

Abstract
The structural covariance network (SCN) has provided a perspective on the large‐scale brain organization impairment in the Alzheimer's Disease (AD) continuum. However, the successive structural impairment across brain regions, which may underlie the disrupted SCN in the AD continuum, is not well understood. In the current study, we enrolled 446 subjects with AD, mild cognitive impairment (MCI) or normal aging (NA) from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database. The SCN as well as a casual SCN (CaSCN) based on Granger causality analysis were applied to the T1‐weighted structural magnetic resonance images of the subjects. Compared with that of the NAs, the SCN was disrupted in the MCI and AD subjects, with the hippocampus and left middle temporal lobe being the most impaired nodes, which is in line with previous studies. In contrast, according to the 194 subjects with records on CSF amyloid and Tau, the CaSCN revealed that during AD progression, the CaSCN was enhanced. Specifically, the hippocampus, thalamus, and precuneus/posterior cingulate cortex (PCC) were identified as the core regions in which atrophy originated and could predict atrophy in other brain regions. Taken together, these findings provide a comprehensive view of brain atrophy in the AD continuum and the relationships among the brain atrophy in different regions, which may provide novel insight into the progression of AD.
Keywords: Alzheimer's disease, granger causality analysis, morphometric MRI, progression, structural covariance network
A casual model combined with structural covariance network (SCN) was provided and applied on the Alzheimer's Disease Neuroimaging Initiative (ADNI) dataset. We found the asynchronism neurodegeneration progress may underlying the disrupted SCN previously reported, and hippocampus, thalamus, and precuneus/posterior cingulate cortex (PCC) were identified as the core regions in which atrophy originated and could predict atrophy in other brain regions.

1. INTRODUCTION
Alzheimer's disease (AD) is one of most common neurodegenerative diseases and has high morbidity and mortality in the aging population worldwide (Li et al., 2015; Petersen, 2004; Weiner et al., 2017). Brain atrophy is a typically reported symptom of AD, and magnetic resonance imaging (MRI) provides an opportunity to investigate brain atrophy in large in vivo samples (Pini et al., 2016). Many previous studies have focused on the structural covariance network (SCN), which describes how structural measures of different brain regions covary across subjects, and found that AD as well as mild cognitive impairment (MCI) patients tend to have a disrupted SCN (Alexander‐Bloch, Giedd, & Bullmore, 2013; Chong et al., 2017; He, Chen, & Evans, 2008; Montembeault et al., 2016; Seeley, Crawford, Zhou, Miller, & Greicius, 2009). These results provide a perspective on the impairment of large‐scale brain organization (He et al., 2008; Li et al., 2019; Montembeault et al., 2016) and the pathology of AD at the regional level, which may also be related to amyloid or Tau pathology (Chang, Huang, Chang, Lee, & Chang, 2018; Voevodskaya et al., 2018).
However, the biological basis of the SCN is still not well understood. It may be related to many different factors such as synchronous GM growth/atrophy and/or effect from shared genetic factors (Alexander‐Bloch et al., 2013), disease pathology (Seeley et al., 2007), white matter fiber connectivity (Gong, He, Chen, & Evans, 2012). Specifically, for AD, it is reasonable to suspect that an enhanced SCN may be related to the synchronous progression of neurodegeneration across regions and that a disrupted SCN may be related to only one affected region but not another. To better understand previous SCN studies as well as AD pathology at the whole‐brain level, it is valuable to investigate the synchrony and succession of brain morphological changes across brain regions in AD. Recently, Granger causality analysis (GCA) (Seth, Barrett, & Barnett, 2015) was utilized to reveal the causal effects of the atrophy of different brain regions across patients with increasing levels of epilepsy severity (Zhang et al., 2017). We anticipate that applying GCA to the sequence of morphometry data from normal aging to mild cognitive impairment (MCI) and finally to AD may reveal the successive effect on the SCN of the atrophy of different brain regions.
Therefore, in the current study, we hypothesize that during AD progression, the disrupted SCN is mainly related to a progression of neurodegeneration that successively affects different brain regions. The causal SCN (CaSCN) (Zhang et al., 2017) may provide a more comprehensive view of brain atrophy patterns in AD and may possibly be enhanced during AD progression. Specifically, we aim to: (a) use source‐based morphology (SBM) (Harenski, Harenski, Calhoun, & Kiehl, 2020; Hopkins et al., 2019; Xu, Groth, Pearlson, Schretlen, & Calhoun, 2009) on AD data to find representative AD‐related spatial components; (b) investigate casual SCN models to find the regions whose atrophy can drive other regions during AD progression; (c) apply a combination of the sliding window method (Allen et al., 2014) with GCA for these components to demonstrate whether and how these effects change across populations with increasing levels of disease severity.
2. MATERIALS AND METHODS
2.1. Participants
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public‐private partnership led by Principal Investigator Michael W. Weiner, MD. The primary goal of the ADNI is to test whether magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer's disease (AD). For more information, please see www.adni-info.org.
Our data cohort consists of 3D T1‐weighted MRI data from normal aging controls (NAs) and MCI and AD subjects obtained from the ADNI‐1, ADNI‐2, ADNI‐3 and ADNI‐GO databases. The inclusion criteria were as follows: (a) Subject had to have “MPRAGE/MP‐RAGE” images acquired by 3.0T scanners, with a thickness of 1.2 mm or 1.0 mm and an in‐plane resolution of 1 mm × 1 mm; (b) the age of the subjects had to be between 60 and 80 years. Furthermore, if the subject had more than one set of MRI data, the first MRI obtained was included in the cohort. Under these criteria, 574 sets of T1‐weighted images were downloaded, each for a different subject. A total of 114 subjects were excluded because of insufficient imaging quality (obvious artifacts and/or failed segmentation in the subsequent voxel‐based morphometry analysis). Another 14 subjects were excluded to match age and gender between groups; therefore, 446 subjects were included in the subsequent data analysis. A total of 194 subjects (85 NA, 59 MCI and 50 AD) also had cerebrospinal fluid (CSF) biomarker records, including Aβ, Tau and P‐Tau measures. All subjects had age, gender data and Mini‐Mental State Examination (MMSE) information in their records. All except four subjects had education records. The four subjects without education information were treated as if they had the average years of education of all other subjects in our data. The demographic, cognitive and CSF biomarker data used in the current study are stated displayed (Table 1). The CSF biomarker is “using the micro‐bead‐based multiplex immunoassay, the INNO‐BIA AlzBio3 RUO test (Fujirebio, Ghent, Belgium), on the Luminex platform,” which provided by ADNI as “UPENN Biomraker.”
TABLE 1.
Demographic and CSF biomarkers
| NA | MCI | AD | F/chi‐squared a | df | p value | |
|---|---|---|---|---|---|---|
| Subjects (all) | 171 | 124 | 151 | |||
| Age (yr.) | 72.6 ± 4.6 | 72.6 ± 5.3 | 72.7 ± 4.9 | <0.0001 | (2,443) | .996 |
| Gender (m:f) | 78:93 | 72:52 | 73:78 | 4.71 | 2 | .095 |
| MMSE score | 29.1 ± 1.2 | 26.8 ± 2.2 | 23.1 ± 2.2 | 423.92 | (2,443) | <.001 |
| Education (yr.) | 16.2 ± 2.6 | 15.4 ± 3.2 | 15.2 ± 3.1 | 5.02 | (2,443) | .007 |
| APOEε4 b | 6/46/113/6 | 26/47/50/1 | 35/70/37/8 | 69.56 | 6 | <.0001 |
| Subjects (with CSF record) | 85 | 59 | 50 | |||
| CSF Aβ42 (ng/l) c | 202 ± 52 | 158 ± 49 | 132 ± 29 | 38.42 | (2,191) | <.001 |
| CSF tau (ng/l) c | 68 ± 37 | 110 ± 74 | 128 ± 47 | 22.67 | (2,191) | <.001 |
| CSF P‐tau (ng/l) c | 30 ± 16 | 37 ± 20 | 47 ± 22 | 13.69 | (2,191) | <.001 |
The chi‐squared test was used for gender and APOE, and ANOVA was applied to all other variables.
The number (a/b/c/d) means there are a of subjects were APOEε4 homozygote; b of subjects were heterozygote; c of subjects were without APOEε4; d of subjects APOE data is missing.
Only those subjects with CSF measures were counted.
2.2. Voxel‐based morphometry
Standard voxel‐based morphometry was first performed for all the images to generate a voxel‐wise gray matter volume map for each subject. The data processing was carried out by using SPM12 (http://www.fil.ion.ucl.ac.uk/spm) with DARTEL method (Ashburner, 2007). The images were transformed into MNI space and resampled to 1.5 × 1.5 × 1.5 mm3. All images were segmented into three tissue classes, representing gray matter (GM), white matter and CSF. Modulated GM maps were generated such that the value in each voxel represents the GM volume. The GM volume maps were further smoothed with 6 mm FWHM, and the results were used for subsequent morphological analyses.
2.3. Source‐based morphometry (SBM)
The SBM analyses in the current study followed the original SBM study by Xu and colleagues (Xu et al., 2009) and were carried out by the GIFT toolbox (http://icatb.sourceforge.net). First, given that not all the images covered the whole cerebellum, a brain mask without the cerebellum was applied on all the GM volume maps. Then, all the GM volume maps were concatenated together into a set of 4D MRI images. During SBM, the spatial dimensions (x, y, z) were converted into one dimension; therefore, the whole dataset was converted to an m × n matrix D, where m is the voxel number within the mask and n is the subject number (therefore, here n is 446). This matrix (D) was then decomposed by independent component analysis (ICA) into two matrices, a mixing matrix (M) and a source matrix (S), where D = M*S. The mixing matrix M is a k × n matrix that expresses the score of each of the n subjects for each of k components. Similarly, the source matrix S is an m × k matrix that expresses the scores of each voxel in each of the k components, which can be mapped back to the brain voxels to generate k spatial maps. For the data used in the current study, k was first estimated by GIFT through Akaike's information criterion, resulting in a value of 20. Therefore, 20 components were extracted, and we marked them as component A to component T.
2.4. Group differences
The mixing matrix was used for statistical analysis of the components among the groups. For every column in the mixing matrix M, the parameters expressed a score measuring the contribution of every component for the 446 subjects. In other words, the score of one component across different subjects represented the individual differences in the volume of this component. Therefore, ANOVA was applied with age, gender, and education as covariates to investigate whether there were significant group differences among the AD, MCI, and NA groups for each component. False discovery rate (FDR) correction was used to control for the 20 applications of ANOVA (one for each component). The significance level was set to p < .05.
2.5. Structural covariance
The SCN was calculated based on SBM components. Namely, instead of typical SCN analysis, we did not calculate the correlation of volume/morphological properties among voxels or predefined ROIs, but between the SBM components. Specifically, SBM gives each subject a score for each component, which represent the volume of this subject on such “distinct regions with common covariation among subjects,” and therefore we can directly apply correlation analysis on these scores to generated SCN results. In detail, for each group of AD, MCI and NA, Pearson's correlation analysis with age, gender and education as covariates was performed on the subject scores between each pair of the 20 components. Then, an interaction analysis was also utilized to evaluate whether the correlation was significantly different among the AD, MCI, and NA groups. FDR correction was applied for all possible component pairs (20 × 19/2 = 190), and the significance level was set to p < .05.
2.6. Causal structural covariance
The CaSCN was generated here by applying Granger's causality analysis (GCA) on the constructed SCN. GCA was first proposed for determining whether the past value of one time course could predict the current value of another time course. If the current value of time course Y could be more accurately estimated by a combination of past values of time courses X and Y than by the past value of Y itself alone, then we define that X has Granger causal influence on Y. In the neuroimaging field, GCA is more often used to investigate the causal relation between functional MRI time courses (Seth et al., 2015). However, it also has potential for evaluating causal relationships between changes in the morphometry of different brain regions if an order among subjects can be identified, as indicated by Zhang et al. (2017).
In the current study, the CaSCN was used to investigate the causal volume changes among components as the severity of AD increased. To define a quantification order of AD severity, CSF Aβ, CSF Tau and CSF P‐Tau were utilized. Specifically, given that the raw numbers of these measures were non‐normally distributed, we first log‐transformed these three measures and then normalized them into Z‐scores. The Z‐scores of Tau and P‐Tau and the inverse Aβ Z‐scores (given that a lower CSF Aβ value indicates AD, not NA, but lower Tau and P‐Tau indicates NA, not AD) were averaged together as the CSF biomarker Z‐score. The subjects were then ordered from smallest to largest average CSF biomarker Z score. In the current study, we assumed that this order represents the progression of AD progressing from least to most sever and used this order in all the following CaSCN steps. Given that only 194 subjects had CSF biomarker data, the CaSCN was based on these subjects only. Additionally, to control the effects of age, gender and education, the residuals of all atrophied component scores after regressing out age, gender and education were used in the CaSCN analysis and corresponding permutation test.
The residuals of each component were reordered by the CSF biomarker Z scores of the subject, and GCA was applied for each pair of “pseudo time series” of the residuals of all components using REST (Song et al., 2011) and following Chen's algorithm (Hamilton, Chen, Thomason, Schwartz, & Gotlib, 2011; Stephan & Roebroeck, 2012). Considering two “pseudo time series,” X and Y. A GCA model can be defined as follows:
The significance of coefficient A i describes the causal relation from X to Y at rank i. In the current study, the rank of GCA, namely, p was set to 1 and we did not considering higher rank as most previous studies (Zhang et al., 2017). A positive GCA coefficient means that the Y values changed in the same direction as X, and a negative GCA coefficient means Y would increase as X decreases and vice versa.
Given that the number of subjects with CSF biomarker records was relatively small, we did not apply the CaSCN for each group (especially for AD and MCI, for which there were only 50/59 subjects. Non‐GCA coefficients reached the significance threshold).
2.7. The driving nodes
To extract the most important causal relations among the structural covariance components from the 20 × 20 network, we calculated the driving scores as in a previous study on fMRI data (Yan & He, 2011) for the components showing significant difference among AD, MCI and NA groups, which means they have atrophied during AD progressing. The driving score of one component is defined as the sum of the GCA coefficients from this component to all other components. Positive and negative driving scores were calculated respectively as sum of all positive/negative GCA coefficients.
To test the significance of these driving scores, a permutation‐based test was applied to validate the significance of all the driving scores. For each permutation, the order of the subjects was randomized, and the CaSCN was analyzed for this randomized new pseudo series and all the GCA coefficients were recorded. After 1,000 permutations, we calculated the driving scores for 1,000 permutations, resulting in 20,000 positive and negative diving scores. We defined scores within the 5% tails of the distribution as significant at p < .05. The components that had significant driving scores were defined as the driving nodes of brain atrophy during AD progression.
2.8. Sliding‐window CaSCN of the driving nodes
Sliding windows were applied to the components that had significant driving scores. Specifically, we computed the casual GCA coefficients from the driving nodes to all other atrophied components for 145 “sliding windows.” Each window consisted of 50 subjects: the 1st window consisted of the 1st subject to the 50th, the second window consisted of the subject from the 2nd to 51st, and so on to the last window, which consisted of the 145th to 194rd subjects. Therefore, if we have x atrophied components and y of them were driving nodes, there would be (x − 1) × y GCA coefficients for each sliding window. The significance of the GCA coefficients was also defined by performing another 1,000 permutation tests. For each permutation, 50 subjects (same as the window length) were randomly selected and ordered, the CaSCN was analyzed, and the GCA coefficients in a sliding window were defined as significant if they were within the 5% tails of the distribution for p < .05. Finally, to assess how these significant GCA coefficients changed from the least to most severe windows, the k‐means clustering algorithm was applied to these windowed GCA coefficient matrices, where the number of clusters was determined by the top 2 to top 15 mean silhouette coefficients.
3. RESULTS
3.1. Source‐based morphometry (SBM)
As shown in Figure 1, SBM decomposed all the variance of the GM volumes across subjects in our data into 20 components. Component A was mainly located in the left middle temporal lobe, while component K was in the right middle temporal lobe. Components D, E and I were all located in the bilateral temporal lobes in different but adjacent regions (superior, middle and inferior, respectively). Component O was in the bilateral temporal poles. Components B and R overlapped with the medial and lateral parts of the sensorimotor regions, respectively. Components L and T were in the medial and lateral middle occipital visual regions, respectively, and J was located in the superior occipital lobe. Components F, M and N were in the inferior, middle, and superior parts of the frontal lobe, respectively. Components C, G and P were in the thalamus, hippocampus, and striatum. Component S was located in the superior parietal lobe, component H was located in the lingual gyrus, and component Q was located in the precuneus and post cingulate cortex (PCC), which is an important hub in the default mode network.
FIGURE 1.
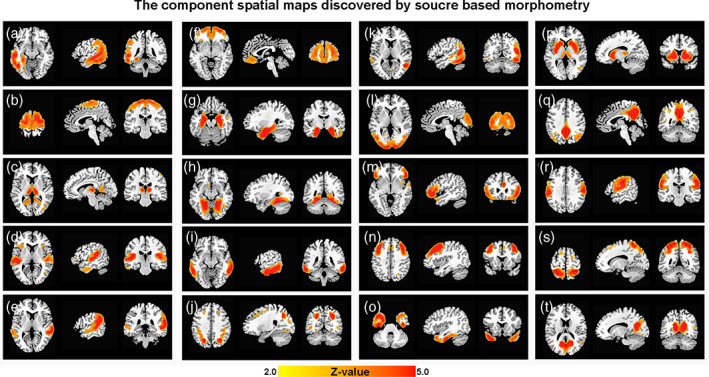
Source‐based morphometry (SBM) component spatial maps. SBM estimated 20 structural covariance components (A–T) from 446 subjects, including those with AD and MCI and NAs. Coronal, sagittal, and axial views of the spatial map for each component are shown here. Images are the z statistics (from yellow to red, 2.0 to 5.0) overlaid on the high‐resolution structural template. Left on the images indicates the left side of the subjects. Except for component A, which was only located on the left hemisphere, all sagittal images show the right hemisphere
In Figure 2, the results of ANOVA after FDR correction (p < .05) revealed that 14 of the 20 components showed significant group differences among the AD, MCI and NA groups: A (F = 20.37, p < .0001), C (F = 3.51, p = .031), E (F = 10.35, p < .0001), F (F = 3.45, p = .033), G (F = 61.86, p < .0001), I (F = 21.61, p < .0001), J (F = 15.46, p < .0001), K (F = 27.63, p < .0001), L (F = 7.02, p ≤ .001), O (F = 20.80, p < .0001), Q (F = 13.13, p < .0001), R (F = 4.23, p = .015), S (4.94, p = .0076) and T (F = 4.60, p = .010). These components are mainly located in temporal and subcortical regions (A, C, E, G, I, K, O) but also involved the parietal (S, J), visual (L, T), frontal (F), sensorimotor (R) and default mode networks (Q). In post hoc analysis, most of these components showed significant differences between each of the three groups. However, MCI showed no significant difference from NA in components C, E, F, L, Q and S. There was no significant difference between AD and MCI in components C and T.
FIGURE 2.
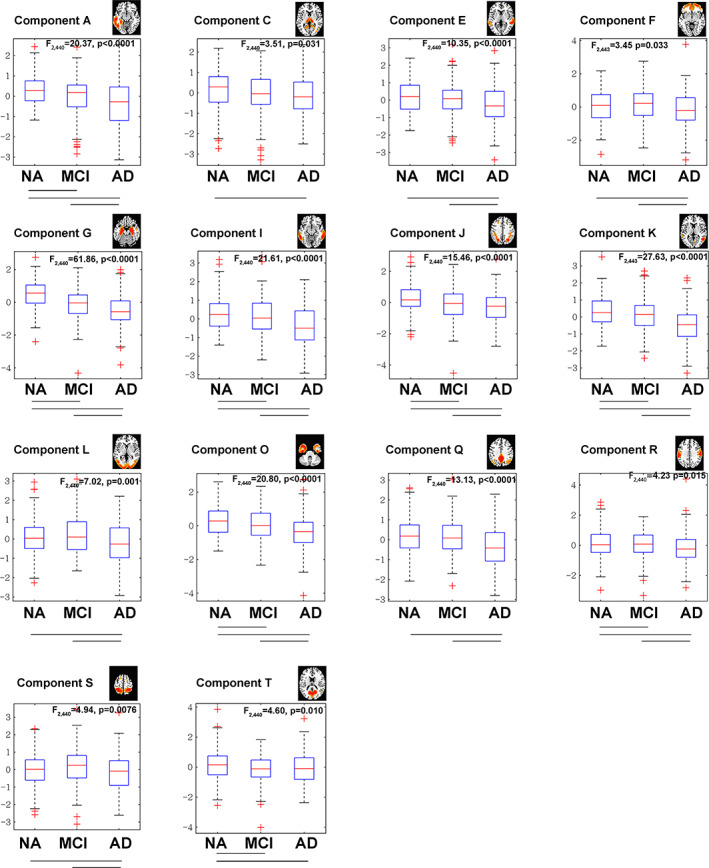
Group differences of the components of the source‐based morphometry (SBM). Fourteen of the 20 components showed significant group differences according to ANOVA among the AD, MCI and NA groups (p < .05, FDR corrected across 20 components). The lines between two groups under each boxplot indicate a significant group difference according to post hoc analysis (p < .05)
3.2. Structural covariance
As illustrated in Figure 3, the SCNs of 11 pairs of components showed significant differences among the three groups. The highest structural covariance was found for the NAs, followed by the MCI group, and the AD group had the lowest. It is also worth noting that all these significant pairs involved component A (left middle temporal lobe) or G (hippocampus), indicating their important role in SCN changes in AD and MCI.
FIGURE 3.
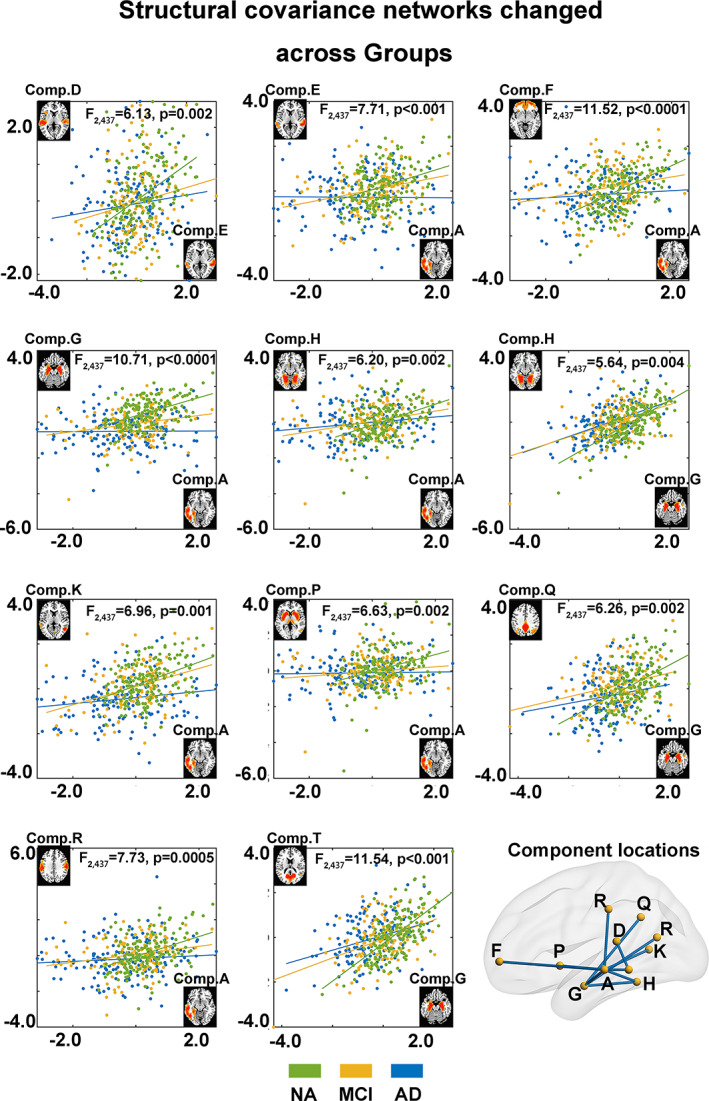
The structural covariance between components was different among the different groups. According to the interaction analysis, there were significant differences in the correlations of the components' Z‐scores across subjects for 11 component pairs (p < .05, FDR corrected across pairs). In the scatter plots, each point represents one subject, and the coordinates are his/her Z‐scores for the two listed components, whose images were shown at the ends of the axes. Subjects from different groups are marked in different colors, and the F‐statistic was used to investigate the interaction effects. The diagram in the bottom right shows the locations for all the component pairs that had significant group differences in structural covariance
3.3. Driving scores of the causal structural covariance
As shown in Figure 4, among all 20 components, three showed a significant positive driving score (permutation test, p < .05), which was defined as the sum of GCA coefficients from the corresponding component to all other components: component C, located in the thalamus, component G, located in the hippocampus, and component Q, located in the PCC/precuneus. None of the negative driving scores reached a significant level. A high positive driving score indicated that during AD progression, the volume alterations in the corresponding component could predict other component volume alterations. These three driving components were further involved in the following sliding window analyses.
FIGURE 4.
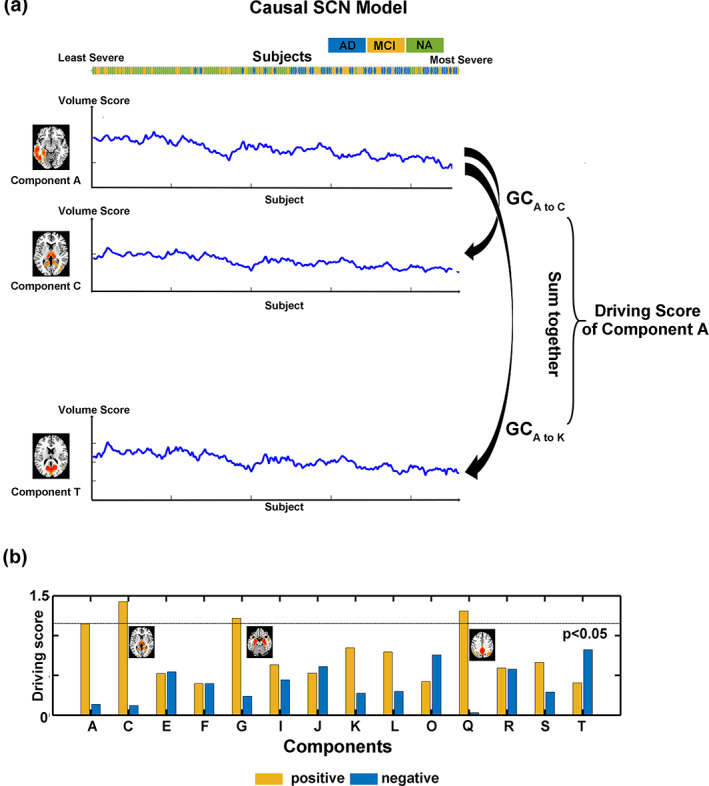
Causal SCN analysis and the “driving nodes.” (a) Schematic plot of Granger causality analysis (GCA). The GCA coefficients were calculated from one to another component by their volume scores after ordering the sequence of AD‐related CSF biomarkers from least to most severe. The driving score of a component was defined as the sum of all GCA coefficients from the corresponding component to all other components. The significance was defined by a permutation test, and the threshold was p < .05. Both positive and negative GCA coefficients were calculated (not shown in the figure). (b) Driving scores of the components that showed significant group differences among AD, MCI and NA. Three of the components (C, thalamus; G, hippocampus; and Q, PCC/precuneus) showed significant positive driving scores, which indicated that during AD progression, their volume alterations can predict other component volume alterations. These significant driving components were further investigated by a sliding window method
3.4. Sliding‐window causal structural covariance
As shown in Figure 5, as the window slid from the least to the most severe subjects, the GCA coefficients from the three driving components to other components also changed. In the less severe windows, the significant GCA coefficients were quite sparse. In more severe windows, the significant GCA coefficients were denser. Moreover, in the severer part, the GCA coefficients patterns were more consistent across windows, and GCA coefficients was higher when closer to the most severe end (Figure 5b).
FIGURE 5.
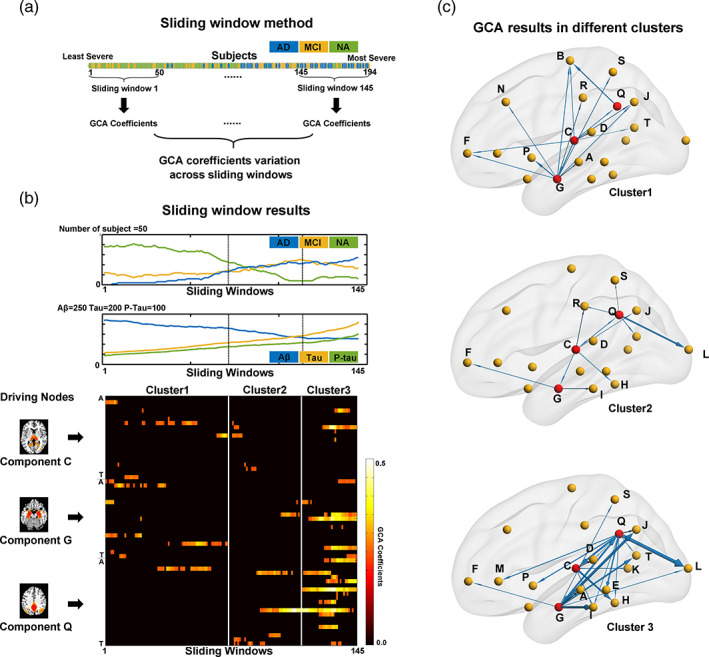
The GCA coefficients from the driving node components vary according to AD progression. (a) The GCA sliding window method. A total of 194 subjects with CSF records were ordered from least to most severe. Each sliding window contained 50 subjects, and 145 such sliding windows were obtained from least to most severe. The GCA coefficients were calculated for each sliding window. (b) The sliding window results of the GCA coefficients for the three driving nodes. The upper plot shows the number of subjects with AD, MCI and NA within each sliding window. The middle plot shows the mean Aβ, Tau and P‐Tau values of all subjects within each sliding window. The bottom panel shows the GCA coefficients from the three driving node components (C, thalamus; G, hippocampus; and Q, PCC/precuneus) to all other components for each sliding window. Only significant GCA coefficients (by permutation test, p < .05) are shown here. The output GCA coefficients from the three driving nodes of each sliding window can be further divided into 3 clusters by k‐means clustering, and the boundaries of the resulting clusters are also marked in all three subfigures in (b). (c) The detailed GCA results from the three driving nodes in each of the k‐mean clusters. The arrow direction indicates the driving direction, and the width represents the proportion of significant sliding windows in the whole cluster
To visualize these progresses of GCA coefficients patterns from least to most severe windows, we used the 57 GCA coefficients from these three driving components (3 driving nodes, each of which has 19 GCA coefficients to all other components) as features, and the k‐means clustering algorithm was utilized to divide the 145 sliding windows into clusters. The mean silhouette coefficient reached its maximum value (0.54) for 3 clusters (from 2 to 20 clusters). Therefore, the 145 sliding windows were divided into 3 clusters: cluster 1 had 71 windows, which mainly included NA subjects, and clusters 2 and 3 contained 42 and 32 windows, which mainly included AD and MCI patients, respectively (Figure 5b).
The average GCA patterns within each cluster in Figure 5c give out a representative visualization of the least to most severe changing progress more clearly. In cluster 1, a relatively low portion of sliding windows had significant GCA coefficients from any of the driving node components. In cluster 2, the small number of edges showed that brain atrophy originated from the driving nodes in the hippocampus, thalamus and PCC/precuneus to the temporal and parietal regions but also spread to the frontal and occipital regions. In cluster 3, which contained the most severe patients, the casual effect from these three driving node components was most significant (nearly 100% of sliding windows in this cluster), and the brain atrophy casual effect radiated from the driving node components into most of the temporal and subcortical components and some of the occipital and parietal components.
4. DISCUSSION
By utilizing and combining source‐based morphometry, structural covariance and Granger causality analysis, the current study investigates brain atrophy during AD progression in a large structural MRI data cohort. Specifically, (a) the individual volume differences between the AD/MCI and NA populations can be decomposed into 20 components. These components show brain atrophy mainly in temporal and subcortical regions but also spread into frontal, parietal and even visual and sensorimotor regions; (b) the structural covariance between these components further indicated that the hippocampus/temporal regions comprised the most important node that showed a disrupted SCN in MCI and AD; (c) by using CaSCN and sliding window, we found that during AD progression, the hippocampus, thalamus and default mode cores have atrophy patterns that can predict atrophy in other components, and such predictions among components may be quite different along the spectrum from normal aging to the most severe manifestation of AD.
According to the results from SBM, the individual differences in brain volume across the AD/MCI/NA population were divided into 20 components. Specifically, the temporal lobe was decomposed into seven components (including the hippocampus), while there were only three components in the frontal and occipital lobes, two components in the sensorimotor areas and only one component in the parietal lobe. This is not surprising because AD pathology first occurs in the temporal regions (Braak & Braak, 1995; Duara et al., 2008; Granadillo, Paholpak, Mendez, & Teng, 2017). The different components obtained with SBM in the temporal lobe may indicate that there are complex and differentiated volume differences in the temporal lobe across subjects, which may suggest that different parts of the temporal lobe are influenced by different pathological, genetic or environmental factors during AD progression (Alexander‐Bloch et al., 2013). In contrast, the frontal, occipital and parietal lobes were spared until late‐stage AD (Apostolova et al., 2007; Middelkoop et al., 2001; Pini et al., 2016); therefore, fewer components can be found by SBM in these regions.
Consistent with previous studies, groupwise differences among the AD/MCI population and the NAs were found in most of the SBM components located in widespread regions in the brain, including temporal, parietal, frontal, default mode, subcortical, and occipital regions (Apostolova et al., 2007; Pini et al., 2016; Prestia et al., 2010). Most of these components showed significant differences between each pair of the three groups, indicating continuous atrophy, while some of the components, such as those in the frontal, parietal, and occipital regions, indicated no significant atrophy in MCI, consistent with previous reports (Braak & Braak, 1995; Ferreira et al., 2016; Fiford et al., 2018; Jagust, 2018; Ting et al., 2015). The default mode network was shown to be a core region of brain alteration during AD progression, and functional alterations of the default mode network at the MCI stage have been previously reported (Franzmeier et al., 2017; Grothe et al., 2016; Ossenkoppele et al., 2015). Our results suggest that atrophy in this region may only be found in the AD group.
The structural covariance among the components also showed alterations in a network whose core regions were components G and A. These two components are located in the hippocampus and middle temporal lobe, respectively. As mentioned above, AD‐related changes in structural covariance may be difficult to interpret. A disrupted SCN may indicate that some independent factors affected one region but not another, and therefore, they lost covariance across subjects. Therefore, this result provides additional evidence that the hippocampus is an important and early impaired region in AD (Albert et al., 2011; Sperling et al., 2011) and may have a more dramatic and earlier atrophy pattern than other components (Alexander‐Bloch et al., 2013). The atrophy in component A, which was located in the left middle temporal lobe, overlapped with the auditory language area. This may be a symbol of logopenic‐variant primary progressive aphasia (Ossenkoppele et al., 2015), which leads to a different atrophy model in these variant patients but not others and therefore leads to attenuated covariance; however, this needs to be verified in a future study.
The CaSCN results further indicated that component G, namely, the hippocampus, was a “driving core,” whose volume alteration pattern was “ahead of” the volume alteration of other components across the sequence from normal aging or less severe AD to more severe AD. Moreover, components C and Q, which are located in the thalamus and PCC/precuneus, respectively, were also identified as “driving cores.” This result indicated that brain atrophy in these three regions may predict atrophy in other regions. The identification of the hippocampus as a driving core is also consistent with previous views of the progression of AD pathology across brain regions (Apostolova et al., 2007; Braak & Braak, 1995; Halliday, 2017; Weiner et al., 2017). Similarly, the PCC/precuneus, as a hub region of the default mode network, has also been identified as an AD‐related region (Franzmeier et al., 2017; Grothe et al., 2016; Ossenkoppele et al., 2015). The thalamus was recently recognized as another early and important pathologically impaired region in AD (Aggleton, Pralus, Nelson, & Hornberger, 2016; Moustafa, McMullan, Rostron, Hewedi, & Haladjian, 2017; Rüb et al., 2016; Štěpán‐Buksakowska et al., 2014). Our current study provided new evidence that macroscopic atrophy in these regions may be a prelude of more widespread brain deterioration. Furthermore, the sliding window results indicated that this a driving effect may be very different in normal aging or less severe AD than in moderately and very severe AD. This may be because of the compensatory effects that appear during very early AD. In the MCI and AD populations, the driving effect of these three core regions is very stable and strong, indicating dramatic volume alterations through the AD pathological pathways from these regions to other temporal regions and then to occipital, parietal and frontal regions. It is also worth noting that sensorimotor areas, such as those where component B is located, are driven in the less severe stages but not in the more severe stages, and component B also did not show significant group difference. This may suggest a “very early impaired but spared until very late stage” pattern but needs further investigation by pathological studies.
It is also worth noting that the SCN was shown to be disrupted in MCI and AD. However, the CaSCN was enhanced during AD progression. This can be expected since neurodegeneration occurs in some regions and then spreads to others; therefore, the covariance between regions decreases. However, the CaSCN can reveal successive atrophy among regions in MCI and AD, which is much weaker and has no uniform pathway in the normal aging population. However, the decreased SCN and enhanced CaSCN did not perfectly overlap, and there may also be some other mechanisms underlying the disrupted SCN seen at different levels of pathological aging, which needs future investigation.
There were some potential factors may affect our results. First is the window length during CaSCN analysis. The window length is set to be 50 in the current study. We also applied window length as 30 or 70 again. The results in sliding windows with length 30 or 70 showed high similarity to those with length 50 if they contained overlapped subjects (see Figure S1). The other factors may be the sites effects. Given ADNI is a multi‐site dataset, the difference of sites may affect our results. However, the 446 subjects including in the current study was from 58 sites and some sites have very few subjects, which may make a regression or other mode control site effect ill‐posed. Here we utilized a recent provided “Combat” method, to validation our results(Fortin et al., 2018; Yu et al., 2018). After excluding 38 subjects which were from those sites only contributed 3 or less subjects, our results were well reproduced using remain data with Combat correction (see Figures S2–S4). Therefore, our results were robust to both site and window length effects.
The current study still has some limitations. The most important one is that in the GCA analysis, the “single steps” are from one subject to another, which may not be identical as they are in fMRI (in which the steps are always the same; one step is one TR, often 2S). Additionally, the CSF biomarker Z score was created as a compromise measure and obtained by combining Aβ, Tau and P‐tau because there is no gold standard to quantitively define which single subject is more severe than another (Jack Jr. et al., 2018). Similarly, the GCA coefficients matrices may not fit all the assumption of K‐means. Therefore, the GCA analysis including the CaSCN, sliding window CaSCN and K‐means analysis in the current study, which is based on a “least to most severe” sequence, is more qualitative than quantitative. The second main limitation is that the across‐subject correlation‐based analyses, including SBM, SCN and CaSCN, were all based on a group of subjects, making it difficult to provide information regarding single‐subject diagnoses or predictions (Alexander‐Bloch et al., 2013), and hard to exclude the individual difference of specific covariance network revealed by some recent study(Cui et al., 2020). The diagnosis of AD and MCI was all provided by ADNI which is based on clinically diagnosis rather than biomarkers, and the data of CSF is not available for all subject and hardly to define if they all meet the new criterial like “ATN” (Jack Jr. et al., 2018) and also lack of information to distinguish subtypes like if they were the late‐onset (LOAD), early‐onset (EOAD) or autosomal dominant AD (ADAD) (Jagust, 2018). Therefore, our results were based on a possible mixture of different neurobiological mechanisms. Additionally, to ensure the homogeneity of the imaging data, we used strict standard for the sequence and resolution, and only small portion of ADNI data can be included. However, this makes there is an insufficient number of subjects especially for those with CSF records, and prevented us from directly comparing the SCN and CaSCN. Finally, the current study used a cross‐sectional data cohort, so these causal effects should be validated in longitudinal data in the future.
In conclusion, the current study provided a comprehensive view of brain atrophy in the AD continuum and the relationships among the brain atrophy in different regions. The hippocampus, default mode network and thalamus were identified as core regions in which brain atrophy originated, and their atrophy may predict subsequent widespread atrophy in other brain regions. Our results provide casual or sequential patterns of brain atrophy across the whole brain, which may provide new insight into understanding the progression of AD.
CONFLICT OF INTEREST
The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript, and all the authors declared no conflict of interest.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (82071904, 81720108022, 81971596); the Fundamental Research Funds for the Central Universities, Nanjing University (2020‐021414380462); the Key Project of Jiangsu Commission of Health (K2019025); The Social Development Project of Science and Technology Project in Jiangsu Province (BE2017707); the Key Medical Talents of Jiangsu Province, the “13th Five‐Year” Health Promotion Project of Jiangsu Province (ZDRCA2016064); Jiangsu Provincial Key Medical Discipline (Laboratory) (ZDXKA2016020); and the Project of the Sixth Peak of Talented People (WSN‐138).
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). The ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; the Alzheimer's Association; the Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Qing, Z. , Chen, F. , Lu, J. , Lv, P. , Li, W. , Liang, X. , Wang, M. , Wang, Z. , Zhang, X. , Zhang, B. , For the Alzheimer's Disease Neuroimaging Initiative (2021). Causal structural covariance network revealing atrophy progression in Alzheimer's disease continuum. Human Brain Mapping, 42(12), 3950–3962. 10.1002/hbm.25531
Funding information Fundamental Research Funds for the Central Universities, Grant/Award Number: 2020‐021414380462; Key Project of Jiangsu Commission of Health, Grant/Award Number: K2019025; Jiangsu Provincial Key Medical Discipline (Laboratory), Grant/Award Number: ZDXKA2016020; National Natural Science Foundation of China, Grant/Award Numbers: 81720108022, 81971596, 82071904; the Key Medical Talents of Jiangsu Province, the “13th Five‐Year” Health Promotion Project of Jiangsu Province, Grant/Award Number: ZDRCA2016064; the Project of the Sixth Peak of Talented People, Grant/Award Number: WSN‐138; The Social Development Project of Science and Technology Project in Jiangsu Province, Grant/Award Number: BE2017707
DATA AVAILABILITY STATEMENT
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public‐private partnership led by Principal Investigator Michael W. Weiner, MD. The primary goal of the ADNI is to test whether magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer's disease (AD). For more information, please see www.adni-info.org.
REFERENCES
- Aggleton, J. P. , Pralus, A. , Nelson, A. J. , & Hornberger, M. (2016). Thalamic pathology and memory loss in early Alzheimer's disease: Moving the focus from the medial temporal lobe to Papez circuit. Brain, 139(Pt 7), 1877–1890. 10.1093/brain/aww083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert, M. S. , DeKosky, S. T. , Dickson, D. , Dubois, B. , Feldman, H. H. , Fox, N. C. , … Phelps, C. H. (2011). The diagnosis of mild cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7(3), 270–279. 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander‐Bloch, A. , Giedd, J. N. , & Bullmore, E. (2013). Imaging structural co‐variance between human brain regions. Nature Reviews. Neuroscience, 14(5), 322–336. 10.1038/nrn3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, E. A. , Damaraju, E. , Plis, S. M. , Erhardt, E. B. , Eichele, T. , & Calhoun, V. D. (2014). Tracking whole‐brain connectivity dynamics in the resting state. Cerebral Cortex, 24(3), 663–676. 10.1093/cercor/bhs352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova, L. G. , Steiner, C. A. , Akopyan, G. G. , Dutton, R. A. , Hayashi, K. M. , Toga, A. W. , … Thompson, P. M. (2007). Three‐dimensional gray matter atrophy mapping in mild cognitive impairment and mild Alzheimer disease. Archives of Neurology, 64(10), 1489. 10.1001/archneur.64.10.1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner, J. (2007). A fast diffeomorphic image registration algorithm. NeuroImage, 38(1), 95–113. 10.1016/j.neuroimage.2007.07.007 [DOI] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1995). Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiology of Aging, 16(3), 271–278 discussion 278–284. 10.1016/0197-4580(95)00021-6 [DOI] [PubMed] [Google Scholar]
- Chang, Y. T. , Huang, C. W. , Chang, W. N. , Lee, J. J. , & Chang, C. C. (2018). Altered functional network affects amyloid and structural covariance in Alzheimer's disease. BioMed Research International, 2018, 8565620. 10.1155/2018/8565620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, J. S. X. , Liu, S. , Loke, Y. M. , Hilal, S. , Ikram, M. K. , Xu, X. , … Zhou, J. (2017). Influence of cerebrovascular disease on brain networks in prodromal and clinical Alzheimer’s disease. Brain, 140(11), 3012–3022. 10.1093/brain/awx224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, Z. , Li, H. , Xia, C. H. , Larsen, B. , Adebimpe, A. , Baum, G. L. , … Satterthwaite, T. D. (2020). Individual variation in functional topography of association networks in youth. Neuron, 106(2), 340–353. e8. 10.1016/j.neuron.2020.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duara, R. , Loewenstein, D. A. , Potter, E. , Appel, J. , Greig, M. T. , Urs, R. , … Potter, H. (2008). Medial temporal lobe atrophy on MRI scans and the diagnosis of Alzheimer disease. Neurology, 71(24), 1986–1992. 10.1212/01.wnl.0000336925.79704.9f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, D. , Cavallin, L. , Granberg, T. , Lindberg, O. , Aguilar, C. , Mecocci, P. , … Westman, E. (2016). Quantitative validation of a visual rating scale for frontal atrophy: associations with clinical status, APOE e4, CSF biomarkers and cognition. European Radiology, 26(8), 2597–2610. 10.1007/s00330-015-4101-9 [DOI] [PubMed] [Google Scholar]
- Fiford, C. M. , Ridgway, G. R. , Cash, D. M. , Modat, M. , Nicholas, J. , Manning, E. N. , … Barnes, J. (2018). Patterns of progressive atrophy vary with age in Alzheimer's disease patients. Neurobiology of Aging, 63, 22–32. 10.1016/j.neurobiolaging.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin, J. , Cullen, N. , Sheline, Y. I. , Taylor, W. D. , Aselcioglu, I. , Cook, P. A. , … Shinohara, R. T. (2018). Harmonization of cortical thickness measurements across scanners and sites. NeuroImage, 167, 104–120. 10.1016/j.neuroimage.2017.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmeier, N. , Göttler, J. , Grimmer, T. , Drzezga, A. , Áraque‐Caballero, M. A. , Simon‐Vermot, L. , … Ewers, M. (2017). Resting‐state connectivity of the left frontal cortex to the default mode and dorsal attention network supports reserve in mild cognitive impairment. Frontiers in Aging Neuroscience, 9. 10.3389/fnagi.2017.00264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, G. , He, Y. , Chen, Z. J. , & Evans, A. C. (2012). Convergence and divergence of thickness correlations with diffusion connections across the human cerebral cortex. NeuroImage, 59(2), 1239–1248. 10.1016/j.neuroimage.2011.08.017 [DOI] [PubMed] [Google Scholar]
- Granadillo, E. , Paholpak, P. , Mendez, M. F. , & Teng, E. (2017). Visual ratings of medial temporal lobe atrophy correlate with CSF tau indices in clinical variants of early‐onset Alzheimer disease. Dementia and Geriatric Cognitive Disorders, 44(1–2), 45–54. 10.1159/000477718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe, M. J. , Teipel, S. J. , & Alzheimers Disease Neuroimaging Initiative . (2016). Spatial patterns of atrophy, hypometabolism, and amyloid deposition in Alzheimer's disease correspond to dissociable functional brain networks. Human Brain Mapping, 37(1), 35–53. 10.1002/hbm.23018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday, G. (2017). Pathology and hippocampal atrophy in Alzheimer's disease. Lancet Neurology, 16(11), 862–864. 10.1016/S1474-4422(17)30343-5 [DOI] [PubMed] [Google Scholar]
- Hamilton, J. P. , Chen, G. , Thomason, M. E. , Schwartz, M. E. , & Gotlib, I. H. (2011). Investigating neural primacy in Major Depressive Disorder: multivariate Granger causality analysis of resting‐state fMRI time‐series data. Molecular Psychiatry, 16(7), 763–772. 10.1038/mp.2010.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harenski, C. L. , Harenski, K. A. , Calhoun, V. D. , & Kiehl, K. A. (2020). Source‐based morphometry reveals gray matter differences related to suicidal behavior in criminal offenders. Brain Imaging and Behavior, 14(1), 1–9. 10.1007/s11682-018-9957-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y. , Chen, Z. , & Evans, A. (2008). Structural insights into aberrant topological patterns of large‐scale cortical networks in Alzheimer's disease. The Journal of Neuroscience, 28(18), 4756–4766. 10.1523/JNEUROSCI.0141-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins, W. D. , Latzman, R. D. , Mareno, M. C. , Schapiro, S. J. , Gomez‐Robles, A. , & Sherwood, C. C. (2019). Heritability of gray matter structural covariation and tool use skills in chimpanzees (Pan troglodytes): A source‐based morphometry and quantitative genetic analysis. Cerebral Cortex, 29(9), 3702–3711. 10.1093/cercor/bhy250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, C. R. , Bennett, D. A. , Blennow, K. , Carrillo, M. C. , Dunn, B. , Haeberlein, S. B. , … Silverberg, N. (2018). NIA‐AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimer's & Dementia, 14(4), 535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust, W. (2018). Imaging the evolution and pathophysiology of Alzheimer disease. Nature Reviews. Neuroscience, 19(11), 687–700. 10.1038/s41583-018-0067-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. J. , Hou, X. , Liu, H. , Yue, C. , He, Y. , & Zuo, X. (2015). Toward systems neuroscience in mild cognitive impairment and Alzheimer's disease: A meta‐analysis of 75 fMRI studies. Human Brain Mapping, 36(3), 1217–1232. 10.1002/hbm.22689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Luo, X. , Zeng, Q. , Huang, P. , Shen, Z. , Xu, X. , … Zhang, M. (2019). Gray matter structural covariance networks changes along the Alzheimer's disease continuum. NeuroImage: Clinical, 23, 101828. 10.1016/j.nicl.2019.101828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelkoop, H. A. M. , van der Flier, W. M. , Burton, E. J. , Lloyd, A. J. , Paling, S. , Barber, R. , … O'Brien, J. T. (2001). Dementia with Lewy bodies and AD are not associated with occipital lobe atrophy on MRI. Neurology, 57(11), 2117–2120. 10.1212/wnl.57.11.2117 [DOI] [PubMed] [Google Scholar]
- Montembeault, M. , Rouleau, I. , Provost, J. S. , Brambati, S. M. , & Alzheimer's Disease Neuroimaging Initiative . (2016). Altered gray matter structural covariance networks in early stages of Alzheimer's disease. Cerebral Cortex, 26(6), 2650–2662. 10.1093/cercor/bhv105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa, A. A. , McMullan, R. D. , Rostron, B. , Hewedi, D. H. , & Haladjian, H. H. (2017). The thalamus as a relay station and gatekeeper: Relevance to brain disorders. Reviews in the Neurosciences, 28(2), 203–218. 10.1515/revneuro-2016-0067 [DOI] [PubMed] [Google Scholar]
- Ossenkoppele, R. , Cohn‐Sheehy, B. I. , La Joie, R. , Vogel, J. W. , Möller, C. , Lehmann, M. , … Rabinovici, G. D. (2015). Atrophy patterns in early clinical stages across distinct phenotypes of Alzheimer's disease. Human Brain Mapping, 36(11), 4421–4437. 10.1002/hbm.22927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, R. C. (2004). Mild cognitive impairment as a diagnostic entity. Journal of Internal Medicine, 256(3), 183–194. 10.1111/j.1365-2796.2004.01388.x [DOI] [PubMed] [Google Scholar]
- Pini, L. , Pievani, M. , Bocchetta, M. , Altomare, D. , Bosco, P. , Cavedo, E. , … Frisoni, G. B. (2016). Brain atrophy in Alzheimer’s Disease and aging. Ageing Research Reviews, 30, 25–48. 10.1016/j.arr.2016.01.002 [DOI] [PubMed] [Google Scholar]
- Prestia, A. , Drago, V. , Rasser, P. E. , Bonetti, M. , Thompson, P. M. , & Frisoni, G. B. (2010). Cortical changes in incipient Alzheimer's disease. Journal of Alzheimer's Disease, 22(4), 1339–1349. 10.3233/JAD-2010-101191 [DOI] [PubMed] [Google Scholar]
- Rüb, U. , Stratmann, K. , Heinsen, H. , Del Turco, D. , Ghebremedhin, E. , Seidel, K. , … Korf, H. (2015). Hierarchical distribution of the tau cytoskeletal pathology in the thalamus of Alzheimer’s disease patients. Journal of Alzheimer's Disease, 49(4), 905–915. 10.3233/jad-150639 [DOI] [PubMed] [Google Scholar]
- Seeley, W. W. , Crawford, R. K. , Zhou, J. , Miller, B. L. , & Greicius, M. D. (2009). Neurodegenerative diseases target large‐scale human brain networks. Neuron, 62(1), 42–52. 10.1016/j.neuron.2009.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley, W. W. , Menon, V. , Schatzberg, A. F. , Keller, J. , Glover, G. H. , Kenna, H. , … Greicius, M. D. (2007). Dissociable intrinsic connectivity networks for salience processing and executive control. Journal of Neuroscience, 27(9), 2349–2356. 10.1523/jneurosci.5587-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth, A. K. , Barrett, A. B. , & Barnett, L. (2015). Granger causality analysis in neuroscience and neuroimaging. The Journal of Neuroscience, 35(8), 3293–3297. 10.1523/JNEUROSCI.4399-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X. W. , Dong, Z. Y. , Long, X. Y. , Li, S. F. , Zuo, X. N. , Zhu, C. Z. , … Zang, Y. F. (2011). REST: A Toolkit for Resting‐State Functional Magnetic Resonance Imaging Data Processing. PLoS ONE, 6(9), e25031. 10.1371/journal.pone.0025031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling, R. A. , Aisen, P. S. , Beckett, L. A. , Bennett, D. A. , Craft, S. , Fagan, A. M. , … Phelps, C. H. (2011). Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7(3), 280–292. 10.1016/j.jalz.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Štěpán‐Buksakowska, I. , Szabó, N. , Hořínek, D. , Tóth, E. , Hort, J. , Warner, J. , … Kincses, Z. T. (2014). Cortical and subcortical atrophy in Alzheimer disease. Alzheimer Disease & Associated Disorders, 28(1), 65–72. 10.1097/wad.0b013e318299d3d6 [DOI] [PubMed] [Google Scholar]
- Stephan, K. E. , & Roebroeck, A. (2012). A short history of causal modeling of fMRI data. NeuroImage, 62(2), 856–863. 10.1016/j.neuroimage.2012.01.034 [DOI] [PubMed] [Google Scholar]
- Ting, W. K. , Fischer, C. E. , Millikin, C. P. , Ismail, Z. , Chow, T. W. , & Schweizer, T. A. (2015). Grey matter atrophy in mild cognitive impairment/early Alzheimer disease associated with delusions: A voxel‐based morphometry study. Current Alzheimer Research, 12(2), 165–172. 10.2174/1567205012666150204130456 [DOI] [PubMed] [Google Scholar]
- Voevodskaya, O. , Pereira, J. B. , Volpe, G. , Lindberg, O. , Stomrud, E. , van Westen, D. , … Hansson, O. (2018). Altered structural network organization in cognitively normal individuals with amyloid pathology. Neurobiology of Aging, 64, 15–24. 10.1016/j.neurobiolaging.2017.11.014 [DOI] [PubMed] [Google Scholar]
- Weiner, M. W. , Veitch, D. P. , Aisen, P. S. , Beckett, L. A. , Cairns, N. J. , Green, R. C. , … Trojanowski, J. Q. (2017). Recent publications from the Alzheimer's Disease Neuroimaging Initiative: Reviewing progress toward improved AD clinical trials. Alzheimer's & Dementia, 13(4), e1–e85. 10.1016/j.jalz.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. , Groth, K. M. , Pearlson, G. , Schretlen, D. J. , & Calhoun, V. D. (2009). Source‐based morphometry: The use of independent component analysis to identify gray matter differences with application to schizophrenia. Human Brain Mapping, 30(3), 711–724. 10.1002/hbm.20540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, C. , & He, Y. (2011). Driving and driven architectures of directed small‐world human brain functional networks. PLoS One, 6(8), e23460. 10.1371/journal.pone.0023460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, M. , Linn, K. A. , Cook, P. A. , Phillips, M. L. , McInnis, M. , Fava, M. , … Sheline, Y. I. (2018). Statistical harmonization corrects site effects in functional connectivity measurements from multi‐site fMRI data. Human Brain Mapping, 39(11), 4213–4227. 10.1002/hbm.24241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Liao, W. , Xu, Q. , Wei, W. , Zhou, H. J. , Sun, K. , … Lu, G. (2017). Hippocampus‐associated causal network of structural covariance measuring structural damage progression in temporal lobe epilepsy. Human Brain Mapping, 38(2), 753–766. 10.1002/hbm.23415 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information
Data Availability Statement
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public‐private partnership led by Principal Investigator Michael W. Weiner, MD. The primary goal of the ADNI is to test whether magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer's disease (AD). For more information, please see www.adni-info.org.