orientation bias, and clustering to specific regions. Such information has been used previously to identify genes that, when integrated into, conferred cellular survival in patients on ART (12,13,35–37). Our analysis identified six genes—BACH2, STAT5B, MKL1, MKL2, IL2RB and MDC1—that met these criteria. These results highlight the fact that the vast majority of genic proviruses in ART-treated patients are unlikely to directly contribute to infected cell survival.
Finally, we applied our RIG-calling methodology to datasets derived from HIV-1-infected wildtype (WT), LKO, and CPSF6 knockout (CKO) HEK293T cells, as well as WT, LKO, and CKD (CPSF6 knockdown) Jurkat T cells. The results of these comparisons highlight a previously underappreciated role for LEDGF/p75 in recurrent HIV-1 integration targeting.
MATERIALS AND METHODS
Reagents
Antibodies for immunoblotting included rabbit anti-CPSF6 (Abcam, Cambridge, UK; catalogue number Ab175237), horse radish peroxidase (HRP)-conjugated anti-rabbit IgG (Agilent Technologies, Inc., Santa Clara, CA; catalogue number P0448) and HRP-conjugated anti-β-actin (Millipore Sigma, Burlington, MA; catalogue number A3854-200UL). Additional immunoblotting reagents included 20X Bolt MOPS SDS running buffer (Thermo Fisher Scientific, Waltham, MA; catalogue number B0001), Bolt 4–12% Bis–Tris Plus gels (Thermo Fisher Scientific catalogue number NW04122BOX), Bolt transfer buffer (20x) (Thermo Fisher Scientific catalogue number BT00061), Immun-Blot(R) PVDF membrane (Bio-Rad Laboratories, Hercules, CA; catalogue number 1620177), PageRuler pre-stained protein ladder (Thermo Fisher Scientific catalogue number PI26616) and complete EDTA-free protease inhibitor cocktail (Millipore Sigma catalogue number 1836170001).
Restriction endonucleases MseI (New England Biolabs, Ipswich, MA; catalogue number R0525L) and BglII (catalogue number R0144L) were used to fragment genomic DNA for integration site sequencing. Sequences of DNA oligonucleotides used for ligation-mediated (LM)-PCR, which were obtained from Integrated DNA Technologies (Coralville, IA), are detailed in Supplementary Table S2. T4 DNA ligase (New England Biolabs catalogue number M0202T) was used to ligate DNA linkers to sheared genomic DNA and Advantage 2 PCR polymerase mix (Takara Bio USA, Inc., Mountain View, CA; catalogue number 639202) was used for subsequent PCR amplification using dNTPs acquired from Qiagen Inc. (Germantown, MD; catalogue number 201901).
Small-interfering RNAs (siRNAs) targeting CPSF6 mRNA (GAAUUGAGUCCAAGUCUUA; catalogue number A-012334-13-0020) and non-targeting (NT) control (UGGUUUACAUGUCGACUAA; catalogue number D-001910–01-05 ) were purchased from Dharmacon, Inc. (Lafayette, CO).
Dulbecco's modified Eagle's medium (DMEM) and RPMI 1640 medium were obtained from Thermo Fisher Scientific (respective catalogue numbers 11965084 and 11875085). Fetal bovine serum (FBS) was also from Thermo Fisher Scientific (catalogue number 10437028). Penicillin-streptomycin was from Corning Life Sciences (Corning, NY; catalogue number 30–002-CI). PolyJet DNA reagent was from Thermo Fisher Scientific (catalogue number 504788) while Cell Line Nucleofector Kit V was from Lonza Group AG (Basal, CH; catalogue number VCA-1003).
HIV-1 concentration was determined via p24 antigen capture kit (Advance Bioscience Laboratories, Rockville, MD; catalogue number 5447). Viruses were treated with TURBO DNase (Thermo Fisher Scientific catalogue number AM2239) prior to infection and infected cells were lysed using Passive Lysis Buffer (Promega Corporation, Madison, WI; catalogue number E1941). d-Luciferin potassium salt was from BD Biosciences (San Jose CA; catalogue number 556878). Protein concentration in cell extracts was determined via Pierce BCA protein assay kit (Thermo Fisher Corporation catalogue number 23225).
Biological resources
HEK293T cells and Jurkat E6-1 T cells were acquired from American Type Culture Collection (respective ATCC catalogue numbers CRL-3216 and TIB-152). Plasmid DNAs pNLX.Luc.R-.ΔAvrII (38), pNLENG1-ES-IRES (39) and pCG-VSV-G (30) were used to make single-round HIV-1 reporter viruses.
Cells and viruses
HEK293T cells maintained in DMEM supplemented to contain 10% FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin were propagated in humidified incubators at 37°C in the presence of 5% CO2. Jurkat E6-1 T cells were cultured in RPMI 1640 medium under otherwise identical conditions.
For transfection with siRNA, 106 Jurkat T cells were resuspended with nucleofection buffer containing the required supplement from Cell Line Nucleofector Kit V. Cells were mixed with 50 nM siRNA and electroporated with nucleofector I according to the manufacturer's instructions. After electroporation, cells were plated in six-well plates containing 2 ml pre-warmed RPMI 1640 media and incubated for 3 days to allow recovery prior to immunoblotting and virus infection.
For immunoblotting, cell pellets were lysed in 1× lysis buffer (50 mM Tris–HCl, pH 8.0, 250 mM NaCl, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate supplemented with complete EDTA-free protease inhibitor cocktail). Total cell protein (5 μg) fractionated through 4–12% polyacrylamide Bis–Tris gels was transferred to polyvinylidene difluoride (PVDF) membranes. CPSF6 protein expression was detected with anti-CPSF6 antibody followed by HRP-conjugated anti-rabbit IgG antibody. Beta-actin was detected using HRP-conjugated anti-beta-actin antibody.
Single-round derivatives of HIV-1NL4-3 carrying firefly luciferase (HIV-Luc) or green fluorescent protein (HIV-GFP) were used to infect cells essentially as previously described (40). In brief, HEK293T cells plated in 10 cm dishes were co-transfected the following day with 7.5 μg pNLX.Luc.R-.ΔAvrII (HIV-Luc) (38) or 13.5 μg pNLENG1-ES-IRES (HIV-GFP) (39) along with 1.5 μg pCG-VSV-G (30) using PolyJet DNA reagent. Virus-containing cell supernatants were concentrated by ultracentrifugation and assessed for p24 content as described (41). Genomic DNAs for integration site libraries were isolated at 5 days post-infection (dpi) for HEK293T cells and 2 dpi for CPSF6 CKD Jurkat T cells and corresponding control siNT cells. Jurkat cell infectivities at 2 dpi quantified from respective luciferase activities were normalized to the total concentration of protein in cell extracts.
Integration site datasets
In silico datasets that mimicked experimental fragmentation methods were constructed using custom Python scripts essentially as previously described (41,42). To simulate random fragmentation (e.g. sonication), fragment lengths were defined by a theoretical normal distribution with a mean length of 400 bp and a standard deviation of 50 bp. For pattern fragmentation (e.g. restriction digestion), fragment length depended on the presence of the relevant enzyme recognition sequence(s) up to a defined maximum distance downstream of the theoretical random integration event. To ensure a representative sampling of the entire genome, the maximum distance for each enzyme cocktail was defined as 20,000 bp. To mimic the size selection protocols in standard next-generation sequencing pipelines, fragments were subsequently filtered to include only those less than or equal to 900 bp and greater than or equal to 20 bp. After filtering, fragments were aligned to human genome (hg19) using STAR (43). Final random integration site libraries for the simulated restriction enzyme digestions contained 13.6 million unique integration sites for MseI/BglII and 7.2 million unique integration sites for NheI/AvrII/SpeI/BamHI (Supplementary Table S2). The final random integration site library for simulated random fragmentation contained 9.1 million unique integration sites (Supplementary Table S2). The relevant Python scripts and example files for rigrag-compatible intersection with gene annotations are available on GitHub (https://github.com/gbedwell/).
Random integration datasets used for model generation were constructed by randomly sampling N number of sites from the relevant master file 10 times. Each final dataset consisted of 10 individual random integration files containing N sites each. N-values used for model generation were 10,000, 15,000, 20,000, 30,000, 40,000, 50,000, 60,000, 70,000, 80,000, 90,000, 100,000, 150,000, 200,000, 300,000, 400,000, 500,000, 600,000, 700,000, 800,000, 900,000 and 1,000,000. Genic integration sites for each file in each dataset were determined by intersecting each file with curated gene coordinates derived from the GENCODE v19 annotation set using BEDtools v2.27.1 (44,45). To construct the curated list of gene coordinates, the original GENCODE annotation file was filtered to contain all annotated protein coding, lincRNA, snoRNA, snRNA, rRNA, Ig variable chain, and TcR genes. This file contained 34,763 annotated coordinates corresponding to 33,579 unique genes. For genes annotated more than once on the same chromosome, the longest annotation was included in the final annotation file. Genes annotated more than once on separate chromosomes or genes containing more than one annotation of the same size were removed from the study. The final annotation file, which contained 33,429 uniquely annotated genes, is available on GitHub. Because of the expanded number and types of annotations included in this genome annotation file, calculated genic integration values for all samples (including random) were ∼6% greater than previously published (20,27,40,41,46).
Of the 10 integration site datasets derived from WT transformed and primary cells infected in vitro in this study, the following 8 were published previously: HEK293T #1 (20,27), HEK293T #3 (31), Jurkat #2 (40), Jurkat #3 (40), peripheral blood mononuclear cells (PBMCs) (47,48), monocyte-derived macrophages (MDM) (46), elite controller (EC) primary CD4+ T cell (49), and HIV negative primary CD4+ T cell (49) (see Supplementary Table S2 for numbers of integration sites as well as genomic DNA fragmentation and library construction methodologies). Matched HEK293T #1, HEK293T LKO, HEK293T CKO, and HEK293T double knockout (DKO) datasets were previously described (20,27), as were matched Jurkat #2, LKO Jurkat #1, and LKO Jurkat #2 datasets (40) (Supplementary Table S2). Remaining integration site datasets were generated herein from genomic DNA isolated from infected HEK293T cells (#2) and Jurkat T cells (NT #1, NT #2, CKD #1, and CKD #2) according to established ligation-mediated PCR protocols (41); these genomic DNAs were fragmented using MseI/BglII restriction endonucleases (Supplementary Table S2). DNA sequencing (150-bp paired end) was performed on the HiSeq Illumina platform at Genewiz.
The ART-treated patient-derived integration site dataset was built by concatenating unique integration sites from three published datasets (13,37,50). One of these datasets was itself a concatenation of several independent datasets (37). The untreated patient-derived integration site dataset was previously published in (37) (Supplementary Table S2). All patient-derived datasets were downloaded from the Retrovirus Integration Database (https://rid.ncifcrf.gov) (51) or obtained from the Microsoft Excel Workbook published in (37). Genic integration sites for all cell- and patient-derived integration site datasets were determined in the same manner as random genic integration sites. In order to accurately assess explicit integration site targeting, all integration sites used in this study were defined as the 5 bp region recognized and cleaved by the integrase enzyme.
Comparison with genomic features
Gene densities across the human genome were calculated using the Homo.sapiens annotation package available through Bioconductor (http://bioconductor.org). Gene expression levels in CD4+ T cells were obtained from the Schmiedel blood cell gene data downloaded from The Human Protein Atlas (https://www.proteinatlas.org/about/download) (52). The SPIN (Spatial Positioning Inference of the Nuclear genome) annotations described in reference (34) were downloaded from GitHub (https://github.com/ma-compbio/SPIN) and converted to hg19 coordinates using LiftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). The SPAD annotation set was constructed as previously described (21,40,53). To facilitate consistent comparisons between SPADs and other files, overlapping features in the SPAD annotation file were combined into a single feature using BEDtools. The final CD4+ T cell super-enhancer (SE) annotation file was built by combining and demultiplexing the annotations from the ‘CD4 Naïve Primary 7Pool’ and ‘CD4 Memory Primary 7Pool’ datasets from dbSUPER (54) (https://asntech.org/dbsuper/). LEDGF/p75 occupancies were calculated using a published LEDGF/p75 ChIP-seq dataset (32). For this dataset and all other ChIP-seq datasets, occupancy was defined as the number of bp occupied in a given region divided by the region size. Occupancy per gene was calculated similarly, with gene lengths used in place of region size. The absolute distance from integration sites to the nearest LEDGF/p75-occupied region in the same gene was calculated in BEDtools (45). For comparison of the overall distributions of LEDGF/p75 occupied regions across RIGs with the overall distribution of integration sites across RIGs, the relative midpoints of each LEDGF/p75-occupied region or integration site, respectively, was calculated. The locations of these midpoints were expressed as a value between 0 and 1, which corresponded to the transcriptional start and end sites of the analyzed RIGs, respectively. Validation of SPIN annotations derived from K562 cells (34) with respect to HIV-1 integration sites was largely performed on publicly available data downloaded from the ENCODE project (55,56) (https://www.encodeproject.org/), 4DN Web Portal (https://www.4dnucleome.org/), UCSC Table Browser (57) (https://genome.ucsc.edu/cgi-bin/hgTables), and dbSUPER (54). The datasets used for these comparisons are listed in Supplementary Table S1. Additional previously published genomic datasets included: HEK293T cell transcriptomics via RNA-seq analysis (27), K562 cell SPADs (53), and HT1080 cell LADs (58).
Computational resources
All data analysis and visualization pertaining to model generation and analysis was performed in R v3.6.3 and RStudio v1.4.869. The following packages and their corresponding dependencies were loaded and used during the analyses: dplyr v1.0.2 (https://dplyr.tidyverse.org/), tidyr v1.1.2 (https://tidyr.tidyverse.org/), ggplot v3.3.2 (https://ggplot2.tidyverse.org/), purrr v0.3.4 (https://purrr.tidyverse.org/), forcats v0.5.0 (https://forcats.tidyverse.org/), data.table v1.13. (https://github.com/Rdatatable/data.table/), ggsignif v0.6.0 (https://github.com/const-ae/ggsignif/), scales v1.1.1 (https://github.com/r-lib/scales/), ggrepel v0.8.2 (https://github.com/slowkow/ggrepel/), broom v0.7.2 (https://broom.tidymodels.org/), minpack.lm v1.2–1 (https://cran.r-project.org/web/packages/minpack.lm/index.html/), cowplot v1.1.0 (https://github.com/wilkelab/cowplot), nls.multstart v.1.2.0 (https://github.com/padpadpadpad/nls.multstart), and rigrag v0.0.0.9. All packages except rigrag are available on CRAN (https://cran.r-project.org); rigrag is available on GitHub (https://github.com/gbedwell/rigrag).
Quantification of duplicate western blots was done in Fiji (https://imagej.net/Fiji).
Statistical analyses
Model generation
For each dataset (10 files of N sites each), the mean integration frequency across all files, and the standard deviation of integration frequency across all files were calculated for each gene. The mean integration frequency and the standard deviation of integration frequency were then plotted as a function of gene length and fit to appropriate functions to obtain expected values for a given gene length. Linear fits were performed using the lm() function in the stats package (https://www.r-project.org/). Nonlinear fits were performed by nonlinear least squares fitting using the nls_multstart() function in the nls.multstart package (https://github.com/padpadpadpad/nls.multstart). As expected for random integration, mean integration frequency increased linearly with gene length. The spread of integration frequency values about the mean for each dataset was calculated by first expressing all integration frequency values as a distance from the corresponding expected mean value. Distance values were then expressed as a ratio of the expected standard deviation. The maximum ratio value for each gene was then multiplied by the corresponding expected standard deviation to summarize the magnitude of spread above the mean. These data were plotted against gene length and fit to a power law of the form  . The fitted curve in this instance represents an expected value but does not account for the maximum spread of possible values. In order to better describe the actual upper limits of the data, we calculated the extreme upper whisker of the residuals distribution, defined as
. The fitted curve in this instance represents an expected value but does not account for the maximum spread of possible values. In order to better describe the actual upper limits of the data, we calculated the extreme upper whisker of the residuals distribution, defined as  . This constant value can be added to the best fit spread above the mean value to generate the final upper boundary above the mean integration frequency. The lower boundary of the model was assumed to be symmetrical to the upper boundary. All individual dataset fits for each fragmentation strategy are shown in Supplementary Figure S1, where best-fit lines are shown in red and the spread upper whisker is shown in blue. Because there is a theoretical lower limit of 0 for integration frequency, for final model construction, negative values generated when calculating the lower spread were taken to be 0. In addition, for each dataset, the gene length distributions for genes harboring 0 integration events were analyzed and the extreme upper whiskers were calculated. This information was incorporated into the final models as the gene length below which the lower limit of integration frequency is 0. The spread upper whisker and zero point fits are shown in Supplementary Figure S2A and B.
. This constant value can be added to the best fit spread above the mean value to generate the final upper boundary above the mean integration frequency. The lower boundary of the model was assumed to be symmetrical to the upper boundary. All individual dataset fits for each fragmentation strategy are shown in Supplementary Figure S1, where best-fit lines are shown in red and the spread upper whisker is shown in blue. Because there is a theoretical lower limit of 0 for integration frequency, for final model construction, negative values generated when calculating the lower spread were taken to be 0. In addition, for each dataset, the gene length distributions for genes harboring 0 integration events were analyzed and the extreme upper whiskers were calculated. This information was incorporated into the final models as the gene length below which the lower limit of integration frequency is 0. The spread upper whisker and zero point fits are shown in Supplementary Figure S2A and B.
Once all of the relevant parameters were calculated for each dataset, parameter values were plotted against the total number of integration sites in each dataset and fit to appropriate functions. In all instances, parameters were found to be well described by simple mathematical functions. Power law fits were performed via nonlinear least squares fitting as described above. The utility of these fits is that, in principle, they allow parameter estimation for any sized integration site dataset. The intercept of the mean integration frequency, the N-parameter for spread estimation, the spread residual upper whisker, and the zero point for each dataset were well-described by a power law of the form  (Supplementary Figure S2A, B, D and E). The slope of the mean integration frequency and the a-parameter for spread estimation was fit by a piecewise combination of two power laws (Supplementary Figure S2C and F). The general form is,
(Supplementary Figure S2A, B, D and E). The slope of the mean integration frequency and the a-parameter for spread estimation was fit by a piecewise combination of two power laws (Supplementary Figure S2C and F). The general form is,
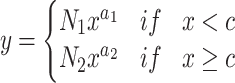 |
where 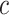 is a breakpoint that was optimized for each fit. All global fits are shown in Supplementary Figure S2. To generate the final models, the fitted equations for each global parameter were combined as appropriate to express all dataset parameters as a function of total sample size. These final equations for parameter estimation were combined as appropriate to generate a complete model of random integration. All of the generated models are incorporated into the R package rigrag. Example usage of rigrag, as well as the package itself, is available on GitHub.
is a breakpoint that was optimized for each fit. All global fits are shown in Supplementary Figure S2. To generate the final models, the fitted equations for each global parameter were combined as appropriate to express all dataset parameters as a function of total sample size. These final equations for parameter estimation were combined as appropriate to generate a complete model of random integration. All of the generated models are incorporated into the R package rigrag. Example usage of rigrag, as well as the package itself, is available on GitHub.
RIG, RAG and non-outlier gene determinations in cell-derived integration site datasets
For each cell-derived integration site dataset, a model of random integration was generated using the appropriate model in rigrag. The genic integration frequencies for each gene represented in a given dataset were then compared to the expected random integration frequency values. Genes integrated into at frequencies greater than the upper boundary of the corresponding random model and genes integrated into at frequencies less than the lower boundary were considered outliers and were called RIGs and RAGs, respectively. Genes integrated into at frequencies within the model boundaries were called non-outliers. Despite differences in cell types and fragmentation methods, values of integration sites/gene correlated reasonably well (r = 0.5–0.9) across all datasets (Supplementary Figure S3), indicating the general comparability of these datasets. Because of these general similarities, RIGs, RAGs, and non-outlier genes from the 10 in vitro datasets derived from infecting WT transformed and primary cell types were pooled to generate the final lists of RIGs, RAGs, and non-outlier genes. These lists are provided in Supplementary Table S3. We note that a small fraction of genes (134 of 21,428 analyzed, 0.6% of total) displayed variable behavior in that they scored as either a RIG or a RAG in different datasets. Owing to the high degree of variability in classification and to avoid categorizing these genes into both RIG and RAG outlier populations simultaneously, we explicitly defined these as non-outlier genes, and removed them from RIG and RAG gene lists in downstream analyses. These genes, along with the number of times they were identified as a RIG, RAG, or non-outlier across datasets, are listed in Supplementary Table S3. For CKO/CKD, LKO and DKO cell comparisons, RIGs, RAGs, and non-outlier genes were determined in a similar manner. The relative gene category distributions between average in vitro, untreated, and ART-suppressed patient datasets were compared using a T-test. Because mean and variance estimates were available for the average in vitro data but not the untreated or ART patient-treated datasets, equal variance was assumed.
Estimation of genome-wide RIGs and RAGs
To estimate the total number of RIGs and RAGs in the human genome, we analyzed the cumulative number of identified RIGs and RAGs versus the cumulative number of genic integration sites. These plots were fit to rectangular hyperbolae of the form,
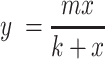 |
where m represents the maximum number of RIGs or RAGs and k is the number of genic sites half-way to maximum.
Comparison with genomic features
The degree of overlap between integration sites and relevant genomic regions, as well as the distance from genic integration sites to the closest relevant genomic region, were determined using BEDtools v2.27.1 (45). Principal component analysis of SPIN annotation overlap frequencies was performed using the prcomp() function in R (https://www.r-project.org/). Significance was determined between gene density distributions, gene expression level distributions, and LEDGF/p75 occupancy distributions using a Wilcoxon rank sum test. Statistical tests on the distance distributions from SPADs and SEs were done by quantifying the number of integration sites in each sample within ± 5 Mb or ± 3 Mb, respectively. Samples were compared to random using the chi-squared test of independence. Calculated P-values were corrected using the Benjamini-Hochberg procedure and a false discovery rate (FDR) of 0.05. Significance levels denoted in figures are as follows: * denotes 5 × 10−2 > P ≥ 10−2; ** denotes 10−2 > P ≥ 10−3; *** denotes P < 10−3, ns denotes P ≥ 5 × 10−2.
Gene ontology (GO) analysis
GO analysis was performed using Panther DB (http://www.pantherdb.org/) (59). Unique gene lists for RIGs, RAGs, non-outliers, and patient-derived integration sites were compared to H. sapiens reference genome for statistical overrepresentation using the Panther GO-Slim Biological Process annotation set. P-values were calculated using Fisher's exact test and corrected for multiple comparisons using an FDR of 0.05. Complete lists of all statistically significant overrepresented and underrepresented GO categories for each gene list are reported in Supplementary Table S4.
Identification of overrepresented genes in patient-derived samples
For comparison of integration targeting frequencies in patient-derived genes to genes derived from in vitro infection, the in vitro integration frequencies were averaged across all 10 datasets. For this calculation, genes present in vitro were stratified into RIG and non-RIG populations, with the non-RIG population comprised of both RAG and non-outlier genes. In vitro samples in which a gene was absent were considered to have an integration frequency value of 0. To determine overrepresentation in patient-derived samples, the expected number of integration events into a given gene was calculated given the in vitro mean integration frequency and the patient-derived dataset sample size. The expected number of integration events was then compared to the observed number of integration events and statistical significance was assessed using Fisher's exact test. Calculated P-values were corrected using the Benjamini-Hochberg procedure and an FDR of 0.05.
Orientation bias of genic integration sites was determined by first counting the number of integration events in the same versus opposite transcriptional orientation as the gene. Statistical significance of the differences between the two numbers for each gene was determined using a binomial test. The clustering of integration sites within overrepresented genes was assessed by first calculating the relative positions of each integration site within a given gene with respect to gene length. The position of each integration site was expressed as a value between 0 and 1, which corresponded to transcription start and end sites, respectively. To compare the patient-derived position distributions to in vitro position distributions, a bootstrapping routine was used. For each overrepresented patient gene, the in vitro position distribution was randomly sampled to extract the same number of integration sites as present in the patient-derived data. This sampling routine was repeated 100 times. For each gene, the shapes of the position distributions in each random sample of the in vitro data were then compared to the shape of the patient-derived distribution using a Kolmogorov-Smirnov test. The mean P-value from the 100 individual comparisons for each gene is reported in Supplemental Table S5.
RESULTS
Model generation
An unbiased model of random integration must accurately describe the mean integration frequency per gene and the range of possible integration frequency values per gene for a given dataset. For general applicability, the model should also account for how these values change across integration site datasets of various sizes. Here, we derived phenomenological models of random integration based on distinct master collections of >7 million unique computational integration sites generated by simulating common genome fragmentation strategies. Our models describe both random fragmentation (e.g. sonication) (60) and pattern-based fragmentation according to specific restriction endonuclease combinations including MseI/BglII and NheI/AvrII/SpeI/BamHI (40,41,61) (Supplementary Table S2). Model construction is described at-length in Materials and Methods. To assess the ability of our models to accurately describe random integration, each master file was iteratively resampled to generate new random datasets containing 15 individual integration site files each. The number of outliers observed above and below upper and lower model boundaries, respectively, were quantified for each file in each dataset. False positive rate (FPR) and outlier number distributions are presented in Supplementary Figure S4 as box plots overlayed with the associated data points. For dataset sizes of ≤∼100,000 sites, the FPRs of upper outliers were greater than corresponding lower outliers, which is likely a combined effect of 1) the lower limit of the lower boundary being strictly defined as 0, while there is no strict upper limit, and 2) a larger fraction of genes harboring zero integration events in smaller as compared to larger datasets (see Supplementary Figure S2B). Even with this disparity, the median FPR for all datasets was <1.6 × 10−3, demonstrating that our approach works well to minimize the introduction of type I errors over a wide range of sample sizes (Supplementary Figure S4).
Application to experimentally-derived data
Analysis of integration sites from cells infected with HIV-1 in vitro has indicated that targeting preferences are largely conserved between transformed and primary cell types (12), though this has not been formally quantified. Accordingly, we used our models to categorize genic integration sites from ten in vitro datasets that were generated by infecting commonly used transformed and primary cell types including HEK293T cells, Jurkat T cells, PBMCs, CD4+ T cells and MDM. These datasets harbored between 18,806 and 960,641 unique intragenic and intergenic integration sites, for a total of 1,736,842 sites across datasets (see Supplementary Table S2 for a study-wide summary of integration site datasets).
Our models comprehensively identified genic integration targeting frequencies that were greater than random (Figure 1A-J, blue data points), less than random (red data points), and indistinguishable from random (non-outlier genes; grey data points). Qualitatively, the overall pattern of genic integration targeting was strikingly similar across all 10 samples, with smaller genes (≤0.5 Mb) comprising the majority of RIGs across samples (blue data points). Recurrent avoided genes (RAGs; red data points) of various sizes were additionally observed across all samples. Overall, the majority of RIGs and RAGs (≥ 80% of total) were identified as such in two or more of the samples (Figure 1K, L). Genes identified as a RIG or RAG in 6 or more samples represented 39% and 19% of each respective gene population (Figure 1K, L).
Figure 1.

Identification of genic subpopulations from in vitro integration site datasets. (A–J) Genic integration frequencies vs. gene length for the indicated samples; total sites include intergenic and intragenic integrations. Upper outliers (RIGs) are depicted in blue, lower outliers (RAGs) are depicted in red, and non-outliers are depicted in gray; data are superimposed onto corresponding random models that are depicted as light gray shaded regions. See Supplementary Table S2 for previously published as well as de novo integration datasets used in this study. (K) Quantification of RIG and (L) RAG cross-sample occurrence (CSO). The boxes in each bar represent the fraction of identified RIGs or RAGs in that sample that were similarly identified as RIGs and RAGs in N other samples. (M) Genic integration targeting frequencies. Bar height represents the total fraction of genic integration, with colored subsections representing relative RIG (light blue), RAG (medium blue), and non-outlier (dark blue) proportions.
As expected (62), genes were predominantly targeted for integration across all datasets (Figure 1M). In some samples (293T #3, Jurkat #3, PBMC and EC CD4+ T), RIGs were the predominant genic fraction and across all samples, RIG fractions significantly outnumbered corresponding RAG fractions. Examination of total cross-sample occurrence (i.e. the total number of samples containing a given gene independent of RIG/RAG classification) further highlighted the targeting bias towards RIGs (Figure 2A). Nearly half of all RIGs (45%) were present in all 10 samples, with 92% present in six or more samples. Cross-sample occurrence of non-outlier genes, by contrast, was less than random. From this we conclude that genic integration targeting preferences during HIV-1 infection in vitro are well represented across transformed and primary cell types. In total, we identified 3718 RIGs, 2150 RAGs and 15,560 non-outlier genes; see Supplementary Table S3 for respective gene lists.
Figure 2.

Genic subpopulations in the human genome. (A) Total cross-sample occurrence (CSO) of RIGs, RAGs, and non-outlier (NO) genes. (B) Plots of cumulative RIGs (blue datapoints) and RAGs (red points) versus cumulative genic integration sites. Solid lines depict the fit of each dataset to a rectangular hyperbola; dotted lines depict respective best-fit m parameters, interpreted as theoretical maximum numbers of RIGs or RAGs in the human genome. (C) Estimated fractions of RIGs (13%), RAGs (14%) and NO genes (73%) in the human genome based on the fits shown in (B). (D) Fractions of genic integration events in RIGs, RAGs, and NO genes in averaged in vitro, untreated patient-derived, and ART-treated datasets. The degree of integration into each gene category is the same across all three samples. (E, F) The number of integration events into RIG-matched or unmatched genes in (E) untreated patient-derived or (F) ART-treated integration datasets. Unmatched histograms (light blue) abut RIG-matched histograms (dark blue).
To estimate the total numbers of RIGs and RAGs in the human genome, cumulative gene numbers in each subpopulation were plotted against cumulative number of genic integration sites. The shapes of these curves leveled off as a function of integration site number, indicating that the number of identified RIGs and RAGs were approaching maximum values (Figure 2B). Fitting these data to rectangular hyperbolae, we estimated maximum RIG and RAG values of 4421 and 4815, respectively (dotted lines in Figure 2B). Based on these fitted values, our analyses identified approximately 84% of all RIGs and 45% of all RAGs. RIGs and RAGs comprise just 13% and 14% of the annotated human genes included in this study, respectively, with non-outliers comprising the remaining 73% (Figure 2C).
Having established similar genic integration targeting and avoidance profiles across in vitro integration datasets, we next investigated the relative contribution of integration into RIGs, RAGs, and non-outliers in samples derived from human patients. Patient data were stratified on the basis of ART treatment. Data derived from patients prior to the start of ART treatment encompassed 13,311 integration sites (11,456 genic sites) (37). Data from ART-treated patients contained 33,451 integration sites (28,085 genic sites) (13,37,50). See Supplementary Table S2 for a summary of integration site datasets and Supplementary Table S3 for gene lists.
To facilitate comparisons between in vitro and patient datasets, the fraction of genic integration sites in RIGs, RAGs, and non-outliers from the 10 in vitro datasets were averaged. Averaged in vitro, untreated patient, and ART-suppressed patient datasets revealed statistically indistinguishable targeting distributions with respect to RIGs, RAGs, and non-outlier genes (Figure 2D). Quantifying numbers of integration events into RIG-matched versus unmatched genes revealed that RIGs were also the predominant targets of recurrent integration in vivo (Figure 2E, F).
To ascertain the types of genes present in the different populations, we performed gene ontology (GO) analysis on RIG, RAG, non-outlier, untreated patient, and ART-suppressed patient genes. In general, RIGs and patient-derived genes were enriched for similar gene types, including genes involved in mRNA synthesis and processing, cell division, and protein modification (Supplementary Table S4). In contrast, RAGs were generally enriched for genes involved in neuronal processes (Supplementary Table S4).
The nuclear landscape of genic integration in vitro and in vivo
Previous studies have shown that HIV-1 integration is biased towards highly expressed genes in gene-dense chromatin regions and biased against heterochromatin (23,62,63). Consistent with these results, we found RIGs and patient-derived genes enriched relative to random in both highly transcribed genes and in gene-dense regions (Figure 3A, B). RAGs showed the opposite phenotype (Figure 3A, B). Non-outlier genes were not significantly different from random with respect to gene expression but were slightly biased towards gene-dense regions (Figure 3A, B).
Figure 3.

Comparison of genic integration sites with genomic features. (A) Gene expression distributions for in vitro RIGs, RAGs, and non-outlier (NO) genes, as well as untreated and ART-treated patient-derived genes. (B) Gene density distributions for the same samples used in (A). Significance values in panels A and B were determined relative to random. (C) Overlap of integration sites from RIG, RAG, NO gene, pooled in vitro, untreated patient, ART-treated patient and random datasets with respect to 10 SPIN genomic regions (34). The degree of overlap with each genomic region is reported as the fraction of total integration in each dataset. Significance signifiers are reported in either light or dark gray to denote significantly less than random or significantly more than random, respectively. (D) Principal component analysis of the data presented in (C). The first two principal components are shown on the X- and Y-axes with the percentage total variance explained by these principal components indicated in the axis titles. Loadings are shown as gray arrows and labeled with their respective feature. In panels A–C, * denotes 5 × 10−2 > P ≥ 10−2; ** denotes 10−2 > P ≥ 10−3; *** denotes P < 10−3; ns denotes non-significant.
Prior studies have shown that SEs tracked significantly with RIGs, although not with bulk sites of HIV-1 integration (21,33). By analyzing the relatively small number of 46 RIGs, we previously concluded that RIGs more closely tracked with SPADs than with SEs (21). To more comprehensively assess the roles of SEs and SPADs in HIV-1 integration targeting, we next analyzed the expanded gene sets determined above. As shown in Supplementary Figure S5A and B, we found that RIGs, non-outlier genes, as well as genes from untreated and ART-suppressed patients significantly tracked with SEs and SPADs, while the association of RAGs with these genomic markers was less than random. Because SPADs are enriched for SEs (53), we hypothesized that these apparent similarities could be due to the convolution of genomic features and that one might be the dominant marker with respect to genic integration targeting. To test this hypothesis, we took the SE annotations from CD4+ T cells and removed all of the annotated regions that overlapped with SPADs. We then repeated the calculations described above with respect to the nearest SPAD-free SE region, which revealed that the previously observed enrichments of RIGs and patient-derived genes for SEs were lost (Supplementary Figure S5A, C). In contrast, the reverse analysis—the distance from each integration site to the nearest SE-free SPAD region—showed the same trends as the original SPAD analysis (Supplementary Figure S5B, D). These data suggest that of the two annotated features, SPADs are the dominant predictive marker for genic HIV-1 integration and that the observed correlation with SEs is likely the result of SE proximity to SPADs.
Along with SPADs, prior studies have additionally highlighted LADs as highly predictive genomic markers of bulk HIV-1 integration site selection (20,21,40). Combined with results of viral imaging (20,21,26), these findings informed the spatial pattern of HIV-1 integration targeting. However, SPADs and LADs provide but two genomic markers for nuclear interior versus peripheral regions, respectively. The spatial positioning inference of the nuclear genome (SPIN) study recently improved spatial resolution of nuclear compartmentalization by incorporating genomic markers for 10 regions, which span from Lamina and Lamina-like at the nuclear periphery to Interior Active regions and Speckles in the nuclear interior (34). To assess the relevance of SPIN markers for HIV-1 integration targeting studies, we first mapped known positive and negative correlates such as gene-density and centromeric repeats, respectively (Supplementary Figure S6) (20–23,27,31,33,40,62–65). As expected, gene density, gene expression, RNA polymerase II occupancy, DNase hypersensitivity, CpG islands, SPADs, and SEs generally correlated with Speckles and Interior Active regions 1–3, while LADs and centromeric repeats mapped to peripheral regions Near Lamina 1, Near Lamina 2, Lamina-like, and Lamina (Supplementary Figure S6A–G). Given these results, we next employed SPIN annotations to define the genomic landscape of genic integration targeting. Integration into RIGs was strongly biased towards interior regions (Speckles and Interior Active region 1) while integration into RAGs was most biased towards the peripheral Near Lamina and Lamina regions (Figure 3C). Integration in non-outlier genes also showed bias towards interior regions, most strongly towards Interior Active region 3, and showed stronger bias than RIGs towards Near Lamina regions 1 and 2. Untreated and ART-suppressed genic integration sites from patients, like RIGs and non-outlier sites, were enriched in Speckle and Interior Active genomic regions (Figure 3C).
To facilitate interpretation, we performed a principal component analysis, the results of which are presented as a single 2D plot. The first two components accounted for 97.4% of the total variance in the SPIN mapped data (Figure 3D). Unsurprisingly, the first principal component was defined primarily by integration frequencies into chromatin near Speckles and near Lamina, respectively, while the second principal component was defined primarily by integration frequencies in Interior Active regions 2–3 and Interior Repressive region 1 (Figure 3D). Notably, the patient-derived data segregated very similarly to the in vitro pooled dataset, further highlighting the similarities in integration targeting in vitro and in vivo (Figure 3D).
Identification of overrepresented genes in patient samples
Previous studies have analyzed integration sites that arose in vivo before the initiation of ART (16,37) and then during ART treatment (12–14,16,37,48,66). Critically, however, the precise interplay among genic integration sites that arise during the initial wave of HIV-1 infection, which was modeled herein using in vitro datasets, with these patient-derived populations is not clearly understood. Several prior studies identified integrations in BACH2, MKL2 and STAT5B that facilitated cell survival in vivo (12–14,35,36). A more recent study additionally highlighted proviral insertions in MKL1, IL2RB, MYB and POU2F1 that likely contributed to cell survival in vivo (37).
Genic proviruses that may confer cell survival in vivo have been defined using the following metrics: (i) overrepresentation of integration frequency relative to in vitro datasets, (ii) a strong bias towards integration in the same transcriptional orientation as the gene and (iii) integration site clustering at particular locations within the gene (12,35,37). Here, we devised a robust method to systematically compare patient-derived and in vitro data with respect to these features. Overrepresentation was determined by comparing the observed number of integration events per gene in patient-derived data to the expected number of integration events in an equally sized dataset based on in vitro data. Gene names, mean integration frequency values, and standard deviations calculated from the in vitro data are provided in Supplementary Table S5.
BACH2, one of the three genes initially reported to confer growth advantage in vivo, was the only gene from untreated patient samples that scored as overrepresented relative to in vitro data (Figure 4A). Importantly, MKL2 and STAT5 additionally scored in ART-treated samples as significantly overrepresented (Figure 4A, Supplemental Table S5). Seven other genes from the ART-treated dataset were additionally significantly overrepresented relative to in vitro data (Figure 4A, Supplemental Table S5). Of these, CEACAM21, KANSL1, PACS1 and STAT3 failed the tests for orientation bias and integration site clustering, suggesting that integrations into these four genes are unlikely to confer cell survival in patients (Figure 4B-C, Supplemental Table S5). In contrast, IL2RB, MKL1 and MDC1 genic proviruses all showed strong orientation bias in patient samples (Figure 4B, Supplemental Table S5). Integration sites in MKL1 and MDC1 were additionally significantly clustered relative to in vitro distributions (Figure 4C, Supplemental Table S5). While the distribution of integration sites in IL2RB in patients did not score as significantly clustered (P = 0.065), this trend was nevertheless evident via visual inspection (Figure 4C). IL2RB harbored comparatively low numbers of in vitro and patient-derived integration sites, indicating that deeper integration site datasets might potentially unveil significant clustering of IL2RB resident proviruses in patient samples as well.
Figure 4.

Identification of genes that can confer cell survival in patients. (A) Plot depicting genes that were found to harbor more integration events than expected based on in vitro integration frequencies. The plot shows –log10(adjusted P-value) for each gene in the ART-treated and untreated patient-derived datasets. The dashed line denotes P = 0.05. RIGs and non-RIGs are colored in dark and light blue, respectively. Overrepresented genes are indicated. (B) Plot depicting the fraction of integration sites in the same transcriptional orientation versus opposite orientation as the indicated gene. (C) Plots showing relative position of every integration site in the indicated gene for both in vitro and patient-derived datasets. Positions are reported relative to gene length, with 0 corresponding to the 5′ end of the gene and 1 corresponding to the 3′ end. For plots with significance indicators, * denotes 5 × 10−2 > P ≥ 10−2; ** denotes 10−2 > P ≥ 10−3; *** denotes P < 10−3; ns denotes non-significant.
Roles of HIV-1 integration targeting cofactors on the genic integration landscape
One of the principal benefits of our methodology is that RIGs and RAGs can be readily identified from a limited number of laboratory-based or clinical integration site databases. Previously, we characterized the top 100 RIGs that arose from infecting HEK293T cells as well as a set of isogenic LEDGF/p75 (LKO), CPSF6 (CKO), and double LEDGF/p75 + CPSF6 (DKO) knockout cells in vitro (20). Here, we used our methodology to expand the depth of RIG calling under these infection conditions and to identify associated RAGs. We moreover included a second cell type, Jurkat T cells, as well as two associated LKO derivatives (40). Unlike our experience with HEK293T cells, we failed to identify Jurkat CKO cell clones despite extensive effort. We therefore transiently knocked down CPSF6 in Jurkat T cells using siRNA. Western blot analyses confirming the CPSF6 knockdown (CKD) phenotype and associated levels of HIV-1 infection are provided in Supplementary Figure S7.
Plots of genic integration frequency versus gene length for WT HEK293T and Jurkat cells alongside factor-depleted cells are shown in Figures 5A–K. Loss of LEDGF/p75 yielded ∼85–90% reductions in recurrent integration (compare Figures 5A to B and I to J and K; Figure 5L), while the extent of recurrent integration in CKO/CKD samples was more similar to WT than the LKO samples (compare Figures 5A–C, E–F and G–H; Figure 5L; Supplementary Table S3). Consistent with our prior report (20), a fraction of HEK293T CKO RIGs were noticeably larger than RIGs observed in WT cells. This trend was also evident in the Jurkat CKD samples, though to a lesser extent. The degree of recurrent integration in DKO HEK293T cells was decreased relative to both WT and CKO cells (compare Figure 5A and C to D; Figure 5L; Supplementary Table S3). Altogether, these data indicate that LEDGF/p75 is a key driver of recurrent integration.
Figure 5.

A dominant role for LEDGF/p75 in recurrent HIV-1 integration targeting. (A–K) Genic integration frequencies vs. gene length for the indicated samples. Upper outliers (RIGs) are depicted in blue, lower outliers (RAGs) are depicted in red, non-outliers (NOs) are depicted in gray. The data are superimposed onto corresponding random models that are depicted as light gray shaded regions. See Supplementary Table S2 for previously published as well as de novo integration datasets used in this study. (L) Sample genic integration targeting frequencies. The bar height represents the total fraction of genic integration, with subsections representing relative proportions of RIGs (light blue), RAGs (medium blue), and NOs (dark blue). Random genic integration targeting is ∼51% (Supplementary Table S2). (M) Relative overlaps of LKO, CKO/CKD, and DKO RIGs with WT RIGs. The RIG-matched fraction of each dataset is depicted in dark blue and the unmatched fraction is depicted in light blue. (N) Cross-sample occurrence (CSO) of CKO/CKD RIGs and LKO RIGs. The percentage of CKO/CKD RIGs with CSO values of 1, 2 and 3 are 43%, 34% and 23%, respectively. In contrast, the percentage of LKO RIGs with CSO values of 1, 2, and 3 are 73%, 26% and 1%, respectively. (O) The relative overlap of LKO, CKO/CKD, and DKO RAGs with WT RAGs. The RAG-matched fraction of each dataset is depicted in dark blue and the unmatched fraction is depicted in light blue. All LKO and CKO/CKD RAGs and 99% of DKO RAGs overlapped with WT RAGs.
To assess host factor roles independent of cell type, the data was combined into master CKO/CKD and LKO sample files; previously-called RIGs, RAGs, and non-outlier genes (provided in Supplementary Table S3) were considered as representative WT cell data. Despite the low overall number of RIGs in LKO cells, the RIGs that were identified extensively overlapped with WT RIGs (83%, Figure 5M). CKO/CKD and DKO cell RIGs overlapped comparatively less well with WT RIGs (52% and 34%, respectively; Figure 5M). CKO/CKD RIGs showed much greater cross-sample occurrence than LKO RIGs, with 55% of CKO/CKD RIGs appearing in two or more of the three samples and 21% in all three (Figure 5N). In contrast, only 29% of LKO RIGs appeared in two or more samples and just 1% appeared in all three (Figure 5M). In stark contrast to RIGs, we found substantial overlap with WT RAGs across all three LKO, CKO/CKD and DKO datasets (Figure 5O).
Previous studies have shown that bulk HIV-1 integration site patterns in DKO cells represent a hybrid between CKO and LKO cell phenotypes (20,27,40). Integration in DKO cells is biased towards transcription start sites, similar to LKO cells, but is additionally biased towards LADs and gene sparse regions, as in CKO/CKD cells (20,27,40). Moreover, the fraction of genic integration in DKO cells is near-random and lower than in either CKO or LKO HEK293T cells (Figure 5L) (20,27,40). As shown in Figures 5D and L, however, there is still a non-negligible degree of genic targeting bias in DKO cells. To better understand the differences in targeting biases between WT, LKO, CKO/CKD, and DKO cells, we next analyzed the nuclear compartmentalization of integration site selection and its relationship to LEDGF/p75 chromatin occupancy. Although the total number of LKO cell RIGs and RAGs was comparatively small compared to other cell types, LKO cell nuclear compartmentalization trends largely mirrored those of WT samples, especially for respective RIG and non-outlier genic integration sites (Figure 6A; see Figure 6B for associated principal component analysis). While CKO/CKD RIGs largely mapped to Lamina-proximal regions, CKO/CKD non-outlier sites, like WT and LKO sample non-outliers, favored Interior Active regions (Figure 6A, B). Overall, integration events in non-outlier genes from all infection conditions were highly enriched in the Interior Active 3 genomic region (Figure 6A, B). DKO cell RIGs were enriched in Near Lamina 2 and Lamina genomic regions (Figure 6A). RAGs from all cell types showed strong bias towards Lamina regions (Figure 6A).
Figure 6.

Sites of LEDGF/p75 occupancy negatively correlate with RIG nuclear compartmentalization. (A) Overlap of integration sites in each of the indicated datasets with SPIN genomic regions (34). The degree of overlap with each genomic region is reported as the fraction of total integration in each dataset. Significance values were determined relative to random. Significance signifiers are reported in either light or dark gray to denote significantly less than random or significantly more than random, respectively. (B) Principal component analysis of the data presented in (A). The first two principal components are shown on the X- and Y-axes with the percentage total variance explained by these principal components indicated in the axis titles. Loadings are shown as gray arrows and labeled with their respective feature. (C) LEDGF/p75 occupancy across SPIN nuclear compartments. (D) The number of annotated LEDGF/p75 regions per RIG. Only RIGs with at least 1 annotated LEDGF/p75 region were included. (E) Absolute distance (in bp) from each integration site in a given RIG to the closest LEDGF/p75-occupied region in the same gene.
Analysis of integration frequencies per gene across cell types indicated that LEDGF/p75 was a principal driver of recurrent integration. To better understand the role of chromatin-bound LEDGF/p75 in integration targeting and recurrent integration, we quantified the occupancy of LEDGF/p75 in each of the 10 SPIN compartments. To our surprise, we found that LEDGF/p75 occupancy in Speckle regions – the nuclear compartment most preferred for integration targeting in vitro and in vivo – was the lowest across all nuclear compartments (Figure 6C). LEDGF/p75 occupancy was highest in Interior Active 3 and Near Lamina 2 regions (Figure 6C, Supplemental Figure S8A). Normalizing LEDGF/p75 occupancies on per gene bases across nuclear compartments revealed a largely compartmental-independent distribution, including Speckle regions (Supplemental Figure S8B, C). To further investigate the relationship between LEDGF/p75 chromatin binding and integration site selection, we compared integration site locations in RIGs to LEDGF/p75-occupied regions in RIGs. Overall, just 1,339 RIGs (36% of all RIGs) contained annotated LEDGF/p75-occupied regions. This RIG subset was found to contain a median value of two LEDGF/p75-occupied regions per gene (Figure 6D). The median distance from a given integration site to the nearest LEDGF/p75-occupied region in the same gene was ∼10 kb (Figure 6E). This finding is consistent with the observation that the overall integration site distribution across genes does not strongly correlate with the overall distribution of LEDGF/p75-occupied regions (Supplemental Figure S8D, E). It should be noted, however, that 3.1% of integration sites in RIGs did overlap with LEDGF/p75-occupied regions of chromatin (Figure 6E).
DISCUSSION
Gene targeting biases in vitro and in vivo
The introduction of the concept of RIGs into the field of HIV-1 integration site mapping has provided a framework for the explicit characterization of repeatedly targeted genes. This is of obvious utility because HIV-1 prefers genes overall for integration and a fraction of initial genic proviruses can persist and clonally expand in patients on ART [reviewed in (67) and (68), (8–12,14,16,66,69)]. In this report, we greatly expand on this idea and explicitly categorized individual genes according to the extent to which they were targeted for integration. We provide a nearly comprehensive list of recurrently targeted genes, as well as approximately half of all genes recurrently avoided for integration. We additionally provide a list of genes consistently targeted for integration at levels indistinguishable from random. Moreover, we demonstrated that the overwhelming majority of genic integration events from both in vitro and in vivo infections take place in the genes that we identify as being recurrently targeted. These results clearly support the idea that not only is HIV-1 integration biased towards genes, but that a specific subset of genes is significantly more likely to be targeted for integration than others. Importantly, we also established operational links between integration targeting biases stemming from initial infection and biases that are present at more advanced phases of infection. In this regard, BACH2 was the only gene from patients prior to ART treatment that harbored more integrations than expected based on in vitro targeting frequency (Figure 4A). As expected (12,13,35,37), BACH2 integrations were further enriched in ART-treated patient samples.
In all, we identified just 6 genes as likely drivers of infected cell survival in vivo. Five of these six genes—STAT5B, BACH2, MKL1, MKL2 and IL2RB—were similarly identified by Coffin et al. (37). That study also reported MYB and POU2F1 as genes linked to cell survival (37). According to our method, MYB was not overrepresented after P-value adjustment for multiple comparisons, and patient integration sites did not significantly cluster relative to the distribution of genic proviruses in vitro (Supplemental Table S5). Integrations in MYB in patients did, however, display pronounced orientation bias (Supplemental Table S5). Potentially, the comparatively small number of patient-derived integration events in MYB contributed to our findings, indicating that deeper integration site datasets could reveal MYB as a significant hit. By contrast, POU2F1 in our hands failed all three tests, including overrepresentation before P-value adjustment (Supplemental Table S5). MDC1, which was uniquely called by our methodology, plays a key role in DNA damage response and can negatively regulate apoptosis (70,71). In this way, MDC1 is similar to STAT5B, whose constitutive expression in peripheral T cell lymphoma has been linked to anti-apoptotic phenotypes (72).
HIV-1 preferentially targets cancer-related genes for integration in vitro (31) and cells harboring proviruses in cancer-related genes can persist and clonally expand in ART-suppressed patients (13). Proviral-mediated deregulation of cellular proto-oncogene expression, which is known to occur in animal cancer models (1–3), has accordingly been proposed to play an important role in homeostasis and expansion of HIV-1-infected reservoir cells (13,73). However, based on our data, it follows that the vast majority of genic proviruses in patients do not directly drive cell survival. Consistent with this interpretation, two recent studies found that clonal expansion of infected cells in vivo is predominantly antigen-driven, rather than driven by genic proviruses (74,75). How many more genes may meet the criteria for integration-linked cell survival is unclear. However, considering our results alongside the recent results from Coffin and colleagues (37), it seems unlikely that the ultimate number will rise significantly above the six or seven genes identified in these studies. Deeper in vitro and patient-derived integration datasets are required to definitively address this issue.
RIG, RAG, and non-outlier genes were strikingly similar between in vitro, untreated, and ART-treated integration site datasets (Figure 2D–F). By applying recently released SPIN annotations, which stratify chromatin into 10 compartmentalized regions, we determined that integration sites in RIGs were predominantly in speckle and speckle-adjacent chromatin regions (Figure 3C, D). Other actively transcribed chromatin regions showed substantially less enrichment for integration sites in RIGs relative to non-outlier genes (compare interior active 2–3 regions to speckle and interior active 1 regions in Figure 3C). We additionally showed that integration site localization in patients very closely resembled the trends observed in vitro (Figure 3C, D). A remaining point of interest is how nuclear compartmentalization might relate to and possibly influence proviral gene expression.
Host factors in integration site targeting
Our analyses of HEK293T and Jurkat T cells depleted for LEDGF/p75 or CPSF6 indicated a dominant role for LEDGF/p75 in recurrent integration. LEDGF/p75 knockout HEK293T cells showed a marked decrease in recurrent integration relative to WT cells. Similarly, HEK293T cells doubly knocked out for both LEDGF/p75 and CPSF6 supported significantly less recurrent integration than both WT and CKO cells. It is notable, however, that DKO cells still supported some degree of recurrent integration despite having near-random levels of overall genic integration.
Current models for the roles of LEDGF/p75 and CPSF6 in HIV-1 integration targeting invoke that the interaction of capsid with CPSF6 licenses the PIC to move beyond the nuclear periphery to interior regions of the nucleus, where the interaction of integrase with LEDGF/p75 helps to guide integration into the interior regions of gene bodies (20,21,26,31,40,76). The strong degree of overlap between RIGs in WT cells and RIGs in LKO cells suggests that in LKO cells, PICs are shuttled to the same general region of the nucleus as in WT cells, which is consistent with results of prior imaging studies (20,77). It is currently unclear why loss of the integrase-LEDGF/p75 interaction would dramatically reduce recurrent integration. LEDGF/p75 can significantly stimulate the strand transfer activity of recombinant HIV-1 integrase protein in vitro (78,79), and we envision this same principle likely applies in cells. In the absence of LEDGF/p75, this stimulatory effect is missing, yielding overall reduced numbers of integration events and hence impaired recurrent integration.
Our analysis of LEDGF/p75 occupancy per nuclear compartment showed lower overall occupancy in Speckle regions relative to all other nuclear compartments. When normalized for gene content, this trend was largely lost, indicating that genic LEDGF/p75 occupancy is relatively constant across nuclear regions. Despite this, RIG targets in WT cells and in LKO cells were highly compartmentalized to Speckles (Figure 6A, B). In addition, integration sites in RIGs predominantly existed several kb away from the closest LEDGF/p75-occupied region (Figure 6E). Our findings appear at least partially consistent with an earlier DamID study that found that not all LEDGF/p75 chromatin binding regions overlapped with sites of HIV-1 integration, and that integration occurred within a ‘wide window’ around sites of chromatin-bound LEDGF/p75 (64). That study additionally reported that 4.65% of 861 analyzed integration sites directly overlapped with ‘LEDGF islands’ and that 30% of integration sites were within 3.5 kb of the center of LEDGF/p75 islands (64). We similarly found that 3.1% of ∼473,000 integration sites overlapped with LEDGF/p75-occupied regions and that 27% were within a 3.5 kb window (Figure 6E). Minimally, these observations suggest that chromatin-bound LEDGF/p75 alone, as identified by either ChIP-seq or DamID, is insufficient to completely account for the well-established roles of LEDGF/p75 in genic integration targeting and recurrent integration. A minor fraction of chromatin-bound LEDGF/p75 does however closely correlate with HIV-1 integration, as evidenced by the 3.1% overlap of integration sites in LEDGF/p75-occupied regions. We note that our conclusions regarding LEDGF/p75 are primarily drawn from a single ChIP-seq dataset published by a different laboratory (32). More rigorous conclusions would likely require additional LEDGF/p75 chromatin binding data.
LEDGF/p75 has long been thought to tether HIV-1 integrase to chromatin via its chromatin-reader PWWP domain and AT-hooks (28,80–84). However, LEDGF/p75 harbors a so-called extended AT-hook, and thus might display binding affinity for RNA as well (85). LEDGF/p75 has additionally been shown to interact with various splicing factors (31). It is possible that in addition to tethering HIV-1 integrase as a statically-bound chromatin element, LEDGF/p75 works as a dynamic RNA and/or splicing factor-bound species to drive recurrent integration into the interior regions of gene bodies. Further investigations are warranted to differentiate the potential roles of primarily static versus dynamic LEDGF/p75 populations in HIV-1 integration targeting.
Genes avoided for integration appeared independent of cellular LEDGF/p75 or CPSF6 content, indicating that RAGs are operationally ‘invisible’ to HIV-1 PICs under these infection conditions. RAGs from various cell types were moreover enriched in Lamina regions of chromatin. Because HIV-1 preferentially targets active genes for integration (62), it seems likely that the comparatively low expression levels of RAGs additionally contributed to their apparent inaccessibility. Due to the predominance of neuronal GO terms among these genes, it is of interest to test if infection of neuronal-specific cell types such as microglia or astrocytes (86) shifts the balance of RIG/RAG targeting.
Integration into non-outlier genes was interestingly enriched in Interior Active 3 region regardless of cellular CPSF6 and LEDGF/p75 content (Figure 6A). Although this region was also enriched for LEDGF/p75 occupancy, the statistical enrichment of integration in non-outlier genes in LKO and DKO cells indicates that LEDGF/p75 occupancy is unlikely to drive this phenotype. Plausibly, Interior Active 3 region is open chromatin between the nuclear pore and preferentially targeted Speckle and Interior Active 1 regions that the PIC can readily target (34).
DATA AVAILABILITY
The R package rigrag, the R Markdown files related to model generation and assessment, and the Python scripts used to generate genome fragments are available on GitHub (https://github.com/gbedwell). Sequencing data for the integration site datasets determined herein are available through the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) under accession number SRP286975.
Supplementary Material
ACKNOWLEDGEMENTS
Authors’ contributions: Conceptualization, G.J.B. and A.N.E.; Methodology, G.J.B; Investigation, G.J.B., S.J., W.L., P.K.S.; Formal analysis, G.J.B.; Software, G.J.B.; Writing – Original and Subsequent Drafts, G.J.B. and A.N.E.; Visualization, G.J.B.; Funding Acquisition, A.N.E. and G.J.B.; Supervision, A.N.E. and G.J.B.
Contributor Information
Gregory J Bedwell, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
Sooin Jang, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
Wen Li, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
Parmit K Singh, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
Alan N Engelman, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01AI052014, U54AI150472 to A.N.E., T32AI007386 to G.J.B.]. The open access publication charge for this paper has been waived by Oxford University Press – NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. A.N.E. has consulted for ViiV Healthcare, Co. No other authors have competing interests to declare.
REFERENCES
- 1. Justice J.F., Morgan R.W., Beemon K.L.. Common viral integration sites identified in avian leukosis virus-induced B-cell lymphomas. mBio. 2015; 6:e01863-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Loyola L., Achuthan V., Gilroy K., Borland G., Kilbey A., Mackay N., Bell M., Hay J., Aiyer S., Fingerman D.et al.. Disrupting MLV integrase:BET protein interaction biases integration into quiescent chromatin and delays but does not eliminate tumor activation in a MYC/Runx2 mouse model. PLoS Pathog. 2019; 15:e1008154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beemon K., Rosenberg N.. Robertson E.S. Mechanisms of oncogenesis by avian and murine retroviruses. Cancer Associated Viruses. 2012; NY: Springer; 677–704. [Google Scholar]
- 4. Seiki M., Eddy R., Shows T.B., Yoshida M.. Nonspecific integration of the HTLV provirus genome into adult T-cell leukaemia cells. Nature. 1984; 309:640–642. [DOI] [PubMed] [Google Scholar]
- 5. Melamed A., Yaguchi H., Miura M., Witkover A., Fitzgerald T.W., Birney E., Bangham C.R.M.. The human leukemia virus HTLV-1 alters the structure and transcription of host chromatin in cis. eLife. 2018; 7:e36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coffin J., Swanstrom R.. HIV pathogenesis: dynamics and genetics of viral populations and infected cells. Cold SpringHarb. Perspect. Med. 2013; 3:a012526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eisele E., Siliciano R.F.. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012; 37:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finzi D., Hermankova M., Pierson T., Carruth L.M., Buck C., Chaisson R.E., Quinn T.C., Chadwick K., Margolick J., Brookmeyer R.et al.. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997; 278:1295–1300. [DOI] [PubMed] [Google Scholar]
- 9. Finzi D., Blankson J., Siliciano J.D., Margolick J.B., Chadwick K., Pierson T., Smith K., Lisziewicz J., Lori F., Flexner C.et al.. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999; 5:512–517. [DOI] [PubMed] [Google Scholar]
- 10. Chun T.-W., Stuyver L., Mizell S.B., Ehler L.A., Mican J.A.M., Baseler M., Lloyd A.L., Nowak M.A., Fauci A.S.. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:13193–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ho Y.-C., Shan L., Hosmane N.N., Wang J., Laskey S.B., Rosenbloom D.I.S., Lai J., Blankson J.N., Siliciano J.D., Siliciano R.F.. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013; 155:540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maldarelli F., Wu X., Su L., Simonetti F.R., Shao W., Hill S., Spindler J., Ferris A.L., Mellors J.W., Kearney M.F.et al.. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014; 345:179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wagner T.A., McLaughlin S., Garg K., Cheung C.Y.K., Larsen B.B., Styrchak S., Huang H.C., Edlefsen P.T., Mullins J.I., Frenkel L.M.. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 2014; 345:570–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cohn L.B., Silva I.T., Oliveira T.Y., Rosales R.A., Parrish E.H., Learn G.H., Hahn B.H., Czartoski J.L., McElrath M.J., Lehmann C.et al.. HIV-1 integration landscape during latent and active infection. Cell. 2015; 160:420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson E.M., Simonetti F.R., Gorelick R.J., Hill S., Gouzoulis M.A., Bell J., Rehm C., Pérez L., Boritz E., Wu X.et al.. Dynamic shifts in the HIV proviral landscape during long term combination antiretroviral therapy: Implications for persistence and control of HIV infections. Viruses. 2020; 12:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coffin J.M., Wells D.W., Zerbato J.M., Kuruc J.D., Guo S., Luke B.T., Eron J.J., Bale M., Spindler J., Simonetti F.R.et al.. Clones of infected cells arise early in HIV-infected individuals. JCI Insight. 2019; 4:e128432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chun T.-W., Fauci A.S.. HIV reservoirs: pathogenesis and obstacles to viral eradication and cure. AIDS. 2012; 26:1261–1268. [DOI] [PubMed] [Google Scholar]
- 18. Craigie R., Bushman F.D.. Host factors in retroviral integration and the selection of integration target sites. Microbiol. Spectr. 2014; 2: 10.1128/microbiolspec.MDNA3-0026-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Engelman A.N., Maertens G.N.. Parent L.J. Virus-host interactions in retroviral integration. Retrovirus-Cell Interactions. 2018; San Diego, CA: Academic Press; 163–198. [Google Scholar]
- 20. Achuthan V., Perreira J.M., Sowd G.A., Puray-Chavez M., McDougall W.M., Paulucci-Holthauzen A., Wu X., Fadel H.J., Poeschla E.M., Multani A.S.et al.. Capsid-CPSF6 interaction licenses nuclear HIV-1 trafficking to sites of viral DNA integration. Cell Host Microbe. 2018; 24:392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Francis A.C., Marin M., Singh P.K., Achuthan V., Prellberg M.J., Palermino-Rowland K., Lan S., Tedbury P.R., Sarafianos S.G., Engelman A.N.et al.. HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat. Commun. 2020; 11:3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marini B., Kertesz-Farkas A., Ali H., Lucic B., Lisek K., Manganaro L., Pongor S., Luzzati R., Recchia A., Mavilio F.et al.. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015; 521:227–231. [DOI] [PubMed] [Google Scholar]
- 23. Wang G.P., Ciuffi A., Leipzig J., Berry C.C., Bushman F.D.. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007; 17:1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee K., Ambrose Z., Martin T.D., Oztop I., Mulky A., Julias J.G., Vandegraaff N., Baumann J.G., Wang R., Yuen W.et al.. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010; 7:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Price A.J., Fletcher A.J., Schaller T., Elliott T., Lee K., KewalRamani V.N., Chin J.W., Towers G.J., James L.C.. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012; 8:e1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chin C.R., Perreira J.M., Savidis G., Portmann J.M., Aker A.M., Feeley E.M., Smith M.C., Brass A.L.. Direct visualization of HIV-1 replication intermediates shows that capsid and CPSF6 modulate HIV-1 intra-nuclear invasion and integration. Cell Rep. 2015; 13:1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sowd G.A., Serrao E., Wang H., Wang W., Fadel H.J., Poeschla E.M., Engelman A.N.. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E1054–E1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J.R., Bushman F.. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005; 11:1287–1289. [DOI] [PubMed] [Google Scholar]
- 29. Marshall H.M., Ronen K., Berry C., Llano M., Sutherland H., Saenz D., Bickmore W., Poeschla E., Bushman F.D.. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One. 2007; 2:e1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shun M.-C., Raghavendra N.K., Vandegraaff N., Daigle J.E., Hughes S., Kellam P., Cherepanov P., Engelman A.. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007; 21:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh P.K., Plumb M.R., Ferris A.L., Iben J.R., Wu X., Fadel H.J., Luke B.T., Esnault C., Poeschla E.M., Hughes S.H.et al.. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 2015; 29:2287–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. LeRoy G., Oksuz O., Descostes N., Aoi Y., Ganai R.A., Kara H.O., Yu J.-R., Lee C.-H., Stafford J., Shilatifard A.et al.. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells. Sci. Adv. 2019; 5:eaay3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lucic B., Chen H.-C., Kuzman M., Zorita E., Wegner J., Minneker V., Wang W., Fronza R., Laufs S., Schmidt M.et al.. Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration. Nat. Commun. 2019; 10:4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y., Zhang Y., Zhang R., van Schaik T., Zhang L., Sasaki T., Peric-Hupkes D., Chen Y., Gilbert D.M., van Steensel B.et al.. SPIN reveals genome-wide landscape of nuclear compartmentalization. Genome Biol. 2021; 22:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cesana D., Santoni de Sio F.R., Rudilosso L., Gallina P., Calabria A., Beretta S., Merelli I., Bruzzesi E., Passerini L., Nozza S.et al.. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat. Commun. 2017; 8:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ikeda T., Shibata J., Yoshimura K., Koito A., Matsushita S.. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. J. Infect. Dis. 2007; 195:716–725. [DOI] [PubMed] [Google Scholar]
- 37. Coffin J.M., Bale M.J., Wells D., Guo S., Luke B., Zerbato J.M., Sobolewski M.D., Sia T., Shao W., Wu X.et al.. Integration in oncogenes plays only a minor role in determining the in vivo distribution of HIV integration sites before or during suppressive antiretroviral therapy. PLoS Pathog. 2021; 17:e1009141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koh Y., Wu X., Ferris A.L., Matreyek K.A., Smith S.J., Lee K., KewalRamani V.N., Hughes S.H., Engelman A.. Differential effects of human immunodeficiency virus type 1 capsid and cellular factors Nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J. Virol. 2013; 87:648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Levy D.N., Aldrovandi G.M., Kutsch O., Shaw G.M.. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:4204–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li W., Singh P.K., Sowd G.A., Bedwell G.J., Jang S., Achuthan V., Oleru A.V., Wong D., Fadel H., Lee K.et al.. CPSF6-dependent targeting of speckle-associated domains distinguishes primate from non-primate lentiviral integration. mBio. 2020; 11:e02254-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Serrao E., Cherepanov P., Engelman A.N.. Amplification, next-generation sequencing, and genomic DNA mapping of retroviral integration sites. J. Vis. Exp. 2016; 53840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Serrao E., Ballandras-Colas A., Cherepanov P., Maertens G.N., Engelman A.N.. Key determinants of target DNA recognition by retroviral intasomes. Retrovirology. 2015; 12:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S.et al.. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Saito A., Henning M.S., Serrao E., Dubose B.N., Teng S., Huang J., Li X., Saito N., Roy S.P., Siddiqui M.A.et al.. Capsid-CPSF6 interaction is dispensable for HIV-1 replication in primary cells but is selected during virus passage in vivo. J. Virol. 2016; 90:6918–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ferris A.L., Wells D.W., Guo S., Del Prete G.Q., Swanstrom A.E., Coffin J.M., Wu X., Lifson J.D., Hughes S.H.. Clonal expansion of SIV-infected cells in macaques on antiretroviral therapy is similar to that of HIV-infected cells in humans. PLoS Pathog. 2019; 15:e1007869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bale M.J., Katusiime M.G., Wells D., Wu X., Spindler J., Halvas E.K., Cyktor J.C., Wiegand A., Shao W., Cotton M.F.et al.. Early emergence and long-term persistence of HIV-infected T cell clones in children. mBio. 2021; 12:e00568-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang C., Lian X., Gao Ce, Sun X., Einkauf KB., Chevalier JM., Chen SM.Y., Hua S., Rhee B., Chang K.et al.. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature. 2020; 585:261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Einkauf K.B., Lee G.Q., Gao C., Sharaf R., Sun X., Hua S., Chen S.M.Y., Jiang C., Lian X., Chowdhury F.Z.et al.. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Invest. 2019; 129:988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shao W., Shan J., Kearney M.F., Wu X., Maldarelli F., Mellors J.W., Luke B., Coffin J.M., Hughes S.H.. Retrovirus Integration Database (RID): a public database for retroviral insertion sites into host genomes. Retrovirology. 2016; 13:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmiedel B.J., Singh D., Madrigal A., Valdovino-Gonzalez A.G., White B.M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J.A., McVicker G.et al.. Impact of genetic polymorphisms on human immune cell gene expression. Cell. 2018; 175:1701–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Y., Zhang Y., Wang Y., Zhang L., Brinkman E.K., Adam S.A., Goldman R., van Steensel B., Ma J., Belmont A.S.. Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J. Cell Biol. 2018; 217:4025–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khan A., Zhang X.. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016; 44:D164–D171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K.et al.. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J.. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004; 32:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meuleman W., Peric-Hupkes D., Kind J., Beaudry J.-B., Pagie L., Kellis M., Reinders M., Wessels L., van Steensel B.. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013; 23:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mi H., Muruganujan A., Ebert D., Huang X., Thomas P.D.. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019; 47:D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gillet N.A., Malani N., Melamed A., Gormley N., Carter R., Bentley D., Berry C., Bushman F.D., Taylor G.P., Bangham C.R.M.. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood. 2011; 117:3113–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Anderson-Daniels J., Singh P.K., Sowd G.A., Li W., Engelman A.N., Aiken C.. Dominant negative MA-CA fusion protein is incorporated into HIV-1 cores and inhibits nuclear entry of viral preintegration complexes. J. Virol. 2019; 93:e01118-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schröder A.R.W., Shinn P., Chen H., Berry C., Ecker J.R., Bushman F.. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002; 110:521–529. [DOI] [PubMed] [Google Scholar]
- 63. Brady T., Agosto L.M., Malani N., Berry C.C., O'Doherty U., Bushman F.. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. AIDS. 2009; 23:1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. De Rijck J., Bartholomeeusen K., Ceulemans H., Debyser Z., Gijsbers R.. High-resolution profiling of the LEDGF/p75 chromatin interaction in the ENCODE region. Nucleic Acids Res. 2010; 38:6135–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Berry C., Hannenhalli S., Leipzig J., Bushman F.D.. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput. Biol. 2006; 2:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McManus W.R., Bale M.J., Spindler J., Wiegand A., Musick A., Patro S.C., Sobolewski M.D., Musick V.K., Anderson E.M., Cyktor J.C.et al.. HIV-1 in lymph nodes is maintained by cellular proliferation during antiretroviral therapy. J. Clin. Invest. 2019; 129:4629–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hughes S.H., Coffin J.M.. What integration sites tell us about HIV persistence. Cell Host Microbe. 2016; 19:588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Anderson E.M., Maldarelli F.. The role of integration and clonal expansion in HIV infection: live long and prosper. Retrovirology. 2018; 15:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Simonetti F.R., Sobolewski M.D., Fyne E., Shao W., Spindler J., Hattori J., Anderson E.M., Watters S.A., Hill S., Wu X.et al.. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl. Acad. Sci. 2016; 113:1883–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nakanishi M., Ozaki T., Yamamoto H., Hanamoto T., Kikuchi H., Furuya K., Asaka M., Delia D., Nakagawara A.. NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. J. Biol. Chem. 2007; 282:22993–23004. [DOI] [PubMed] [Google Scholar]
- 71. Solier S., Pommier Y.. MDC1 cleavage by caspase-3: a novel mechanism for inactivating the DNA damage response during apoptosis. Cancer Res. 2011; 71:906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Simpson H.M., Furusawa A., Sadashivaiah K., Civin C.I., Banerjee A.. STAT5 inhibition induces TRAIL/DR4 dependent apoptosis in peripheral T-cell lymphoma. Oncotarget. 2018; 9:16792–16806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu R., Yeh Y.-H.J., Varabyou A., Collora J.A., Sherrill-Mix S., Talbot C.C., Mehta S., Albrecht K., Hao H., Zhang H.et al.. Single-cell transcriptional landscapes reveal HIV-1–driven aberrant host gene transcription as a potential therapeutic target. Sci. Transl. Med. 2020; 12:eaaz0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Simonetti F.R., Zhang H., Soroosh G.P., Duan J., Rhodehouse K., Hill A.L., Beg S.A., McCormick K., Raymond H.E., Nobles C.L.et al.. Antigen-driven clonal selection shapes the persistence of HIV-1–infected CD4+ T cells in vivo. J. Clin. Invest. 2021; 131:e145254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gantner P., Pagliuzza A., Pardons M., Ramgopal M., Routy J.-P., Fromentin R., Chomont N.. Single-cell TCR sequencing reveals phenotypically diverse clonally expanded cells harboring inducible HIV proviruses during ART. Nat. Commun. 2020; 11:4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Burdick R.C., Li C., Munshi M., Rawson J.M.O., Nagashima K., Hu W.-S., Pathak V.K.. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. U.S.A. 2020; 17:5486–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Burdick R.C., Delviks-Frankenberry K.A., Chen J., Janaka S.K., Sastri J., Hu W.S., Pathak V.K.. Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 2017; 13:e1006570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cherepanov P., Maertens G., Proost P., Devreese B., Van Beeumen J., Engelborghs Y., De Clercq E., Debyser Z.. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003; 278:372–381. [DOI] [PubMed] [Google Scholar]
- 79. Hare S., Shun M.-C., Gupta S.S., Valkov E., Engelman A., Cherepanov P.. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 2009; 5:e1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Eidahl J.O., Crowe B.L., North J.A., McKee C.J., Shkriabai N., Feng L., Plumb M., Graham R.L., Gorelick R.J., Hess S.et al.. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013; 41:3924–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shun M.-C., Botbol Y., Li X., Di Nunzio F., Daigle J.E., Yan N., Lieberman J., Lavigne M., Engelman A.. Identification and characterization of PWWP domain residues critical for LEDGF/p75 chromatin binding and human immunodeficiency virus type 1 infectivity. J. Virol. 2008; 82:11555–11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hare S., Cherepanov P.. The interaction between lentiviral integrase and LEDGF: Structural and functional insights. Viruses. 2009; 1:780–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Engelman A., Cherepanov P.. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008; 4:e1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Llano M., Saenz D.T., Meehan A., Wongthida P., Peretz M., Walker W.H., Teo W., Poeschla E.M.. An essential role for LEDGF/p75 in HIV integration. Science. 2006; 314:461–464. [DOI] [PubMed] [Google Scholar]
- 85. Filarsky M., Zillner K., Araya I., Villar-Garea A., Merkl R., Längst G., Németh A.. The extended AT-hook is a novel RNA binding motif. RNA Biol. 2015; 12:864–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gray L.R., Roche M., Flynn J.K., Wesselingh S.L., Gorry P.R., Churchill M.J.. Is the central nervous system a reservoir of HIV-1. Curr. Opin. HIV AIDS. 2014; 9:552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R package rigrag, the R Markdown files related to model generation and assessment, and the Python scripts used to generate genome fragments are available on GitHub (https://github.com/gbedwell). Sequencing data for the integration site datasets determined herein are available through the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) under accession number SRP286975.