

Abstract
While the surface charge state of co-catalysts plays a critical role for boosting photocatalysis, studies on surface charge regulation via their precise structure control remain extremely rare. Herein, metal-organic framework (MOF) stabilized bimetallic Pd@Pt nanoparticles, which feature adjustable Pt coordination environment and a controlled structure from core-shell to single-atom alloy (SAA), have been fabricated. Significantly, apart from the formation of a Mott-Schottky junction in a conventional way, we elucidate that Pt surface charge regulation can be alternatively achieved by changing its coordination environment and the structure of the Pd@Pt co-catalyst, where the charge between Pd and Pt is redistributed. As a result, the optimized Pd10@Pt1/MOF composite, which involves an unprecedented SAA co-catalyst, exhibits exceptionally high photocatalytic hydrogen production activity, far surpassing its corresponding counterparts.
Keywords: metal-organic framework (MOF), single-atom alloy (SAA), co-catalyst, surface charge state, photocatalysis
The MOF-stabilized Pd10@Pt1 single-atom alloy was fabricated, featuring optimized Pt surface charge state and greatly boosted photocatalysis, thanks to the charge redistribution regulated by Pt coordination environment and Pd@Pt microstructure.
INTRODUCTION
Photocatalysis, converting solar to chemical energy, represents a very promising solution to current energy and environmental issues [1–4]. Photocatalytic performance depends largely on the surface charge state of a catalyst, as it directly correlates with charge transfer from the catalyst surface to reactant molecules as well as adsorption and activation of the latter [5–7]. Various strategies have been developed to control the surface charge states of photocatalytic materials [6,7]. Amongst them, the construction of a Schottky junction by introducing co-catalyst is one of the most common and effective strategies via increasing the active sites, promoting charge separation, accelerating charge transfer and minimizing reaction over-potentials [8–12]. Recent studies on co-catalysts are mostly related to the development of non-noble metal co-catalysts, the control of particle sizes, particle distribution, exposed crystal facets as well as their interface contact with photocatalysts, and even synergistic effect among various co-catalysts [6,8–12]. Nonetheless, specialized regulation on the surface charge state of co-catalysts, especially by changing their microstructure, remains largely unexplored, though this would be highly desired to boost the photocatalysis [6,10,12].
To achieve surface charge regulation, the introduction of bimetallic nanoparticles (NPs) as co-catalysts, in which one metal behaves as the active site and the other as charge regulator, would be a promising solution. Of the common alloyed or core-shell bimetallic NPs, the core-shell architecture featuring active metal shell would be preferred, for ease of access to active sites. The gradually decreased content of active metal would cause its varying coordination environment and surface charge. Particularly, atomically dispersed active metal atoms will be formed when their content goes down to a certain amount. The microstructure of active metal in its atomically dispersed form on the other metal support is called single-atom alloy (SAA) [13–17]. Compared with other single-atom catalysts (SACs) [18,19], SAA catalysts present geometric/strain, electronic and synergistic effects originating from bimetallic components [13–17,20,21], causing the d-band center change of the surface active metals and thereby affecting their catalytic performance [13–17]. Surprisingly, the related exploration on SAA catalysts toward their photocatalysis has not yet been reported. In this context, it is imperative to investigate the structural evolution of bimetallic NPs from core-shell to SAA, which might be able to systematically regulate the charge state of surface active metal atoms, for boosting photocatalysis.
Meanwhile, the ideal photosensitizers, which not only have intimate contact with but are also able to stabilize SAA co-catalysts, are highly desired. In this regard, metal-organic frameworks (MOFs) [22–27], a class of crystalline porous materials featuring well-defined and tailored structures, high surface area, as well as semiconductor-like behavior [28–31], would be ideal candidates. Given the well-tuned conjugation of linkers, MOFs are able to harvest solar light in a broad spectral range and have shown their great potential as photosensitizers and photocatalysts [32–45]. Various co-catalysts, particularly the most classical Pt NPs with low over-potential, have been introduced and well stabilized by MOFs, greatly accelerating the charge separation and photocatalysis, mainly via the formation of a Pt-MOF ‘Schottky junction’ [9–11,46–49]. Therefore, MOFs would be very promising in stabilizing single atomic Pt-based bimetallic NPs for photocatalysis.
Bearing the above in mind, bimetallic core-shell Pd@Pt NPs have been in situ fabricated and stabilized by a representative robust MOF with good light harvesting capability, UiO-66-NH2, to afford Pd10@Ptx/UiO-66-NH2 composite (the 10 and x mean the mass permillage, at wt‰, of Pd and Pt to the MOF in synthesis, respectively). Remarkably, the Pt component on the Pd surface can be precisely controlled from the shell-form to single-atom dispersion. Along with structural evolution of Pd@Pt NPs from core-shell to SAA, the Pt coordination environment changes and the regulation of its surface charge state is accordingly achieved. The obtained Pd10@Ptx/UiO-66-NH2 exhibits excellent photocatalytic activity toward hydrogen production by water splitting (Fig. 1). Particularly, thanks to the charge redistribution, the optimized Pd10@Pt1/UiO-66-NH2 with SAA structure possesses superb activity, far surpassing the monometallic NPs and their physical mixture stabilized by the MOF. Though both size and location of Pt NPs have been reported to be crucial for charge separation efficiency and activity [9,11], to the best of our knowledge, there has not yet been explored on regulating the Pt coordination environment and its surface charge state by controlling co-catalyst structure in the photocatalytic system. Moreover, this is the first work investigating the SAA catalyst for photocatalysis.
Figure 1.

Schematic illustration. Illustration showing the synthetic strategy for Pd10@Ptx/UiO-66-NH2 toward photocatalytic hydrogen production, highlighting the much higher activity of Pd10@Pt1 SAA than Pd10@Pt10 core-shell NPs.
RESULTS
The fabrication and characterizations of Pd10@Ptx/UiO-66-NH2
The Pd precursor was impregnated to UiO-66-NH2 and subsequently reduced by ammonia borane (NH3BH3) to give Pd10/UiO-66-NH2. Upon the exhaustion of NH3BH3, different amounts of Pt precursor are added into the synthetic system. The surface Pd-H species, generated during NH3BH3 hydrolysis, would serve as both nucleation seed and reducing agent for the Pt precursor, generating core-shell-structured Pd10@Ptx (x = 0.3, 1, 5, 10) NPs stabilized by UiO-66-NH2 (Fig. 1) [50–55].
Powder X-ray diffraction (PXRD) patterns indicate that the high crystallinity of UiO-66-NH2 is well maintained during the introduction of Pd@Pt NPs (Supplementary Fig. 1). The actual contents of Pd and Pt are determined by the inductively coupled plasma atomic emission spectrometer (ICP-AES) (Supplementary Table 1). All Pd10@Ptx/UiO-66-NH2 have similar Pd loadings, while the Pt steadily increases as more Pt precursors are introduced. Transmission electron microscopy (TEM) observation shows that Pd NPs in Pd10/UiO-66-NH2 are well dispersed with average sizes of 4.01 nm (Supplementary Fig. 2). Upon Pt deposition, the Pd@Pt sizes slightly increase with higher Pt contents (Fig. 2a and b, Supplementary Figs 3–5 and Supplementary Table 2), in good agreement with the expectation of the epitaxial Pt shell growth on Pd NPs. Meanwhile, scanning electron microscope (SEM) images of Pd10@Ptx/UiO-66-NH2 indicate the maintained morphology of UiO-66-NH2 after introducing Pd@Pt NPs (Supplementary Figs 6 and 7).
Figure 2.
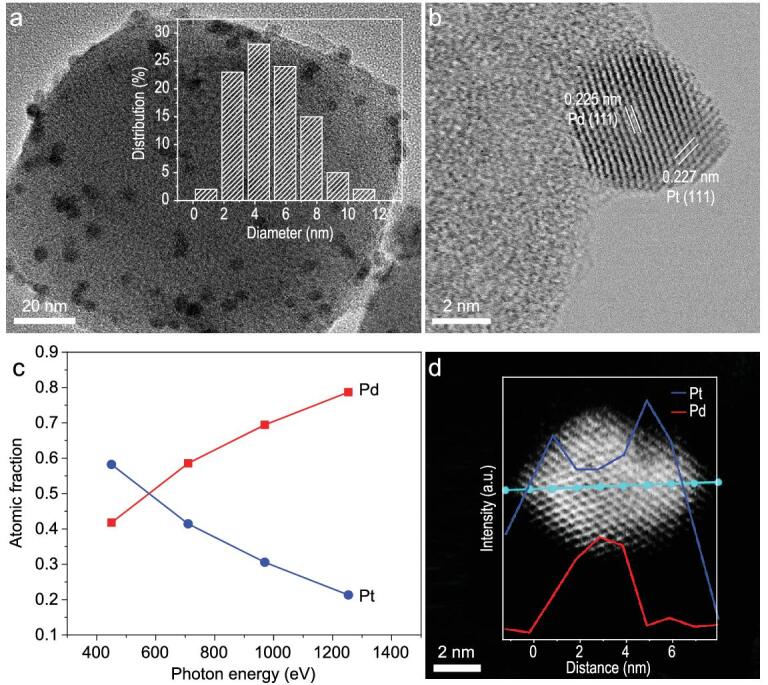
Structural characterizations of Pd10@Pt10 core-shell NPs. (a) TEM (inset: size distribution for Pd10@Pt10 NPs) and (b) HRTEM images of Pd10@Pt10/UiO-66-NH2. (c) Dependence of Pd and Pt atomic fractions for the Pd10@Pt10/UiO-66-NH2 (treated with H3PO4) as a function of photon energy. (d) Elemental line-scanning spectra across a single Pd10@Pt10 particle (stabilized by UiO-66-NH2) along the direction marked by a cyan line.
To verify whether the Pd@Pt core-shell structure has been successfully constructed, multiple characterizations have been adopted by taking Pd10@Pt10/UiO-66-NH2 as a representative. A high-resolution TEM (HRTEM) image reveals the Pd core is covered by a Pt shell, as evidenced by their exposed {111} facets with clear lattice fringes (Fig. 2b). The spatial distribution of the constituent elements of Pd@Pt NPs is detected by synchrotron radiation photoemission spectroscopy (SRPES) spectra for Pd 3d and Pt 4f, at photon energies of 450, 710, 970 and 1253.6 eV, respectively (Supplementary Figs 8–10) [56,57]. The peaks at 340.3 and 335.0 eV can be assigned to Pd 3d3/2 and Pd 3d5/2 of metallic Pd0, respectively [56], whereas the peaks at 74.3 and 70.9 eV clearly correspond to Pt0 (Supplementary Fig. 10) [58,59]. The outside-in atomic fractions of Pd and Pt in Pd@Pt NPs can be obtained from their peak areas calibrated by the beam flux and photoionization cross-section at the corresponding photon energy. As a direct result, a decreased atomic fraction of Pt and an increased atomic fraction of Pd unambiguously suggest that Pt stays on the external surface of Pd, pointing to a Pd10@Pt10 core-shell structure (Fig. 2c). More intuitively, the core-shell structure can also be clearly identified by elemental line-scanning spectra for a single Pd10@Pt10 particle (Fig. 2d and Supplementary Fig. 11).
Furthermore, the thickness of the Pt shell can be precisely controlled by varying the ratio of Pd/Pt precursors. As the Pt content decreases to give Pd10@Pt1/UiO-66-NH2, strikingly, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image indicates that Pt is atomically dispersed on the Pd surface (Fig. 3a). Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) and X-ray absorption near-edge structure (XANES) analyses provide more detailed structural information on Pd@Pt NPs as well as the Pt coordination environment (Fig. 3b, Supplementary Fig. 12 and Supplementary Tables 3 and 4). The absence of Pt–Pt peak in the Pt L3-edge FT-EXAFS spectrum of Pd10@Pt1/UiO-66-NH2 further demonstrates that the Pt, in its single-atom form, locates on the Pd surface (Fig. 3b) [58,59]. The weak peak at 1.5 Å is ascribed to the Pt-O bonding, which is possibly caused by partial surface oxidation. The slightly oxidized Pt also gives rise to a minor increase in white line intensity in the Pt L3-edge XANES spectrum for Pd10@Pt1/UiO-66-NH2 (Supplementary Fig. 13) [58,59]. All the HAADF-STEM, FT-EXAFS and XANES results unambiguously confirm that the SAA structure is formed in Pd10@Pt1/UiO-66-NH2. In sharp contrast to Pd10@Pt1/UiO-66-NH2, the Pd10@Pt10/UiO-66-NH2 apparently shows the presence of a Pt-Pd/Pt coordination bond. Moreover, the Pt-Pd coordination number in Pd10@Pt10/UiO-66-NH2 is much lower than that in Pd10@Pt1/UiO-66-NH2 (Supplementary Tables 3 and 4), indicating that Pt possibly presents a cumulative atomic layer-like distribution on the Pd surface in the former sample. Therefore, the Pd@Pt NPs are successfully stabilized by UiO-66-NH2, and the coordination environment of Pt is adjustable along with precise regulation of Pt shells to single Pt atoms on the surface of Pd NPs.
Figure 3.

Structural characterizations of Pd10@Pt1 SAA. (a) HAADF-STEM image of Pd10@Pt1/UiO-66-NH2. The single Pt atoms are highlighted by the red circles and the lattice fringes are from Pd while the MOF is shaded in black background. (b) The Pt L3-edge FT-EXAFS spectra for Pd10@Pt1/UiO-66-NH2 and Pd10@Pt10/UiO-66-NH2.
Charge redistribution effect
Given the different working functions of Pd and Pt (Supplementary Fig. 14), to balance the Fermi distribution of electrons in Pd@Pt NPs, electrons tend to leave Pd (the lower work function and electrons being less tightly bound) and travel to Pt (the higher work function) at their interface, causing an altered surface charge state of Pt. It is assumed that, as the structure of Pd@Pt NPs evolves from core-shell to SAA, charge redistribution (charge density change between the core and the surface metals) would take place and this affects the Pt surface charge state [12,60]. The work function calculations for Pd@Pt with SAA and core-shell structure support the above point (Supplementary Fig. 14). To further verify this hypothesis, X-ray photoelectron spectroscopy (XPS) measurements have been conducted (Fig. 4). Compared to the Pt0 4f7/2 peak at 70.9 eV for Pt1/UiO-66-NH2, Pd10@Pt1/UiO-66-NH2 presents lower binding energy to varying degrees, reflecting that the Pt surface in Pd@Pt NPs becomes electron-rich and charge redistribution effect between Pd and Pt exists. Moreover, when less Ptx (x value from 10 to 1) is covered, the Pt coordination number increases via surrounding Pd-Pt bonds (Supplementary Tables 2–4) and the Pt binding energy decreases (Fig. 4), further manifesting gradually enhanced charge redistribution effect and the most electron-rich Pt in Pd10@Pt1 NPs. In fact, from the aforementioned SRPES spectra of Pt 4f (Supplementary Fig. 10), when the probe depth gets close to the Pd core as photon energy increases, the Pt binding energy appears as a negative shift, which further supports the above conclusion.
Figure 4.

XPS results. The Pt 4f XPS spectra for Pt1/UiO-66-NH2, Pd10@Pt1/UiO-66-NH2 and Pd10@Pt10/UiO-66-NH2 (the purple solid and dashed lines represent slightly oxidized Pt species).
In addition, the differential charge density of Pd@Pt NPs (from core-shell structure with different Pt thicknesses to SAA) based on density-functional theory (DFT) calculations affords consistent results, manifesting the enhanced charge accumulation on the Pt surface along with the decrease of Pt layer numbers (i.e. from core-shell to SAA structure, finally) (Fig. 5, red color). From the differential charge density, the Bader charge of each Pt atom and the binding energy shift of Pt 4f7/2 show similar slopes along with the increased Pt layer thickness, indicating the consistent charge redistribution trend in simulation and experiment (Supplementary Fig. 15, Supplementary Tables 5 and 6).
Figure 5.

Charge redistribution indicated by differential charge density. Differential charge density of Pd@Pt NPs with SAA and core-shell structure featuring different Pt layer thicknesses. The red and blue colors represent the increase and decrease in electron density, respectively.
Photocatalysis and photoelectrochemical measurements
Encouraged by the electron-rich Pt surface in Pd10@Ptx/UiO-66-NH2, we set out to investigate the influence of charge redistribution for photocatalytic H2 production (Fig. 6a). Prior to the measurement, UV-Vis absorption spectra of Pd10@Ptx/UiO-66-NH2 have been examined to demonstrate their similar light absorption (Supplementary Fig. 16). As expected, as the Pt loading in Pd@Pt NPs decreases (x from 10 to 1), the Pt-Pd coordination number increases, charge redistribution effect strengthens and Pt becomes more electron-rich. Accordingly, the activity of Pd10@Ptx/UiO-66-NH2 exhibits gradual increase. Strikingly, Pd10@Pt1/UiO-66-NH2 featuring SAA structure achieves the highest photocatalytic H2 production rate, 1200.5 μmol · g−1 · h−1, among all investigated samples, which is 25, 30 and ∼4.5 times higher than that of Pd10/UiO-66-NH2, Pt1/UiO-66-NH2, Pd10 + Pt1/UiO-66-NH2 with a mixture of monometallic NPs, and Pd10Pt1/UiO-66-NH2, respectively (Fig. 6a and Supplementary Figs 17–20). When the Pt content continues to decline (x from 1 to 0.3), unexpectedly, the photocatalytic hydrogen production activity slightly decreases, possibly due to the decreased Pt sites in the catalyst.
Figure 6.

Photocatalytic results and photoelectrochemical measurements. (a) Photocatalytic hydrogen production rate of: A, Pt1/UiO-66-NH2; B, Pd10/UiO-66-NH2; C, Pd10 + Pt1/UiO-66-NH2; D, Pd10Pt1/UiO-66-NH2 and E–H, Pd10@Ptx/UiO-66-NH2 (x = E, 0.3; F, 1; G, 5; H, 10). (b) Recycling performance of Pd10@Pt1/UiO-66-NH2. (c) Photoluminescence spectra under excitation at λ = 360 nm. (d) Photocurrent response.
The calculated Gibbs free energy (ΔGH*) of Pt and Pd atoms in the Pd10@Pt1 SAA structure indicates that Pt is the more favorable active site than Pd for hydrogen production (Supplementary Fig. 21). Together with the overwhelmingly higher activity of Pd10@Pt1/UiO-66-NH2 than Pd10/UiO-66-NH2, the H2 generation taking place on Pd should be negligible and turnover frequencies of Pt (TOFPt) are a reasonable descriptor for the activity for the catalysts in SAA structure. The significantly higher TOFPt for Pd10@Pt0.3/UiO-66-NH2 and Pd10@Pt1/UiO-66-NH2 than all other counterparts further indicates the superiority of SAA structure in photocatalysis (Supplementary Table 7).
In addition, this optimized Pd10@Pt1/UiO-66-NH2 catalyst shows good recyclability and its activity does not present obvious decay in five consecutive runs and a continued test for 25 h (Fig. 6b, Supplementary Fig. 25). Thanks to the good stability, PXRD patterns confirm that the structural integrity and crystallinity of the MOF in all samples are well retained after catalysis (Supplementary Fig. 22). Especially, both the SAA structure and the dispersion of Pd10@Pt1 NPs are almost maintained, thanks to the excellent stabilization effect by the MOF (Supplementary Figs 23 and 24).
The activity promotion by surface charge regulation based on Pd10@Ptx/UiO-66-NH2 for photocatalysis has also been demonstrated by enhanced charge transfer. Photoluminescence (PL) emission spectra for all samples show that Pd10@Pt1/UiO-66-NH2 has the weakest fluorescence intensity, indicating its best charge separation efficiency, and the peak intensity order is in perfect reverse correlation with that of photocatalytic activity (Fig. 6c). The photocurrent and electrochemical impedance spectroscopy (EIS) measurements also manifest such a trend (Fig. 6d and Supplementary Fig. 26), further confirming the discriminative charge separation efficiency and being in good agreement with the results by XPS, SRPES spectra and DFT calculations, and well explaining the activity trend. Moreover, the cathodic polarization curves indicate Pd10@Pt1/UiO-66-NH2 has lower over-potential for hydrogen evolution (Supplementary Fig. 27). Therefore, the regulation of Pt surface charge state by charge redistribution at the interface of Pd and Pt plays an important role in optimizing photocatalytic performance. Base on the results above and previous reports [9,48,49], the photocatalytic process of Pd@Pt/UiO-66-NH2 is clear that the MOF is excited by visible light to produce photogenerated electron-hole pairs. Due to the Schottky junction, the electrons transfer to Pd, and finally to Pt (catalytic center), promoted by the charge redistribution effect between Pd and Pt (Fig. 1). The charge redistribution effect is influenced by the Pt coordination environment and microstructure, which causes distinct charge accumulation on the Pt surface in Pd10@Ptx/UiO-66-NH2, resulting in different H2 production activity. The results unambiguously manifest that, in addition to the well-established Schottky junction, the microstructure regulation of a co-catalyst reported herein might be another effective and novel strategy to enhance charge separation and thus photocatalysis.
DISCUSSION
In summary, a bimetallic NPs/MOF system, Pd@Pt/UiO-66-NH2, has been rationally fabricated to investigate the structural influence of a Pd@Pt co-catalyst on its surface charge state and resulting photocatalytic activity. Remarkably, the Pt shell on Pd surface can be precisely controlled and the structure evolves from core-shell to SAA, so as to change the Pt coordination environment. Consequently, the surface charge on Pd and Pt is redistributed, which regulates the electronic state of Pt active sites and is found to be crucial for the resulting activity. As a result, the photocatalytic hydrogen production activity of Pd10@Ptx/UiO-66-NH2 can be optimized to exponentially increase by precisely fabricating the Pd10@Pt1 SAA co-catalyst, based on the rational control of Pt loading. This work not only presents SAA behaving as a very promising co-catalyst for photocatalysis for the first time, but also opens a novel avenue to the surface charge regulation of co-catalysts for boosting photocatalysis.
METHODS
Preparation of Pd10@Ptx/UiO-66-NH2
Typically, 100 mg UiO-66-NH2 was put in a porcelain crucible and a certain amount of 50 mg/mL Pd(NO3)2 aqueous solution (Pd/UiO-66-NH2 = 1 wt%) was added. Then, 200 μL MeOH was added into the mixture and rapidly stirred with a glass rod and heated at 80°C to evaporate the solution. This procedure was repeated twice and the obtained solid was dried in a 85°C drying oven for 15 min to give Pd2+/UiO-66-NH2. After that, a 15 mg sample of Pd2+/UiO-66-NH2 was dispersed in 5 mL MeOH by ultrasonication, and 5 mg NH3BH3 was added during stirring. After H2 bubbling was completed, a certain amount of H2PtCl6 aqueous solution (the mass ratio of Pt/Pd = 0.3, 1, 5, 10/10, respectively) was quickly added and stirred for 1 h. The solid was collected by centrifugation and washed by MeOH several times, and dried at 60°C under vacuum overnight. The product was named Pd10@Ptx/UiO-66-NH2 (the 10 and x mean the mass permillage, at wt‰, of Pd and Pt to the MOF in the synthesis, respectively).
Synchrotron radiation photoemission spectroscopy (SRPES)
The SRPES spectra were measured with photon energies of 450 eV, 710 eV, 970 eV and 1253.6 eV at the BL10B beamline in the National Synchrotron Radiation Laboratory (NSRL, Hefei, China) [56,57]. Before the SRPES test, 30 mg Pd10@Pt10/UiO-66-NH2 was dispersed in 1 mL H3PO4 and stirred for 2 h at room temperature to dissolve the MOF to improve the electrical conductivity of the sample.
The X-ray absorption spectra
The Pt L3-edge X-ray absorption spectra (XAS) data were collected at 1W1B beam line of the Beijing Synchrotron Radiation Facility (BSRF, Beijing) operated at 2.5 GeV at room temperature. The Si (111) double crystal monochromator was calibrated via Pt foil and then the Pt foil XAS data was collected as the reference spectrum. The XAS measurement for the samples was performed in the fluorescence mode using a Lytle detector. Data analysis was performed with Athena and Artemis included in the IFEFFIT version 0.8.012. A first-shell single scattering path was used in fitting the FT-EXAFS data.
Photocatalytic experiments
Typically, 3 mg photocatalyst was dispersed in 27 mL acetonitrile containing 0.2 mL deionized water and 3 mL triethylamine (as a sacrificial reagent). Then the suspension was transferred into an optical reaction vessel (160 mL) and purged with nitrogen for 15 min to remove air. The reaction solution was irradiated by the 300 W Xe lamp (LX-300F, Japan) equipped with a UV cut-off filter (>380 nm). Hydrogen gas was measured by gas chromatography (Shimadzu GC-2014) using a thermal conductivity detector.
Supplementary Material
Acknowledgements
We acknowledge the XAS support from BSRF and simulation support from the Supercomputing Center of University of Science and Technology of China.
Contributor Information
Yating Pan, Hefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Soft Matter Chemistry, Department of Chemistry, University of Science and Technology of China, Hefei 230026, China.
Yunyang Qian, Hefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Soft Matter Chemistry, Department of Chemistry, University of Science and Technology of China, Hefei 230026, China.
Xusheng Zheng, National Synchrotron Radiation Laboratory (NSRL), University of Science and Technology of China, Hefei 230029, China.
Sheng-Qi Chu, Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China.
Yijun Yang, Key Laboratory of Luminescence and Optical Information, Ministry of Education, Department of Physics, School of Science, Beijing Jiaotong University, Beijing 100044, China.
Chunmei Ding, Dalian National Laboratory for Clean Energy, State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China.
Xi Wang, Key Laboratory of Luminescence and Optical Information, Ministry of Education, Department of Physics, School of Science, Beijing Jiaotong University, Beijing 100044, China.
Shu-Hong Yu, Hefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Soft Matter Chemistry, Department of Chemistry, University of Science and Technology of China, Hefei 230026, China.
Hai-Long Jiang, Hefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Soft Matter Chemistry, Department of Chemistry, University of Science and Technology of China, Hefei 230026, China.
FUNDING
This work was supported by the National Natural Science Foundation of China (21725101, 21673213, 21871244 and 21521001), Dalian National Laboratory Cooperation Fund, Chinese Academy of Sciences (DNL201911), Fundamental Research Funds for the Central Universities (WK2060030029), and Fujian Institute of Innovation (CAS).
AUTHOR CONTRIBUTIONS
H.-L.J. conceived and designed the project. Y.P. performed the experiments and collected the data. Y.Q. conducted the DFT calculations. X.Z. performed the measurements and analysis of SRPES and XPS. S.-Q.C. performed the measurements and analysis of FT-EXAFS and XANES. Y.Y. and X.W. conducted the HRTEM and elemental line-scanning spectra characterization. H.-L.J. and Y.P. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Conflict of interest statement. None declared.
REFERENCES
- 1. Li XB, Tung CH, Wu LZ. Semiconducting quantum dots for artificial photosynthesis. Nat Rev Chem 2018; 21: 60–173. 10.1038/s41570-018-0024-8 [DOI] [Google Scholar]
- 2. Wang Z, Li C, Domen K. Recent developments in heterogeneous photocatalysts for solar-driven overall water splitting. Chem Soc Rev 2019; 48: 2109–25. 10.1039/C8CS00542G [DOI] [PubMed] [Google Scholar]
- 3. Schultz DM, Yoon TP. Solar synthesis: prospects in visible light photocatalysis. Science 2014; 343: 1239176. 10.1126/science.1239176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang F, Li Q, Xu D. Recent progress in semiconductor-based nanocomposite photocatalysts for solar-to-chemical energy conversion. Adv Energy Mater 2017; 7: 1700529. 10.1002/aenm.201700529 [DOI] [Google Scholar]
- 5. Pacchioni G, Freund HJ. Controlling the charge state of supported nanoparticles in catalysis: lessons from model systems. Chem Soc Rev 2018; 47: 8474–502. 10.1039/C8CS00152A [DOI] [PubMed] [Google Scholar]
- 6. Liu L, Corma A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem Rev 2018; 118: 4981–5079. 10.1021/acs.chemrev.7b00776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tong H, Ouyang S, Bi Yet al. Nano-photocatalytic materials: possibilities and challenges. Adv Mater 2012; 24: 229–51. 10.1002/adma.201102752 [DOI] [PubMed] [Google Scholar]
- 8. Ran J, Zhang J, Yu Jet al. Earth-abundant cocatalysts for semiconductor-based photocatalytic water splitting. Chem Soc Rev 2014; 43: 7787–812. 10.1039/C3CS60425J [DOI] [PubMed] [Google Scholar]
- 9. Xiao JD, Shang Q, Xiong Yet al. Boosting photocatalytic hydrogen production of a metal-organic framework decorated with platinum nanoparticles: the platinum location matters. Angew Chem Int Ed 2016; 55: 9389–93. 10.1002/anie.201603990 [DOI] [PubMed] [Google Scholar]
- 10. Li X, Yu J, Jaroniec Met al. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem Rev 2019; 119: 3962–4179. 10.1021/acs.chemrev.8b00400 [DOI] [PubMed] [Google Scholar]
- 11. Xiao JD, Han L, Luo Jet al. Integration of plasmonic effects and Schottky junctions into metal-organic framework composites: steering charge flow for enhanced visible-light photocatalysis. Angew Chem Int Ed 2018; 57: 1103–7. 10.1002/anie.201711725 [DOI] [PubMed] [Google Scholar]
- 12. Chen F, Huang H, Guo Let al. The role of polarization in photocatalysis. Angew Chem Int Ed 2019; 58: 10061–73. 10.1002/anie.201901361 [DOI] [PubMed] [Google Scholar]
- 13. Pei GX, Liu XY, Wang Aet al. Ag alloyed Pd single-atom catalysts for efficient selective hydrogenation of acetylene to ethylene in excess ethylene. ACS Catal 2015; 5: 3717–25. 10.1021/acscatal.5b00700 [DOI] [Google Scholar]
- 14. Wrasman CJ, Boubnov A, Riscoe ARet al. Synthesis of colloidal Pd/Au dilute alloy nanocrystals and their potential for selective catalytic oxidations. J Am Chem Soc 2018; 140: 12930–9. 10.1021/jacs.8b07515 [DOI] [PubMed] [Google Scholar]
- 15. Marcinkowski MD, Darby MT, Liu Jet al. Pt/Cu single-atom alloys as coke-resistant catalysts for efficient C-H activation. Nat Chem 2018; 10: 325–32. 10.1038/nchem.2915 [DOI] [PubMed] [Google Scholar]
- 16. Giannakakis G, Flytzani-Stephanopoulos M, Sykes ECH. Single-atom alloys as a reductionist approach to the rational design of heterogeneous catalysts. Acc Chem Res 2019; 52: 237–47. 10.1021/acs.accounts.8b00490 [DOI] [PubMed] [Google Scholar]
- 17. Sun G, Zhao ZJ, Mu Ret al. Breaking the scaling relationship via thermally stable Pt/Cu single atom alloys for catalytic dehydrogenation. Nat Commun 2018; 9: 4454. 10.1038/s41467-018-06967-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang A, Li J, Zhang T. Heterogeneous single-atom catalysis. Nat Rev Chem 2018; 2: 65–81. 10.1038/s41570-018-0010-1 [DOI] [Google Scholar]
- 19. Jiao L, Jiang HL. Metal-organic-framework-based single-atom catalysts for energy applications. Chem 2019; 5: 786–804. 10.1016/j.chempr.2018.12.011 [DOI] [Google Scholar]
- 20. Luo M, Guo S. Strain-controlled electrocatalysis on multimetallic nanomaterials. Nat Rev Mater 2017; 2: 17059. 10.1038/natrevmats.2017.59 [DOI] [Google Scholar]
- 21. Wang X, Zhu Y, Vasileff Aet al. Strain effect in bimetallic electrocatalysts in the hydrogen evolution reaction. ACS Energy Lett 2018; 3: 1198–204. 10.1021/acsenergylett.8b00454 [DOI] [Google Scholar]
- 22. Furukawa H, Cordova KE, O’Keeffe Met al. The chemistry and applications of metal-organic frameworks. Science 2013; 341: 1230444. 10.1126/science.1230444 [DOI] [PubMed] [Google Scholar]
- 23. Zhou HC, Kitagawa S. Meta-organic frameworks MOFs. Chem Soc Rev 2014; 43: 5415–8. 10.1039/C4CS90059F [DOI] [PubMed] [Google Scholar]
- 24. Li B, Wen HM, Cui Yet al. Emerging multifunctional metal-organic framework materials. Adv Mater 2016; 28: 8819–60. 10.1002/adma.201601133 [DOI] [PubMed] [Google Scholar]
- 25. Jiao L, Wang Y, Jiang HLet al. Metal-organic frameworks as platforms for catalytic applications. Adv Mater 2018; 30: 1703663. 10.1002/adma.201703663 [DOI] [PubMed] [Google Scholar]
- 26. Dhakshinamoorthy A, Li Z, Garcia H. Catalysis and photocatalysis by metal organic frameworks. Chem Soc Rev 2018; 47: 8134–72. 10.1039/C8CS00256H [DOI] [PubMed] [Google Scholar]
- 27. Islamoglu T, Goswami S, Li Zet al. Postsynthetic tuning of metal-organic frameworks for targeted applications. Acc Chem Res 2017; 50: 805–13. 10.1021/acs.accounts.6b00577 [DOI] [PubMed] [Google Scholar]
- 28. Silva CG, Luz I, Xamena Fet al. Water stable Zr-benzenedicarboxylate metal-organic frameworks as photocatalysts for hydrogen generation. Chem Eur J 2010; 16: 11133–8. 10.1002/chem.200903526 [DOI] [PubMed] [Google Scholar]
- 29. Wu ZL, Wang CH, Zhao Bet al. A semi-conductive copper-organic framework with two types of photocatalytic activity. Angew Chem Int Ed 2016; 55: 4938–42. 10.1002/anie.201508325 [DOI] [PubMed] [Google Scholar]
- 30. Xu C, Liu H, Li Det al. Direct evidence of charge separation in a metal-organic framework: efficient and selective photocatalytic oxidative coupling of amines via charge and energy transfer. Chem Sci 2018; 9: 3152–8. 10.1039/C7SC05296K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu G, Huang J, Zang Yet al. Porous field-effect transistors based on a semiconductive metal-organic framework. J Am Chem Soc 2017; 139: 1360–3. 10.1021/jacs.6b08511 [DOI] [PubMed] [Google Scholar]
- 32. Zhou T, Du Y, Borgna Aet al. Post-synthesis modification of a metal-organic framework to construct a bifunctional photocatalyst for hydrogen production. Energy Environ Sci 2013; 6: 3229–34. 10.1039/c3ee41548a [DOI] [Google Scholar]
- 33. Kim D, Whang DR, Park SY. Self-healing of molecular catalyst and photosensitizer on metal-organic framework: robust molecular system for photocatalytic H2 evolution from water. J Am Chem Soc 2016; 138: 8698–701. 10.1021/jacs.6b04552 [DOI] [PubMed] [Google Scholar]
- 34. Zhang T, Lin W. Metal-organic frameworks for artificial photosynthesis and photocatalysis. Chem Soc Rev 2014; 43: 5982–93. 10.1039/C4CS00103F [DOI] [PubMed] [Google Scholar]
- 35. An Y, Liu Y, An Pet al. NiII coordination to an Al-based metal-organic framework made from 2-aminoterephthalate for photocatalytic overall water splitting. Angew Chem Int Ed 2017; 56: 3036–40. 10.1002/anie.201612423 [DOI] [PubMed] [Google Scholar]
- 36. Yang SZ, Pattengale B, Kovrigin ELet al. Photoactive zeolitic imidazolate framework as intrinsic heterogeneous catalysts for light-driven hydrogen generation. ACS Energy Lett 2017; 2: 75–80. 10.1021/acsenergylett.6b00540 [DOI] [Google Scholar]
- 37. Fu Y, Sun D, Chen Yet al. An amine-functionalized titanium metal-organic framework photocatalyst with visible-light-induced activity for CO2 reduction. Angew Chem Int Ed 2012; 51: 3364–7. 10.1002/anie.201108357 [DOI] [PubMed] [Google Scholar]
- 38. Xu HQ, Hu J, Wang Det al. Visible-light photoreduction of CO2 in a metal-organic framework: boosting electron-hole separation via electron trap states. J Am Chem Soc 2015; 137: 13440–3. 10.1021/jacs.5b08773 [DOI] [PubMed] [Google Scholar]
- 39. Zhang H, Wei J, Dong Jet al. Efficient visible-light-driven carbon dioxide reduction by a single-atom implanted metal-organic framework. Angew Chem Int Ed 2016; 55: 14310–4. 10.1002/anie.201608597 [DOI] [PubMed] [Google Scholar]
- 40. Wang Y, Huang NY, Shen JQet al. Hydroxide ligands cooperate with catalytic centers in metal-organic frameworks for efficient photocatalytic CO2 reduction. J Am Chem Soc 2018; 140: 38–41. 10.1021/jacs.7b10107 [DOI] [PubMed] [Google Scholar]
- 41. Wu LY, Mu YF, Guo XXet al. Encapsulating perovskite quantum dots in iron-based metal-organic frameworks (MOFs) for efficient photocatalytic CO2 reduction. Angew Chem Int Ed 2019; 58: 9491–5. 10.1002/anie.201904537 [DOI] [PubMed] [Google Scholar]
- 42. Li N, Liu J, Liu JJet al. Adenine components in biomimetic metal-organic frameworks for efficient CO2 photoconversion. Angew Chem Int Ed 2019; 58: 5226–30. 10.1002/anie.201814729 [DOI] [PubMed] [Google Scholar]
- 43. Xia Z, He C, Wang Xet al. Modifying electron transfer between photoredox and organocatalytic units via framework interpenetration for β-carbonyl functionalization. Nat Commun 2017; 8: 361. 10.1038/s41467-017-00416-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Y, Zhang Y, Guo Jet al. Tunable chiral metal organic frameworks toward visible light-driven asymmetric catalysis. Sci Adv 2017; 3: e1701162. 10.1126/sciadv.1701162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiao JD, Jiang HL. Metal-organic frameworks for photocatalysis and photothermal catalysis. Acc Chem Res 2019; 52: 356–66. 10.1021/acs.accounts.8b00521 [DOI] [PubMed] [Google Scholar]
- 46. Wang C, deKrafft KE, Lin W. Pt nanoparticles@photoactive metal-organic frameworks: efficient hydrogen evolution via synergistic photoexcitation and electron injection. J Am Chem Soc 2012; 134: 7211–4. 10.1021/ja300539p [DOI] [PubMed] [Google Scholar]
- 47. Shen L, Luo M, Huang Let al. A clean and general strategy to decorate a titanium metal-organic framework with noble-metal nanoparticles for versatile photocatalytic applications. Inorg Chem 2015; 54: 1191–3. 10.1021/ic502609a [DOI] [PubMed] [Google Scholar]
- 48. Fang X, Shang Q, Wang Yet al. Single Pt atoms confined into a metal-organic framework for efficient photocatalysis. Adv Mater 2018; 30: 1705112. 10.1002/adma.201705112 [DOI] [PubMed] [Google Scholar]
- 49. Zuo Q, Liu T, Chen Cet al. Ultrathin metal-organic framework nanosheets with ultrahigh loading of single Pt atoms for efficient visible-light-driven photocatalytic H2 evolution. Angew Chem Int Ed 2019; 58: 10198–203. 10.1002/anie.201904058 [DOI] [PubMed] [Google Scholar]
- 50. Hu Z, Peng Y, Kang Zet al. A modulated hydrothermal (MHT) approach for the facile synthesis of UiO-66-type MOFs. Inorg Chem 2015; 54: 4862–8. 10.1021/acs.inorgchem.5b00435 [DOI] [PubMed] [Google Scholar]
- 51. Hu P, Morabito JV, Tsung CK. Core-shell catalysts of metal nanoparticle core and metal-organic framework shell. ACS Catal 2014; 4: 4409–19. 10.1021/cs5012662 [DOI] [Google Scholar]
- 52. Yang Q, Xu Q, Jiang HL. Metal-organic frameworks meet metal nanoparticles: synergistic effect for enhanced catalysis. Chem Soc Rev 2017; 46: 4774–808. 10.1039/C6CS00724D [DOI] [PubMed] [Google Scholar]
- 53. Chen L, Huang B, Qiu Xet al. Seed-mediated growth of MOF-encapsulated Pd@Ag core-shell nanoparticles: toward advanced room temperature nanocatalysts. Chem Sci 2016; 7: 228–33. 10.1039/C5SC02925B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li X, Zhang B, Tang Let al. Cooperative multifunctional catalysts for nitrone synthesis: platinum nanoclusters in amine-functionalized metal-organic frameworks. Angew Chem Int Ed 2017; 56: 16371–4. 10.1002/anie.201710164 [DOI] [PubMed] [Google Scholar]
- 55. Zhao M, Yuan K, Wang Yet al. Metal-organic frameworks as selectivity regulators for hydrogenation reactions. Nature 2016; 539: 76–80. 10.1038/nature19763 [DOI] [PubMed] [Google Scholar]
- 56. Ge J, He DS, Chen Wet al. Atomically dispersed Ru on ultrathin Pd nanoribbons. J Am Chem Soc 2016; 138: 13850–3. 10.1021/jacs.6b09246 [DOI] [PubMed] [Google Scholar]
- 57. Tao F, Grass ME, Zhang Yet al. Reaction-driven restructuring of Rh-Pd and Pt-Pd core-shell nanoparticles. Science 2008; 322: 932–4. 10.1126/science.1164170 [DOI] [PubMed] [Google Scholar]
- 58. Duchesne PN, Li ZY, Deming CPet al. Golden single-atomic-site platinum electrocatalysts. Nat Mater 2018; 17: 1033–9. 10.1038/s41563-018-0167-5 [DOI] [PubMed] [Google Scholar]
- 59. Chao T, Luo X, Chen Wet al. Atomically dispersed copper-platinum dual sites alloyed with palladium nanorings catalyze the hydrogen evolution reaction. Angew Chem Int Ed 2017; 56: 16047–51. 10.1002/anie.201709803 [DOI] [PubMed] [Google Scholar]
- 60. Tedsree K, Li T, Jones Set al. Hydrogen production from formic acid decomposition at room temperature using a Ag-Pd core-shell nanocatalyst. Nat Nanotechnol 2011; 6: 302–7. 10.1038/nnano.2011.42 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.