

Abstract
Hydrazone is a bioactive pharmacophore that can be used to design antitumor agents. We synthesised a series of hydrazones (compounds 4–24) incorporating a 4-methylsulfonylbenzene scaffold and analysed their potential antitumor activity. Compounds 6, 9, 16, and 20 had the most antitumor activity with a positive cytotoxic effect (PCE) of 52/59, 27/59, 59/59, and 59/59, respectively, while compounds 5, 10, 14, 15, 18, and 19 had a moderate antitumor activity with a PCE of 11/59–14/59. Compound 20 was the most active and had a mean 50% cell growth inhibition (GI50) of 0.26 µM. Compounds 9 and 20 showed the highest inhibitory activity against COX-2, with a half-maximal inhibitory concentration (IC50) of 2.97 and 6.94 μM, respectively. Compounds 16 and 20 significantly inhibited EGFR (IC50 = 0.2 and 0.19 μM, respectively) and HER2 (IC50 = 0.13 and 0.07 μM, respectively). Molecular docking studies of derivatives 9, 16, and 20 into the binding sites of COX-2, EGFR, and HER2 were carried out to explore the interaction mode and the structural requirements for antitumor activity.
Keywords: Hydrazones synthesis, antitumor activity, cell cycle analysis, enzymatic assay, COX-2 inhibition, EGFR inhibition, HER2 inhibition, molecular docking
Graphical Abstract
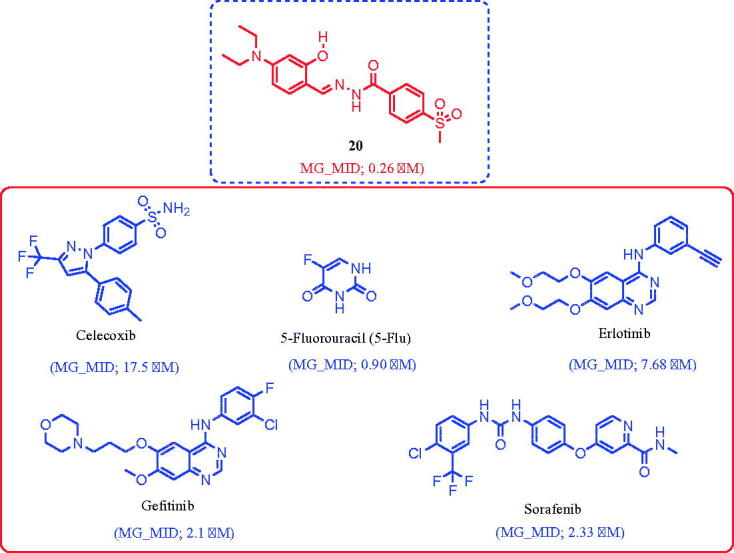
Compound 20 (MG_MID = 0.26 µM) is nearly 65-fold more potent than celecoxib (MG_MID = 17.5 µM), 3-fold more potent than 5-Fu (MG_MID = 0.90 µM), 30-fold more potent than erlotinib (MG_MID = 7.68 µM), and 9-fold more potent than gefitinib (MG_MID = 2.1 µM) and sorafenib (MG_MID = 2.33 µM).
1. Introduction
Cancer is the most dangerous disease and a leading cause of death worldwide1. Also, cancer cells have evolved to become resistant to already used therapeutic agents2–4. Therefore, novel and effective antitumor agents are in high demand, and their development remains a challenge for pharmaceutical chemists5–21. The use of more than one drug in combination with cancer therapy has several side effects22–24. These effects can be diminished by using a single compound with multiple molecular mechanisms, which is currently the preferred therapeutic strategy25–30. EGFR and the structurally related human epidermal growth factor receptor 2 (HER2) are members of the tyrosine kinase receptor family31–35. EGFR and HER2 overexpression has been found in various cancers, such as prostate, breast, colon, and ovarian cancers, and their inhibition results in apoptotic-inducing activity in lung and breast cancers36–39. Therefore, EGFR and HER2 are important targets for antitumor agent design and development27,32,33,39–42. Several tyrosine kinase inhibitors, such as imatinib (I), gefitinib (II), lapatinib (III), sorafenib (IV), afatinib (V), and sunitinib (VI), are used to treat various cancers (Figure 1)43–50. In contrast, COX-2 isozyme is overexpressed in several cancers, such as colon, hepatocellular, gastric, breast, lung, prostate, and ovarian cancers, indicating that COX-2 is a target for cancer treatment51,52. These findings show that celecoxib (VII) and other selective COX-2 inhibitors can be used for cancer treatment and prevention (Figure 2)53,54. The anticancer mechanism through COX-2 inhibition might proceed via proliferation inhibition or apoptotic induction55.
Figure 1.

The reported anticancer agents with EGFR and HER2 inhibition activities.
Figure 2.

The reported anticancer agents bearing hydrazone, sulphonyl, and benzamide fragments.
Hugo Schiff first prepared Schiff’s bases or azomethines with an imine core skeleton (RHC = N-R) in 1864 through the reaction of carbonyl compounds and a primary amine56. The chemical and biological behaviours of Schiff’s bases are due to the lone pair of electrons on the imine core, which are responsible for their chelating properties57,58. Especially, hydrazone is an important, highly bioactive pharmacophore that can be used to design various antitumor agents, such as arylhydrazone (VIII), quinazolinylhydrazone (IX), and PAC-1 (X), and PAC-1 selectively induces apoptosis in cancer cells (Figure 2)21,59–63. Other compounds with hydrazones show the tumour-associated carbonic anhydrase IX and COX-2 inhibition activities64–66. Interestingly, compounds with methylsulfonylbenzene, such as vismodegib (XI), and hydrazine derivative–linked sulphonyl fragments, such as arylsulfonylhydrazone (XII), are potential antitumor agents against skin, hepatocellular, lung, and colon cancers and melanoma (Figure 2)66–69. The mechanism underlying the anticancer activity of a few hydrazones has been investigated through selective COX-2 inhibition, EGFR, and HER2 inhibition, in addition to apoptosis induction39,60,66,69.
In this study, we synthesised a series of hydrazones (compounds 4–24) incorporating a 4-methylsulfonylbenzene scaffold (Figure 3). The chemical structure of the designed compounds was based on some fragments of compounds shown in Figures 1 and 2. A 4-methylsulfonylbenzene core was linked with a hydrazone moiety and connected with various arylidenes with or without hydroxyl and N,N-diethylamine fragments (Figure 3). We also evaluated the in vitro antitumor activities of the synthesised compounds using 59 human cancer cell lines and investigated the structure–activity relationship (SAR) of the compounds with various substituents, depending on their antitumor activities. Next, we performed a cell cycle analysis and apoptotic induction assay of the most active compounds using the HL-60 cell line. We performed an enzymatic assay of the EGFR and HER2 inhibitory activity of the most promising compounds and evaluated the COX-2 inhibitory activity of the most active derivatives. Finally, we used molecular docking to predict the interaction mode of the biologically active compounds in the binding pockets of COX-2 isozyme, and EGFR, and HER2 tyrosine kinases.
Figure 3.
The designed target arylhydrazones 4–24 based on the chemical structure of compounds I-XII.
2. Results and discussion
2.1. Chemistry
4-(Methylsulfonyl)-N'-(substituted-benzylidene)benzohydrazides 4–24 were obtained with a yield of 85–95% by stirring an appropriate aldehyde and 4-(methylsulfonyl)benzohydrazide (3) in methanol containing a catalytic amount of acetic acid at room temperature (Scheme 1). Multiple spectral analyses were performed to confirm the structures of target compounds 4–24. The amide fragment of the benzylidene benzohydrazide moiety (PhCONH=CHPh) was verified by 1H NMR spectra with peaks at 12.35–11.81 ppm for the amidic proton and by 13 C NMR spectra with characteristic peaks at 163.0–161.3 ppm for the carbonyl group. In addition, the imine fragment of the benzylidene benzohydrazide moiety (PhCONH = CHPh) was verified by 1H NMR spectra with peaks at 9.12–8.36 ppm and by 13 C NMR spectra with characteristic peaks at 151.4–143.9 ppm. The methyl group of the 4-methylsulfonyl moiety (SO2CH3) was verified by 1H NMR and 13 C NMR spectra with peaks at 3.32–3.29 and 43.8–43.7 ppm, respectively.
Scheme 1.

Synthesis of the designed hydrazones 4–24.
2.2. Antitumor activity and SAR study
2.2.1. Growth inhibition percentage (GI) at a single dose concentration of 10 µM
Compounds 4–24 were selected by the National Cancer Institute (Bethesda, MD, USA) for evaluation of their in vitro antitumor activity against a full panel of 59 cancer cell lines (Tables 1 and 2) taken from nine human tissue (i.e. blood, lung, colon, brain, skin, ovary, kidney, prostate, and breast)17,40,70. We performed the initial antitumor evaluation at a single dose concentration of 10 µM and calculated the GI in the 59 cell lines. The GI results were compared with those of imatinib, gefitinib, and 5-fluorouracil (5-FU) as reference drugs. Tables 1 and 2 summarise the GI activity of compounds 4–24, which showed significant antitumor activity against the 59 cell lines at a 10 µM concentration with PCE (ratio between the number of cell lines with percentage growth inhibition from 10 to 100 and the total number of cell lines) of 5/59–59/59 and a percentage mean growth (MG) of 100.00%–20.59%. Compounds 5, 6, 9, 10, 14–16, and 18–20 showed the highest PCE of 11/59–59/59 (MG = 99.74%–20.95%), while compounds 4, 7, 8, 11–13, 17, and 21–24 showed the lowest PCE of ≤10/59 (MG = 100.00%–95.96%) compared to imatinib (PCE = 20/55 and MG = 92.62%). Interestingly, compounds 6, 9, 16, and 20 were the most active antitumor agents (PCE = 52/59, 27/59, 59/59, and 59/59, respectively, and MG = 71.75%, 88.01%, 39.27%, and 20.59%, respectively), while compounds 5, 14, 15, 18, and 19 showed moderate activity (PCE = 13/59, 12/59, 14/59, 14/59, and 13/59, respectively, and MG = 98.37%, 95.16%, 95.92%, 94.23%, and 96.65%, respectively).
Table 1.
Antitumor activity of the designed hydrazones at 10 µM concentration.
| Compd No | 60 cell lines assay in one dose 10.0 µM concentration (GI%)a |
|
|---|---|---|
| PCEb | Most sensitive cell lines | |
| 4 | 10/59 | Leukaemia (HL-60 (TB): 60.3, K-562: 11.3, MOLT-4: 25.5), NSC Lung Cancer (A549/ATCC: 28.5, HOP-62: 12.0, NCI-H522: 42.1), CNS Cancer (U251: 20.4), Melanoma (SK-MEL-2: 21.5, UACC-257: 44.2), Ovarian Cancer (OVCAR-8: 15.1). |
| 5 | 13/59 | Leukaemia (CCRF-CEM: 11.1, HL-60(TB): 20.5, K-562: 18.8), NSC Lung Cancer (A549/ATCC: 10.6, HOP-62: 16.5, NCI-H226: 11.9, NCI-H522: 15.1), Colon Cancer (HT29: 13.3), Ovarian Cancer (SK-OV-3: 16.9), Renal Cancer (A498: 11.2, UO-31: 16.3), Prostate Cancer (PC-3: 17.9), Breast Cancer (T-47D: 13.7). |
| 6 | 52/59 | Leukaemia (CCRF-CEM: 22.4, HL-60(TB): 62.5, K-562: 72.2, MOLT-4: 53.2, PRMI-8226: 12.3, SR: 67.5), NSC Lung Cancer (A549/ATCC: 58.7, EKVX: 12.6,HOP-62: 25.1, NCI-H226: 16.3, NCI-H23: 10.4, NCI-H460: 33.2, NCI-H522: 95.5), Colon Cancer (COLO 205: 14.3, HCC-2998: 15.5, HCT-116: 38.2, HCT-15: 50.2, HT29: 50.5, KM12: 49.2, SW-620: 63.8), CNS Cancer (SF-268: 14.3, SF-295: 24.0, SF-539: 10.7, SNB-19: 20.2, SNB-75: 10.3, U251: 23.7), Melanoma (LOX IMVI: 27.6, MALME-3M: 30.3 , M14: 33.2, MDA-MB-435: 82.9, SK-MEL-2: 52.3, SK-MEL-28: 10.6, SK-MEL-5: 32.8, UACC-257: 58.9, UACC-62: 38.4), Ovarian Cancer (IGROV1: 36.8, OVCAR-3: 32.2, OVCAR-8: 16.2, NCI/ADR-RES: 26.5, SK-OV-3: 21.9), Renal Cancer (A498: 30.9, CAKI-1: 32.8, SN12C: 12.6, TK-10: 14.9, UO-31: 21.5), Prostate Cancer (PC-3: 21.2, DU-145: 11.2), Breast Cancer (MCF7: 55.1, HS 578 T: 14.9, BT-549: 12.4, T-47D: 14.7, MDA-MB-468: 25.8). |
| 7 | 7/59 | Leukaemia (HL-60 (TB): 52.1, K-562: 14.0, MOLT-4: 29.4), NSC Lung Cancer (NCI-H522: 15.7), Melanoma (SK-MEL-2: 12.8), Renal Cancer (A498: 12.3), Prostate Cancer (PC-3: 22.8). |
| 8 | 9/59 | Leukaemia (CCRF-CEM: 11.8, HL-60(TB): 23.1, K-562: 13.6, MOLT-4: 23.9, SR: 20.2), NSC Lung Cancer ( A549/ATCC: 11.1, NCI-H522: 15.9), Colon Cancer (HCT-116: 10.8), Melanoma (UACC-257: 18.4). |
| 9 | 27/59 | Leukaemia (CCRF-CEM: 20.8, HL-60(TB): 24.3, K-562: 44.1, MOLT-4: 22.6, SR: 43.8), NSC Lung Cancer (A549/ATCC: 12.7, NCI-H522: 53.9), Colon Cancer (HCT-116: 13.9, HCT-15: 16.3, HT29: 17.7, KM12: 25.1, SW-620: 15.2), CNS Cancer (U251: 13.7), Melanoma (MALME-3M: 15.2 , MDA-MB-435: 47.5, SK-MEL-2: 19.8, SK-MEL-5: 13.5, UACC-257: 28.4, UACC-62: 19.1), Ovarian Cancer (IGROV1: 12.3, NCI/ADR-RES: 14.6), Renal Cancer (A498: 17.9, CAKI-1: 25.0, UO-31: 12.3), Prostate Cancer (PC-3: 26.9), Breast Cancer (MCF7: 21.4, MDA-MB-468: 21.5) |
| 10 | 11/59 | Leukaemia (CCRF-CEM: 10.6, HL-60(TB): 42.9, MOLT-4: 21.1), NSC Lung Cancer (A549/ATCC: 29.8 , NCI-H226: 13.4, NCI-H522: 20.8), Colon Cancer (HT29: 10.8), Melanoma (SK-MEL-2: 11.8, UACC-257: 29.0), Ovarian Cancer (OVCAR-8: 11.3), Prostate Cancer (PC-3: 13.8). |
| 11 | 5/59 | Leukaemia (HL-60(TB): 50.5, K-562: 26.2, MOLT-4: 32.5), NSC Lung Cancer (NCI-H522: 11.7), Prostate Cancer (PC-3: 10.9). |
| 12 | 9/59 | Leukaemia (HL-60(TB): 38.0, K-562: 17.2, MOLT-4: 29.8), NSC Lung Cancer (A549/ATCC: 28.4, HOP-62: 11.4, NCI-H522: 40.4), Colon Cancer (HT29: 20.5), Melanoma (UACC-257: 39.2), Renal Cancer (UO-31: 11.2). |
| 13 | 10/59 | Leukaemia (HL-60(TB): 47.7, K-562: 32.5, MOLT-4: 32.3), NSC Lung Cancer (A549/ATCC: 21.6, HOP-62: 12.7), CNS Cancer (SNB-75: 15.1), Melanoma ( UACC-257: 35.5), Ovarian Cancer (SK-OV-3: 11.7), Renal Cancer (UO-31: 15.7), Breast Cancer (T-47D: 12.5) |
| 14 | 12/59 | Leukaemia (HL-60(TB): 36.0, K-562: 25.7, MOLT-4: 26.4, SR: 49.2), NSC Lung Cancer (A549/ATCC: 29.3, HOP-62: 15.8, NCI-H522: 26.0), Colon Cancer (HT29: 18.6), Melanoma ( UACC-257: 42.4), Ovarian Cancer (OVCAR-8: 14.5), Renal Cancer ( CAKI-1: 11.7), Breast Cancer (T-47D: 11.8). |
| 15 | 14/59 | Leukaemia (CCRF-CEM: 16.1, HL-60(TB): 36.5, K-562: 23.4, MOLT-4: 11.6, SR: 15.6), NSC Lung Cancer (A549/ATCC: 11.9, HOP-62: 18.9, NCI-H226: 10.3), Colon Cancer (HCT-116: 10.7), Melanoma (UACC-257: 10.1), Ovarian Cancer (SK-OV-3: 17.1), Renal Cancer (UO-31: 14.4), Prostate Cancer (PC-3: 19.1), Breast Cancer (T-47D: 18.2) |
| 16 | 59/59 | Leukaemia (CCRF-CEM: 84.7, HL-60(TB): 93.7, K-562: 59.5, MOLT-4: 90.8, PRMI-8226: 51.3, SR: 79.6), NSC Lung Cancer (A549/ATCC: 61.2, EKVX: 74.2,HOP-62: 71.8, NCI-H226: 44.8, NCI-H23: 45.2, NCI-H322M: 61.6, NCI-H460: 85.9, NCI-H522: 84.9), Colon Cancer (COLO 205: 68.8, HCC-2998: 52.7, HCT-116: 70.9, HCT-15: 82.0, HT29: 56.4, KM12: 65.9, SW-620: 56.7), CNS Cancer (SF-268: 61.6, SF-295: 62.0, SF-539: 44.7, SNB-19: 54.6, SNB-75: 22.3, U251: 72.2), Melanoma (LOX IMVI: 78.6, MALME-3M: 36.5 , M14: 67.5, MDA-MB-435: 40.3, SK-MEL-2: 60.0, SK-MEL-28: 44.1, SK-MEL-5: 67.8, UACC-257: 77.4, UACC-62: 83.6), Ovarian Cancer (IGROV1: 59.0, OVCAR-3: 77.2, OVCAR-4: 56.3, OVCAR-5: 35.3, OVCAR-8: 72.1, NCI/ADR-RES: 70.1, SK-OV-3: 60.1), Renal Cancer (786-0: 67.0, A498: 24.8, ACHN: 71.7, CAKI-1: 65.1, RXF 393: 47.0, SN12C: 41.4, TK-10: 54.2, UO-31: 82.0), Prostate Cancer (PC-3: 45.7, DU-145: 59.6), Breast Cancer (MCF7: 77.5, MDA-MB-231/ATCC: 52.6, HS 578 T: 18.5, BT-549: 47.0, T-47D: 47.6, MDA-MB-468: 35.3). |
| 17 | 9/59 | NSC Lung Cancer (A549/ATCC: 13.7, HOP-62: 10.2, NCI-H226: 13.6, NCI-H522: 25.0), Colon Cancer (HT29: 11.3), CNS Cancer (SNB-75: 10.9), Melanoma (UACC-257: 29.4), Renal Cancer (UO-31: 12.7), Breast Cancer (MDA-MB-468: 18.1). |
| 18 | 14/59 | Leukaemia (HL-60(TB): 30.6, K-562: 17.7, MOLT-4: 31.7, SR: 24.5), NSC Lung Cancer (A549/ATCC: 20.1, NCI-H522: 17.2), Colon Cancer (HCT-116: 12.7), Melanoma (MALME-3M: 12.2 , UACC-257: 21.8, UACC-62: 11.2), Ovarian Cancer (IGROV1: 15.0), Renal Cancer (A498: 16.0, CAKI-1: 10.1), Prostate Cancer (PC-3: 10.3). |
| 19 | 13/59 | Leukaemia (HL-60(TB): 67.3, K-562: 27.2, MOLT-4: 30.9, SR: 18.5), NSC Lung Cancer (A549/ATCC: 29.3, HOP-62: 12.1, NCI-H522: 37.7), Colon Cancer (HT29: 17.5), CNS Cancer (U251: 13.0), Melanoma (SK-MEL-2: 13.9, UACC-257: 48.4), Ovarian Cancer (OVCAR-8: 19.6), Renal Cancer (UO-31: 12.8). |
| 20 | 59/59 | Leukaemia (CCRF-CEM: 93.1, HL-60(TB): 100, K-562: 87.6, MOLT-4: 97.3, PRMI-8226: 79.0, SR: 91.0), NSC Lung Cancer (A549/ATCC: 87.5, EKVX: 84.5, HOP-62: 87.1, NCI-H226: 77.4, NCI-H23: 67.2, NCI-H322M: 75.0, NCI-H460: 96.7, NCI-H522: 90.1), Colon Cancer (COLO 205: 91.4, HCC-2998: 60.4, HCT-116: 81.6, HCT-15: 71.5, HT29: 78.2, KM12: 74.1, SW-620: 67.0), CNS Cancer (SF-268: 78.2, SF-295: 78.6, SF-539: 96.0, SNB-19: 68.0, SNB-75: 44.8, U251: 79.4), Melanoma (LOX IMVI: 90.3, MALME-3M: 86.0 , M14: 96.6, MDA-MB-435: 62.8, SK-MEL-2: 80.5, SK-MEL-28: 72.88, SK-MEL-5: 100, UACC-257: 100, UACC-62: >100), Ovarian Cancer (IGROV1: 78.8, OVCAR-3: 66.6, OVCAR-4: 65.3, OVCAR-5: 67.5, OVCAR-8: 83.2, NCI/ADR-RES: 82.2, SK-OV-3: 61.0), Renal Cancer (786-0: 84.6, A498: 54.7, ACHN: 82.9, CAKI-1: 79.6, RXF 393: 71.5, SN12C: 68.8, TK-10: 69.5, UO-31: 100), Prostate Cancer (PC-3: 50.8, DU-145: 75.5), Breast Cancer (MCF7: 91.2, MDA-MB-231/ATCC: 56.8, HS 578 T: 32.0, BT-549: 69.3, T-47D: 70.0, MDA-MB-468: 98.8). |
| 21 | 8/59 | Leukaemia (HL-60(TB): 45.9, K-562: 19.1, MOLT-4: 34.1), NSC Lung Cancer (NCI-H522: 15.7), Colon Cancer (HCT-116: 11.0), CNS Cancer (SNB-75: 14.2), Renal Cancer (CAKI-1: 11.0), Prostate Cancer (PC-3: 11.5). |
| 22 | 9/59 | Leukaemia (CCRF-CEM: 13.0, HL-60(TB): 43.0, K-562: 14.5, MOLT-4: 29.9), NSC Lung Cancer (A549/ATCC: 32.3, NCI-H522: 14.0), Colon Cancer (HT29: 11.0), Melanoma (SK-MEL-2: 10.9, UACC-257: 40.9). |
| 23 | 9/59 | Leukaemia (HL-60(TB): 44.2, K-562: 37.6, MOLT-4: 32.6), NSC Lung Cancer (A549/ATCC: 22.8), Colon Cancer (COLO 205: 14.6), CNS Cancer (SNB-75: 11.3), Melanoma (UACC-257: 28.6), Ovarian Cancer (SK-OV-3: 14.2), Renal Cancer (UO-31: 10.2). |
| 24 | 8/59 | Leukaemia (MOLT-4: 21.4), NSC Lung Cancer (A549/ATCC: 17.1, NCI-H522: 39.8), Colon Cancer (HT29: 17.9), Melanoma (K-MEL-2: 11.7, UACC-257: 21.6), Renal Cancer (UO-31: 10.5), Prostate Cancer (PC-3: 18.6). |
| Imatinib | 20/54 | Leukaemia (MOLT-4: 18.0, PRMI-8226: 12.6, SR: 14.6), NSC Lung Cancer (EKVX: 15.7, NCI-H226: 10.6, NCI-H23: 17.1), Colon Cancer (HCT-116: 18.6, HCT-15: 11.5, HT29: 47.1), CNS Cancer (SF-295: 15.1, SF-539: 24.5, U251: 10.6), Melanoma (LOX IMVI: 11.6, SK-MEL-5: 22.3), Renal Cancer (A498: 13.7), Prostate Cancer (PC-3: 10.6, DU-145: 14.4), Breast Cancer (MDA-MB-231/ATCC: 11.2, T-47D: 18.6, MDA-MB-468: 29.1). |
aGrowth Inhibition percentages (GI%) lower than 10% are not shown.
bPCE: Positive cytotoxic effect (ratio between the number of cell lines with percentage growth inhibition from 10 to 100 and the total number of cell lines).
Table 2.
Growth inhibition percentage (GI%) of the most active hydrazones against individual cell lines.
| Growth inhibition percentage (GI%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Subpanel tumour cell lines | 6 | 9 | 16 | 20 | Imatinib | Gefitinib | 5-Fu | |
| Leukaemia | ||||||||
| CCRF-CEM | 22.4 | 20.8 | 84.7 | 93.1 | – | 96.0 | 42.9 | |
| HL-60(TB) | 62.5 | 24.3 | 93.7 | >100 | – | 100 | 52.1 | |
| K-562 | 72.2 | 44.1 | 59.5 | 87.6 | NT | NT | 57.7 | |
| MOLT-4 | 53.2 | 22.6 | 90.8 | 97.3 | 18.0 | >100 | 56.9 | |
| PRMI-8226 | 12.3 | – | 51.3 | 79.0 | 12.6 | 18.0 | 58.6 | |
| SR | 67.5 | 43.8 | 79.6 | 91.0 | 14.6 | 44.7 | 75.2 | |
| Non-Small Cell Lung Cancer | ||||||||
| A549/ATCC | 58.7 | 12.7 | 61.2 | 87.5 | – | 87.0 | 65.8 | |
| EKVX | 12.6 | – | 74.2 | 84.5 | 15.7 | 92.3 | 41.6 | |
| HOP-62 | 25.1 | – | 71.8 | 87.1 | NT | NT | 52.2 | |
| NCI-H226 | 16.3 | – | 44.8 | 77.4 | 10.6 | 25.2 | 30.5 | |
| NCI-H23 | 10.4 | – | 45.2 | 67.2 | 17.1 | 86.2 | 61.0 | |
| NCI-H322M | – | – | 61.6 | 75.0 | NT | NT | 40.5 | |
| NCI-H460 | 33.2 | – | 85.9 | 96.7 | – | 42.7 | 87.0 | |
| NCI-H522 | 95.5 | 53.9 | 84.9 | 90.1 | NT | NT | 42.0 | |
| Colon Cancer | ||||||||
| COLO 205 | 14.3 | – | 68.8 | 91.4 | – | 49.6 | 59.8 | |
| HCC-2998 | 15.5 | – | 52.7 | 60.4 | – | 55.3 | >100 | |
| HCT-116 | 38.2 | 13.9 | 70.9 | 81.6 | 18.6 | 72.1 | 82.2 | |
| HCT-15 | 50.2 | 16.3 | 82.0 | 71.5 | 11.5 | 71.5 | 73.5 | |
| HT29 | 50.5 | 17.7 | 56.4 | 78.2 | 47.1 | 50.3 | 72.9 | |
| KM12 | 49.2 | 25.1 | 65.9 | 74.1 | – | 36.9 | 59.3 | |
| SW-620 | 63.8 | 15.2 | 56.7 | 67.0 | – | 29..3 | 49.9 | |
| CNS Cancer | ||||||||
| SF-268 | 14.3 | – | 61.6 | 78.2 | – | 64.1 | 41.0 | |
| SF-295 | 24.0 | – | 62.0 | 78.6 | 15.1 | 15.8 | 30.9 | |
| SF-539 | 10.7 | – | 44.7 | 96.0 | 24.5 | 15.1 | >100 | |
| SNB-19 | 20.2 | – | 54.6 | 68.0 | – | 73.8 | 34.1 | |
| SNB-75 | 10.3 | – | 22.3 | 44.8 | – | 61.5 | 34.1 | |
| U251 | 23.7 | 13.7 | 72.2 | 79.4 | 10.6 | 56.5 | 49.7 | |
| Melanoma | ||||||||
| LOX IMVI | 27.6 | – | 78.6 | 90.3 | 11.6 | 46.0 | 69.6 | |
| MALME-3M | 30.3 | 15.2 | 36.5 | 86.0 | – | 22.1 | 41.8 | |
| M14 | 33.2 | – | 67.5 | 96.6 | – | 89.7 | NT | |
| MDA-MB-435 | 82.9 | 47.5 | 40.3 | 62.8 | – | 63.3 | 63.4 | |
| SK-MEL-2 | 52.3 | 19.8 | 60.0 | 80.5 | NT | NT | – | |
| SK-MEL-28 | 10.6 | – | 44.1 | 72.8 | – | 27.3 | NT | |
| SK-MEL-5 | 32.8 | 13.5 | 67.8 | >100 | 22.3 | 58.1 | 66.3 | |
| UACC-257 | 58.9 | 28.4 | 77.4 | >100 | – | 24.7 | 80.5 | |
| UACC-62 | 38.4 | 19.1 | 83.6 | >100 | – | 28.3 | 60.3 | |
| Ovarian Cancer | ||||||||
| IGROV1 | 36.8 | 12.3 | 59.0 | 78.8 | – | 57.2 | 48.8 | |
| OVCAR-3 | 32.2 | – | 77.2 | 66.6 | – | 55.8 | 52.6 | |
| OVCAR-4 | – | – | 56.3 | 65.3 | – | 11.5 | 40.6 | |
| OVCAR-5 | – | – | 35.3 | 67.5 | – | 54.5 | 55.7 | |
| OVCAR-8 | 16.2 | – | 72.1 | 83.2 | – | 78.2 | NT | |
| NCI/ADR-RES | 26.5 | 14.6 | 70.1 | 82.2 | – | 86.3 | 52.4 | |
| SK-OV-3 | 21.9 | – | 60.1 | 61.0 | – | 83.0 | 22.5 | |
| Renal Cancer | ||||||||
| 786-0 | – | – | 67.0 | 84.6 | – | 80.7 | 51.3 | |
| A498 | 30.9 | 17.9 | 24.8 | 54.7 | 13.7 | 65.9 | >100 | |
| ACHN | – | – | 71.7 | 82.9 | – | 91.5 | 60.7 | |
| CAKI-1 | 32.8 | 25.0 | 65.1 | 79.6 | – | 98.8 | 60.6 | |
| RXF 393 | – | – | 47.0 | 71.5 | – | 36.0 | 65.7 | |
| SN12C | 12.6 | – | 41.4 | 68.8 | – | 85.5 | 46.0 | |
| TK-10 | 14.9 | – | 54.2 | 69.5 | – | 75.0 | 33.1 | |
| UO-31 | 21.5 | 12.3 | 82.0 | >100 | – | 89.7 | 58.7 | |
| Prostate Cancer | ||||||||
| PC-3 | 21.2 | 26.9 | 45.7 | 50.8 | 10.6 | 49.6 | 41.8 | |
| DU-145 | 11.2 | – | 59.6 | 75.5 | 14.4 | 69.6 | 64.5 | |
| Breast Cancer | ||||||||
| MCF7 | 55.1 | 21.4 | 77.5 | 91.2 | – | 56.0 | 88.5 | |
| MDA-MB-231/ATCC | – | – | 52.6 | 56.8 | 11.2 | 31.4 | 21.9 | |
| HS 578 T | 14.9 | – | 18.5 | 32.0 | – | 10.0 | >100 | |
| BT-549 | 12.4 | – | 47.0 | 69.3 | – | 61.2 | 62.2 | |
| T-47D | 14.7 | – | 47.6 | 70.0 | 18.6 | 26.8 | 43.3 | |
| MDA-MB-468 | 25.8 | 21.5 | 35.3 | 98.8 | 29.1 | >100 | NT | |
NT: Not tested.
Structure correlation analysis gave the following results:
2-hydroxyphenyl derivatives, such as compounds 16 and 20 (PCE = 59/59), had significant and potent antitumor activity compared to unsubstituted phenyl and 4-hydroxyphenyl derivatives, such as compounds 4, 11, and 19 (PCE = 10/59, 5/59, and 13/59, respectively).
Derivatives incorporating the 2-hydroxyphenyl moiety, such as compounds 16 and 20 (PCE = 59/59), were potent antitumor agents compared to the corresponding 2-substituted compounds 13–15 (PCE = 10/59–14/59).
Derivatives based on a naphthalene scaffold, such as compound 6, showed a sharp increase in antitumor activity (PCE = 52/59) compared to phenyl and pyridyl compounds 4 and 5 (PCE = 10/59 and 13/59, respectively).
Replacement of the phenyl moiety of compound 4 with a 4-tolyl fragment, such as in compound 9, increased antitumor activity (PCE = 10/59 and 27/59, respectively).
The 4-tolyl compound 9 showed significant antitumor activity (PCE = 27/59) compared to the corresponding halogenated derivatives, such as compounds 7 and 8 (PCE = 7/59 and 9/59, respectively), and derivatives incorporating 4-methoxyphenyl (compound 10), 4-hydroxyphenyl (compound 11), and 4-(N,N-dimethylamino)phenyl (compound 12) fragments (PCE = 11/59, 5/59, and 9/59 respectively).
Introduction of one or more methoxy group at the phenyl fragment, such as in compounds 10 and 21–24, did not improve antitumor activity (PCE = 11/59, 8/59, 9/59, 9/59, and 8/59, respectively) compared to the unsubstituted phenyl compound 4 (PCE = 10/59).
Insertion of a methoxy group into compound 11 (PCE = 5/59) at position three significantly increased the antitumor activity of compound 19 (PCE = 13/59).
Insertion of a halogen atom into a phenyl derivative, such as compound 4 (PCE = 10/59), produced compounds 7, 8, 13, and 14 with retention of antitumor activity (PCE = 7/59, 9/59, 10/59, and 12/59, respectively).
Replacement of the 3,4-dimethoxyphenyl moiety, such as compound 21, with a 3,4-dichlorophenyl derivative, such as compound 18, increased antitumor activity (PCE = 8/59 and 14/59, respectively).
The broad-spectrum and selectivity of compounds 4–24 (Tables 1 and 2) against the 59 cell lines showed that compounds 6, 9, 16, and 20 had significant GI (>10–100%) against most of the cancer cell lines tested (leukaemia, non–small cell lung cancer [NSCLC], melanoma, and colon, CNS, ovarian, renal, prostate, and breast cancer) compared to imatinib (GI < 10%–47.1%). Compounds 6, 9, 16, and 20 showed significant antitumor activity against leukaemia (GI = 12.3–100%), NSCLC (GI = 10.4%–96.7%), colon cancer (GI = 13.9%–91.4%), CNS cancer (GI = 10.3%–96.0%), melanoma (GI = 10.6–100%), ovarian cancer (GI = 12.3%–83.2%), renal cancer (GI = 12.3–100%), prostate cancer (GI = 11.2%–75.5%), and breast cancer (GI = 12.4%–98.8%). In contrast, the antitumor activity of imatinib was moderate against leukaemia (GI = 12.6%–18.0%), NSCLC (GI = 10.6%–17.1%), colon cancer (GI = 11.5%–47.1%), CNS cancer (GI = 10.6%–24.5%), melanoma (GI = 11.6%–22.3%), ovarian cancer (GI < 10.0%), renal cancer (GI < 10.0%–13.7%), prostate cancer (GI = 10.6%–14.4%), and breast cancer (GI = 11.2%–29.1%).
2.2.2. Gi50, TGI, and LC50 of compound 20 and a comparative study
Compound 20 was the most active broad-spectrum antitumor agent among the 21 compounds tested, so we evaluated its potencies (50% cell growth inhibition [GI50]) in an advanced assay against a panel of 60 cancer cell lines (Figure S1 and Tables 3 and 4) at tenfold dilution of five different concentrations (100, 10, 1, 0.1, and 0.01 µM). We also comparatively compared the assay results to the potencies of celecoxib, erlotinib, gefitinib, sorafenib, vismodegib, and 5-Flu as reference drugs (Tables 3 and 4). Accordingly, we listed three dose–response parameters of antitumor activity for compound 20 and the reference drugs against each cell line in Table 4, including GI50, total cell growth inhibition (TGI), and median lethal concentration (LC50). In addition, we calculated the mean GI50 graph midpoints (GI50 MG_MID) for these parameters in order to obtain an average activity parameter over all cell lines for each molecule. Tables 3 and 4 list the GI50 values, which show that compound 20 had significantly potent antitumor activity, with GI50 = 0.063–11.7 µM. We compared this value to celecoxib, erlotinib, gefitinib, sorafenib, and vismodegib (GI50 = 3.98–63.09, 0.10–100.0, 0.0125–10.0, 1.26–3.98, and 19.95–100.0 µM, respectively (Table 4). With regard to individual human organs, compound 20 showed significantly potent antitumor activity against leukaemia (mean GI50 = 0.23 µM), NSCLC (mean GI50 = 0.26 µM), colon cancer (mean GI50 = 0.27 µM), CNS cancer (mean GI50 = 0.22 µM), melanoma (mean GI50 = 0.31 µM), ovarian cancer (mean GI50 = 1.10 µM), renal cancer (mean GI50 = 1.40 µM), prostate cancer (mean GI50 = 0.23 µM), and breast cancer (mean GI50 = 2.20 µM) (Tables 3 and 4). In contrast, celecoxib, 5-Fu, erlotinib, gefitinib, and sorafenib had the following mean GI50: leukaemia (12.35, 2.82, 27.85, 2.56, and 1.91 µM, respectively), NSCLC (25.09, 13.59, 13.11, 2.05, and 2.34 µM, respectively), colon cancer (20.73, 0.27, 51.68, 5.23, and 2.19 µM, respectively), CNS cancer (17.22, 14.38, 16.99, 5.64, and 2.33 µM, respectively), melanoma (22.46, 7.83, 23.74, 3.68, and 1.87 µM, respectively), ovarian cancer (17.14, 5.17, 5.52, 3.05, and 2.89 µM, respectively), renal cancer (19.87, 0.93, 2.46, 1.41, and 2.86 µM, respectively), prostate cancer (14.22, 1.46, 20.90, 3.29, and 2.58 µM, respectively), and breast cancer (17.49, 6.87, 24.72, 4.67, and 2.17 µM, respectively) (Table 4). Comparing the antitumor activity of compound 20 with the reference drugs, we found that compound 20 (GI50 MG_MID = 0.26 µM) is nearly 65-fold more potent than celecoxib (GI50 MG_MID =17.5 µM), 3-fold more potent than 5-Fu (GI50 MG_MID = 0.90 µM), 30-fold more potent than erlotinib (GI50 MG_MID = 7.68 µM), and 9-fold more potent than gefitinib (GI50 MG_MID = 2.1 µM) and sorafenib (GI50 MG_MID = 2.33 µM).
Table 3.
Influence of compound 20, and reference drugs on the growth of the individual tumour cell lines; median growth inhibitory (GI50, µM).
| Subpanel tumour cell lines | GI50 (µM) |
|||||
|---|---|---|---|---|---|---|
|
20 (799156/1)a |
Celecoxib (719627)a |
Erlotinib (718781)a |
Gefitinib (759856)a |
Sorafenib (747971)a |
Vismodegib (755986)a |
|
| Leukaemia | ||||||
| CCRF-CEM | 0.078 | 12.58 | 50.12 | 0.398 | 1.99 | 31.62 |
| HL-60(TB) | 0.164 | 15.85 | 25.12 | 1.0 | 1.58 | 31.62 |
| K-562 | 0.604 | 10.00 | 31.62 | 2.51 | 3.16 | 31.62 |
| MOLT-4 | 0.164 | 15.85 | 25.12 | 1.995 | 3.16 | 31.62 |
| PRMI-8226 | 0.297 | 3.98 | 25.12 | 6.31 | 1.58 | 31.62 |
| SR | 0.063 | 15.85 | 10.00 | 3.16 | 3.16 | 39.81 |
| Non-Small Cell Lung Cancer | ||||||
| A549/ATCC | 0.402 | 15.85 | 10.00 | 2.51 | 3.16 | 39.81 |
| EKVX | 0.162 | 19.95 | 0.199 | 0.0501 | 2.51 | 39.81 |
| HOP-62 | 0.085 | 63.09 | 15.85 | 3.16 | 1.99 | 79.43 |
| HOP-92 | 0.491 | NTb | 3.98 | 0.316 | 1.58 | 25.12 |
| NCI-H226 | 0.073 | NTb | 39.81 | 3.98 | 1.99 | 50.12 |
| NCI-H23 | 0.446 | 15.85 | 31.62 | 2.51 | 1.99 | 50.12 |
| NCI-H322M | 0.228 | 19.95 | 0.10 | 0.063 | 2.51 | 100 |
| NCI-H460 | 0.113 | 15.85 | 6.31 | 3.16 | 2.51 | 39.81 |
| NCI-H522 | 0.395 | NTb | 1.00 | 1.0 | 1.99 | 25.12 |
| Colon Cancer | ||||||
| COLO 205 | 0.269 | 15.85 | 50.12 | 7.94 | 1.99 | 39.81 |
| HCC-2998 | 0.301 | 15.85 | 79.43 | 2.51 | 3.16 | 39.81 |
| HCT-116 | 0.121 | 50.01 | 6.31 | 3.16 | 1.58 | 50.12 |
| HCT-15 | 0.092 | 15.85 | 3.98 | 3.98 | 2.51 | 31.62 |
| HT29 | 0.297 | 15.85 | 63.09 | 3.98 | 1.99 | 25.12 |
| KM12 | 0.404 | 15.85 | 79.43 | 10.0 | 1.58 | 31.62 |
| SW-620 | 0.387 | 15.85 | 79.43 | 5.011 | 2.51 | 63.09 |
| CNS Cancer | ||||||
| SF-268 | 0.0789 | 15.85 | 15.84 | 2.51 | 2.51 | 39.81 |
| SF-295 | 0.200 | 15.85 | 15.84 | 10.0 | 1.58 | 31.62 |
| SF-539 | 0.244 | 19.95 | 19.95 | 10.0 | 1.58 | 50.12 |
| SNB-19 | 0.428 | 19.95 | 12.58 | 3.16 | 3.16 | 63.09 |
| SNB-75 | 0.228 | 15.85 | 12.58 | 5.011 | 3.16 | 25.12 |
| U251 | 0.137 | 15.85 | 25.12 | 3.16 | 1.99 | 50.12 |
| Melanoma | ||||||
| LOX IMVI | 0.0997 | 19.95 | 6.31 | 3.16 | 1.58 | 39.81 |
| MALME-3M | 0.44 | 19.95 | 1.58 | 3.98 | 1.99 | 39.81 |
| M14 | 0.171 | 63.09 | 6.31 | 1.995 | 1.99 | 31.62 |
| MDA-MB-435 | 0.623 | 15.85 | 19.95 | 3.16 | 1.58 | 31.62 |
| SK-MEL-2 | 0.307 | 15.85 | 6.31 | 10.0 | 1.99 | 39.81 |
| SK-MEL-28 | 0.340 | 15.85 | 50.12 | 3.16 | 2.51 | 39.81 |
| SK-MEL-5 | 0.223 | 15.85 | 19.5 | 3.16 | 1.58 | 25.12 |
| UACC-257 | 0.340 | 19.95 | 100 | 2.51 | 1.99 | 50.12 |
| UACC-62 | 0.234 | 15.85 | 3.16 | 1.995 | 1.58 | 31.62 |
| Ovarian Cancer | ||||||
| IGROV1 | 0.196 | 15.85 | 0.316 | 3.16 | 2.51 | 100 |
| OVCAR-3 | 0.125 | 15.85 | 3.98 | 5.011 | 3.16 | 50.12 |
| OVCAR-4 | 0.063 | 12.58 | 10.00 | 6.31 | 3.16 | 39.81 |
| OVCAR-5 | 6.48 | 19.95 | 7.94 | 3.98 | 3.16 | 100 |
| OVCAR-8 | 0.25 | 19.95 | 7.94 | 0.631 | 3.16 | 31.62 |
| NCI/ADR-RES | 0.324 | 15.85 | 7.94 | 1.26 | 2.51 | 31.62 |
| SK-OV-3 | 0.251 | 19.95 | 0.50 | 1.0 | 2.51 | NTb |
| Renal Cancer | ||||||
| 786-0 | 0.182 | 39.81 | 10.00 | 3.16 | 3.16 | NTb |
| A498 | 9.81 | 15.85 | 2.51 | 0.631 | 2.51 | 31.62 |
| ACHN | 0.117 | 15.85 | 0.199 | 0.158 | 2.51 | 39.81 |
| CAKI-1 | 0.104 | 19.95 | 0.199 | 0.5012 | 3.16 | 79.43 |
| RXF 393 | 0.271 | 15.85 | 3.98 | 3.16 | 2.51 | 25.12 |
| SN12C | 0.354 | 15.85 | 1.58 | 1.258 | 2.51 | 39.81 |
| TK-10 | 0..207 | 19.95 | 0.251 | 0.794 | 3.98 | 63.09 |
| UO-31 | 0.139 | 15.85 | 1.00 | 1.584 | 2.51 | 31.62 |
| Prostate Cancer | ||||||
| PC-3 | 0.319 | 12.58 | 39.81 | 5.011 | 1.99 | 25.12 |
| DU-145 | 0.144 | 15.85 | 1.99 | 1.584 | 3.16 | 50.12 |
| Breast Cancer | ||||||
| MCF7 | 0.253 | 15.85 | 100.00 | 5.011 | 2.51 | 31.62 |
| MDA-MB-231/ATCC | 0.308 | 15.85 | 6.31 | 3.98 | 1.26 | 19.95 |
| HS 578 T | 11.7 | 19.95 | 6.31 | 7.94 | 2.51 | 79.43 |
| BT-549 | 0.346 | 19.95 | 31.62 | 3.16 | 3.16 | NTb |
| T-47D | 0.39 | 15.85 | 3.98 | 7.94 | 1.58 | 31.62 |
| MDA-MB-468 | 0.203 | NTb | 0.126 | 0.01258 | 1.99 | 19.95 |
bNT: Not tested.
Table 4.
Average antitumor activity of compound 20, and reference drugs against tumour cell lines from nine different organs at 10-fold dilution of five concentrations; median growth inhibitory (GI50, µM), total growth inhibitory (TGI, µM) and median lethal (LC50, µM)a.
| Subpanel tumour cell lines |
MG-MIDb | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd. No. (NSC)c |
Activity | leukaemia | NSC lung cancer |
colon cancer | CNS cancer | melanoma | ovarian cancer | renal cancer | prostate cancer | breast cancer | |
|
20 (799156) |
GI50 | 0.23 | 0.26 | 0.27 | 0.22 | 0.31 | 1.10 | 1.40 | 0.23 | 2.20 | 0.26 |
| TGI | 83.5 | 65.0 | c | 71.7 | 45.6 | 70.0 | 80.3 | c | 69.5 | 46.8 | |
| LC50 | c | c | c | c | c | c | c | c | c | c | |
|
Celecoxib (719627) |
GI50 | 12.35 | 25.09 | 20.73 | 17.22 | 22.46 | 17.14 | 19.87 | 14.22 | 17.49 | 17.5 |
| TGI | 29.27 | 41.93 | 41.39 | 31.62 | 38.49 | 34.20 | 41.19 | 28.37 | 34.89 | 34.0 | |
| LC50 | 67.09 | 67.08 | 66.51 | 58.76 | 64.31 | 65.91 | 61.22 | 57.01 | 67/03 | 63.3 | |
|
5-Flu (19893) |
GI50 | 2.82 | 13.59 | 0.27 | 14.38 | 7.83 | 5.17 | 0.93 | 1.46 | 6.87 | 0.90 |
| TGI | 68.37 | 80.45 | 51.41 | 73.70 | 61.57 | 38.36 | 46.25 | c | 68.01 | 43.4 | |
| LC50 | c | c | 89.30 | c | 95.42 | 92.87 | c | c | c | 95.6 | |
|
Erlotinib (718781) |
GI50 | 27.85 | 13.11 | 51.68 | 16.99 | 23.74 | 5.52 | 2.46 | 20.90 | 24.72 | 7.68 |
| TGI | 96.57 | 73.76 | c | 82.11 | 77.89 | 74.41 | 42.59 | c | 70.53 | 66.3 | |
| LC50 | c | 97.71 | c | c | 93.31 | 97.06 | 89.15 | c | 96.43 | 95.6 | |
|
Gefitinib (759856) |
GI50 | 2.56 | 2.05 | 5.23 | 5.64 | 3.68 | 3.05 | 1.41 | 3.29 | 4.67 | 2.1 |
| TGI | 12.07 | 13.86 | 18.47 | 19.62 | 12.49 | 33.29 | 12.50 | 31.62 | 18.62 | 14.3 | |
| LC50 | 93.85 | 94.68 | 51.74 | 50.56 | 36.40 | 83.58 | 52.82 | 89.72 | 52.47 | 51.9 | |
|
Sorafenib (747971) |
GI50 | 1.91 | 2.34 | 2.19 | 2.33 | 1.87 | 2.89 | 2.86 | 2.58 | 2.17 | 2.33 |
| TGI | 42.12 | 7.99 | 7.94 | 7.64 | 6.29 | 14.43 | 10.86 | 10.27 | 9.35 | 9.11 | |
| LC50 | c | 49.15 | 38.06 | 27.77 | 28.57 | 71.34 | 54.75 | 69.91 | 57.29 | 43.1 | |
aGI50, molar concentration of the compound that inhibits 50% net cell growth; TGI, molar concentration of the compound leading to total inhibition; and LC50, molar concentration of the compounds leading to 50% net cell death.
bFull panel mean-graph midpoint (µM).
c: Compounds showed values ≥ 100 µM. Bold values used only for more precise comparison.
2.3. Apoptosis assay
2.3.1. Annexin V–FITC apoptosis assay
Apoptosis induction is the most important mechanism by which major chemotherapeutics kill cancer cells71. Apoptosis causes cellular changes whereby the translocation of phosphatidylserine (PS) occurs through the plasma membrane from the inside to the outside71. Annexin-V can bind to PS, which can be used as a sensitive probe for PS on the outer side of the plasma membrane72. We performed cytometric analysis to distinguish apoptosis from the necrosis mode of HL60 cell death induced by the most active compounds 6, 9, 16, and 20 using annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (AV/PI) dual-staining assay with the BD FACSCalibur (BD Biosciences, San Jose, CA, USA) (Table 5). The AV/PI staining of HL60 cells was performed at a mixed molar concentration of 10 µM with compounds 6, 9, 16, and 20 for 24 h. Figure 4 and Table 5 show the results of treating HL60 cells with compounds 6, 9, 16, and 20 for 24 h. We found an increase in the early apoptosis ratio (Figure 4, lower-right quadrant of the cytogram) from 0.57% in the control sample (DMSO) to 3.52%–8.42% and a sharp increase in the late apoptosis ratio (Figure 4, upper-right quadrant of the cytogram) from 0.22% to 7.64%–13.41%. These data support the apoptotic mechanism underlying programmed cell death induced by compounds 6, 9, 16, and 20 rather than the necrotic pathway.
Table 5.
Effect of compounds 6, 9, 16, and 20 and DMSO on the percentage of HL60 cells stained positive for annexin V-FITC
| Apoptosis | ||||
|---|---|---|---|---|
| Sample/cell line | Total | Early | Late | Necrosis |
| 6 | 12.57 | 3.52 | 7.64 | 1.41 |
| 9 | 16.89 | 5.66 | 9.04 | 2.19 |
| 16 | 22.41 | 7.34 | 13.41 | 1.66 |
| 20 | 24.31 | 8.42 | 12.66 | 3.23 |
| DMSO | 1.71 | 0.57 | 0.22 | 0.92 |
Figure 4.

Effect of DMSO (upper left panel), and compounds 6 (upper right panel), 9 (middle left panel), 16 (middle right panel), and 20 (lower panel) on the percentage of annexin V-FITC-positive staining in HL60 cells.).
2.3.2. In vitro cell cycle analysis
Antitumor agents can induce apoptosis by activating signalling pathways, leading to G2/M phase arrest73,74. Flow cytometry is used to measure cell growth in different cell cycle phases (pre-G1, G1, S, and G2/M)73,74. We selected the most active compounds 6, 9, 16, and 20 for further analysis of their effects on cell cycle progression in the HL60 cell line (Figure 5 and Table 6). We used the solvent DMSO as a negative control. Briefly, we incubated HL60 cells with 10 µM compounds 6, 9, 16, and 20 for 24 h. Compounds 6, 9, 16, and 20 interfered with the normal cell cycle of HL60 cells. There was a significant effect on the percentage of apoptotic cells, as indicated by an increase in cells in the pre-G1 phase (12.57%–24.31%) and the G2/M phase (23.27%–38.09%) compared to the control (1.71% and 12.03% cells, respectively). In contrast, the percentage of cells in S and G0/G1 phases significantly decreased (22.49%–29.43% and 36.28%–48.31%, respectively) compared to the control (35.3% and 52.67%, respectively), causing cell cycle arrest. These results clearly indicated that compounds 6, 9, 16, and 20 arrests the G2/M phase of the cell cycle (Figure 5 and Table 6).
Figure 5.

Cell cycle analysis of HL60 cells treated with DMSO (upper left panel) and compounds 6 (upper right panel), 9 (middle left panel), 16 (middle right panel), and 20 (lower panel).
Table 6.
Effect of compounds 6, 9, 16, and 20 and DMSO on the cell cycle of HL60 cells.
| Sample |
Conc.
(µM) |
%G0-G1 | %S | %G2-M | %Pre-G1 |
|---|---|---|---|---|---|
| 6 | 10.0 | 48.31 | 28.42 | 23.27 | 12.57 |
| 9 | 10.0 | 43.15 | 27.41 | 29.44 | 16.89 |
| 16 | 10.0 | 39.42 | 22.49 | 38.09 | 22.41 |
| 20 | 10.0 | 36.28 | 29.43 | 34.29 | 24.31 |
| DMSO | 0.0 | 52.67 | 35.3 | 12.03 | 1.71 |
2.4. Enzymatic inhibition assay
2.4.1. Cox-2 inhibition activity
COX-2 is overexpressed in several cancer cell lines during cell proliferation. Its inhibition is used as a target for cancer treatment and prevention51,52,75. Therefore, we performed COX-2 inhibition assays (kit catalogue no. 560101; Cayman Chemicals Inc., Ann Arbour, MI, USA) using compounds 6, 9, 16, and 20, which showed the highest antitumor activity, in addition to the reference drug celecoxib15,76. The results were expressed as IC50 (µM) as the mean of three acquired determinations (Table 7). The IC50 of celecoxib as a COX-2 inhibitor was 2.79 µM. Compounds 9 and 20 were the most active COX-2 inhibitors (IC50 = 2.97 and 6.94 µM, respectively). In contrast, compounds 6 and 16 showed significantly low COX-2 inhibition (IC50 = 36.27 and 82.45 µM, respectively). Compounds with the 4-alkylphenyl moiety, such as the 4-tolyl fragment in compound 9 and the N,N-diethylaminophenyl fragment in compound 20, have high COX-2 inhibition compared to compounds devoid of the 4-alkylphenyl moiety, such as compounds 6 and 16.
Table 7.
In vitro inhibitory effects of COX-2, EGFR, and HER2 of the antitumor agents 6, 9, 16, and 20.a
| Compound No. | IC50 (µM)a |
||
|---|---|---|---|
| COX-2 Inhibition |
EGFR Inhibition |
HER2 Inhibition |
|
| 6 | 36.27 ± 2.55 | 0.26 ± 0.09 | 0.35 ± 0.01 |
| 9 | 2.97 ± 0.07 | 0.77 ± 0.03 | 0.41 ± 0.01 |
| 16 | 82.45 ± 3.61 | 0.20 ± 0.07 | 0.13 ± 0.04 |
| 20 | 6.94 ± 0.41 | 0.19 ± 0.01 | 0.07 ± 0.02 |
| Celecoxib | 2.79 ± 0.07 | – | – |
| Erlotenib | – | 0.11 ± 0.04 | 0.09 ± 0.03 |
| Sorafenib | – | 0.10 ± 0.04 | 0.05 ± 0.02 |
| Gefitinib | – | 0.055 ± 0.99 | 0.079 ± 1.42 |
aIC50 value is the compound concentration required to produce 50% inhibition.
2.4.2. Kinase inhibition activity
We tested the inhibitory effects of the most active compounds 6, 9, 16, and 20 against EGFR and HER240. We also tested the reference drugs erlotinib, sorafenib, and gefitinib. Table 7 summarises the inhibitory activities of compounds 6, 9, 16, and 20, and the reference drugs. The IC50 of erlotinib, sorafenib, and gefitinib against EGFR was 0.11, 0.10, and 0.055 µM, respectively, and against HER2 was 0.09, 0.05, and 0.079 µM, respectively. The IC50 of compounds 6, 9, 16, and 20 against EGFR and HER2 were in the submicromolar range of 0.07–0.77 µM. Compounds 16 and 20 showed the highest and most potent inhibitory activity against EGFR (IC50 = 0.20 and 0.19, respectively) and HER2 (IC50 = 0.13 and 0.07 µM, respectively) compared to erlotinib (EGFR-IC50 = 0.11 and HER2-IC50 = 0.09 µM), sorafenib (EGFR- IC50 = 0.10 and HER2-IC50 = 0.05 µM), and gefitinib (EGFR-IC50 = 0.055 and HER2-IC50 = 0.079 µM). Compound 9 was the least active against EGFR and HER2 (IC50 = 0.77 and 0.41 µM, respectively), while compound 6 was more effective against EGFR (IC50 = 0.26 µM) compared to HER2 (IC50 = 0.35 µM). Compound 20 showed inhibitory activity approximately similar to the reference drugs against EGFR and HER2. Compounds with 2-hydroxyphenyl fragments, such as compounds 16 and 20, are more potent than corresponding compounds with the 4-tolyl moiety, such as compound 9, or the 1-naphthyl fragment, such as compound 6. In the enzymatic assay, the 2-hydroxyphenyl moiety plays a major role in the inhibitory activity against EGFR and HER2.
2.5. Molecular docking study
Molecular modelling is an important tool for studying the biological activity and SARs of bioactive compounds and exploring the binding mode of ligand molecules within the receptor- or putative enzyme-binding sites77–80. We performed molecular docking using the MOE 2008.10 program protocol obtained from Chemical Computing Group Inc. (Montreal, QC, Canada) 81. We subjected the selected compounds and the co-crystallized bound inhibitors to molecular docking into the putative active site of the protein to ensure docking accuracy and generate an appropriate binding orientation27,40,41,82,83.
2.5.1. Molecular docking of compound 9 with COX-2
Molecular docking was performed to study the mode of interaction between the most active compound 9 and the COX-2 pocket–binding site (Figure 6). We derived the crystallographic binding site on the COX-2 isozyme in a complex with the SC-558 ligand, a celecoxib analogue, from the Protein Data Bank (PDB code: 1CX2) (Figure 6, left panel). We used the interaction energy and hydrogen bond formation among compound 9 and the amino acids within the putative active pocket of the COX-2 isozyme to predict the mode of interaction. Figure 6 shows the molecular docking results for compound 9. Compound 9 was placed into the catalytic site of the COX-2 isozyme, where the pharmacophoric 4-methylsulfonylbenzene group can interact with amino acid residues Ile-517, Phe-518, His-90, Gln-192, and Arg-513 through a network of classical and nonclassical hydrogen bonds. These binding interactions were approximately similar to those of the SC-558 inhibitor co-crystallized in the COX-2-binding site. The stability of the docked complex of the target compound 9 in the COX-2-binding site depended on the methylsulfonyl pharmacophore (–SO2CH3) by forming classical and nonclassical hydrogen bonds with the key amino acid residues Arg-513 (3.06 Å), Gln-192 (3.26 Å), Ile-517 (3.64 Å), and Phe-518 (3.29 Å) (Figure 6, right panel), while the phenyl ring attached to the methylsulfonyl pharmacophore formed two nonclassical hydrogen bonds by binding with amino acid residues His-90 (3.33 Å) and Leu-352 (2.78 Å) and undergoing an additional CH–π interaction with amino acid residue Ser-353 (3.87 Å). Also, the benzylidenehydrazine fragment of compound 9 interacted with amino acid residues Ala-527, Val-349, Val-116, Leu-359, and Leu-531 through hydrophobic interactions. The hydrazino fragment of benzylidenehydrazine formed three classical hydrogen bonds with amino acid residues Arg-120 (2.79 Å), Tyr-355 (3.15, and 3.15 Å), while the phenyl part of the benzylidenehydrazine moiety interacted with the amino acid residue Arg-120 by forming a nonclassical H-bond (2.99 Å) and CH–π interaction (3.98 Å) (Figure 6, right panel).
Figure 6.

Binding mode of co-crystallized inhibitor (upper panel), compounds 9 (middle panel), and 20 (lower panel) within COX-2 binding site (PDB ID: 1CX2).
2.5.2. Molecular docking of compound 20 with EGFR
The results of the antitumor activity and enzymatic assay of compound 20 against EGFR prompted us to perform molecular docking studies of the ATP-binding site of EGFR, along with the reference drug erlotinib to predict the binding interactions of the target compound (Figure 7). We retrieved the ligand erlotinib from the PDB as a co-crystallized ligand in a complex with EGFR (PDB code: 1M17) (Figure 7, left panel). Both phenyl rings of compound 20 surrounded and interacted with amino acid residues lining the hydrophobic pocket in EGFR-TK, such as Gly-772, Leu-768, Pro-770, Leu-694, Leu-820, and Val-702 (Figure 7, right panel). Also, the –OH group of compound 20 formed triple hydrogen bonds with amino acid residues Met-769 (3.19 Å), Pro-770 (3.49 Å), and Gly-772 (3.73 Å). The methylsulfonyl (CH3-SO2–) moiety showed significant interactions, where the oxygen atom of the CH3-SO2– group directly formed hydrogen bonds with the amino acid residue Thr-766 (3.26 Å) and the Thr-830 side chain (3.00 Å) and showed additional binding with a water molecule (HOH-10)-mediated hydrogen bonding with Thr-766 (2.78, and 3.10 Å). Also, the methyl moiety of the CH3-SO2– group interacted with amino acid residue Met-742 by a nonclassical hydrogen bond of 3.91 Å with the sulphur (S-) part of Met-742, while the phenyl part attached to the methylsulfonyl moiety interacted with amino acid residue Leu-820 through CH–π interaction (4.34 Å). These binding interactions indicated that both 2-hydroxyphenyl and 4-methylsulfonylbenzene fragments are important for binding and subsequent inhibitory effects (Figure 7).
Figure 7.

Binding mode of co-crystallized inhibitor (left panel) and compounds 20 (right panel) within EGFR binding site (PDB ID: 1M17).
2.5.3. Molecular docking of compound 20 with HER2
We retrieved the crystal 3 D structure of HER2 co-crystallized with its bound inhibitor 03Q from the PDB (PDB code: 3PP0) (Figure 8, left panel). Molecular docking of compound 20 into the HER2-binding cavity showed that the 2-hydroxyphenyl fragment forms four hydrogen bonds with amino acid residues Leu-796 (2.95 Å), Thr-798 (3.81 Å), Ala-751 (3.65 Å), and Lys-753 (3.61 Å), while the hydrazide fragment forms two hydrogen bonds with a water molecule (HOH-22)-mediated hydrogen bonding with Thr-862 (2.45, and 3.05 Å) (Figure 8, right panel). In addition, we observed one more hydrogen bond between one oxygen of the sulphonyl group and the amino acid residue Cys-805 (2.50 Å). In contrast, the N,N-diethylaminophenyl moiety of compound 20 shows hydrophobic interactions with the side chains of amino acid residues Glu-770, Ser-783, Leu-785, Met-774, Phe-864, Leu-796, Thr-798, Asp-863, and Lys-753, while the methylsulfonylbenzene moiety showed hydrophobic interactions with amino acid residues Leu-852, Met-801, Leu-800, Val-734, Leu-826, and Gly-804. The binding modes of compound 20 are approximately similar to the co-crystallized bound inhibitor with HER2 kinase.
Figure 8.

Binding mode of co-crystallized inhibitor (left panel) and compounds 20 (right panel) within HER2 binding site (PDB ID: 3PP0).
2.6. Physicochemical and pharmacokinetic predictions
We predicted the pharmacokinetic and physicochemical properties of the most active compounds 6, 9, 16, 20, and reference drugs celecoxib, erlotinib, gefitinib, and vismodegib using the automated SwissADME online calculation system (Table 8)84. Compounds 6, 9, 16, 20 showed high gastrointestinal absorption, while compound 20 was predicted as an inhibitor of CYP2C19, CYP2C9, and CYP3A4 isoforms and a non-inhibitor of CYP1A2 and CYP2D6 isoforms (Table 8). In contrast, compound 16 was predicted as a non-inhibitor of all CYP isoforms. In addition, compounds 6 and 9 were predicted as inhibitors of CYP2C19 and CYP1A2 isoforms and non-inhibitors of CYP2C9, CYP3A4, and CYP2D6 isoforms. In addition, we calculated the drug-likeness properties, as indicated by major Lipinski’s (Pfizer), Ghose’s (Amgen), Veber’s (GSK), and Egan’s (Pharmacia) pharmaceutical rules85–87. Compounds 6, 9, 16, 20 successfully passed all filters (Table 8). The BOILED-Egg graph88 of the WlogP/tPSA (topological polar surface area) showed that compounds 6, 9, 16, 20, together with celecoxib and vismodegib, are located in the human intestinal absorption (HIA) region with no BBB permeation, indicating few CNS side effects (Figure 9). Indeed, compounds 6, 9, 16, 20 are not P-glycoprotein (P-gp−) substrates, suggesting that they are not susceptible to the efflux mechanism carried out by P-gp that is used by many cancer cell lines as a drug resistance mechanism (Figure 9)89–91. In addition, the bioavailability radar chart of compounds 6, 9, 16, 20, and reference drugs are shown in Figures 10 and 11. These charts were drawn as six axes for six key properties of oral bioavailability: polarity (POLAR), solubility (INSOLU), lipophilicity (LIPO), flexibility (FLEX), saturation (INSATU), and size (SIZE) 89–91. The optimal property ranges are shown as a pink area, while the red line represents predicted properties for the examined molecule. The SwissADME tool calculation of compounds 6, 9, 16, and 20 predicts that they possess appropriate physicochemical and pharmacokinetic properties.
Table 8.
Predictions of the physicochemical and pharmacokinetic properties for target compounds 6, 9, 16, and 20 together with reference drugsa
| Compounds No. | 6 | 9 | 16 | 20 | celecoxib | Erlotinib | Gefitinib | Vismodegib |
|---|---|---|---|---|---|---|---|---|
| Properties | ||||||||
| BBBb | NO | NO | NO | NO | NO | Yes | Yes | NO |
| GIAb | High | High | High | High | High | High | High | High |
| P-gpbsubstrate | No | No | No | No | No | No | No | No |
| CYP1A2 inhibitorb | Yes | Yes | No | No | No | Yes | No | No |
| CYP2C19 inhibitorb | Yes | Yes | No | Yes | No | Yes | Yes | Yes |
| CYP2C9 inhibitorb | No | No | No | Yes | Yes | Yes | Yes | Yes |
| CYP2D6 inhibitorb | No | No | No | No | No | Yes | Yes | No |
| CYP3A4 inhibitorb | No | No | No | Yes | No | Yes | Yes | Yes |
| Log S (Water Solubility) | −6.96 (Poorly soluble) | −5.69 (Moderately soluble) | −4.73 (Moderately soluble) | −5.60 (Moderately soluble) | −6.22 (Poorly soluble) | −7.26 (Poorly soluble) | −7.94 (Poorly soluble) | −8.51 (Poorly soluble) |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Lipinski #violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ghose #violations | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| Veber #violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Egan #violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
aAll calculations were performed using SwissADME.
bGIA: gastrointestinal absorption; BBB: blood-brain barrier permeation; P-gp: permeability glycoprotein; CYP1A2, CYP2C9, CYP2C19, CYP3A4 and CYP2D6 are isoforms of cytochromes P450.
Figure 9.

Boiled-Egg plot predicted by swissADME online web tool for target molecules 6, 9, 16, 20, and the reference drugs (celecoxib, erlotinib, gefitinib, and vismodegib).
Figure 10.

Bioavailability radar charts as predicted by swissADME online web tool for target molecules 6 (upper left panel), 9 (upper right panel), 16 (lower left panel), and 20 (lower right panel).
Figure 11.

Bioavailability radar charts as predicted by swissADME online web tool for reference drugs celecoxib (upper left panel), erlotinib (upper right panel), gefitinib (lower left panel), and vismodegib (lower right panel).
3. Conclusion
We synthesised a series of hydrazones 4–24 based on a 4-methylsulfonylbenzene scaffold starting from 4-methylsulfonylphenylhydrazide. We also analysed the potential antitumor activities of the 21 hydrazones using 59 human cell lines and performed enzyme inhibition assays using EGFR, HER2, and COX-2. Compounds 6, 9, 16, and 20 possess the highest broad-spectrum and potent antitumor activity with a GI of >10–100% and a PCE of 52/59, 27/59, 59/59, and 59/59, respectively, for nine human tissue compared to imatinib (GI = >10%–47.1%; PCE = 20/55). The antitumor activity of compounds 6, 9, 16, and 20 against individual organs is GI = 20.8–100% for leukaemia, 12.7%–96.7% for NSCLC, 13.9%–91.4% for colon cancer, 10.3%–96.0% for CNS cancer, 10.6–100% for melanoma, 12.3%–83.2% for ovarian cancer, 12.3–100% for renal cancer, 11.2%–75.5% for prostate cancer, and 12.4%–98.8% for breast cancer. Compound 20 has the highest GI and shows significant antitumor activity (GI50 = 0.063–11.7 µM) compared to celecoxib, erlotinib, gefitinib, sorafenib, and vismodegib (GI50 = 3.98–63.09, 0.10–100.0, 0.0125–10.0, 1.26–3.98, and 19.95–100.0 µM, respectively). Cell cycle analysis showed that the apoptotic mechanism underlying programmed cell death is the main mechanism rather than the necrotic pathway, and compounds 6, 9, 16, and 20 induce apoptosis by inhibiting cell growth at the G2/M phase. In addition, compounds 9 and 20 are the most active inhibitory agents against COX-2 (IC50 = 2.97 and 6.94 µM, respectively) compared celecoxib (IC50 = 2.79 µM), while compounds 16 and 20 have the highest inhibition activity against EGFR (IC50 = 0.2 and 0.19 µM, respectively) and HER2 (IC50 = 0.13 and 0.07 µM, respectively) compared to erlotinib (EGFR-IC50 = 0.11 and HER2-IC50 = 0.09 µM), sorafenib (EGFR-IC50 = 0.10 and HER2-IC50 = 0.05 µM), and gefitinib (EGFR-IC50 = 0.055 and HER2-IC50 = 0.079 µM). The results as mentioned above indicated that these compounds are potential multitarget agents as COX-2, EGFR, and HER2 inhibitors. Compound 9 was subjected to molecular docking into binding sites of COX-2, while compound 20 was subjected to molecular docking into the putative binding sites of EGFR and HER2 to find the binding mode and molecular models required for interaction of these compounds with respective enzymes or receptors. Molecular docking showed that the respective molecules bound approximately similar to the co-crystallized inhibitors in COX-2-, EGFR-, and HER2-binding sites. SwissADME and drug-likeness prediction showed that compounds 6, 9, 16, and 20 possess good physicochemical, pharmacokinetic, and drug-likeness properties.
4. Experimental
4.1. Chemistry
Melting points (uncorrected) were recorded on a Barnstead 9100 Electrothermal melting apparatus (APS Water Services Corporation, Van Nuys, CA, USA), while the IR spectra were recorded on a FT-IR Perkin-Elmer spectrometer (PerkinElmer Inc., Waltham, MA, USA). The 1H NMR and 13 C NMR were measured in DMSO-d6 or CDCl3, on Bruker 700 or 500 and 176 or 125 MHz instruments, respectively (Bruker, Billerica, MA, USA). Chemical shifts are reported in δ ppm. Mass spectra were recorded on an Agilent 6320 Ion Trap mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). C, H, and N were analysed at the Research Centre, College of Pharmacy, King Saud University, Saudi Arabia. The results were within ± 0.4% of the theoretical values. Compounds 3, 11, 16, 19, and 20 were prepared according to a previous report65.
4.1.1. General procedure for the synthesis of hydrazones 4–24 (Scheme 1)
A mixture of 4-(methylsulfonyl)benzohydrazide 3 (10 mmol) and an appropriate aromatic aldehyde (10 mmol) was stirred in methanol (10 ml) containing a catalytic amount of acetic acid (0.5 ml) at room temperature for 24 h. The obtained solid was filtered, dried, and recrystallized from absolute ethanol.
4.1.1.1. N'-Benzylidene-4-(methylsulfonyl)benzohydrazide (4)
M.p 273–275ο; 95% yield; IR (KBr, cm−1) ν: 3215 (NH), 1656 (C = O), 1284, 1148 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.10 (s, 1H), 8.48 (s, 1H), 8.16 (d, 2H, J = 7.77 Hz), 8.11 (d, 2H, J = 7.77 Hz), 7.77 (d, 2H, J = 7.14 Hz), 7.48 (dd, 3H, J = 19.95 & 7.14 Hz), 3.31 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.3, 149.2, 143.7, 138.3, 134.5, 130.8, 129.3, 129.1, 127.7, 127.6, 43.7; C15H14N2O3S: m/z (302.1).
4.1.1.2. 4-(Methylsulfonyl)-N'-(pyridin-3-ylmethylene)benzohydrazide (5)
M.p 339–341ο; 89% yield; IR (KBr, cm−1) ν: 3221 (NH), 1640 (C=O), 1339, 1147 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.26 (s, 1H), 8.89 (s, 1H), 8.64 (d, 1H, J = 4.55 Hz), 8.53 (s, 1H), 8.16 (d, 3H, J = 10.57, 8.33 Hz), 8.11 (d, 2H, J = 8.12 Hz), 7.51 (dd, 1H, J = 12.53, 2.80 Hz), 3.31 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.4, 151.4, 149.3, 146.4, 143.8, 138.1, 134.0, 130.4, 129.1, 127.6, 124.5, 43.7; C14H13N3O3S: m/z (303.3).
4.1.1.3. 4-(Methylsulfonyl)-N'-(naphthalen-1-ylmethylene)benzohydrazide (6)
M.p 235–237ο; 88% yield; IR (KBr, cm−1) ν: 3222 (NH), 1651 (C=O), 1295, 1144 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.18 (s, 1H), 9.12 (s, 1H), 8.05 (dd, 1H, J = 15.47 & 8.19 Hz), 8.21 (d, 2H, J = 7.63 Hz), 8.14 (d, 2H, J = 7.63 Hz), 8.05 (dd, 2H, J = 15.47 & 8.19 Hz), 7.97 (d, 1H, J = 7.07 Hz), 7.70 (t, 1H, J = 15.12 Hz), 7.63 (dd, 2H, J = 7.09 Hz), 3.32 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.3, 149.1, 143.8, 138.3, 134.0, 131.3, 130.6, 129.7, 129.3, 129.1, 128.6, 127.9, 127.7, 126.8, 126.0, 124.7, 43.7; C19H16N2O3S: m/z (352.1).
4.1.1.4. N'-(4-Chlorobenzylidene)-4-(methylsulfonyl)benzohydrazide (7)
M.p 268–270ο; 92% yield; IR (KBr, cm−1) ν: 3239 (NH), 1656 (C=O), 1284, 1145 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.16 (s, 1H), 8.46 (s, 1H), 8.16 (d, 2H, J = 8.26 Hz), 8.11 (d, 2H, J = 8.33 Hz), 7.78 (d, 2H, J = 8.40 Hz), 7.54 11 (d, 2H, J = 8.33 Hz), 3.30 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.4, 147.8, 143.8, 138.2, 135.2, 133.4, 129.4, 129.3, 129.1, 127.6, 43.7; C15H13ClN2O3S m/z: 336.0, (M + 2; 338.0).
4.1.1.5. N'-(4-Fluorobenzylidene)-4-(methylsulfonyl)benzohydrazide (8)
M.p 278–280ο; 90% yield; IR (KBr, cm−1) ν: 3239 (NH), 1655 (C=O), 1285, 1146 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.17 (s, 1H), 8.48 (s, 1H), 8.16 (d, 2H, J = 7.98 Hz), 8.11 (d, 2H, J = 5.39 Hz), 7.82 (t, 2H, J = 13.09 Hz), 7.32 (t, 2H, J = 8.43 Hz), 3.31 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 164.4, 163.0, 162.3, 148.0, 143.7, 138.3, 131.2, 131.1, 130.7, 129.9, 129.8, 129.1, 127.6, 116.5, 116.3, 43.7; C15H13FN2O3S: m/z: 320.2.
4.1.1.6. N'-(4-Methylbenzylidene)-4-(methylsulfonyl)benzohydrazide (9)
M.p 271–272ο; 94% yield; IR (KBr, cm−1) ν: 3199 (NH), 1652 (C=O), 1292, 1150 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.04 (s, 1Η), 8.44 (s, 1Η), 8.16 (d, 2H, J = 8.12 Hz), 8.11 (d, 2H, J = 8.19 Hz), 7.65 (d, 2H, J = 7.84 Hz), 7.29 (d, 2H, J = 7.84 Hz), 3.30 (s, 3H), 2.35 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.3, 149.2, 143.7, 140.7, 138.4, 131.8, 129.9, 129.1, 127.7, 127.6, 43.7, 21.5; C16H16N2O3S m/z: 316.4.
4.1.1.7. N'-(4-Methoxybenzylidene)-4-(methylsulfonyl)benzohydrazide (10)
M.p 249–251ο; 88% yield; IR (KBr, cm−1) ν: 3212 (NH), 1698 (C=O), 1282, 1149 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.08 (s, 0.25Η), 11.97 (s, 0.75 H), 8.84 (s, 0.25H), 8.42 (s, 0.75H), 8.18 (d, 0.58H, J = 8.12 Hz), 8.15 (d, 1.42H, J = 8.12 Hz), 8.10 (d, 1.77H, J = 8.12 Hz), 8.01 (d, 0.23 H, J = 7.91 Hz), 7.90 (d, 0.3H, J = 7.56 Hz), 7.71 (d, 1.7H, J = 8.12 Hz), 7.13 (d, 0.3H, J = 8.33 Hz), 7.04 (d, 1.7H, J = 8.33 Hz), 3.88 (s, 0.8H), 3.82 (s, 2.2H), 3.30 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.1, 161.4, 158.3, 149.0, 144.9, 144.6, 143.7, 143.6, 138.5, 138.3, 129.3, 129.1, 129.0, 127.6, 127.0, 114.8, 56.1, 55.7, 43.75; C16H16N2O4S m/z: 332.3.
4.1.1.8. N'-(4-(Dimethylamino)benzylidene)-4-(methylsulfonyl)benzohydrazide (12)
M.p 295–297ο; 85% yield; IR (KBr, cm−1) ν: 3488 (NH), 1655 (C=O), 1278, 1148 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 11.81 (s, 1H), 8.34 (s, 1H), 8.14 (d, 2H, J = 7.77 Hz), 8.09 (d, 2H, J = 7.91 Hz), 7.57 (d, 2H, J = 8.12 Hz), 7.63 (d, 2H, J = 8.19 Hz), 3.30 (s, 3H), 2.97 (s, 5.3H), 2.92 (s, 0.7H); 13 C NMR (176 MHz, DMSO-d6): δ 161.8, 152.1, 150.0, 143.5, 138.7, 130.7, 129.1, 129.0, 127.6, 121.7, 112.2, 49.0, 43.7; C17H19N3O3S m/z: 345.1.
4.1.1.9. N'-(2-Chlorobenzylidene)-4-(methylsulfonyl)benzohydrazide (13)
M.p 346–348ο; 85% yield; IR (KBr, cm−1) ν: 3278 (NH), 1684 (C=O), 1271, 1142 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.31 (s, 1H), 8.89 (s, 1H), 8.19 (s, 1H), 8.18 (d, 2H, J = 8.12 Hz), 8.11 (d, 2H, J = 8.19 Hz), 8.05 (d, 2H, J = 7.14 Hz), 7.55 (d, 1H, J = 7.56 Hz), 7.47 (p, 2H, J = 16.31, 7.14 & 7.0 Hz), 3.31 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.4, 145.0, 143.9, 138.0, 133.8, 132.2, 131.8, 130.4, 129.2, 128.1, 127.6, 127.4, 43.7; m/z: C15H13ClN2O3S m/z 336.0, (M + 2; 338.0).
4.1.1.10. N'-(2-Fluorobenzylidene)-4-(methylsulfonyl)benzohydrazide (14)
M.p 300–302ο; 88% yield; IR (KBr, cm−1) ν: 3211 (NH), 1654 (C=O), 1280, 1144 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.21 (s, 1H), 8.72 (s, 1H), 8.18 (d, 2H, J = 8.12 Hz), 8.11 (d, 2H, J = 8.12 Hz), 7.98 (t, 1H, J = 7.31 Hz), 7.52 (dd, 1H, J = 6.72, 6.93 Hz), 7.33 (d, 2H, J = 7.98 Hz), 3.31 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.3, 162.0, 160.6, 143.9, 141.9, 141.8, 138.1, 132.8, 132.7, 129.1, 127.7, 126.8, 125.4, 122.1, 122.0, 116.5, 43.7; C15H13FN2O3S m/z: 320.2.
4.1.1.11. N'-(2-Methoxybenzylidene)-4-(methylsulfonyl)benzohydrazide (15)
M.p 333–335ο; 91% yield; IR (KBr, cm−1) ν: 3198 (NH), 1679 (C=O), 1276, 1145 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.08 (s, 1H), 8.84 (s, 1H), 8.18 (d, 2H, J = 7.56 Hz), 8.09 (d, 2H, J = 7.63 Hz), 7.90 (d, 1H, J = 7.70 Hz), 7.44 (t, 1H, J = 7.73 Hz), 7.13 (d, 1H, J = 8.33 Hz), 7.04 (t, 1H, J = 7.42 Hz), 3.82 (s, 3H), 3.31 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.1, 158.3, 144.6, 143.7, 138.3, 132.3, 129.1, 127.6, 126.0, 122.5, 121.2, 112.3, 56.1, 43.7; C16H16N2O4S m/z: 332.1.
4.1.1.12. N'-(2,4-Dichlorobenzylidene)-4-(methylsulfonyl)benzohydrazide (17)
M.p > 350ο; 93% yield; IR (KBr, cm−1) ν: 3227 (NH), 1677 (C=O), 1294, 1150 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.35 (s, 1H), 8.83 (s, 1H), 8.18 (d, 2H, J = 7.84 Hz), 8.11 (d, 2H, J = 7.77 Hz), 8.05 (d, 1H, J = 8.47 Hz), 7.75 (s, 1H), 7.55 (d, 1H, J = 8.47 Hz), 3.31 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.4, 143.9, 137.9, 135.8, 134.5, 130.9, 129.9, 129.2, 128.6, 127.7, 43.7; C15H12Cl2N2O3S: m/z 371.0, (M + 2; 373).
4.1.1.13. N'-(3,4-Dichlorobenzylidene)-4-(methylsulfonyl)benzohydrazide (18)
M.p 242–244ο; 87% yield; IR (KBr, cm−1) ν: 3282 (NH), 1688 (C=O), 1274, 1146 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.29 (s, 1H), 8.44 (s, 1H), 8.15 (d, 2H, J = 8.05 Hz), 8.11 (d, 2H, J = 8.05 Hz), 7.99 (s, 1H), 7.76 (dd, 2H, J = 8.12 & 7.84 Hz), 3.30 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.5, 146.4, 143.9, 138.0, 135.4, 132.9, 132.2, 131.6, 129.2, 129.1, 127.6, 127.4, 43.7; C15H12Cl2N2O3S: m/z 371.0, (M + 2; 373).
4.1.1.14. N'-(3,4-Dimethoxybenzylidene)-4-(methylsulfonyl)benzohydrazide (21)
M.p 245–247ο; 90% yield; IR (KBr, cm−1) ν: 3212 (NH), 1657 (C=O), 1269, 1140 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 11.97 (s, 1H), 8.39 (s, 1H), 8.14 (d, 2H, J = 8.19 Hz), 8.09 (d, 2H, J = 8.19 Hz), 7.37 (s, 1H), 7.24 (d, 1H, J = 8.05 Hz), 7.05 (d, 1H, J = 8.26 Hz), 3.83 (s, 3H), 3.82 (s, 3H), 3.30 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 172.5, 162.2, 151.4, 149.5, 149.3, 143.6, 138.5, 129.0, 127.6, 122.6, 111.9, 108.6, 56.0, 55.9, 43.7; C17H18N2O5S m/z: 362.3.
4.1.1.15. N'-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(methylsulfonyl)benzohydrazide (22)
M.p 289–291ο; 86% yield; IR (KBr, cm−1) ν: 3220 (NH), 1655 (C=O), 1275, 1145 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.00 (s, 1H), 8.38 (s, 1H), 8.14 (d, 2H, J = 8.19 Hz), 8.09 (d, 2H, J = 8.19 Hz), 7.33 (s, 1H), 7.20 (d, 1H, J = 7.91 Hz), 7.02 (d, 1H, J = 7.91 Hz), 6.11 (s, 2H), 3.30 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 162.2, 149.7, 148.9, 148.5, 143.7, 138.4, 129.0, 128.9, 127.6, 124.1, 108.9, 105.6, 102.1, 43.7; C16H14N2O5S m/z: 346.2.
4.1.1.16. 4-(Methylsulfonyl)-N'-(3,4,5-trimethoxybenzylidene)benzohydrazide (23)
M.p 228–230ο; 95% yield; IR (KBr, cm−1) ν: 3199 (NH), 1668 (C=O), 1272, 1132 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 12.09 (s, 1H), 8.40 (s, 1H), 8.14 (d, 2H, J = 8.19 Hz), 8.09 (d, 2H, J = 8.05 Hz), 7.06 (s, 2H), 3.85 (s, 6H), 3.72 (s, 3H), 3.30 (s, 3H), 13 C NMR (176 MHz, DMSO-d6): δ 162.4, 153.6, 149.1, 143.7, 139.8, 138.4, 130.0, 129.1, 127.6, 1104.8, 60.6, 56.4, 43.7; C18H20N2O6S m/z: 392.1.
4.1.1.17. (4-(Methylsulfonyl)-N'-(2,4,5-trimethoxybenzylidene)benzohydrazide (24)
M.p 277–279ο; 94% yield; IR (KBr, cm−1) ν: 3209 (NH), 1667 (C=O), 1275, 1140 (O = S=O); 1H NMR (700 MHz, DMSO-d6): δ 11.96 (s, 1H), 8.77 (s, 1H), 8.17 (d, 2H, J = 8.12 Hz), 8.09 (d, 2H, J = 8.12 Hz), 7.38 (s, 1H), 6.76 (s, 1H), 3.88 (s, 3H), 3.87 (s, 3H), 3.77 (s, 3H), 3.30 (s, 3H), 13C NMR (176 MHz, DMSO-d6): δ 161.8, 154.0, 152.7, 144.8, 143.7, 143.6, 138.4, 129.0, 127.5, 113.7, 107.9, 98.2, 56.9, 56.3, 56.2, 43.7; C18H20N2O6S m/z: 392.1.
4.2. Biological evaluation
4.2.1. In vitro antitumor assay
The antitumor assay was performed for 59 human tumour cell lines obtained from nine human tissue under the protocol of the Drug Evaluation Branch, National Cancer Institute, Bethesda, MD. Three dose-response parameters; GI50, TGI, and LC50; were calculated for each compound17,40,70.
4.2.2. Apoptosis assay
According to our previous report, apoptosis induction was performed using the Leukaemia HL-60 cell line and well-established Annexin 5-FITC/PI detection kit. The cell line samples were analysed using FACSCalibur flow cytometer40,72.
4.2.3. Cell cycle analysis
Cell cycle analysis was carried out similar to our previous report using the Leukaemia HL-60 cell line stained with the DNA fluorochrome PI and analysed by FACSCalibur flow cytometer40,74.
4.2.4. In vitro cyclooxygenase (COX) inhibition assay
The colorimetric COX-2 inhibition assay (kit catalogue number 560101, Cayman Chemical, Ann Arbour, MI) was used to measure the ability of the tested derivatives and celecoxib to inhibit COX-2 isozyme under the manufacturer’s instructions15,76 .
4.2.5. Egfr and HER2 tyrosine kinases assay
In vitro luminescent EGFR tyrosine kinase assay using Kinase-Glo® MAX as a detection reagent, and In vitro HER2 tyrosine kinase assay using DP‐Glo™ reagent that measures ADP formed from a kinase reaction, this luminescent signal positively correlates with ADP amount and kinase activity40.
4.3. Molecular docking and ADME methodology
Molecular docking protocols were carried out using the MOE 2008.10 software from Chemical Computing Group Inc. (Montreal, QC, Canada) following established methods40,81. The crystal structures of COX-2 (PDB code: 1CX2), EGFR (PDB Code: 1M17), and HER2 (PDB Code: 3PP0) were retrieved from the protein data bank. The Swiss Target Prediction and the Swiss ADME online tools were used to predict the physicochemical, pharmacokinetic, and drug-likeness properties of the test compounds and used reference drugs84.
Supplementary Material
Acknowledgements
The authors thank the Deanship of Scientific Research and RSSU at King Saud University for their technical support.
Funding Statement
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this project through the Research Group Project No. RGP-163.
Disclosure statement
The authors declare that they have no conflict of interest.
References
- 1.Avendaño C, Menendez JC.. Medicinal chemistry of anticancer drugs. Amsterdam, The Netherlands: Elsevier; 2015. [Google Scholar]
- 2.Vainshelboim B, Müller J, Lima RM, et al. Cardiorespiratory fitness, physical activity and cancer mortality in men. Prevent Med 2017;100:89–94. [DOI] [PubMed] [Google Scholar]
- 3.Varmus H. The new era in cancer research. Science 2006;312:1162–5. [DOI] [PubMed] [Google Scholar]
- 4.Li Q, Xu W.. Novel anticancer targets and drug discovery in post genomic age. Curr Med Chem 2005;5:53–63. [DOI] [PubMed] [Google Scholar]
- 5.El-Sherbeny MA, Abdel-Aziz AA, Ahmed MA.. Synthesis and antitumor evaluation of novel diarylsulfonylurea derivatives: molecular modeling applications. Euro J Med Chem 2010;45:689–97. [DOI] [PubMed] [Google Scholar]
- 6.El-Azab AS, Alanazi AM, Abdel-Aziz NI, et al. Synthesis, molecular modeling study, preliminary antibacterial, and antitumor evaluation of N-substituted naphthalimides and their structural analogues. Med Chem Res 2013;22:2360–75. [Google Scholar]
- 7.El-Deeb IM, Bayoumi SM, El-Sherbeny MA, Abdel-Aziz AA.. Synthesis and antitumor evaluation of novel cyclic arylsulfonylureas: ADME-T and pharmacophore prediction. Euro J Med Chem 2010;45:2516–30. [DOI] [PubMed] [Google Scholar]
- 8.Alanazi AM, Abdel-Aziz AA, Shawer TZ, et al. Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: in silico studies. J Enzyme Inhibition Med Chem 2016;31:721–35. [DOI] [PubMed] [Google Scholar]
- 9.Alanazi AM, Abdel-Aziz AA, Al-Suwaidan IA, et al. Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents. Euro J Med Chem 2014;79:446–54. [DOI] [PubMed] [Google Scholar]
- 10.El-Azab AS, Abdel-Aziz AA, Ghabbour HA, Al-Gendy MA.. Synthesis, in vitro antitumour activity, and molecular docking study of novel 2-substituted mercapto-3-(3,4,5-trimethoxybenzyl)-4(3H)-quinazolinone analogues. J Enzyme Inhib Med Chem 2017;32:1229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohamed MA, Ayyad RR, Shawer TZ, et al. Synthesis and antitumor evaluation of trimethoxyanilides based on 4(3H)-quinazolinone scaffolds. Euro J Med Chem 2016;112:106–13. [DOI] [PubMed] [Google Scholar]
- 12.El-Sayed MA, El-Husseiny WM, Abdel-Aziz NI, et al. Synthesis and biological evaluation of 2-styrylquinolines as antitumour agents and EGFR kinase inhibitors: molecular docking study. J Enzyme Inhib Med Chem 2018;33:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdel-Aziz AA, El-Azab AS, El-Subbagh HI, et al. Design, synthesis, single-crystal and preliminary antitumor activity of novel arenesulfonylimidazolidin-2-ones. Bioorg Med Chem Lett 2012;22:2008–14. [DOI] [PubMed] [Google Scholar]
- 14.Al-Suwaidan IA, Alanazi AM, Abdel-Aziz AA, et al. Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: molecular docking study. Bioorg Med Chem Lett 2013;23:3935–41. [DOI] [PubMed] [Google Scholar]
- 15.El-Husseiny WM, El-Sayed MA, Abdel-Aziz NI, et al. Structural alterations based on naproxen scaffold: Synthesis, evaluation of antitumor activity and COX-2 inhibition, and molecular docking. Euro J Med Chem 2018;158:134–43. [DOI] [PubMed] [Google Scholar]
- 16.Alanazi AM, Al-Suwaidan IA, Alaa A-M, et al. Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents. Med Chem Res 2013;22:5566–77. [Google Scholar]
- 17.Abdel-Aziz AA, El-Azab AS, Alanazi AM, et al. Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzyme Inhib Med Chem 2016;31:796–809. [DOI] [PubMed] [Google Scholar]
- 18.El-Husseiny WM, El-Sayed MA, Abdel-Aziz NI, et al. Synthesis, antitumour and antioxidant activities of novel α,β-unsaturated ketones and related heterocyclic analogues: EGFR inhibition and molecular modelling study. J Enzyme Inhib Med Chem 2018;33:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alanazi AM, El-Azab AS, Al-Swaidan IA, et al. Synthesis, single-crystal, in vitro antitumor evaluation and molecular docking of 3-substitued 5, 5-diphenylimidazolidine-2, 4-dione derivatives. Medicinal Chemistry Research 2013;22:6129–42. [Google Scholar]
- 20.Al-Suwaidan IA, Abdel-Aziz AA, Shawer TZ, et al. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4(3H)quinazolinone analogues. J Enzyme Inhib Med Chem 2016;31:78–89. [DOI] [PubMed] [Google Scholar]
- 21.El-Azab AS, Al-Dhfyan A, Abdel-Aziz AA, et al. Synthesis, anticancer and apoptosis-inducing activities of quinazoline-isatin conjugates: epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies. J Enzyme Inhib Med Chem 2017;32:935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stratikopoulos EE, Dendy M, Szabolcs M, et al. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015;27:837–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayat Mokhtari R, Homayouni TS, Baluch N, et al. Combination therapy in combating cancer. Oncotarget 2017;8:38022–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szakács G, Paterson JK, Ludwig JA, et al. Targeting multidrug resistance in cancer. Nature Reviews. Drug Discovery 2006;5:219–34. [DOI] [PubMed] [Google Scholar]
- 25.Fu RG, Sun Y, Sheng WB, Liao DF.. Designing multi-targeted agents: An emerging anticancer drug discovery paradigm. Euro J Med Chem 2017;136:195–211. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Dewan V, Yin H.. Discovery of small molecules as multi-toll-like receptor agonists with proinflammatory and anticancer activities. J Med Chemistry 2017;60:5029–44. [DOI] [PubMed] [Google Scholar]
- 27.Alkahtani HM, Abdalla AN, Obaidullah AJ, et al. Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: Dual inhibitors of EGFR/HER2. Bioorg Chem 2020;95:103461. [DOI] [PubMed] [Google Scholar]
- 28.Daydé-Cazals B, Fauvel B, Singer M, et al. Rational design, synthesis, and biological evaluation of 7-azaindole derivatives as potent focused multi-targeted kinase inhibitors. J Med Chem 2016;59:3886–905. [DOI] [PubMed] [Google Scholar]
- 29.El-Azab AS, Abdel-Aziz AA, Abou-Zeid LA, et al. Synthesis, antitumour activities and molecular docking of thiocarboxylic acid ester-based NSAID scaffolds: COX-2 inhibition and mechanistic studies. J Enzyme Inhib Med Chem 2018;33:989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdel-Aziz AA, Angeli A, El-Azab AS, et al. Synthesis and anti-inflammatory activity of sulfonamides and carboxylates incorporating trimellitimides: Dual cyclooxygenase/carbonic anhydrase inhibitory actions. Bioorg Chem 2019;84:260–8. [DOI] [PubMed] [Google Scholar]
- 31.Kolibaba KS, Druker BJ.. Protein tyrosine kinases and cancer. Biochim Biophys Acta 1997;1333:F217–48. [DOI] [PubMed] [Google Scholar]
- 32.Antonello A, Tarozzi A, Morroni F, et al. Multitarget-directed drug design strategy: a novel molecule designed to block epidermal growth factor receptor (EGFR) and to exert proapoptotic effects. J Med Chem 2006;49:6642–5. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa T, Seto M, Banno H, et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J Med Chem 2011;54:8030–50. [DOI] [PubMed] [Google Scholar]
- 34.Salomon DS, Brandt R, Ciardiello F, Normanno N.. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995;19:183–232. [DOI] [PubMed] [Google Scholar]
- 35.Black JD, Brattain MG, Krishnamurthi SA, et al. ErbB family targeting. Curr Opin Invest Drugs 2003;4:1451–4. [PubMed] [Google Scholar]
- 36.Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br Med Bull 1991;47:87–98. [DOI] [PubMed] [Google Scholar]
- 37.Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Therapeutics 1999;82:241–50. [DOI] [PubMed] [Google Scholar]
- 38.Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Therapeutics 2001;1:85–94. [PubMed] [Google Scholar]
- 39.Xu G, Abad MC, Connolly PJ, et al. 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors. Bioorganic Med Chem Lett 2008;18:4615–9. [DOI] [PubMed] [Google Scholar]
- 40.El-Azab AS, Abdel-Aziz AA, AlSaif NA, et al. Antitumor activity, multitarget mechanisms, and molecular docking studies of quinazoline derivatives based on a benzenesulfonamide scaffold: Cell cycle analysis. Bioorg Chem 2020;104:104345. [DOI] [PubMed] [Google Scholar]
- 41.Alkahtani HM, Alanazi MM, Aleanizy FS, et al. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: Molecular docking studies. Saudi Pharmaceutical J 2019;27:682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gundla R, Kazemi R, Sanam R, et al. Discovery of novel small-molecule inhibitors of human epidermal growth factor receptor-2: combined ligand and target-based approach. J Med Chem 2008;51:3367–77. [DOI] [PubMed] [Google Scholar]
- 43.Danchev N, Nikolova I, Momekov G. A new era in anticancer therapy/imatinib—a new era in anticancer therapy. Biotechnol Biotechnol Equip. 2008;22(3):769–70. [Google Scholar]
- 44.Iqbal N, Iqbal N.. Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Prac. 2014;2014:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madhusudan S, Ganesan TS.. Tyrosine kinase inhibitors in cancer therapy. Clin Biochem. 2004;37:618–35. [DOI] [PubMed] [Google Scholar]
- 46.Barker AJ, Gibson KH, Grundy W, et al. Studies leading to the identification of ZD1839 (Iressa™): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001;11:1911–4. [DOI] [PubMed] [Google Scholar]
- 47.Frampton JE. Lapatinib: a review of its use in the treatment of HER2-overexpressing, trastuzumab-refractory, advanced or metastatic breast cancer. Drugs 2009;69:2125–48. [DOI] [PubMed] [Google Scholar]
- 48.Takimoto CH, Awada A.. Safety and anti-tumor activity of sorafenib (Nexavar) in combination with other anti-cancer agents: a review of clinical trials. Cancer Chemother Pharmacol. 2008;61:535–48. [DOI] [PubMed] [Google Scholar]
- 49.Dungo RT, Keating GM.. Afatinib: first global approval. Drugs 2013;73:1503–15. [DOI] [PubMed] [Google Scholar]
- 50.Christensen JG. A preclinical review of sunitinib, a multitargeted receptor tyrosine kinase inhibitor with anti-angiogenic and antitumour activities. Ann Oncol 2007;18: x3–10. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh N, Chaki R, Mandal V, Mandal SC.. COX-2 as a target for cancer chemotherapy. Pharmacol Rep PR 2010;62:233–44. [DOI] [PubMed] [Google Scholar]
- 52.Blanke C. Role of COX-2 inhibitors in cancer therapy. Cancer Invest 2004;22:271–82. [DOI] [PubMed] [Google Scholar]
- 53.Dai ZJ, Ma XB, Kang HF, et al. Antitumor activity of the selective cyclooxygenase-2 inhibitor, celecoxib, on breast cancer in Vitro and in Vivo. Cancer Cell Inter 2012;12:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vosooghi M, Amini M.. The discovery and development of cyclooxygenase-2 inhibitors as potential anticancer therapies. Exp Opin Drug Disc 2014;9:255–67. [DOI] [PubMed] [Google Scholar]
- 55.Elder DJ, Halton DE, Hague A, Paraskeva C.. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res 1997;3:1679–83. [PubMed] [Google Scholar]
- 56.Hodnett EM, Dunn WJ. 3rd., Structure-antitumor activity correlation of some Schiff bases. J Med Chem 1970;13:768–70. [DOI] [PubMed] [Google Scholar]
- 57.Petrović ZD, Đorović J, Simijonović D, et al. Experimental and theoretical study of antioxidative properties of some salicylaldehyde and vanillic Schiff bases. RSC Advances 2015;5:24094–100. [Google Scholar]
- 58.el-Ayaan U, Abdel-Aziz AA.. Synthesis, antimicrobial activity and molecular modeling of cobalt and nickel complexes containing the bulky ligand: bis[N-(2,6-diisopropylphenyl)imino] acenaphthene. Euro J Med Chem 2005;40:1214–21. [DOI] [PubMed] [Google Scholar]
- 59.Zineddine Z, Suat T, Hasan K, et al. Synthesis and anticancer properties of novel hydrazone derivatives incorporating pyridine and isatin moieties. Arch Pharm 2020;354:e2000377. [DOI] [PubMed] [Google Scholar]
- 60.Vogel S, Kaufmann D, Pojarová M, et al. Aroyl hydrazones of 2-phenylindole-3-carbaldehydes as novel antimitotic agents. Bioorg Med Chem 2008;16:6436–47. [DOI] [PubMed] [Google Scholar]
- 61.Rollas S, Küçükgüzel SG.. Biological activities of hydrazone derivatives. Molecules 2007;12:1910–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang Z, Zhang D, Ai J, et al. Identification and synthesis of N'-(2-oxoindolin-3-ylidene)hydrazide derivatives against c-Met kinase. Bioorg Med Chem Lett 2011;21:3749–54. [DOI] [PubMed] [Google Scholar]
- 63.Peterson QP, Hsu DC, Goode DR, et al. Procaspase-3 activation as an anti-cancer strategy: Structure − activity relationship of procaspase-activating compound 1 (PAC-1) and Its cellular co-localization with caspase-3. J Med Chem 2009;52:5721–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abdel-Aziz AA, El-Azab AS, Abu El-Enin MA, et al. Synthesis of novel isoindoline-1,3-dione-based oximes and benzenesulfonamide hydrazones as selective inhibitors of the tumor-associated carbonic anhydrase IX. Bioorg Chem 2018;80:706–13. [DOI] [PubMed] [Google Scholar]
- 65.El-Azab AS, Abdel-Aziz AA, Bua S, et al. Synthesis and comparative carbonic anhydrase inhibition of new Schiff's bases incorporating benzenesulfonamide, methanesulfonamide, and methylsulfonylbenzene scaffolds. Bioorg Chem 2019;92:103225. [DOI] [PubMed] [Google Scholar]
- 66.Şenkardeş S, Han M, Kulabaş N, et al. Synthesis, molecular docking and evaluation of novel sulfonyl hydrazones as anticancer agents and COX-2 inhibitors. Mol Diversity 2020;24:673–89. [DOI] [PubMed] [Google Scholar]
- 67.Bánvölgyi A, Anker P, Lőrincz K, et al. Smoothened receptor inhibitor vismodegib for the treatment of basal cell carcinoma: a retrospective analysis of efficacy and side effects. J Dermatol Treat 2020;31:387–98. [DOI] [PubMed] [Google Scholar]
- 68.Gould SE, Low JA, Marsters JC, Jr, et al. Discovery and preclinical development of vismodegib. Exp Opin Drug Disc 2014;9:969–84. [DOI] [PubMed] [Google Scholar]
- 69.George RF. Facile synthesis of simple 2-oxindole-based compounds with promising antiproliferative activity. Fut Med Chem 2018;10:269–82. [DOI] [PubMed] [Google Scholar]
- 70.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006;6:813–23. [DOI] [PubMed] [Google Scholar]
- 71.Pfeffer CM, Singh AT.. Apoptosis: a target for anticancer therapy. Inter J Mol Sci 2018;19:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C.. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods 1995;184:39–51. [DOI] [PubMed] [Google Scholar]
- 73.Ormerod MG. Investigating the relationship between the cell cycle and apoptosis using flow cytometry. J Immunol Methods 2002;265:73–80. [DOI] [PubMed] [Google Scholar]
- 74.Kim KH, Sederstrom JM.. Assaying cell cycle status using flow cytometry. Curr Protoc Mol Biol 2015;111:28.6. 1–.6. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gurpinar E, Grizzle WE, Piazza GA.. COX-Independent mechanisms of cancer chemoprevention by anti-inflammatory drugs. Front Oncol 2013;3:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abdel-Aziz AA, El-Azab AS, Abou-Zeid LA, et al. Synthesis, anti-inflammatory, analgesic and COX-1/2 inhibition activities of anilides based on 5,5-diphenylimidazolidine-2,4-dione scaffold: Molecular docking studies. Euro J Med Chem 2016;115:121–31. [DOI] [PubMed] [Google Scholar]
- 77.Abdel-Aziz AA, Asiri YA, Al-Agamy MH.. Design, synthesis and antibacterial activity of fluoroquinolones containing bulky arenesulfonyl fragment: 2D-QSAR and docking study. Euro J Med Chem 2011;46:5487–97. [DOI] [PubMed] [Google Scholar]
- 78.Goda FE, Abdel-Aziz AA, Ghoneim HA.. Synthesis and biological evaluation of novel 6-nitro-5-substituted aminoquinolines as local anesthetic and anti-arrhythmic agents: molecular modeling study. Bioorg Med Chem 2005;13:3175–83. [DOI] [PubMed] [Google Scholar]
- 79.El-Azab AS, Mary YS, Panicker CY, et al. DFT and experimental (FT-IR and FT-Raman) investigation of vibrational spectroscopy and molecular docking studies of 2-(4-oxo-3-phenethyl-3, 4-dihydroquinazolin-2-ylthio)-N-(3, 4, 5-trimethoxyphenyl) acetamide. J Mol Struct 2016;1113:133–45. [Google Scholar]
- 80.El-Gamal MI, Bayomi SM, El-Ashry SM, et al. Synthesis and anti-inflammatory activity of novel (substituted)benzylidene acetone oxime ether derivatives: molecular modeling study. Euro J Med Chem 2010;45:1403–14. [DOI] [PubMed] [Google Scholar]
- 81.MOE 2008.10 of Chemical Computing Group. Inc. Available from : http://www.chemcomp.com/press_releases/2008-11-04.htm.
- 82.Abdel-Aziz AA, Abou-Zeid LA, ElTahir KE, et al. Design, synthesis of 2,3-disubstitued 4(3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg Med Chem 2016;24:3818–28. [DOI] [PubMed] [Google Scholar]
- 83.Abdel-Sayed MA, Bayomi SM, El-Sherbeny MA, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibition activities and molecular docking study of pyrazoline derivatives. Bioorg Med Chem 2016;24:2032–42. [DOI] [PubMed] [Google Scholar]
- 84.Swiss Institute of Bioinformatics. http://www.swissadme.ch/index.php.
- 85.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ.. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Del Rev 2001;46:3–26. [DOI] [PubMed] [Google Scholar]
- 86.Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45:2615–23. [DOI] [PubMed] [Google Scholar]
- 87.Egan WJ, Merz KM, Jr., Baldwin JJ.. Prediction of drug absorption using multivariate statistics. J Med Chem 2000;43:3867–77. [DOI] [PubMed] [Google Scholar]
- 88.Daina A, Zoete V.. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. Chem Med Chem 2016;11:1117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Daina A, Michielin O, Zoete V.. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Szakács G, Váradi A, Ozvegy-Laczka C, Sarkadi B.. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Disc Today 2008;13:379–93. [DOI] [PubMed] [Google Scholar]
- 91.Daina A, Michielin O, Zoete V.. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inform Model 2014;54:3284–301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.