


Abstract
Integrating omics data with quantification of biological traits provides unparalleled opportunities for discovery of genetic regulators by in silico inference. However, current approaches to analyze genetic-perturbation screens are limited by their reliance on annotation libraries for prioritization of hits and subsequent targeted experimentation. Here, we present iTARGEX (identification of Trait-Associated Regulatory Genes via mixture regression using EXpectation maximization), an association framework with no requirement of a priori knowledge of gene function. After creating this tool, we used it to test associations between gene expression profiles and two biological traits in single-gene deletion budding yeast mutants, including transcription homeostasis during S phase and global protein turnover. For each trait, we discovered novel regulators without prior functional annotations. The functional effects of the novel candidates were then validated experimentally, providing solid evidence for their roles in the respective traits. Hence, we conclude that iTARGEX can reliably identify novel factors involved in given biological traits. As such, it is capable of converting genome-wide observations into causal gene function predictions. Further application of iTARGEX in other contexts is expected to facilitate the discovery of new regulators and provide observations for novel mechanistic hypotheses regarding different biological traits and phenotypes.
Graphical Abstract
Graphical Abstract.

iTARGEX is an analytical tool that accounts for heterogeneity in transcriptional responses to single-gene deletion mutants to facilitate the discovery of new regulators and novel mechanistic hypotheses regarding different biological traits and phenotypes.
INTRODUCTION
One of the broad challenges in molecular biology is to identify the genetic factors responsible for certain cellular functions and phenotypic traits. In the past two decades, vast amounts of transcriptomic data have been generated, facilitating the mapping of intricate molecular interactions the underlie phenotypic traits (1,2). Two major strategies have been established to analyze transcriptome data in studies of how genetic regulation influences traits. Using forward genetics, approximate loci of genetic variants can be associated with particular phenotypes, and methods such as expression quantitative trait loci (eQTLs) have been utilized for genome-wide mapping by statistical correlation (3–5). Alternatively, reverse genetics screens examine phenotype-genotype associations by inducing genetic perturbations across the genome, including mutations (6), gene knockdowns (7) or genome editing with CRISPR–Cas9 (8). These screening methods provide rich molecular datasets that can reveal novel insights into the consequences of transcriptomic regulation; however, the methods of analysis are often limited.
Although both forward and reverse genetics approaches have been successfully applied in many studies, identification of phenotypic determinants by these strategies has typically involved prioritizing candidate genes through a combination of prior knowledge and targeted experimentation. In addition, the application of vast genomic data is often limited to the specific questions addressed in a study or the conditions under which the data are gathered. Importantly, novel analyses of previously generated genome-wide transcriptome data may reveal unexpected causal connections and allow for inter-study comparisons to uncover broad regulatory mechanisms. Herein, we describe the application of one such new method, which analyzes transcriptome data from a reverse genetics screen and can be used to infer novel regulators of biological traits (Figure 1A).
Figure 1.

iTARGEX flowchart. (A) Strategy for identification of candidate regulators by analyzing associations between gene expression changes and traits. (B) iTARGEX analysis proceduRes. The fold change (FC) values of gene expression for 1484 single-gene deletion mutants were log-transformed, and the magnitudes of biological traits of interest were scaled using scale() function in R. In the first step of association analysis, iTARGEX obtains the value of  by performing a generalized linear mode analysis. Second, the given
by performing a generalized linear mode analysis. Second, the given  is used as an initial value for the EM algorithm to estimate the parameters in the regression mixture model. Then, the mixture weights of each gene are estimated as the probabilities for two regression components reported in the following steps. Third, each gene is assigned to either regression component according to the mixture weights, according to a threshold of 0.5; the associations of two distinct subpopulations are examined using linear regression. The weights for the significant associated components are then used to estimate the weighted Pearson's correlation. Finally, the significance threshold for a high-confidence regulator is set as q-value of weighted Pearson's correlation coefficient (WPCC) < 10–3.
is used as an initial value for the EM algorithm to estimate the parameters in the regression mixture model. Then, the mixture weights of each gene are estimated as the probabilities for two regression components reported in the following steps. Third, each gene is assigned to either regression component according to the mixture weights, according to a threshold of 0.5; the associations of two distinct subpopulations are examined using linear regression. The weights for the significant associated components are then used to estimate the weighted Pearson's correlation. Finally, the significance threshold for a high-confidence regulator is set as q-value of weighted Pearson's correlation coefficient (WPCC) < 10–3.
The dataset we chose to analyze comprises microarray results from Saccharomyces cerevisiae single-gene deletion mutants (9). This dataset was especially attractive because expression patterns in isogenic populations tend to exhibit relatively low variability; cellular systems often maintain phenotypic stability in the face of environmental or genetic perturbations through complex regulatory networks and genetic capacitors (10,11). Additionally, cell–cell variability in this dataset is masked because gene expression levels were measured in bulk populations of isogenic strains. Hence, the expression profiles between different populations with the same genetic background should exhibit only negligible variations, and these stochastic variations would not be associated with any biological traits. Another important aspect of the dataset is that deletion of a particular gene may affect the expression of many other genes either directly or indirectly. As a result, significant correlations between a mutant and phenotype can be understood as the effect of the gene mutation on the entire cellular system, and a correlation between the expression level of a particular gene and a given biological trait may be observable in strains with mutations in several different genes.
In this work, we leveraged these features of the dataset to establish a statistical framework that incorporates a mixture regression model, and we implemented it as an analytical tool called identification of Trait-Associated Regulatory Genes via mixture regression using Expectation maximization (iTARGEX). Since biological data are usually noisy, global correlation across all data points (genes) may lack power due to the blurred signals, especially in the case of marginal regulators that only affect the expression of a limited number of genes. Our mixture regression model can account for this potential heterogeneity in transcriptional responses to single-gene deletion mutants. As such, we assume that the data are a mixture of two Gaussian distributions (correlated and uncorrelated groups), and we incorporate the covariates (gene effects of interest) into the model of mixture regression.
To illustrate the utility of predictions made by iTARGEX, we associated gene expression in a set of 1,484 budding yeast deletion mutants (9) with two biological traits, gene expression buffering during S phase and global protein turnover rate. After identifying gene candidates with highly significant associations, we conducted biological experiments to further evaluate the phenotypes of those mutants. Our results revealed that Elg1 is associated with transcriptional buffering during S phase. Moreover, four mutants of mrn1, upf3, ram1 and ssn3 showed significantly delayed protein turnover when compared to the wild type (WT). Thus, iTARGEX is a robust analytical tool that can be used to identify novel determinants of wide-ranging biological traits, and it can thus convert genome-wide observations into specific predictions of causal gene functions.
MATERIALS AND METHODS
Data resources
The Deletome collection of mRNA expression profiles for individual gene deletions in yeast S. cerevisiae was created by Holstege's group (9). The Deletome contains the transcriptional responses of 1,484 single-gene deletion mutants in S. cerevisiae that each lack a single nonessential gene, as measured by microarray. The targets of this genetic perturbation assay covered 25% of the entire yeast transcriptome and include most of the genes with high potential to exhibit regulatory functions. As described in the original article, mutant lines with at least one significant differentially expressed gene (P < 0.05 and log2FC > 1.7) were considered to be responsive mutants.
Statistical framework of iTARGEX
Estimation of mixture regression models using EM algorithm
The transcriptomic changes caused by a genetic perturbation can be associated with a given phenotype of interest in two scenarios, depending on the role of the disrupted gene. iTARGEX considers models of both association scenarios with a statistical framework implemented in R/mixtools package (12). For each deletion mutant, the n-vector corresponds to the number of genes for the given phenotype of interest.  and
and 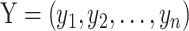 , respectively denoting the vector of phenotype of interest and the vector of gene expression changes. Each variable is centered, such that it has a zero mean across the samples. For vector X, the variables are scaled using the scale function in R. For vector Y, the variables are log-transformed fold changes in gene expression for the given mutant, as obtained from the original paper (9). In the first association scenario, ‘global’ association represents a general pattern across the whole transcriptome and the phenotype of interest. In this case, the likelihood function given the n observed data is defined as
, respectively denoting the vector of phenotype of interest and the vector of gene expression changes. Each variable is centered, such that it has a zero mean across the samples. For vector X, the variables are scaled using the scale function in R. For vector Y, the variables are log-transformed fold changes in gene expression for the given mutant, as obtained from the original paper (9). In the first association scenario, ‘global’ association represents a general pattern across the whole transcriptome and the phenotype of interest. In this case, the likelihood function given the n observed data is defined as
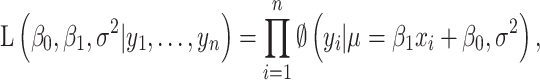 |
where  is the pdf of normal distribution,
is the pdf of normal distribution,  is the slope,
is the slope,  is the intercept,
is the intercept, 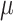 is the mean of the normal density, and
is the mean of the normal density, and  is the random error.
is the random error.
In the second scenario, an association only exists along a subset of genes, and it is defined as a ‘partial’ association. The probability density function of the observed data would be a mixture of two regression models; one contributes to the association and the other does not. An additional parameter is needed to account for the proportion of data points that are genuinely associated with each other. The likelihood function is thus modified as
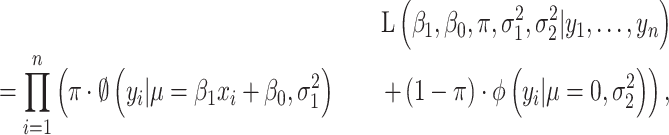 |
where  represents the ratio of associated genes.
represents the ratio of associated genes.
For each independent dataset, iTARGEX accounts for the scenario of partial association and estimates the parameters (i.e.  ) by maximizing the likelihood through an iterative Expectation Maximization (EM) algorithm (13). Of note, we provide an initial
) by maximizing the likelihood through an iterative Expectation Maximization (EM) algorithm (13). Of note, we provide an initial  for the EM algorithm from the slope generated by performing generalized linear model (glm) with identity link function on the dataset. When the EM algorithm converges, a set of mixture weights
for the EM algorithm from the slope generated by performing generalized linear model (glm) with identity link function on the dataset. When the EM algorithm converges, a set of mixture weights for each gene are reported, representing the probabilities of belonging to either estimated component (regression models). We then used the set of mixture weights for all genes to select candidate regulators in the following steps.
for each gene are reported, representing the probabilities of belonging to either estimated component (regression models). We then used the set of mixture weights for all genes to select candidate regulators in the following steps.
Significant regression component selection
In iTARGEX, a selection procedure is applied to identify which regression component shows a strong association relationship in the single-gene deletion mutants. First, every gene in a given mutant is assigned as either component 1 or component 2 based on a threshold of its mixture weight (set at 0.5). Because the sum of mixture weights for each gene equals 1, the two regression components include distinct sets of genes with higher probabilities of belonging to the assigned group. Then, the association of the two components with distinct subpopulations of genes is estimated by performing linear regression and separately obtaining the significance levels of the regression slopes. In order to avoid inflation of type I error due to application of multiple tests, all P-values are adjusted by a Bonferroni correction to obtain q-values. An association for a given component in each mutant is considered significant if the q-values of regression slopes of the assigned component pass a stringent cutoff value of <10–3. If the slopes of the two components in a given mutant are both significant, the component with a smaller significant q-value is used to represent the significant association of the mutant. As a result, iTARGEX identifies single-gene deletion mutants with a significant association of the selected component as candidate regulators.
Correlation test for ranking high-confidence regulators
In order to evaluate the overall correlation between the gene expression changes and the phenotype of interest, iTARGEX performs a weighted Pearson's correlation test, wherein each data point has a corresponding weight determined by the EM algorithm and selection procedures outlined above. In order to avoid inflation of type I errors due to multiple correlation tests, a Bonferroni correction is also applied to adjust the P-values for the weighted Pearson's correlation coefficients (WPCC). Finally, iTARGEX sorts high-confidence regulators by their q-values after applying a stringent cutoff value of <10–3 for significant correlations.
Bioinformatics analysis pipeline for RNA-seq
The raw sequencing reads (FASTQ files) were trimmed using Trimmomatic (14) to remove illumine adapter sequences and reads with low quality. The quality of the trimmed reads was then evaluated using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and after passing quality control, the transcript level abundance was quantified against a reference sequence of S. cerevisiae S288c genome (SGD, R64-2-1) using salmon v0.13.1 (15). For all comparisons in this work, TPM counts were used to normalize the differences in sequencing depth.
Quantification of expression homeostasis during DNA duplication
In order to investigate gene expression buffering, we adopted a quantification method from a previous study (16) with slight modifications (Supplementary Figure S1). Briefly, 500 early and 500 late replicated genes (17) were selected for quantification of transcript level abundance. For RNA-seq data from different time-points, the regulated genes, which were defined by Voichek et al. (2016), and the gene expression values with TPM ⩽ 1 were excluded in subsequent steps. Next, the expression level of each gene at every time-point was divided by its signal in G1 phase and then log2 transformed. To calculate the average signal, the expression levels were averaged separately over early and late replicated genes for each time-point. The ‘% Signal early vs. late’ value was calculated by dividing the average early and late signals at each time-point and multiplying by 100.
Yeast strain and growth conditions for expression homeostasis
Experiments were conducted using S. cerevisiae haploid strains. Strains used in this article are derivatives of BY4741 (RNA experiments) or BY4742 (proteomic experiments), unless otherwise indicated. For gene disruptions, the indicated gene was replaced with the KanMX gene or disrupted through a PCR-based strategy. All yeast cells were supplemented with 2% (vol/vol) glucose in YPD medium.
G1 arrest and release
To synchronize cells from G1 to S phase, cells were arrested at G1 by α-factor, using a standard protocol (18).
Nascent RNA labeling by 4-thiouracil (4tU)
The protocol for labeling nascent RNA with 4tU was described previously (16). Cells were harvested at –10 min (before G1 release) and at 6 min intervals up to 1 h after release.
Biotinylation and purification of labeled RNA
Yeast RNA was prepared by extraction with hot acidic phenol (19). Total RNA was extracted, and 4tU-containing mRNA was biotinylated and isolated as previously described (20,21). To obtain high quality RNA, samples were further subjected to the clean-up protocol with the RNeasy Mini Kit (Qiagene, Cat. #74104), and quality was checked with gel electrophoresis. Nascent RNA was then biotinylated as described (20,21), using 400 μl (∼40 μg) total RNA and 4 μg MTSEA biotin-XX (Biotium) and purified by MyOne Streptavidin C1 Dynabeads (Invitrogen). The isolated RNA was further purified and concentrated using Qiagen miRNeasy columns (#217084), according to the manufacturer's protocol. The isolated RNA was then prepared for sequencing using the Ovation SoLo or Ovation Universal RNA-seq System kits (Tecan), according to the manufacturer's instructions. Libraries were sequenced on the Illumina NextSeq high output platform using single reads 75 in Institute of Molecular Biology of Academia Sinica
Flow cytometry
To assess cell cycle synchronization efficiency and progression of the cell cycle, a standard protocol was followed (18). Samples taken throughout the experiment were stained for DNA, and the DNA content was quantified by flow cytometry in the instrument core facility of the Institute of Cellular and Organismic Biology at Academia Sinica.
Yeast growth conditions for protein turnover
S. cerevisiae strains were grown in synthetic medium containing 6.7 g/l yeast nitrogen base, 2 g/l dropout mix (US Biological) containing all amino acids except lysine and 2% glucose. For heavy pre-labeling, heavy [13C6/15N2] l-lysine (Cambridge Isotope Labs) was added to a final concentration of 30 mg/l. Cells were precultured in 5 ml medium containing heavy lysine overnight at 30°C; the procedure was repeated twice.
Pulse SILAC
The experiment was adapted from a previously described protocol (22). Briefly, after pre-culture or nSILAC labeling, cells were cultured in biological duplicates up to optical density at 600 nm = 0.4. After three washes at 4°C with cold SILAC medium without lysine, cells were transferred to SILAC medium containing light lysine and harvested at 0, 60 and 180 min. For each time-point, ∼3 OD units of cells were harvested by centrifugation. Cell lysates were prepared, digested, and fractionated as described (22). Eluted peptides from the column were directly electrosprayed into the mass spectrometer located in the MS facility of Academia Sinica. Mass spectra were acquired and protein half-lives were determined following a procedure described previously (23).
RESULTS
Overview of iTARGEX
To identify candidate regulators associated with biological traits, we developed a novel tool, iTARGEX, for the identification of Trait-Associated Regulators via mixture regression using EXpectation maximization (Figure 1B). iTARGEX is written in R with embedded code in C for accelerating EM and can be easily executed by users. The statistical framework of iTARGEX primarily considers partial associations between the trait of interest and gene expression changes in the single-gene deletion mutants by a generalization of multiple linear regression. Association analyses are then iteratively performed for each individual deletion mutant. A key step in this approach is use of an EM algorithm to consider partial associations, in order to identify genuine association in subpopulations. Next, a stringent selection procedure is applied to identify the significant association in the subpopulation of data points in a given mutant using linear regression analysis. The candidate regulators are called as hits if the regression slope of either of two components is significant (q-value < 10–3). Consequently, the weights of each gene from the significant regression component are integrated in the calculation of weighted Pearson's correlation coefficients (WPCC). Finally, the high-confidence regulators are reported based on the significance levels of WPCC, using a threshold of q-value < 10–3. The output of iTARGEX includes the gene names of candidate regulators with their WPCC, -log10(q-value) and  of the associated component.
of the associated component.
Use of iTARGEX to identify regulators of gene dosage buffering during DNA replication
To demonstrate the effectiveness of iTARGEX, we performed a screen to identify candidate genes needed for buffering transcription during the replication process. The transcriptional profiles of 1,484 deletion mutants (9) were used to identify genes required for maintaining expression homeostasis during DNA replication (28). In this in silico screen, candidates that appear to play a role in expression homeostasis were deduced using a simple correlation between the time of replication per gene and gene expression changes in the mutants. Of note, there is no statistical procedure that can be used to validate their candidate selection, and the candidates were instead verified directly by empirical analyses. In contrast, the statistical framework of iTARGEX provides a reasonable computational procedure for candidate selection. For each mutant, the q values of correlation coefficient were generated (Table 1 and Figure 2) and were then used to select candidate genes that may regulate the buffering process. In comparison with the previous literature (28), iTARGEX identified seven out of nine mutants that were empirically shown to be required for expression homeostasis. Several of the candidates with highly ranked q values in our list were also identified and verified previously, including genes involved in cell-cycle control and DNA replication (MRC1 and CLB5), and chromatin assembly or modification (ASF1, RTT109 and HOS4). Interestingly, we found that genes encoding components of the CTF18 complex (CTF8, CTF18 and DCC1) were clustered together with high q-value ranks. As one of the three RLC (replication factor C-like) complexes (25), Ctf18-RLC has been shown to interact with PCNA (the sliding replication clamp for DNA polymerases) during both loading and unloading (26,27). In addition, the deletion of ELG1, which encodes a subunit of an alternative replication factor C complex (28), exhibited a very high rank based on q-value in the iTARGEX analysis. With regard to chromatin-related genes, the rank of ELG1 was only lower than that of ASF1 and even higher than the value for RTT109. These results suggested that PCNA loading and unloading activities may be important for buffering gene dosage during S phase. Taken together, these results showed that iTARGEX (assisted by statistical power) was able to identify known regulators and also potentially reveal novel factors for a given genetic trait.
Table 1.
Summary of iTARGEX results from the association analysis for replication time of genes and regulators that play a role in expression homeostasis during DNA replication. Stars indicate genetic mutants with q-values of correlation below the threshold of 10–3 in the iTARGEX but that did not pass in the simple correlation. PCC: Pearson's correlation coefficient; WPCC: Weighted Pearson's correlation coefficient; q-values are log-transformed;  : the proportion of genes were estimated to be significant association by EM
: the proportion of genes were estimated to be significant association by EM
| iTARGEX | Simple correlation | ||||||
|---|---|---|---|---|---|---|---|
| Regulators | WPCC | –log10(q-value) | π | PCC | –log10(q-value) | Reported in literatures | Validation in this study |
| MRC1 | −0.232 | 71.929 | 0.879 | −0.132 | 21.752 | Voichek et al., 2018 | v |
| CLB5 | −0.154 | 30.318 | 0.895 | −0.114 | 15.505 | Voichek et al., 2018 | |
| ASF1 | −0.138 | 23.853 | 0.857 | −0.120 | 17.480 | Voichek et al., 2016 | |
| CKB1 | −0.129 | 20.710 | 0.909 | −0.068 | 4.116 | ||
| CKB2* | −0.093 | 9.736 | 0.910 | ||||
| ELG1 | −0.093 | 9.650 | 0.917 | −0.075 | 5.468 | v | |
| RTR1* | 0.091 | 9.278 | 0.896 | ||||
| DUN1 | −0.089 | 8.681 | 0.892 | −0.075 | 5.437 | ||
| RTT109 | −0.085 | 7.762 | 0.786 | −0.071 | 4.621 | Voichek et al., 2016 | v |
| CTF8* | −0.082 | 7.149 | 0.897 | Voichek et al., 2018 | v | ||
| CTF18* | −0.082 | 7.007 | 0.837 | ||||
| DCC1* | −0.078 | 6.223 | 0.850 | −0.060 | 2.682 | ||
| HHT1 | −0.078 | 6.193 | 0.915 | −0.066 | 3.788 | ||
| HOS4* | −0.073 | 5.243 | 0.918 | Voichek et al., 2018 | |||
| SWI6 | −0.072 | 4.916 | 0.801 | −0.071 | 4.765 | Voichek et al., 2018 | |
| NPT1* | −0.070 | 4.504 | 0.924 | ||||
| RNR4 | −0.068 | 4.126 | 0.853 | −0.066 | 3.699 | ||
Figure 2.

iTARGEX predicts candidate regulators involved in expression homeostasis during S phase. (A) Volcano-like plot shows the candidate regulators associated with DNA replication time predicted by iTARGEX. (B) Representative flow cytometry profiles of WT, rtt109Δ, mrc1Δ, ctf8Δ after factor arrest and release reveal defects in postponement of the cell cycle in mutants, as indicated by red arrows. (C) Expression of nascent RNAs during S phase. The mRNA synthesis rate of early replicating genes relative to late replicating genes was increased during S phase in the rtt109Δ mutant cells compared to WT cells. (D) Similar effects of lost transcriptional buffering were observed in the mrc1Δ and ctf8Δ mutants.
Verification of mutants that lose expression homeostasis during S phase
RTT109 and ASF1 were previously shown to suppress transcription from replicated DNA depending on the acetylation of histone H3 on lysine 56 (H3K56ac) (16). As a first step to establish the role of PCNA loading/unloading in expression homeostasis during S phase, we decided to re-examine the role of RTT109 in expression homoeostasis. Yeast cells were collected, and nascent mRNAs were profiled from synchronized cell cultures during S phase progression. (Figure 2B). WT and RTT109-deleted (rtt109Δ) cells were analyzed at a time resolution of 6 min. The DNA content analysis indicated that the WT cells entered S phase at 18 min after release from G1 and reached G2/M phase at 42 min after release. The rtt109Δ cells exhibited a slower cell cycle, with entry into S phase at 24 min after G1 release and arrival at G2/M phase at 48 min post-release. Despite these differences in cell cycle progression, the temporal dynamics of transcription profiles through the cell cycle were highly similar for WT and rtt109Δ cells. However, the expression of replicated genes in rtt109Δ cells was increased at 18 min after release, which was 6 min earlier than the onset of S phase (Figure 2C), consistent with the notion that expression homeostasis is lost in these cells (24). In addition, we also examined the transcription profiles and cell cycle progressions of MRC1-deleted and CTF8-deleted cells. We confirmed a pronounced loss of transcription buffering in mrc1Δ cells (24). The effect of ctf8Δ cells was similar with rtt109Δ cells, confirming that the activity of Ctf18-RLC likely plays a role in transcriptional buffering during S phase.
Elg1 is a novel regulator of transcriptional buffering during S phase
One of the candidate genes, ELG1, stood out from our iTARGEX analysis for several reasons. First, ELG1 gene was not identified in previous screens (16,24), so it was important to verify its role in transcriptional buffering during S phase. Furthermore, Elg1 is thought function as a PCNA unloader (28), and this unloading function appears to be conserved in humans; as such, ATAD5 (the human Elg1 homolog) is required for removal of PCNA from chromatin in human cells (29,30). Finally, the identification of Elg1 as a regulator of transcriptional buffering suggested that the timely unloading of PCNA from chromatin is required for expression homeostasis during DNA replication. Because the molecular function of Elg1-RLC differs from previously identified buffering regulators, such as Asf1-Rtt109-H3K56ac (16) and PAF1-SET1-H3K4me (24), we suspected Elg1-RLC may be an unexpected and novel regulator of gene dosage buffering.
To test this possibility, we analyzed the contribution of Elg1 to expression homeostasis by profiling cell cycle progression and temporal gene expression of WT and elg1Δ cells. Similar to rtt109Δ cells, elg1Δ cells exhibited delays in entry to and exit from S phase (Figure 3A). However, we surprisingly found a significant reduction in the expression of early replicated genes at 6 min, almost immediately after G1 release. Moreover, the transcriptional repression in elg1Δ cells started to recover at 24 min, concomitant with entry into S phase, and the level of transcription continued to increase as the cells progressed to G2/M phase (Figure 3B). To confirm this unexpected finding, we repeated the same transcription and cell cycle profiling experiments in a genetic background (31), other than the conventional BY4741. Remarkably, we obtained very similar results for both gene expression and DNA content measurements on the new background (Figure 3D).
Figure 3.

Elg1 controls genetic buffering during S phase. (A) Representative flow cytometry profiles of WT (BY4741), elg1Δ, pol30-D150E, and double mutants after factor arrest and release reveal defects in the ability of elg1Δ mutant cells to postpone the cell cycle, as indicated by red arrows. (B) Expression of nascent RNAs during S phase. The mRNA synthesis rate of early-replicating genes relative to late-replicating genes was increased during S phase in the elg1Δ mutant cells compared to WT cells. (C) Transcriptional buffering was rescued in the double-mutant cells. (D) Representative flow cytometry profiles of elg1Δ mutant and double mutant (elg1Δ and pol30-D150E) on the RDKY5964 genetic background after factor arrest and release. (E) Expression of nascent RNAs during S phase shows loss of transcriptional buffering in the elg1Δ mutant on RDKY5964 background. (F) Rescue of phenotype in the double-mutant cells. (G) The proposed model for the role of Elg1 in genetic buffering during DNA replication. The unloading of PCNA by ELG1 protein is critical for pol II to transcribe the mRNA after DNA replication.
Because Elg1 is an PCNA unloader (28), we considered the possibility that it regulates expression homeostasis during S phase by modulating PCNA occupancy on replicating chromatin. To test this idea, we took advantage of a disassembly-prone PCNA mutant with a substitution of aspartic acid-150 to glutamic acid (PCNA-D150E) (31). This mutant has been shown to rescue PCNA-mediated DNA repair (31) and replication-coupled nucleosome assembly (32) that are disrupted in the absence of Elg1. To our surprise, the PCNA-D150E mutation completely restored the expression homeostasis of elg1Δ cells to a level similar to WT (Figure 3C and F). Of note, the PCNA-D150E mutation alone did not exhibit any observable defect in S phase transcription buffering (Supplementary Figure S2).
The effect of Elg1 on PCNA loading during S phase has been carefully detailed (33,34). Normally, PCNA is loaded close to the replication origin in early S phase, and it is unloaded behind the replication forks (12). However, in the absence of Elg1, PCNA is retained on replicated chromatin (33). Thus, the timely removal of PCNA during DNA replication is disrupted in elg1Δ cells, causing accumulation of PCNA at origins, and this phenomenon is likely due to a delay in unloading (33). Even without Elg1, PCNA is unloaded eventually; however, unloading in the absence of Elg1 occurs in late S phase (33,35).
The results of our transcriptional profiling combined with the putative role of Elg1 in modulating PCNA dynamics during S phase (33,34) suggest that PCNA may function to limit RNA pol II loading during replication. In WT cells (Figure 3G, left panel), PCNA is loaded at early replicated genes and may be quickly unloaded by Elg1; this unloading of PCNA is required for the reloading of RNA pol II, which has been evicted by the replication fork (36) and allows the resumption of transcription at these regions. PCNA unloading and RNA pol II reloading also occurs at late firing origins during late S phase. Therefore, transcriptional homeostasis is maintained by the efficient PCNA loading/unloading and RNA pol II reloading throughout the S phase. In elg1Δ cells (Figure 3G, right panel), the accumulation of PCNA at the early replication chromatin would inhibit the reloading of RNA pol II, consistent with the observed reduction of mRNA synthesis at early S phase (Figure 3B and E). At late S phase, the reloading of the RNA pol II at late replicated genes is prevented by the accumulation of PCNA, but the early replicated genes have resumed transcription on both newly synthesized sister DNA strands due to unloading of PCNA through an Elg1-independent mechanism (33,35). The delay in PCNA unloading may thus introduce a strong imbalance in transcription of early and late replicating genes (Figure 3B and E) at late S phase. Overall, iTARGEX identified ELG1 as a novel regulator of transcriptional buffering during S phase, which is probably based on its PCNA unloading activity.
iTARGEX predicts novel factors involved in protein turnover
To further assess the applicability of iTARGEX, we performed a similar analysis for another biological trait. This time, we chose protein turnover, the balance between synthesis and degradation (37) because dysfunction in protein turnover is associated with aging and diseases, such as Parkinson's and Alzheimer's diseases (38). Despite the clear importance of protein turnover, our knowledge of what drives proteostasis is still limited.
We reasoned that the deletion of protein turnover regulators (either negative or positive regulators) may trigger compensatory feedback responses on gene expression to buffer the impact on protein abundance. Thus, we applied iTARGEX to examine the correlations between protein half-lives (22) and expression changes in the single-gene deletion mutants. The top 20 candidate regulators predicted by iTARGEX are listed in Table 2, along with their association test results and their biological functions. Notably, a few candidate regulators, such as SSN3 and YDR306C, were reported to regulate protein stabilization processes, while other candidate regulators are associated with the transcriptional regulation (e.g. UPF3, SRB8, SSN8, AZF1, CAF40) and post-transcriptional regulation (e.g. MRN1, UTP30). Thus, processes upstream of protein biosynthesis may also act as determinants of protein turnover rate. The candidate regulators were ranked based on their q-values (Figure 4A), and we selected the MRN1- and UPF1-deletion strains for further analysis, as these strains were two top-ranked candidates exhibiting positive correlation with protein half-lives. The deletions of RAM1 and SSN3 were the top-ranked negative correlations, so we also determined the protein turnover rates in those strains. We metabolically labeled the WT and mutant yeast cells with heavy isotope lysine and then replaced the culture medium with one containing normal lysine. We then analyzed the decay of the heavy lysine signal in the proteome over time by high-resolution mass spectrometry-based proteomics, as reported previously (22). We found that all mutants of interest exhibited higher total retention rates of heavy lysine at 60 min after release into normal lysine medium, indicating that the average rates of protein turnover in mutant cells were all slower than that in WT cells (Figure 4B, upper panel). This trend was further exacerbated in all tested mutants at 180 min after release (Figure 4B, lower panel), with the exception of mrn1Δ cells. We next analyzed the contribution of each protein to the overall protein turnover rate by calculating their degradation constant (Kdp), using a protocol adapted from a previous study (23) (Figure 4C). The results indicated that all tested gene deletions influenced turnover of the majority of yeast proteins, suggesting that these genes encode general regulators to protein half-life. In sum, these results demonstrate that iTARGEX is capable of identifying novel regulators of protein turnover.
Table 2.
Summary of iTARGEX results from the association analysis for protein turnover rate and regulators of protein turnover. WPCC: Weighted Pearson's correlation coefficient; q-values are log-transformed;  : the proportion of genes estimated to be significantly associated by the EM algorithm. Biological functions were taken from the Saccharomyces Genome Database (SGD) (39)
: the proportion of genes estimated to be significantly associated by the EM algorithm. Biological functions were taken from the Saccharomyces Genome Database (SGD) (39)
| iTARGEX | |||||
|---|---|---|---|---|---|
| Regulators | WPCC | –log10(q-value) | π | Biological functions | Validation in this study |
| MRN1 | 0.225 | 35.444 | 0.921 | Involved in translational regulation | v |
| UPF3 | 0.210 | 29.528 | 0.796 | Involved in decay of mRNA decay pathway | v |
| PDE2 | 0.201 | 29.028 | 0.924 | High-affinity cyclic AMP phosphodiesterase | |
| YPR153W | 0.191 | 25.323 | 0.896 | ER chaperone for nutrient permeases | |
| CAC2 | 0.182 | 24.203 | 0.907 | Involved in DNA replication-dependent nucleosome assembly | |
| SSN3 | −0.179 | 22.991 | 0.904 | Involved in protein destabilization | v |
| YDR306C | −0.177 | 21.054 | 0.947 | Involved in ubiquitin-dependent protein catabolic process | |
| MEI5 | 0.173 | 20.731 | 0.931 | Involved in meiotic DNA recombinase assembly | |
| SRB8 | −0.172 | 20.668 | 0.908 | Involved in regulation of transcription by RNA polymerase II | |
| RAM1 | −0.181 | 20.616 | 0.896 | Involved in protein farnesylation | v |
| SSN8 | −0.168 | 19.636 | 0.909 | Involved in regulation of transcription by RNA polymerase II | |
| ELP4 | 0.167 | 19.002 | 0.917 | Required for Elongator structural integrity | |
| ATG23 | 0.169 | 18.314 | 0.913 | Involved in positive regulation of macroautophagy | |
| AZF1 | 0.161 | 18.053 | 0.935 | Involved in regulation of transcription by RNA polymerase II | |
| CAF40 | −0.166 | 17.581 | 0.886 | Involved in positive regulation of transcription elongation | |
| IRA2 | 0.160 | 16.808 | 0.815 | Involved in negative regulation of Ras protein signal transduction | |
| DDR48 | 0.159 | 16.471 | 0.945 | Involved in DNA repair | |
| UTP30 | 0.156 | 16.365 | 0.924 | Involved in ribosomal small subunit biogenesis | |
| SAM4 | 0.155 | 16.135 | 0.940 | Cause abnormal vacuole, which is main site for protein turnover | |
Figure 4.

iTARGEX predicts novel factors involved in protein turnover. (A) Volcano-like plot shows the candidate regulators associated with protein turnover rate predicted by iTARGEX. (B) Density distributions of the synthesized proteins that incorporate the heavy label at 60 min and 180 min. Overall, the new protein synthesis was postponed in four mutant strains compared to wild type. (C) The degradation constants (Kdp) for each protein were calculated from log2(H/H+L) ratios at two time points using linear regression. The dashed lines denote the mean Kdp values, derived by averaging all proteins. In comparison with the mean Kdp of the wild-type strain, the P-values of strains showing significant differences were 10–28, 0, 0, and 0 for mrn1Δ, upf3Δ, ram1Δ and ssn3Δ mutant strains, respectively, according to the Mann–Whitney U test.
DISCUSSION
In this study, we introduce iTARGEX, a bioinformatics tool that predicts genes involved in the regulation of the biological traits by using association tests with fine statistical inference. Previously, researchers identified regulators that maintain expression homeostasis during DNA replication (16,24) by performing simple correlations on a published dataset of transcriptional profiles for deletion mutants in the budding yeast (9). However, the major weakness of the previous analysis was that it lacked a rigorous procedure to select plausible candidates from the association tests, and therefore, the logical inferences are difficult to recapitulate. For instance, the authors selected two regulators for further experiments, SET1 (PCC = −0.053, –log10(q-value) = 1.56) and SWD3 (PCC = −0.022, -log10(q-value) = 0.00), which were not significantly associated using the simple correlation test (24). In this study, iTARGEX was used to consider not only simple correlations but also partial correlations, providing an effective and automated approach to identify associations among sub-populations, using a mixture model with EM search algorithm. Notably, iTARGEX could identify seven out of nine important regulators (except for SET1 and SWD3) reported by previous studies (16,24) in addition to novel regulators, such as ELG1, with promising levels of statistical significance (Table 1). In addition, iTARGEX estimated the association patterns of mutant regulators with slightly lower correlation coefficients and smaller q-values compared to simple correlations (Supplementary Figure S3). Therefore, iTARGEX is not expected to overestimate correlations. We further examined how Elg1 might function in gene expression buffering during DNA replication by a series of genetic experiments that involved crossing different mutants. With these experiments, we demonstrated that unloading PCNA from duplicated chromosomes is probably critical for maintaining expression homeostasis in the budding yeast (Figure 3). Importantly, the involvement of Elg1 as a regulator of expression homeostasis was discovered solely based on statistical inference from heterogeneous data and not prior biological knowledge. The use of an explicit statistical framework allows our method to account for associated relationships while accommodating noisy expression data.
In addition, we used iTARGEX to measure associations between gene expression profiles and protein turnover rate, assuming the effect of transcriptional changes is linked to the translation or/and degradation for the continual renewal of proteins. According to our follow-up SILAC experiments, the top-ranked candidate regulators indeed affected the synthesis of most yeast proteins (Figure 4). These observations further suggest that iTARGEX is reliable enough to predict novel regulators of various cellular functions. Notably, negative or positive correlations between expression profile and protein turnover rate did not imply the direction of regulation for degradation or synthesis of proteins. Since our approach utilizes correlation tests, the correlation outputs cannot carry any causative inferences. Therefore, understanding the potential roles of candidate genes requires researchers to rely on current knowledge and experience to design further experiments that reveal the mechanisms underlying identified correlations.
Other phenotypes of interest (shown in Figure 1A) are also worthy of exploration. We have provided lists of potential candidates for all the given phenotypes on GitHub (https://github.com/bio-it-station/iTARGEX), which will allow yeast biologists to generate novel hypotheses and investigate molecular mechanisms beyond the list of regulators. For example, gene expression noise is the outcome of intrinsically stochastic complex molecular interactions (40), so it is not easily analyzed to pinpoint potential regulators without performing large-scale biological screening experiments. We ran iTARGEX to analyze associations between the Deletome data and gene expression noise, which is represented as the coefficient of variation of gene expression from four independent microarray experiments (41). Such an associative study can identify potential regulators that affect the variability of expression and the intrinsic noise levels for other genes.
Our work has some limitations related to the development of a novel bioinformatics tool based on an EM algorithm. As a likelihood-based method, the stochastic convergence procedure of the EM approach can handle the approximation of two mixture models to efficiently estimate the regression coefficients of the subgroups (42). However, a major documented drawback of the EM algorithm is the need for good initial values to avoid trapping in local optima (43). We compared random initialization with alternative procedures by independently running the stochastic EM algorithm 20 times, using a  estimated from the slope of linear regression on the replication dataset or constraining the slope of another component as zero. In doing so, we found that the lists of candidate regulators were the same when using random assignments or initial
estimated from the slope of linear regression on the replication dataset or constraining the slope of another component as zero. In doing so, we found that the lists of candidate regulators were the same when using random assignments or initial  across 20 runs of implementations (Supplementary Table S1). According to the similarities between estimated proportions of subpopulations for the component with significant association in all the candidate regulators, use of the estimated
across 20 runs of implementations (Supplementary Table S1). According to the similarities between estimated proportions of subpopulations for the component with significant association in all the candidate regulators, use of the estimated  for EM initialization is most likely to reach the global optima. Furthermore, the convergence time was slightly reduced when providing an initial
for EM initialization is most likely to reach the global optima. Furthermore, the convergence time was slightly reduced when providing an initial  . In addition to this test, we also assumed that one component of the subpopulation of data points was significantly associated and the rest of the points were not in our proposed statistical scenario. If we forced the fitting procedures of the EM algorithm to constrain the slope of non-associated component as zero, the outputs were inconsistent among the 20 experimental runs. In particular, the regulator RTT109, which has been documented to play a role in expression homeostasis (16), did not always pass the selection procedures of iTARGEX under these conditions. Finally, we expect that iTARGEX may not reliably find significant results when assessing partial associations based on small proportions of data points (e.g. 0.1), which are usually hard to estimate correctly. Hence, we propose that validation experiments should first be performed for regulators with significant correlations based on more than half of the data points.
. In addition to this test, we also assumed that one component of the subpopulation of data points was significantly associated and the rest of the points were not in our proposed statistical scenario. If we forced the fitting procedures of the EM algorithm to constrain the slope of non-associated component as zero, the outputs were inconsistent among the 20 experimental runs. In particular, the regulator RTT109, which has been documented to play a role in expression homeostasis (16), did not always pass the selection procedures of iTARGEX under these conditions. Finally, we expect that iTARGEX may not reliably find significant results when assessing partial associations based on small proportions of data points (e.g. 0.1), which are usually hard to estimate correctly. Hence, we propose that validation experiments should first be performed for regulators with significant correlations based on more than half of the data points.
Imbalanced research efforts focusing on a minority of known human genes have resulted in an incomplete catalog of gene characteristics and features (44). Since the inference method presented in this study is not limited to use in budding yeast studies, our iTARGEX tool and its conceptual design can be easily extended to perform association tests with other organisms and perturbations, such as RNA-mediated interference or CRISPR-CAS9 screen systems, with transcriptome data (45,46). To facilitate the pace of discovery for further exploration, iTARGEX is sufficiently flexible to analyze input datasets of transcriptional profiles and trait data by researchers with diverse interests. In summary, the development of iTARGEX opens a window of opportunity for future work to identify novel regulators without the limitation of prior gene annotations.
DATA AVAILABILITY
The results published here are available on the BioProject with the accession number PRJNA701566 and PRIDE with the accession number PXD024258.
BioProject web site: https://www.ncbi.nlm.nih.gov/bioproject/.
PRIDE web site https://www.ebi.ac.uk/pride/.
The source code for the iTARGEX is available on GitHub (https://github.com/bio-it-station/iTARGEX).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Takashi Kubota for giving us yeast strains, including RDKY5964 pol30::POL30(TRP1), RDKY5964 pol30::pol30-D150E(TRP1), RDKY5964 pol30::POL30(TRP1) elg1Δ::kanMX4, and RDKY5964 pol30::pol30-D150E(TRP1) elg1Δ::kanMX4. We also thank the Institute of Cellular and Organismic Biology (ICOB) for providing the BD LSRII system, Shy-Yun Tung at the Institute of Molecular Biology for RNA-sequencing, and Lin ShuYu of Academia Sinica common MS Facilities.
Contributor Information
Jia-Hsin Huang, Institute of Information Science, Academia Sinica, Taipei 115, Taiwan.
You-Rou Liao, Institute of Cellular and Organismic Biology, Academia Sinica, Taipei 115, Taiwan.
Tzu-Chieh Lin, Institute of Information Science, Academia Sinica, Taipei 115, Taiwan.
Cheng-Hung Tsai, Institute of Information Science, Academia Sinica, Taipei 115, Taiwan.
Wei-Yun Lai, Institute of Information Science, Academia Sinica, Taipei 115, Taiwan.
Yang-Kai Chou, Institute of Information Science, Academia Sinica, Taipei 115, Taiwan.
Jun-Yi Leu, Institute of Molecular Biology, Academia Sinica, Taipei 115, Taiwan.
Huai-Kuang Tsai, Institute of Information Science, Academia Sinica, Taipei 115, Taiwan.
Cheng-Fu Kao, Institute of Cellular and Organismic Biology, Academia Sinica, Taipei 115, Taiwan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Academia Sinica [AS-TP-107-ML06 to J.-Y.L., H.-K.T., C.-F.K.]; Taiwan Ministry of Science and Technology [MOST108-2221-E-001-014-MY3 to H.-K.T.]. Funding for open access charge: Academia Sinica [AS-TP-107-ML06].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ritchie M.D., Holzinger E.R., Li R., Pendergrass S.A., Kim D.. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015; 16:85–97. [DOI] [PubMed] [Google Scholar]
- 2. Chen H., Wu C.I., He X.. The genotype–phenotype relationships in the light of natural selection. Mol. Biol. Evol. 2018; 35:525–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kita R., Venkataram S., Zhou Y., Fraser H.B.. High-resolution mapping of cis-regulatory variation in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E10736–E10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brem R.B., Yvert G., Clinton R., Kruglyak L.. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002; 296:752–755. [DOI] [PubMed] [Google Scholar]
- 5. Brem R.B., Storey J.D., Whittle J., Kruglyak L.. Genetic interactions between polymorphisms that affect gene expression in yeast. Nature. 2005; 436:701–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scherens B., Goffeau A.. The uses of genome-wide yeast mutant collections. Genome Biol. 2004; 5:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drinnenberg I.A., Weinberg D.E., Xie K.T., Mower J.P., Wolfe K.H., Fink G.R., Bartel D.P.. RNAi in budding yeast. Science. 2009; 326:544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dicarlo J.E., Norville J.E., Mali P., Rios X., Aach J., Church G.M.. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013; 41:4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kemmeren P., Sameith K., van de Pasch L.A.L., Benschop J.J., Lenstra T.L., Margaritis T., O’Duibhir E., Apweiler E., van Wageningen S., Ko C.W.et al.. Large-Scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell. 2014; 157:740–752. [DOI] [PubMed] [Google Scholar]
- 10. Kitano H. Biological robustness. Nat. Rev. Genet. 2004; 5:826–837. [DOI] [PubMed] [Google Scholar]
- 11. Masel J., Siegal M.L.. Robustness: mechanisms and consequences. Trends Genet. 2009; 25:395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Benaglia T., Chauveau D., Hunter D.R., Young D.S.. Mixtools: an R package for analyzing finite mixture models. J. Stat. Softw. 2009; 32: 10.18637/jss.v032.i06. [DOI] [Google Scholar]
- 13. Moon T.K. The expectation-maximization algorithm. IEEE Signal Process. Mag. 1996; 13:47–60. [Google Scholar]
- 14. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Voichek Y., Bar-Ziv R., Barkai N.. Expression homeostasis during DNA replication. Science. 2016; 351:1087–1090. [DOI] [PubMed] [Google Scholar]
- 17. Yabuki N., Terashima H., Kitada K.. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells. 2002; 7:781–789. [DOI] [PubMed] [Google Scholar]
- 18. Rosebrock A.P. Synchronization and arrest of the budding yeast cell cycle using chemical and genetic methods. Cold Spring Harb. Protoc. 2017; 2017:47–52. [DOI] [PubMed] [Google Scholar]
- 19. Collart M.A., Oliviero S.. Preparation of yeast RNA. Curr. Protoc. Mol. Biol. 2001; Chapter 13:Unit13.12. [DOI] [PubMed] [Google Scholar]
- 20. Bonnet J., Wang C.-Y., Baptista T., Vincent S.D., Hsiao W.-C., Stierle M., Kao C.-F., Tora L., Devys D.. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 2014; 28:1999–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duffy E.E., Simon M.D.. Enriching s4U-RNA using methane thiosulfonate (MTS) chemistry. Curr. Protoc. Chem. Biol. 2016; 8:234–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Christiano R., Nagaraj N., Fröhlich F., Walther T.C.. Global proteome turnover analyses of the yeasts S. cerevisiae and S. pombe. Cell Rep. 2014; 9:1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M.. Global quantification of mammalian gene expression control. Nature. 2011; 473:337–342. [DOI] [PubMed] [Google Scholar]
- 24. Voichek Y., Mittelman K., Gordon Y., Bar-Ziv R., Smit Lifshitz, Shenhav D., Barkai N.. Epigenetic control of expression homeostasis during replication is stabilized by the replication checkpoint. Mol. Cell. 2018; 70:1121–1133. [DOI] [PubMed] [Google Scholar]
- 25. Mayer M.L., Gygi S.P., Aebersold R., Hieter P.. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol. Cell. 2001; 7:959–970. [DOI] [PubMed] [Google Scholar]
- 26. Bylund G.O., Burgers P.M.J.. Replication protein A-directed unloading of PCNA by the Ctf18 cohesion establishment complex. Mol. Cell. Biol. 2005; 25:5445–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Farina A., Shin J.-H., Kim D.-H., Bermudez V.P., Kelman Z., Seo Y.-S., Hurwitz J.. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J. Biol. Chem. 2008; 283:20925–20936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parnas O., Zipin-Roitman A., Pfander B., Liefshitz B., Mazor Y., Ben-Aroya S., Jentsch S., Kupiec M.. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010; 29:2611–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shiomi Y., Nishitani H.. Alternative replication factor C protein, Elg1, maintains chromosome stability by regulating PCNA levels on chromatin. Genes Cells Devoted Mol. Cell. Mech. 2013; 18:946–959. [DOI] [PubMed] [Google Scholar]
- 30. Lee K., Fu H., Aladjem M.I., Myung K.. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 200:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson C., Gali V.K., Takahashi T.S., Kubota T.. PCNA retention on DNA into G2/M phase causes genome instability in cells lacking Elg1. Cell Rep. 2016; 16:684–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Janke R., King G.A., Kupiec M., Rine J.. Pivotal roles of PCNA loading and unloading in heterochromatin function. Proc. Natl. Acad. Sci. 2018; 115:E2030–E2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kubota T., Katou Y., Nakato R., Shirahige K., Donaldson A.D.. Replication-coupled PCNA unloading by the Elg1 complex occurs genome-wide and requires okazaki fragment ligation. Cell Rep. 2015; 12:774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu C., Gan H., Han J., Zhou Z.-X., Jia S., Chabes A., Farrugia G., Ordog T., Zhang Z.. Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol. Cell. 2014; 56:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kubota T., Nishimura K., Kanemaki M.T., Donaldson A.D.. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell. 2013; 50:273–280. [DOI] [PubMed] [Google Scholar]
- 36. Bar-Ziv R., Brodsky S., Chapal M., Barkai N.. Transcription factor binding to replicated DNA. Cell Rep. 2020; 30:3989–3995. [DOI] [PubMed] [Google Scholar]
- 37. Martin-Perez M., Villén J.. Determinants and regulation of protein turnover in yeast. Cell Syst. 2017; 5:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Balch W.E., Morimoto R.I., Dillin A., Kelly J.W.. Adapting proteostasis for disease intervention. Science. 2008; 319:916–919. [DOI] [PubMed] [Google Scholar]
- 39. Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R.et al.. Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res. 2012; 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elowitz M.B., Levine A.J., Siggia E.D., Swain P.S.. Stochastic gene expression in a single cell. Science. 2002; 297:1183–1186. [DOI] [PubMed] [Google Scholar]
- 41. Newman J.R.S., Ghaemmaghami S., Ihmels J., Breslow D.K., Noble M., DeRisi J.L., Weissman J.S.. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006; 441:840–846. [DOI] [PubMed] [Google Scholar]
- 42. Dempster A.P., Laird N.M., Rubin D.B.. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B Methodol. 1977; 39:1–22. [Google Scholar]
- 43. Baudry J.-P., Celeux G.. EM for mixtuRes. Stat. Comput. 2015; 25:713–726. [Google Scholar]
- 44. Stoeger T., Gerlach M., Morimoto R.I., Amaral L.A.N.. Large-scale investigation of the reasons why potentially important genes are ignored. PLOS Biol. 2018; 16:e2006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Datlinger P., Rendeiro A.F., Schmidl C., Krausgruber T., Traxler P., Klughammer J., Schuster L.C., Kuchler A., Alpar D., Bock C.. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods. 2017; 14:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gasperini M., Hill A.J., McFaline-Figueroa J.L., Martin B., Kim S., Zhang M.D., Jackson D., Leith A., Schreiber J., Noble W.S.et al.. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell. 2019; 176:377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The results published here are available on the BioProject with the accession number PRJNA701566 and PRIDE with the accession number PXD024258.
BioProject web site: https://www.ncbi.nlm.nih.gov/bioproject/.
PRIDE web site https://www.ebi.ac.uk/pride/.
The source code for the iTARGEX is available on GitHub (https://github.com/bio-it-station/iTARGEX).