


Abstract
Technological advances in rare DNA mutations detection have revolutionized the diagnosis and monitoring of tumors, but they are still limited by the lack of supersensitive and high-coverage procedures for identifying low-abundance mutations. Here, we describe a single-tube, multiplex PCR-based system, A-Star, that involves a hyperthermophilic Argonaute from Pyrococcus furiosus (PfAgo) for highly efficient detection of rare mutations beneficial from its compatibility with DNA polymerase. This novel technique uses a specific guide design strategy to allow PfAgo selective cleavage with single-nucleotide resolution at 94°C, thus mostly eliminating wild-type DNA in the denaturation step and efficiently amplifying rare mutant DNA during the PCR process. The integrated single-tube system achieved great efficiency for enriching rare mutations compared with a divided system separating the cleavage and amplification. Thus, A-Star enables easy detection and quantification of 0.01% rare mutations with ≥5500-fold increase in efficiency. The feasibility of A-Star was also demonstrated for detecting oncogenic mutations in solid tumor tissues and blood samples. Remarkably, A-Star achieved simultaneous detection of multiple oncogenes through a simple single-tube reaction by orthogonal guide-directed specific cleavage. This study demonstrates a supersensitive and rapid nucleic acid detection system with promising potential for both research and therapeutic applications.
INTRODUCTION
The detection of rare polymorphic alleles is becoming increasingly relevant for the early diagnosis and monitoring of a variety of tumors (1,2). Clinical detection methods currently available for rare mutations, including single nucleotide variations (SNVs) and insertion/deletion (indel) mutations, involve either sequencing or PCR (3,4). Compared with authentication by sequencing DNA fragments, allele-specific diagnostic PCR, using methods such as the amplification refractory mutation system (ARMS), blocker displacement amplification (BDA), co-amplification at lower denaturation temperature polymerase chain reaction (COLD-PCR), restriction endonuclease-mediated selective PCR, nuclease-assisted minor-allele enrichment with probe-overlap (NaME-PrO) and digital PCR, is not only simpler and time-saving but also practical and effective (5–12). For these methods, the detection of extremely rare variant alleles within a complex mixture of DNA molecules has attracted increasing attention with the primary aim at solving the technical problems associated with the strict requirements for both precise single-nucleotide resolution and simple multiplex detection, especially for detecting cancer-relevant DNA biomarkers in patients (13).
Endonucleases with sequence-specific catalytic capabilities are powerful tools for recognizing nucleic acid targets and informing on subsequent detection design (4,14,15). However, conventional restriction endonucleases are known to be limited by their specific target sequence. Recently, programmable CRISPR-Cas systems have been developed as next-generation diagnostic platforms (16–18). These strategies are primarily based on Cas9′s cleavage activity (19–21), or the collateral cleavage activity of the Cas12 or Cas13a effectors (22,23). When combined with the isothermal amplification process to provide higher detection sensitivity, CRISPR-Cas systems detected SNV levels down to 1% (e.g. BRAF target) or 0.1% (e.g. EGFR target) (24–26). However, all these approaches are hampered by the strict requirement of the cleavage site preference being in the vicinity of the target sequence and by the designed reporter system (21,24–28). In addition, when multiplex methods are used to target multiple genomic loci simultaneously, the need to tediously screen for orthogonal CRISPR enzymes remains (28). Another impediment of the conventional methodology is that most Cas effectors are used in combination with an isothermal amplification step to boost the detection sensitivity, such as the recombinase polymerase amplification (RPA), which although boosts the detection sensitivity from picomolar to attomolar concentrations, but the target RNA is converted from the pre-amplified circulating tumour DNA (ctDNA) by RT-RPA, therefore resulting in high costs and extra laboriousness (24–25,28).
Thermophilic Argonaute (Ago) proteins provide a potential means of circumventing the above mentioned limitations through their unique properties, including no requirement for a specific sequence motif, such as the protospacer adjacent motif (PAM) used by Cas9 and Cas12 for cleavage. Instead, several identified Agos can specifically cleave target nucleic acids via base-pairing with small guide nucleic acids between positions 10 and 11 of the guide (29–34). Accordingly, the Ago of the hyperthermophile Pyrococcus furiosus (PfAgo) was designed as a programmable DNA-guided artificial restriction enzyme that can recognize and cleave target DNA (tDNA) with a pair of optimal 5′-phosphorylated guide DNA (gDNA) at temperatures as high as 95°C (35). In addition, the position of the mutation site within the gDNA markedly affects the target cleavage efficiency (32). Recently, a method to detect SNV derived from Thermus thermophilus Ago (TtAgo) was developed, in which the enriched SNV products were generated from PCR amplification and then treated by discrimination cleavage dependent on mismatched nucleotide positions of 5′-phosphorylated gDNA between tDNAs (36). The Ago protein in these methods is always used as an efficient nuclease to recognize the target; but its tolerance at extremely high temperatures and programmable DNA recognition ability have been less thoroughly explored. Therefore, developing a new detection strategy that combines the high specificity and thermostability of Ago with the great efficiency of PCR amplification is merited. The catalytic ability of PfAgo to cleave single-stranded DNA (ssDNA) at 95°C suggests that it is capable of cleaving unwound double-stranded DNA (dsDNA) during the first step of PCR denaturation. Hence, the combination of precise cleavage, as directed by the gDNA, and the ability to function at the high temperatures required in PCR should grant PfAgo the capability to enrich rare mutations by precisely discriminating the wild-type (WT) DNA from its rare variants, leading to a new tool to detect rare mutations with greater precision and efficiency.
In the present work, we report the development of A-Star (Ago-directed Specific Target enrichment and detection), a simple but supersensitive single-tube PCR system. In brief, a rationally designed gDNA with an extra introduced mismatch and diphosphorylation modification directs specific cleavage of PfAgo on WT sequences in the denaturation step of PCR, resulting in progressive and rapid enrichment of rare variant alleles in the ensuing amplification step. The amplified products can subsequently be used for various downstream detections, including Sanger sequencing and TaqMan quantitative real-time PCR (TaqMan qRT-PCR), which were performed in this study. Our results demonstrate the greater efficiency, by orders of magnitude, of the single-tube system compared to a divided system separating the process of cleaving WT DNA from the process of variant allele amplification. We selected five mutations identified as highly relevant to diagnosis and monitoring of clinical cancers, such as non-small cell lung cancer and lung adenocarcinoma (Supplementary Table S1), and then verified reliable detection of A-Star for target enrichment of these rare mutations. In addition, A-Star illustrated the detection sensitivity of testing single-digit copies with mock cell-free DNA (cfDNA) and clinical human samples. To the best of our knowledge, this study presents the first instance where a readily available and programmable endonuclease with high sequence specificity has been integrated into a broadly used PCR procedure, which, we believe, could soon become a super-sensitive, rapid and inexpensive method for rare SNV detection for a wide array of applications.
MATERIALS AND METHODS
Nucleic acid preparation
The ssDNA target, gDNA and primers were synthesized commercially (Sangon Biotech, China). The ∼600 bp dsDNA target was synthesized by GenScript (Nanjing, China) in the form of a pET-28a-derived plasmid. The plasmid DNA was extracted using a Plasmid DNA MiniPreps Kit (Generay, China). The 130–160 bp dsDNA target was obtained by amplification of the corresponding plasmid with the primers designed using NCBI Primer-BLAST with the following parameters: amplicon size between 130 and 160 nt, primer melting temperatures between 50°C and 60°C, and primer sizes between 18 and 25 nt. For PCR amplification, 2× PCR Precision™ MasterMix (Abm, Canada) was used. The target dsDNA fragments for the titration experiments were quantified using a PikoGreen dsDNA Quantitative Kit (Life iLab Biotech, China). All of the nucleic acids used in this study are listed in Supplementary Table S2–S5.
PfAgo expression and purification
A codon-optimized version of the PfAgo gene was synthesized by GenScript (Nanjing, China), and was cloned into the pET28a plasmid to construct pEX-PfAgo with an N-terminal His-tag. The expression plasmid was transformed into Escherichia coli BL21(DE3) cells. A 5 ml seed culture was grown at 37°C in LB medium with 50 μg/ml kanamycin and was subsequently transferred to 1 l of LB in a shaker flask containing 50 μg/ml kanamycin. The cultures were incubated at 37°C until the OD600 reached 0.8–1.0, and protein expression was then induced by the addition of isopropyl β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, followed by incubation for 16 h at 20°C. Cells were harvested by centrifugation for 20 min at 6000 rpm, and the cell pellet was collected for later purification. Cell pellets were resuspended in lysis buffer (20 mM Tris/HCl, 1 M NaCl, pH 8.0) and then disrupted using a High-Pressure Homogenizer at 600–800 bar for 3 min (Gefran, Italy). The lysates were centrifuged for 30 min at 12 000 rpm at 4°C, after which the supernatants were subjected to Ni-NTA affinity purification with elution buffer (20 mM Tris/HCl, 1 M NaCl, 200 mM imidazole, pH 8.0). Further gel filtration purification using a Superdex 200 (GE Tech, USA) was carried out with elution buffer (20 mM Tris/HCl, 1 M NaCl, pH 8.0). The fractions resulting from gel filtration were analyzed by SDS-PAGE, and fractions containing the protein were flash frozen at −80°C in storage buffer (20 mM Tris–HCl, pH 8.0, 250 mM NaCl, 15% (v/v) glycerol).
DNA cleavage assays
Generally, PfAgo-mediated cleavage assays were carried out in reaction buffer (15 mM Tris/HCl pH 8.0, 250 mM NaCl, and 0.5 mM MnCl2). For ssDNA cleavage, 0.2 μM PfAgo, 2 μM gDNA and 0.8 μM ssDNA target were mixed in reaction buffer and then incubated for 15 min at 95°C in a thermocycler (Eppendorf, Germany). Following high-temperature incubation, the samples were cooled down by slowly lowering the temperature at a rate of 0.1°C/s until it reached 10°C. The addition of loading buffer (95% formamide, 0.5 mmol/l EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol FF) at a 1:1 ratio (v/v) stopped the reactions. The samples were then separated in 16% denaturing polyacrylamide gels and analyzed by staining with GelRed (Biotium, USA). The nucleic acids were visualized using a Tanon 3500BR (Shanghai, China), and data were analyzed using Quantity One (Bio-Rad, USA). For the dsDNA cleavage assays, 0.16 μM PfAgo, 2 μM gDNAs and 58 nM dsDNA target were mixed in reaction buffer before incubation for 15 min at 95°C in a thermocycler (Eppendorf, Germany). Following this high-temperature incubation step, the samples were cooled down by slowly lowering the temperature at a rate of 0.1°C/s until it reached 10°C. The reactions were quenched with 5 × DNA loading buffer (Generay, China) and analyzed using 2% agarose gels. The gels were visualized with a Tanon 3500BR (Shanghai, China), and the data were analyzed using Quantity One (Bio-Rad, USA). The cleavage (%) was defined as the ssDNA target cleavage percentage calculated from gels quantitative analysis. The specificity index of WT/SNV is the ratio of the cleavage (%) of WT to that of SNV directed by the mismatched gDNAs.
PfAgo activity at different temperatures
The effects of temperature on PfAgo activity mediated by different guides were tested across a range of temperatures from 60 to 99°C. Generally, 0.2 μM PfAgo, 2 μM ssDNA guide and 0.8 μM ssDNA target were mixed in reaction buffer, and the mixture was then incubated for 15 min at 60.0, 64.7, 70.0, 75.0, 80.0, 86.0, 90.7, 95.4, 98.7 or 99.0°C. The samples were resolved in 16% denaturing polyacrylamide gels. The gels were stained using GelRed (Biotium, USA). The nucleic acids were visualized using a Tanon 3500BR (Shanghai, China), and the data were analyzed using Quantity One (Bio-Rad, USA).
Preparation of variant allele fraction (VAF) samples
Approximately 160 bp WT and mutant DNA fragments for PIK3CA E545K, KRAS G12D, EGFR L861Q, EGFR S768I, and EGFR delE746-A750 were obtained by PCR amplification. The PCR products were purified using a GeneJET Gel Extraction Kit (Thermo Scientific, USA) and quantified with PikoGreen dsDNA quantitative kits (Life iLab Bio, China). These wild-type and mutant DNA fragments were mixed to obtain VAFs of 1%, 0.1%, 0.01% for each SNV allele in a total DNA concentration of 10 nM to evaluate the performance of A-Star in enriching mutant DNA.
Coupling of cleavage and enrichment, and uncoupling A-Star
For the PfAgo-coupled PCR process, 30 nM PfAgo, 600 nM gDNA, 10 nM target DNA with 0.1% and 1% VAF of KRAS G12D, 200 nM forward and reverse primers, and 500 μM Mn2+ were mixed in 2 × PCR Taq MasterMix (Abm, Canada). Enrichment proceeded in a Mastercycler® RealPlex instrument (Eppendorf, Germany) with a temperature profile of 94°C for 3 min, followed by 25 cycles of amplification (94°C for 30 s, 55°C for 30 s and 72°C for 20 s) and a final 72°C extension for 1 min. For the uncoupling process, the reaction mixture was the same as that of the PfAgo-coupled PCR process. Enrichment proceeded in the same instrument with a temperature profile of 94°C for 15 min, then the cleavage products were cleaned up, followed by 25 cycles of amplification as in the procedure described above.
A-Star for variant alleles enrichment
For the PfAgo-coupled PCR system, 30 nM PfAgo, 600 nM gDNA, 10 nM target DNA with different VAFs, 200 nM each of forward and reverse primers and 500 μM Mn2+ were mixed in 2× PCR Taq MasterMix (Abm, Canada). Enrichment proceeded in a Mastercycler® RealPlex instrument (Eppendorf, Germany) with a temperature profile of 94°C for 3 min, followed by 25 cycles of amplification (94°C for 30 s, 55°C for 30 s and 72°C for 20 s) and a final 72°C extension for 1 min. For the limit of detection (LOD) assays, 1%, 0.1% and 0.01% VAFs samples prepared by mixing WT amplicons and SNV amplicons (WT plus SNV at a final concentration of 10 nM) were served as input.
For variant allele enrichment in mock cell cfDNA, mock cfDNA standards simulating actual patient cfDNA samples were purchased from a commercial vendor (Horizon Discovery Group, UK). These standards were provided for each target (PIK3CA E545K, KRAS G12D and EGFR del) with different VAFs (0.1%, 1% and 5%) at a concentration of 20 ng/μl. Then, 1.67 μl of each of these standards was used as input to perform pre-amplification and the products were subsequently examined using A-Star. The pre-amplification procedure was carried out in a 25 μl reaction volume using 33.3 ng of mock genomic DNA, 2 × PCR PCR Precision™ MasterMix (Abm, Canada), and 250 nM of the forward and reverse primers. Samples were placed in a Mastercycler® RealPlex instrument (Eppendorf, Germany) set at a temperature profile of 94°C for 3 min, followed by 30 cycles of amplification (94°C for 10 s, 55°C for 30 s and 72°C for 20 s) and a final 72°C extension for 1 min. Two microliters of the pre-amplification products were used as input to perform A-Star. The A-Star system consisted of 20–30 nM PfAgo, gDNAs at a concentration 20-fold higher than that of PfAgo, 200 nM of the forward and reverse primers, and 500 μM Mn2+, which were mixed in 2× PCR Taq MasterMix (Abm, Canada). Enrichment proceeded in a Mastercycler® RealPlex instrument (Eppendorf, Germany) as described above.
For variant allele enrichment in tissue samples, 33.3 ng of extracted genomic DNA from each tissue sample was used as the input to perform pre-amplification, and the product was then examined using A-Star as described above for the variant allele enrichment of mock cfDNA. For variant allele enrichment in cfDNA samples, 3.3 ng of extracted genomic DNA (due to the low concentration of extracted cfDNA) from each sample was used as the input to perform pre-amplification. Then 2 μl of the pre-amplification products were used as the input to perform A-Star. The A-Star system consisted of 10–20 nM PfAgo, gDNAs at a concentration 20-fold higher than that of PfAgo, 200 nM of the forward and reverse primers, and 500 μM Mn2+, which were mixed in 2× PCR Taq MasterMix (Abm, Canada). Enrichment proceeded in a Mastercycler® RealPlex instrument (Eppendorf, Germany) as described above.
Combined A-Star and TaqMan qRT-PCR approach in a single PCR
In this coupled system of A-Star with TaqMan qRT-PCR, the following components: 25 nM PfAgo, 500 nM gDNA, 1 nM target DNA with 1% KRAS G12D, 100 μM Mn2+, 250 nM primers and 200 nM probes were mixed with the AceQ® qPCR Probe Master Mix (Vazyme Biotech Co., Ltd, China). The reaction was carried out in a QuantStudio 5 Real-Time PCR System (Thermo Fisher, USA) with a temperature profile of 95°C for 8 min, followed by 40 cycles of amplification (95°C for 15 s, 61.5°C for 40 s).
Triplex variant alleles enrichment of A-Star
Triplex enrichment was performed in 25 μl reaction volumes with 200 nM PfAgo, each gDNA at 4,000 nM, 500 μM Mn2+, 200 nM of each forward and reverse primer, 0.4 mM dNTPs (Sangon, China), 0.5 μl of Taq DNA Polymerase (Abm, Canada), and three VAF targets each at a concentration of 10 nM in 10× PCR buffer (Abm, Canada). The reactions were carried out in a Mastercycler® RealPlex instrument (Eppendorf, Germany) with a temperature profile of 94°C for 5 min, followed by 25 cycles of amplification (94°C for 30 s, 55°C for 30 s, 72°C for 20 s) and a final 72°C extension for 1 min.
For the triplex SNV enrichment of mock cfDNA, cfDNA standards purchased from a commercial vendor (Horizon Discovery Group, UK) were used as the three targets (PIK3CA E545K, KRAS G12D and EGFR del) with different VAFs (0.1%, 1% and 5%) at concentrations of 20 ng/μl. Then 1.67 μl of these standards was used as the input to perform pre-amplification, followed by analysis using A-Star. The pre-amplification process was carried out in a 25 μl reaction volume using 33.3 ng of mock cfDNA, 2 × PCR Precision™ MasterMix (Abm, Canada), and 250 nM of each corresponding forward and reverse primer for the three targets. The Mastercycler® RealPlex instrument (Eppendorf, Germany) was programmed as follows: 94°C for 3 min, followed by 30 cycles of amplification (94°C for 10 s, 55°C for 30 seconds, and 72°C for 20 s), and a final 72°C extension for 1 minute. Then 2 μl of the pre-amplification product were used as the input to perform A-Star as described above for the SNV enrichment of mock cfDNA.
Sanger sequencing and TaqMan qRT-PCR verify enriched products
The A-Star enriched products were checked for quality and yield by running 5 μl of the products in 2.0% agarose gels, which were visualized on a Tanon 3500BR (Shanghai, China) before processing directly for Sanger sequencing (Sangon, China). The primers and probes for the targets (PIK3CA E545K, KRAS G12D, and EGFR del) were designed using Beacon Designer's standard assay design pipeline and ordered as individual primers and probes from Life Technologies (Thermo Fisher, USA).
To validate the enriched mutant products, assays were performed using AceQ® qPCR Probe Master Mix (Vazyme Biotech Co., Ltd, China), and the results were quantified in a StepOnePlus™ Real-Time PCR System (Thermo Fisher, USA) with the primer and probe sets (Supplementary Table S5) at final concentrations of 250 nM primers and 200 nM probes. The temperature profile for amplification consisted of an activation step at 95°C for 8 min, followed by 40 cycles of amplification (95°C for 15 s and 60°C for 40 s). To quantify the enriched mutant product, we prepared a series of standard solutions containing WT and SNV amplicon with concentrations ranging from 100 pM to 100 aM, which were quantified using the PikoGreen dsDNA quantitative kits (Life iLab Bio, China). Then the standard curves were generated by ploting the threshold cycle (Ct) values against the logarithm of the copy numbers. The enriched sample was analyzed by TaqMan qRT-PCR and then the copy number of WT or SNV is calculated using its Ct value based on the standard curve. The enrichment percentage and fraction of mutant alleles mentioned in study is calculated as the ratio of the copy number of SNV/(WT + SNV).
Collection and DNA extraction of patient samples
Patients with cancers of the pancreas, colorectum, lung, or breast were recruited from Shanghai General Hospital under methods approved by the Human Research Committee of Shanghai General Hospital. The ‘healthy donor’ samples consisted of peripheral blood samples obtained from two individuals with no history of cancer. The cancer and healthy control samples listed in Supplementary Table S6 were processed identically. DNA extraction from formalin-fixed, paraffin-embedded (FFPE) tumour blocks was performed using a QIAamp® DNA Blood Mini Kit (Qiagen, Germany) according to the kit's recommended protocol. Five millilitres of peripheral venous blood were collected in Streck Cell-Free DNA BCT (Streck Inc., USA). Samples were then centrifuged at 1600 g for 10 min at 10°C, and the resulting supernatant was clarified by additional centrifugation. Clarified plasma was transferred to a fresh tube, and DNA was immediately extracted using a QIAamp® DNA Blood Mini Kit (Qiagen, Germany). Finally, exome sequencing and data processing to produce a BAM file were performed using established next-generation sequencing (NGS) analytical pipelines at Shanghai General Hospital to validate the SNV alleles of the samples.
Phylogenetic and alignment analyses of thermophilic Agos
To screen for active thermophilic Agos, a similarity search for the PfAgo amino acid sequence was performed using BLAST in the NCBI database, and sequences were selected and analyzed by the MEGA 7.0 software for phylogenetic analysis (37). The sequence alignments of the proteins with the functional DEDX motif were carried out using ClustalW (38).
Statistics and reproducibility
Each experiment contains independent replicates for three times as indicated in figure legends. Sample sizes for clinic detection were set according to experiment experience and referred literature. Gel band intensities were visualized using a Tanon 3500BR (Shanghai, China), and the data were analyzed using Quantity One (Bio-Rad, USA). Data are expressed as mean ± S.E.M. and analyzed by ordinary one-way ANOVA using GraphPad Prism 7.0. P-value <0.05 was considered statistically significant.
RESULTS
Design of the A-Star platform
A flow chart of the A-Star method is shown in Figure 1. During the DNA denaturation step at 94°C, the optimized gDNAs extend the precise complementation of WT ssDNA but not of the variant alleles, consequently directing Ago-specific cleavage of unwound dsDNA and enabling the elimination of a large fraction of the WT background. The subsequent annealing and strand-extension steps initiated using standard PCR primers and DNA polymerase favour the template amplification of the retained variant alleles rather than the cleaved WT alleles. Thus, the catalytic elimination of WT accompanied by the exponential amplification of the variant alleles in each cycle of PCR led to specific enrichment of the mutant target in an efficient and simple manner. TaqMan qRT-PCR or Sanger sequencing following the A-Star approach allowed for detection of the enriched rare variant alleles.
Figure 1.

Schematic representation of the A-Star approach for rare mutation enrichment. Black lines represent gDNA and blue and green lines represent forward and reverse strands of target dsDNA, respectively. The coordinating-colored lines with arrows above or below the blue and green lines represent the forward and reverse primers. Red dots indicate the mutated nucleotides of SNV targets.
To find a possible candidate Ago, we first screened and tested the feasibility of hyperthermophile Agos by examining the temperature dependence and thermostability of their cleavage activity. Through the phylogenetic and alignment analyses, candidate proteins were chosen according to two requirements: ability to predict endonuclease activity (DEDX motif) and thermostability (at cultivation temperatures over 80°C). Only three proteins, PfAgo, ThAgo from Thermococcus thioreducens and ToAgo from Thermococcus onnurineus, were competent for use in further cleavage assays, while the other proteins failed due to insoluble expression or inactivation at 95°C. Among the successful candidates, we found high cleavage activity from PfAgo at temperatures ranging from 85 to 99°C and thermostability at 95°C for over 20 min, signifying its superiority to ThAgo and ToAgo (Supplementary Figure S1). This result indicated that PfAgo can not only function at the 94°C temperature of the PCR denaturation step but may also be stable during the whole PCR thermal cycling process. When PfAgo was incubated with synthesized gDNAs of varied lengths at 95°C for 15 min, we found the 16-nt gDNA was sufficient for efficient cleavage (Supplementary Figure S2A, B). We also verified that PfAgo can be active in several PCR buffers and selected the buffer that delivered the best performance (Supplementary Figure S2C). Moreover, to avoid nonspecific extension initiated by the gDNA in subsequent PCR amplification, we modified each gDNA by adding an extra 3′-phosphate on canonical 5′-phosphate gDNA and then verified its effectiveness of being primers used in PCR (Supplementary Figure S3). Taken together, the results indicate that conditions needed for PfAgo cleavage are potentially compatible with PCR amplification in a single reaction when used with a 16 nt gDNA diphosphorylated at both the 5′- and 3′-terminal ends.
gDNA design precisely discriminates between WT and SNV
Next, we investigated the design and selection of gDNA for accurate discrimination of tDNAs at single-nucleotide resolution to achieve precise cleavage of WT alleles. We optimized the gDNA using the synthesized KRAS ssDNA fragment of the WT or G12D allele (a well-studied oncogene, c.35G>A) as a representative cleavage target. As previously reported, mismatch position and nucleotide type on gDNA would both potentially affect tDNA cleavage percentage (32), so we applied a systematic design to select the best gDNA hit to achieve the highly specific cleavage of WT sequence. After identifying diphosphorylated gDNA sequences that precisely matched the WT sequence but had a mismatch with the SNV mutant at the position 11 of the gDNA, they were incubated with PfAgo and KRAS ssDNA at 95°C for 15 min. Both WT KRAS and mutant KRAS containing the SNV encoding a G12D substitution were cleaved indiscriminately at 95°C (Supplementary Figure S4). This result suggests that a single-nucleotide difference in the gDNA for the WT and SNV alleles was not sufficient to precisely differentiate the undesired allele for DNA cleavage under the evaluated conditions. The single nucleotide mismatch between the gDNA to target has been systematically evaluated in our previous study, and the result showed some positions of mismatch affected the cleavage activity but in dependence of target sequences (39).
To achieve discrimination between the WT and SNV tDNA sequences, we introduced an additional mismatched nucleotide of the gDNA at a certain position because the particular mismatch may lead to a ‘bubble’ in gDNA–tDNA pairing and impair recognition and cleavage. When a mismatch was introduced at nucleotide positions 7, 10 or 11 of the gDNA, PfAgo preferentially cleaved the WT KRAS ssDNA, resulting in the lowest cleavage percentage of the SNV allele. (Figure 2A and Supplementary Figure S5). The same phenomenon was observed on the WT and SNV mutations of tested oncogene PIK3CA ssDNA (Supplementary Figure S6). Therefore, the three positions (7,10 and 11) were defined as the hot spots for introducing mismatches when designing gDNAs. Due to the importance of the flanking positions of the cutting site of the gDNA and to further optimize our gDNA, we set the introduced mismatches that correspond to two adjacent nucleotides between positions 10 and 11 of the gDNA and SNV sequence. We also observed that the mismatched nucleotide identity of the gDNA affected the discriminatory behaviour of PfAgo, indicating a high dependency on the target sequence, instead of the specific nucleotides (G/C or A/T) near the SNV site, as tested for four oncogenes, including KRAS, PIK3CA and EGFR tDNA (Supplementary Figures S7A–D, S8A–D, S9A–D and S10A–D). Collectively, these results suggested that introducing mismatches in gDNA at corresponding cutting sites conferring extra mismatches in the SNV sequence can direct the preferential cleavage of WT sequences, thereby achieving more precise selection for SNVs. Here, we suggest the importance of these hot spots positions for gDNA design, which may contribute to conformational changes between the guide to WT or SNV target thus lead to the discrimination effect. In consistent with our previous results, the adjacent mismatches at 10–11 position of gDNA significantly affect cleavage activity of PfAgo in a sequence-independent manner (39). Therefore, we establish the rule for gDNA design: a 16 nt gDNA with introduced single mismatch located at position adjunct with corresponding SNV site, to gain the continuous mismatches at 10–11 position for SNV target, and refer to single mismatch at position either 10 or 11 for WT target, to achieve the best discrimination.
Figure 2.

Discriminant DNA cleavage by designed gDNAs and suitability of PfAgo use in PCR in a single-tube reaction for SNV enrichment. (A) Schematic representation of designed gDNAs for KRAS G12D ssDNA cleavage and heat map analysis of the gel detection results for the designed mismatched gDNA directed cleavage on WT and SNV targets. Of the designed gDNAs, the introduced mismatched nucleotides are indicated by red capital letters, the nucleotides corresponding to the SNV site are indicated by black capital letters, and the remaining nucleotides pairing to the ssDNA target are represented by grey circles. The black triangles on the WT ssDNA represent the specific cleavage sites. The cleavage (red heatmaps) was quantified by measuring the electropherograms of the cleavage products with the ssDNA target of KRAS G12D WT or SNV directed by the mismatched gDNAs. The specificity index of WT/SNV (blue heatmap) is the ratio of the cleavage percentage (%) of WT to that of SNV directed by the mismatched gDNAs. (B) Gel electrophoresis analysis for the PfAgo-PCR coupled reaction to cleave WT sequences and enrich SNVs. Mx: a mixture of WT and SNV target at equal molar concentrations. (C) TaqMan qRT-PCR evaluation of the enrichment results tested with a mixture of WT and SNV targets at equal molar concentrations treated by PCR or PfAgo-PCR coupled reaction. Error bars represent standard deviations of the means, n = 3. (D) Sanger sequencing evaluation of the enrichment results tested with a mixture of WT and SNV targets at equal molar concentrations treated by PCR or PfAgo-PCR coupled reaction.
After optimizing the gDNA candidates for each single strand of the dsDNA sequence, we further evaluated the effect of the paired gDNA which consisted each single strand's gDNA in a random combination mode on cleavage discrimination between the WT and SNV dsDNA targets. The gDNA pair with the highest specificity index of WT/SNV ratio were chosen as the best-hit gDNAs. When we used the best-hit gDNAs, the SNV variants of three targets remained uncleaved, whereas their corresponding WT counterparts were almost completely cleaved during incubation with PfAgo at 95°C for 15 min (Supplementary Figures S7E–F, S8E–F, S9E–F and S10E–F). When using a pair of semi-best hit gDNA, in which a best hit gDNA was combined with a poorer hit gDNA, we did not observe clearly discriminant cleavage of the dsDNA target. This result shows that the pair of best-hit gDNAs successfully directed PfAgo to discriminately cleave each strand of unwound dsDNA. Notably, a pair of gDNA could be used to discriminate three variations at the same position of the WT sequence, as shown by the dsDNA targets of mutations of KRAS-G12A, KRAS-G12V and KRAS-G12D, respectively (Supplementary Figures S11).
Additionally, considering that indel mutations are commonly found in oncogenes, we first examined whether a pair of gDNAs matching the WT sequence could discriminate between the WT and mutant dsDNA target using EGFR and its 15 nt deletion mutant. Results confirmed that PfAgo can efficiently cleave WT dsDNA with minimal effect on the mutant allele at 95°C (Supplementary Figure S12). To determine whether a shorter sequence with deletions can be discriminated from the WT sequence, the synthetic sequences with 1–3 deleted bp compared EGFR WT fragment were investigated (Supplementary Figure S13). Using the gDNAs that completely paired with WT sequences, the deletion mutations with at least two consecutive bases pairing of mismatches were spared, and the WT dsDNA was completely cleaved. Then we also introduced additional mismatched gDNAs using principle like the SNV gDNA design described above, and observed a clear discriminant cleavage of the 1 bp deletion target directed by our designed gDNAs. Taken together, these results demonstrate that PfAgo is capable of effectively discriminating mutated dsDNA from WT alleles when directed by a pair of gDNA where each gDNA contains introduced mismatches from the SNV templates at nucleotide positions 10 and 11.
A-Star specifically enriches rare SNVs in a single tube
The crucial concern in developing the A-Star technique was whether gDNA-directed PfAgo-targeting could function well under a standard PCR amplification program to achieve discriminatory cleavage of the undesired allele and amplification of the desired allele simultaneously in a single reaction tube. We tested the effect of PfAgo on the cleavage of tDNA using KRAS G12D WT and SNV alleles in the presence of their respective pair of best hit gDNAs. The PCR was performed with 25 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 20 s after pre-incubation for activation of the polymerase, as suggested by the protocol for PCR amplification. The gel electrophoresis results indicated discriminatory cleavage occurred because the products from WT reactions were almost completely digested into two small fragments, whereas SNV alleles appeared enriched as single, full-length amplicons (Figure 2B). We further evaluated SNV allele enrichment quantitatively using TaqMan qRT-PCR. The results showed that more than 90% of the amplicons from the mixed template were of the SNV allele when in the presence of PfAgo-gDNA. In comparison, ∼50% of the amplicons were of the SNV allele following standard PCR (Figure 2C). Sanger sequencing also provided support for the enrichment of SNV alleles: a major peak of adenine nucleotide treated by PfAgo-gDNA was present, which differed from the two distinct peaks of adenine and guanine at equivalent levels following standard PCR (Figure 2D). Altogether, we hypothesized that the WT amplicon accumulates in the extension step and serves as a cleavage target for PfAgo in the following annealing step during the A-Star process, highlighting the cycled process incorporated the repressed WT amplification and specific cleavage. Collectively, the results suggest that the PCR approach combined with the use of gDNA-directed PfAgo leads to specific enrichment of SNV alleles and elimination of WT alleles simultaneously and feasibly in a single tube.
To increase detection sensitivity for rare SNV alleles, we used a 1% VAF of KRAS G12D and systematically titrated the amount of PfAgo added as well as the molar ratio between PfAgo and gDNA, and also altered the PCR thermal cycling conditions, then evaluated SNV enrichment efficiency. PfAgo concentration greatly affected SNV enrichment (Supplementary Figure S14). As the PfAgo concentration increased to 30 nM, with a ratio of 3:1 PfAgo to gDNA, the majority of WT alleles were removed, resulting in efficient enrichment of rare SNVs. When the concentration rose above 30 nM, the SNV enrichment efficiency decreased. The decrease might have been caused by the strong binding affinity of PfAgo toward unspecific DNA which reduced the concentration of valid tDNA selected for catalytic cleavage. We also found that an increased ratio of 20:1 gDNA to PfAgo enhanced SNV enrichment (Supplementary Figure S15), implying that gDNA at a high concentration could competitively bind PfAgo and release the bound tDNA for further specific cleavage. Moreover, during the PCR procedure, 25–30 thermal cycles efficiently enriched the rare SNV alleles (Supplementary Figure S16). Taken together, the results showed that the optimal conditions to enrich rare SNV alleles were adding 30 nM PfAgo to a PfAgo:gDNA ratio of 1:20 and using a 25 cycle PCR program.
A-Star has high efficiency of enrichment of rare SNVs
To evaluate the LOD of A-Star, different levels of VAFs of KRAS G12D at 0.01%, 0.1% and 1% were used under conditions optimized above. A-Star can enrich these rare SNV alleles efficiently, ranging from 55 to 75% enrichment, as detected by TaqMan qRT-PCR (Figure 3A), although the initial tested SNV VAFs varied by several orders of magnitude. Samples at VAF of 1% and 0.1% were enriched to over 60%, corresponding to a 75- and 600-fold increase, respectively, compared to the control in the absence of PfAgo. In a sample with a VAF as low as 0.01%, we detected a 5500-fold enrichment of SNV products (Figure 3B). These enriched products were also readily detected by Sanger sequencing, showing a clear adenine nucleotide signal representing the SNV allele, in contrast with only the guanine nucleotide representing the WT allele in the control (Figure 3C). Similar results were obtained at a VAF of 0.01% for samples containing three SNVs of PIK3CA and EGFR or an EGFR indel mutation (Supplementary Figure S17).
Figure 3.
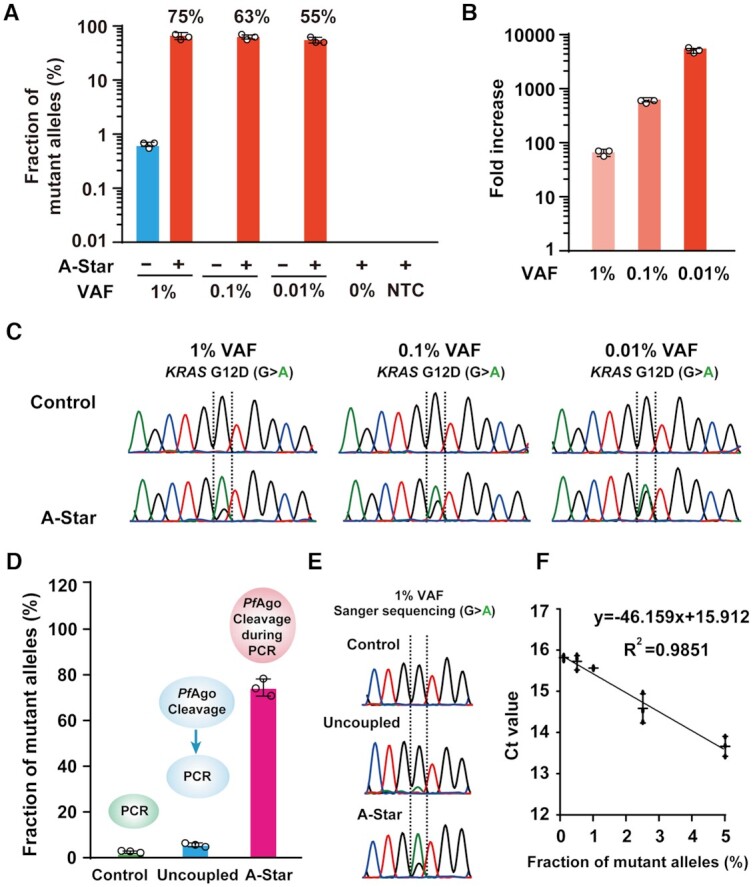
Detection sensitivity of A-Star for rare SNV enrichment. (A) Evaluation of enrichment efficiency of A-Star with KRAS G12D samples of varying VAFs by TaqMan qRT-PCR. (B) The values of fold increases were calculated from the data in (A). (C) Evaluation of enrichment efficiency of A-Star with KRAS G12D samples of varying VAFs by Sanger sequencing. (D, E) Comparison enrichment efficiency of A-Star with the uncoupled PfAgo-PCR tested with 1% VAF samples of KRAS G12D by TaqMan qRT-PCR (D) and Sanger sequencing (E). (F) Correlation of the threshold cycle (Ct) value and KRAS G12D samples of varying VAFs by TaqMan qRT-PCR. The no-template control (NTC) contained only water. Error bars represent the mean ± S.D., n = 3.
Further, we demonstrated the advantage of A-Star over the uncoupled-PfAgo PCR technique by comparing their efficiencies in the enrichment of rare variant alleles using 1% KRAS G12D (Figure 3D). The latter technique was set up as a two-step reaction: after an initial 15 min incubation with the PfAgo-gDNA complex at 94°C to cleave WT DNA, reactions underwent 25 PCR cycles with purified and cleaved DNA products to ensure the absence of PfAgo. We observed a marked difference in the enrichment efficiency of rare mutations between our single-tube PfAgo-PCR and the uncoupled process. The A-Star technique increased the percentage of KRAS G12D SNV sequences in the final PCR products from 1% to 75%, which was far greater than the increase in percentage for the uncoupled strategy which rose to 6% (Figure 3D). Consistently, an obvious peak of adenine nucleotide treated by A-Star was detected, while the major peak of guanine nucleotide was observed following treatment with the uncoupled strategy (Figure 3E). This demonstrates that integrating the two processes of PfAgo cleavage and PCR cycles in a single tube provides simple operation but also results in the progressive and efficient elimination of the WT allele and enrichment of the rare mutant allele, compared with the uncoupled methods of enzyme catalytic cleavage and PCR. Also, we evaluated the mean cycle threshold (Ct) values of the samples of different VAFs and found a clear linear relationship (Figure 3F).
Although we demonstrated the efficient enrichment of rare SNVs could combine various downstream detection methods, we also demonstrated the success of combining two serial PCR reactions, A-Star and Taqman qRT-PCR reactions into a single PCR reaction. Using the DNA fragment of the KRAS G12D mutation, we tested the practicality of this combined PCR method (Supplementary Figure S18). After optimizing conditions, the 1% VAF of KRAS G12D at 1 nM concentration could be enriched and detected by WT and SNV fluorescence signals in the single PCR process. Moreover, the 1% SNV target was enriched to 65% with 65-fold enrichment efficiency by this simplified A-Star method. The cleavage activity of PfAgo is compatible and programmable with both the standard PCR and Taqman qRT-PCR procedures, indicating great potential in analysis of rare mutations in oncogenes in the early stage of diagnosis or in monitoring oncogenes.
To explore the clinical utility of A-Star, we investigated the LOD of A-Star using a set of commercial cfDNA standards containing genomic DNA at three available VAFs of 0.1%, 1% and 5% for KRAS G12D, PIK3CA E545K and the EGFR indel. Because PfAgo non-specifically binds DNA template in PCR, we found that a low amount of cfDNA mock samples with 33 ng input was not directly detectable using the A-Star method (Supplementary Figure S19) (31,39). We surmise that PfAgo bound the DNA sample to some extent and thus decreased the available amount of template for PCR amplification when the initial template concentration was low (Supplementary Figure S20), Thus, we added a pre-amplification step to increase the amount of cfDNA for both the WT and variant alleles before conducting the A-Star process. We also determined the number of cycles needed in the pre-amplification step to achieve sufficient enrichment and found that pre-amplification of 30 cycles was adequate for the cfDNA mock samples (Supplementary Figure S21). The inclusion of this pre-amplification step enabled the detection sensitivity of 0.1% SNVs to achieve over 40% enrichment from samples containing only 33 ng of cfDNA for all tested targets (Figure 4). To further verify the specificity of the A-Star method, we also tested the technique using water alone and WT genomic DNA samples in a panel with varying VAF samples, and no false-positive results were detected (Figure 4). Therefore, the sensitivity and specificity of A-Star for the mock cfDNA samples emphasize its potential for analyzing samples with high levels of background genomic DNA.
Figure 4.

Evaluation of the A-Star enrichment results for KRAS G12D,PIK3CA E545K and EGFR del of Horizon cfDNA standard samples with different VAFs analyzed by TaqMan qRT-PCR. All controls were processed in the absence of a pair of gDNAs. NTC contained only water. Error bars represent standard deviations of the mean, n = 3.
A-Star applied to the clinical analysis of tissue and blood samples
We also tested A-Star on DNA extracted from tissue (33.3 ng of genomic DNA) and blood samples (3.3 ng of cfDNA) from patients with different cancer types who carried the KRAS G12D SNV and healthy donors that were identified by deep sequencing (Figure 5A). Here, we performed a pre-amplification step before conducting the A-Star procedure as mentioned above for mock cfDNA samples. We observed that SNVs were successfully enriched and detected by TaqMan qRT-PCR (Figure 5B). The VAFs of tissue samples processed by A-Star directly increased to 60–90% from the original VAFs of <20%. In particular, the lung adenocarcinoma (LUAD) sample with an extremely low VAF (below 1%) was enriched by more than 50%, representing an improvement of over 60-fold. For blood samples, the VAFs increased to 40–85% from the original VAFs of 3–20%. These enriched products can be easily detected by Sanger sequencing with a greatly enhanced adenine nucleotide signal represented as a mutant allele (Supplementary Figure S22). The above results show that A-Star offers impressive specificity and sensitivity for detecting rare SNVs in clinical applications.
Figure 5.
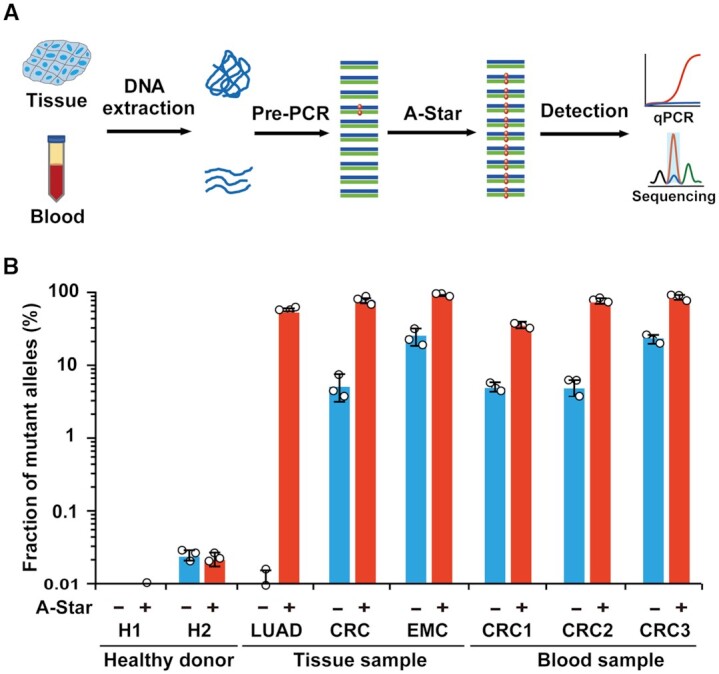
Analysis of A-Star with human clinical samples. (A) Flow chart of the A-Star procedure for the identification of cancer-associated SNVs in human samples. (B) Evaluation of A-Star by TaqMan qRT-PCR using clinical samples from diverse cancer types, as exemplified by KRAS G12D-containing samples from patients with lung adenocarcinoma (LUAD), colorectal cancer (CRC), or endometrial cancer (EMC). The control reactions did not contain gDNA pairs. Error bars represent standard deviations of the mean, n = 3.
Multiplexed detection of oncogenes
Given that the sequence recognition capacity of Ago is mediated by gDNA, we envisioned that A-Star in combination with multiple designed-gDNAs and primers might be suitable for multiplexed mutation detection (Figure 6A and Supplementary Figure S23). First, we tested the enrichment efficiency of triplex assays using different combinations of variant allele targets among KRAS G12D, PIK3CA E545K, and a deletion mutation for EGFR, each at a 1% VAF. Each combination of samples was mixed with all the required primers, designed gDNAs, and PfAgo in one tube. The degree of enrichment of all targets analyzed by subsequent TaqMan qRT-PCR analysis was of similar magnitudes to the values detected in the non-multiplex A-Star assays (Figure 6B). The triplex assays conducted in single tubes showed that all three targets were >50-fold enriched (Supplementary Figure S24), implying orthogonal activity of PfAgo when simply supplied with multiple gDNA pairs.
Figure 6.
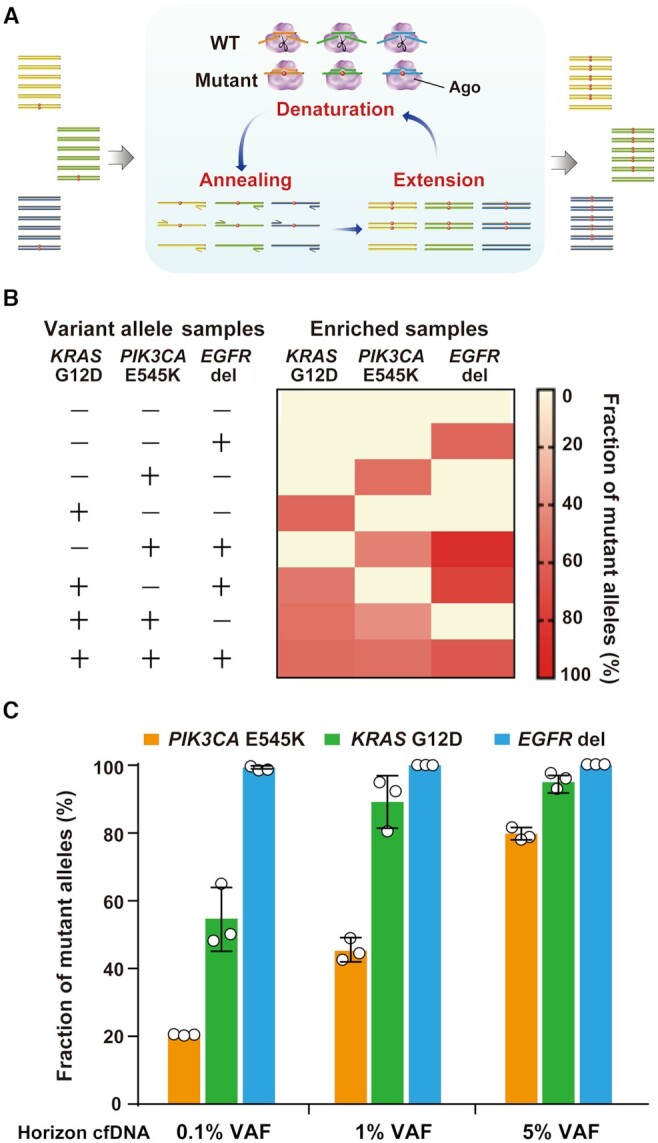
Multiplex detection by A-Star. (A) Schematic representation of multiplex mutation detection in a single-tube reaction containing multiple pairs of primers and gDNAs. (B) Triplex detection by A-Star of the 1% VAF samples of targets KRAS G12D, PIK3CA E545K and EGFR del. (C) A-Star triplex enrichment of KRAS G12D, PIK3CA E545K and EGFR del using standard cfDNA samples with different VAFs of 0.1%, 1% and 5%. The enrichment output was detected using TaqMan qRT-PCR. Error bars represent standard deviations of the mean, n = 3 for each variant.
To mimic clinical multiplex detection with samples containing a complicated DNA background, we also conducted triplex A-Star assays using mock cfDNA standards of three VAFs (0.1%, 1% and 5%) following the multiplex procedure as described above but with an initial pre-PCR amplification step. The quantitative analysis revealed over 50% enrichment of variant alleles for each KRAS and PIK3CA sample with an original range of 0.1% to 5% SNV alleles (Figure 6C and Supplementary Figure S25). Excepting the 0.1% PIK3CA sample, A-Star increased VAF to 21% (a 210-fold increase), which was a sufficient amount for the subsequent analyses. Enrichment efficiency of the deletion mutation of EGFR in the triplex assay achieved the highest level of nearly 100% for a wide range of initial mutant alleles. Altogether, the results indicate that the orthogonal activity of PfAgo directed by corresponding gDNAs is an effective yet simple technique using a single-tube reaction for a multiplex enrichment.
DISCUSSION
In this study, we described a PfAgo-directed PCR method for the rapid and efficient enrichment and super-sensitive detection of rare variant alleles. Our technique, dubbed A-Star, substantially improves the sensitivity of detecting rare variant alleles compared to the prevalent methods. This method enables specific amplification of SNV allele fractions as low as 0.01% with an over 5500-fold enrichment efficiency and detection at a limit of single-digit copies of rare SNV using the mock cfDNA sample of 0.1% VAF (Figure 3A, B, Figure 4 and Supplementary Figure S17). Importantly, A-Star enhanced sensitivity of detection relies on a very easy-to-implement design and is ideal for targeting virtually any mutant sequences, presenting great potential for diagnostic applications. Sensitive PCR-based methods are currently being developed for the enrichment of mutations present at low to extremely low levels. New approaches, either exploring new nucleic acid-editing enzymes or PCR procedures, have appeared in past years (5–9,12); however, these approaches are unsatisfactory in terms of detection sensitivity, cost, time and simplicity (Table 1). Conventional PCR methods, including real-time PCR, cannot detect minor mutated ctDNA <20% (40). The ARMS method can selectively amplify variant sequences but does not permit a sequence-based verification of the mutated allele because the mutant base sequence is included in the primer sequence (9). Other methods, such as BDA and COLD-PCR, need to consider Tm value or require strict temperature control (5,6,11). The restriction endonuclease-mediated selective PCR assay allows the simultaneous amplification of mutant signal and inhibition of WT amplification, but is constrained by the limited availability of thermostable restriction enzymes (12). The dPCR method, currently the most advanced technology for analysis of ctDNA, may achieve detection at 0.01% VAF and has multiple advantages over conventional approaches (8). Unfortunately, the high cost of dPCR equipment hinders the widespread adoption of this approach.
Table 1.
Major characteristics of general methods for rare allele enrichment and detection
| Method | Nuclease | Guide/blocker | Sensitivity | Specificity | Enrichment efficiency | Multiplex | Target motif requirement | Tight temperature control | Operations |
|---|---|---|---|---|---|---|---|---|---|
| COLD-PCR ( 6,11) | - | - | 0.01% with NGS | Medium | 500-fold | Yes | No | Yes | PCR |
| BDA ( 5 ) | - | DNA blockers | 0.01% with qPCR | Medium | 10000-fold | Yes | No | No | PCR |
| HOLMESv2 ( 26 ) | Cas12 | ∼120 nt sgRNA | 0.1% with fluorescence detection | High | - | No | PAM | No | LAMP + Nuclease reaction |
| SHERLOCKv2 ( 28 ) | Cas12, Cas13 and Csm6 | ∼120 nt sgRNA | 0.6% with fluorescence detection | High | - | Yes | PFS/PAM | No | RPA + Nuclease reaction |
| PAND ( 41 ) | PfAgo | 16 nt gDNA | 0.1% with fluorescence detection | High | - | Yes | No | No | PCR/tHDA + Nuclease reaction |
| Cut-PCR ( 27 ) | Cas9 | ∼120 nt sgRNA | 0.01% with NGS | High | 18-fold | No | PAM | No | Nuclease reaction + PCR |
| NAVIGATER ( 36 ) | TtAgo | 16 nt gDNA | 0.01% with XNA-PCR | High | 60-fold (0.5% VAF) | Yes | No | No | Nuclease reaction + dPCR |
| A-Star | PfAgo | 16 nt gDNA | 0.01% with TaqMan qRT-PCR | High | 5500-fold | Yes | No | No | Nuclease reaction in PCR |
The major advantages of our approach are the combination of programmable gDNA–tDNA pairing, effective discriminatory DNA cleavage by the PfAgo endonuclease at DNA melting temperatures, and variant allele amplification by PCR in a single-tube experiment. A-Star exhibited high efficiency in variant allele enrichment, which was likely due to the properties of PfAgo and its suitability for use in PCR (Figure 3B–D). The advantage of combining WT tDNA elimination with variant-allele amplification in a single tube makes A-Star an improved technique than other amplification methods, such as the two- or three-round PCR enrichment methods of CUT-PCR (Cas9-based) and NAVIGATER (TtAgo-based) (Table 1) (27,36). In these two methods, the procedures include PCR for DNA enrichment and target DNA cleavage that are performed in a separated manner and repeatedly to obtain high detection sensitivity as well as efficiency. However, only 20-fold and 60-fold enrichment, respectively, can be obtained after three rounds of CUT-PCR or two rounds of NAVIGATER. Recently, the PAND (PfAgo-based) was developed by utilizing PfAgo's second round of cleavage initialled by the ssDNA product for SNV detection but lacking the capability to enrich the rare SNVs (41). In the present study, the single-tube A-Star method achieved more efficient enrichment than the uncoupled cleavage-and-PCR procedure achieved in enrichment of rare variant alleles (75-fold versus 6-fold enrichment from the 1% VAF sample) (Figure 3D, E), demonstrating the superiority of the coupled cleavage and PCR processes in the A-Star method. Besides, the coupling of programmable WT cleavage and standard PCR amplification of variant alleles in A-Star can reduce the use of RPA, which is limited by its non-stringent control of the rate of amplification, low efficiency for amplifying highly structured regions, and challenges in primer design. Derived from broadly used conventional PCR, the A-Star method uses short DNA fragments as guides rather than RNA in a CRISPR system, and thus provides an inexpensive way to synthesize guide oligos that can be conveniently conducted in a laboratory. For A-Star, we needed to screen at most six mismatched gDNAs (three variations at either the 10 or 11 position), which was relatively less laboriousness in comparison to NAVIGATER and PAND, which considered the mismatch position and was used to screen eleven and sixteen guides, respectively. Moreover, we also demonstrated that the A-Star and Taqman qRT-PCR assays could be combined into a single assay, which allowed for more direct signal readouts while simplifying the procedure. This simplified A-Star method showed 65-fold enrichment efficiency of SNV target, a considerable level with that of 75-fold enrichment efficiency attained by separated process of A-Star and Taqman qRT-PCR. This joint strategy using a single-PCR reaction would be beneficial to applications that are time-sensitive, high-throughput, or have less contamination risk.
Moreover, A-Star can be used in multiplex assays where PfAgo is solely directed by the corresponding pool of gDNAs present in a single-tube reaction. Considering the possibility that multiple gene mutations, such as EGFR, KRAS or PIK3CA, exist in ctDNA samples from lung carcinomas, multiplex detection is necessary to identify the mutated genotype more precisely when used in targeted therapies. Here, we demonstrated that multiplexing can be performed with A-Star and successfully detected multiple variant alleles with the orthogonal activity of PfAgo (Supplementary Figure S24). Moreover, robust evidence was provided by the non-interfering signals observed using the duplex and triplex samples (Figure 6B). We also demonstrated A-Star-multiplex detection of three oncogenes in the same reaction with the input of a mock cfDNA standard of 0.1% VAF (Figure 6C). Therefore, A-Star is a simple, single-tube, low-cost, and rapid reaction method that can be used to simultaneously analyze multiple targets and classes of somatic mutations, notably, by adding multiple gDNA fragments and primers in an orthogonal manner. A multiplexed A-Star system markedly differs from the CRISPR-based multiplex detection system SHERLOCK v2, because the latter system requires both the selection of multiple enzymes and the optimization of reaction parameters (Table 1) (28).
All the advantages of A-Star, especially its sensitivity in detection, make it a valuable tool for clinical detection of rare variant alleles in cancer diagnoses. Oncogene analyses have revealed that the cellular genome consists of pools of SNVs and indel mutations (42), which have been shown to affect normal cell metabolism and signal transduction. For example, a KRAS mutation can affect the Ras signalling pathway, which in turn affects the lung and colorectal carcinoma onset and development (43). Notably, many patients’ ctDNA levels are present at considerably low levels (<0.5%), which typically further decline following therapy (44,45). Thus, the super-sensitivity of A-Star has great potential in cancer detection along with therapy. Although we focused on applications related to oncogenes, our method can be applied to other situations that require genotyping of rare alleles from complex DNA samples, in which rare variants display disproportionate effects, such as infectious diseases caused by antibiotic-resistant subpopulations. We predict that A-Star can facilitate a much broader spectrum of basic scientific and clinical research and applications.
Despite all the advantages of A-Star, we note that there were some differences observed in the enrichment efficiencies of the different rare variant alleles in the multiplex system. The differences were likely caused by rules governing the precise discrimination of the WT and variant alleles with varied sequences (Supplementary Figures S7–S10). Thus, there is still room for improvement of A-Star's performance through more rigorous and sophisticated bioinformatic analyses and further empirical optimization. For example, our lab is continuing to optimize the thermodynamic parameters of designed gDNAs paired to various targets, aiming to provide the probable general rule for any detection target.
DATA AVAILABILITY
The authors declare that all data supporting the findings in this study are available within the paper and its Supplementary Information files.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Ming Yu and Xiaojing Ma from Shanghai Jiao Tong University for helpful comments on the manuscript. We also thank staffs of National Facility for Translational Medicine (Shanghai) for their assistance in data analysis.
Contributor Information
Qian Liu, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Xiang Guo, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Guanhua Xun, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Zhonglei Li, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Yuesheng Chong, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Litao Yang, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Hongxia Wang, Department of Oncology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080, China.
Fengchun Zhang, Department of Oncology, Shanghai Ruijin Hospital, Shanghai Jiao Tong University School ofMedicine, Shanghai 200025, China.
Shukun Luo, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Li Cui, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Pengshu Zhao, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Xingyu Ye, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Heshan Xu, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Hui Lu, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Xiao Li, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Zixin Deng, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Kai Li, GeneTalks Biotechnology Inc., Changsha, Hunan 410013, China.
Yan Feng, State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Science Foundation of China [31770078]; Ministry of Science and Technology [2020YFA0907700]. Funding for open access charge: Natural Science Foundation of China [31770078].
Conflict of interest statement. Shanghai Jiao Tong University has applied for a patent (application no. CN109880891A, PCT/CN2019/115151) for A-Star with Y.F., Q.L., G.X., X.G., Z.L. and Y.C. listed as co-inventors.
REFERENCES
- 1. Bettegowda C., Sausen M., Leary R.J., Kinde I., Wang Y., Agrawal N., Bartlett B.R., Wang H., Luber B., Alani R.M.et al.. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014; 6:224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abbosh C., Birkbak N.J., Wilson G.A., Jamal-Hanjani M., Constantin T., Salari R., Le Quesne J., Moore D.A., Veeriah S., Rosenthal R.et al.. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017; 545:446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salk J.J., Schmitt M.W., Loeb L.A.. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018; 19:269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Milbury C.A., Li J., Makrigiorgos G.M.. PCR-based methods for the enrichment of minority alleles and mutations. Clin. Chem. 2009; 55:632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu L.R., Chen S.X., Wu Y., Patel A.A., Zhang D.Y.. Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat Biomed Eng. 2017; 1:714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mauger F., How-Kit A., Tost J.. COLD-PCR technologies in the area of personalized medicine: methodology and applications. Mol. Diagn. Ther. 2017; 21:269–283. [DOI] [PubMed] [Google Scholar]
- 7. Song C., Liu Y., Fontana R., Makrigiorgos A., Mamon H., Kulke M.H., Makrigiorgos G.M.. Elimination of unaltered DNA in mixed clinical samples via nuclease-assisted minor-allele enrichment. Nucleic Acids Res. 2016; 44:e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taly V., Pekin D., Benhaim L., Kotsopoulos S.K., Le Corre D., Li X., Atochin I., Link D.R., Griffiths A.D., Pallier K.et al.. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 2013; 59:1722–1731. [DOI] [PubMed] [Google Scholar]
- 9. Newton C.R., Graham A., Heptinstall L.E., Powell S.J., Summers C., Kalsheker N., Smith J.C., Markham A.F.. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989; 17:2503–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vogelstein B., Kinzler K.W.. Digital PCR. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:9236–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J., Wang L., Mamon H., Kulke M.H., Berbeco R., Makrigiorgos G.M.. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat. Med. 2008; 14:579–584. [DOI] [PubMed] [Google Scholar]
- 12. Ward R., Hawkins N., O’Grady R., Sheehan C., O’Connor T., Impey H., Roberts N., Fuery C., Todd A.. Restriction endonuclease-mediated selective polymerase chain reaction - a novel assay for the detection of K-ras mutations in clinical samples. Am. J. Pathol. 1998; 153:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heitzer E., Haque I.S., Roberts C.E.S., Speicher M.R.. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019; 20:71–88. [DOI] [PubMed] [Google Scholar]
- 14. Ladas I., Fitarelli-Kiehl M., Song C., Adalsteinsson V.A., Parsons H.A., Lin N.U., Wagle N., Makrigiorgos G.M.. Multiplexed elimination of wild-type DNA and high-resolution melting prior to targeted resequencing of liquid biopsies. Clin. Chem. 2017; 63:1605–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xiao L., Luo D.X., Pu W., Luo W., Pan Y., Xu H., Li W., Zhang R., Wang H., Wang F.et al.. Primerless amplification for circulating tumor DNA assays. J. Biomed. Nanotechnol. 2019; 15:1052–1060. [DOI] [PubMed] [Google Scholar]
- 16. Huang C.H., Lee K.C., Doudna J.A.. Applications of CRISPR-Cas enzymes in cancer therapeutics and detection. Trends Cancer. 2018; 4:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chiu C. Cutting-edge infectious disease diagnostics with CRISPR. Cell Host Microbe. 2018; 23:702–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Y., Li S., Wang J., Liu G.. CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol. 2019; 37:730–743. [DOI] [PubMed] [Google Scholar]
- 19. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anders C., Niewoehner O., Duerst A., Jinek M.. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014; 513:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gu W., Crawford E.D., O’Donovan B.D., Wilson M.R., Chow E.D., Retallack H., DeRisi J.L.. Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016; 17:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B., Shmakov S., Makarova K.S., Semenova E., Minakhin L.et al.. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016; 353:aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H., Tjian R., Doudna J.A.. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016; 538:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A.et al.. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017; 356:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen J.S., Ma E., Harrington L.B., Da Costa M., Tian X., Palefsky J.M., Doudna J.A.. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018; 360:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li L.X., Li S.Y., Wu N., Wu J.C., Wang G., Zhao G.P., Wang J.. HOLMESv2: a CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synthetic Biology. 2019; 8:2228–2237. [DOI] [PubMed] [Google Scholar]
- 27. Lee S.H., Yu J., Hwang G.H., Kim S., Kim H.S., Ye S., Kim K., Park J., Park D.Y., Cho Y.K.et al.. CUT-PCR: CRISPR-mediated, ultrasensitive detection of target DNA using PCR. Oncogene. 2017; 36:6823–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F.. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018; 360:439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Swarts D.C., Makarova K., Wang Y., Nakanishi K., Ketting R.F., Koonin E.V., Patel D.J., van der Oost J.. The evolutionary journey of Argonaute proteins. Nat. Struct. Mol. Biol. 2014; 21:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hegge J.W., Swarts D.C., van der Oost J.. Prokaryotic Argonaute proteins: novel genome-editing tools. Nat. Rev. Microbiol. 2018; 16:5–11. [DOI] [PubMed] [Google Scholar]
- 31. Swarts D.C., Hegge J.W., Hinojo I., Shiimori M., Ellis M.A., Dumrongkulraksa J., Terns R.M., Terns M.P., van der Oost J.. Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 2015; 43:5120–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y., Juranek S., Li H., Sheng G., Tuschl T., Patel D.J.. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature. 2008; 456:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Willkomm S., Oellig C.A., Zander A., Restle T., Keegan R., Grohmann D., Schneider S.. Structural and mechanistic insights into an archaeal DNA-guided Argonaute protein. Nat Microbiol. 2017; 2:17035. [DOI] [PubMed] [Google Scholar]
- 34. Chong Y.S., Liu Q., Huang F., Song D., Feng Y.. Characterization of a recombinant thermotolerant argonaute protein as an endonuclease by broad guide utilization. Bioresour. Bioprocess. 2019; 6:21. [Google Scholar]
- 35. Enghiad B., Zhao H.. Programmable DNA-guided artificial restriction enzymes. ACS Synth Biol. 2017; 6:752–757. [DOI] [PubMed] [Google Scholar]
- 36. Song J., Hegge J.W., Mauk M.G., Chen J., Till J.E., Bhagwat N., Azink L.T., Peng J., Sen M., Mays J.et al.. Highly specific enrichment of rare nucleic acid fractions using Thermus thermophilus argonaute with applications in cancer diagnostics. Nucleic Acids Res. 2020; 48:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kumar S., Stecher G., Tamura K.. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016; 33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thompson J.D., Higgins D.G., Gibson T.J.. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994; 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xun G., Liu Q., Chong Y., Li Z., Guo X., Li Y., Fei H., Li K., Feng Y.. The stepwise endonuclease activity of a thermophilic Argonaute protein. 2019; bioRxiv doi:29 October 2019, preprint: not peer reviewed 10.1101/821280. [DOI] [PMC free article] [PubMed]
- 40. Das J., Kelley S.O.. High-Performance Nucleic Acid Sensors for Liquid Biopsy Applications. Angew. Chem. Int. Ed. Engl. 2020; 59:2554–2564. [DOI] [PubMed] [Google Scholar]
- 41. He R.Y., Wang L.Y., Wang F., Li W.Q., Liu Y., Li A.T., Wang Y., Mao W.X., Zhai C., Ma L.X.. Pyrococcus furiosus Argonaute-mediated nucleic acid detection. Chem. Commun. 2019; 55:13219–13222. [DOI] [PubMed] [Google Scholar]
- 42. Zehir A., Benayed R., Shah R.H., Syed A., Middha S., Kim H.R., Srinivasan P., Gao J., Chakravarty D., Devlin S.M.et al.. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017; 23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gautschi O., Huegli B., Ziegler A., Gugger M., Heighway J., Ratschiller D., Mack P.C., Gumerlock P.H., Kung H.J., Stahel R.A.et al.. Origin and prognostic value of circulating KRAS mutations in lung cancer patients. Cancer Lett. 2007; 254:265–273. [DOI] [PubMed] [Google Scholar]
- 44. Diehl F., Li M., Dressman D., He Y., Shen D., Szabo S., Diaz L.A. Jr, Goodman S.N., David K.A., Juhl H.et al.. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:16368–16373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taniguchi K., Uchida J., Nishino K., Kumagai T., Okuyama T., Okami J., Higashiyama M., Kodama K., Imamura F., Kato K.. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin. Cancer Res. 2011; 17:7808–7815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings in this study are available within the paper and its Supplementary Information files.