

Abstract
Here we determine the complete genomic sequence of the Gram negative, γ-Proteobacterium Vibrio cholerae El Tor N16961 to be 4,033,460 base pairs (bp). The genome consists of two circular chromosomes of 2,961,146 bp and 1,072,314 bp that together encode 3,885 open reading frames. The vast majority of recognizable genes for essential cell functions (such as DNA replication, transcription, translation and cell-wall biosynthesis) and pathogenicity (for example, toxins, surface antigens and adhesins) are located on the large chromosome. In contrast, the small chromosome contains a larger fraction (59%) of hypothetical genes compared with the large chromosome (42%), and also contains many more genes that appear to have origins other than the γ-Proteobacteria. The small chromosome also carries a gene capture system (the integron island) and host ‘addiction’ genes that are typically found on plasmids; thus, the small chromosome may have originally been a megaplasmid that was captured by an ancestral Vibrio species. The V. cholerae genomic sequence provides a starting point for understanding how a free-living, environmental organism emerged to become a significant human bacterial pathogen.
Supplementary information
The online version of this article (doi:10.1038/35020000) contains supplementary material, which is available to authorized users.
Main
Vibrio cholerae is the aetiological agent of cholera, a severe diarrhoeal disease that occurs most frequently in epidemic form1. Cholera has been epidemic in southern Asia for at least 1,000 years, but also spread worldwide to cause seven pandemics since 1817 (ref. 1). When untreated, cholera is a disease of extraordinarily rapid onset and potentially high lethality. Although clinical management of cholera has advanced over the past 40 years, cholera remains a serious threat in developing countries where sanitation is poor, health care limited, and drinking water unsafe.
Vibrio cholerae as a species includes both pathogenic and nonpathogenic strains that vary in their virulence gene content2. This bacterium contains a wide variety of strains and biotypes, receiving and transferring genes for toxins3, colonization factors4,5, antibiotic resistance6, capsular polysaccharides that provide resistance to chlorine7 and new surface antigens, such as the 0139 lipopolysaccharide and O antigen capsule8,9. The lateral or horizontal transfer of these virulence genes by phage3, pathogenicity islands10,11 and other accessory genetic elements12 provides insights into how bacterial pathogens emerge and evolve to become new strains.
Vibrio species represent a significant portion of the culturable heterotrophic bacteria of oceans, coastal waters and estuaries13,14. Environmental studies show that these bacteria strongly influence nutrient cycling in the marine environment. Various species of this genus are also devastating pathogens for finfish, shellfish and mammals. There is still much to be learned about the aquatic ecology and natural history of V. cholerae including its autochthonous (native) existence in endemic locales during cholera-free, interepidemic periods, which environmental factors, such as climate13,15, aided its re-emergence in Latin America, and which environmental factors are associated with its habitat in cholera endemic regions. For example, V. cholerae, during interepidemic periods, is an inhabitant of brackish and estuarine waters, and in these environments is associated with zooplankton and other aquatic flora and fauna16. The organism also enters a “viable but nonculturable”17 state under certain conditions. Roles for these environmental interactions and this dormant physiological state in the emergence and persistence of pathogenic V. cholerae have been proposed14,17.
Here we report the determination and analysis of the Vibrio cholerae genome sequence. This analysis represents an important step toward the complete molecular description of how this free-living environmental organism emerged to become a human pathogen by horizontal gene transfer.
Genome analysis
The genome of V. cholerae was sequenced by the whole genome random sequencing method18. The genome consists of two circular chromosomes19,20 of 2,961,146 (chromosome 1) and 1,072,314 (chromosome 2) base pairs, with an average G+C content of 46.9% and 47.7%, respectively. There are a total of 3,885 predicted open reading frames (ORFs) and 792 predicted Rho-independent terminators; with 2,770 and 1,115 ORFs and 599 and 193 Rho-independent terminators on the individual chromosomes (Table 1, Figs 1 (see the one page PDF file (900K or the larger, printable three page PDF file (2622K))and 2). Most genes required for growth and viability are located on chromosome 1, although some genes found only on chromosome 2 are also thought to be essential for normal cell function (for example, dsdA, thrS and the genes encoding ribosomal proteins L20 and L35). Additionally, many intermediaries of metabolic pathways are encoded only on chromosome 2 (Fig. 3).
Table 1.
General features of the Vibrio cholerae genome
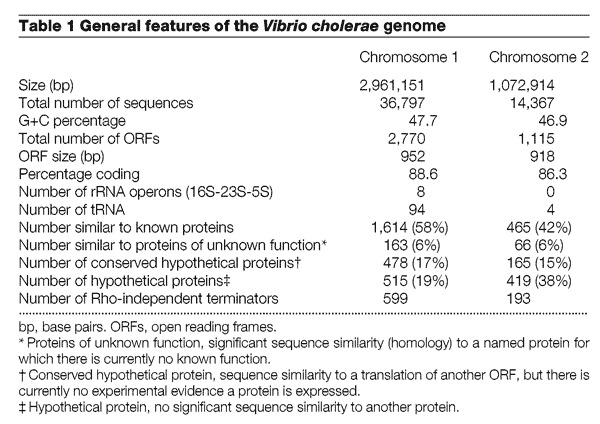
Figure 1. Linear representation of the V. cholerae chromosomes.

The location of the predicted coding regions, colour-coded by biological role, RNA genes, tRNAs, other RNAs, Rho-independent terminators and Vibrio cholerae repeats (VCRs) are indicated (see the larger, printable PDF file (2622K). Arrows represent the direction of transcription for each predicted coding region. Numbers next to the tRNAs represent the number of tRNAs at a locus. Numbers next to GES represent the number of membrane-spanning domains predicted by the Goldman, Engleman and Steitz scale calculated by TopPred for that protein. Gene names are available at the TIGR web site (www.tigr.org) and as Supplementary Information .
Figure 2. Circular representation of the V. cholerae genome.
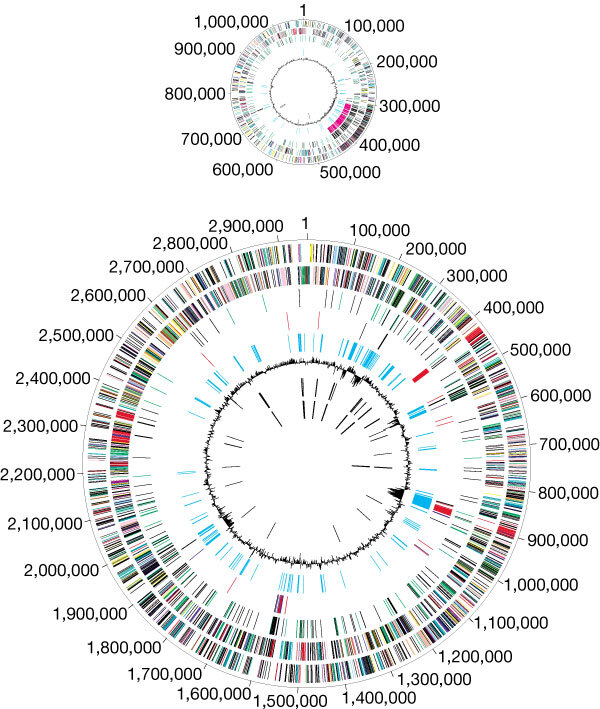
The two chromosomes, large and small, are depicted. From the outside inward: the first and second circles show predicted protein-coding regions on the plus and minus strand, by role, according to the colour code in Fig. 1 (unknown and hypothetical proteins are in black). The third circle shows recently duplicated genes on the same chromosome (black) and on different chromosomes (green). The fourth circle shows transposon-related (black), phage-related (blue), VCRs (pink) and pathogenesis genes (red). The fifth circle shows regions with significant χ2 values for trinucleotide composition in a 2,000-bp window. The sixth circle shows percentage G+C in relation to mean G+C for the chromosome.The seventh and eighth circles are tRNAs and rRNAs, respectively.
Figure 3. Overview of metabolism and transport in V. cholerae.
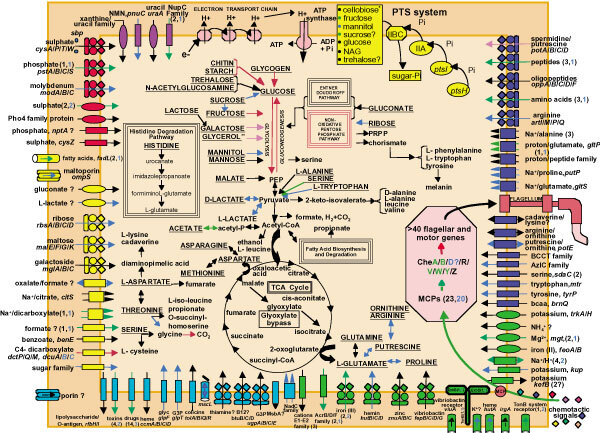
Pathways for energy production and the metabolism of organic compounds, acids and aldehydes are shown. Transporters are grouped by substrate specificity: cations (green), anions (red), carbohydrates (yellow), nucleosides, purines and pyrimidines (purple), amino acids/peptides/amines (dark blue) and other (light blue). Question marks associated with transporters indicate a putative gene, uncertainty in substrate specificity, or direction of transport. Permeases are represented as ovals; ABC transporters are shown as composite figures of ovals, diamonds and circles; porins are represented as three ovals; the large-conductance mechanosensitive channel is shown as a gated cylinder; other cylinders represent outer membrane transporters or receptors; and all other transporters are drawn as rectangles. Export or import of solutes is designated by the direction of the arrow through the transporter. If a precise substrate could not be determined for a transporter, no gene name was assigned and a more general common name reflecting the type of substrate being transported was used. Gene location on the two chromosomes, for both transporters and metabolic steps, is indicated by arrow colour: all genes located on the large chromosome (black); all genes located on the small chromosome (blue); all genes needed for the complete pathway on one chromosome, but a duplicate copy of one or more genes on the other chromosome (purple); required genes on both chromosomes (red); complete pathway on both chromosomes (green). (Complete pathways, except for glycerol, are found on the large chromosome.) Gene numbers on the two chromosomes are in parentheses and follow the colour scheme for gene location. Substrates underlined and capitalized can be used as energy sources. PRPP, phosphoribosyl-pyrophosphate; PEP, phosphoenolpyruvate; PTS, phosphoenolpyruvate-dependant phosphotransferase system; ATP, adenosine triphosphate; ADP, adenosine diphosphate; MCP, methyl-accepting chaemotaxis protein; NAG, N-acetylglucosamine; G3P, glycerol-3-phosphate; glyc, glycerol; NMN, nicotinamide mononucleotide. Asterisk, because V. cholerae does not use cellobiose, we expect this PTS system to be involved in chitobiose transport.
The replicative origin in chromosome 1 was identified by similarity to the Vibrio harveyi and Escherichia coli origins, co-localization of genes (dnaA, dnaN, recF and gyrA) often found near the origin in prokaryotic genomes, and GC nucleotide skew (G-C/G+C) analysis21. Based on these, we designated base-pair 1 in an intergenic region that is located in the putative origin of replication. Only the GC skew analysis was useful in identifying a putative origin on chromosome 2.
This genomic sequence of V. cholerae confirmed the presence of a large integron island (a gene capture system) located on chromosome 2 (125.3 kbp)22,23. The V. cholerae integron island contains all copies of the V. cholerae repeat (VCR) sequence and 216 ORFs (Fig. 1 (see the one page PDF file (900K or the larger, printable three page PDF file (2622K))). However, most of these ORFs have no homology to other sequences. Among the recognizable integron island genes are three that encode gene products that may be involved in drug resistance (chloramphenicol acetyltransferase, fosfomycin resistance protein and glutathione transferase), several DNA metabolism enzymes (MutT, transposase, and an integrase), potential virulence genes (haemagglutinin and lipoproteins) and three genes which encode gene products similar to the ‘host addiction’ proteins (higA, higB and doc), which are used by plasmids to select for their maintenance by host cells.
Comparative genomics
The two-chromosome structure of V. cholerae allows for comparisons, both between the two chromosomes of this organism and between either of the V. cholerae chromosomes and the chromosomes of other microbial species. There is pronounced asymmetry in the distribution of genes known to be essential for growth and virulence between the two chromosomes. Significantly more genes encoding DNA replication and repair, transcription, translation, cell-wall biosynthesis and a variety of central catabolic and biosynthetic pathways are encoded by chromosome 1. Similarly, most genes known to be essential in bacterial pathogenicity (that is, those encoding the toxin co-regulated pilus, cholera toxin, lipopolysaccharide and the extracellular protein secretion machinery) are also located on chromosome 1. In contrast, chromosome 2 contains a larger fraction (59%) of hypothetical genes and genes of unknown function, compared with chromosome 1 (42%) (Fig. 4). This partitioning of hypothetical proteins on chromosome 2 is highly localized in the integron island (Fig. 2). Chromosome 2 also carries the 3-hydroxy-3-methylglutaryl CoA reductase, a gene apparently acquired from an archaea (Y. Boucher and W. F. Doolittle, personal communication).
Figure 4. Percentage of total Vibrio cholerae open reading frames (ORFs) in biological roles compared with other γ-Proteobacteria.
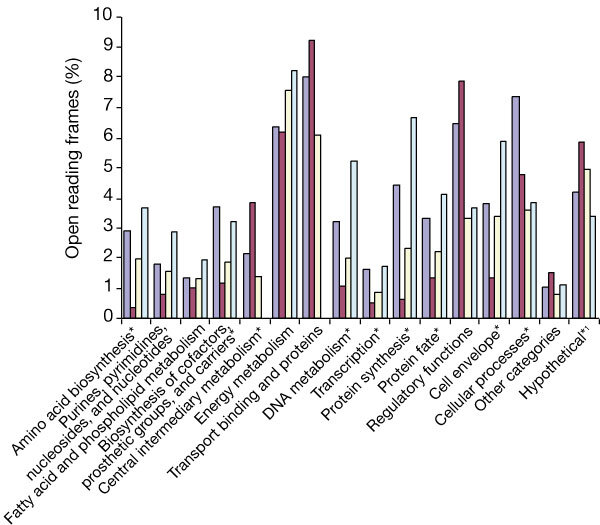
These were V. cholerae, chromosome 1 (blue); V. cholerae, chromosome 2 (red); Escherichia coli (yellow); Haemophilus influenzae (pale blue). Significant partitioning (P < 0.01) of biological roles between V. cholerae chromosomes is indicated with an asterisk, as determined with a χ2 analysis. 1, Hypothetical contains both conserved hypothetical proteins and hypothetical proteins, and is at 1/10 scale compared with other roles.
The majority of the V. cholerae genes were very similar to E. coli genes (1,454 ORFs), but 499 (12.8%) of the V. cholerae ORFs showed highest similarity to other V. cholerae genes, suggesting recent duplications (Figs 5 and 6). Most of the duplicated ORFs encode products involved in regulatory functions (59), chemotaxis (50), transport and binding (42), transposition (18), pathogenicity (13) or unknown functions encoded by conserved hypothetical (62) and hypothetical proteins (113). There are 105 duplications with at least one of each ORF on each chromosome indicating there have been recent crossovers between chromosomes. The extensive duplication of genes involved in scavenging behaviour (chemotaxis and solute transport) suggests the importance of these gene products in V. cholerae biology, notably its ability to inhabit diverse environments. These environments, in turn, may have selected the duplication and divergence of genes useful for specialized functions. Additionally, whereas El Tor strain N16961 carries only a single copy of the cholera toxin prophage, other V. cholerae strains carry several copies of this element24,25, and strains of the classical biotype have a second copy of the prophage that is localized on chromosome 2 (ref. 20). Thus, virulence genes are presumed to be subject to selective pressure, affecting copy number and chromosomal location.
Figure 5. Comparison of the V. cholerae ORFs with those of other completely sequenced genomes.
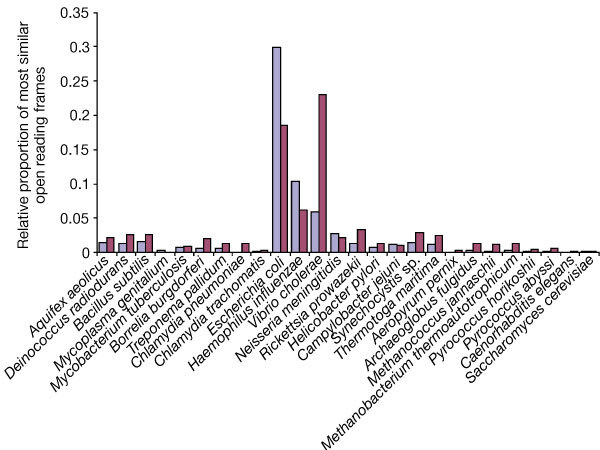
The sequence of all proteins from each completed genome were retrieved from NCBI, TIGR and the Caenorhabditis elegans (wormpep16) databases. All V. cholerae ORFs (large chromosome, blue; small chromosome, red) were searched against all other genomes with FASTA3. The number of V. cholerae ORFs with greatest similarity (E ≤ 10-5) are shown in proportion to the total number of ORFs in that genome. There were no ORFs that were most similar to a Mycoplasma pneumoniae ORF.
Figure 6. Phylogenetic tree of methyl-accepting chemotactic proteins (MCP) homologues in completed genomes.
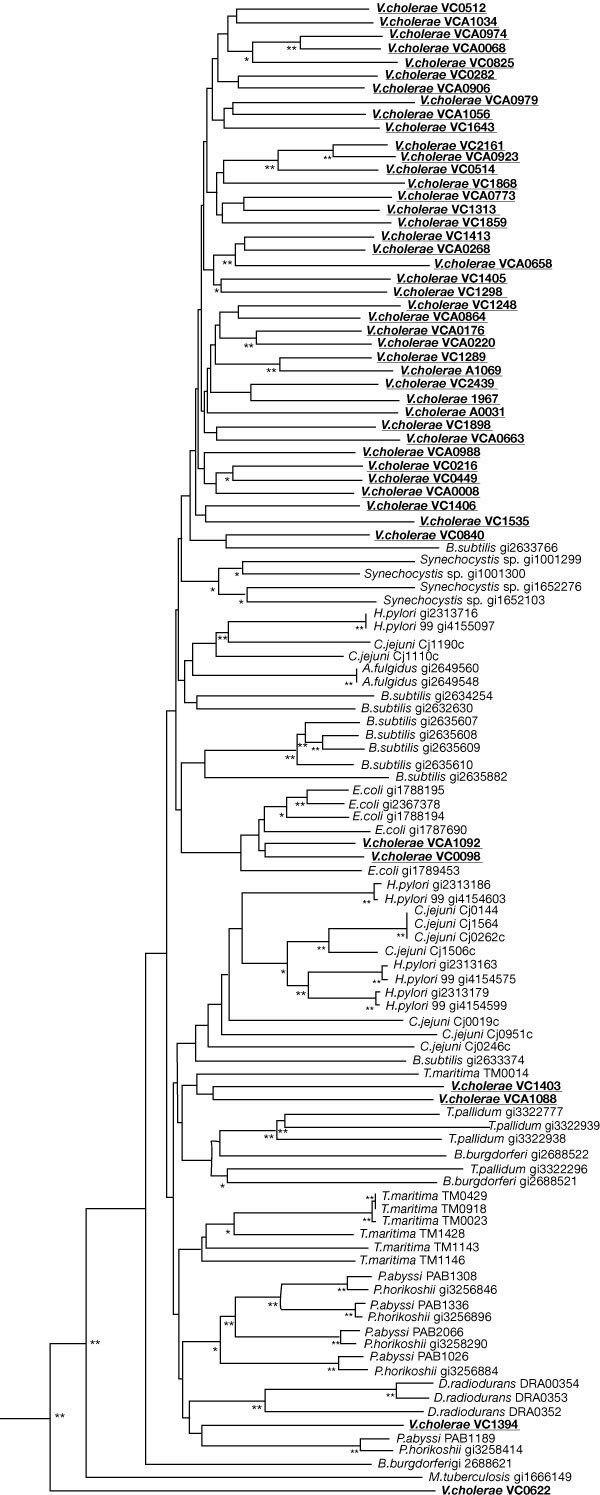
Homologues of MCP were identified by FASTA3 searches of all available complete genomes. Amino-acid sequences of the proteins were aligned using CLUSTALW, and a neighbour-joining phylogenetic tree was generated from the alignment using the PAUP* program (using a PAM-based distance calculation). Hypervariable regions of the alignment and positions with gaps in many of the sequences were excluded from the analysis. Nodes with significant bootstrap values are indicated: two asterisks, >70%; asterisk, 40–70%.
Several ORFs with apparently identical functions exist on both chromosomes which were probably acquired by lateral gene transfer. For example, glyA (encoding serine hydroxymethyl transferase) is found once on each chromosome but the phylogenetic analysis suggest the glyA copy on chromosome 1 branches with the α-Proteobacteria, whereas the copy on chromosome 2 branches with the γ-Proteobacteria (see Supplementary Information). The chromosome 2 glyA is flanked by genes encoding transposases, suggesting that this gene was acquired through a transposition event.
Origin and function of the small chromosome of V. cholerae
Several lines of evidence suggest that chromosome 2 was originally a megaplasmid captured by an ancestral Vibrio species. The phylogenetic analysis of the ParA homologues located near the putative origin of replication of each chromosome shows chromosome 1 ParA tending to group with other chromosomal ParAs, and the ParA from chromosome 2 tending to group with plasmid, phage and megaplasmid ParAs (see Supplementary Information). In general, genes on chromosome 2, with an apparently identical functioning copy on chromosome 1, appear less similar to orthologues present in other γ-Proteobacteria species (see Supplementary Information). Also, chromosome 1 contains all the ribosomal RNA operons and at least one copy of all the transfer RNAs (four tRNAs are found on chromosome 2, but there are duplicates on chromosome 1). In addition, chromosome 2 carries the integron region, an element often found on plasmids26. Finally, the bias in the functional gene content is more easily explained, if chromosome 2 was originally a megaplasmid (Fig. 4). The megaplasmid presumably acquired genes from diverse bacterial species before its capture by the ancestral Vibrio. The relocation of several essential genes from chromosome 1 to the megaplasmid completed the stable capture of this smaller replicon. Apparently this capture of the megaplasmid occurred long enough ago that the trinucleotide composition and percentage G+C content between the two chromosomes has become similar (except for laterally moving elements such as the integron island, bacteriophage genomes, transposons, and so on). The two chromosome structure is found in other Vibrio species19 suggesting that the gene content of the megaplasmid continues to provide Vibrio with an evolutionary advantage, perhaps within the aquatic ecosystem where Vibrio species are frequently the dominant microorganisms14,16.
It is unclear why chromosome 2 has not been integrated into chromosome 1. Perhaps chromosome 2 plays an important specialized function that provides the evolutionary selective pressure to suppress integration events when they do occur. For example, if under some environmental condition there is a difference in copy number between the chromosomes, then chromosome 2 may have accumulated genes that are better expressed at higher or lower copy number than genes on chromosome 1. A second possibility is that, in response to environmental cues, one chromosome may partition to daughter cells in the absence of the other chromosome (aberrant segregation). Such single-chromosome-containing cells would be replication-defective but still maintain metabolic activity (‘drone’ cells), and, therefore, be a potential source of “viable, but nonculturable (VBNC)” cells observed to occur in V. cholerae 17. Such ‘drone’ cells may also play a role in V. cholerae biofilms7,27,28 by, for example, producing extracellular chitinase, protease and other degradative enzymes that enhance survival of cells in a biofilm, retaining two chromosomes without directly competing with these cells for nutrients.
Transport and energy metabolism
Vibrio cholerae has a diverse natural habitat that includes association with zooplankton in a sessile stage, a planktonic state in the water column, and the capacity to act as a pathogen within the human gastrointestinal tract. It is, therefore, no surprise that this organism maintains a large repertoire of transport proteins with broad substrate specificity and the corresponding catabolic pathways to enable it to respond efficiently to these different and constantly changing ecosystems (Fig. 4). Many of the sugar transporter systems and their corresponding catabolic pathway enzymes are localized on a single chromosome (that is, ribose and lactate transport and degradation enzymes are contained on chromosome 2, whereas the trehalose systems reside on chromosome 1). However, many of the other energy metabolism pathways are split (that is, chitin, glycolysis, and so on) between the chromosomes (Fig. 3).
In aquatic environments, chitin often represents a source of both carbon and nitrogen. This energy source is important for V. cholerae as it is associated with zooplankton, which have a chitinous exoskeleton13,15,29. Vibrio cholerae degrades chitin by a pathway that is very similar to that of Vibrio furnissii30. Sequence analysis suggests a phosphenolpyruvate phosphotransferase system (PTS) for cellobiose transport, but as V. cholerae does not use cellobiose, it is more likely that this PTS is involved in transport of the structurally similar compound, chitobiose, analogous to the situation proposed for Bourrelia burgdorferi18.
The three anions that are transported by ABC transport systems in V. cholerae are molybdenum, phosphate and sulphate. Molybdenum transport genes (modA/B/C) are all located on chromosome 2, and most of the sulphate transport genes are on the large molecule. However, copies of the genes for phosphate transporters are found in both chromosomes. The genes in these two phosphate transport operons are different from each other and do not represent a recent duplication; instead, this suggests that one may be an acquired operon.
Interchromosomal regulation
Several of the regulatory pathways, both for regulation in response to environmental and pathogenic signals, are divided between the two chromosomes. These included pathways for starvation survival, ‘quorum sensing’ and expression of the entertoxigenic haemolysin, HlyA.
During periods of nutrient starvation, V. cholerae, and other Gram-negative bacteria, enter the stationary phase and, later, the viable but nonculturable (VBNC) state14,17. The alternative sigma factor σ38 (rpoS) is required for survival of V. cholerae in the environment but not for pathogenicity31, and therefore probably plays an important role in the initiation of the VBNC state. There is one copy of rpoS, located on chromosome 1, near the oriC. The RpoS regulates expression of several other proteins, including catalase, cyclopropane-fatty-acyl-phospholipid synthase and HA/protease, which are found on both chromosomes31.
Genes involved in ‘quorum sensing’, or cell-density-dependent regulation, also exist on both chromosomes of V. cholerae. In bioluminescent Vibrio species (notably Vibrio fischeri and V. harveyi), quorum sensing is used to control light production. Although this strain of V. cholerae lacks the genes for bioluminescence, it does have the genes required for the autoinducer-2 (AI-2) quorum-sensing mechanism32 (luxOPQSU) but this pathway is split between the chromosomes with luxOSU on chromosome 1 and luxPQ on chromosome 2. Similarly, another transcriptional regulatory gene, hlyU (ref. 33), is located on chromosome 1, while the gene it regulates, hlyA, is located on chromosome 2.
DNA repair
Vibrio cholerae has genes encoding several DNA-repair and DNA-damage-response pathways, including nucleotide-excision repair, mismatch-excision repair, base-excision repair, AP endonuclease, alkylation transfer, photoreactivation, DNA ligation, and all the major components of recombination and recombinational repair, including initiation, recombination and resolution34. In addition, homologues of many of the genes involved in the SOS response in E. coli are found. The presence of three photolyase homologues, more than have been found in other bacterial species, probably allows for the ability to photoreactivate the two major forms of ultraviolet-induced DNA damage (cyclobutane pyrimidine dimers and 6-4 photoproducts), and may also allow use of a range of wavelengths of light used for the energy required for photoreactivation. It is also of interest that many of the repair genes are on chromosome 2 (alkA, ada1, ada2, phr3, mutK, sbcCD, dcm, mutT3), indicating that this chromosome is probably required for full DNA repair capability.
Pathogenicity
Toxins
. The genome sequence of V. cholerae El Tor N16961 revealed a single copy of the cholera toxin (CT) genes, ctxAB, located on chromosome 1 within the integrated genome of CTXφ, a temperate filamentous phage3. The receptor for entry of CTXφ into the cell is the toxin-coregulated pilus (TCP)3, and the TCP gene cluster (see below) also resides on chromosome 1. Like the structural genes for CT and TCP, the regulatory gene, toxR, which controls their expression in vivo35, is also located on chromosome 1.
On the other side of CTXφ prophage is a region encoding an RTX toxin (rtxA), and its activator (rtxC) and transporters (rtxBD )36. A third transporter gene has been identified that is a paralogue of rtxB, and is transcribed in the same direction as rtxBD. Downstream of this gene are two genes encoding a sensor histidine kinase and response regulator. Trinucleotide composition analysis suggests that the RTX region was horizontally acquired along with the sensor histidine kinase/response regulator, suggesting these regulators effect expression of the closely linked RTX transcriptional units.
Also present are genes encoding numerous potential toxins, including several haemolysins, proteases and lipases. These include hap, the haemagluttinin protease, a secreted metalloprotease that seems to attack proteins involved in maintaining the integrity of epithelial cell tight junctions37, and hlyA, encoding a secreted haemolysin that displays enterotoxic activity38. In contrast to CTX, RTX and all known intestinal colonization factors, the hap and hlyA genes virulence factors reside on chromosome 2.
Vibrio cholerae has been reported to produce shiga-like toxins39; however, the sequence did not reveal genes encoding specific homologues of the A or B subunits of shiga toxin. Also not detected were genes encoding homologues of E. coli heat stable toxin (ST), which have been detected in other pathogenic strains of V. cholerae40.
Colonization factors
. The critical intestinal colonization factor of V. cholerae is the TCP, a type IV pilus5,41. The genome sequence confirmed that the genes involved in TCP assembly (tcpABCDEFGHIJNQRST) reside on chromosome 1 (ref. 20) as part of a proposed ‘pathogenicity island’ (also referred to as VPI) composed of recently acquired DNA that encodes not only TCP, but also other genes associated with the ToxR regulatory cascade, such as acfABCD, toxT, aldA and tagAB10,11. Trinucleotide composition analysis suggest that this 45.3-kb segment begins at a 20-bp site upstream of aldA , and encompasses a helicase-related protein and a transcriptional activator which both share homology with bacteriophage proteins. At the other end of the segment of atypical trinucleotide composition is a phage family integrase and the other copy of the 20-bp site, which is presumably the target for integration of the island onto the chromosome10,11. It has been proposed that the TCP/ACF island corresponds to the genome of a filamentous phage that uses TCP pilin as a coat protein4. However, other than the three genes encoding phage-related proteins (that is, the helicase, transcriptional activator and integrase) we could find no other genes on the island that encoded products with significant homology to the conserved gene products of other filamentous phages or the structural proteins of nonfilamentous phages.
The maltose-sensitive haemagglutinin (MSHA) is unique to the El Tor biotype of V. cholerae. Initially characterized as a haemagglutinin, it was later found to be a type IV pilus42,43. The MSHA biogenesis (MshHIJKLMNEGF) and structural (MshBACD) proteins are all clustered on chromosome 1. There are no apparent integrases or transposases that might define this region as a pathogenicity island or suggest an origin for it other than V. cholerae. In support of this conclusion, trinucleotide composition analysis shows that this region has similar composition to the rest of the chromosome, suggesting that if these genes were acquired it was very early in the Vibrio phylogenetic history. Recently, several investigators have reported that MSHA is not required for intestinal colonization, nor does it seem to appreciably affect the efficiency of colonization44,45,46, but instead plays a role in biofilm formation27,28. Accordingly, this pilus may be important for the environmental fitness of Vibrio species rather than for pathogenic potential.
The pilA region of V. cholerae genome apparently encodes a third type IV pilus, although it has not been visualized47. This gene cluster includes a gene encoding a prepilin peptidase (PilD) that is required for the efficient processing of protein complexes with type IV prepilin-like signal sequences including TCP, MSHA and EPS47,48. The EPS system of V. cholerae encodes a type II secretion system involved in extracellular export of CT and other proteins. The EPS system is encoded by chromosome 1 but, like the MSHA genes, trinucleotide analysis suggests that the EPS genes of V. cholerae have not been recently acquired. In contrast, trinucleotide composition analysis suggests that the pilA gene cluster was acquired by horizontal transfer. Thus, analysis of the V. cholerae genome sequence provides some evidence that older gene clusters, like MSHA and EPS, have become dependent on newly acquired genes such as PilD.
Conclusions
The Vibrio cholerae genome sequence provides a new starting point for the study of this organism's environmental and pathobiological characteristics. It will be interesting to determine the gene expression patterns that are unique to its survival and replication during human infection35 as well as in the environment13,14,16. Additionally, the genomic sequence of V. cholerae should facilitate the study of this model multi-chromosomal prokaryotic organism. Comparative genomics between several species in the genus Vibrio will provide a better understanding of the origin of the new small chromosome and the role that it plays in Vibrio biology. The genome sequence may also provide important clues to understanding the metabolic and regulatory networks that link genes on the two chromosomes. Finally, V. cholerae clearly represents a promising genetic system for studying how several horizontally acquired loci located on separate chromosomes can still efficiently interact at the regulatory, cell biology and biochemical levels.
Methods
Whole-genome random sequencing procedure.
Vibrio cholerae N16961 was grown from a single isolated colony. Cloning, sequencing and assembly were as described for genomes sequenced by TIGR18. One small-insert plasmid library (2–3 kb) was generated by random mechanical shearing of genomic DNA. One large insert library was ligated into λ-DASHII/EcoRI vector (Stratagene). In the initial sequence phase, approximately sevenfold sequence coverage was achieved with 49,633 sequences from plasmid clones. Sequences from both ends of 383 λ-clones served as a genome scaffold, verifying the orientation, order and integrity of the contigs. The plasmid and λ sequences were jointly assembled using TIGR Assembler. Sequence gaps were closed by editing the end sequences and/or primer walking on plasmid clones. Physical gaps were closed by direct sequencing of genomic DNA, or combinatorial polymerase chain reaction (PCR) followed by sequencing the PCR product. The final genome sequence is based on 51,164 sequences.
ORF prediction and gene family identification. An initial set of ORFs, likely to encode proteins, was identified with GLIMMER49, and those shorter than 30 codons were eliminated. ORFs that overlapped were visually inspected, and in some cases removed. ORFs were searched against a non-redundant protein database18. Frameshifts and point mutations were detected and corrected where appropriate. Remaining frameshifts and point mutations are considered to be authentic and were annotated as ‘authentic frameshift’ or ‘authentic point mutation’. ORFs were also analysed with two sets of hidden Markov models (HMMs) constructed for a number of conserved protein families (1,313 from Pfam v3.1 (ref. 50) and 476 from the TIGRFAM) by use of the HMMER package. TopPred was used to identify membrane-spanning domains in proteins.
Paralogous gene families were constructed by searching the ORFs against themselves using BLASTX, identifying matches with E ≤ 10-5 over 60% of the query search length, and subsequently clustering these matches into multigene families. Multiple alignments for these protein families were generated with the CLUSTALW program and the alignments scrutinized.
Distribution of all 64 trinucleotides (3-mers) for each chromosome was determined, and the 3-mer distribution in 2,000-bp windows that overlapped by half their length (1,000 bp) across the genome was computed. For each window, we computed the χ2 statistic on the difference between its 3-mer content and that of the whole chromosome. A large value of this statistic indicates that the 3-mer composition in this window is different from the rest of the chromosome. Probability values for this analysis are based on the assumption that the DNA composition is relatively uniform throughout the genome. Because this assumption may be incorrect, we prefer to interpret high χ2 values merely as indicators of regions on the chromosome that appear unusual and demand further scrutiny.
Homologues of the genes of interest were identified using the BLASTP and FASTA3 search programs. All homologues were then aligned to each other using the CLUSTALW program with default settings. Phylogenetic trees were generated from the alignments using the neighbour-joining algorithm as implemented by the PAUP* program (with a PAM matrix based distance calculation). Regions of the alignment that were hypervariable or were of low confidence were excluded from the phylogenetic analysis. All alignments are available upon request.
Supplementary information
Supplementary tables and figures (DOC 355 kb)
Acknowledgements
This work was supported by the National Institutes of Health, National Institute of Allergy and Infectious Disease. We thank M. Heaney, V. Sapiro, B. Lee, M. Holmes and B. Vincent for database and software support.
References
- 1.Wachsmuth K, Olsvik Ø, Evins GM, Popovic T. Vibrio Cholerae And Cholera: Molecular To Global Perspective. 1994. pp. 357– 370. [Google Scholar]
- 2.Faruque SM, Albert MJ, Mekalanos JJ. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 1998;62: 1301–1314. doi: 10.1128/MMBR.62.4.1301-1314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272: 1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 4.Karaolis DK, Somara S, Maneval DR , Jr, Johnson JA, Kaper JB. A bacteriophage encoding a pathogenicity island, a type-IV pilus and a phage receptor in cholera bacteria. Nature. 1999;399:375– 379. doi: 10.1038/20715. [DOI] [PubMed] [Google Scholar]
- 5.Brown RC, Taylor RK. Organization of tcp, acf, and toxT genes within a ToxT-dependent operon. Mol. Microbiol. 1995; 16:425–439. doi: 10.1111/j.1365-2958.1995.tb02408.x. [DOI] [PubMed] [Google Scholar]
- 6.Hochhut B, Waldor MK. Site-specific integration of the conjugal Vibrio cholerae SXT element into prfC. Mol. Microbiol. . 1999;32:99–110. doi: 10.1046/j.1365-2958.1999.01330.x. [DOI] [PubMed] [Google Scholar]
- 7.Yildiz FH, Schoolnik GK. Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. . Proc. Natl Acad. Sci. USA. 1999;96:4028– 4033. doi: 10.1073/pnas.96.7.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bik EM, Bunschoten AE, Gouw RD, Mooi FR. Genesis of the novel epidemic Vibrio cholerae O139 strain: evidence for horizontal transfer of genes involved in polysaccharide synthesis. EMBO J. 1995;14:209–216. doi: 10.1002/j.1460-2075.1995.tb06993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waldor MK, Colwell R, Mekalanos JJ. The Vibrio cholerae O139 serogroup antigen includes an O-antigen capsule and lipopolysaccharide virulence determinants. . Proc. Natl Acad. Sci. USA. 1994;91:11388– 11392. doi: 10.1073/pnas.91.24.11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovach ME, Shaffer MD, Peterson KM. A putative integrase gene defines the distal end of a large cluster of ToxR-regulated colonization genes in Vibrio cholerae . Microbiology. 1996;142:2165– 2174. doi: 10.1099/13500872-142-8-2165. [DOI] [PubMed] [Google Scholar]
- 11.Karaolis DK. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc. Natl Acad. Sci. USA. 1998; 95:3134–3139. doi: 10.1073/pnas.95.6.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mekalanos JJ, Rubin EJ, Waldor MK. Cholera: molecular basis for emergence and pathogenesis. . FEMS Immunol. Med. Microbiol. 1997;18:241– 248. doi: 10.1111/j.1574-695X.1997.tb01052.x. [DOI] [PubMed] [Google Scholar]
- 13.Colwell RR. Global climate and infectious disease: the cholera paradigm. Science . 1996;274:2025–2031. doi: 10.1126/science.274.5295.2025. [DOI] [PubMed] [Google Scholar]
- 14.Colwell RR, Spira WM. Cholera. 1992. pp. 107–127. [Google Scholar]
- 15.Lobitz B. Climate and infectious disease: use of remote sensing for detection of Vibrio cholerae by indirect measurement. Proc. Natl Acad. Sci. USA. 2000;97:1438–1443. doi: 10.1073/pnas.97.4.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colwell RR, Huq A. Environmental reservoir of Vibrio cholerae. The causative agent of cholera. Ann. NY Acad. Sci. 1994;740:44–54. doi: 10.1111/j.1749-6632.1994.tb19852.x. [DOI] [PubMed] [Google Scholar]
- 17.Roszak DB, Colwell RR. Survival strategies of bacteria in the natural environment. Microbiol. Rev. 1987;51: 365–379. doi: 10.1128/mr.51.3.365-379.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraser CM. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580– 586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 19.Yamaichi Y, Iida T, Park KS, Yamamoto K, Honda T. Physical and genetic map of the genome of Vibrio parahaemolyticus: presence of two chromosomes in Vibrio species. . Mol. Microbiol. 1999;31:1513– 1521. doi: 10.1046/j.1365-2958.1999.01296.x. [DOI] [PubMed] [Google Scholar]
- 20.Trucksis M, Michalski J, Deng YK , Kaper JB. The Vibrio cholerae genome contains two unique circular chromosomes. . Proc. Natl Acad. Sci. USA. 1998;95:14464– 14469. doi: 10.1073/pnas.95.24.14464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- 22.Rowe-Magnus DA, Guerout AM, Mazel D. Super-integrons. Res. Microbiol. 1999; 150:641–651. doi: 10.1016/S0923-2508(99)00127-8. [DOI] [PubMed] [Google Scholar]
- 23.Hall RM, Brookes DE, Stokes HW. Site-specific insertion of genes into integrons: role of the 59-base element and determination of the recombination cross-over point. Mol. Microbiol. 1991;5:1941– 1959. doi: 10.1111/j.1365-2958.1991.tb00817.x. [DOI] [PubMed] [Google Scholar]
- 24.Davis BM, Kimsey HH, Chang W, Waldor MK. The Vibrio cholerae O139 Calcutta bacteriophage CTXphi is infectious and encodes a novel repressor. J. Bacteriol. 1999;181:6779 –6787. doi: 10.1128/JB.181.21.6779-6787.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mekalanos JJ. Duplication and amplification of toxin genes in Vibrio cholerae. Cell. 1983;35:253–263. doi: 10.1016/0092-8674(83)90228-3. [DOI] [PubMed] [Google Scholar]
- 26.Mazel D, Dychinco B, Webb VA , Davies J. A distinctive class of integron in the Vibrio cholerae genome. Science. 1998;280:605–608. doi: 10.1126/science.280.5363.605. [DOI] [PubMed] [Google Scholar]
- 27.Watnick PI, Kolter R. Steps in the development of a Vibrio cholerae El Tor biofilm. Mol. Microbiol. 1999; 34:586–595. doi: 10.1046/j.1365-2958.1999.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watnick PI, Fullner KJ, Kolter R. A role for the mannose-sensitive hemagglutinin in biofilm formation by Vibrio cholerae El Tor. J. Bacteriol. 1999; 181:3606–3609. doi: 10.1128/JB.181.11.3606-3609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huq A, et al. Ecological relationships between Vibrio cholerae and planktonic crustacean copepods. Appl. Environ. Microbiol. 1983;45: 275–283. doi: 10.1128/aem.45.1.275-283.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bassler BL, Yu C, Lee YC, Roseman S. Chitin utilization by marine bacteria. Degradation and catabolism of chitin oligosaccharides by Vibrio furnissii. J. Biol. Chem. 1991;266: 24276–24286. doi: 10.1016/S0021-9258(18)54225-3. [DOI] [PubMed] [Google Scholar]
- 31.Yildiz FH, Schoolnik GK. Role of rpoS in stress survival and virulence of Vibrio cholerae. J. Bacteriol. 1998; 180:773–784. doi: 10.1128/JB.180.4.773-784.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bassler BL, Greenberg EP, Stevens AM. Cross-species induction of luminescence in the quorum-sensing bacterium Vibrio harveyi. J. Bacteriol. 1997;179: 4043–4045. doi: 10.1128/jb.179.12.4043-4045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams SG, Attridge SR, Manning PA. The transcriptional activator HlyU of Vibrio cholerae : nucleotide sequence and role in virulence gene expression. Mol. Microbiol. 1993;9:751–760. doi: 10.1111/j.1365-2958.1993.tb01735.x. [DOI] [PubMed] [Google Scholar]
- 34.Eisen JA, Hanawalt PC. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat. Res. 1999; 435:171–213. doi: 10.1016/S0921-8777(99)00050-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SH, Hava DL, Waldor MK , Camilli A. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell. 1999;99:625– 634. doi: 10.1016/S0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- 36.Lin W, et al. Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc. Natl Acad. Sci. USA. 1999;96:1071–1076. doi: 10.1073/pnas.96.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Z, Milton D, Nybom P, Sjo A, Magnusson KE. Vibrio cholerae hemagglutinin/protease (HA/protease) causes morphological changes in cultured epithelial cells and perturbs their paracellular barrier function. Microb. Pathog. 1996;21: 111–123. doi: 10.1006/mpat.1996.0047. [DOI] [PubMed] [Google Scholar]
- 38.Alm RA, Stroeher UH, Manning PA. Extracellular proteins of Vibrio cholerae: nucleotide sequence of the structural gene (hlyA) for the haemolysin of the haemolytic El Tor strain 017 and characterization of the hlyA mutation in the non- haemolytic classical strain 569B. Mol. Microbiol. 1988; 2:481–488. doi: 10.1111/j.1365-2958.1988.tb00054.x. [DOI] [PubMed] [Google Scholar]
- 39.O'Brien AD, Chen ME, Holmes RK , Kaper J, Levine MM. Environmental and human isolates of Vibrio cholerae and Vibrio parahaemolyticus produce a Shigella dysenteriae 1 (Shiga)-like cytotoxin. Lancet. 1984;1: 77–78. doi: 10.1016/S0140-6736(84)90006-0. [DOI] [PubMed] [Google Scholar]
- 40.Ogawa A, Kato J, Watanabe H, Nair BG, Takeda T. Cloning and nucleotide sequence of a heat-stable enterotoxin gene from Vibrio cholerae non-O1 isolated from a patient with traveler's diarrhea. Infect. Immun. 1990;58:3325– 3329. doi: 10.1128/iai.58.10.3325-3329.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manning PA. The tcp gene cluster of Vibrio cholerae. Gene. 1997; 192:63–70. doi: 10.1016/S0378-1119(97)00036-X. [DOI] [PubMed] [Google Scholar]
- 42.Jonson G, Holmgren J, Svennerholm AM. Identification of a mannose-binding pilus on Vibrio cholerae El Tor. Microb. Pathog. 1991;11: 433–441. doi: 10.1016/0882-4010(91)90039-D. [DOI] [PubMed] [Google Scholar]
- 43.Jonson G, Lebens M, Holmgren J. Cloning and sequencing of Vibrio cholerae mannose-sensitive haemagglutinin pilin gene: localization of mshA within a cluster of type 4 pilin genes. Mol. Microbiol. 1994;13:109– 118. doi: 10.1111/j.1365-2958.1994.tb00406.x. [DOI] [PubMed] [Google Scholar]
- 44.Attridge SR, Manning PA, Holmgren J, Jonson G. Relative significance of mannose-sensitive hemagglutinin and toxin-coregulated pili in colonization of infant mice by Vibrio cholerae El Tor. Infect. Immun. 1996;64:3369–3373. doi: 10.1128/iai.64.8.3369-3373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tacket CO. Investigation of the roles of toxin-coregulated pili and mannose- sensitive hemagglutinin pili in the pathogenesis of Vibrio cholerae O139 infection. Infect. Immun. 1998;66:692– 695. doi: 10.1128/IAI.66.2.692-695.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thelin KH, Taylor RK. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. . 1996;64:2853–2856. doi: 10.1128/iai.64.7.2853-2856.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fullner KJ, Mekalanos JJ. Genetic characterization of a new type IV-A pilus gene cluster found in both classical and El Tor biotypes of Vibrio cholerae. Infect. Immun. 1999;67: 1393–1404. doi: 10.1128/IAI.67.3.1393-1404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marsh JW, Taylor RK. Identification of the Vibrio cholerae type 4 prepilin peptidase required for cholera toxin secretion and pilus formation. Mol. Microbiol. 1998;29: 1481–1492. doi: 10.1046/j.1365-2958.1998.01031.x. [DOI] [PubMed] [Google Scholar]
- 49.Salzberg SL, Delcher AL, Kasif S , White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998;26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bateman, A. et al. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res.27, 260–262 (1999). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary tables and figures (DOC 355 kb)