

Abstract
Directed leukocyte migration is a hallmark of inflammatory immune responses. Leukotrienes are derived from arachidonic acid and represent a class of potent lipid mediators of leukocyte migration. In this review, we summarize the essential steps leading to the production of LTB4 in leukocytes. We discuss the recent findings on the exosomal packaging and transport of LTB4 in the context of chemotactic gradients formation and regulation of leukocyte recruitment. We also discuss the dynamic roles of the LTB4 receptors, BLT1 and BLT2, in mediating chemotactic signaling in leukocytes and contrast them to other structurally related leukotrienes that bind to distinct GPCRs. Finally, we highlight the specific roles of the LTB4-BLT1 axis in mediating signal-relay between chemotaxing neutrophils and its potential contribution to a wide variety of inflammatory conditions including tumor progression and metastasis, where LTB4 is emerging as a key signaling component.
Keywords: leukotriene B4, neutrophils, chemotaxis, inflammation, migration
INTRODUCTION
Cell migration is a fundamental process conserved from bacteria to humans. Migration of cells towards a chemical cue, referred to as directed cell migration or chemotaxis, contributes to a number of fundamental biological responses in different organisms. For example, bacteria use chemotaxis to interact with chemoattractant-releasing plant roots to establish a symbiotic relationship [1] and in the case of H. pylori, which causes stomach ulcers and gastric cancers, chemotactic signaling is required to establish stomach colonization by the bacteria [2]. The social amoeba, Dictyostelium discoideum, upon nutrient depletion chemotax to form cellular aggregates that then differentiate into multicellular slugs and fruiting bodies [3]. In higher organisms including flies, zebrafish and mouse, directed migration of primordial germ cells is observed very early during development [4]. Finally, leukocyte migration in response to inflammation is another striking example of chemotaxis in higher mammals, including humans [3].
The chemical nature of chemoattractants varies widely from small molecules and lipids to peptides and proteins depending on the model system. E. coli chemotax to many chemicals including glucose and amino acids such as aspartate and serine [5]. For Dictyostelium, both folic acid and cyclic adenosine monophosphate (cAMP) act as physiological chemoattactants [3]. Not surprisingly, in higher eukaryotes, the complexity of chemoattractants is impressive and ranges from formylated peptides secreted by bacteria (such as N-formylmethionyl-leucyl-phenylalanine - fMLP), to products of the complement cascade (such as C5a) and phospholipid metabolites (such as leukotriene B4 (LTB4)) as well as the large family of chemokines that are derived from endothelial, epithelial, and stromal cells [6, 7]. Chemoattractants are further considered to be either end-point/primary (fMLP, C5a etc.) or intermediate/secondary (LTB4, IL8 etc.), depending on their chemotactic potential in competing gradients and signaling specificity [8], an aspect that will be discussed later. Remarkably, despite their diverse nature, the vast majority of chemoattractants bind and activate specific G-protein coupled receptors (GPCRs) to mediate their effects [3, 6].
For cells to chemotax in complex in vivo environments, directional cues need to be sustained for significant period of time over long distances. One of the ways cells accomplish sustained long-range chemotactic behavior is by ‘signal-relay’. For example, in Dictyostelium cAMP induces further production of cAMP through activation of Adenyl cyclase A or ACA, which then promotes and sustains a head-to-tail chemotactic response of migrating cells [9–11]. In the case of immune cells such as neutrophils, signal-relay occurs when cells chemotax toward a primary chemoattractant source, where they then actively produce and secrete a secondary chemoattractant that is required to drive the directional movement of both the cells that produce it (autocrine signaling) as well as the cells that follow it (paracrine signaling) in a spatio-temporal manner [9, 12–15]. In this review, we will specifically focus on the production, gradient formation and regulation of signal-relay by the interaction of LTB4 with its GPCRs - BLT1 and BLT2, with an emphasis on the role of such an axis in regulating chemotactic responses of immune cells in a variety of inflammatory scenarios, including cancer.
LTB4 SYNTHESIS, REGULATION AND SECRETION
Before we dwell on the functional consequences of LTB4-BLT interactions, it is essential to understand the sequence of events leading to synthesis, packaging and secretion/distribution of LTB4 in the context of leukocyte activation by inflammatory agents.
Tissue distribution of LTB4
In 1979, Borgeat and Samuelsson described the release of C20 dihydroxy fatty acids with three conjugated double bonds, first from rabbit [16, 17] and then from human morphonuclear leukocytes [17] in response to arachidonic acid (AA) and the calcium ionophore A23187. This class of eicosanoid was called LTB4 to reflect the nature of the compounds as well as their source [18]. Since then, LTB4 is known to be produced by tissues under various conditions, most notably during inflammatory responses.
LTB4 production by immune cells
Leukotriene release from human mature myeloid cells occurs in response to stimulation with molecules including endotoxin, C5a, IL-1, TNFα, GMCSF, LIF and PAF. However, the quantity released varies among myeloid cell types. Basophils, for example, do not produce LTB4, but can release a related leukotriene, LTC4, upon stimulation with anti-IgE or A23187 [19]. Similarly, A23187-stimulated eosinophil primarily secrete LTC4 and only very low quantities of LTB4 [20]. In contrast, neutrophils primarily release LTB4 upon stimulation [20]. On the other hand, human monocytes can produce both LTB4 or LTC4 in response to physiologically relevant stimuli such as unopsonized zymosan particles or IgG-sensitized sheep erythrocytes or to non-physiological stimulus like A23187, with a slight preference towards LTB4 [21]. Similar observations have been reported for macrophages and mast cells [22]. However, mucosal mast cells isolated from the intestine of Nippostrongylus brasiliensis-infected rats and activated with anti-IgE show higher LTC4 production rates compared to LTB4 [23].
The production of LTB4 from lymphocytes has been mottled in controversy. Although the production and release of LTB4 from human T cells were initially reported [24], further work on highly purified T-lymphocytes and monoclonal lymphoblastoid cells failed to detect LTB4, either in the presence or absence of AA or calcium ionophores [25]. Studies on T and NK cell lines such as Jurkat and RNK-16 also showed no evidence of LTB4 release upon stimulation [26]. Similar reports on the absence of LTB4 in B cells were first published in late 1980s and early 1990s but were quickly reconsidered after the demonstration of in vitro synthesis of LTB4 from B cell lysates [27]. Later, B cells activated by the protein A-positive Staphylococcal strain, Cowan 1, and a mixture of recombinant IL-2/IL-6 were shown to possess high capacity to generate LTB4 [27].
Elevated leukotriene secretion in myeloid cells, especially cells of myeloblastic origin, has important implications in the role of LTB4 during immune surveillance and immune cell chemotactic responses. These cells either reside in tissues or actively survey injury and infection sites by detecting chemoattractant gradients. In later sections, we will describe how LTB4 is especially well suited to amplify primary chemotactic signals by recruiting cells of lymphoid origin to inflammation sites.
LTB4 production by non-immune cells
The other major source of LTB4 in systemic circulation comes from non-immune sources associated with inflammatory pathophysiology. Asthma and upper respiratory airway inflammation represent the best-studied cases of LTB4-related inflammatory pathophysiology. While neutrophils represent the major source of LTB4 in bronchoalveolar lavage in bronchial asthma [28], a proportion of leukotrienes is produced by allergic lung alveoli that is involved in recruiting macrophages [29]. Also, LTB4 release from tracheal epithelial cells during asthma has been shown to contribute to bronchial constriction and vasodilation [30]. Surprisingly, the brain is another major source of LTB4 in non-immune tissues and LTB4 synthesis positively correlates with aging in both rats [31] and humans [32]. The expression of 5-Lipoxygenase (5-LO), the enzyme responsible for LTB4 production, in different parts of the rat brain, including the cerebellum and hippocampus, has been reported and significant changes in LTB4 levels have been associated with age-dependent cognitive impairment [33]. The expression of LTB4 synthesizing enzymes in neurons and glial cells and the involvement of LTB4 in brain ischemia, trauma, astrocytoma and other inflammatory pathophysiology of the brain provides indirect evidence for a role of LTB4 in such conditions [34]. In fact, inhibition of LTB4 production has been shown to be neuroprotective in cases of brain injury [35]. Finally, elevated LTB4 levels have been found in a disparate array of tissues including corneal and conjunctival tissues of the eye [36], loose connective tissues such as adipocytes [37], endocrinal glands such as thyroid [38] and by mucosal epithelial cells in pathological conditions such as during ulcerative colitis [39] and type C gastritis [40].
Biosynthesis of LTB4
The synthesis of LTB4 begins with the mobilization of AA on the nuclear membrane, where free AA is released by the cleavage of the arachidonoyl ester from a membrane glycerophospholipid by the hydrolytic action of a calcium sensitive phospholipase A2 (cPLA2) [41]. The released AA then binds to an integral nuclear membrane protein called 5-Lipoxygenase activating protein (FLAP) [42]. Sequestration of AA by FLAP increases local AA concentration which changes membrane anisotropy and fluidity, a key step for the recruitment of 5-LO to the nuclear membrane [43, 44]. Once recruited, 5-LO interacts with FLAP, most likely through the interaction of the α2 domain of FLAP [45] and the dimerization interface of 5-LO [46], to further anchor it to the membrane. FLAP then delivers the substrate AA to 5-LO and through the action of 5-LO, AA then undergoes hydrogen abstraction at the C7 position followed by the addition of an oxygen to give rise to 5-hydroperoxyeicosatetraenoic acid (5-HpETE). This step is followed by the removal of the hydrogen atom from the C10 position to form a conjugate triene epoxide LTA4 [47]. LTA4 is then acted upon by LTA4 hydrolase (LTA4H) to give rise to LTB4 by the addition of a water molecule at the C12 position [48]. Apart from the catalytic processes involved, another level of regulation of leukotriene synthesis occurs via post translation modifications, calcium binding and, more importantly, by the subcellular partitioning of the enzymes involved in LTB4 synthesis, which will be discussed further below.
Regulation of cPLA2
The key enzyme that connects cell stimulation with the initiation of leukotriene synthesis is cPLA2, which can be activated by binding of growth factors, mitogens, cytokines, formylated peptides to their cognate receptors, as well as non–receptor mediated triggers such as oxidative and shear stress. The common factor between these disparate processes is the increase in cytosolic calcium either from emptying of internal stores or through extracellular calcium influx. The increase in cytosolic calcium is required for cPLA2 to bind to membranes [49] and causes cPLA2 to rapidly redistribute from the cytosol to the endoplasmic reticulum (ER) and the nuclear envelope where it hydrolyzes nuclear phospholipids to release AA. Additionally, cell activation leads to rapid phosphorylation of cPLA2 resulting in its increased lipase activity (Fig.1) [50] [51]. While phosphorylation by MAPK [52] and CAM kinases [53] are thought to mediate translocation, other reports suggest effects on enzyme activity alone [54]. Hence, the precise role of cPLA2 phosphorylation in its nuclear translocation remains controversial. Interestingly, cPLA2 phosphorylation is dependent on the type of stimuli: in neutrophils both ERK and p38MAPK phosphorylate cPLA2 in response to PAF, but not fMLP [55].
Figure 1. LTB4 synthesis and transport.
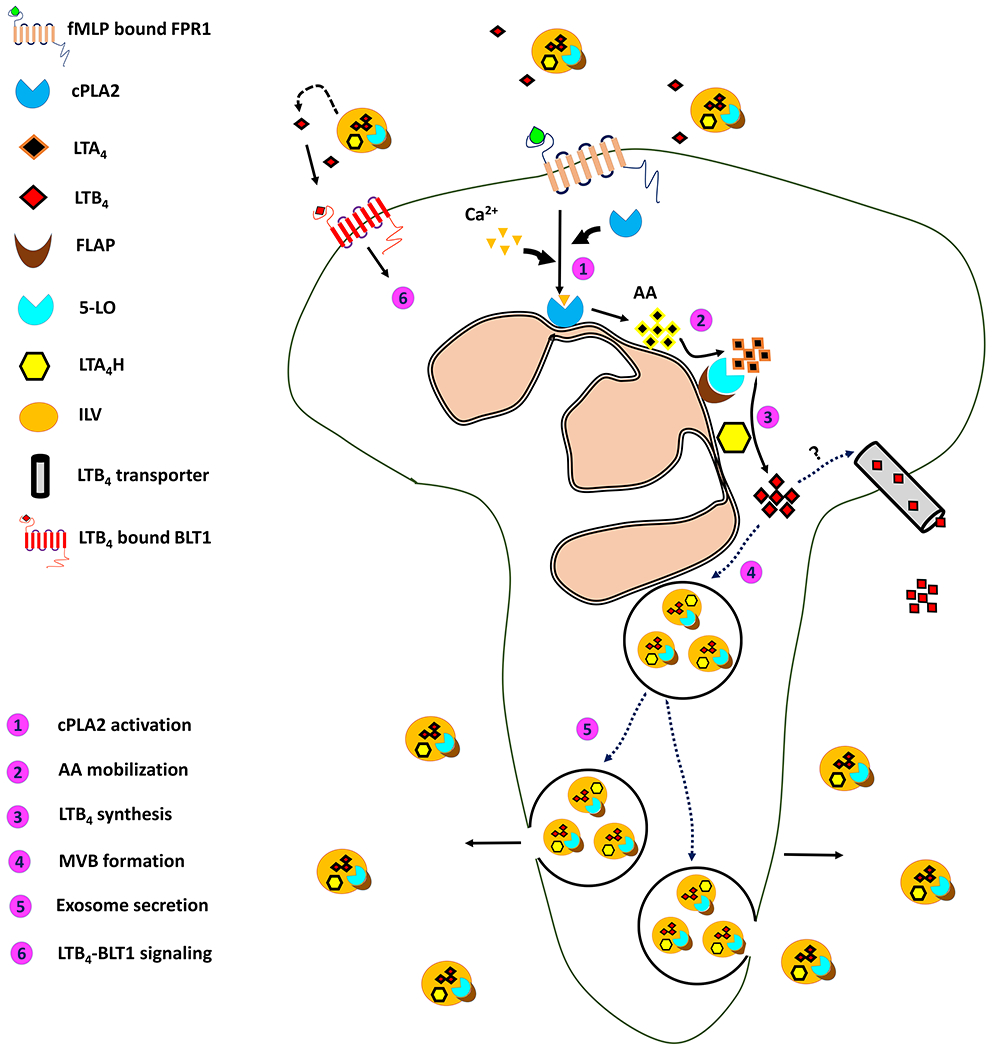
A schematic depiction of the various steps that lead to the synthesis of LTB4, it’s packaging into MVBs and secretion as exosomes from neutrophils chemotaxing towards a primary chemoattractant.
Regulation of FLAP
The modulation of AA availability by FLAP is one of the least understood steps in the leukotriene synthesis cascade. FLAP belongs to a protein superfamily called membrane-associated proteins involved in eicosanoid and glutathione metabolism, or MAPEG. But unlike other members of the same family, such as LTC4 synthase, FLAP does not possess any inherent enzymatic activity [56] and the mechanisms by which FLAP binds to AA in the nuclear lipid bilayer and how it transfers AA to 5-LO are not well understood. It is well established that 5-LO nuclear translocation and AA catalysis are independent of FLAP. Also, there is no literature to suggest that calcium binding to the C2-like domain of 5-LO in anyway mediates the direct interaction of FLAP with 5-LO. However, FLAP is essential for the transfer of AA to 5-LO, as LTB4 synthesis fails to occur when cells are treated with a specific competitive inhibitor of FLAP, MK886. The regulation of LTB4 synthesis by FLAP is perhaps mediated by a “catch and release” mechanism of AA to 5-LO [57]. According to the recently solved inhibitor bound crystal structure of FLAP [45], free AA molecules laterally diffuse within the plane of the membrane and bind in the three hydrophobic surface grooves formed by the FLAP trimer. AA-bound FLAP then undergoes a conformational change in its positively charged cytosolic loops allowing interaction with 5-LO. The mechanism by which the “caught” AA is now “released” to 5-LO is not understood. Interestingly, mutations in the cytosolic loops of FLAP, the regions that are proposed as 5-LO interaction sites, have marked effect on binding affinities of FLAP to AA, suggesting that these flexible loops are central to the “capture” and “release” of AA. Moreover, FLAP has been shown to exist as a homodimer [58] and a trimer [45] or even as a multimer [59], but whether FLAP multimer formation is ligand-induced and whether such arrangement is required for its function remains unresolved.
Regulation of 5-LO
5-LO is a non-heme iron-containing enzyme that catalyzes the conversion of AA to LTA4. 5-LO activity is mostly regulated through its subcellular localization within the cell. 5-LO contains nuclear transport signals at its C- and N-terminal regions and is found in the cytosol or nucleus depending on the cell type [60, 61]. Translocation of 5-LO from the cytoplasm to the nucleus is calcium dependent and is thought to be mediated through N-terminal β-barrel which resembles a C2 domain [62, 63]. Tryptophan residues in C2-like domain are required for binding to phosphatidylcholine in a calcium-dependent manner [63] and is further augmented by AA itself [64]. However, nuclear import may also take place before cell stimulation - as in the case of alveolar macrophages [65] - or may be enhanced by priming of the cell or by cell adhesion as in the case of neutrophils [66]. Interestingly, the nuclear presence of 5-LO in resting neutrophils has been reported to be gender specific and is ERK-dependent in response to androgens [67]. Nuclear import also appears to depend on MAPK pathways other than ERK [68] and has been shown to be facilitated by 5-LO interaction with a coactosin-like protein [69]. Upon stimulation, cytosolic and nuclear 5-LO translocate to the inner and outer nuclear membrane in a calcium-dependent manner, although transport of the enzyme across the perinuclear space has not yet been ruled out [70]. The localization of 5-LO with either the inner or outer nuclear membrane leads to the generation of HpETE and LTA4 in distinct cellular compartments, which presumably account for the distinct functions of LTB4 in a cell type specific manner [71].
Regulation of LTA4H
LTA4H is a zinc containing metalloenzyme with intrinsic aminopeptidase activity and an exquisite stereospecificity to substrates containing 5,6-trans-epoxide, like 5-HpETE [72]. LTA4H is expressed in wide variety of tissue, even those that lack significant 5-LO activity e.g. endothelial cells, erythrocytes, and lymphocytes. LTA4 is, hence, thought to be exported from a producer cell to a LTA4H-containing recipient cell where it can convert LTA4 to LTB4 [73]. This example of transcellular biosynthesis of LTB4 provides a key regulatory mechanism for LTB4 biosynthesis and tissue availability. The other key regulation mediated by LTA4H is its suicide inactivation by its own substrate LTA4. LTA4 covalently modifies LTA4H to irreversibly inactivate the enzyme, thereby shutting down LTB4 biosynthesis. The final regulatory step mediating LTA4H function is the subcellular localization of the enzyme [74]. Unlike 5-LO, LTA4H has been purified as a soluble protein and its activity is not expected to depend on membrane association. However, LTA4H localizes in the nucleus in a cell-type dependent manner. For example, LTA4H resides in the nucleus of alveolar macrophages and rat basophilic leukemia cells, but is predominantly cytoplasmic in neutrophils [75]. Interestingly, neutrophils show higher vesicular, punctate cytoplasmic localization of LTA4H upon adhesion compared to cells in suspension [75]. This observation will become important when we discuss vesicular packaging of LTB4.
Distinct roles of intracellular and extracellular LTB4
Nuclear LTB4
The direct action of LTB4 in the nucleus occurs in conjunction with the nuclear receptor PPARα [76]. In silico docking studies suggests that LTB4 and other AA metabolites, such as 5-HpETE and LTA4, can directly bind to PPARα and can mediate PPARα action on the transcription of target genes [77]. More importantly, binding of LTB4 to PPARα has been shown to represent an alternate level of control of the downstream effects of LTB4 during inflammatory responses. Indeed, β- and ω-fatty acid oxidation mediated by LTB4-induced PPARα activation leads to the breakdown of LTB4 and thereby act as a potential feedback mechanism during inflammation resolution [78].
Cellular transport of LTB4 and formation of chemotactic gradients in tissues
Once synthesized in the cytosol, LTB4 is released into the extracellular environment, where it acts in an autocrine and paracrine fashion by binding to surface receptors (see below). The mechanism by which this occurs is not obvious given the low solubility of eicosanoids and the requirement for a specific transport mechanism. It is known that LTB4 is rapidly released following cellular activation [79] and that free cytosolic intracellular LTB4 does not accumulate in cells [80, 81]. Moreover, the release of LTB4 is a saturable process that is dependent on temperature and involves a carrier [82]. MRP4, a member of the ATP binding cassette subfamily C, has been proposed as a candidate carrier as membrane vesicles containing ABCC4 (MRP4) are capable of transporting LTB4 in the presence of glutathione [83]. Neutrophils treated with an inhibitor of MRP4 show reduced chemotaxis and LTB4 release in response to IL8. However, inhibition of MRP4 itself stimulated the release of LTB4, suggesting that MRP4 may only be involved in the ligand induced synthesis or transport of factors required for LTB4 synthesis [84]. Nevertheless, a transporter-mediated process would not explain how a hydrophobic molecule such as LTB4 can maintain its stability in circulation and form stable gradients in tissue, which are required for immune cell chemotaxis. Stable gradients of biomolecules have been proposed to be maintained by reaction-diffusion systems, where the buildup of the molecule is prevented by degradation/inactivation by ectoenzymes [9]. Maintenance of LTB4 gradients through specific inactivation by β-oxidation although possible, does not seem to be efficient given the very short half-life of LTB4 in vivo (less than half minute), which is very much like the β-oxidation resistant LTB4 analogs [85]. In fact, when compared to primary chemoattractants like formyl peptides, the release and the subsequent diffusion of AA, the synthetic precursor for LTB4, is extremely transient and does not create effective gradients [86]. While the stability of LTB4 may be enhanced in systemic circulation by binding to serum albumin [87], this would not explain its stability in interstitial spaces. Alternatively, stable and long acting LTB4 gradients could be maintained by facilitated diffusion through transcytosis, vesicle-mediated transport or packaging in extracellular vesicles such as exosomes [9]. Vesicular packaging represents an especially efficient process for dissemination of hydrophobic molecules. For example, lipid adduct morphogens in Drosophila such as Wnt are known to be released from cells in extracellular vesicles called argosomes [88]. Even non-lipid chemoattractants such as chemokines have been shown to be associated with extracellular vesicles [88]. In the social amoeba Dictyostelium cAMP, the primary chemoattractant that governs its development, has been proposed to be released through vesicular packaging [11].
Vesicular packaging of LTB4 - implication in chemotaxis and signal relay
Extracellular vesicles (EV) are small membrane bound organelles that are released from cells to the extracellular milieu. EVs derived from the plasma membrane (PM) are typically larger (~ 200 nm- 2 μm) and are called secretory vesicles, whereas smaller vesicles (~ 50-150 nm) derived intracellularly from multivesicular bodies are called exosomes. These EVs, initially thought to be cellular trash have been shown to mediate varied immune processes [89]. For example, exosomes from bronchoalveolar lavage fluid of patients with sarcoidosis contain FLAP, LTA4H and 5-LO and increase leukotriene production by bronchial epithelial cells [90]. Similarly, exosomes isolated from macrophages and dendritic cells contain the three LTB4 synthesizing enzymes [91] and are thus capable of LTB4 synthesis. Active cPLA2 and other eicosanoids are also released from exosomes derived from monocytic RBL-2H3 cells [92]. Recently, vesicular packaging of LTB4 was directly demonstrated in migrating neutrophils. Remarkably, the vesicular redistribution of 5-LO from the nuclear envelope into MVBs was readily observed in these cells upon fMLP-treatment. Furthermore, the secreted exosomes released LTB4 in a time-dependent manner, which was poised to activate neutrophils through autocrine or paracrine mechanisms. Further, the inhibition of exosome production lead to a decrease in exosomal LTB4 release and a loss in directed migration [14]. Together, these reports suggest that LTB4 and its synthesizing enzymes are packaged in exosomes, which upon secretion causes further release of LTB4 in a time-dependent manner to form a secondary chemoattractant gradient. This secondary gradient of LTB4 becomes the ‘bread-crumbs’ for recruiting and guiding incoming follower cells, as the primary chemoattractant gradient becomes too shallow to be effectively perceived. Exosomes may therefore amplify primary chemoattractant gradients by relaying their signals through LTB4. Our current understanding on the production and exosomal packaging of LTB4 in neutrophils is summarized in the Figure 1.
THE BIOLOGY OF LTB4 RECEPTORS
LTB4 receptor properties
LTB4 can bind and activate two distinct GPCRs - BLT1 and BLT2. BLT1 was cloned from the leukemic cell line HL-60 upon differentiation with retinoic acid [93]. It has a high affinity for LTB4, with a Kd of ~ 0.1-2 nM depending on the cell type [93, 94]. BLT2 was identified using the BLT1 sequence as a template and binds LTB4 with lower affinity (~20 nM) [95]. LTB4 binding to BLT1 requires the specific carbon backbone arrangement and the hydroxyl groups of LTB4. Importantly, LTB4-BLT1 interaction cannot be competed out by the presence of related lipids such as 12(S)-hydroxyeicosatetraenoic acid (12(S)-HETE), 12(S)-hydroperoxyeicosatetraenoic acid (12(S)-HPETE) or 15(S)-HETE. However, the binding of LTB4 to BLT2 can be competed out by 12(S)-HETE, 12(S)-HPETE or 15(S)-HETE [96]. In fact, 12(S)-hydroxyheptadecatrienoic acid (12-HHT), which is abundantly produced by platelets during blood coagulation, competes out LTB4 at a much lower concentration and has been described as the naturally occurring high-affinity in vivo ligand for BLT2 [94, 97]. Interestingly, the BLT1 promoter is also a part of the BLT2 open reading frame (ORF), which is upstream of BLT1 in the genome, raising the possibility of their co-regulation under certain conditions [95]. BLT1 and BLT2 belong to the family A of GPCRs consisting of Rhodopsin/β2-adrenergic-like receptors [98]. They are further subcategorized under the “non-chemokine chemoattractant receptors” group that includes formyl peptide receptors (FPR1 and FPR2), platelet activating factor receptor (PAFR), and complement-3/-5a receptors (C3aR and C5aR) [6].
Cellular distribution of LTB4 receptors
The expression of BLT1 and BLT2 has been investigated in a variety of tissues and cell types under a plethora of conditions. Generally, BLT2 is more widely expressed in tissues such as heart, lung, liver, kidney, pancreas, prostate gland, and ovary than is BLT1 [93–95, 98]. Innate immune cells such as neutrophils, monocytes, macrophages, eosinophils, basophils, mast cell and dendritic cells represent the most prominent group of cells that express BLT1. Interestingly, BLT2 is expressed in most of the above-mentioned innate immune cells except for basophils and macrophages. The ability of lymphocytes - T and B cells - as well as endothelial cells to specifically express BLT1, but not BLT2, is also intriguing [98].
G protein specificity of BLTs
BLT1 and BLT2 are characterized by the presence of a DRY motif within their second intracellular domain, which is known to interact with the Gαi subunit of the heterotrimeric G protein complex [6]. Initial studies showed that BLT1 can couple to both Gαi and Gq subunits [93]. However, it was subsequently established that the LTB4-dependent BLT1 receptor activation is sensitive to pertussis toxin and therefore a Gαi-mediated event [93, 99–101]. Interestingly, and unlike BLT1, BLT2 can associate with different G proteins, such as Gα14 and to a lesser extent with Gαq and Gα11 [97].
The role of helix8 and GRKs in BLT trafficking
The binding of a ligand to its cognate GPCR leads to a conformational change in the receptor resulting in loading of GTP to the Gα subunit of the heterotrimeric G protein complex [101]. This stage represents the sensitized state of the GPCR. Once the receptor is activated, the GTP-loaded G protein is released from the receptor and the Gα and βγ subunits then dissociate to regulate downstream signaling pathways [98, 102]. The G protein-dissociated receptors then become prone to phosphorylation of Ser/Thr residues in their intracellular C-terminal tail by GPCR regulated kinases (GRKs), protein kinase C (PKC) or cAMP-dependent protein kinases (PKA). This step marks the desensitization of the receptor. Once phosphorylated, the tail of the GPCR becomes susceptible to loading of arrestin molecules, which then recruit the adapter AP2 molecule to help dock clathrin, a mediator of receptor endocytosis [101, 102]. Among the different intracellular domains, the C-terminal tail from the seventh transmembrane domain harbors the intracellular helix 8, which is critical for a subset of GPCRs responses, including desensitization and internalization [94]. Surprisingly, BLTs lack the conserved cysteine residue within helix 8, which upon palmitoylation anchors the helix 8 of Class A GPCRs to the PM. Instead, based on modeling studies, hydrophobic di-leucine residues (Leu304-Leu305) within helix 8 are predicted to anchor helix 8 of BLT1 to the PM [101]. However, conversion of the di-leucine residues to alanine residues (LL/AA) had little impact on the ability of the mutant to localize to the PM. Yet, upon LTB4 stimulation, while the LL/AA mutant exhibits resistance to internalization when expressed in COS-7 cells [103], the same mutant shows higher internalization compared to wild-type (WT) receptor in CHO cells [104]. Interestingly, the ability of the LL/AA mutant to bind LTB4 and subsequently induce inositol phosphate (IP) accumulation is better than that of WT BLT1 in both of these studies. In terms of trafficking, unlike WT BLT1, the LL/AA mutant strongly co-localizes with clathrin upon stimulation with LTB4 [104]. BLT1 internalization is dependent on dynamin activity, but surprisingly independent of β-arrestin-2 that is generally involved in clathrin-mediated endocytosis of GPCRs [105].
The major drivers of BLT1 internalization are GRK2, GRK5 and GRK6 [105]. In cells that express low endogenous GRK2 amounts, such as HEK293 and COS-7, GRK6 regulates BLT1 endocytosis by phosphorylating Thr308 residue in helix 8 [105, 106]. In contrast, in cells that express high amounts of GRK2, BLT1 endocytosis is dependent on the interaction of GRK2 with the C-terminal tail of BLT1 upon LTB4 stimulation [105]. Therefore, it is possible that multiple GRKs can interact with BLT1 simultaneously to affect its internalization in a dynamin-dependent manner. With regards to BLT2, helix 8 is required for efficient transport of BLT2 from the ER to the PM. Also, addition of agonists for BLT2, such as 12-HHT, promote the re-localization of the LL/AA mutant of BLT2 from the ER to the PM, exhibiting a chaperon-like effect of the ligand [107]. The trafficking of WT and LL/AA BLT1 and BLT2 mutants remains to be studied during neutrophil migration, where the LTB4-BLT1 signaling axis is required for efficient chemotaxis in vitro and in vivo [12, 13].
Regulation of signaling pathways upon BLT activation
The activation of BLT1 and BLT2 regulates a plethora of downstream signaling events that impact on a wide array of cellular functions. However, in the context of this review we will restrict our discussion to the specific signaling cascades that contribute to BLT-induced leukocyte migration. BLT1 is known to mediate migration of several cell types in vitro and in vivo, including vascular smooth muscle cells [108], neutrophils [109], eosinophils [110], macrophages [111], mast cells [112], CD8+ T cells [113], Th17 cells [114], Treg cells [115] and γδ T cells [116]. On the other hand, BLT2 has been shown to regulate migration only in neutrophils [109], mast cells [112] and dendritic cells [117]. It is also important to point out that LTB4-BLT1/BLT2-mediated signaling events in leukocytes have not been completely deciphered, compared to the events mediated by fMLP or IL8.
Upstream phosphatidylinositol signaling
In the context of leukocyte migration, GPCRs activate a variety of upstream signaling events including phospholipase β (PLCβ), Ras and other small GTPases that dictate polarity and directional migration [3]. Once dissociated from the G protein heterotrimeric complex, Gβγ subunits activate PLCβ, which in turn catalyzes the breakdown of phosphotidylinositol(4,5)bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). Both DAG and IP3 then act as second messengers to transduce downstream signals [102]. In neutrophils, activation of Ras is dependent on PLCβ2 and PLCβ3, which activate a Ras guanine exchange factor (RasGRP4) to promote Ras activation [118]. Chemoattractants such as fMLP and LTB4 induce the production of phosphotidylinositol(3,4,5)trisphosphate (PIP3) in neutrophils [119]. This occurs following Ras binding and activation of the Class IB phosphatidylinositol 3-kinase (PI3K), p110γ [120]. Indeed, PI3Kγ-dependent production of PIP3 contributes to neutrophil chemokinesis and chemotaxis [120–122].
Small GTPase activation and actin regulation
The dynamic actin polymerization present at the leading edge of migrating leukocytes is dependent on the presence of a branched actin network that is regulated by the Arp2/3 complex [3] [123]. The activation of the Arp2/3 complex is in turn dependent on Rac driven SCAR/WAVE and Cdc42 driven WASP activation [3]. Filamentous actin (F-actin) production at the leading edge of neutrophils in response to stimulation with chemoattractant is in part dependent on the activity of PI3K/AKT/PKC [124]. PIP3 binds and activates the Rac GEF DOCK2, thereby activating Rac and in turn F-actin assembly, neutrophil polarization and chemotaxis [125]. Importantly, activated Rac feeds back to further increase the production of PIP3 during neutrophil chemotaxis [126]. Apart from Rac, PIP3 is required for the activation of another small GTPase Cdc42 [126] and LTB4 is known to activate both these small GTPases during dendritic cell migration [127]. Although Cdc42 does promote actin nucleation through WASP and Arp2/3 complex, Cdc42 is perhaps more important for the maintenance of polarity and suppression of lateral protrusions during neutrophil chemotaxis [126, 128, 129]. Overall, specific signaling cascades activated downstream of GPCR stimulation are dedicated to promote actin dynamics at the front to drive pseudopod extension and thereby leukocyte migration. However, it remains unclear as to why neutrophils require the LTB4-BLT1 signaling axis to efficiently enhance cAMP production, F-actin assembly and to maintain neutrophil polarity in response to a primary chemoattractant such as fMLP [12].
Molecular players in back retraction
For efficient chemotaxis, the signals that regulate back retraction of a migrating leukocyte are just as important as the events dedicated to regulate front protrusions. Just as PIP3, Rac and Cdc42 regulate actin polymerization at the leading edge, PIP2, Rho, Rho-associated kinase (ROCK), Myosin II light chain kinase (MLCK) and Myosin II dictate posterior dynamics during neutrophil chemotaxis [3, 130]. Importantly, in response to fMLP much of the active Ras, Rac and Cdc42 preferentially localize to the front while RhoA is restricted to the center and back in chemotaxing neutrophils [131]. The restricted activity of RhoA is important, as dominant-positive RhoA mutants inhibit neutrophil polarity and dominant-negative or inhibition of Rho leads to the formation of multiple front protrusions accompanied with elevated Rac2 activity and delayed back retraction [132]. The interplay of specific molecules is required for the spatio-temporal regulation of Myosin II light chain phosphorylation, which in turn regulates back retraction. For ROCK-dependent MLCK to phosphorylate myosin light chain, calcium from intracellular stores released upon IP3 production in response to GPCR stimulation is required [133]. Another signaling cascade that is important for Myosin II-mediated back retraction is the mammalian Target of Rapamycin Complex 2 (mTORC2)-dependent cAMP generation, which feeds into the RhoA pathway [134, 135]. In the context of neutrophil signal-relay, LTB4 acts in a paracrine as well as autocrine manner to facilitate polarity and persistence of chemotactic neutrophils to a primary attractant such as fMLP. This is explained in part by the lack of sustained phosphorylated myosin light chain when the LTB4-BLT1 axis is inhibited in response to fMLP, which most-likely impacts back retraction [12]. However, the molecular dependence of FPR1 on BLT1-mediated myosin light chain phosphorylation for back retraction is an open area of research.
LTB4 and leukocyte adhesion
Adhesion to and detachment from the substrate have a central role in the migration of a cell. In the case of neutrophils as well as other leukocytes, the ability to maintain low-adhesion and detachment occurs in the back, and, not surprisingly, several adhesion molecules such as P-Selectin ligand 1 (PSGL1), CD11a and intercellular adhesion molecule 1 (ICAM1) are found in the back of migrating neutrophils [130, 136]. Integrins are a class of adhesion molecules expressed by mammalian cells, including leukocytes, that are rapidly activated by chemoattractants to signal ‘inside-out’ and thereby mediate adhesion and leukocyte migration [137]. This is in contrast to observed endothelial and fibroblast cell migration, which require ‘outside-in’ integrin signals accompanied by focal adhesion kinase activation and downstream signaling to drive motility [138]. The ability of LTB4 and other chemoattractants to induce adhesion of neutrophils to endothelial cells depends on β2 integrins – CD11a, CD11b, CD11c - and the common β chain CD18 [139, 140]. Importantly, perturbation of integrin molecules, directly or indirectly, impacts leukocyte migration behavior [141, 142]. Although chemoattractants in general induce β2-integrins clustering, mobility and ‘inside-out’ activation in neutrophils, LTB4 induces a modest yet significant increase in integrin affinity when compared to fMLP [143]. Also, unlike fMLP, LTB4 fails to promote the translocation of β1-integrins to the PM and migration through fibrin [144]. Interestingly, while LTB4 and other chemoattractants promote β2-integrin expression on the PM, the adhesion molecule gp100MEL-14 is actively shed from the surface following stimulation [145].
The small GTPase Rap1 contributes significantly to adhesion, polarity and migration of leukocytes including neutrophils. Rap1 is activated downstream of Ras upon GPCR stimulation in neutrophils [118] and activation of β1-, β2- and β3-integrins depends on the activity of Rap1, which in turn is dependent on the exchange factor CalDAG-GEFI. In the absence of CalDAG-GEFI, impaired actin arrangement at the leading edge of neutrophils is accompanied by defective chemotaxis towards LTB4 [146]. In fact, the constitutively active Rap1aQ63E mutant or overexpression of the Rap1 effector Radil, induces greater adhesion and reduced back retraction in neutrophil-like cells [147]. Although, integrins are not necessary for neutrophil chemotaxis in tissues, it is important to point out that cells lacking Talin, a regulator of integrin activation, of BLT1 have strikingly similar phenotypes in terms of their inability to penetrate collagen-free zone upon injury in vivo [13], suggesting that LTB4 signaling is required for integrin activation. The signaling events driving BLT1 trafficking, neutrophil polarity and adhesion in response to LTB4 stimulation is pictorially depicted in Figure 2.
Figure 2. BLT1-mediated signaling in leukocyte.
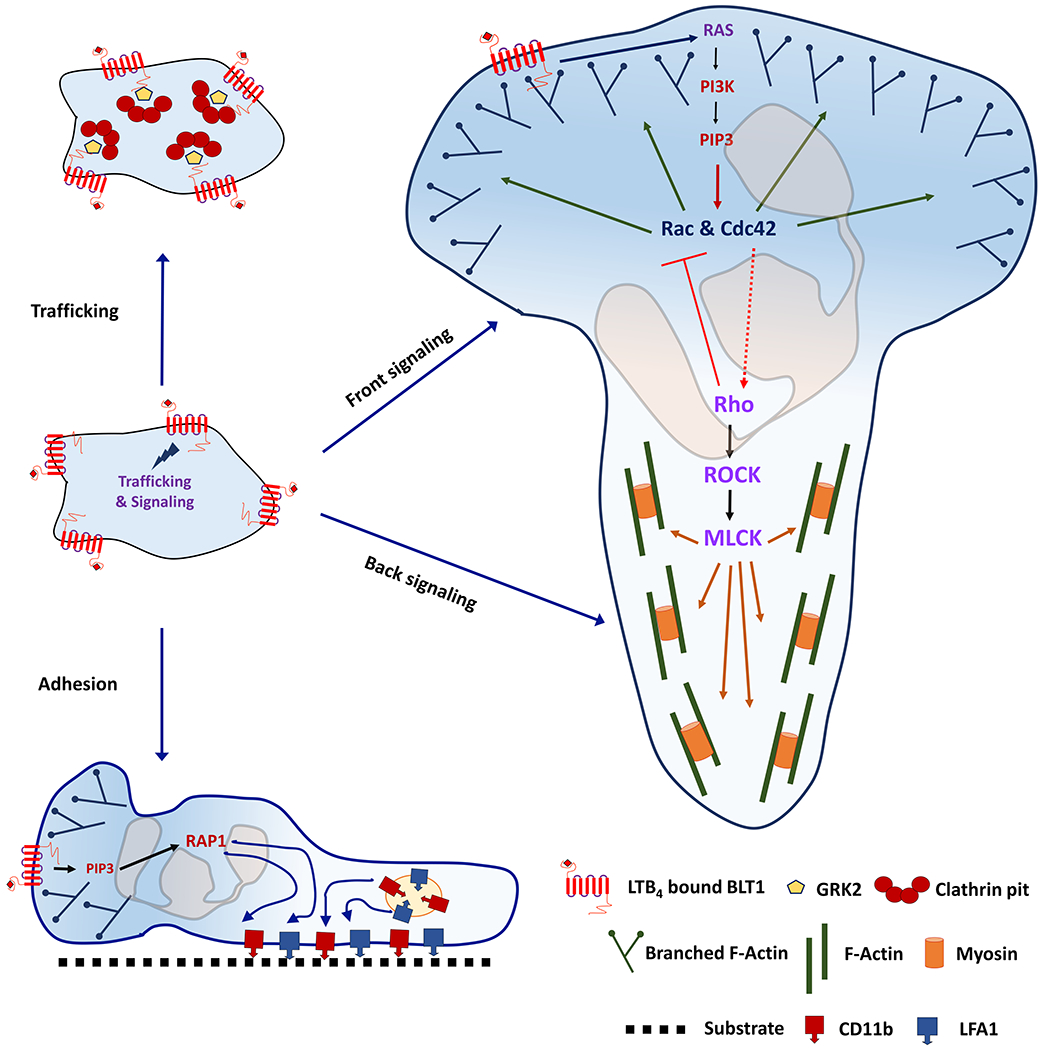
A schematic representation of the signaling events that occur upon the activation of BLT1 by LTB4. Clathrin-mediated endocytosis of BLT1, front and back signaling as well as inside-out integrin activation in a polarized neutrophil are shown.
LTB4 and MAPK activation
The other signal transduction pathway downstream of GPCR activation is the mitogen associated protein kinases (MAPKs). LTB4 can activate MAPKs – ERK, p38MAPK and JNK - in multiple cell types such as fibroblasts [148], neutrophils [149] [150] and pancreatic cancer cells [151]. In neutrophils, the activation of ERK and p38MAPK, but not JNK, are dependent on PI3K activity upon fMLP stimulation [152]. Although evidence for the role of ERK during chemotaxis is yet to be clearly established, it is required for the survival and anti-apoptotic effects of LTB4 on neutrophils [150]. Similarly, while LTB4-induced JNK activation has been shown to regulate chemokine production in monocytes, the role of JNK activation during neutrophil chemotaxis is yet to be clearly established [153]. On the other hand, p38MAPK activation is required for efficient chemotaxis to a number of chemoattractants including LTB4, fMLP and C5a [122]. Although competition of p38MAPK with GRK2 to bind and phosphorylate FPR1 has been proposed [154], the exact mechanism by which activated p38MAPK impacts neutrophil chemotaxis to different chemoattractants remains unclear.
Contrasting the actions of LTB4 to related lipid mediators
Other LTB4-related ecosianoids that bind to specific GPCRs have very different effects from those of LTB4 on target cells and thereby immune responses. For example, intraperitoneal injection of LTC4, unlike LTB4, fails to induce neutrophil recruitment in mice [155]. While LTB4 is produced by fMLP-stimulated neutrophils and is essential for the persistence of directionality during chemotaxis [12], the addition of a BLT2 agonist, 12-HETE, suppresses fMLP-induced calcium flux and migration of neutrophils [156]. Also, Lipoxin A4 (LXA4) that binds to LXA4R or FPR2, a receptor highly homologous to FPR1, inhibits fMLP-induced CD11b up-regulation and aggregation of neutrophils and thus exhibits anti-inflammatory properties [157]. In fact, pre-treatment of neutrophils with LXA4 or Lipoxin B4 (LXB4), a structurally distinct member of the lipoxin family that signals differently from that of LXA4, inhibits responses such as calcium flux and chemotaxis to LTB4 as well as to fMLP [158]. Interestingly, LXA4 treatment blocks the binding of Jurkat cells to endothelial cells in response to the cytokines IL1β and TNFα and inhibits E-Selectins, ICAM1 and vascular cell adhesion molecule 1, VCAM1 [159]. Similarly, LXB4 inhibits degranulation of mast cells and eosinophils, reduces eosinophil chemotaxis and represses type 2 cytokine receptors in a murine airway allergic inflammation model [160]. Overall, LTB4 and structurally related ligands bind to distinct GPCRs and while some of them appear to promote leukocyte function during inflammation, others actively suppress activation and effector functions of leukocytes.
THE PHYSIOLOGICAL ACTIONS OF THE LTB4-BLT1 AXIS
Competing chemoattractant gradients in leukocyte migration
The release of cellular LTB4 is usually a consequence of tissue damage, infection or other insults. LTB4 is critical to amplify chemotactic responses to a primary chemoattractant [12]. However, immune cells, such as leukocytes, encounter mixed and opposing gradients of chemoattractants in vivo. For example, during endotoxemia, a pathophysiological global activation of inflammatory responses due to bacterial endotoxins, leukocytes encounter both bacterial-derived formylated peptides as well as host-derived chemoattractants such as LTB4 and IL8 [161]. Interestingly, under these conditions, the response of neutrophils to host-derived chemoattractants is reduced, whereas chemotaxis towards bacterial peptides remains unaffected [162]. Hence, a hierarchical preference for certain chemoattractants has been proposed for leukocytes. Indeed, it has been shown in vitro that leukocytes respond preferentially to end-target chemoattractants, such as bacterial peptides, over other host-derived factors or intermediate chemoattractants, such as LTB4 [163]. This ability to distinguish exogenous cues enables leukocytes to differentiate immediate threats over host-derived endogenous chemoattractant gradients.
A signaling preference in chemoattractant hierarchy
The terms end-target and intermediate chemoattractants are also relevant in terms of the signaling pathways they elicit. Neutrophils predominantly migrate to end target chemoattractants in a p38MAPK-dependent manner, whereas migration to intermediate chemoattractant such as LTB4 and IL8, is more PI3K/AKT-dependent [8]. The negative bias of neutrophils towards LTB4 in the presence of end-target chemoattractants is most likely because of the inhibitory effects of p38MAPK on the molecule PTEN as PTEN−/− neutrophils can be distracted by an intermediate chemoattractant gradient while migrating towards fMLP [164]. Interestingly, cells moving towards a very shallow concentration gradient of fMLP show higher chemotactic response time and speed in the presence of LTB4, as compared to gradients of only LTB4 or fMLP [165]. Similarly, monocyte migration to fMLP increases when cells are pretreated with LTB4, although the reverse, i.e. migration to LTB4 when pretreated with fMLP, remains unaffected [166]. Moreover, neutrophils treated with LTB4 receptor antagonists show decreased motility towards fMLP, indicating that the fMLP-induced release of LTB4 is important for neutrophil motility towards an end-target chemoattractant [14, 167]. Taken together, these findings would suggest that although the p38MAPK pathway has an overriding effect on the PI3K-AKT pathway, the reverse may not be true and may be dependent on the intensity of its activation. Although leukocyte migration in competing gradient is expected to be frequent, if not common during infection and inflammation, only a handful of studies have addressed such a phenomenon. Establishment of signaling hierarchy in competing gradients requires further investigations involving parameters such as signal strength, time and stochastic variation within cellular population. Single cell measurements of key signaling pathways in a spatio-temporal manner will also help further resolved these issues.
The IL8-LTB4 power struggles
Migration of cells towards opposing intermediate chemoattractant, e.g. LTB4 versus IL8, seems to be less obvious and assay dependent. In under agarose assays, leukocyte migration is not dependent on either LTB4 or IL8, but gradient shape and distance between the two sources [168]. Indeed, it has been established that neutrophils migrate toward the more distant attractant source and away from the more proximal one, independently of the chemoattractant species, by integrating signals from both species [169]. Microfluidic chambers that create linear competing gradients of LTB4 and IL8 show a weak hierarchical preference of LTB4 over IL8 in directing neutrophil chemotaxis [170]. This observation is in agreement with in vivo conditions found in chronic obstructive pulmonary disease patients where LTB4 levels correlate strongly with neutrophil accumulation compared to IL8 levels [171]. On the other hand, competing gradients created by microfluidic platforms utilizing shear flow, suggest a reversal of the hierarchical order, with IL8 eliciting preferential neutrophil chemotaxis compared to LTB4 [172]. The discrepancy in these results may be due to the inherent hydrophobicity of LTB4 and the calculation of apparent diffusivity of LTB4. Notwithstanding the order of preference, intermediate chemoattractants such as LTB4 can contribute to the “chemotactic memory” of the cell that promotes sequential chemotaxis from one chemoattractant to another without losing sensitivity of migration. The lack of ordered preference enables a leukocyte that is migrating towards an IL-8 gradient to turn away and chemotax towards LTB4 irrespective of the concentration gradients of the individual chemoattractant [173]. The mechanisms that control how such disparate molecules elicit similar physiological responses remain unknown.
The LTB4-BLT axis in infection and inflammation
In a relatively simple model of injecting LTB4 intra-peritoneally, BLT1 is required for neutrophil, but not macrophage, infiltration. However, unlike injection of LTB4 alone, intra-peritoneal injection of a more physiological stimulant such as zymosan or thioglycollate leads to recruitment of neutrophils, macrophages and eosinophils in a BLT1-dependent manner, most likely requiring LTB4-BLT1 signal-relay downstream to primary inflammatory agents [174, 175]. Similarly, LTB4 production is known to elicit the recruitment of neutrophils and other leukocytes in several mouse disease models including endotoxemia-induced multiple organ injury [176], diabetic retinopathy [177], abdominal aortic aneurysms [178], sterile inflammation in diabetes [179], contact dermatitis [180], cecal ligation and puncture model of sepsis [181], traumatic spinal cord injury model [182], experimental autoimmune encephalomyelitis [183] and T. cruzi infection [184]. However, in some in vivo models, cytokine action precedes and induces LTB4 production and function. For example, in a mouse model of ovalbumin-induced intra-peritoneal recruitment of neutrophils, LTB4 production is dependent on the secretion of TNFα by CD4+ T cells [185]. In another mouse model, the production of IL1β and TNFα by mast cells as well as LTB4 production by macrophages dictates the IL1β-induced recruitment of neutrophils to the peritoneum [186]. The LTB4-BLT1 axis also promotes the migration of smooth muscle cells during atherosclerosis and intimal hyperplasia in vivo and the IL1β-induced expression of BLT1 in these cells appears to be NFκB-dependent [187]. In vivo, hemolytic events are known to induce LTB4 production and the recruitment of neutrophils. Indeed, heme alone can act upon resident macrophages and induce LTB4 production, which in turn can recruit neutrophils to the peritoneal cavity in mice [188]. The LTB4-BLT1 axis can also function to mediate much more complex inflammatory scenarios. For example, in the murine inflammatory arthritis model deposition of auto-antibody-antigen complex to the joints triggers complement-mediated reaction and the recruitment of neutrophils in a C5aR-dependent manner. The activation of C5aR precedes LTB4 release by neutrophils to recruit more neutrophils to the joint [189]. On the other hand, FcγR is activated on the recruited neutrophils that signals through the LTB4-BLT1 axis to produce IL1, which then promotes further neutrophil recruitment in a CXCR1- and CXCR2-dependent manner [190]. Therefore, although multiple cytokines might be essential for initial immune response in variety of different models, they all converge in activating LTB4-BLT1 mediated signal-relay for efficient leukocyte recruitment.
In several disease models, LTB4-BLT1 interaction on specific leukocyte cell types contributes to the observed phenotype. In a viral peptide-induced colitis mouse model, BLT1 promotes the recruitment of specific cytotoxic CD8+ T cells [191]. In mouse models of airway hyper-responsiveness, allergen-sensitized and challenged mice display BLT1-dependent recruitment of neutrophils, eosinophils, dendritic cells, CD4+ and CD8+ effector T cells, which together promote TH2 type responses in the lungs [192–196]. In humans, BLT1 is up-regulated on T cells from lung transplant recipients and in a mouse lung transplantation model, BLT1 mediates CD8+ T cell recruitment and thereby transplant rejection [197]. In another mouse model, the LTB4-BLT1 axis mediates the activation of autoreactive T cells and effector leukocyte function to cause autoimmune uveitis [198]. LTB4-BLT1 interactions in general promote inflammation under most circumstances. However, in a mouse model of Lyme arthritis induced by B. burgdorferi, the lack of 5-LO resulted in faster joint swelling, a defective clearance of apoptotic neutrophils by macrophages and lesser uptake of opsonized spirochete by neutrophils [199]. Therefore, the LTB4-BLT1 axis contributes to inflammatory responses in a context-specific manner.
The LTB4-BLT axis: extravasation versus tissue migration
It is pertinent to differentiate the effects of LTB4-BLT1-mediated signal-relay on purely chemotactic responses with that on other leukocyte responses in the context of inflammation. In fact, BLT1 is required for efficient leukocyte arrest in response to LTB4 in the cremaster muscle venule in mice [174]. Therefore, in most, if not all, of the above mentioned mouse infection and inflammation model systems, the LTB4-BLT1 axis most likely contributes to inflammation at multiple levels including rolling, arrest/adhesion to the endothelium, extravasation of activated leukocytes from blood vessels and interstitial chemotaxis. However, in the case of a laser induced ear injury model in mice, neutrophils were labeled and injected in the ear prior to laser injury. In this model, where WT and mutant neutrophils are directly compared in the same tissue, long distance neutrophil recruitment and directionality were 5-LO- and BLT1-dependent [13]. The primary attractants are predicted to be FPR2 ligands and necrosis of the first responding neutrophils, which then use the LTB4-BLT1 axis to relay signals to neighboring neutrophils to be recruited over longer distances. Interestingly, apart from functioning primarily as a chemoattractant, LTB4 can also induce neutrophils to deliver elastase in a CD11b-dependent manner to endothelial cells expressing junctional adhesion molecule and thereby promote neutrophil’s reverse-transmigration [200]. These and other such examples highlight the adaptation of the LTB4-BLT1 axis for extravasation, tissue migration and reverse migration of leukocytes in vivo. It, however, remains to be determined if exosomal packaging of LTB4 and its synthesizing enzymes is involved in mediating all of the LTB4-BLT1 signaling events. Further studies in different immune cell types and under multiple inflammatory conditions are required to determine if such packaging in exosomes applies to LTB4 and other small signaling molecules that direct leukocyte migration.
LTB4-related eicosanoids and their distinct chemoattractant properties
Unlike 5-LO products, products of 15-LO, such as LXA4, function distinctly different from LTB4 under many circumstances. For example, 15-LO is required for efficient epithelial wound healing in the mouse cornea, most likely by inhibiting KC production and promoting timed neutrophil recruitment to the corneal epithelium [201]. Second, in certain sputum samples from chronic bronchitis patients, amounts of 15-LO and 15(S)-HETE inversely correlate with LTB4 amounts, suggesting an inhibitory nature of one on the other. Third, treatment of neutrophils with 15(S)-HETE inhibits their ability to produce LTB4 in response to A23187 or fMLP in vitro [202]. Therefore, LTB4 and structurally related eicosanoids can differentially signal to leukocytes to induce distinct and opposing chemotactic responses.
Unlike BLT1, BLT2 has an anti-inflammatory role in mice as BLT2−/− mice are more sensitive to dextran sodium sulfate-induced colitis. In this case, BLT2 appears to regulate barrier functions of colonic epithelial cells [203]. Similarly, the 12-HHT/BLT2 axis is important for skin wound closure and healing, most probably by inducing keratinocytes to produce TNFα and MMPs during the process [204]. Importantly, the expression of BLT2 is significantly reduced in CD4+ T cells from the bronchiolar lavage in human asthma patients compared to healthy control subjects. Not surprisingly, unlike BLT1, the lack of BLT2 specifically promotes higher IL-13 producing CD4+ T cells and thereby contributes to eosinophilia in the lungs of a airway hyper-responsive mouse model [205].
PERSPECTIVES
Given that most chemoattractant GPCRs are Gαi-coupled and are expected to activate similar pathways, it will be important to understand how similar signaling cascades activated by structurally distinct ligands can bring about different phenotypic responses in the same target cells. In this review, we largely focused on the role of the LTB4-BLT1 axis in directing leukocyte migration in a variety of physiologically relevant conditions. We emphasized the intricate events leading to LTB4 synthesis and potential transport within exosomes (Fig. 1). Following the production of LTB4, we discussed aspects of BLT1-mediated downstream signaling essential for efficient leukocyte polarization and chemotaxis (Fig. 2). We also discuss the context in which the LTB4-BLT1 axis functions in a complex and competing gradient environment (Fig. 3). However, it remains to be determined how the LTB4-BLT1 axis regulates specific signaling pathways in response to primary chemoattractants during immune cell migration. Dissecting such intricate and complex signaling networks will undoubtedly lead to a clearer understanding of leukocyte function and a better ability to design drugs that have specific impact on a variety of inflammatory diseases.
Figure 3. Functional roles of the LTB4-BLT1 axis in diverse cell migration scenarios.
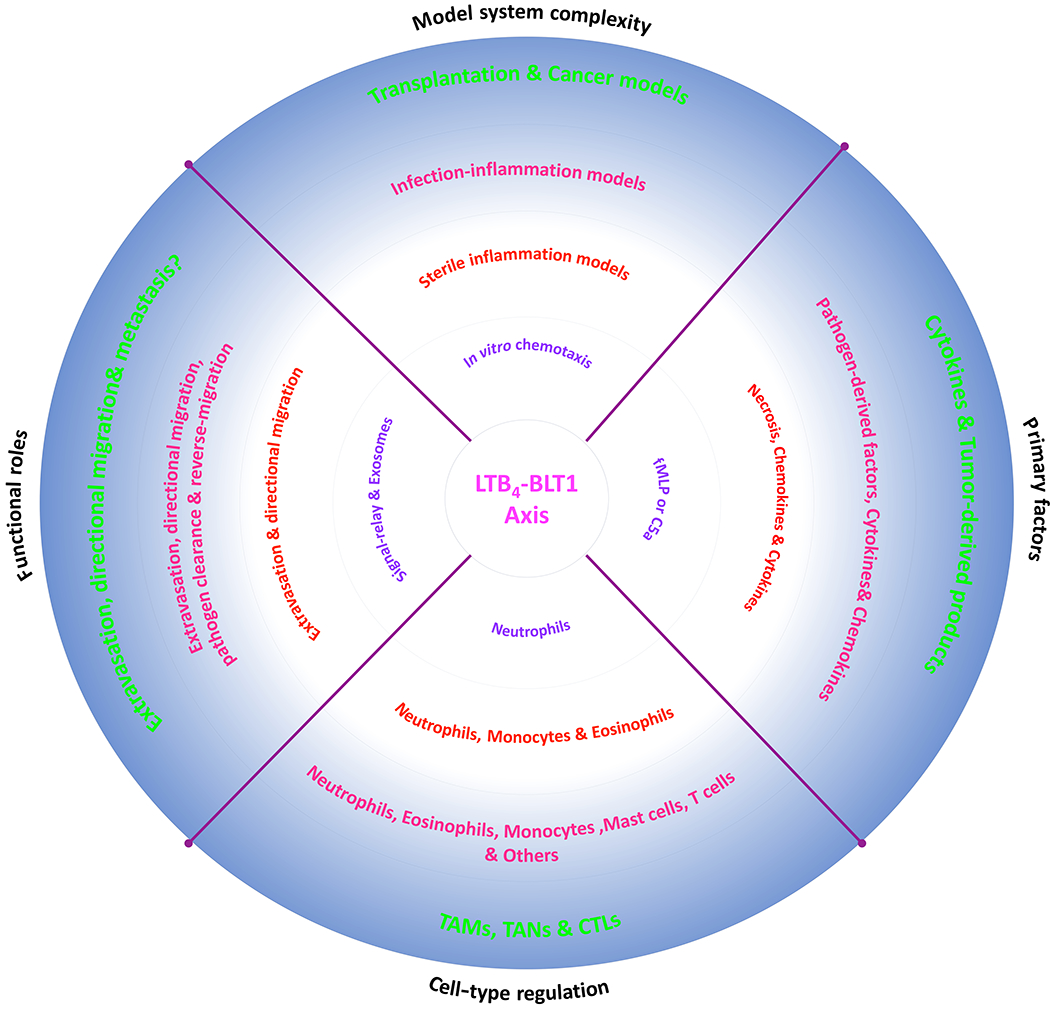
A pie chart depicting the diverse roles of the LTB4-BLT1 axis in distinct situations that involve increased complexities, factors and cell-types.
Overall, there is no doubt about or on the contribution of the LTB4-BLT1 axis as a relay module in the recruitment of several different cell types in a physiological setting (Fig. 3). However, other small molecules, such as 5-oxo-ETE [206], H2O2 [207] and Redox signaling [208], can also potentially function as drivers of signal-relay among leukocytes in the presence or absence of the LTB4-BLT1 axis. Although the packaging/transport of such small molecules needs further characterization, the above examples highlight the distinct advantage of small molecules that are produced rapidly, in robust amounts and propagated using efficient diffusion mechanisms to form spatio-temporal gradients that are required for the long-distance recruitment of leukocytes during inflammation.
ACKNOWLEDGEMENTS
We thank Christina Stuelten for helpful discussions. We apologize to authors whose recent work could not be included in this review. This effort was supported by the Intramural Research Program of the Center for Cancer Research, NCI, National Institutes of Health.
REFERENCES
- 1.Scharf BE, Hynes MF, and Alexandre GM, Chemotaxis signaling systems in model beneficial plant–bacteria associations. Plant Molecular Biology, 2016. 90(6): p. 549–559. [DOI] [PubMed] [Google Scholar]
- 2.Keilberg DO, K. M, How Helicobacter pylori senses, targets and interacts with the gastric epithelium. Environ Microbiol., 2016. 18(3): p. 791–806. [DOI] [PubMed] [Google Scholar]
- 3.Artemenko Y, Lampert TJ, & Devreotes PN, Moving towards a paradigm: common mechanisms of chemotactic signaling in Dictyostelium and mammalian leukocytes. Cell Mol Life Sci, 2014. 71(19): p. 3711–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molyneaux KW, C., Primordial germ cell migration. Int J Dev Biol., 2004. 48(5-6): p. 537–44. [DOI] [PubMed] [Google Scholar]
- 5.Son K, Brumley DR, and Stocker R, Live from under the lens: exploring microbial motility with dynamic imaging and microfluidics. Nature Reviews Microbiology, 2015. 13(12): p. 761–775. [DOI] [PubMed] [Google Scholar]
- 6.Zabel BA, Rott A, & Butcher EC, Leukocyte chemoattractant receptors in human disease pathogenesis. Annu Rev Pathol, 2015. 10: p. 51–81. [DOI] [PubMed] [Google Scholar]
- 7.Sadik CD and Luster AD, Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. Journal of Leukocyte Biology, 2011. 91(2): p. 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heit B, et al. , An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol, 2002. 159(1): p. 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majumdar R, Sixt M, and Parent CA, New paradigms in the establishment and maintenance of gradients during directed cell migration. Current Opinion in Cell Biology, 2014. 30: p. 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kriebel PW, Barr VA, & Parent CA, Adenylyl cyclase localization regulates streaming during chemotaxis. Cell, 2003. 112(4): p. 549–60. [DOI] [PubMed] [Google Scholar]
- 11.Kriebel PW, et al. , Collective cell migration requires vesicular trafficking for chemoattractant delivery at the trailing edge. The Journal of Cell Biology, 2008. 183(5): p. 949–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Afonso PV, Janka-Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, Losert W, Cicerone MT, & Parent CA, LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev Cell, 2012. 22(5): p. 1079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, & Germain RN, Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature, 2013. 498(7454): p. 371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majumdar R, Tavakoli Tameh A, and Parent CA, Exosomes Mediate LTB4 Release during Neutrophil Chemotaxis. PLoS Biol, 2016. 14(1): p. e1002336. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Sumida H, et al. , Interplay between CXCR2 and BLT1 facilitates neutrophil infiltration and resultant keratinocyte activation in a murine model of imiquimod-induced psoriasis. J Immunol, 2014. 192(9): p. 4361–9. [DOI] [PubMed] [Google Scholar]
- 16.Borgeat P and Samuelsson B, Transformation of arachidonic acid by rabbit polymorphonuclear leukocytes. Formation of a novel dihydroxyeicosatetraenoic acid. J Biol Chem, 1979. 254(8): p. 2643–6. [PubMed] [Google Scholar]
- 17.Borgeat P and Samuelsson B, Arachidonic acid metabolism in polymorphonuclear leukocytes: effects of ionophore A23187. Proc Natl Acad Sci U S A, 1979. 76(5): p. 2148–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuelsson B, et al. , Introduction of a nomenclature: leukotrienes. Prostaglandins, 1979. 17(6): p. 785–7. [DOI] [PubMed] [Google Scholar]
- 19.Warner JA, et al. , Purified human basophils do not generate LTB4. Biochem Pharmacol, 1987. 36(19): p. 3195–9. [DOI] [PubMed] [Google Scholar]
- 20.Weller PF, et al. , Generation and metabolism of 5-lipoxygenase pathway leukotrienes by human eosinophils: predominant production of leukotriene C4. Proc Natl Acad Sci U S A, 1983. 80(24): p. 7626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams JD, Czop JK, and Austen KF, Release of leukotrienes by human monocytes on stimulation of their phagocytic receptor for particulate activators. J Immunol, 1984. 132(6): p. 3034–40. [PubMed] [Google Scholar]
- 22.Schonfeld W, et al. , Leukotriene generation and metabolism in isolated human lung macrophages. Immunology, 1988. 65(4): p. 529–36. [PMC free article] [PubMed] [Google Scholar]
- 23.Heavey DJ, et al. , Generation of leukotriene C4, leukotriene B4, and prostaglandin D2 by immunologically activated rat intestinal mucosa mast cells. The Journal of Immunology, 1988. 140(6): p. 1953–7. [PubMed] [Google Scholar]
- 24.Atluru D and Goodwin JS, Leukotriene B4 causes proliferation of interleukin 2-dependent T cells in the presence of suboptimal levels of interleukin 2. Cell Immunol, 1986. 99(2): p. 444–52. [DOI] [PubMed] [Google Scholar]
- 25.Goldyne ME and Rea L, Stimulated T cell and natural killer (NK) cell lines fail to synthesize leukotriene B4. Prostaglandins, 1987. 34(6): p. 783–95. [DOI] [PubMed] [Google Scholar]
- 26.Goldyne ME and Rea L, Stimulated T cell and natural killer (NK) cell lines fail to synthesize leukotriene B4. Prostaglandins, 1987. 34(6): p. 783–795. [DOI] [PubMed] [Google Scholar]
- 27.Jakobsson PJ, Odlander B, and Claesson HE, Effects of monocyte-lymphocyte interaction on the synthesis of leukotriene B4. Eur J Biochem, 1991. 196(2): p. 395–400. [DOI] [PubMed] [Google Scholar]
- 28.Shindo K, Koide K, and Fukumura M, Enhancement of leukotriene B4 release in stimulated asthmatic neutrophils by platelet activating factor. Thorax, 1997. 52(12): p. 1024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva RC, et al. , Leukotrienes produced in allergic lung inflammation activate alveolar macrophages. Cell Physiol Biochem, 2010. 26(3): p. 319–26. [DOI] [PubMed] [Google Scholar]
- 30.Holtzman MJ, et al. , Uptake, release and novel species-dependent oxygenation of arachidonic acid in human and animal airway epithelial cells. Biochim Biophys Acta, 1988. 963(3): p. 401–13. [DOI] [PubMed] [Google Scholar]
- 31.Uz T, et al. , Aging-associated up-regulation of neuronal 5-lipoxygenase expression: putative role in neuronal vulnerability. FASEB J, 1998. 12(6): p. 439–49. [DOI] [PubMed] [Google Scholar]
- 32.Waki H, et al. , Excessive Leukotriene B4 in Nucleus Tractus Solitarii Is Prohypertensive in Spontaneously Hypertensive Rats. Hypertension, 2013. 61(1): p. 194-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manev H, et al. , Putative role of neuronal 5-lipoxygenase in an aging brain. FASEB J, 2000. 14(10): p. 1464–9. [DOI] [PubMed] [Google Scholar]
- 34.Tomimoto H, et al. , A comparative study on the expression of cyclooxygenase and 5-lipoxygenase during cerebral ischemia in humans. Acta Neuropathol, 2002. 104(6): p. 601–7. [DOI] [PubMed] [Google Scholar]
- 35.Arai K, Nishiyama N, Matsuki N, & Ikegaya Y, Neuroprotective effects of lipoxygenase inhibitors against ischemic injury in rat hippocampal slice cultures. Brain Res, 2001. 904(1): p. 167–72. [DOI] [PubMed] [Google Scholar]
- 36.Sahin A, et al. , Regulation of leukotriene B4 secretion by human corneal, conjunctival, and meibomian gland epithelial cells. Arch Ophthalmol, 2012. 130(8): p. 1013–8. [DOI] [PubMed] [Google Scholar]
- 37.Mothe-Satney I, et al. , Adipocytes Secrete Leukotrienes. Contribution to Obesity-Associated Inflammation and Insulin Resistance in Mice, 2012. 61(9): p. 2311–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sellmayer A, et al. , Increased leukotriene B4 synthesis in patients with primary hyperparathyroidism is normalized after parathyroidectomy: a study comparing parathyroidectomy to thyroid adenomectomy. J Clin Endocrinol Metab, 1987. 64(2): p. 387–90. [DOI] [PubMed] [Google Scholar]
- 39.Hawthorne AB, et al. , Colorectal leukotriene B4 synthesis in vitro in inflammatory bowel disease: inhibition by the selective 5-lipoxygenase inhibitor BWA4C. Gut, 1992. 33(4): p. 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudson N, et al. , Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non-steroidal anti-inflammatory drugs. Gut, 1993. 34(6): p. 742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burke JE and Dennis EA, Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res, 2009. 50 Suppl: p. S237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woods JW, et al. , 5-lipoxygenase and 5-lipoxygenase-activating protein are localized in the nuclear envelope of activated human leukocytes. J Exp Med, 1993. 178(6): p. 1935–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pande AH, Qin S, and Tatulian SA, Membrane fluidity is a key modulator of membrane binding, insertion, and activity of 5-lipoxygenase. Biophys J, 2005. 88(6): p. 4084–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pande AHM, D.; Nemec KN; Qin S; Tan S; Tatulian SA, Modulation of human 5-lipoxygenase activity by membrane lipids. Biochemistry, 2004. 43(46): p. 14653–66. [DOI] [PubMed] [Google Scholar]
- 45.Ferguson AD, et al. , Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science, 2007. 317(5837): p. 510–2. [DOI] [PubMed] [Google Scholar]
- 46.Hafner AK, et al. , Characterization of the interaction of human 5-lipoxygenase with its activating protein FLAP. Biochim Biophys Acta, 2015. 1851(11): p. 1465–72. [DOI] [PubMed] [Google Scholar]
- 47.Ford-Hutchinson AW, Gresser M, & Young RN, 5-Lipoxygenase. Annual Review of Biochemistry, 1994. 63(1): p. 383–417. [DOI] [PubMed] [Google Scholar]
- 48.Haeggström JZ, Tholander F, and Wetterholm A, Structure and catalytic mechanisms of leukotriene A 4 hydrolase. Prostaglandins & other lipid mediators, 2007. 83(3): p. 198–202. [DOI] [PubMed] [Google Scholar]
- 49.Gijon MA and Leslie CC, Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J Leukoc Biol, 1999. 65(3): p. 330–6. [DOI] [PubMed] [Google Scholar]
- 50.Gijón MAS, D. M; Kaiser AL; Leslie CC, Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J Cell Biol., 1999. 145(6): p. 1219–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Das S, et al. , Mechanism of group IVA cytosolic phospholipase A(2) activation by phosphorylation. J Biol Chem, 2003. 278(42): p. 41431–42. [DOI] [PubMed] [Google Scholar]
- 52.Lin LLW, M.; Lin AY; Knopf JL; Seth A; Davis RJ, cPLA2 is phosphorylated and activated by MAP kinase. Cell, 1993. 72(2): p. 269–78. [DOI] [PubMed] [Google Scholar]
- 53.Fatima SY, F. A; Ahmed A; Khandekar Z; Malik KU, CaM kinase IIalpha mediates norepinephrine-induced translocation of cytosolic phospholipase A2 to the nuclear envelope. Journal of Cell Science, 2002. 116(2): p. 353–365. [DOI] [PubMed] [Google Scholar]
- 54.Schievella ARR, M. K; Smith WL; Lin LL, Calcium-mediated translocation of cytosolic phospholipase A2 to the nuclear envelope and endoplasmic reticulum. J Biol Chem, 1995. 270(51): p. 30749–54. [DOI] [PubMed] [Google Scholar]
- 55.Syrbu SI, et al. , Phosphorylation of Cytosolic Phospholipase A2 and the Release of Arachidonic Acid in Human Neutrophils. The Journal of Immunology, 1999. 162(4): p. 2334–2340. [PubMed] [Google Scholar]
- 56.Jakobsson PJ, et al. , Membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG). A widespread protein superfamily. Am J Respir Crit Care Med, 2000. 161(2 Pt 2): p. S20–4. [DOI] [PubMed] [Google Scholar]
- 57.Ferguson AD, Structure-based drug design on membrane protein targets: human integral membrane protein 5-lipoxygenase-activating protein. Methods Mol Biol, 2012. 841: p. 267–90. [DOI] [PubMed] [Google Scholar]
- 58.Plante H, et al. , 5-Lipoxygenase-activating protein homodimer in human neutrophils: evidence for a role in leukotriene biosynthesis. Biochem J, 2006. 393(Pt 1): p. 211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mandal AK, et al. , The membrane organization of leukotriene synthesis. Proc Natl Acad Sci U S A, 2004. 101(17): p. 6587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Healy AM, et al. , Identification of a bipartite nuclear localization sequence necessary for nuclear import of 5-lipoxygenase. J Biol Chem, 1999. 274(42): p. 29812–8. [DOI] [PubMed] [Google Scholar]
- 61.Luo M, et al. , Multiple nuclear localization sequences allow modulation of 5-lipoxygenase nuclear import. Traffic, 2004. 5(11): p. 847–54. [DOI] [PubMed] [Google Scholar]
- 62.Chen XS and Funk CD, The N-terminal “beta-barrel” domain of 5-lipoxygenase is essential for nuclear membrane translocation. J Biol Chem, 2001. 276(1): p. 811–8. [DOI] [PubMed] [Google Scholar]
- 63.Kulkarni S, et al. , Molecular basis of the specific subcellular localization of the C2-like domain of 5-lipoxygenase. J Biol Chem, 2002. 277(15): p. 13167–74. [DOI] [PubMed] [Google Scholar]
- 64.Flamand N, et al. , Arachidonic acid regulates the translocation of 5-lipoxygenase to the nuclear membranes in human neutrophils. J Biol Chem, 2006. 281(1): p. 129–36. [DOI] [PubMed] [Google Scholar]
- 65.Woods JW, et al. , 5-Lipoxygenase is located in the euchromatin of the nucleus in resting human alveolar macrophages and translocates to the nuclear envelope upon cell activation. J Clin Invest, 1995. 95(5): p. 2035–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brock TG, et al. , Rapid import of cytosolic 5-lipoxygenase into the nucleus of neutrophils after in vivo recruitment and in vitro adherence. J Biol Chem, 1997. 272(13): p. 8276–80. [DOI] [PubMed] [Google Scholar]
- 67.Pergola C, et al. , ERK-mediated regulation of leukotriene biosynthesis by androgens: a molecular basis for gender differences in inflammation and asthma. Proc Natl Acad Sci U S A, 2008. 105(50): p. 19881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Flamand N, et al. , Phosphorylation of serine 271 on 5-lipoxygenase and its role in nuclear export. J Biol Chem, 2009. 284(1): p. 306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rakonjac M, et al. , Coactosin-like protein supports 5-lipoxygenase enzyme activity and up-regulates leukotriene A4 production. Proceedings of the National Academy of Sciences, 2006. 103(35): p. 13150–13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hammarberg T, et al. , The N-terminal domain of 5-lipoxygenase binds calcium and mediates calcium stimulation of enzyme activity. J Biol Chem, 2000. 275(49): p. 38787–93. [DOI] [PubMed] [Google Scholar]
- 71.Mandal AK, et al. , The nuclear membrane organization of leukotriene synthesis. Proc Natl Acad Sci U S A, 2008. 105(51): p. 20434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thunnissen MM, Nordlund P, and Haeggström JZ, Crystal structure of human leukotriene A4 hydrolase, a bifunctional enzyme in inflammation. Nature Structural & Molecular Biology, 2001. 8(2): p. 131–135. [DOI] [PubMed] [Google Scholar]
- 73.Fabre J-E, et al. , Transcellular biosynthesis contributes to the production of leukotrienes during inflammatory responses in vivo. The Journal of Clinical Investigation, 2002. 109(10): p. 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mueller MJ, et al. , Leukotriene A4 hydrolase: mapping of a henicosapeptide involved in mechanism-based inactivation. Proc Natl Acad Sci U S A, 1995. 92(18): p. 8383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brock TG, et al. , Co-localization of Leukotriene A4Hydrolase with 5-Lipoxygenase in Nuclei of Alveolar Macrophages and Rat Basophilic Leukemia Cells but Not Neutrophils. Journal of Biological Chemistry, 2001. 276(37): p. 35071–35077. [DOI] [PubMed] [Google Scholar]
- 76.Fiedler J, et al. , Effect of peroxisome proliferator-activated receptor alpha activation on leukotriene B4 metabolism in isolated rat hepatocytes. J Pharmacol Exp Ther, 2001. 299(2): p. 691–7. [PubMed] [Google Scholar]
- 77.Narala VR, et al. , Leukotriene B4 is a physiologically relevant endogenous peroxisome proliferator-activated receptor-alpha agonist. J Biol Chem, 2010. 285(29): p. 22067–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Devchand PR, et al. , The PPARalpha-leukotriene B4 pathway to inflammation control. Nature, 1996. 384(6604): p. 39–43. [DOI] [PubMed] [Google Scholar]
- 79.Leslie CC, Properties and regulation of cytosolic phospholipase A2. J Biol Chem, 1997. 272(27): p. 16709–12. [DOI] [PubMed] [Google Scholar]
- 80.Williams JD, et al. , Intracellular retention of the 5-lipoxygenase pathway product, leukotriene B4, by human neutrophils activated with unopsonized zymosan. J Immunol, 1985. 134(4): p. 2624–30. [PubMed] [Google Scholar]
- 81.Mita H, et al. , Isocratic determination of arachidonic acid 5-lipoxygenase products in human neutrophils by high-performance liquid chromatography. J Chromatogr, 1988. 430(2): p. 299–308. [DOI] [PubMed] [Google Scholar]
- 82.Lam BK, et al. , The mechanism of leukotriene B4 export from human polymorphonuclear leukocytes. Journal of Biological Chemistry, 1990. 265(23): p. 13438–13441. [PubMed] [Google Scholar]
- 83.Rius M, Hummel-Eisenbeiss J, and Keppler D, ATP-dependent transport of leukotrienes B4 and C4 by the multidrug resistance protein ABCC4 (MRP4). J Pharmacol Exp Ther, 2008. 324(1): p. 86–94. [DOI] [PubMed] [Google Scholar]
- 84.Capannolo M, et al. , The atypical antipsychotic clozapine selectively inhibits interleukin 8 (IL-8)-induced neutrophil chemotaxis. Eur Neuropsychopharmacol, 2015. 25(3): p. 413–24. [DOI] [PubMed] [Google Scholar]
- 85.Marleau SDN; Poubelle PE; Borgeat P, Metabolic disposition of leukotriene B4 (LTB4) and oxidation-resistant analogues of LTB4 in conscious rabbits. Br J Pharmacol, 1994. 112(2): p. 654–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Uden AM, Hafstrom I, and Palmblad J, Relation to chemotactic factor gradients to neutrophil migration and orientation under agarose. J Leukoc Biol, 1986. 39(1): p. 27–35. [DOI] [PubMed] [Google Scholar]
- 87.Zsila F, Bikádi Z, and Lockwood SF, In vitro binding of leukotriene B 4 (LTB 4) to human serum albumin: evidence from spectroscopic, molecular modeling, and competitive displacement studies. Bioorganic & medicinal chemistry letters, 2005. 15(16): p. 3725–3731. [DOI] [PubMed] [Google Scholar]
- 88.Greco V, Hannus M, and Eaton S, Argosomes: a potential vehicle for the spread of morphogens through epithelia. Cell, 2001. 106(5): p. 633–645. [DOI] [PubMed] [Google Scholar]
- 89.Robbins PD and Morelli AE, Regulation of immune responses by extracellular vesicles. Nat Rev Immunol, 2014. 14(3): p. 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Torregrosa Paredes P, et al. , Bronchoalveolar lavage fluid exosomes contribute to cytokine and leukotriene production in allergic asthma. Allergy, 2012. 67(7): p. 911–9. [DOI] [PubMed] [Google Scholar]
- 91.Esser J, et al. , Exosomes from human macrophages and dendritic cells contain enzymes for leukotriene biosynthesis and promote granulocyte migration. J Allergy Clin Immunol, 2010. 126(5): p. 1032–40, 1040 e1-4. [DOI] [PubMed] [Google Scholar]
- 92.Subra C, et al. , Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J Lipid Res, 2010. 51(8): p. 2105–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yokomizo T, Izumi T, Chang K, Takuwa Y, & Shimizu T, A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature, 1997. 387(5): p. 620–624. [DOI] [PubMed] [Google Scholar]
- 94.Yokomizo T, Two distinct leukotriene B4 receptors, BLT1 and BLT2. J Biochem, 2015. 157(2): p. 65–71. [DOI] [PubMed] [Google Scholar]
- 95.Yokomizo T, Kato K, Terawaki K, Izumi T, & Shimizu T, A Second Leukotriene B4 Receptor, BLT2: A New Therapeutic Target in Inflammation and Immunological Disorders. J. Exp. Med, 2000. 192(3): p. 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yokomizo T, et al. , Hydroxyeicosanoids bind to and activate the low affinity leukotriene B4 receptor, BLT2. J Biol Chem, 2001. 276(15): p. 12454–9. [DOI] [PubMed] [Google Scholar]
- 97.Okuno T, Iizuka Y, Okazaki H, Yokomizo T, Taguchi R, & Shimizu T, 12(S)-Hydroxyheptadeca-5Z, 8E, 10E-trienoic acid is a natural ligand for leukotriene B4 receptor 2. J Exp Med, 2008. 205(4): p. 759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gether U, Uncovering Molecular Mechanisms Involved in Activation of G Protein-Coupled Receptors. Endocrine Reviews, 2000. 21(1): p. 90–113. [DOI] [PubMed] [Google Scholar]
- 99.Haribabu B, Zhelev DV, Pridgen CV, Richardson RM, and Ali H, & Snyderman R, Chemoattractant Receptors Activate Distinct Pathways for Chemotaxis and Secretion. J Biol Chem, 1999. 274(52): p. 37087–37092. [DOI] [PubMed] [Google Scholar]
- 100.Ito N, Yokomizo T, Sasaki T, Kurosu H, Penninger J, Kanaho Y, Katada T, Hanaoka K, & Shimizu T, Requirement of phosphatidylinositol 3-kinase activation and calcium influx for leukotriene B4-induced enzyme release. J Biol Chem, 2002. 277(47): p. 44898–904. [DOI] [PubMed] [Google Scholar]
- 101.Okuno T, Yokomizo T, Hori T, Miyano M, & Shimizu T, Leukotriene B4 receptor and the function of its helix 8. J Biol Chem, 2005. 280(37): p. 32049–52. [DOI] [PubMed] [Google Scholar]
- 102.Ritter SL, & Hall RA, Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Biol, 2009. 10(12): p. 819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gaudreau R, Beaulieu ME, Chen Z, Le Gouill C, Lavigne P, Stankova J, & Rola-Pleszczynski M, Structural determinants regulating expression of the high affinity leukotriene B4 receptor: involvement of dileucine motifs and alpha-helix VIII. J Biol Chem, 2004. 279(11): p. 10338–45. [DOI] [PubMed] [Google Scholar]
- 104.Aratake Y, Okuno T, Matsunobu T, Saeki K, Takayanagi R, Furuya S, & Yokomizo T, Helix 8 of leukotriene B4 receptor 1 inhibits ligand-induced internalization. FASEB J, 2012. 26(10): p. 4068–78. [DOI] [PubMed] [Google Scholar]
- 105.Chen Z, Gaudreau R, Le Gouill C, Rola-Pleszczynski M, & Stanková J, Agonist-Induced Internalization of Leukotriene B4 Receptor 1 requires G-Protein-Coupled Receptor Kinase 2 but not Arrestins. Mol Pharmacol, 2004. 66(3): p. 377–386. [DOI] [PubMed] [Google Scholar]
- 106.Gaudreau R, Le Gouill C, Venne MH, Stankova J, & Rola-Pleszczynski M, Threonine 308 within a putative casein kinase 2 site of the cytoplasmic tail of leukotriene B(4) receptor (BLT1) is crucial for ligand-induced, G-protein-coupled receptor-specific kinase 6-mediated desensitization. J Biol Chem, 2002. 277(35): p. 31567–76. [DOI] [PubMed] [Google Scholar]
- 107.Yasuda D, Okuno T, Yokomizo T, Hori T, Hirota N, Hashidate T, Miyano M, Shimizu T, & Nakamura M, Helix 8 of leukotriene B4 type-2 receptor is required for the folding to pass the quality control in the endoplasmic reticulum. FASEB J, 2009. 23(5): p. 1470–81. [DOI] [PubMed] [Google Scholar]
- 108.Moraes J, Assreuy J, Canetti C, & Barja-Fidalgo C, Leukotriene B4 mediates vascular smooth muscle cell migration through alphavbeta3 integrin transactivation. Atherosclerosis, 2010. 212(2): p. 406–13. [DOI] [PubMed] [Google Scholar]
- 109.Mathis SP, Jala VR, Lee DM, & Haribabu B, Nonredundant roles for leukotriene B4 receptors BLT1 and BLT2 in inflammatory arthritis. J Immunol, 2010. 185(5): p. 3049–56. [DOI] [PubMed] [Google Scholar]
- 110.Gladue RP, Carroll LA, Milici AJ, Scampoli DN, , Stukenbrok HA, Pettipher EI~, Salter ED, Contillo L, & Showell HJ , Inhibition of Leukotriene B4-Receptor Interaction Suppresses Eosinophil Infiltration and Disease Pathology in a Murine Model of Experimental Allergic Encephalomyelitis. J Exp Med. , 1996. 183(4): p. 1893–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Mayoral R, Maris M, Ofrecio JM, Taguchi S, Lu M, Olefsky JM, LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med, 2015. 21(3): p. 239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lundeen KA, Sun B, Karlsson L, & Fourie AM, Leukotriene B4 Receptors BLT1 and BLT2: Expression and Function in Human and Murine Mast Cells. The Journal of Immunology, 2006. 177(5): p. 3439–3447. [DOI] [PubMed] [Google Scholar]
- 113.Sharma RK, Chheda Z, Jala VR, & Haribabu B, Expression of leukotriene B(4) receptor-1 on CD8(+) T cells is required for their migration into tumors to elicit effective antitumor immunity. J Immunol, 2013. 191(6): p. 3462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee W, Su Kim H, & Lee GR, Leukotrienes induce the migration of Th17 cells. Immunol Cell Biol, 2015. 93(5): p. 472–9. [DOI] [PubMed] [Google Scholar]
- 115.Wang L, Zhao L, Lv J, Yin Q, Liang X, Chu Y, & He R, BLT1-dependent alveolar recruitment of CD4(+)CD25(+) Foxp3(+) regulatory T cells is important for resolution of acute lung injury. Am J Respir Crit Care Med, 2012. 186(10): p. 989–98. [DOI] [PubMed] [Google Scholar]
- 116.Costa MF, de Souza-Martins R, de Souza MC, Benjamim CF, Piva B, Diaz BL, Peters-Golden M, Henriques Md, Canetti C, & Penido C, Leukotriene B4 mediates gammadelta T lymphocyte migration in response to diverse stimuli. J Leukoc Biol, 2010. 87(2): p. 323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shin EH, Lee HY, & Bae YS, Leukotriene B4 stimulates human monocyte-derived dendritic cell chemotaxis. Biochem Biophys Res Commun, 2006. 348(2): p. 606–11. [DOI] [PubMed] [Google Scholar]
- 118.Suire S, Lecureuil C, Anderson KE, Damoulakis G, Niewczas I, Davidson K, Guillou H, Pan D, Jonathan, Clark, Phillip T, Hawkins & Stephens L, GPCR activation of Ras and PI3Kc in neutrophils depends on PLCb2/b3 and the RasGEF RasGRP4. EMBO J, 2012. 31(14): p. 3118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Traynor-Kaplan AE, Thompson BL, Harris AL, Taylor P, Omann GM, & Sklar LA, Transient increase in phosphatidylinositol 3,4-bisphosphate and phosphatidylinositol trisphosphate during activation of human neutrophils. J Biol Chem., 1989. 264(26): p. 15668–73. [PubMed] [Google Scholar]
- 120.Suire S, Condliffe AM, Ferguson GJ, Ellson CD, Guillou H, Davidson K, Welch H, Coadwell J, Turner M, Chilvers ER, Hawkins PT, & Stephens L, Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol, 2006. 8(11): p. 1303–9. [DOI] [PubMed] [Google Scholar]
- 121.Ferguson GJ, Milne L, Kulkarni S, Sasaki T, Walker S, Andrews S, Crabbe T, Finan P, Jones G, Jackson S, Camps M, Rommel C, Wymann M, Hirsch E, Hawkins P, & Stephens L, PI(3)Kgamma has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol, 2007. 9(1): p. 86–91. [DOI] [PubMed] [Google Scholar]
- 122.Heit B, Tavener S, Raharjo E, & Kubes P, An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol, 2002. 159(1): p. 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weiner OD, Servant G, Welch MD, Mitchison TJ, Sedat JW, & Bourne HR, Spatial control of actin polymerization during neutrophil chemotaxis. Nat Cell Biol., 1999. 1(2): p. 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chodniewicz DZ, D. V, Chemoattractant receptor-stimulated F-actin polymerization in the human neutrophil is signaled by 2 distinct pathways. Blood, 2003. 101(3): p. 1181–4. [DOI] [PubMed] [Google Scholar]
- 125.Kunisaki Y, Nishikimi A, Tanaka Y, Takii R, Noda M, Inayoshi A, Watanabe K, Sanematsu F, Sasazuki T, Sasaki T, & Fukui Y, DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol, 2006. 174(5): p. 647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Srinivasan S, Wang F, Glavas S, Ott A, Hofmann F, Aktories K, Kalman D, & Bourne HR, Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol, 2003. 160(3): p. 375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sawada Y, Honda T, Hanakawa S, Nakamizo S, Murata T, Ueharaguchi-Tanada Y, Ono S, Amano W, Nakajima S, Egawa G, Tanizaki H, Otsuka A, Kitoh A, Dainichi T, Ogawa N, Kobayashi Y, Yokomizo T, Arita M, Nakamura M, Miyachi Y, & Kabashima K, Resolvin E1 inhibits dendritic cell migration in the skin and attenuates contact hypersensitivity responses. J Exp Med, 2015. 212(11): p. 1921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Glogauer M, Hartwig J, & Stossel T, Two pathways through Cdc42 couple the N-formyl receptor to actin nucleation in permeabilized human neutrophils. J Cell Biol., 2000. 150(4): p. 785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Szczur K, Zheng Y, & Filippi MD, The small Rho GTPase Cdc42 regulates neutrophil polarity via CD11b integrin signaling. Blood, 2009. 114(20): p. 4527–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hind LE, Vincent WJ, & Huttenlocher A, Leading from the Back: The Role of the Uropod in Neutrophil Polarization and Migration. Dev Cell, 2016. 38(2): p. 161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yang HW, Collins SR, & Meyer T, Locally excitable Cdc42 signals steer cells during chemotaxis. Nat Cell Biol, 2016. 18(2): p. 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, Takuwa Y, Sugimoto N, Mitchison T, & Bourne HR, Divergent Signals and Cytoskeletal Assemblies Regulate Self-Organizing Polarity in Neutrophils. Cell, 2003. 114(2): p. 201–214. [DOI] [PubMed] [Google Scholar]
- 133.Eddy RJ, Pierini LM, Matsumura F, Maxfield FR, Ca2+-dependent myosin II activation is required for uropod retraction during neutrophil migration. J Cell Sci, 2000. 113(7): p. 1287–98. [DOI] [PubMed] [Google Scholar]
- 134.Liu L, Das S, Losert W, & Parent CA, mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev Cell, 2010. 19(6): p. 845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Surve CR, To JY, Malik S, Kim M, & Smrcka AV, Dynamic regulation of neutrophil polarity and migration by the heterotrimeric G protein subunits Gαi-GTP and Gβγ. Sci Signal, 2016. 9(416): p. ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sánchez-Madrid FS, J. M, Bringing up the rear: defining the roles of the uropod. Nat Rev Mol Cell Biol, 2009. 10(5): p. 353–9. [DOI] [PubMed] [Google Scholar]
- 137.Kinashi T, Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol, 2005. 5(7): p. 546–59. [DOI] [PubMed] [Google Scholar]
- 138.Zhao X and Guan JL, Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev, 2011. 63(8): p. 610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zimmerman GAM, T. M, Neutrophil adherence to human endothelium in vitro occurs by CDw18 (Mo1, MAC-1/LFA-1/GP 150,95) glycoprotein-dependent and -independent mechanisms. J Clin Invest, 1988. 81(2): p. 531–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tonnesen MG, Anderson DC, Springer TA, Knedler A, Avdi N, & Henson PM, Adherence of neutrophils to cultured human microvascular endothelial cells. Stimulation by chemotactic peptides and lipid mediators and dependence upon the Mac-1, LFA-1, p150,95 glycoprotein family. J Clin Invest, 1989. 83(2): p. 637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pouwels J, De Franceschi N, Rantakari P, Auvinen K, Karikoski M, Mattila E, Potter C, Sundberg JP, Hogg N, Gahmberg CG, Salmi M, & Ivaska J, SHARPIN regulates uropod detachment in migrating lymphocytes. Cell Rep, 2013. 5(3): p. 619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, Reichel CA, Krombach F, Pardi R, & Dejana E, JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci, 2009. 122(Pt 2): p. 268–77. [DOI] [PubMed] [Google Scholar]
- 143.Patcha V, Wigren J, Winberg ME, Rasmusson B, Li J, & Sarndahl E, Differential inside-out activation of beta2-integrins by leukotriene B4 and fMLP in human neutrophils. Exp Cell Res, 2004. 300(2): p. 308–19. [DOI] [PubMed] [Google Scholar]
- 144.Loike JD, Cao L, Budhu S, Marcantonio EE, El Khoury J, Hoffman S, Yednock TA, & Silverstein SC, Differential regulation of beta1 integrins by chemoattractants regulates neutrophil migration through fibrin. J Cell Biol., 1999. 144(5): p. 1047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kishimoto TK, Jutila MA, Berg EL, & Butcher EC, Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science, 1989. 245(4923): p. 1238–41. [DOI] [PubMed] [Google Scholar]
- 146.Carbo C, Duerschmied D, Goerge T, Hattori H, Sakai J, Cifuni SM, White GC 2nd, Chrzanowska-Wodnicka M, Luo HR, & Wagner DD, Integrin-independent role of CalDAG-GEFI in neutrophil chemotaxis. J Leukoc Biol, 2010. 88(2): p. 313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liu L, Aerbajinai W, Ahmed SM, Rodgers GP, Angers S, & Parent CA, Radil controls neutrophil adhesion and motility through beta2-integrin activation. Mol Biol Cell, 2012. 23(24): p. 4751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Woo CH, You HJ, Cho SH, Eom YW, Chun JS, Yoo YJ, & Kim JH, Leukotriene B(4) stimulates Rac-ERK cascade to generate reactive oxygen species that mediates chemotaxis. J Biol Chem, 2002. 277(10): p. 8572–8. [DOI] [PubMed] [Google Scholar]
- 149.Gaudreault EG, J., Leukotriene B4 potentiates CpG signaling for enhanced cytokine secretion by human leukocytes. J Immunol, 2009. 183(4): p. 2650–8. [DOI] [PubMed] [Google Scholar]
- 150.Petrin D, Turcotte S, Gilbert AK, Rola-Pleszczynski M, & Stankova J, The anti-apoptotic effect of leukotriene B4 in neutrophils: a role for phosphatidylinositol 3-kinase, extracellular signal-regulated kinase and Mcl-1. Cell Signal, 2006. 18(4): p. 479–87. [DOI] [PubMed] [Google Scholar]
- 151.Tong WG, Ding XZ, Talamonti MS, Bell RH, & Adrian TE, LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem Biophys Res Commun, 2005. 335(3): p. 949–56. [DOI] [PubMed] [Google Scholar]
- 152.Rane MJ, Carrithers SL, Arthur JM, Klein JB, & McLeish KR, Formyl peptide receptors are coupled to multiple mitogen-activated protein kinase cascades by distinct signal transduction pathways: role in activation of reduced nicotinamide adenine dinucleotide oxidase. J Immunol., 1997. 159(10): p. 5070–8. [PubMed] [Google Scholar]
- 153.Huang L, Zhao A, Wong F, Ayala JM, Struthers M, Ujjainwalla F, Wright SD, Springer MS, Evans J, & Cui J, Leukotriene B4 strongly increases monocyte chemoattractant protein-1 in human monocytes. Arterioscler Thromb Vasc Biol, 2004. 24(10): p. 1783–8. [DOI] [PubMed] [Google Scholar]
- 154.Liu X, Ma B, Malik AB, Tang H, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y, Ye RD, & Xu J, Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nat Immunol, 2012. 13(5): p. 457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Griswold DE, Webb EF, & Hillegass LM, Induction of plasma exudation and inflammatory cell infiltration by leukotriene C4 and leukotriene B4 in mouse peritonitis. Inflammation, 1991. 15(4): p. 251–8. [DOI] [PubMed] [Google Scholar]
- 156.Yoshino H, Kitayama S, Morita K, Okamoto H, Tsujimoto A, & Dohi T, 12-Lipoxygenase product as an inhibitor of the action of chemoattractant peptide fMet-Leu-Phe in rat neutrophils. Gen Pharmacol, 1993. 24(5): p. 1249–51. [DOI] [PubMed] [Google Scholar]
- 157.Fiore SS, C. N, Lipoxin A4 receptor activation is distinct from that of the formyl peptide receptor in myeloid cells: inhibition of CD11/18 expression by lipoxin A4-lipoxin A4 receptor interaction. Biochemistry, 1995. 34(51): p. 16678. [DOI] [PubMed] [Google Scholar]
- 158.Lee TH, Horton CE, Kyan-Aung U, Haskard D, Crea AE, & Spur BW, Lipoxin A4 and lipoxin B4 inhibit chemotactic responses of human neutrophils stimulated by leukotriene B4 and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Clin Sci (Lond), 1989. 77(2): p. 195–203. [DOI] [PubMed] [Google Scholar]
- 159.Chinthamani S, Odusanwo O, Mondal N, Nelson J, Neelamegham S, & Baker OJ, Lipoxin A4 inhibits immune cell binding to salivary epithelium and vascular endothelium. Am J Physiol Cell Physiol, 2012. 302(7): p. C968–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Karra L, Haworth O, Priluck R, Levy BD, & Levi-Schaffer F, Lipoxin B(4) promotes the resolution of allergic inflammation in the upper and lower airways of mice. Mucosal Immunol, 2015. 8(4): p. 852–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.VanderMeer TJ, et al. , Acute lung injury in endotoxemic pigs: role of leukotriene B4. J Appl Physiol (1985), 1995. 78(3): p. 1121–31. [DOI] [PubMed] [Google Scholar]
- 162.Wagner JG and Roth RA, Neutrophil migration during endotoxemia. J Leukoc Biol, 1999. 66(1): p. 10–24. [DOI] [PubMed] [Google Scholar]
- 163.Phillipson M and Kubes P, The neutrophil in vascular inflammation. Nat Med, 2011. 17(11): p. 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Heit B, et al. , PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol, 2008. 9(7): p. 743–752. [DOI] [PubMed] [Google Scholar]
- 165.Cho H, et al. , On-demand, competing gradient arrays for neutrophil chemotaxis. Lab Chip, 2014. 14(5): p. 972–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ternowitz T, Herlin T, and Fogh K, Human monocyte and polymorphonuclear leukocyte chemotactic and chemokinetic responses to leukotriene B4 and FMLP. Acta Pathol Microbiol Immunol Scand C, 1987. 95(2): p. 47–54. [DOI] [PubMed] [Google Scholar]
- 167.Afonso PV, et al. , LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev Cell, 2012. 22(5): p. 1079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Foxman EF, Kunkel EJ, and Butcher EC, Integrating conflicting chemotactic signals The role of memory in leukocyte navigation. The Journal of cell biology, 1999. 147(3): p. 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Foxman EF, Campbell JJ, and Butcher EC, Multistep navigation and the combinatorial control of leukocyte chemotaxis. The Journal of cell biology, 1997. 139(5): p. 1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lin F, et al. , Neutrophil migration in opposing chemoattractant gradients using microfluidic chemotaxis devices. Ann Biomed Eng, 2005. 33(4): p. 475–82. [DOI] [PubMed] [Google Scholar]
- 171.Woolhouse IS, Bayley DL, and Stockley RA, Sputum chemotactic activity in chronic obstructive pulmonary disease: effect of alpha(1)-antitrypsin deficiency and the role of leukotriene B(4) and interleukin 8. Thorax, 2002. 57(8): p. 709–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim D and Haynes CL, Neutrophil Chemotaxis within a Competing Gradient of Chemoattractants. Analytical Chemistry, 2012. 84(14): p. 6070–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Foxman EF, Kunkel EJ, and Butcher EC, Integrating Conflicting Chemotactic Signals. The Role of Memory in Leukocyte Navigation, 1999. 147(3): p. 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tager AM, Dufour JH, Goodarzi K, Bercury SD, von Andrian UH, & Luster AD, BLTR mediates leukotriene B(4)-induced chemotaxis and adhesion and plays a dominant role in eosinophil accumulation in a murine model of peritonitis. J Exp Med, 2000. 192(3): p. 439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Haribabu B, Verghese MW, Steeber DA, Sellars DD, Bock CB, & Snyderman R, Targeted disruption of the leukotriene B(4) receptor in mice reveals its role in inflammation and platelet-activating factor-induced anaphylaxis. J Exp Med, 2000. 192(3): p. 433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Collin M, Rossi A, Cuzzocrea S, Patel NS, Di Paola R, Hadley J, Collino M, Sautebin L, & Thiemermann C, Reduction of the multiple organ injury and dysfunction caused by endotoxemia in 5-lipoxygenase knockout mice and by the 5-lipoxygenase inhibitor zileuton. J Leukoc Biol, 2004. 76(5): p. 961–70. [DOI] [PubMed] [Google Scholar]
- 177.Gubitosi-Klug RA, et al. , 5-Lipoxygenase, but Not 12/15-Lipoxygenase, Contributes to Degeneration of Retinal Capillaries in a Mouse Model of Diabetic Retinopathy. Diabetes, 2008. 57(5): p. 1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bhamidipati CM, et al. , 5-Lipoxygenase Pathway in Experimental Abdominal Aortic Aneurysms. Arteriosclerosis, Thrombosis, and Vascular Biology, 2014. 34(12): p. 2669–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Filgueiras LR, Brandt SL, Wang S, Wang Z, Morris DL, Evans-Molina C, Mirmira RG, Jancar S, & Serezani CH, Leukotriene B4-mediated sterile inflammation promotes susceptibility to sepsis in a mouse model of type 1 diabetes. Sci Signal, 2015. 8(361): p. ra10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lv J, et al. , Leukotriene B(4)-leukotriene B(4) receptor axis promotes oxazolone-induced contact dermatitis by directing skin homing of neutrophils and CD8(+) T cells. Immunology, 2015. 146(1): p. 50–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rios-Santos F, Benjamim CF, Zavery D, Ferreira SH, & Cunha Fde Q, A critical role of leukotriene B4 in neutrophil migration to infectious focus in cecal ligaton and puncture sepsis. Shock, 2003. 19(1): p. 61–5. [DOI] [PubMed] [Google Scholar]
- 182.Saiwai H, Ohkawa Y, Yamada H, Kumamaru H, Harada A, Okano H, Yokomizo T, Iwamoto Y, & Okada S, The LTB4-BLT1 axis mediates neutrophil infiltration and secondary injury in experimental spinal cord injury. Am J Pathol, 2010. 176(5): p. 2352–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kihara Y, et al. , The leukotriene B4 receptor, BLT1, is required for the induction of experimental autoimmune encephalomyelitis. Biochem Biophys Res Commun, 2010. 394(3): p. 673–8. [DOI] [PubMed] [Google Scholar]
- 184.Pavanelli WR, et al. , 5-Lipoxygenase is a key determinant of acute myocardial inflammation and mortality during Trypanosoma cruzi infection. Microbes and Infection, 2010. 12(8-9): p. 587–597. [DOI] [PubMed] [Google Scholar]
- 185.Canetti C, Silva JS, Ferreira SH, & Cunha FQ, Tumour necrosis factor-alpha and leukotriene B(4) mediate the neutrophil migration in immune inflammation. Br J Pharmacol, 2001. 134(8): p. 1619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Oliveira SH, Canetti C, Ribeiro RA, & Cunha FQ, Neutrophil migration induced by IL-1beta depends upon LTB4 released by macrophages and upon TNF-alpha and IL-1beta released by mast cells. Inflammation, 2008. 31(1): p. 36–46. [DOI] [PubMed] [Google Scholar]
- 187.Back M, Bu DX, Branstrom R, Sheikine Y, Yan ZQ, & Hansson GK, Leukotriene B4 signaling through NF-kappaB-dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc Natl Acad Sci U S A, 2005. 102(48): p. 17501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Monteiro AP, Pinheiro CS, Luna-Gomes T, Alves LR, Maya-Monteiro CM, Porto BN, Barja-Fidalgo C, Benjamim CF, Peters-Golden M, Bandeira-Melo C, Bozza MT, & Canetti C, Leukotriene B4 mediates neutrophil migration induced by heme. J Immunol, 2011. 186(11): p. 6562–7. [DOI] [PubMed] [Google Scholar]
- 189.Sadik CD, Kim ND, Iwakura Y, & Luster AD, Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcγR signaling. Proc Natl Acad Sci U S A, 2012. 109(46): p. E3177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chou RC, et al. , Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity, 2010. 33(2): p. 266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nancey S, Boschetti G, Hacini F, Sardi F, Durand PY, Le Borgne M, Furhmann L, Flourie B, & Kaiserlian D, Blockade of LTB(4) /BLT(1) pathway improves CD8(+) T-cell-mediated colitis. Inflamm Bowel Dis, 2011. 17(1): p. 279–88. [DOI] [PubMed] [Google Scholar]
- 192.Miyahara N, et al. , Leukotriene B4 Receptor-1 Is Essential for Allergen-Mediated Recruitment of CD8+ T Cells and Airway Hyperresponsiveness. The Journal of Immunology, 2005. 174(8): p. 4979–4984. [DOI] [PubMed] [Google Scholar]
- 193.Miyahara N, et al. , Requirement for leukotriene B4 receptor 1 in allergen-induced airway hyperresponsiveness. Am J Respir Crit Care Med, 2005. 172(2): p. 161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Medoff BD, et al. , Antibody-antigen interaction in the airway drives early granulocyte recruitment through BLT1. Am J Physiol Lung Cell Mol Physiol, 2006. 290(1): p. L170–8. [DOI] [PubMed] [Google Scholar]
- 195.Terawaki K, et al. , Absence of Leukotriene B4 Receptor 1 Confers Resistance to Airway Hyperresponsiveness and Th2-Type Immune Responses. The Journal of Immunology, 2005. 175(7): p. 4217–4225. [DOI] [PubMed] [Google Scholar]
- 196.Miyahara N, et al. , Leukotriene B4 Receptor 1 Expression on Dendritic Cells Is Required for the Development of Th2 Responses and Allergen-Induced Airway Hyperresponsiveness. The Journal of Immunology, 2008. 181(2): p. 1170–1178. [DOI] [PubMed] [Google Scholar]
- 197.Medoff BD, et al. , BLT1-mediated T cell trafficking is critical for rejection and obliterative bronchiolitis after lung transplantation. J Exp Med, 2005. 202(1): p. 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Liao T, et al. , Blockade of the interaction of leukotriene b4 with its receptor prevents development of autoimmune uveitis. Invest Ophthalmol Vis Sci, 2006. 47(4): p. 1543–9. [DOI] [PubMed] [Google Scholar]
- 199.Blaho VA, et al. , 5-Lipoxygenase-Deficient Mice Infected with Borrelia burgdorferi Develop Persistent Arthritis. The Journal of Immunology, 2011. 186(5): p. 3076–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Colom B, et al. , Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity, 2015. 42(6): p. 1075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gronert K, Maheshwari N, Khan N, Hassan IR, Dunn M, & Laniado Schwartzman M, A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J Biol Chem, 2005. 280(15): p. 15267–78. [DOI] [PubMed] [Google Scholar]
- 202.Profita M, Sala A, Riccobono L, Pace E, Paternò A, Zarini S, Siena L, Mirabella A, Bonsignore G, & Vignola AM, 15(S)-HETE modulates LTB(4) production and neutrophil chemotaxis in chronic bronchitis. Am J Physiol Cell Physiol, 2000. 279(4): p. C1249–58. [DOI] [PubMed] [Google Scholar]
- 203.Iizuka Y, Okuno T, Saeki K, Uozaki H, Okada S, Misaka T, Sato T, Toh H, Fukayama M, Takeda N, Kita Y, Shimizu T, Nakamura M, & Yokomizo T, Protective role of the leukotriene B4 receptor BLT2 in murine inflammatory colitis. FASEB J, 2010. 24(12): p. 4678–90. [DOI] [PubMed] [Google Scholar]
- 204.Liu M, Saeki K, Matsunobu T, Okuno T, Koga T, Sugimoto Y, Yokoyama C, Nakamizo S, Kabashima K, Narumiya S, Shimizu T, & Yokomizo T, 12-Hydroxyheptadecatrienoic acid promotes epidermal wound healing by accelerating keratinocyte migration via the BLT2 receptor. J Exp Med, 2014. 211(6): p. 1063–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Matsunaga Y, et al. , Leukotriene B4 receptor BLT2 negatively regulates allergic airway eosinophilia. FASEB J, 2013. 27(8): p. 3306–14. [DOI] [PubMed] [Google Scholar]
- 206.Enyedi B, et al. , Tissue damage detection by osmotic surveillance. Nat Cell Biol, 2013. 15(9): p. 1123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Niethammer P, et al. , A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature, 2009. 459(7249): p. 996–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tauzin S, et al. , Redox and Src family kinase signaling control leukocyte wound attraction and neutrophil reverse migration. J Cell Biol, 2014. 207(5): p. 589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]