

Abstract
We have previously shown that podocyte injury increases the glomerular filtration of liver-derived angiotensinogen (Agt) and the generation of intrarenal angiotensin (Ang) II and that the filtered Agt is reabsorbed by proximal tubules in a manner dependent on megalin. In the present study, we aimed to study the role of megalin in the generation of renal Ang II and sodium handling during nephrotic syndrome. We generated proximal tubule-specific megalin knockout (KO) mice and crossed these animals with NEP25 mice, in which podocyte-specific injury can be induced by injection of the immunotoxin LMB2. Without podocyte injury, renal Agt staining was markedly diminished and urinary Agt increased in KO mice. However, renal Ang II was similar between KO and control mice on average: 117 (95% CI 101–134) vs. 101 (68–133) fmol/g tissue. We next tested the effect of megalin KO on intrarenal Ang II generation with podocyte injury. Control NEP25 mice showed markedly increased renal Agt staining and renal Ang II levels: 450 (336–565) fmol/g tissue. Megalin KO/NEP25 mice showed markedly diminished Agt reabsorption and attenuated renal Ang II: 199 (156–242) fmol/g tissue (p < 0.001). Compared with control NEP25 mice, megalin KO/NEP25 mice excreted 5-fold more sodium in the urine. Western blot analysis showed that megalin KO decreased NHE3 and the cleaved α and γ forms of ENaC. These data indicate that Agt reabsorbed by proximal tubules via megalin in nephrotic syndrome is converted to Ang II, which may contribute to sodium retention and edema formation by activating NHE3 and ENaC.
Summary
This study shows that in nephrotic syndrome, plasma angiotensinogen leaked into the renal tubular lumen is reabsorbed by proximal tubules via megalin and converted to angiotensin II, which may contribute to sodium retention and edema formation by activating NHE3 and ENaC.
Keywords: angiotensinogen, angiotensin II, megalin, podocyte injury, NHE3, ENaC
Graphical abstract
Introduction
Using kidney- and liver-specific Agt knockout (KO) mice, we previously demonstrated that the liver is the major source of Agt protein and Ang II in the kidney. On the other hand, proximal tubular cells transcribe a large amount of Agt mRNA, but this does not significantly contribute to renal Ang II.1 Furthermore, we demonstrated that podocyte injury increases glomerular filtration of liver-derived Agt, which was accompanied by increased renal Ang II generation independently of renal Agt mRNA.2 Filtered Agt is reabsorbed by proximal tubular cells in a manner dependent on megalin.1 Megalin is a multiligand receptor that is intensely expressed on the apical membrane of proximal tubules. Agt protein is one of the ligands of megalin and is taken up by proximal tubules via megalin-dependent endocytosis.3 These observations led us to speculate that the Agt reabsorbed via megalin may contribute to the production of intrarenal Ang II.
Severe podocyte injury causes nephrotic syndrome, in which the urinary excretion of sodium is suppressed.2 Intrarenal Ang II may work on transporters and channels in the regulation of blood pressure and urinary concentration through the type 1 (AT1) receptor expressed in renal tubules.4,5 We hypothesized that intrarenal Ang II contributes to sodium retention in podocyte injury.
In the present study, we tested whether megalin is involved in intrarenal Ang II generation with or without podocyte injury using proximal tubular cell-specific megalin KO mice. We also studied the effect of megalin KO on urinary sodium excretion.
Methods
Disclosure
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal experiments
The protocols for animal experiments were approved by the Animal Experimentation Committee of Tokai University School of Medicine, in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
In the present study, mice carrying megalin-loxP, KAP-Cre, Ndrg1-CreERT26 and NEP25 mice7 were used. Detailed information about methods for the animal experiments was shown in the supplement.
Assays for renal Ang II content and renal and urinary Agt protein
The renal Ang II content was determined by radioimmunoassay (RIA) as previously described.1 Western blot analysis for Agt was performed with whole kidney samples. Detailed information about its methods was shown in the supplement.
Urinary Agt was determined by ELISA (IBL, Fujioka, Japan) as previously described.2
Assays for sodium channels and transporters
Western blot analysis for sodium channels and transporters was performed with membrane protein samples. Detailed information about its methods was shown in the supplement.
We confirmed that the amount loaded was in the linear range of detection for each protein (Figure S1).
The following antibodies were used at the indicated dilutions: rabbit polyclonal antibodies against α (1:5000)8, β (1:40000)9, and γ ENaC (1:60000)9 (gifts from Johannes Loffing at the University of Zurich); rabbit polyclonal antibodies against phosphorylated (p) NKCC2 at threonine 96 and 101 (1:1000)10 and pNCC at threonine 53 (1:5000)11 (gifts from David H. Ellison at Oregon Health & Science University); rabbit polyclonal anti-NHE3 (1:1000) (27190–1-AP, Proteintech, Rosemont, IL); mouse monoclonal antibody against pNHE3 at serine 552 (1:800) (14D5, Novus, Centennial, CO); rabbit polyclonal anti-β-actin (1:1000) (#4967, Cell Signaling Technology, Danvers, MA); and HRP-conjugated mouse anti-rabbit IgG antibody (1:5000) (NA9340V, GE Healthcare, Chicago, IL). There are two cleavage sites in αENaC that give rise to two cleavage products.12
Histology and immunostaining
PAS staining and immunostaining were performed on 2-μm serial paraffin sections. Rabbit polyclonal anti-mouse/rat Agt antibody (1:50, IBL, Fujioka, Japan) and rabbit polyclonal anti-rat megalin antibody (1:1000, generated by A. Saito) were used at the indicated dilution. Detailed information about its methods was shown in the supplement.
Statistical analysis
Detailed information about methods for the statistical analyses was shown in the supplement.
Results
Generation of proximal tubule-specific megalin knockout mice
To delete the megalin gene in the proximal tubule, we first generated MegalinloxP/loxP: Ndrg1-CreERT2TG/WT mice. Initial trials revealed that the megalin gene was most effectively deleted when male mice were treated with tamoxifen (100 μg/g BW) 5 times starting at 3 weeks of age. However, megalin staining remained in the S3 segment, which showed a compensatory increase in the reabsorption of Agt protein (data not shown). To delete the megalin gene in the S3 segment, we further mated mice with KAP-Cre transgenic mice. Later, we found that two cycles of tamoxifen treatment deleted the megalin gene in MegalinloxP/loxP:KAP-Cre WT /WT:Ndrg1-CreERT2TG/WT mice as efficiently as in MegalinloxP/loxP:KAP-Cre TG /WT:Ndrg1-CreERT2TG/WT mice. In addition, these two types of mice showed similar phenotypes. We therefore combined these animals, designated them as megalin KO mice and compared these mice with MegalinloxP/loxP:KAP-CreWT /WT:Ndrg1-CreERT2WT/WT mice (megalin control).
Megalin has no impact on Ang II in the kidney without podocyte injury
Renal megalin mRNA and protein were markedly decreased in megalin KO mice (Figure 1A and B). Agt protein was stained in a granular pattern in the proximal tubule of S1 and S2 segments of megalin control mice. In contrast, Agt staining was undetectable in the proximal tubule of megalin KO mice (Figure 1B). Western blot analysis showed that the kidneys of megalin KO mice contained less Agt protein than those of megalin control mice (Figure 2).
Figure 1.

Effect of megalin knockout (KO) on Agt and Ang II in the kidney without podocyte injury. Inactivation of the megalin gene was confirmed by RT-PCR (A) and immunohistochemistry (B, top). Both megalin control (Cont) and megalin KO mice showed normal renal histology (B, middle). While Agt protein was stained in a granular pattern in the S1 and S2 segments of the proximal tubule of megalin Cont mice, Agt staining was undetectable in megalin KO mice (B, bottom). Urinary Agt excretion was markedly increased in megalin KO mice (C). There was no significant difference in renal Ang II between megalin Cont and KO mice (D). Horizontal bars represent geometric means in A and C and arithmetic means in B.
Figure 2.

Western blot analysis of renal Agt protein. When podocytes were intact, the renal Agt content in megalin KO mice was lower than that in megalin control (Cont) mice. With podocyte injury, the renal Agt content was increased in NEP25/megalin Cont mice and further increased in NEP25/megalin KO mice. Error bars represent means ± 95% confidence intervals. Note that the sample in the third lane from the left in the megalin KO group (arrowhead) was obtained from a mouse with abnormally high albuminuria and proteinaceous casts. This mouse was therefore excluded from all analyses.
The urinary albumin/creatinine (Cr) ratio (ACR) in megalin KO mice was, on average, 0.56 (95% CI 0.50–0.63), which was greater than that in megalin control mice at 0.079 (0.069–0.090) (p < 0.0001). In addition, megalin KO mice showed a markedly higher urinary Agt/Cr ratio than in megalin control mice: 21.6 (19.5–23.7) vs. 0.0998 (0.0850–0.115) μg/mg (p < 0.0001) (Figure 1C). These data indicate that the majority of renal Agt protein detected by immunostaining was reabsorbed by proximal tubules via megalin.
We next measured Ang II in the renal homogenate. Renal Ang II content in megalin KO mice was, on average, 117 (101–134) fmol/g tissue, which was comparable to that in megalin control mice at 101 (68–133) fmol/g tissue (Figure 1D). This result indicates that the Agt protein reabsorbed via megalin does not contribute to renal Ang II when podocytes are intact.
Megalin control and KO mice showed similar systolic blood pressure (113 (108–118) vs. 104 (99–109) mmHg) (Figure S2), urine volume (1380 (1130–1620) vs. 1590 (1370–1820) ml/day), and urinary sodium (0.58 (0.52–0.64) vs. 0.57 (0.53–0.61) mEq/mg) and chloride (0.74 (0.65–0.83) vs. 0.73 (0.65–0.80) mEq/mg) excretion. Both types of mice showed normal renal histology (Figure 1B). Renal renin, Ace, and Agt mRNAs were similar between the two groups (data not shown).
Megalin KO decreases Ang II in kidneys with podocyte injury
We next studied the effect of megalin KO on Ang II in kidneys with podocyte injury. Podocyte injury was induced in NEP25/megalin control and NEP25/megalin KO mice by injecting LMB2 (Figure 3). At seven days after the induction of podocyte injury, both types of mice showed similar massive proteinuria, with urinary ACRs of 155.4 (84.9–226.0) and 125.8 (94.9–156.6), respectively (Figure S3).
Figure 3.

Effect of megalin knockout (KO) on Agt and Ang II in the kidney with podocyte injury. Inactivation of the megalin gene was confirmed by RT-PCR (A) and immunohistochemistry (B, top). Agt staining in proximal tubular cells was markedly increased in NEP25/megalin control (Cont) mice, whereas no Agt staining was observed within proximal tubular cells of NEP25/megalin KO mice (B, bottom). NEP25/megalin KO mice showed massive proteinaceous casts, which were intensely stained for Agt (B, middle and bottom). NEP25/megalin Cont and KO mice showed similarly high urinary Agt/creatinine ratios (C). Renal Ang II was markedly increased in NEP25/megalin Cont mice and significantly decreased in NEP25/megalin KO mice (D). Horizontal bars represent geometric means in A and C and arithmetic means in B.
Immunostaining revealed that the Agt protein reabsorbed in proximal tubular cells was markedly increased in NEP25/megalin control mice, whereas no Agt staining was observed within the proximal tubular cells of NEP25/megalin KO mice (Figure 3B). Notably, NEP25/megalin KO mice showed massive proteinaceous casts, which were intensely stained for Agt. Western blot analysis confirmed that renal Agt content was markedly increased in NEP25/megalin control mice compared to megalin control mice without injury (Figure 2). NEP25/megalin KO mice showed a larger amount of Agt protein in Western blot analysis than NEP25/megalin control mice, indicating that a large amount of Agt protein was contained in the proteinaceous casts of NEP25/megalin KO mice. The urinary Agt/Cr ratio was dramatically increased in both NEP25/megalin control mice (56.6 (42.0–71.2) μg/mg) and NEP25/megalin KO mice (72.4 (56.2–88.6) μg/mg) (Figure 3C).
Renal Ang II content was markedly increased in NEP25/megalin control mice, as previously reported.2 The increase in renal Ang II was significantly attenuated in NEP25/megalin KO mice (450 (336–565) vs. 199 (156–242) fmol/g tissue, p < 0.001) (Figure 3D). Agt and Renin mRNA levels were increased in both NEP25/megalin control mice and NEP25/megalin KO mice. Agt and Ace mRNAs were slightly higher in NEP25/megalin KO mice than in NEP25/megalin control mice (Figure S4). None of these changes in mRNA explained the attenuation of renal Ang II in NEP25/megalin KO mice.
Systolic blood pressure was comparable between NEP25/megalin control and NEP25/megalin KO mice (Figure S5A). Urine volume was decreased in only NEP25/megalin control mice (Figure S6A). Urinary Na/Cr and Cl/Cr ratios were markedly decreased in NEP25/megalin control mice, but this decrease was attenuated in NEP25/megalin KO mice (Figure S7).
In contrast to the mild morphological changes in tubules of NEP25/megalin control mice, NEP25/megalin KO mice developed severe and extensive cast nephropathy with tubular dilatation and the flattening of tubular cells (Figure 3B). The observed attenuation of sodium retention in NEP25/megalin KO mice on day 7 can potentially be attributed to tubular injury. To rule out this possibility, we analyzed sodium retention at an earlier time point on day 4.
Megalin KO attenuated the suppression of urinary excretion of NaCl in podocyte injury
Four days after the induction of podocyte injury, NEP25/megalin KO mice showed only mild morphological changes in the tubules. Megalin KO mice excreted more urinary Agt on day 4; this result was similar to that obtained before LMB2 injection but different from that observed on day 7 (Figure S8A). Importantly, similar to the results on day 7, the renal Ang II content in NEP25/megalin KO mice on day 4 was less than that in NEP25/megalin control mice (Figure S8B). Furthermore, as was the case with the mice on day 7, the urinary excretion of sodium (0.057 (0.042–0.074) vs. 0.21 (0.15–0.26) mEq/mg, p < 0.0001) and chloride (0.59 (0.52–0.66) vs. 0.83 (0.67–0.99) mEq/mg, p < 0.05) was markedly suppressed in NEP25/megalin control mice, and this suppression was attenuated in NEP25/megalin KO mice (Figure 4). Systolic blood pressure was comparable between the two groups (Figure S5B). Urine volume was suppressed in NEP25/megalin control mice compared with that in NEP25/megalin KO mice (Figure S6B).
Figure 4.

Effect of megalin KO on urinary sodium and chloride excretion. When podocytes were intact, there was no significant difference between megalin control (Cont) and KO mice. Podocyte injury markedly suppressed urinary sodium and chloride excretion. Compared to NEP25/megalin Cont mice, this suppression was attenuated in NEP25/megalin KO mice at 4 days after podocyte injury. Horizontal bars represent geometric means.
To elucidate the molecular mechanisms of sodium retention induced by podocyte injury, we analyzed ENaC, NHE3, pNKCC2 and pNCC by Western blot analysis. Without podocyte injury, no difference was observed in the amount of cleaved forms of the α or γ subunit (35 kD or 70 kD, respectively) or the β subunit of ENaC in the membrane fraction between megalin control and megalin KO mice (data not shown). After the induction of podocyte injury, the amounts of the cleaved forms of α and γ subunits were markedly increased in NEP25/megalin control mice, whereas the noncleaved forms of the α and γ subunits (90 kD and 85 kD, respectively) and the β subunit were not changed (data not shown). The increases in the cleaved forms of α and γ subunits were significantly attenuated in NEP25/megalin KO mice treated with LMB2 (both p < 0.001) (Figure 5A–C). The administration of amiloride, an ENaC inhibitor, to NEP25 mice at 3 days after LMB2 injection significantly increased urinary sodium excretion during the subsequent 15 hours, confirming that ENaC plays an important role in sodium retention in nephrotic syndrome in NEP25 mice (Figure S9). Next, we measured plasma aldosterone and found no difference between NEP25/megalin control and NEP25/megalin KO mice (Figure S10). We also measured urinary aprotinin-dependent plasmin-like activity and observed no significant difference between NEP25/megalin control and NEP25/megalin KO mice on day 4 (Figure S11A). Additionally, the change in activity from day 0 to day 4 was not different between the two types of mice (Figure S11B).
Figure 5.

Effect of megalin KO on ENaC and NHE3 in the kidney with podocyte injury. Compared to NEP25/megalin control (Cont) mice, NEP25/megalin KO mice showed fewer cleaved α and γ subunits of ENaC and NHE3 (A, C, D). There was no significant difference in the β subunit of ENaC (B) and pNHE3 (E). Bars represent the means ± 95% confidence intervals.
After the induction of podocyte injury, the amount of NHE3 was significantly increased in NEP25/megalin control mice and was significantly attenuated in NEP25/megalin KO mice treated with LMB2 (Figure 5D). In contrast, there was no difference in pNHE3, the inactive form of NHE3 (Figure 5E).
Quantitative analysis of pNKCC2 and pNCC showed high variability among the samples, and no significant difference was detected (Figure S12).
Discussion
In the present study, we demonstrated that intrarenal Ang II was increased by podocyte injury and that this increase was attenuated by the genetic inactivation of megalin in the proximal tubules. This result suggests that a portion of plasma Agt filtered through glomeruli and reabsorbed by proximal tubules is converted to Ang II in the nephrotic state. In contrast, when podocytes were intact, megalin knockout showed no impact on renal Ang II. Since our previous study indicated that the disruption of the Agt gene in the liver markedly decreases renal Ang II, 1 baseline renal Ang II is probably generated from liver-derived Agt in the capillary lumen or the interstitium, which was not detected by our immunostaining.
Podocyte injury increases glomerular leakage of renin as well as Agt, and the leaked renin is also reabsorbed by proximal tubular cells via megalin.3 In addition, a large amount of ACE is expressed in the brush border and basolateral membrane of proximal tubular cells.3,13 Giani et al. reported that tubular ACE contributes to intrarenal Ang II production and sodium retention using a hypertensive mouse model induced by L-NAME.14,15 These data collectively suggest that Ang II is likely formed in proximal tubules. However, the compartment (within the cell, tubular lumen, or peritubular interstitium) in which Ang II formation occurs is not known. It is also not known how Ang II is delivered to other tubular segments.
Studies using collecting duct-specific renin KO mice revealed that high Ang II increases renin production in the collecting duct and that the increased renin augments local Ang II activity.16,17 This mechanism may also function in NEP25 mice, which have very high renal Ang II levels. This possibility is supported by the results of the present study, in which the activation of ENaC but not NKCC2 or NCC was detected. Theoretically, more Agt protein is delivered to the collecting duct in NEP25/megalin KO mice, but NEP25/megalin KO mice actually showed lower renal Ang II than NEP25/megalin control mice. This result indicates that the amount of Agt delivered to the collecting duct is not the rate-limiting factor for Ang II generation.
Sodium retention is involved in the development of systemic edema in nephrotic syndrome. Two opposing mechanisms are proposed for its pathogenesis. The underfill mechanism puts forth that sodium retention is an adaptive response to volume depletion. The overfill mechanism explains that sodium retention is primarily caused by an intrinsic inability of the nephrotic kidney to normally excrete sodium. Supporting the latter hypothesis, a previous micropuncture study using unilateral nephrotic rats showed that sodium retention occurred in the collecting duct of PAN-perfused kidneys.18 The two mechanisms likely contribute to pathogenesis to a variable degree in individual patients. As observed in human patients with nephrotic syndrome, NEP25 mice showed sodium retention and edema formation after induction of podocyte injury. NEP25/megalin KO mice showed attenuated sodium retention compared to NEP25/megalin control mice. Urinary sodium excretion was greater in NEP25/megalin KO mice at 4 days after the induction of podocyte injury, before the kidney showed severe tubular injury. Our study suggests that renal Ang II induced by podocyte injury may be involved in sodium retention in nephrotic syndrome, accounting for the overfill mechanism.
Consistent with urinary sodium excretion, NEP25/megalin KO also showed less activated NHE3 and α, γ ENaCs. Since the proximal tubule reabsorbs 60–75% of the filtered sodium, mainly via NHE3,19–21 and Ang II, increased NHE3 protein and activity in renal proximal tubular cells,22–27 this result may suggest that increased NHE3 is involved in sodium retention in nephrotic syndrome. However, the interpretation of the NHE3 data is not simple. First, NHE3 is more intensely expressed in the thick ascending limb than the proximal tubule in mice. Second, not all NHE3 proteins exert the function of sodium reabsorption. A significant portion of NHE3 is complexed with megalin in the nonendosomal dense granules of proximal tubular cells and has no Na+-H+ exchange activity.28,29 Other studies indicated that inactive and active forms of NHE3 are located in different parts of proximal tubule microvilli and that high blood pressure induces movement from inactive dense to active light membrane segments.30,31Kocinsky et al. reported that NHE3 phosphorylated at serine 552 is localized to the coated pit of the brush border membrane and reflects the inactive form of NHE3.32 In the present study, there was no difference in pNHE3 between NEP25/megalin control and KO mice. We therefore speculate that NHE3 contributes to sodium retention activated by renal Ang II.
In a previous study, the activation of ENaC was reported to contribute to sodium retention in nephrotic rats induced by puromycin aminonucleoside33 or by HgCl2.34 We also confirmed that amiloride markedly augmented urinary sodium excretion in NEP25 mice. Several lines of evidence have indicated that Ang II regulates ENaC activity independently of aldosterone. First, ENaC expression was observed in the absence of the adrenal gland35 or mineralocorticoid receptor.36 Second, the α subunit of ENaC was decreased in AT1a receptor knockout mice, in spite of the higher plasma aldosterone.37 Third, a functional study using isolated perfused CCD showed that Ang II increased apical sodium transport, which was completely inhibited by an ENaC blocker.38 Fourth, Mamenko et al.39 demonstrated that Ang II directly increased the open probability of the ENaC channel via the AT1 receptor and induced apical translocation of the α subunit of ENaC in an additive manner with aldosterone.39 These findings collectively support the possibility that renal Ang II induced by podocyte injury is involved in sodium retention. In this study, there was no difference in plasma aldosterone between NEP25/megalin control and KO mice. This finding indicates that the difference in sodium reabsorption cannot be ascribed to plasma aldosterone and suggests that increased renal Ang II does not act on the adrenal gland.
Svenningsen et al.40 proposed that proteolytic processing by urinary serine protease plays an important role in the activation of ENaC40 in the nephrotic state. Thus, plasminogen filtered aberrantly through glomeruli is converted to plasmin by the plasminogen activator in the tubular lumen. This serine protease cleaves an inhibitory peptide segment from the γ subunit, which increases the open channel probability. Aprotinin, a serine protease inhibitor, can abolish γENaC cleavage and sodium retention,41 suggesting that ENaC activation by serine protease is the major molecular mechanism explaining the overfill phenomenon. However, this mechanism cannot explain the increased cleavage of the γ subunit of ENaC in NEP25/megalin control mice compared to NEP25/megalin KO mice because both mice showed heavy proteinuria and therefore likely show similar high activity of urinary serine protease. In addition, we could not detect differences in urinary plasmin activity between NEP25/megalin control and NEP25/megalin KO mice.
The proposed mechanism of renal Ang II generation and its potential function on sodium homeostasis in nephrotic syndrome is illustrated in Figure 6. Our study indicates that Ang II is generated from Agt reabsorbed by proximal tubular cells rather than from Agt in the tubular lumen. However, it is not known why reabsorption via megalin is necessary to generate renal Ang II and why NKCC2 and NCC were not affected by megalin KO. The detailed mechanism and precise location of Ang II generation and transport need to be clarified to solve this conundrum.
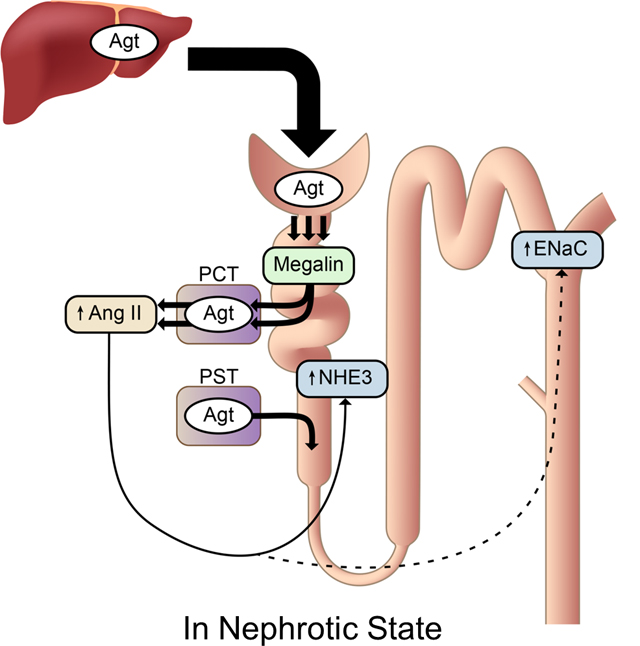
Figure 6.

The proposed mechanism of renal Ang II generation and its function. (A) In normal conditions, a portion of plasma Agt is filtered through glomeruli and reabsorbed by proximal convoluted tubules (PCT) via megalin. However, the reabsorbed Agt is not converted to Ang II. Agt is synthesized in proximal straight tubules (PST), but this Agt makes little contribution to renal Ang II. Renal Ang II is probably generated from liver-derived Agt in the capillary lumen or interstitium. (B) In nephrotic syndrome, podocyte injury increases glomerular leakage of plasma Agt. The filtered Agt is absorbed by proximal tubules via megalin and thereafter converted to Ang II. The increased renal Ang II may contribute to sodium retention possibly by activating NHE3 and ENaC, although direct action on these molecules has not been proven.
Perspectives
The present study revealed the novel concept that in podocyte injury, a large amount of Agt protein absorbed by proximal tubular cells via megalin after filtration through the glomeruli is converted to intrarenal Ang II, which may activate NHE3 and ENaC. This notion may explain the pathogenesis of edema formation in nephrotic syndrome. To understand the precise pathogenesis in more detail, further elucidation of the location in which Agt is converted to intrarenal Ang II and the pathway through which intrarenal Ang II acts on NHE3 and ENaC would be necessary.
Supplementary Material
Novelty and Significance.
What Is New?
Our study demonstrated for the first time that glomerular filtered-plasma angiotensinogen can be a source of renal angiotensin II.
What Is Relevant?
Our findings explain the “overfill” mechanism of sodium retention in nephrotic syndrome.
Acknowledgments
We acknowledge Dr. Thomas Willnow at the Max Delbruck Center for providing megalin KO mice, Duncan J Campbell for advice on Ang II measurement by RIA, Johannes Loffing at University Zurich for supplying the ENaC antibody, and David H. Ellison at Oregon Health Science University for supplying the pNKCC2 and pNCC antibodies. We thank Ms. Shiho Imai and Ms. Chie Sakurai for excellent technical assistance and Ms. Yukiko Tanaka for administrative assistance. We are also grateful to the Support Center for Medical Research and Education, Tokai University for instructive advice about Western blot analysis.
Sources of Funding
This study was supported by a Grant-in-Aid for Scientific Research of the Japan Society for the Promotion of Science of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (17K16100, 22249033) and the MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2015-2019).
T.M. received research funding from Teijin Pharmaceutical.
Footnotes
Disclosures
All other authors declare no competing interests.
References
- 1.Matsusaka T, Niimura F, Pastan I, Shintani A, Nishiyama A, Ichikawa I. Podocyte injury enhances filtration of liver-derived angiotensinogen and renal angiotensin II generation. Kidney Int. 2014;85(5):1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, Nishiyama A, Ichikawa I. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol. 2012;23(7):1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pohl M, Kaminski H, Castrop H, Bader M, Himmerkus N, Bleich M, Bachmann S, Theilig F. Intrarenal renin angiotensin system revisited: role of megalin-dependent endocytosis along the proximal nephron. J Biol Chem. 2010;285(53):41935–41946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13(4):469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stegbauer J, Gurley SB, Sparks MA, Woznowski M, Kohan DE, Yan M, Lehrich RW, Coffman TM. AT1 receptors in the collecting duct directly modulate the concentration of urine. J Am Soc Nephrol. 2011;22(12):2237–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Endo T, Nakamura J, Sato Y, Asada M, Yamada R, Takase M, Takaori K, Oguchi A, Iguchi T, Higashi AY, Ohbayashi T, Nakamura T, Muso E, Kimura T, Yanagita M. Exploring the origin and limitations of kidney regeneration. J Pathol. 2015;236(2):251–263. [DOI] [PubMed] [Google Scholar]
- 7.Matsusaka T, Xin J, Niwa S, Kobayashi K, Akatsuka A, Hashizume H, Wang QC, Pastan I, Fogo AB, Ichikawa I. Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol. 2005;16(4):1013–1023. [DOI] [PubMed] [Google Scholar]
- 8.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013;83(5):811–824. [DOI] [PubMed] [Google Scholar]
- 9.Wagner CA, Loffing-Cueni D, Yan Q, Schulz N, Fakitsas P, Carrel M, Wang T, Verrey F, Geibel JP, Giebisch G, Hebert SC, Loffing J. Mouse model of type II Bartter’s syndrome. II. Altered expression of renal sodium- and water-transporting proteins. Am J Physiol Renal Physiol. 2008;294(6):F1373–1380. [DOI] [PubMed] [Google Scholar]
- 10.Reiche J, Theilig F, Rafiqi FH, Carlo AS, Militz D, Mutig K, Todiras M, Christensen EI, Ellison DH, Bader M, Nykjaer A, Bachmann S, Alessi D, Willnow TE. SORLA/SORL1 functionally interacts with SPAK to control renal activation of Na(+)-K(+)-Cl(−) cotransporter 2. Mol Cell Biol. 2010;30(12):3027–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCormick JA, Nelson JH, Yang CL, Curry JN, Ellison DH. Overexpression of the sodium chloride cotransporter is not sufficient to cause familial hyperkalemic hypertension. Hypertension. 2011;58(5):888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem. 2004;279(18):18111–18114. [DOI] [PubMed] [Google Scholar]
- 13.Alhenc-Gelas F, Baussant T, Hubert C, Soubrier F, Corvol P. The angiotensin converting enzyme in the kidney. J Hypertens Suppl. 1989;7(7):S9–S13; discussion S14. [DOI] [PubMed] [Google Scholar]
- 14.Giani JF, Janjulia T, Kamat N, Seth DM, Blackwell WL, Shah KH, Shen XZ, Fuchs S, Delpire E, Toblli JE, Bernstein KE, McDonough AA, Gonzalez-Villalobos RA. Renal angiotensin-converting enzyme is essential for the hypertension induced by nitric oxide synthesis inhibition. J Am Soc Nephrol. 2014;25(12):2752–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giani JF, Eriguchi M, Bernstein EA, Katsumata M, Shen XZ, Li L, McDonough AA, Fuchs S, Bernstein KE, Gonzalez-Villalobos RA. Renal tubular angiotensin converting enzyme is responsible for nitro-L-arginine methyl ester (L-NAME)-induced salt sensitivity. Kidney Int. 2017;91(4):856–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez AA, Lara LS, Prieto MC. Role of Collecting Duct Renin in the Pathogenesis of Hypertension. Curr Hypertens Rep. 2017;19(8):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramkumar N, Stuart D, Rees S, Hoek AV, Sigmund CD, Kohan DE. Collecting duct-specific knockout of renin attenuates angiotensin II-induced hypertension. Am J Physiol Renal Physiol. 2014;307(8):F931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichikawa I, Rennke HG, Hoyer JR, Badr KF, Schor N, Troy JL, Lechene CP, Brenner BM. Role for intrarenal mechanisms in the impaired salt excretion of experimental nephrotic syndrome. J Clin Invest. 1983;71(1):91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN, Shull GE. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet. 1998;19(3):282–285. [DOI] [PubMed] [Google Scholar]
- 20.Du Z, Yan Q, Duan Y, Weinbaum S, Weinstein AM, Wang T. Axial flow modulates proximal tubule NHE3 and H-ATPase activities by changing microvillus bending moments. Am J Physiol Renal Physiol. 2006;290(2):F289–296. [DOI] [PubMed] [Google Scholar]
- 21.Wang T, Yang CL, Abbiati T, Schultheis PJ, Shull GE, Giebisch G, Aronson PS. Mechanism of proximal tubule bicarbonate absorption in NHE3 null mice. Am J Physiol. 1999;277(2 Pt 2):F298–302. [DOI] [PubMed] [Google Scholar]
- 22.Li XC, Zhuo JL. Selective knockdown of AT1 receptors by RNA interference inhibits Val5-ANG II endocytosis and NHE-3 expression in immortalized rabbit proximal tubule cells. Am J Physiol Cell Physiol. 2007;293(1):C367–C378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li XC, Hopfer U, Zhuo JL. AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through a microtubule-dependent endocytic pathway. Am J Physiol Renal Physiol. 2009;297(5):F1342–F1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karim ZG, Chambrey R, Chalumeau C, Defontaine N, Warnock DG, Paillard M, Poggioli J. Regulation by PKC isoforms of Na(+)/H(+) exchanger in luminal membrane vesicles isolated from cortical tubules. Am J Physiol. 1999;277(5 Pt 2):F773–F778. [DOI] [PubMed] [Google Scholar]
- 25.Banday AA, Lokhandwala MF. Oxidative stress causes renal angiotensin II type 1 receptor upregulation, Na+/H+ exchanger 3 overstimulation, and hypertension. Hypertension. 2011;57(3):452–459. [DOI] [PubMed] [Google Scholar]
- 26.Xu L, Dixit MP, Nullmeyer KD, Xu H, Kiela PR, Lynch RM, Ghishan FK. Regulation of Na+/H+ exchanger-NHE3 by angiotensin-II in OKP cells. Biochim Biophys Acta. 2006;1758(4):519–526. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen MT, Han J, Ralph DL, Veiras LC, McDonough AA. Short-term nonpressor angiotensin II infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiol Rep. 2015;3(9):e12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biemesderfer D, Nagy T, DeGray B, Aronson PS. Specific association of megalin and the Na+/H+ exchanger isoform NHE3 in the proximal tubule. J Biol Chem. 1999;274(25):17518–17524. [DOI] [PubMed] [Google Scholar]
- 29.Biemesderfer D, DeGray B, Aronson PS. Active (9.6 s) and inactive (21 s) oligomers of NHE3 in microdomains of the renal brush border. J Biol Chem. 2001;276(13):10161–10167. [DOI] [PubMed] [Google Scholar]
- 30.Yang LE, Maunsbach AB, Leong PK, McDonough AA. Differential traffic of proximal tubule Na+ transporters during hypertension or PTH: NHE3 to base of microvilli vs. NaPi2 to endosomes. Am J Physiol Renal Physiol. 2004;287(5):F896–906. [DOI] [PubMed] [Google Scholar]
- 31.Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol. 2010;298(1):F177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kocinsky HS, Girardi AC, Biemesderfer D, Nguyen T, Mentone S, Orlowski J, Aronson PS. Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am J Physiol Renal Physiol. 2005;289(2):F249–258. [DOI] [PubMed] [Google Scholar]
- 33.Kim SW, Wang W, Nielsen J, Praetorius J, Kwon T-H, Knepper MA, Frokier J, Nielsen S. Increased expression and apical targeting of renal ENaC subunits in puromycin aminonucleoside-induced nephrotic syndrome in rats. Am J Physiol Renal Physiol. 2004;286:F922–F935. [DOI] [PubMed] [Google Scholar]
- 34.SdS Soo Wan Kim, Sassen Martin C., Lee JongUn, Kim Jin, Knepper Mark A., Frokier Jorgen, Nielsen Soren. Increased apical targeting of renal ENaC subunits and decreased expression of 11βHSD2 in HgCl2-induced nephrotic syndrome in rats. Am J Physiol Renal Physiol. 2006;290:F674–F687. [DOI] [PubMed] [Google Scholar]
- 35.Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD. Aldosterone-independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci U S A. 2012;109(25):10095–10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, Kriz W, Warth R, Greger R, Schutz G. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A. 1998;95(16):9424–9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooks HL, Allred AJ, Beutler KT, Coffman TM, Knepper MA. Targeted proteomic profiling of renal Na(+) transporter and channel abundances in angiotensin II type 1a receptor knockout mice. Hypertension. 2002;39(2 Pt 2):470–473. [DOI] [PubMed] [Google Scholar]
- 38.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol. 2002;13(5):1131–1135. [DOI] [PubMed] [Google Scholar]
- 39.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem. 2012;287(1):660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C, Skott O. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20(2):299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bohnert BN, Menacher M, Janessa A, Worn M, Schork A, Daiminger S, Kalbacher H, Haring HU, Daniel C, Amann K, Sure F, Bertog M, Haerteis S, Korbmacher C, Artunc F. Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome. Kidney Int. 2017;93(1):159–172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.