

Abstract
Adaptive thermogenesis has attracted much attention because of its ability to raise systemic energy expenditure and counter obesity and diabetes1,2,3. Recent data have indicated that thermogenic fat cells utilize creatine to stimulate futile substrate cycling, dissipating chemical energy as heat4,5. This model was based on the super-stoichiometric relationship between creatine added to mitochondria and O2 consumed. Here we provide direct evidence for the molecular basis of this futile creatine cycling (FCC) activity. Thermogenic fat cells contain robust phosphocreatine phosphatase activity, attributable to tissue-nonspecific alkaline phosphatase (TNAP). TNAP hydrolyzes phosphocreatine to initiate a futile cycle of creatine dephosphorylation and phosphorylation. Remarkably, unlike in other cells, TNAP is localized to mitochondria of thermogenic fat cells, where FCC occurs. TNAP expression is powerfully induced when animals are subjected to cold exposure. Moreover, the essential role of TNAP in the FCC is illustrated by the loss of this cycle when TNAP is inhibited in isolated mitochondria. Finally, genetic ablation of TNAP in adipocytes reduces whole body energy expenditure and causes rapid-onset obesity, with no change in movement or feeding behavior of the animals. These data illustrate the critical role of TNAP as a phosphocreatine phosphatase in the FCC.
Main
Adipose tissues have gained enormous interest because of the tight linkage between obesity and metabolic disorders such as type 2 diabetes, cardiovascular diseases, and many cancers1. Two special types of fat cells, brown and beige adipocytes, are notable for their exceptional ability to oxidize biological fuels and to support adaptive thermogenesis2,3. These two types of thermogenic fat cells utilize futile cycles to dissipate chemical energy in the form of heat without performing mechanical or chemical work4. Accordingly, they both serve as a physiological defense against hypothermia, obesity, and obesity-linked metabolic disorders. Importantly, the presence of functional thermogenic adipocytes in human adults is now well-established6–8.
Despite the important metabolic functions of adaptive thermogenesis, its molecular mechanisms are not fully understood. While uncoupling protein 1 (UCP1) is the most recognized thermogenic effector9,10, it is required for defense of body temperature only on a pure genetic background in mice11,12. Ucp1 knockout mice on a mixed genetic background can tolerate cold exposure with no loss of core body temperature. Moreover, recent work demonstrated that even within inbred strains of mice, brown fat without UCP1 suffers severe loss/damage to the entire electron transport chain of mitochondria upon cold exposure13. Hence, at least some portion of the loss of defense against hypothermia due to the genetic absence of UCP1 must now be reconsidered mechanistically.
New pathways of thermogenesis in adipose tissues have been discovered in the last several years14,15. Previously, we described a pathway in which creatine (Cr) and phosphocreatine (PCr) are in a futile cycle, dissipating the high energy charge of PCr, without performing any mechanical or chemical work5. We have thus named this the futile creatine cycle (FCC). Furthermore, biochemical, pharmacological and genetic evidence indicates that this pathway is a very important component of adipose tissue thermogenesis5,16–18. Proteomic and transcriptomic data also suggest that this pathway is important for thermogenesis in human fat19,20, while cultured human brown/beige fat cells show a profound suppression of thermogenic respiration when creatine metabolism is inhibited5.
Despite the apparent physiological importance of the FCC, critical questions concerning how PCr and Cr are futilely cycled remain to be answered. PCr is isoenergetic with ATP and serves both as a reservoir and shuttle of the high energy phosphate21. In theory, the FCC could involve a direct enzymatic hydrolysis of PCr, but such an enzymatic activity has never been described in the biomedical literature from cellular sources. In this study, we report the identification of tissue-nonspecific alkaline phosphatase, TNAP (also known as Alpl), as a robust PCr phosphatase (PCr’ase) in thermogenic fat. Importantly, TNAP in thermogenic fat cells localizes within mitochondria, a highly atypical subcellular location. Chemical and genetic ablation of adipose TNAP activity demonstrates that this enzyme plays a novel and critical role in supporting the FCC and adaptive thermogenesis.
PCr’ase activity in thermogenic fat
To determine if thermogenic fat displays PCr’ase activity, we incubated PCr with mitochondrial protein extracts prepared from the interscapular brown adipose tissue (BAT) of mice that were chronically exposed to cold (4 °C for 2 weeks). 31P nuclear magnetic resonance (NMR) was used to simultaneously monitor the consumption of PCr and the production of inorganic phosphate (Pi) (Fig. 1a–b). Using this approach, we found mitochondrial protein extracts of interscapular BAT and inguinal WAT (containing beige adipocytes) to exhibit a robust PCr’ase activity (Extended Data Fig. 1a). To further characterize this activity, mitochondrial protein extracts from BAT were separated into soluble and insoluble fractions by ultracentrifugation. The insoluble fraction was gently solubilized with the non-ionic detergent NP-40; a cold-inducible PCr’ase activity was found in the previously insoluble fraction, and was further purified by FPLC-based size-exclusion chromatography (SEC) (Fig. 1c–d). This procedure was done on mitochondrial protein extracts from brown fat of mice that were kept at either room temperature (RT) or at 4 °C. Equivalent fractions were also included from mitochondria isolated from the hearts of the same mice that were kept at 4 °C. As shown in Fig. 1d, a distinctive activity peak at elution volume 10 mL was observed only in material derived from brown fat mitochondria isolated from cold-acclimated mice.
Figure 1. Isolation and identification of TNAP as a cold-inducible PCr’ase from mitochondria of thermogenic fat.
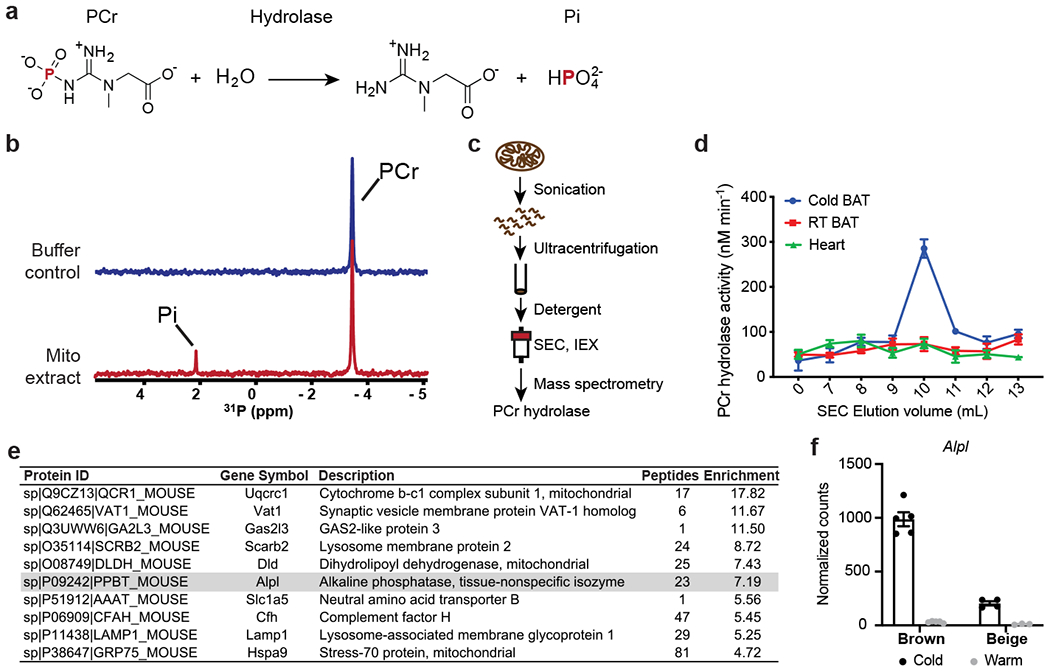
(a) Chemical equation for hydrolysis reaction of phosphocreatine, with phosphorus-containing chemical species indicated on top. PCr: phosphocreatine; and Pi: inorganic phosphate.
(b) Stacked 1-dimensional traces of 31P NMR frequency-domain spectra showing the occurrence of hydrolysis reaction of phosphocreatine with addition of mitochondrial protein extract (1.0 mg/ml) from brown adipose tissue (BAT) of cold-acclimated mice in 6 hours. All activities shown in rest of the figure were measured by 31P NMR, if not otherwise indicated.
(c) Scheme of isolation and identification of PCr phosphatase(s) from sucrose-gradient purified mitochondria preparation from murine tissues.
(d) PCr phosphatase activities of different fractions of size-exclusion chromatography. Error bars represent the standard errors of non-linear regression that fits a straight-line model to the initial linear phase of PCr hydrolysis kinetics measured by 31P NMR over 4 time points (shown in Source Data).
(e) List of top 10 proteins enriched in the active fraction versus a nearby less active fraction of IEX. TNAP (gene name: Alpl) is highlighted in grey.
(f) UCP1-TRAP (Translating Ribosome Affinity Purification) data showing the cold-sensitivity of Alpl expression in brown and beige adipocytes. Data were adapted from Roh, H. C. et al.23 and normalized with DESeq241; and are presented as means ± SEM; n=5 for cold and warm brown, 4 for cold beige, and 3 for warm beige (all are biologically independent samples).
TNAP is the cold-inducible PCr’ase
We further fractionated the active fraction from SEC by ion exchange chromatography (IEX), and found that the enzymatic activity eluted as a single tight peak (Extended Data Fig. 1b). Quantitative Mass Spectrometry was performed on an active fraction (fraction a) and a nearby, much less active fraction (fraction b). Alkaline phosphatase, liver form (Alpl) was 7-fold enriched in the active versus less active fraction (Fig. 1e), in agreement with the detected hydrolytic activities. Note that the protein product of this gene has been termed tissue-nonspecific alkaline phosphatase (TNAP). The enrichment of TNAP in the active fractions was validated by the western blots using an antibody that detects endogenous TNAP (Extended Data Fig. 1c). In addition, the PCr’ase activity detected in the total mitochondrial protein extract derived from BAT was blunted by treatment with a highly specific inhibitor of TNAP, SBI-42522 (Extended Data Fig. 1d). Finally, the cold sensitivity of Alpl expression in adipocytes was examined using a published adipocyte-specific ribosomal profiling dataset23. As shown in Fig. 1f, Alpl mRNA is expressed in a cold-sensitive manner in both brown (31.4-fold) and beige fat (18.8-fold).
Alpl mRNA is broadly expressed in many tissues, especially in liver, kidney and bone24. In bone, TNAP is best known for its function in facilitating the harvest of Pi from many substrates, including PPi, for the synthesis of mineralized bone matrix25,26. We next asked whether purified TNAP protein can hydrolyze PCr in our assays. This data shows that recombinant human TNAP hydrolyzes PCr robustly (Extended Data Fig. 1e–g).
TNAP is localized to mitochondria
TNAP is annotated as a cell surface and secreted enzyme that does not contain an apparent mitochondrial targeting sequence (Uniprot: P09242). However, as shown in Extended Data Fig. 1c and 3e, it is certainly present within our mitochondrial preparations from BAT. We next investigated the intracellular distribution of TNAP by immunocytochemistry (ICC). We first ectopically expressed full-length TNAP (Uniprot: P09242-1) in brown adipocytes; and observed a very substantial amount of this expressed TNAP in mitochondria, colocalizing with the well-known mitochondrial protein HSP60 (Fig. 2a). Importantly, this mitochondrial localization is essentially absent when we expressed the same TNAP cDNA in non-thermogenic fat cell types (Fig. 2a and Extended Data Fig. 2).
Figure 2. TNAP targets mitochondria in brown adipocytes.
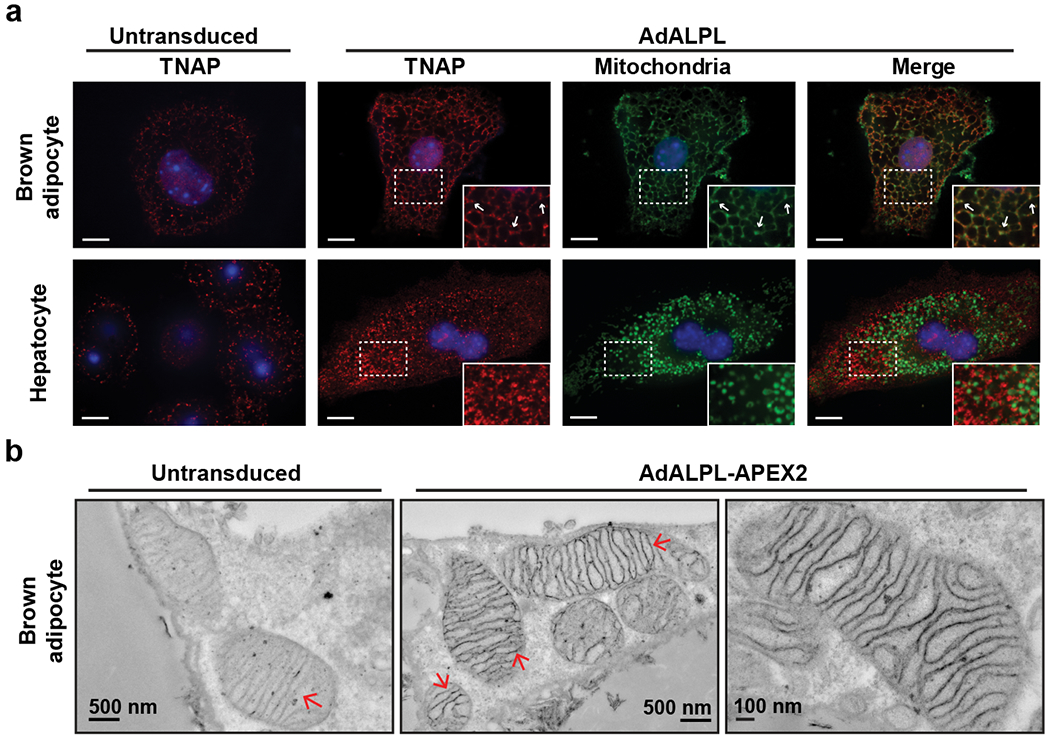
(a) Confocal fluorescence images showing subcellular localization of ectopically expressed TNAP in immortalized brown adipocytes and primary hepatocytes. The insets show an enlarged region of the image outlined by the dotted box. Arrows denote selected signals of TNAP that colocalize with mitochondria signal. Anti-TNAP (Red) and anti-HSP60 (Green) were used to visualize TNAP and mitochondria, respectively. Scale bar: 10 μm.
(b) Transmission electron microscope images showing the detailed localization of TNAP-APEX2 in primary mature brown adipocytes. Arrows denote the structures of mitochondrial inner membrane.
We next studied the subcellular location of endogenous TNAP, comparing wild-type cells with cells from a genetic knock-out of Alpl. As shown in Extended Data Fig. 3a, a large fraction of the fluorescence signals marked by the anti-TNAP antibody colocalizes with mitochondria in these brown adipocytes. In the control, genetic depletion of Alpl eliminated nearly all the mitochondria-colocalized signals (Extended Data Fig. 3a–c). Moreover, we failed to detect endogenous TNAP in mitochondria of non-thermogenic fat cell types (Extended Data Fig. 3a and d), in agreement with what we observed for ectopic TNAP, and in agreement with the extensive literature on this protein. These data together provide strong evidence that TNAP is localized to mitochondria and that this localization appears to be specific to thermogenic adipocytes, at least within the cell types examined.
As canonical TNAP is tethered to biological membranes via a glycosylphosphatidylinositol anchor (GPI-anchor) that is installed within the lumen of endoplasmic reticulum (ER), we examined whether this is also true for mitochondrial TNAP. To this end, the insoluble fraction of mitochondrial protein extract from BAT was treated with a phospholipase that is specific to phosphatidylinositol (PI-PLC). As shown in Extended Data Fig. 3f, TNAP was readily dissociated from the mitochondrial membranes by this treatment, whereas VDAC1, a mitochondrial transmembrane protein, remained associated with membranes. These data suggest that mitochondrial TNAP is indeed tethered to mitochondrial membranes via a GPI-anchor.
Ultrastructural location of TNAP
As mitochondrial localization of TNAP is highly unusual, an APEX2/TNAP-fusion protein that is useful for ultrastructural studies27 was prepared (Extended Data Fig. 4b). We ectopically expressed TNAP-APEX2 in brown adipocytes that stably express a mitochondrial fluorescent reporter (Extended Data Fig. 4a), and first confirmed the mitochondrial localization and peroxidase activity of TNAP-APEX2 by confocal microscopic imaging (Extended Data Fig. 4c). Critically, this mitochondrial localization of TNAP-APEX2 was not observed in hepatocytes (Extended Data Fig. 4c).
We next performed DAB/H2O2 staining followed by OsO4 fixing on brown adipocytes that expressed TNAP-APEX2 for electron microscopic examination. As shown in Fig. 2b, we found that the TNAP-APEX2 strongly labeled the inner mitochondrial membrane (IMM) and intermembrane space (IMS), with many dense areas coincident with the cristae structures. These images strongly resemble published staining patterns displayed by an IMS-localized protein fused to APEX or APEX227,28. Additionally, protease-protection assays of BAT mitochondria derived from cold-acclimated mice demonstrated that TNAP displayed a similar profile as known IMS-localized proteins such as glycerol-3-phosphate dehydrogenase (GPD2) and cytochrome c (Cyt C) (Extended Data Fig. 4d–e), suggesting again that TNAP is likely to reside in the IMS.
Role of TNAP in FCC and thermogenesis
Our findings suggest that TNAP might participate in adaptive thermogenesis by supporting the FCC. To test this, we silenced Alpl expression in primary brown adipocytes and found that it specifically decreased norepinephrine-stimulated respiration (Extended Data Fig. 5a–c). To assess the metabolic function of TNAP in a more acute manner, we utilized SBI-425 to ablate its enzymatic activity (Fig. 3a). Mitochondria were prepared from the iWAT fat depot from cold-acclimated mice and creatine-dependent respiration was measured. As shown in Fig. 3b and Extended Data Fig. 6a, SBI-425 treatment eliminated creatine-stimulated respiration only under limiting ADP conditions, indicating that blocking TNAP activity abolishes the FCC5. Consistent with this idea, creatine stimulated mitochondrial respiration was not observed in mitochondria derived from Adipo-Alpl KO mice (Extended Data Fig. 6a). Notably, TNAP inhibition had no effect on ADP-dependent mitochondrial respiration in the absence of creatine supplementation (Extended Data Fig. 6b). This data effectively excludes the possibility that TNAP works as an ATPase to affect respiration, despite of its high ATPase activity in vitro29. Creatine also elicited the release of a molar excess of ADP from mitochondria of brown adipocytes18; and this effect was eliminated by SBI-425 treatment (Fig. 3c). These data suggest that TNAP promotes the FCC in thermogenic adipocytes.
Figure 3. Ablation of TNAP activity abolishes the futile creatine cycle by hydrolysis of phosphocreatine.
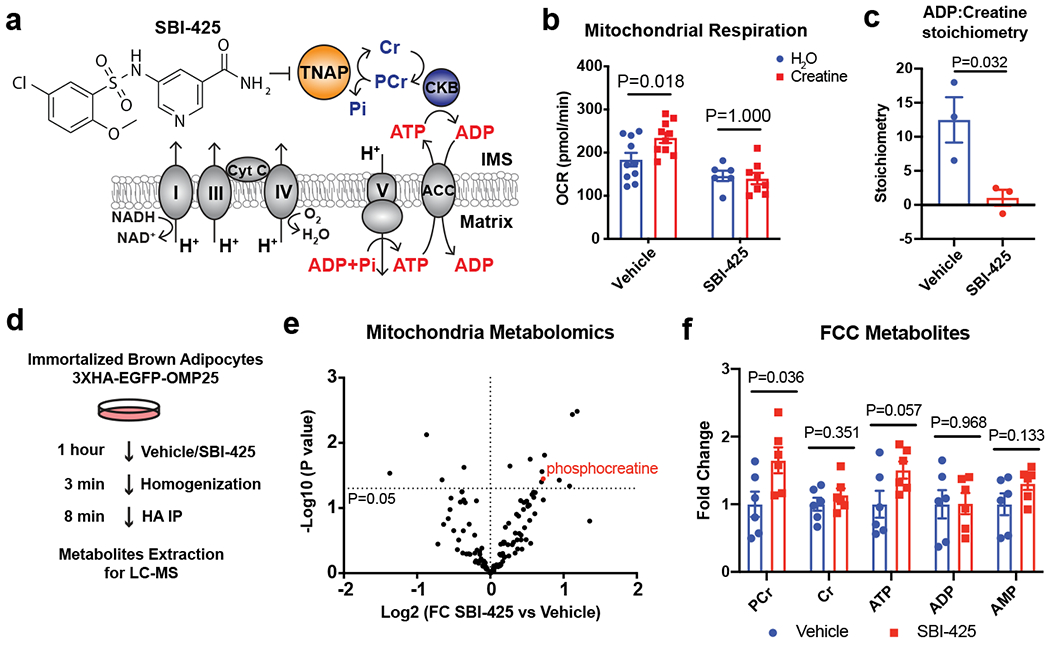
(a) Model of inhibition of FCC by the TNAP-selective inhibitor, SBI-425.
(b) Effect of SBI-425 treatment (10 μM) on creatine-dependent stimulation of oxygen consumption rate (OCR) of beige fat-derived mitochondria in the presence of 0.1 mM ADP, as measured by a Seahorse XF24 Extracellular Flux Analyzer; n=10 independent measurements for the vehicle group, 6 independent measurements for the SBI-425 group supplemented with H2O, and 8 independent measurements for the SBI-425 group supplemented with creatine.
(c) Calculated ADP:creatine stoichiometry based on the mitochondrial oxygen consumption during futile creatine cycle in presence of 0.01 mM creatine and 0.1 mM ADP, as measured by a Clark-type oxygen electrode; n=3 biologically independent samples per group.
(d) Experimental scheme of rapid mitochondrial metabolites extraction from immortalized brown adipocytes for metabolomics profiling.
(e) Volcano plot of mitochondrial metabolites of cells treated with SBI-425 (10 μM) vs Vehicle.
(f) Changes in mitochondrial levels of major metabolites involved in futile creatine cycle upon SBI-425 treatment; n=6 biologically independent samples.
Data are presented as means ± SEM. Statistical significance was calculated by either two-way ANOVA with Bonferroni’s multiple comparisons test (b) or unpaired Student’s two-sided t-test (c, e, and f).
To determine directly whether acute inhibition of TNAP was blocking PCr hydrolysis within mitochondria, we utilized a rapid mitochondrial purification scheme (MITO-Tag) recently developed to enable metabolite measurements30,31 (Fig. 3d). The successful enrichment of mitochondria by this approach was confirmed by the elevated levels of several mitochondrial metabolites and proteins (Extended Data Fig. 7a–b). Importantly, mass spectrometric profiling of the mitochondrial metabolites revealed that the PCr level was among the most quantitatively and significantly altered metabolites (65% increase) due to TNAP inhibition (Fig. 3e–f). As expected, this blockade of PCr hydrolysis led to some (non-significant) elevation of ATP levels. These data are consistent with a role for TNAP as a mitochondrial PCr phosphatase in thermogenic fat cells.
To examine the role of TNAP on energy expenditure in vivo, mice were subjected to the TNAP inhibitor SBI-425 simultaneous with stimulation of adaptive thermogenesis by injection with CL 316,243, a β3 adrenergic receptor agonist. The treatment with the TNAP inhibitor caused a large (30%) reduction in whole-body energy expenditure elicited by this β3 adrenergic stimulation (Fig. 4a–b); importantly, this occurred without changes in basal respiration, physical movement, or food intake (Fig. 4b and Extended Data Fig. 8a). In contrast, the drug-dependent suppression of respiration was completely lost in mice bearing a fat-specific deletion of TNAP (Adipo-Alpl KO) (Fig. 4c–d, and Extended Data Fig. 8b). These data together provide very strong evidence that TNAP activity in adipose tissues contributes to adaptive thermogenesis.
Figure 4. Ablation of TNAP in fat represses adaptive thermogenesis and stimulates obesity.
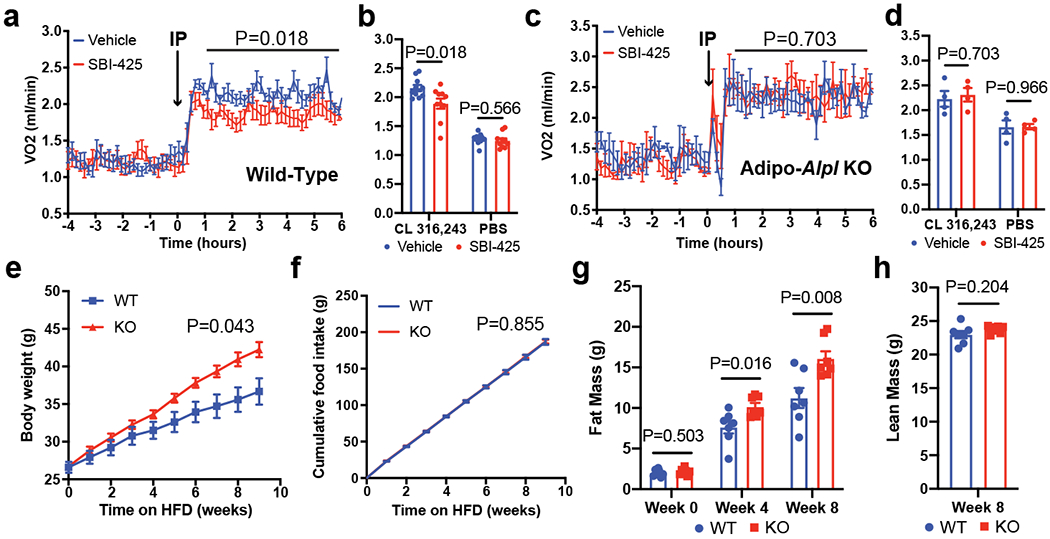
(a) Indirect calorimetric measurement on wild-type mice kept in metabolic cages at 22 °C, showing effect of SBI-425 administration (25 mg/kg/day) on CL 316,243 (1.0 mg/kg/day) stimulated respiration. Arrow denotes the time point of drug administration; n=10 mice per group; all mice were pre-treated with CL 316,243 (1.0 mg/kg/day) for 5 days before SBI-425 administration.
(b) Averaged respiration rates measured in (a) over the time span from 1-hour to 6-hour after drug administration along with the administration of either CL 316,243 or PBS; n=10 mice per group.
(c) Indirect calorimetric measurement on Adipo-Alpl KO mice kept in metabolic cages at 22 °C, showing effect of SBI-425 administration (25 mg/kg/day) on CL 316,243 (1.0 mg/kg/day) stimulated respiration. Arrow denotes the time point of drug administration; n=4 mice per group; all mice were pre-treated with CL 316,243 (1.0 mg/kg/day) for 5 days before SBI-425 administration.
(d) Averaged respiration rates measured in (c) over the time span from 1-hour to 6-hour after drug administration along with the administration of either CL 316,243 or PBS; n=4 mice per group.
(e-h) Body mass (e), cumulative food intake (f), fat mass (g) and lean mass (h) of Adipo-Alpl KO mice and their littermate controls over high-fat feeding at 22 °C; n=7 mice per group.
Data are presented as means ± SEM. Statistical significance was calculated by either two-way ANOVA (a, c, e, and f) or unpaired Student’s two-sided t-test (b, d, g, and h).
Ablation of TNAP stimulates obesity
The FCC has been shown to play an important role in diet-induced thermogenesis (DIT)16–18. We therefore studied the role of TNAP in whole body energy balance by feeding a high-fat diet (HFD) to Advpo-Alpl KO mice and their littermate controls. Fig. 4e shows that the KO mice had a strikingly faster increase in body weight compared to the control animals. The KO mice also gained more fat mass than their littermate controls (Fig. 4g). Critically, this increased weight and fat mass in the KOs occurred with no changes in lean mass, food intake or physical movement (Fig. 4f, 4h, and Extended Data Fig. 9c). Moreover, indirect calorimetry showed a lower whole-body energy expenditure for HFD-fed KO mice compared with their littermate controls (Extended Data Fig. 9a–b). These data indicate an important role of TNAP in protecting mice from diet-induced obesity. Surprisingly, Adipo-Alpl KO mice did not exhibit an apparent deficiency in CL 316,243-induced thermogenesis (Extended Data Fig. 10a); this might be due to an upregulation of UCP1, as well as other proteins involved in oxidative metabolism, in a likely compensatory pathway (Extended Data Fig. 10b–d). This has also been observed when the FCC is inhibited in mice via fat-specific gene knockouts for GATM16 or CKB18. Interestingly, this upregulation of UCP1 protein occurs without increase in its mRNA abundance, suggesting it is governed by post-transcriptional regulation (Extended Fig. 10e–f).
Discussion
We previously provided stoichiometric evidence that futile cycling between creatine and phosphocreatine was part of a novel thermogenic pathway5, but there was no direct demonstration of phosphocreatine dephosphorylation. Here we demonstrate for the first time the existence of an enzyme that hydrolyses phosphocreatine in a biological context. Surprisingly, this enzyme is the very well-studied phosphatase TNAP, and it plays a previously unrecognized role in thermogenesis. TNAP in thermogenic fat cells takes up a localization in mitochondria, where it has never been previously observed. The cellular mechanisms by which TNAP becomes localized in mitochondria in a cell-selective manner are not clear but are likely to involve one or more factors that allow TNAP to diverge from its usual ER-Golgi pathway32. The role for TNAP in adipose thermogenesis is demonstrated by: first, the loss of O2 consumption in isolated thermogenic fat mitochondria and cells upon loss of TNAP by chemical inhibition and shRNA-mediated knock-down, respectively; second, the reduced energy expenditure in vivo upon chemical inhibition of TNAP. That this drug was acting “on target” was demonstrated with mice lacking TNAP in fat. Lastly, mice with fat-selective ablation of TNAP develop rapid-onset obesity without any change in food intake or physical movement. It is important to note that this obesity on a high fat diet far exceeds anything that has been shown with loss of UCP111,33,34. It is worth noting that the portion of adrenergic thermogenesis attributable to FCC is not fully determined due to the secondary effects of the genetic disrupt of TNAP. Nevertheless, we believe that the reduced energy expenditure in vivo caused by the chemical inhibition of TNAP represents a rough estimation of the contribution of FCC; this is in agreement with what has been shown for mice bearing fat-specific deletion of CKB, which interferes with the FCC prior to the phosphorylation of creatine18. The data from the TNAP-APEX2/EM study and trypsin protection assay suggest that TNAP is attached to the IMM and resides in the IMS. This is interesting because the mitochondrial CKB was also likely to reside in the IMS18. Whether there is a physical association between TNAP and CKB in thermogenic adipocytes remains to be determined. Although TNAP is active on both PCr and ATP and both are present in IMS, ATP isn’t substantially hydrolyzed, evidenced by that State 3 respiration is not affected by the inhibition of TNAP in the absence of creatine. The reason why ATP isn’t a substantial substrate of TNAP is not clear but seems most likely because that the effective transfer of ATP might be spatially compartmentalized within the mitochondrial CK and ANT35. Finally, the FCC must operate without compromising the energetic requirements of normal cellular functions in thermogenic adipocytes. This is in agreement with our mitochondrial metabolomics data, which shows that, with the operation of FCC, a substantial fraction of the mitochondrial PCr remains unhydrolyzed at steady state. This remaining PCr is likely to be exported from mitochondria and utilized to allow normal cellular processes to proceed.
Our previous genetic studies demonstrated the beneficial metabolic functions of the FCC in fat cells and on whole-body energy expenditure by altering the supplies of creatine or phosphocreatine5,16,17. This suggested that at least some human populations, especially the obese, might benefit from dietary creatine supplementation. Our current work elucidates a direct molecular target that participates in the major heat-generating step of FCC. Whether the mitochondrial TNAP undergoes any cell-selective modifications that render it subject to drugging in a more specific way remains to be determined. In addition to adaptive thermogenesis and anti-obesity actions, it is possible that the FCC might have some involvement in pathological thermic events that are poorly understood, such as in during hyperthyroidism, burn injury, anesthesia-induced hyperthermia, and cancer cachexia36–40. These possibilities deserve further study.
Methods
Mitochondria Isolation and Purification
Mitochondria isolation and fractionation were conducted at 4°C. Murine tissues were excised, washed with ice-cold SHE buffer (250 mM sucrose, 10 mM HEPES, 0.1 mM EGTA, and pH 7.2), minced to homogeneity, and further homogenized with a motorized teflon pestle in complete SHE buffer (5 ml g−1, SHE buffer supplemented with 2% essentially fatty acid-free BSA (Sigma Aldrich) and 1X EDTA-free Protease Inhibitor Cocktail (Roche)). Homogenates were passed through 2 layers of cheesecloth and centrifuged at 8,500 g for 10 min to remove lipids in the supernatant and to pellet debris, nuclei and mitochondria. The pellet was resuspended in complete SHE buffer, transferred to a clean tube, and centrifuged at 700 g for 10 min to spin down nuclei and cell debris. The supernatant was transferred to a clean tube again and centrifuged at 8,500 g for 10 min to pellet crude mitochondria. The mitochondrial pellet was then washed with complete SHE buffer two times and either pelleted for long-term storage in −80°C or resuspended to yield a chosen concentration of mitochondrial protein with an appropriate buffer (10 mg ml−1 in complete SHE buffer if not otherwise indicated).
For sucrose gradient purification, sucrose was dissolved to a concentration of 1M and 1.5M in sucrose gradient buffer (10 mM HEPES, 5 mM EDTA, 2 mM DTT, and pH 7.8) and gently layered in a 5 ml polyallomer centrifuge tube (first 2 ml of 1.5 M sucrose solution for bottom layer, and then 1.5 ml of 1.0 M sucrose solution for top layer). 5 mg of crude mitochondria resuspension (10 mg ml−1 in complete SHE buffer) were loaded on top of the sucrose solution layers and centrifuged at 28,000 rpm for 30 min at 4°C using the SW55Ti rotor. Purer mitochondria that banded at the interface of the sucrose cushion were carefully extracted, washed twice in complete SHE buffer, and stored at −80°C in pellet form till further protein extraction for either activity assays or biochemical fractionations.
Fractionations of Mitochondrial Protein Extract
Sucrose gradient-purified mitochondria pellets were resuspended in ice-cold resuspension buffer (100 mM Tris, 0.1 mM EGTA, and pH 7.8), fragmented using a cold H2O-bath ultrasonicator at high power-level for 15 min, and centrifuged at 9,000 g for 10 min to spin down unfragmented mitochondria. The supernatant was transferred to a 5 ml polypropylene centrifuge tube, topped with resuspension buffer, and centrifuged at 32,4000 rpm for 1 hr at 4°C using a SW55Ti rotor. The resultant supernatant that contains the “soluble” fraction was quantified using BCA assay and re-concentrated to 2.0 mg ml−1 of protein using an Amicon centrifugal filter unit (3k MWCO); the pellet that contains the “insoluble” fraction was resuspended with a small volume of solubilization buffer (100 mM Tris, 0.1 mM EGTA, 0.5% NP-40, and pH 7.8), ultrasonicated till the solution turned clear, quantified using BCA assay, and diluted with solubilization buffer to yield a protein concentration of 2.0 mg ml−1. Both “soluble” and “insoluble” fractions were then assayed for PCr phosphatase activity using 31P NMR methods (see below).
For further fractionation by size-exclusion chromatography (SEC) on FPLC, 1.0 mg of solubilized “insoluble” fraction was loaded on a Superdex 200 Increase 10/300 GL column (GE Healthcare) pre-equilibrated with solubilization buffer. The eluents were collected in 1.0 ml fractions and assayed for PCr phosphatase activity using the 31P NMR assay (see below). The active fractions were next fractionated by ion-exchange chromatography (IEX) on FPLC, using a Mono Q 5/50 GL column (GE Healthcare). Both the unbound and NaCl-eluted analytes were collected in 0.5 ml fractions and assayed for PCr phosphatase activity using enzyme-coupled phosphatase assay (see below); the activity of the most active fractions was verified by 31P NMR.
Phosphatase Activity Assays
31P NMR spectra were acquired on Bruker 500 MHz Avance III Spectrometer equipped with a Smart probe for multi-nuclear detection and SampleJet for automated sample-handling. Each spectrum was recorded on 0.5 ml of aqueous solution sample in a 5 mm NMR tube at 298 K; with a spectral width of 50 kHz, relaxation delay of 2 seconds, and scan number of either 64 or 256. 10% D2O was included in each sample for the external field-frequency lock. TopSpin v4.0.7 was used for NMR data processing and analysis. The phosphorus in PCr and Pi resonates at −3.2 and 2.2 ppm, respectively. For continuous PCr phosphatase activity measurement, each reaction was initiated by mixing 10 mM PCr with a protein sample in an NMR tube right before the recording of spectrum for the first time point. The progressing reaction was kept in the NMR tube held in SampleJet at room temperature between measurements for the entire time course. Before addition of PCr for activity measurements, mitochondrial protein extract was passed through PD-10 Desalting column (GE Healthcare) for removal of endogenous metabolites.
Enzyme-coupled phosphatase assay was performed using EnzChek Phosphate Assay Kit (Molecular Probes) that couples the generation of Pi to an enzymatic conversion of purine riboside to purine that causes a spectrophotometric shift in maximal absorbance from 330 nm to 360 nm. To assay the PCr phosphatase activity of IEX fractions, 90 ul of solution from each fraction was mixed with 10 ul of 10X PCr stock solution (100 mM, dissolved in H2O), and incubated at room temperature for 48 hours. Endpoint measurements were performed in duplicate by mixing 20 ul of the phosphatase reaction with 80 ul of coupling-reaction components to make a final solution containing 0.2 mM purine ribose (MESG), 1 unit of purine nucleoside phosphorylase (PNPase), and 1X reaction buffer (50 mM Tris-HCl, 1 mM MgCl2, 0.01 mM sodium azide, and pH 7.5). After 10 min of equilibration, absorbance was measured at 360 nm using a FLUOstar Omega plate reader at room temperature.
To derive the Michaelis-Menten curves of TNAP toward different substrates, continuous enzyme-coupled assay was performed in duplicate at 37°C on a reaction mixture containing 0.05 μM of recombinant human TNAP (R&D Systems, 2909-AP) and a serial concentration of substrate in assay buffer (50 mM Tris-HCl, 1 mM MgCl2, 20 μM ZnCl2, and pH 7.8) supplemented with 0.2 mM MESG and 1 unit of PNPase. Non-linear regression of Michaelis-Menten model to measured activities was performed with GraphPad Prism version 8 for Mac (GraphPad Software, La Jolla California USA).
Quantitative Mass Spectrometry
Proteins in selected IEX fractions were precipitated by 25% ice-cold HPLC-grade TCA overnight in cold and were washed three times with 100% ice-cold HPLC-grade MeOH to remove detergent. Dried protein precipitates were trypisinized, TMT-labeled, and subjected to orthogonal bpHrp fractionation as previously described5. The subsequent LC-MS/MS experiments, data processing, and quantitative analysis were all performed as previously described5.
Gene Expression Analysis by Quantitative PCR (qRT-PCR)
Total RNA was prepared from frozen tissue using TRIzol (Invitrogen) extraction and RNeasy Mini purification kit (QIAGEN); and reverse transcribed using HighCapacity cDNA Reverse Transcription kit (Applied Biosystems). The resultant cDNA was analyzed by qRT-PCR. Briefly, 10 ng cDNA and 150 nmol of each primer were mixed with SYBR GreenERTM qPCR SuperMix (Applied Biosystems). Reactions were performed in a 384-well format using an ABI PRISM 7900HT real time PCR system (Applied Biosystems). Relative mRNA levels were referenced to mRNA level of Rplp0, if not otherwise indicated. A complete list of primers and sequences can be found below.
qRT-PCR Primers
Alpl Fwd: TCA ACA CCA ATG TAG CCA AGA, Rev: GTA GCT GGC CCT TAA GGA TTC; Rplp0 Fwd: GGA GTG ACA TCG TCT TTA AAC CCC, Rev: TCT GCT CCC ACA ATG AAG CA; Pparg Fwd: TCG CTG ATG CAC TGC CTA TG, Rev: GAG AGG TCC ACA GAG CTG ATT; Pgc1a Fwd: CCC TGC CAT TGT TAA GAC C, Rev: TGC TGC TGT TCC TGT TTT C; Prdm16 Fwd: CAG CAC GGT GAA GCC ATT C, Rev: GCG TGC ATC CGC TTG TG; Adipoq Fwd: GCA CTG GCA AGT TCT ACT GCA A, Rev: GTA GGT GAA GAG AAC GGC CTT GT; Ucp1 Fwd: CAC CTT CCC GCT GGA CAC T, Rev: CCC TAG GAC ACC TTT ATA CCT AAT GG; Leptin Fwd: ATG TGC TGG AGA CCC CTG TG, Rev: TCA GCA TTC AGG GCT AAC ATC C; Ckb Fwd: GCC TCA CTC AGA TCG AAA CTC, Rev: GGC ATG TGA GGA TGT AGC CC; Gatm Fwd: GAC CTG GTC TTG TGC TCT CC, Rev: GGG ATG ACT GGT GTT GGA GG; Enpp1 Fwd: GGC CCG AAA TCA GAC AGG AA, Rev: CTG ACA AAA CCA GCG ACA GC; Phospho1 Fwd: AAG CAC ATC ATC CAC AGT CCC TC, Rev: TTG GTC TCC AGC TGT CAT CCA G.
Western Blotting
Protein concentration was determined using the bicinchoninic acid assay (Pierce). 7.5 μg of protein sample was loaded in each lane for western blot analysis if not otherwise indicated. Protein samples were denatured in 1X NuPAGE LDS Sample Buffer (Invitrogen) without boiling, resolved by 4-12% NuPAGE BisTris SDS–PAGE (Invitrogen), and transferred to a polyvinylidene difluoride (PVDF) membrane. Primary antibody incubations were performed in TBS containing 0.05% Tween (TBST) and 5% milk, for either 4 hr at room temperature or overnight at 4°C; secondary antibody incubations were performed using either horseradish peroxidase (HRP)-conjugated or IRDye secondary antibodies in TBST with 5% milk for 2 hr at room temperature, for visualization using Immobilon Crescendo Western HRP substrate (Millipore) or fluorescence signal respectively. A complete list of antibodies can be found below.
Antibodies
The following commercially available antibodies were used for Western blotting: TNAP (R&D Systems, AF2910), OXPHOS (Abcam, ab110413), UCP1 (Abcam, ab10983), VDAC (Abcam, ab34726), COX1 (Abcam, ab14705), HSP60 (Cell Signaling, 12165), and VCL (Sigma-Aldrich, V9264). The following commercially available antibodies were used for immunocytochemistry: TNAP (R&D Systems, MAB29092), HSP60 (Cell Signaling, 12165), OXPHOS (Abcam, ab110413), Gt-anti-Ms Alexa Fluor 568 (Invitrogen, A-11004), and Gt-anti-Rb Alexa Fluor 647 (Invitrogen, A-21245).
Phospholipase-C, phosphatidylinositol specific (PI-PLC) Treatments
The insoluble fraction of mitochondria (1.0 mg) derived from BAT of cold-acclimated mice was subjected to 1 unit of PI-PLC (Sigma-Aldrich) for 10 min at 30°C. The reaction product was centrifuged at 17,000 g for 10 min at 4°C to remove protein precipitates and subjected to ultracentrifugation at 32,400 rpm for 1 hour at 4°C using a SW55Ti rotor. The pellet was resuspended with solubilization buffer (100 mM Tris, 0.1 mM EGTA, 0.5% NP-40, and pH 7.8) and volume-matched to supernatant. Both the solubilized pellet and supernatant were then subjected to western-blot analysis.
Brown Adipocyte Cultures
Primary brown preadipocytes were prepared from stromal vascular fraction (SVF) of interscapular brown adipose tissue as described previously45. In general, brown preadipocytes were grown to 100% confluency and induced to differentiate with 1 μM rosiglitazone, 0.5 mM IBMX, 5 μM dexamethasone, 0.114 μg ml−1 insulin, 1 nM T3, and 125 μM Indomethacin, for 2 days. Cells were re-fed every 2 days with 1 μM rosiglitazone, 0.5 μg ml−1 insulin, and 1 mM T3, and were fully differentiated by day 8 post-induction. For ICC, preadipocytes were seeded on 12 mm PDL-coverslip (Corning, 354086) coated with 10 μg ml−1 laminin (Sigma-Aldrich, L2020) and grown till 100% confluency before induced to differentiate. Cells were subjected to adenoviral transduction on day 4 and imaging studies on day 8 post-induction.
Immortalized brown preadipocytes were prepared using 3T3 immortalization protocol as described previously46. For differentiation, immortalized brown preadipocytes were grown to 100% confluency and induced to differentiate with 1 μM rosiglitazone, 0.5 mM IBMX, 5 μM dexamethasone, 0.114 μg ml−1 insulin, 1 nM T3, and 125 μM Indomethacin for 4 days. Cells were re-fed every 2 days with 1 μM rosiglitazone, 0.5 μg ml−1 insulin, and 1 mM T3, and were fully differentiated by day 10 post differentiation. For ICC, preadipocytes were seeded on 12 mm PDL-coverslip (Corning, 354086) coated with 10 μg ml−1 laminin (Sigma-Aldrich, L2020) and grown till 100% confluency before induced to differentiate. Cells were subjected to adenoviral transduction on day 8 and imaging studies on day 10 post-induction. Stable integration of mitochondrial reporter, 3XHA-EGFP-OMP2530, was mediated by retroviral transduction in preadipocytes right before initiation of differentiation.
Primary mature brown adipocytes were collected and attached to coverslips as described previously9. Aclar 12 mm coverslips pre-coated with 50 μg ml−1 Poly-D-Lysine (Sigma-Aldrich, P4707) and 10 μg ml−1 laminin (Sigma-Aldrich, L2020) were used. The attached cells were subjected to adenoviral transduction for 48 hours before electron microscopic imaging studies.
Hepatocyte, Myotube, and PTEC Cultures
Primary hepatocytes were prepared from perfused livers dissected from 8- to 12-week-old male C57BL/6J mice, as described previously47. Hepatocytes were seeded on PDL-coverslips coated with 10 μg ml−1 laminin at a density of 20,000/cm2, in maintenance medium (DMEM with 0.2% BSA, 2 mM sodium pyruvate, 1X Penicillin/streptomycin, 0.1 μM dexamethasone, and 1 nM insulin). The attached hepatocytes were subjected to adenoviral transduction for 2 days before imaging studies. Primary myoblasts were prepared from limb muscle dissected from 8- to 12-week-old male C57BL/6J mice as described previously48. To initiate fusion, myoblasts were seeded on PDL-coverslips coated with 10 μg ml−1 laminin at a density of 50,000/cm2 and were induced next day by switching to fusion medium (5% horse serum in DMEM, 1X PenStrep). The resultant myotubes were subjected to adenoviral transduction on day 2 and imaging studies on day 4 post-induction. AML12 and PTEC cells were obtained from ATCC and cultured based on manufacturer’s instruction.
Confocal Fluorescent Microscopic Imaging and Analysis
Cells attached on PDL-coverslips (Corning, 354086) coated with 10 μg ml−1 laminin (Sigma-Aldrich, L2020) were fixed by 4% paraformaldehyde (1 volume of 16% paraformaldehyde (Electron Microscopy Sciences) diluted with 3 volumes of PBS) for 10 min at RT, washed 3 times with PBS, permeabilized by permeabilization solution (PBS supplemented with 0.1% Triton x-100) for 10 min at RT, and blocked by blocking solution (PBS supplemented with 0.1% Triton x-100 and 5% FBS) for 10 min at RT. For immunofluorescence staining, anti-TNAP mouse monoclonal antibody (R&D Systems, MAB29092) at 1:100 and anti-HSP60 rabbit monoclonal antibody (Cell Signaling, 12165) at 1:500 in blocking solution were used for primary labeling of TNAP and mitochondria, respectively; and anti-mouse Alexa Fluor 568 and anti-rabbit Alexa Fluor 647 at 1:1000 (Invitrogen) were used for secondary labeling. 3 μM DAPI (BioLegend) was used to stain nuclei. Coverslips were mounted in ProLong Diamond Antifade Mountant (Invitrogen). Fluorescent images were acquired using MetaMorph v7.8.13.0 on a Nikon Ti motorized inverted microscope equipped with Yokogawa spinning disk confocal, and stored and managed using OME Remote Objects (OMERO)49. Pearson’s Correlation Coefficient was calculated using Coloc2 plugin in Fiji50 on individual cells that were outlined manually.
APEX2-Assisted Fluorescence and Electron Microscopic Imaging
Immortalized brown adipocytes stably integrated with 3XHA-EGFP-OMP25 were attached to laminin-coated PDL-coverslips prior to initiation of differentiation. On day 6 of differentiation, cells were subjected to adenoviral transduction to express TNAP-APEX2, and, on day 10 of differentiation, subjected to fluorescent staining using Alexa Fluor 647 Tyramide SuperBoost Kit according to the manufacturer’s instruction (Invitrogen, B40936). Confocal fluorescent imaging was performed as described above. Immortalized hepatocytes (AML12) integrated with 3XHA-EGFP-OMP25 were attached to PDL-coverslips and subjected to adenoviral transduction next day to express TNAP-APEX2. Fluorescent staining using Alexa Fluor 647 Tyramide SuperBoost Kit was perfomed 4 days after transduction.
Primary mature brown adipocytes described above were subjected to adenoviral transduction to express TNAP-APEX2. After 48 hours, the localization of TNAP-APEX2 was detected by electron microscopy as described previously27. In detail, after 48 hours of ectopic expression, cells were fixed by freshly prepared 2% glutaraldehyde for 30 min, quenched with 20 mM glycine, washed with PBS, and incubated in solution containing 0.7 mg ml−1 diaminobenzidine (DAB) and 0.7 mg ml−1 H2O2 in Tris buffer (Sigma, D4293) for 6 hr. After extensive washing, samples were post-fixed with 1% osmium tetroxide, dehydrated and embedded in EPON resin. 48 h later, coverslips were removed and areas containing cells were randomly selected and mounted. Ultrathin sections (70 nm) were cut and examined on a JOEL electron microscope.
Protease Protection Assay
Differential centrifugation-purified mitochondria derived from cold-acclimated (7 days) mice, were incubated with trypsin in presence of varying concentrations of digitonin (see figure legends for details) for 30 minutes at room temperature. Mitochondria were treated rapidly with 1% SDS and boiled at 95 °C for 5 min to denature proteins and kept at −20 °C before Western blot analysis.
Adenovirus Preparations and Use
pAd-DEST expression clones were prepared using Gateway recombination cloning technology (Invitrogen). For AdALPL, the open reading frame (ORF) of mouse Alpl was amplified from a cDNA clone (Origene, MR208392), inserted into pDONR221 vector via BP recombination reaction, and subcloned into pAd/CMV/V5-DEST vector via LR recombination reaction. For AdALPL-APEX2, site-directed mutagenesis was utilized to insert the APEX2 DNA sequence (Addgene) into the pDONR221-ALPL construct, between nucleotide c1494 and t1495 of the ORF of Alpl (APEX2 peptide will thus be expressed between amino acid residue A498 and W499 of wild-type TNAP (Uniprot: P09242)). The resultant pDONR221-ALPL-APEX2 was subcloned into pAd/CMV/V5-DEST vector via LR recombination reaction.
Adenovirus was produced using ViraPower™ Adenoviral Expression System (Invitrogen). In brief, pAd-DEST expression clones were digested by Pac I restriction enzyme (New England Biolabs), gel purified, and packed by Lipofectamine 2000 (Invitrogen) to transfect 293A cells. The cells were then grown for 10-13 days to allow appearance of 80% cytopathic effect before being harvested. The titer numbers were determined by Adeno-X™ Rapid Titer Kit (Takara Bio), and an MOI of 500 was used to transduce adipocytes.
Mitochondria Respiration
XF24 Extracellular Flux Analyzer (Seahorse Bioscience) was used to measure mitochondrial respiration as described previously5. In brief, 5-10 μg mitochondria isolated from iWAT of cold-acclimated mice were plated in each well of the XF24 V7 cell culture microplate, in a cold respiration buffer (125 mM sucrose, 20 mM K-TES at pH 7.2, 2mM MgCl2, 1 mM EDTA, 4 mM KH2PO4, 4% fatty acid-free BSA, 10 mM pyruvate, 5 mM malate, and 1 mM GDP); and adhered to the bottom by centrifugation at 2,000 g for 20 min at 4 °C. To inhibit TNAP, adhered mitochondria were treated with 10 μM final concentration of SBI-425 (100 mM stock in 100% DMSO) in respiration buffer for 8 min at 37 °C, prior to respiration measurement. Creatine was added to a final concentration of 0.01 mM, and coupled respiration was initiated by addition of 0.1 mM of ADP to detect the creatine-dependent stimulation.
To determine the ADP:creatine stoichiometry during the futile creatine cycle (FCC), mitochondria respiration was monitored by a Clark-type oxygen electrode (Rank Brothers) as described18. Mitochondria were isolated from differentiated immortalized brown preadipocytes described above by differential centrifugation using an MSH buffer (210 mM mannitol, 70 mM sucrose, 20 mM HEPES, and pH 7.8). For each respiration measurement, 0.25 mg mitochondria in respiration buffer (125 mM sucrose, 20 mM K-TES at pH 7.2, 2mM MgC2, 1 mM EDTA, 4 mM KH2PO4, 4% fatty acid-free BSA, 10 mM pyruvate, 5 mM malate, and 1 mM GDP) was used, and final concentrations of 0.01 mM creatine and 0.1 mM ADP were added to stimulate FCC. Pilot experiments were conducted to determine the length of time required to reach state 4 respiration. State 4 respiration was defined as the respiratory rate, following ADP addition, that closely matched the respiratory rate prior to ADP addition. Once these parameters were established, total nmoles of oxygen consumed was measured over this time, based on the oxygen consumption rate (measured as nmoles/min). The excess of oxygen consumed by creatine supplementation was used to quantify ADP liberation based on a P/O ratio of 2.7, which is then used to calculate the ADP:creatine stoichiometry.
Adenoviral shRNA Cloning, Construction, and Use
Complementary DNA oligos for shRNA generation were annealed and cloned into the pENTRTM/U6 entry vector according to the manufacturer’s instructions (Invitrogen; K4944). Sequences for shRNA targeting Alpl were: sense, CAC CGC CTG GAT CTC ATC AGT ATT TCG AAA AAT ACT GAT GAG ATC CAG GC; antisense, AAA AGC CTG GAT CTC ATC AGT ATT TTT CGA AAT ACT GAT GAG ATC CAG GC. Cloned shRNA was shuttled into the pAD/BLOCK-iTTM-DEST vector and transfected into 293A cells. Adenovirus was produced according to the manufacturer’s instructions (Invitrogen; K4941), and purified using the Fast Trap Adenovirus Purification and Concentration Kit (EMD Millipore), according to the manufacturer’s instructions. Primary adipocytes were transduced with purified adenovirus in the evening of day 1 and then again in the evening at day 3 of differentiation, with medium replacement the following morning each time.
Brown Adipocyte Respiration
Cellular respiration of primary brown adipocytes was measured using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience). Preadipocytes were plated at 6,000 cells per well of an XF24 V7 cell culture microplates, and differentiation was induced 12 hours later. On day 6 of differentiation, adipocyte culture medium was changed to respiration medium consisting of DMEM lacking NaHCO3 (Sigma), 1.85 g/L NaCl, 3 mg/L phenol red, 2% fatty-acid-free BSA, and 1 mM sodium pyruvate, at pH 7.4. Uncoupled respiration was determined by addition of 2.5 μM oligomycin. Maximal respiration was determined by addition of 0.5 mM 2,4-dinitrophenol (DNP). Rotenone and antimycin (5 μM each) were used to inhibit Complex 1- and Complex 3-dependent respiration.
Rapid Mitochondria Extraction
This was performed as per the procedures of Chen, W. W., et al30. Immortalized brown preadipocytes stably expressing 3XHA-EGFP-OMP25 described above were plated on 6-well plates and grown till 100% confluency before induced to differentiate. On day 12 of differentiation, cells were treated with 10 μM SBI-425 (as described above) vs vehicle for 1 hour, prior to rapid immunoprecipitations of 3XHA-tagged mitochondria as described previously30 with minor changes. In detail, cells from each well were washed once with 0.5 ml cold PBS, once with 0.5 ml cold KPBS (136 mM KCl, 10 mM KH2PO4, pH 7.25), harvested in 1 ml cold KPBS, transferred to a pre-chilled 2 ml homogenizer glass vessel (VWR, 89026-386), and disrupted by 20 stokes of a plain plunger (VWR, 89026-398) on ice. The resultant homogenate was poured into one 1.5 ml tube and centrifuged at 1,000 g for 2 min at 4 °C, and the supernatant was collected and loaded onto Pierce anti-HA magnetic beads (Thermo Fisher Scientific) prewashed with cold KPBS. For cell lysate from one well of a 6-well dish, 20 μl slurry of anti-HA beads was used. The lysate/anti-HA beads mixture was incubated on a rotator for 3.5 min in cold and pulse spun to collect any residual liquid on the lid. The anti-HA beads were then collected on a magnet strip and the liquid was aspirated. The beads were quickly resuspended with 1 ml cold KPBS, split into 0.8 ml for metabolite extraction and 0.2 ml for western-blot analysis, and collected again on a magnet strip before the liquid was aspirated. Metabolites were eluted with 55 ul pre-chilled 80% MeOH, and proteins were eluted with 50 ul 1X NuPAGE LDS Sample Buffer (Invitrogen) supplemented with 1% BME. The time that each step takes were strictly controlled among different purifications to minimize the variations in IMS metabolites levels introduced by the leakage over time across the permeable outer mitochondrial membrane during the isolation. Metabolites extraction was centrifuged at 17,000 g for 10 min at 4 °C, and 50 ul of supernatant was collected, flash-frozen and stored in liquid nitrogen till further processing.
Polar Metabolomics Profiling
The mitochondrial metabolites extraction in 50 ul 80% MeOH was lyophilized using a SpeedVac system (Labconco) for 15 min at 4 °C. The resultant lyophilized samples were resuspended in 12 ul LC-MS grade water and kept on dry ice till subjected to polar metabolomics profiling using the AB/SCIEX 5500 QTRAP triple quadrupole instrument as described previously51. Peak integration was performed using MultiQuant v2.0. The whole-cell metabolites were extracted as described previously51 from fully differentiated immortalized brown preadipocytes described above.
Animals
All mice for mitochondria isolation and physiological studies were male C57BL/6J (6 – 8 weeks of age) obtained from The Jackson Laboratory if not otherwise indicated. To breed Adipo-Alpl KO mice, Alpfl/fl mice were obtained from Sanford Burnham Prebys Medical Discovery Institute52 and crossed with adiponectin-cretg/0 line maintained by Beth Israel Deaconess Medical Center to generate Alplfl/−+adiponectin-cretg/0 mice; these were then back-crossed with Alplfl/fl mice to generate Alplfl/fl+adiponectin-cretg/0 (Homo Adipo-Alpl KO), Alplfl/−+adiponectin-cretg/0 (Hetero Adipo-Alpl KO), and their littermate control mice. All mice were housed at 22°C under a 12 hr light/dark cycle with free access to food and water. Animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee (IACUC) of Beth Israel Deaconess Medical Center.
Indirect Calorimetry
Whole body metabolic rate was measured using the Promethion Metabolic Cage System (Sable Systems, Las Vegas, NV). Animals were housed individually in metabolic chambers maintained at 22°C under a 12 hr light/dark cycle with free access to food and water. Cold exposure or intraperitoneal injection of CL316,243 (Sigma-Aldrich; 1 mg kg−1) into mice started at the indicated time.
Body Temperature
Mice were either housed at room temperature or pre-acclimatized at thermoneutrality (28–30 °C) for 1 week before being shifted to 4°C. Body temperature was measured with and a microprobe thermometer (Physitemp, BAT-12) equipped with a rectal probe (Physitemp, RET3).
High Fat Feeding
Adipo-Alpl KO mice and age-matched littermate controls were housed at 22°C and fed a chow diet till 10 weeks of age. Mice were then singly caged and switched to high fat diet (Research Diets, D12492) containing 60% kcal% fat, 20% kcal% carbohydrate, and 20% kcal% protein. Mice have free access to food and water. Food was changed every week.
Body Composition Analysis
Fat and lean mass of mice were analyzed with Echo MRI (Echo Medical Systems) using the 3-in-1 Echo MRI Composition Analyzer.
Statistics and Reproducibility
Data were expressed as mean± SEM. P values were calculated using two-tailed Student’s t-test for pairwise comparison of variables, one-way ANOVA for multiple comparison of variables, and two-way ANOVA for multiple comparisons involving two independent variables. Sample sizes were determined on the basis of previous experiments using similar methodologies. For all cellular and animal experiments, all stated replicates are biological replicates. For SBI-425 treatment studies, mice were randomly assigned to treatment groups. For mass spectrometry analyses, samples were processed in random order and experimenters were blinded to experimental conditions. All experiments have been successfully repeated with similar results at least two to three times.
Data Reporting
No statistical methods were used to predetermine sample size. For in vivo experiments where grouping was based on genotype, mice in each genotype were randomly picked for the experiments; for in vivo experiments where grouping was based on pharmacological treatments, mice of the same genotype were randomly allocated to treatment groups. The investigators were not blinded to allocation during experiments and outcome assessment.
Extended Data
Extended Data Figure 1. PCr’ase activities of thermogenic fat and TNAP.
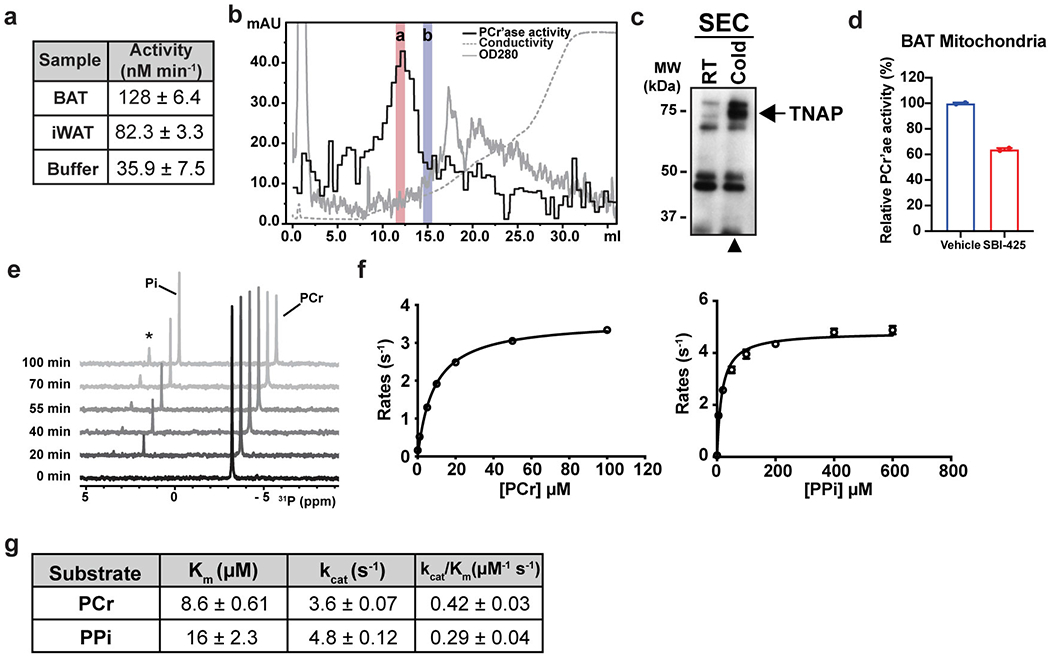
(a) PCr’ase activities of total mitochondrial protein extracts from different tissues of cold-acclimated mice. BAT, interscapular brown adipose tissue; and iWAT, inguinal white adipose tissue. Mitochondrial protein extract was prepared from tissues excised from 10 mice for BAT or 20 mice for iWAT. Each reaction contains 10 mM of PCr and 0.4 mg/mL of mitochondrial protein extract, except the buffer control. Data are presented as the estimated parameters ± uncertainties. Uncertainties are represented by the standard errors of non-linear regression that fits a straight-line model to the initial linear phase of PCr hydrolysis kinetics measured by 31P NMR over 11 time points for BAT and iWAT and 6 time points for the buffer control (shown in Source Data).
(b) Ion-exchange chromatography of the active fraction of SEC. The PCr’ase activity of each fraction was measured by enzyme-coupled assay. The activity of the most active fraction was also verified by 31P NMR. Red and blue bars denote the fractions used for isobaric labeling (TMT) and quantitative mass spectrometric analysis.
(c) Western-blot analysis of the active SEC fraction prepared from cold-acclimated mice (Cold), compared with the equivalent fraction prepared from room-temperature housed mice (RT).
(d) PCr’ase activities of total mitochondrial protein extracts from BAT of cold-acclimated mice treated with vehicle or SBI-425 (10 μM), measured by 31P NMR. n=2 technical replicates per group. Data are presented as means ± SEM.
(e) Stacked traces of 31P NMR spectra recorded at indicated time points, demonstrating the kinetics of PCr hydrolysis catalyzed by recombinant TNAP. The minor peak marked with an asterisk on top is from glycerol-3-phosphate, a side-product of the phospho-transferase activity of TNAP42, that transfers the phosphoryl-group from PCr to glycerol present in the reaction buffer.
(f) Km curves of hydrolysis of PCr (left) and PPi (right) catalyzed by recombinant TNAP. Activities were measured by the enzyme-coupled assay; n=2 technical replicates. Data are presented as means ± SEM.
(g) Comparison of the Michaelis-Menten parameters extrapolated from (f). Data are presented as the estimated parameters ± uncertainties. Uncertainties are represented by standard errors derived from the non-linear regression fit of Michaelis-Menten model to the data in (f).
Extended Data Figure 2. Mitochondrial localization of ectopically expressed TNAP in non-thermogenic fat cell types.
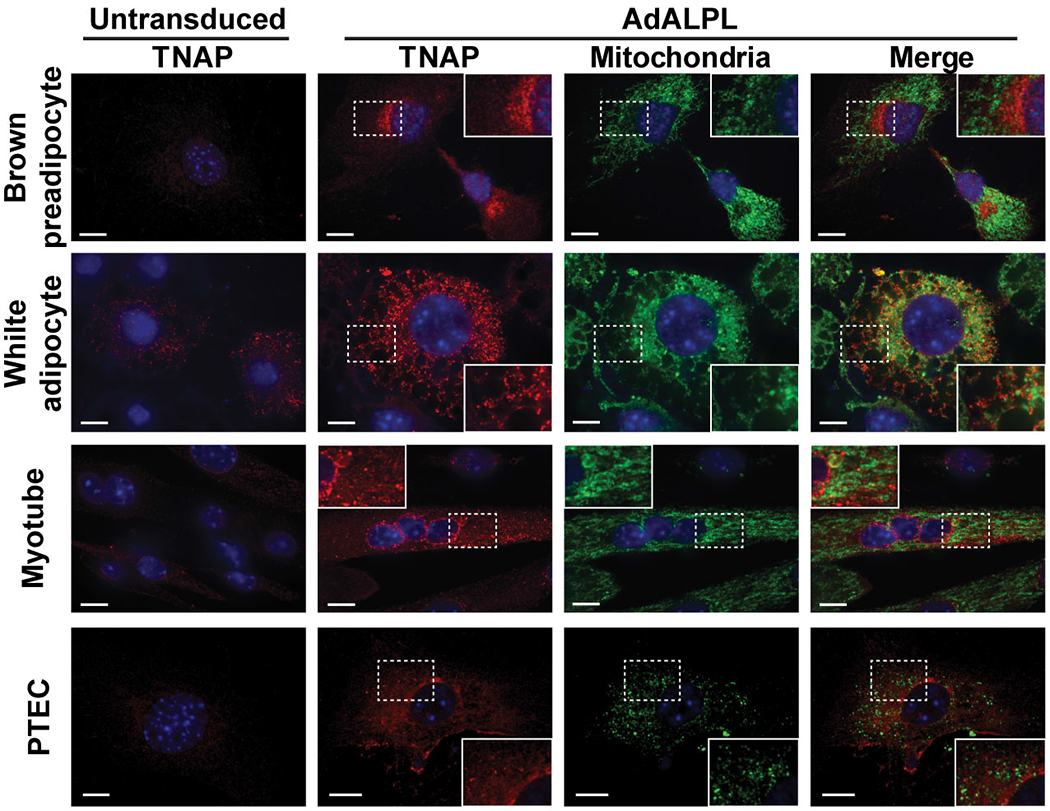
Confocal fluorescence microscopic images showing subcellular localization of ectopically expressed TNAP in different cell types. PTEC stands for kidney proximal tubule epithelial cells. The insets show a zoomed-in region of the image outlined by a dotted box. Anti-TNAP and anti-HSP60 were used to visualize TNAP and mitochondria, respectively. Scale bar: 10 μm.
Extended Data Figure 3: Mitochondrial localization of endogenous TNAP in BAT and non-thermogenic fat cells.
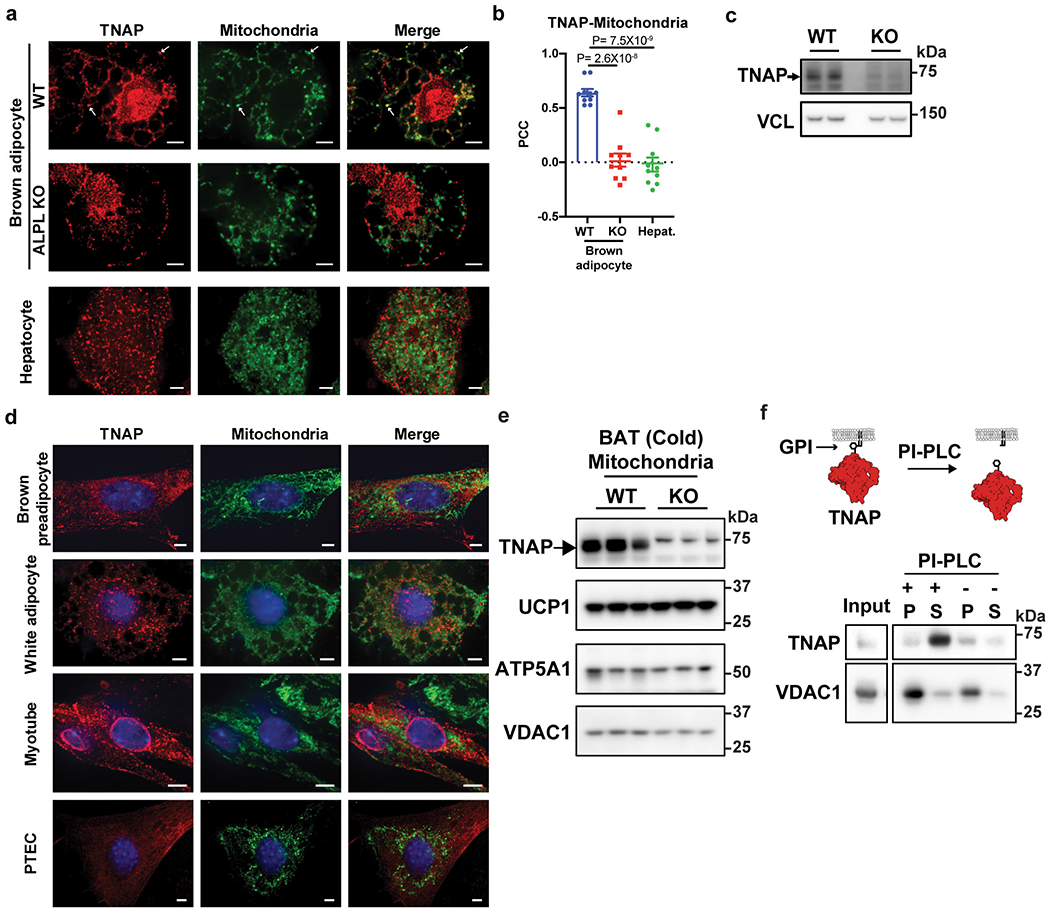
(a) Confocal fluorescence images showing subcellular localization of endogenous TNAP in brown adipocytes (upper and middle panels) and hepatocytes (lower panels). Primary brown preadipocytes were prepared from Alpl fl/fl mice, transduced with either AdGFP (WT) or AdCRE (ALPL KO) on day 4 of differentiation, and fixed for imaging on day 8. Arrows denote selected peri-nuclear areas of TNAP signal that colocalize with mitochondria signal. Antibodies for TNAP (Red) and HSP60 (Green) were used to visualize TNAP and mitochondria. Scale bar: 5 μm.
(b) Pearson’s Correlation Coefficient (PCC) analysis showing the extent of colocalization of TNAP with mitochondria in indicated cell types; n=10 cells per group; data are presented as means ± SEM; statistical significance was calculated by one-way ANOVA with Bonferroni’s multiple comparisons test.
(c) Western-blot analysis on TNAP in WT versus KO cells. Vinculin (VCL) blot was used as a sample preparation control.
(d) Confocal fluorescence microscopic images showing subcellular localization of endogenous TNAP in different cell types. PTEC stands for kidney proximal tubule epithelial cells. Anti-TNAP and anti-HSP60 were used to visualize TNAP and mitochondria, respectively. Scale bar: 5 μm.
(e) Western-blot analysis on TNAP and mitochondrial markers in mitochondria preparations from BAT of cold-acclimated, WT vs Adipo-Alpl KO mice. Blots were processed in parallel with samples derived from the same experiment.
(f) Western-blot analysis of the insoluble fraction of mitochondria extract treated with Phospholipase-C, phosphatidylinositol-specific (PI-PLC), followed by ultracentrifugation, showing that PLC treatment releases TNAP from membranes. P: pellet; and S: supernatant. Mitochondria preparation was fragmented by sonication before treatment. Blots were processed in parallel with samples derived from the same experiment.
Extended Data Figure 4. Proximity-based fluorescent labeling by TNAP-APEX2 and trypsin protection assay on mitochondria from BAT.
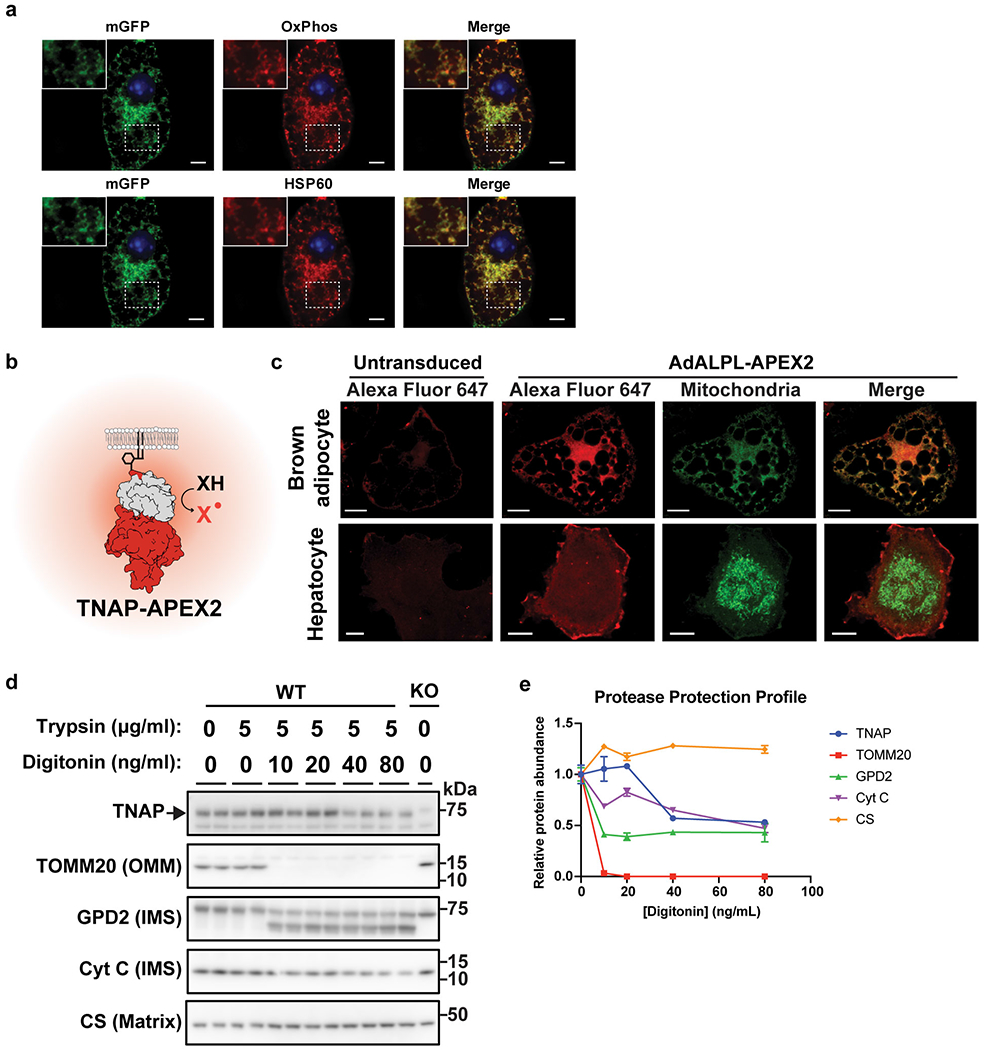
(a) Confocal fluorescence microscopic images of immortalized brown adipocytes showing colocalization of the GFP signal from 3XHA-EGFP-OMP25 construct (mGFP, channel: 488 nm) with different mitochondria markers. Endogenous antibodies, OxPhos (Upper red, channel: 561 nm) and HSP60 (Lower red, channel: 640 nm), were used to visualize mitochondria. The insets show a zoomed-in region of the image outlined by the dotted box. Scale bar: 5 μm.
(b) Graphic illustration of how APEX2 reports subcellular localization of TNAP by its peroxidase activity. X stands for either Alexa Fluor 647 conjugated tyramide for confocal or 3,3’-Diaminobenzidine TEM studies.
(c) Confocal fluorescence analysis of immortalized brown adipocytes (upper panels) and hepatocytes (lower panels) ectopically expressing a TNAP-APEX2 construct. Cells were fixed and treated with Alexa Fluor 647-Tyramide/H2O2 for proximity-based fluorescent labeling facilitated by the peroxidase activity of APEX2. Stably expressed 3XHA-EGFP-OMP25 was used as mitochondria reporter. Scale bar: 10 μm.
(d) Western-blot analysis of the Trypsin protection assay on mitochondria derived from BAT of cold-acclimated mice. TOMM20, GPD2 (glycerol-3-phosphate dehydrogenase), Cyt C (cytochrome c) and CS (citrate synthase) are shown as markers of outer mitochondrial membrane (OMM), intermembrane space (IMS) and mitochondrial matrix. Blots were processed in parallel with samples derived from the same experiment.
(e) Relative protein abundances in trypsin-digested mitochondria derived from band intensities of intact protein quantified from (d); n=2 technical replicates. Data are presented as means ± SEM.
Extended Data Figure 5. Effect of Alpl silencing on cellular respiration.
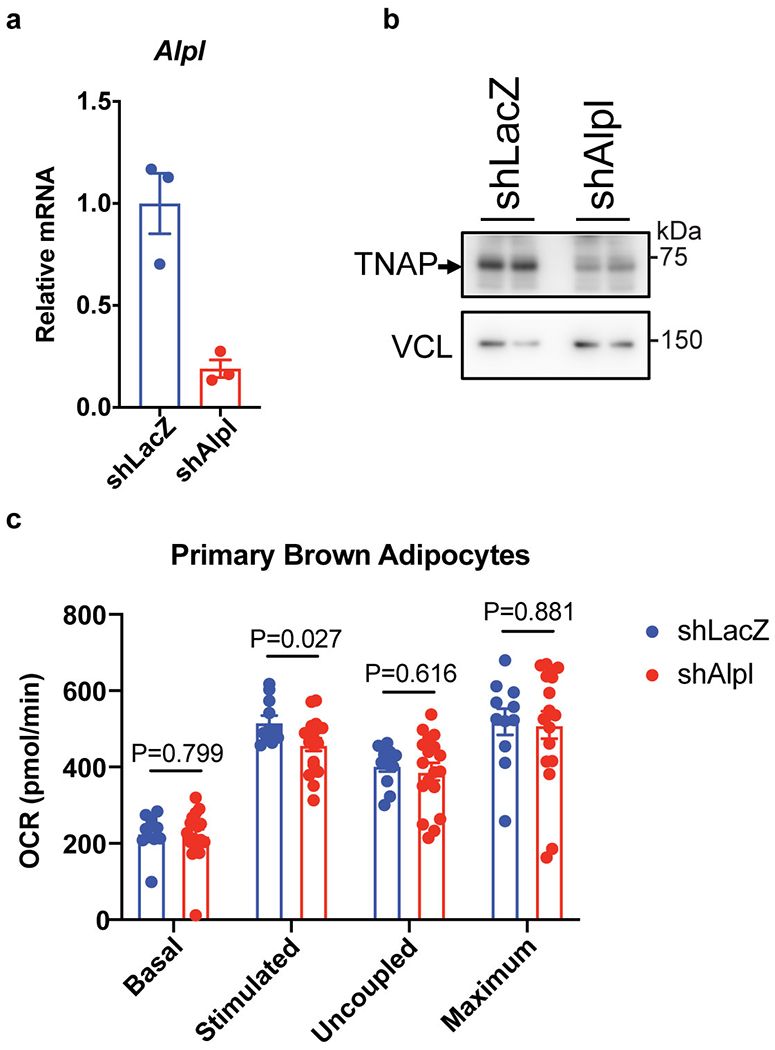
(a) qRT-PCR of differentiated primary brown preadipocytes treated with shLacZ or shAlpl; n=3 biologically independent samples per group.
(b) Western-blot analysis on TNAP in cells treated with shLacZ or shAlpl. Vinculin (VCL) blot was used as a sample processing control.
(c) Effect of Alpl knockdown (adenoviral shAlpl) on OCR of primary brown adipocytes. Treatments to initiate different respiration states: Stimulated, norepinephrine; Uncoupled, oligomycin; Maximum, carbonyl cyanide m-chlorophenyl hydrazone; n=11 biologically independent samples for the shLacZ group and 18 biologically independent samples for the shAlpl group. Data are presented as means ± SEM. Statistical significance was calculated by unpaired Student’s two-sided t-test.
Extended Data Figure 6. Effect of TNAP inhibition on the futile creatine cycle.
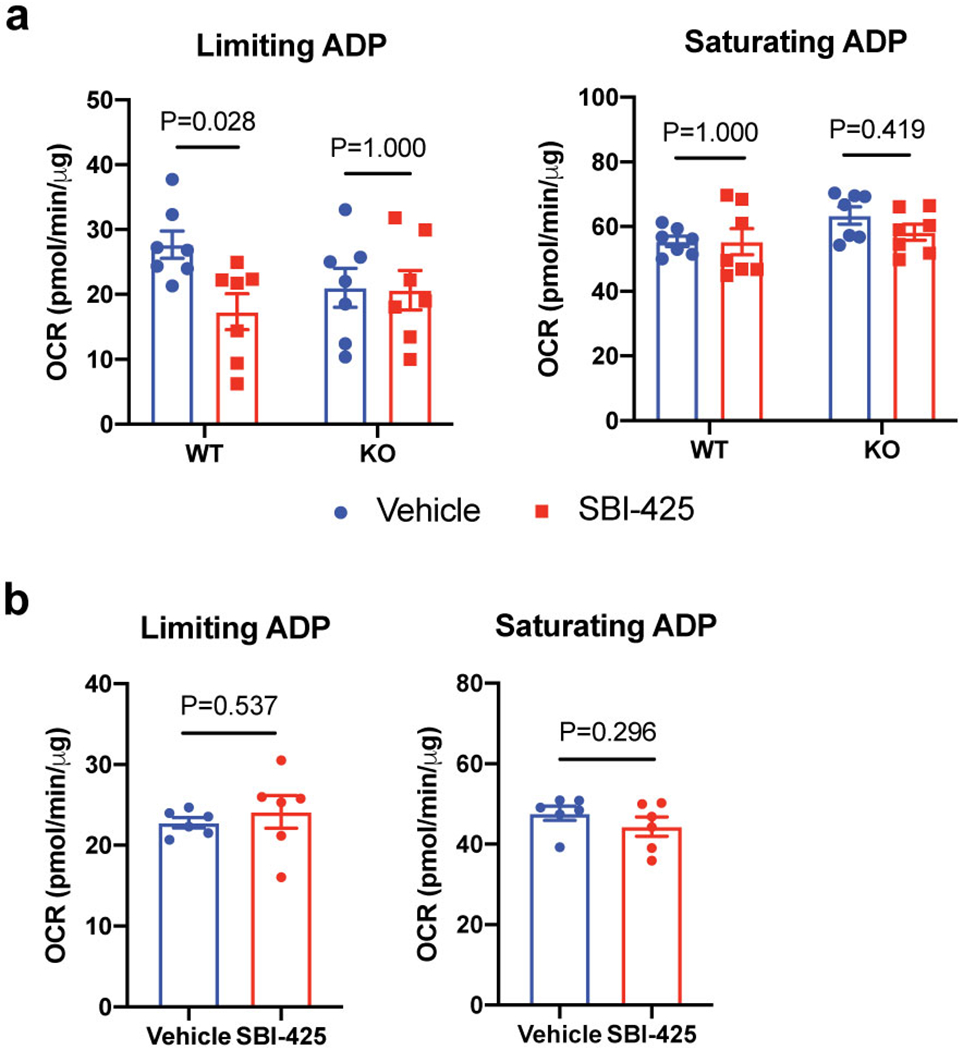
(a) Effect of SBI-425 treatment (10 μM) on oxygen consumption rate (OCR) of beige fat-derived mitochondria from WT vs Adipo-Alpl KO mice in the presence of 0.01 mM creatine and 0.1 mM ADP (limiting ADP) or 1 mM ADP (saturating ADP), as measured by a Seahorse XF24 Extracellular Flux Analyzer; n=7 independent measurements per group.
(b) Effect of SBI-425 treatment (10 μM) on oxygen consumption rate (OCR) of beige fat-derived mitochondria in the presence of 0.1 mM ADP (limiting ADP) or 1 mM ADP (saturating ADP), but in the absence of creatine, as measured by a Seahorse XF24 Extracellular Flux Analyzer; n=6 independent measurements per group.
Data are presented as means ± SEM. Statistical significance was calculated by either two-way ANOVA with Bonferroni’s multiple comparisons test (a) or unpaired Student’s two-sided t-test (b).
Extended Data Figure 7. Rapid mitochondria purification enriched mitochondrial metabolites and proteins.
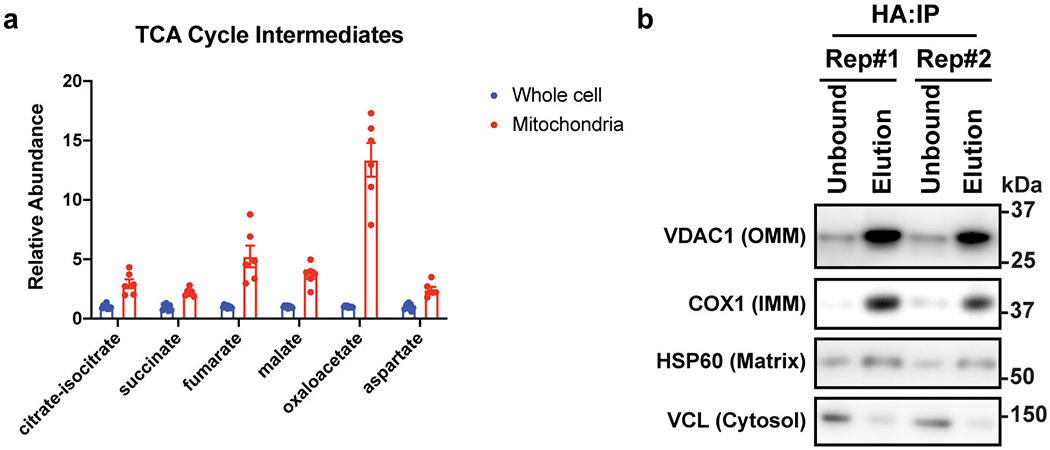
(a) Relative abundances of citric acid cycle intermediates in mitochondria vs whole-cell metabolomics; n=6 biologically independent samples. Data are presented as means ± SEM.
(b) Western-blot analysis on mitochondrial markers in immunoprecipitated mitochondria for metabolomics study. OMM: outer mitochondrial membrane; IMM: inner mitochondrial membrane. Blots were processed in parallel with samples derived from the same experiment.
Extended Data Figure 8. Movement and food intake of mice upon SBI-425 treatment.
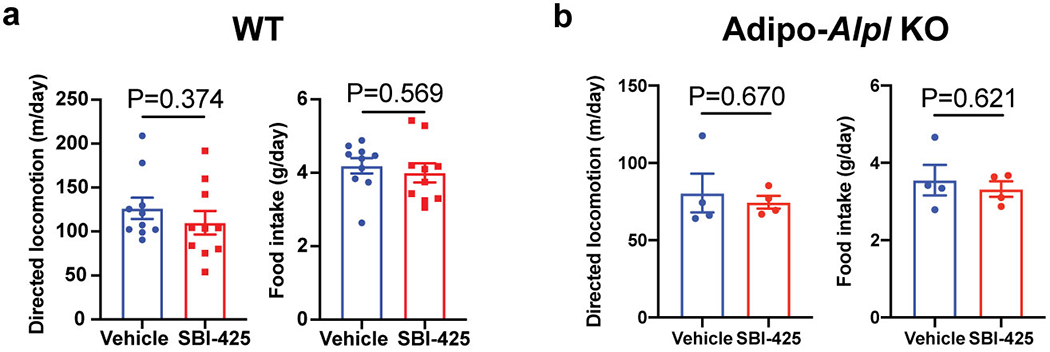
(a and b) Cumulative movement and food intake of wild-type (a) and Adipo-Alpl KO mice (b) for 24 hours after treatment of SBI-425 versus vehicle; n=10 mice for wild-type and 4 mice for Adipo-Alpl KO.
Data are presented as means ± SEM. Statistical significance was calculated by unpaired Student’s two-sided t-test.
Extended Data Figure 9. Energy expenditure and movement of wild-type versus Adipo-Alpl KO mice upon HFD.
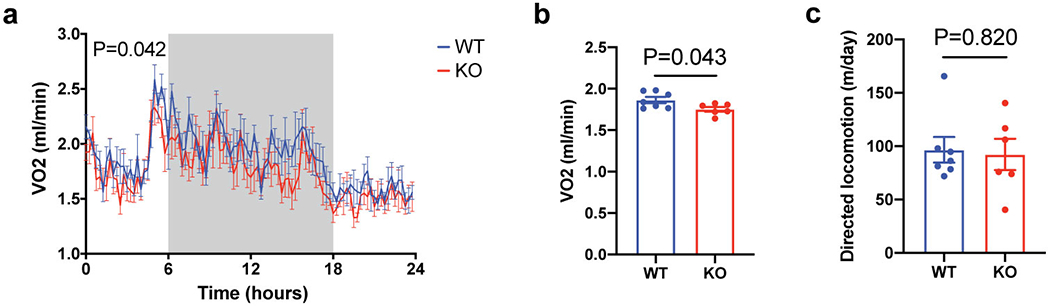
(a) Indirect calorimetric measurement on wild-type vs Adipo-Alpl KO mice that had been on HFD for 4 weeks at 22 °C; n=7 mice for wild-type and 6 mice for Adipo-Alpl KO; the grey area indicates the dark period.
(b) Averaged respiration rates over 24 hours as measured in (a); n=7 mice for wild-type and 6 mice for Adipo-Alpl KO.
(c) Cumulative movement of mice over 24 hours; n=7 mice for wild-type and 6 mice for Adipo-Alpl KO.
Data are presented as means ± SEM. Statistical significance was calculated by either two-way ANOVA (a) or unpaired Student’s two-sided t-test (b and c).
Extended Data Figure 10. Compensatory thermogenesis in Adipo-Alpl KO mice.
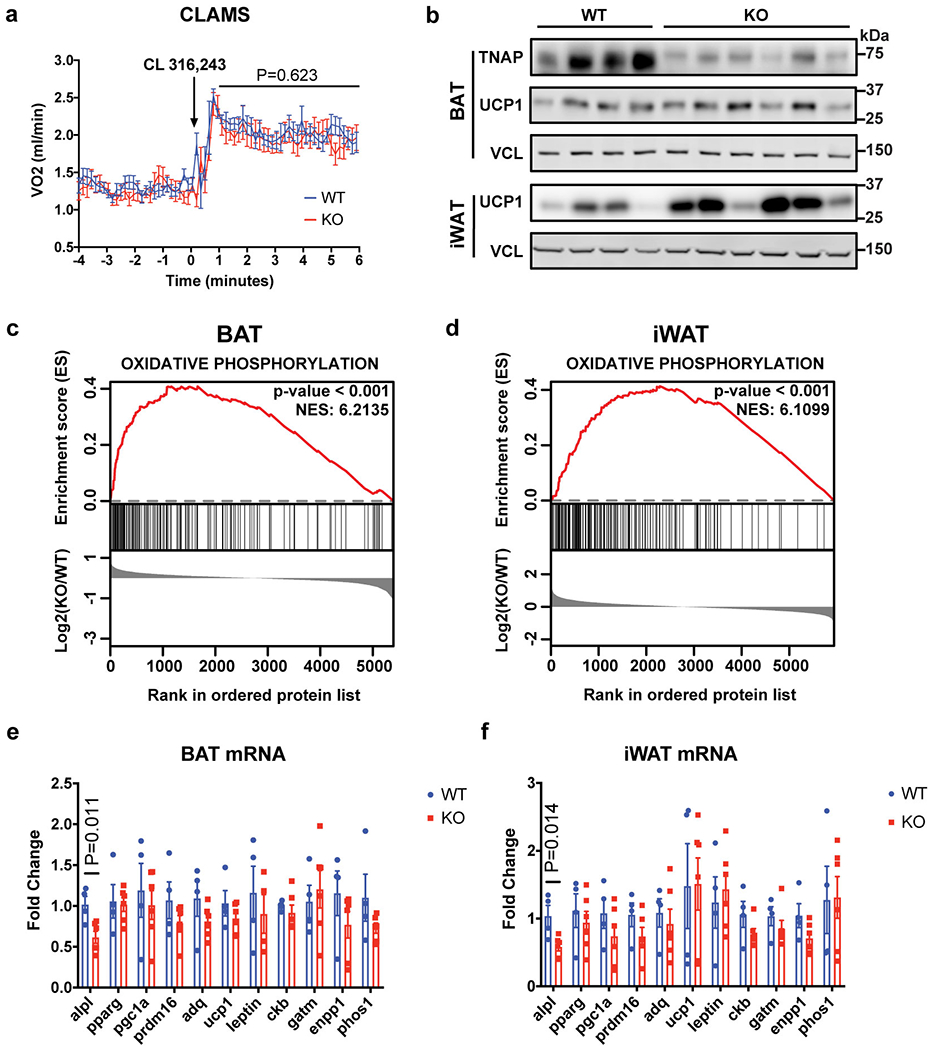
(a) Indirect calorimetric measurement on wild-type versus Adipo-Alpl KO mice kept in metabolic cages at 22 °C, showing stimulation of mice respiration by CL 316,243 administration (1.0 mg/kg). Arrow denotes the time point of drug administration; n=12 mice for wild-type and 9 mice for Adipo-Alpl KO; all mice were pre-treated with CL 316,243 (1.0 mg/kg/day) for 5 days.
(b) Western-blot analysis of BAT and iWAT from wild-type or Adipo-Alpl KO mice pre-treated with CL 316,243 (1.0 mg/kg/day) for 5 days.
(c and d) Gene set enrichment plot of quantitative mass spectrometric analyses of BAT (c) and iWAT (d) from wild-type or Adipo-Alpl KO mice treated with CL 316,243 (1.0 mg/kg/day) for 5 days; n = 4 mice for wild-type and 6 mice for Adipo-Alpl KO. Enrichment analysis was performed with GSEA 4.1.043,44. The hallmark gene sets were surveyed, and oxidative phosphorylation is the top hit for both BAT and iWAT. Family-wise error rate (FWER) p-value was presented for statistical significance (number of permutations=1000) and NES stands for normalized enrichment score (enrichment statistics = “classic”).
(e and f) qRT-PCR of BAT (e) and iWAT (f) from wild-type vs Adipo-Alpl KO mice treated with CL 316,243 (1.0 mg/kg/day) for 5 days; n=4 mice for wild-type and 6 mice for Adipo-Alpl KO.
Data are presented as means ± SEM. Statistical significance was calculated by either two-way ANOVA (a) or unpaired Student’s two-sided t-test (e and f).
Supplementary Material
Acknowledgements
We thank Clifford J. Rosen for sharing the Alplfl/fl mice strain with permission of J.L.M.; Ryan Garrity for help with CLAMS studies; the NMR Core jointly operated by Harvard Medical School and Dana-Farber Cancer Institute for the help with NMR data acquisition; Nikon Imaging Center at Harvard Medical School for the help with fluorescent imaging studies; the EM Core at Harvard Medical School for APEX2/EM imaging studies; the Mass Spectrometry Facility at Beth Israel Deaconess Medical Center for targeted metabolomics studies. Y.S. and C.L.R. are supported by the American Heart Association postdoctoral fellowship. J.F.R is supported by the Charlotte and Leo Karassik Fellowship. B.H. was a Cancer Research Institute/Leonard Kahn Foundation Fellow. P.A.D. is supported by a Damon Runyon Cancer Research Foundation Fellowship. This study was supported by NIDCR grant DE12889 to J.L.M., NIH grant DK 123095 to E.T.C, Canadian Institutes of Health Research (CIHR) grant PJT-159529 to L.K., and JPB Foundation 6293803 and NIH grant DK123228 to B.M.S.
Footnotes
Competing Interests Statement
The authors declare no competing financial interests.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD025032. The published adipocyte specific ribosomal profiling dataset could be downloaded at this link (https://ars.els-cdn.com/content/image/1-s2.0-S1550413118301839-mmc2.xlsx). Full scans for all western blots are provided in Supplementary Information. Source data for all biochemical, cellular, and animal experiments are provided. All other data are available from the corresponding author on reasonable request.
References
- 1.Rosen ED & Spiegelman BM What we talk about when we talk about fat. Cell 156, 20–44, doi: 10.1016/j.cell.2013.12.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harms M & Seale P Brown and beige fat: development, function and therapeutic potential. Nat Med 19, 1252–1263, doi: 10.1038/nm.3361 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Carobbio S, Guénantin AC, Samuelson I, Bahri M & Vidal-Puig A Brown and beige fat: From molecules to physiology and pathophysiology. Biochim Biophys Acta Mol Cell Biol Lipids 1864, 37–50, doi: 10.1016/j.bbalip.2018.05.013 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Chouchani ET, Kazak L & Spiegelman BM New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab 29, 27–37, doi: 10.1016/j.cmet.2018.11.002 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Kazak L et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 163, 643–655, doi: 10.1016/j.cell.2015.09.035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cypess AM et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med 19, 635–639, doi: 10.1038/nm.3112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cypess AM et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab 21, 33–38, doi: 10.1016/j.cmet.2014.12.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leitner BP et al. Mapping of human brown adipose tissue in lean and obese young men. Proc Natl Acad Sci U S A 114, 8649–8654, doi: 10.1073/pnas,1705287114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachman ES et al. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science 297, 843–845, doi: 10.1126/science.1073160 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Zeng X et al. Innervation of thermogenic adipose tissue via a calsyntenin 3β-S100b axis. Nature 569, 229–235, doi: 10.1038/s41586-019-1156-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enerback S et al. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387, 90–94, doi: 10.1038/387090a0 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Hofmann WE, Liu X, Bearden CM, Harper ME & Kozak LP Effects of genetic background on thermoregulation and fatty acid-induced uncoupling of mitochondria in UCP1-deficient mice. J Biol Chem 276, 12460–12465, doi: 10.1074/jbc.Ml00466200 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Kazak L et al. UCP1 deficiency causes brown fat respiratory chain depletion and sensitizes mitochondria to calcium overload-induced dysfunction. Proc Natl Acad Sci U S A 114, 7981–7986, doi: 10.1073/pnas.1705406114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda K et al. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nature medicine 23, 1454–1465, doi: 10.1038/nm.4429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long JZ et al. The Secreted Enzyme PM20D1 Regulates Lipidated Amino Acid Uncouplers of Mitochondria. Cell 166, 424–435, doi: 10.1016/j.cell.2016.05.071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kazak L et al. Genetic Depletion of Adipocyte Creatine Metabolism Inhibits Diet-Induced Thermogenesis and Drives Obesity. Cell Metab 26, 660–671 e663, doi: 10.1016/j.cmet.2017.08.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kazak L et al. Ablation of adipocyte creatine transport impairs thermogenesis and causes diet-induced obesity. Nat Metab 1, 360–370, doi: 10.1038/s42255-019-0035-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rahbani JF et al. Creatine kinase B controls futile creatine cycling in thermogenic fat. Nature 590, 480–485, doi: 10.1038/s41586-021-03221-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller S et al. Proteomic Analysis of Human Brown Adipose Tissue Reveals Utilization of Coupled and Uncoupled Energy Expenditure Pathways. Sci Rep 6, 30030, doi: 10.1038/srep30030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Svensson PA et al. Gene expression in human brown adipose tissue. Int J Mol Med 27, 227–232, doi: 10.3892/ijmm.2010.566 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Bessman SP & Carpenter CL The creatine-creatine phosphate energy shuttle. Annu Rev Biochem 54, 831–862, doi: 10.1146/annurev.bi.54.070185.004151 (1985). [DOI] [PubMed] [Google Scholar]
- 22.Pinkerton AB et al. Discovery of 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorg Med Chem Lett 28, 31–34, doi: 10.1016/j.bmcl.2017.11.024 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roh HC et al. Warming Induces Significant Reprogramming of Beige, but Not Brown, Adipocyte Cellular Identity. Cell Metab 27, 1121–1137.e1125, doi: 10.1016/j.cmet.2018.03.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchet R, Millan JL & Magne D Multisystemic functions of alkaline phosphatases. Methods Mol Biol 1053, 27–51, doi: 10.1007/978-1-62703-562-0_3 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Hessle L et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A 99, 9445–9449, doi: 10.1073/pnas.142063399 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson HC et al. Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. Am J Pathol 164, 841–847, doi: 10.1016/s0002-9440(10)63172-0 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam SS et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12, 51–54, doi: 10.1038/nmeth.3179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhee HW et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331, doi: 10.1126/science.1230593 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kiffer-Moreira T et al. Catalytic signature of a heat-stable, chimeric human alkaline phosphatase with therapeutic potential. PLoS One 9, e89374, doi: 10.1371/journal.pone.0089374 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen WW, Freinkman E, Wang T, Birsoy K & Sabatini DM Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1337.e1311, doi: 10.1016/j.cell.2016.07.040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen WW, Freinkman E & Sabatini DM Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat Protoc 12, 2215–2231, doi: 10.1038/nprot.2017.104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haarhaus M, Brandenburg V, Kalantar-Zadeh K, Stenvinkel P & Magnusson P Alkaline phosphatase: a novel treatment target for cardiovascular disease in CKD. Nat Rev Nephrol 13, 429–442, doi: 10.1038/nrneph.2017.60 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Liu X et al. Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J Clin Invest 111, 399–407, doi: 10.1172/JCI15737 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feldmann HM, Golozoubova V, Cannon B & Nedergaard J UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab 9, 203–209, doi: 10.1016/j.cmet.2008.12.014 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Vendelin M, Lemba M & Saks VA Analysis of functional coupling: mitochondrial creatine kinase and adenine nucleotide translocase. Biophys J 87, 696–713, doi: 10.1529/biophysj.103.036210 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shulman GI, Ladenson PW, Wolfe MH, Ridgway EC & Wolfe RR Substrate cycling between gluconeogenesis and glycolysis in euthyroid, hypothyroid, and hyperthyroid man. J Clin Invest 76, 757–764, doi: 10.1172/JCI112032 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolfe RR, Herndon DN, Jahoor F, Miyoshi H & Wolfe M Effect of severe burn injury on substrate cycling by glucose and fatty acids. N Engl J Med 317, 403–408, doi: 10.1056/NEJM198708133170702 (1987). [DOI] [PubMed] [Google Scholar]
- 38.Clark MG et al. Letter: Accelerated substrate cycling of fructose-6-phosphate in the muscle of malignant hyperthermic pigs. Nature 245, 99–101, doi: 10.1038/245099a0 (1973). [DOI] [PubMed] [Google Scholar]
- 39.Newsholme EA & Crabtree B Substrate cycles in metabolic regulation and in heat generation. Biochem Soc Symp, 61–109 (1976). [PubMed] [Google Scholar]
- 40.Klein S & Wolfe RR Whole-body lipolysis and triglyceride-fatty acid cycling in cachectic patients with esophageal cancer. J Clin Invest 86, 1403–1408, doi: 10.n72/JCI114854 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gettins P, Metzler M & Coleman JE Alkaline phosphatase. 31P NMR probes of the mechanism. J Biol Chem 260, 2875–2883 (1985). [PubMed] [Google Scholar]
- 43.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550, doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mootha VK et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273, doi: 10.1038/ng1180 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Klein J et al. beta(3)-adrenergic stimulation differentially inhibits insulin signaling and decreases insulin-induced glucose uptake in brown adipocytes. J Biol Chem 274, 34795–34802, doi: 10.1074/jbc.274.49.34795 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Wu J et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150, 366–376, doi: 10.1016/jcell.2012.05.016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharabi K et al. Selective Chemical Inhibition of PGC-1α Gluconeogenic Activity Ameliorates Type 2 Diabetes. Cell 169, 148–160.e115, doi: 10.1016/j.cell.2017.03.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Springer ML, Rando TA & Blau HM Gene delivery to muscle. Curr Protoc Hum Genet Chapter 13, Unit 13.14, doi: 10.1002/0471142905.hgl304s31 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Allan C et al. OMERO: flexible, model-driven data management for experimental biology. Nat Methods 9, 245–253, doi: 10.1038/nmeth,1896 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682, doi: 10.1038/nmeth.2019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan M, Breitkopf SB, Yang X & Asara JM A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7, 872–881, doi: 10.1038/nprot.2012.024 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foster BL et al. Conditional Alpl Ablation Phenocopies Dental Defects of Hypophosphatasia. J Dent Res 96, 81–91, doi: 10.1177/0022034516663633 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD025032. The published adipocyte specific ribosomal profiling dataset could be downloaded at this link (https://ars.els-cdn.com/content/image/1-s2.0-S1550413118301839-mmc2.xlsx). Full scans for all western blots are provided in Supplementary Information. Source data for all biochemical, cellular, and animal experiments are provided. All other data are available from the corresponding author on reasonable request.