

Abstract
Objective
The COVID‐19 pandemic poses an immense need for accurate, sensitive and high‐throughput clinical tests, and serological assays are needed for both overarching epidemiological studies and evaluating vaccines. Here, we present the development and validation of a high‐throughput multiplex bead‐based serological assay.
Methods
More than 100 representations of SARS‐CoV‐2 proteins were included for initial evaluation, including antigens produced in bacterial and mammalian hosts as well as synthetic peptides. The five best‐performing antigens, three representing the spike glycoprotein and two representing the nucleocapsid protein, were further evaluated for detection of IgG antibodies in samples from 331 COVID‐19 patients and convalescents, and in 2090 negative controls sampled before 2020.
Results
Three antigens were finally selected, represented by a soluble trimeric form and the S1‐domain of the spike glycoprotein as well as by the C‐terminal domain of the nucleocapsid. The sensitivity for these three antigens individually was found to be 99.7%, 99.1% and 99.7%, and the specificity was found to be 98.1%, 98.7% and 95.7%. The best assay performance was although achieved when utilising two antigens in combination, enabling a sensitivity of up to 99.7% combined with a specificity of 100%. Requiring any two of the three antigens resulted in a sensitivity of 99.7% and a specificity of 99.4%.
Conclusion
These observations demonstrate that a serological test based on a combination of several SARS‐CoV‐2 antigens enables a highly specific and sensitive multiplex serological COVID‐19 assay.
Keywords: COVID‐19, IgG, multiplex, SARS‐CoV‐2, serological assay
Five selected antigens show different patterns when used for detection of IgG antibodies in samples from 331 COVID‐19 patients and convalescents, and 2090 negative controls. Analysing multiple antigens simultaneously can generate data that can be used for diagnostic purposes as well as to further understand the responses of the immune system. The multifaceted data that our method delivers furthermore allow for an adaptation of the outcome to the individual analysis, giving as accurate answers as possible regardless of time passed since infection onset.

Introduction
The coronavirus disease 2019 (COVID‐19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), emerged during late 2019. 1 Since then, the virus has spread rapidly and developed into a global pandemic within just a few months from its initial detection. Today, almost four million individuals have lost their lives because of the disease. 2 Since the symptoms from the disease range from asymptomatic to severe respiratory illnesses, clinical diagnosis is challenging. 3 , 4 Further, because of the manifestations that largely overlap with other respiratory infections, such as seasonal influenza and common colds, confirmation of the disease largely depends on laboratory detection of the viral genome by reverse transcriptase–polymerase chain reaction (RT‐PCR), 4 , 5 which requires timing with the active infection. In order to enable epidemiological studies, to understand the frequency of asymptomatic infections, to select the most appropriate convalescent blood donors for plasma treatment and to trace the virus spread in households and communities, serological tests are of utmost importance. These tests are also necessary for evaluation of the different vaccines as well as the duration of remaining antibodies post‐infection. A more comprehensive knowledge regarding the spread of the virus and the vaccine‐induced immunity is needed for the world to be able to revert to the pre‐pandemic way of living.
The envelope of the coronavirus SARS‐CoV‐2 consists of a lipid bilayer in which three different surface proteins are anchored, the membrane (M), the envelope (E) and the spike glycoprotein 6 , 7 (Figure 1a). M and E are both key players in the assembly of the virus particle and also crucial in maintaining its structural shape. The spike protein mediates infection of host target cells. 8 It consists of a heavily glycosylated homotrimer with the ability to bind to the ACE2 receptor, 9 thereby enabling fusion and entry of the virus into host cells. Furthermore, an additional interaction between the spike protein and the cell surface receptor neuropilin 1 was recently reported by Daly et al. 10 Each spike monomer is composed of two functional subunits, S1 and S2. S1 forms the outer part, including the receptor binding domain (RBD) and the N‐terminal domain (NTD), whereas S2 is anchored in the bilipid layer of the virus envelope. 9 Another immunogenic protein is the nucleocapsid (NC) protein, which is found inside the virus particle where it encapsulates the genomic RNA. 7 , 11 The spike protein and NC have been shown to be the main immunogens of SARS‐CoV‐2. 12
Figure 1.
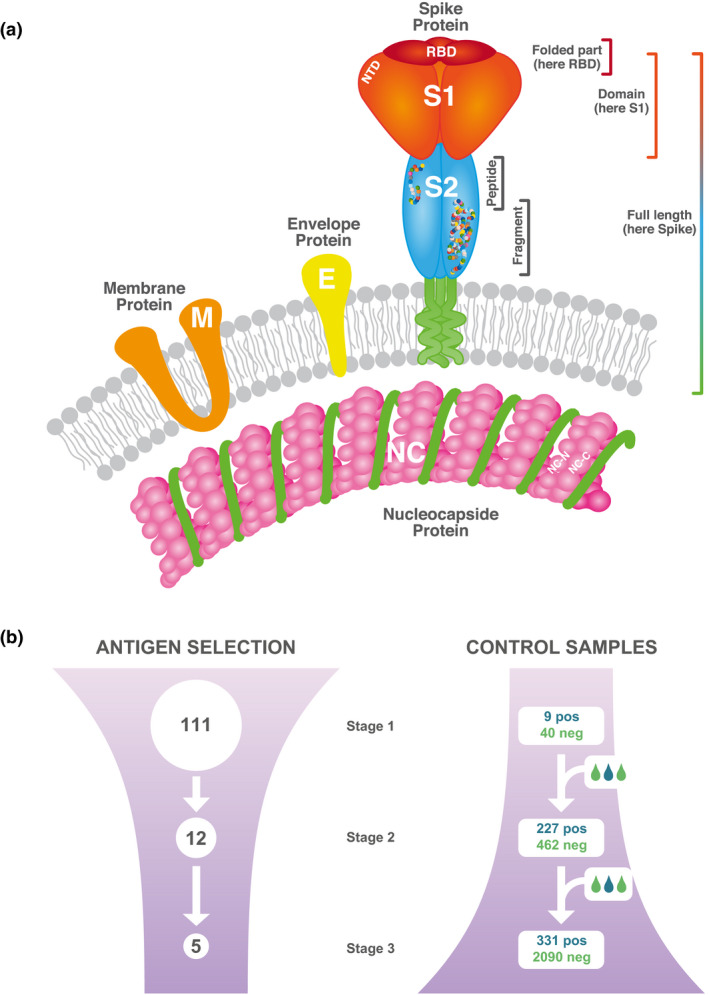
Study design. (a) Overview of the viral antigens included in our study and evaluated for performance in antibody detection. (b) Overview of the antigen evaluation. In the first stage, antigens providing antibody detection in a majority of a small set of positive samples were selected. This was followed by the second stage where 12 selected antigens were evaluated for their sensitivity and specificity. In an intermediate set of eight antigens, we also investigated the influence of different protein production systems in relation to the outcome of the analysis. In the final stage, five antigens were further assessed for sensitivity and specificity in the full sample set, both separate and combined in panels.
To date, the Joint Research Center and Food and Drug Administration have listed approximately 700 commercialised serological assays for SARS‐CoV‐2 13 , 14 , and many more have been developed in academic research centres and hospitals worldwide. The most common assay formats are lateral flow assays (LFA), classical enzyme‐linked immunosorbent assays (ELISAs) and chemiluminescent assays (CLIAs). Most of these tests measure the presence of IgG and IgM, separate or combined, against the spike protein, NC or domains thereof. 15 , 16 However, the precision and the degree of the validations differ substantially because of the character and number of samples used. LFA has the advantage of being a rapid test that can be applied at point of care but shows in general lower sensitivity and specificity compared with ELISA and CLIA. The laboratory‐based assays are often limited by the requirement of a venous blood sample and low multiplexing possibility. Current and future mass screenings and surveillances demand the availability of sensitive and specific high‐throughput serological tests. By utilising methods with multiplexing capability, it is possible to follow and compare the development and duration of antibodies against diverse antigens representing many different variants of the viral proteins. In this context, bead‐based assays represent useful platforms with broad dynamic range, low sample consumption and a possibility for high throughput 17 , 18 enabling large screenings of SARS‐CoV‐2 viral protein representations to evaluate and select the best‐performing combination of antigens.
Here, we present the development and validation of a multiplex, bead‐based serologic assay for detection of antibodies able to bind to different SARS‐CoV‐2 virus proteins including the soluble pre‐fusion stabilised spike glycoprotein (Spike‐f) as well as various versions of spike: S1, RBD and S2, and also different representations of NC. A set of 331 serum samples from SARS‐CoV‐2 patients and convalescents was used as positive controls, while 2090 negative controls were selected among pre‐pandemic collected samples including samples from individuals with PCR‐confirmed seasonal coronavirus infections and autoimmune conditions. The compiled data show that the developed assay is highly stringent and, because of its multiplexity, delivers accurate evaluation of exposure to SARS‐CoV‐2.
Results
The development and evaluation of a serological test for detection of IgG antibodies towards SARS‐CoV‐2 proteins using a multiplex bead‐based assay are here described. Through a three‐stage study design, we explored the potential for antibody detection in a collection of 111 in‐house designed and produced as well as commercially available antigens representing the spike, NC, M and E proteins of the SARS‐CoV‐2 virus (Figure 1a and Supplementary table 1). Although it is preconceived that correctly folded full‐length proteins would function best in a serological assay, we decided to screen a large variety of antigens to enable a comprehensive understanding of the antigenicity of different parts of the proteins. Various representations, such as full‐length versions, domains, folded chains, fragments and peptides of the different proteins, were explored together with different expression hosts and purification formats (Figure 1a and Supplementary table 1). Here, we present the results obtained for antibody detection when using these different protein versions in a cohort of 331 positive and 2090 negative control samples. The antigens enabling the best combination of sensitivity and specificity were further characterised and subsequently combined in panels to create a high performing assay.
Stage 1 ‐ Exploring viral antigens for antibody detection
To establish a method with high performance in terms of both sensitivity and specificity, a large number of proteins and peptides were evaluated during the initial assay development (Supplementary table 1). The results were compared in terms of background levels and intensity signals obtained for a first set of available confirmed positive (n = 9 convalescent blood donors) and negative (n = 40 blood donors from 2019) samples (Figure 1b and Supplementary table 2). This first small set of samples allowed us to perform an initial screening of the capability of each antigen to classify confirmed positive and negative samples, and hence exclude the ones with no or poor separation between the groups from further evaluation. Many of the E. coli produced proteins (Supplementary figure 1a) and the B‐cell predicted peptides (Supplementary figure 1b) yielded a low degree of antibody binding in most positive controls and were therefore excluded from further evaluation. Besides these, various other antigens were defined as less informative than desired in this context and were thus also excluded from further analysis (Supplementary figure 1a and b). From this evaluation, 12 antigens were ranked as best performing in terms of classification and therefore selected for further detailed assessment regarding assay performance (Figure 1b and Supplementary table 1). These included eight different representations of the spike protein, three NC‐based antigens and one antigen representing the membrane protein (Table 1 and Supplementary table 1).
Table 1.
Performance based on binary data from the analysis of 227 positive controls and 442 negative samples collected before 2020
| Antigen | Host | Positive controls | Negative controls | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sensitivity [%] | 95% CI | Positive result | Negative result | Specificity [%] | 95% CI | Positive result | Negative result | ||
| Spike‐f | HEK | 99.6 | 97.6–100 | 226 | 1 | 98.9 | 97.4–99.6 | 5 | 437 |
| Spike‐f | CHO | 99.6 | 97.6–100 | 226 | 1 | 99.3 | 98.0–99.9 | 3 | 439 |
| S1 | HEK | 97.8 | 94.9–99.3 | 222 | 5 | 98.9 | 97.4–99.6 | 5 | 437 |
| S1 | CHO | 99.1 | 96.9–99.9 | 225 | 2 | 99.1 | 97.7–99.8 | 4 | 438 |
| S2 | E. coli | 98.7 | 96.2–99.7 | 224 | 3 | 91.0 | 87.9–93.5 | 40 | 402 |
| RBD | HEK | 99.6 | 97.6–100 | 226 | 1 | 84.6 | 80.9–87.9 | 68 | 374 |
| RBD | CHO | 100 | 98.4–100 | 227 | 0 | 82.1 | 78.2–85.6 | 79 | 363 |
| NTD | HEK | 89.9 | 85.2–93.5 | 204 | 23 | 97.7 | 95.9–98.9 | 10 | 432 |
| NC | E. coli | 96.0 | 92.6–98.2 | 218 | 9 | 98.4 | 96.8–99.4 | 7 | 435 |
| NC‐N | E. coli | 72.2 | 65.9–78.0 | 164 | 63 | 99.3 | 98.0–99.9 | 3 | 439 |
| NC‐C | E. coli | 99.6 | 97.6–100 | 226 | 1 | 88.5 | 85.1–91.3 | 51 | 391 |
| Membrane | E. coli | 87.2 | 82.2–91.3 | 198 | 29 | 97.7 | 95.9–98.9 | 10 | 432 |
Samples with signals that are higher than the cut‐off for antibody reactivity, based on the 6 × SD + mean of the negative controls included in the analysis, are regarded as positive.
Stage 2 ‐ Evaluating 12 selected antigens
The 12 antigens found to provide the most informative profiles of antibody detection were further analysed using an extended set of control samples, including individuals infected by other coronaviruses. To aid in decision making and assess assay performance as well as the inter‐assay variability, 12 carefully selected negative reference samples were included in each assay. These reference samples were selected to provide a broad representation of signal distributions among a long range of pre‐pandemic samples (Supplementary figure 2) and were used to calculate antigen‐wise cut‐off levels in each assay, thereby converting the intensities obtained into binary positive/negative scores.
As a first step, the 12 antigens (Table 1) were characterised in a sample set composed of 301 individuals sampled either once (mild‐to‐moderate disease) or every second day during hospital stay (admitted COVID‐19 patients), with the time of sampling spanning 1–53 days after symptom onset or a positive PCR test. The aim of this analysis was to define the earliest time point at which, by using these 12 antigens, the biological variation of antibody development between individuals and antigens reached a plateau (Figure 2). Spike‐f, RBD and NC‐C reached a plateau at about 12 days while others took longer. Based on this data, we established that, in order for a sample to be considered as a positive control in our assay, the sampling had to be performed at least 17 days after symptom onset or a positive PCR test result. Hence, the presented performance of the assay does not reflect the detectability during early antibody development.
Figure 2.
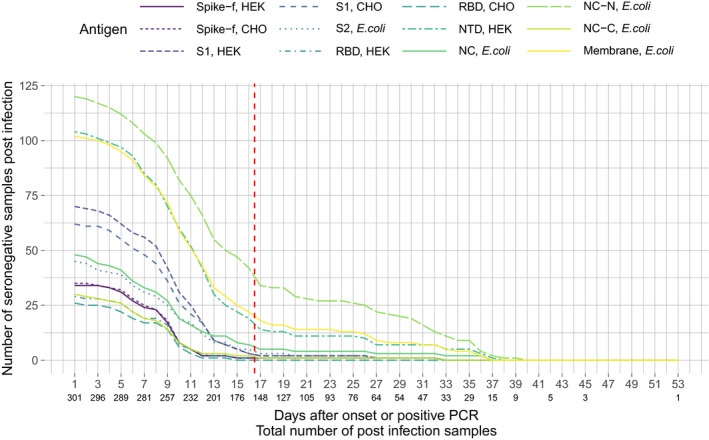
Evaluation of antibody development after symptom onset or a positive PCR test in a total of 301 individuals, shown as number of seronegative post‐infection samples per antigen and day. The red dashed line indicates the selected cut‐off for the definition of a positive control.
Using this definition, the performance of each of the 12 selected antigens to detect previous SARS‐CoV‐2 infections was evaluated by analysing 227 positive and 442 negative control samples. The absolute majority of positive control samples obtained a positive binary score for all 12 antigens (Figure 3a). However, some of the antigens showed lower performance in detecting the positive controls, such as NC‐N and NTD. Interestingly, 116 of the 442 negative controls were classified as positive for one or more of the antigens (Figure 3b), with RBD showing the lowest specificity using this set of control samples. Summarising the binary data, the calculated sensitivities ranged from 72.2 to 100% and specificities from 82.1 to 99.3% for the different antigens (Table 1). The best performance was observed for different representations of the spike and the NC protein, while M showed low sensitivity (87.2%). With regard to specificity, the highest number of false positives was observed for RBD (82.1% and 84.6% of CHO‐ and HEK‐produced RBD, respectively).
Figure 3.
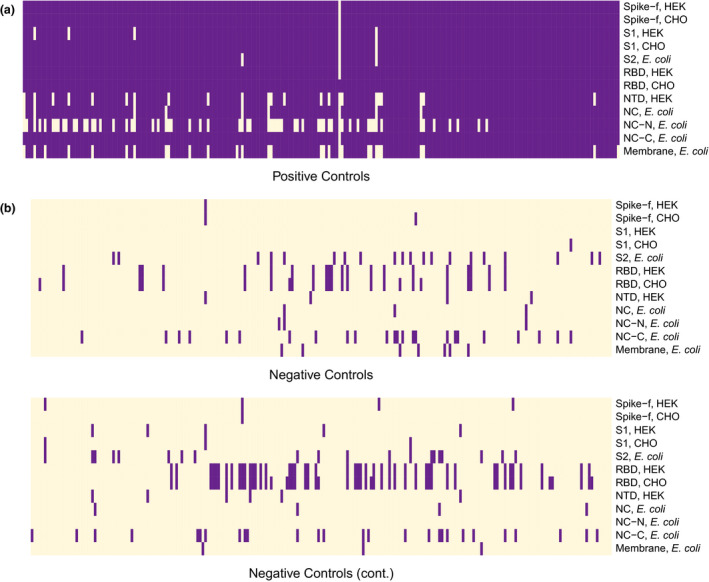
Antibody profiles presented as binary data over the 12 selected antigens in positive (a) (n = 227) and negative (b) (n = 442) controls. The negative controls are divided into two panels with 221 samples each. Purple cells represent signals above the cut‐off for antibody reactivity, based on the mean + 6 × SD of the 12 negative reference samples included in each analysis.
Stage 3 ‐ Comparing the performance of the finally selected antigens using 2421 samples
The eight antigens with best performance in terms of sensitivity and specificity were further characterised using 331 positive and 2090 negative controls. In order to evaluate potential false‐positive samples from individuals within chronic inflammatory systemic diseases, samples from patients with multiple sclerosis, rheumatoid arthritis and systemic lupus erythematosus were also included. An extra sample set representing infections by other coronaviruses as well as CMV and EBV were also included (Supplementary table 2). At this stage, we also aimed to characterise the influence on antibody detection played by the antigen production in different cellular systems. We, therefore, compared the assay output of RBD, Spike‐f, and S1 produced in CHO and HEK. The compiled data for the former two antigens looked very similar (R = 1 and 0.98, respectively, Supplementary figure 3a and b), while for the S1 protein a small discrepancy could be seen (R = 0.97, Supplementary figure 3c). When comparing the binary data, the most similar results were observed for Spike‐f with 19 out of 2421 samples classified differently when using the CHO‐ or HEK‐produced antigen. For RBD produced in different hosts, the data also showed a high concordance with 23 out of 2421 samples classified differently. For S1 produced in different hosts, the discrepancy was larger, with 34 out of 2421 samples classified differently, with CHO‐produced protein as the best performing. Based on the antigen performance in combination with protein production parameters, five of the eight tested antigens were selected for further characterisation. These included HEK‐produced Spike‐f and RBD, CHO‐produced S1, as well as NC and NC‐C produced in E. coli.
The five best‐performing antigens yielded sensitivities ranging from 96.7 to 99.7% and specificities from 92.0 to 98.7%. The top‐performing antigens in terms of sensitivity were Spike‐f, RBD and NC‐C (all 99.7%) while the best in terms of specificity was S1 (98.7%) followed by NC (98.3%) and the Spike‐f (98.1%) (Table 2). Further, the inter‐assay reproducibility was shown to be high for all five antigens, with mean CVs between 7.2 and 13, ranging from 3.1 to 20 (Supplementary table 3). However, Spike‐f, S1, and NC‐C showed the best reproducibility with mean CVs of 7.2 to 8.9 and a maximum of 12.6.
Table 2.
Performance based on binary data from the analysis of 331 positive controls and 2090 negative samples collected before 2020
| Antigen | Host | Positive controls | Negative controls | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sensitivity [%] | 95% CI | Positive result | Negative result | Specificity [%] | 95% CI | Positive result | Negative result | ||
| Spike‐f* | HEK | 99.7 | 98.3–100 | 330 | 1 | 98.1 | 97.4–98.6 | 40 | 2050 |
| Spike‐f | CHO | 99.7 | 98.3–100 | 330 | 1 | 97.9 | 97.2–98.5 | 43 | 2047 |
| S1 | HEK | 98.2 | 96.1–99.3 | 325 | 6 | 98.6 | 98.0–99.0 | 30 | 2060 |
| S1* | CHO | 99.1 | 97.4–99.8 | 328 | 3 | 98.7 | 98.1–99.1 | 27 | 2063 |
| RBD* | HEK | 99.7 | 98.3–100 | 330 | 1 | 92.0 | 90.7–93.1 | 168 | 1922 |
| RBD | CHO | 100 | 98.9–100 | 331 | 0 | 91.8 | 90.5–92.9 | 172 | 1918 |
| NC* | E. coli | 96.7 | 94.1–98.3 | 320 | 11 | 98.3 | 97.6–98.8 | 36 | 2054 |
| NC‐C* | E. coli | 99.7 | 98.3–100 | 330 | 1 | 95.7 | 94.8–96.6 | 89 | 2001 |
A sample is regarded as positive if the signal exceeds 6 x SD from the means of the negative controls included in the analysis. The five antigens that were selected for the final panel are marked with an asterisk.
Comparison of the binary outcomes for the 331 positive and 2090 negative control samples revealed that 319 of the 331 (96.0%) positive controls showed IgG reactivity towards all the five viral antigens. Another nine of the positive control samples showed antibodies to all antigens but NC. One positive control sample did not contain detectable antibodies binding to S1, and one positive control sample had no detectable antibodies to either NC or S1. Notably, one of the positive control samples had no detectable antibodies to any of the five selected viral antigens (Figure 4a). Among the negative controls, the majority of samples (1782/2090, 85.0%) did not show antibodies binding to any of the five antigens. The detectable antibodies were seemingly stochastically distributed over the antigens and only 48 of the negative controls showed antibodies binding towards more than one of the five antigens, with a maximum of 3 reactive antigens per sample seen in four samples (Figure 4b). We also explored the performance of these five targets utilising receiver operating characteristic (ROC) curves and they all display AUC values between 0.994 and 0.999 (Supplementary figure 4).
Figure 4.
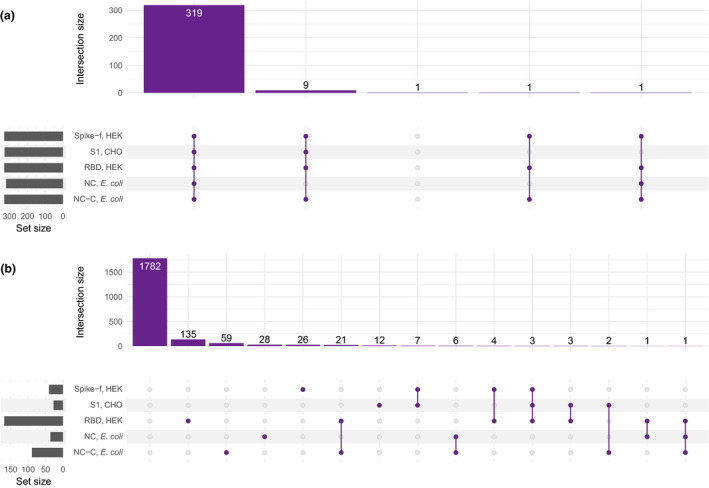
Summary of the antibody profiles in positive and negative controls. The plot indicates the frequency of antibody reactivity towards one or different combinations of viral antigens in (a) positive controls (n = 331) and (b) negative controls (n = 2090).
When evaluating the continuous data from the five best‐performing antigens, we observed a wide distribution of signals among the positive controls for all antigens. Also, some of the negative controls were classified as positive in the binary analysis because of signals just above the cut‐off levels and up to levels comparable with the positive controls (Figure 5a). Interestingly, while IgG levels detected from the different spike antigens showed generally good concordance between the protein representations (Figure 5b), there was a larger discrepancy between the results for the spike versions vs the NC versions (Figure 5c).
Figure 5.
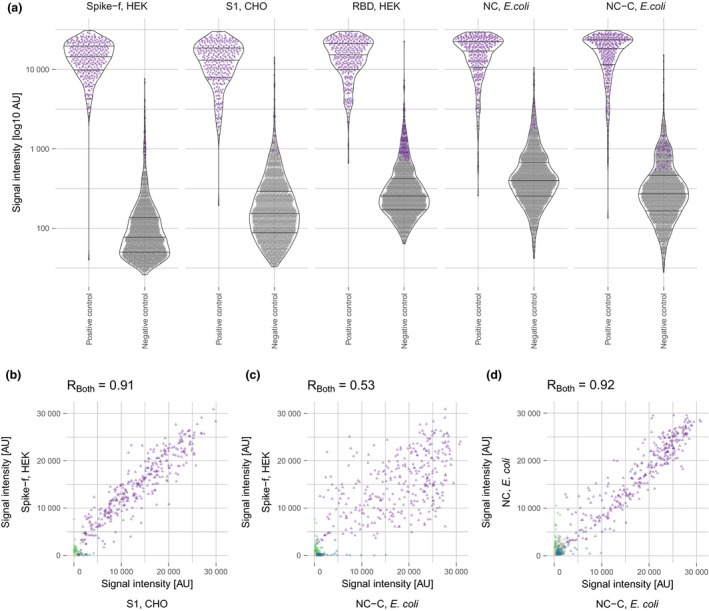
(a) Antibody levels for the five best‐performing antigens in positive (n = 331) and negative (n = 2090) controls. Purple dots represent signals classified as positive. Antibody correlation using 331 positive (triangles) and 290 negative (dots) control samples for (b) Spike‐f and S1, (c) Spike‐f and NC‐C, and (d) NC and NC‐C. (b –d): purple, seropositive against both antigens, used for calculating the Pearson correlation coefficient; green, seropositive against Spike‐f; blue, seropositive against S1/NC‐C; grey, seronegative.
Evaluation of a multiplex serological test
Following the observation of false‐positive results detected for some of the antigens, we investigated combinations of the binary results to increase the performance of the assay. The top three performing antigens, Spike‐f, S1 and NC‐C, were combined into all possible two‐protein panels, and their performance in classifying the 331 positive and 2090 negative controls was evaluated. The sensitivities for the different combinations ranged from 99.1 to 99.7% (1–3 false negative out of 331, Table 3), in line with the singleplex assay performance, while the specificities were improved, with performances between 99.4% and 100.0% (0–12 false‐positive samples out of 2090, Table 3). The best combination, for both sensitivity and specificity, was shown to be Spike‐f and NC‐C, which gave 99.7% sensitivity and 100.0% specificity. Considering the ongoing vaccination efforts focusing on the spike protein 19 and the potential faster decline of anti‐NC antibodies compared with anti‐Spike, 20 , 21 the combination of all three but requiring at least two was also evaluated, resulting in a specificity of 99.7% and a sensitivity of 99.4% (Table 3). All pairwise combinations using the five antigens in stage 3, and panels requiring two out of three antigens, were also evaluated, resulting in a wide range of sensitivities and specificities (Supplementary tables 4 and 5).
Table 3.
Performance when requiring reactivity towards two specific antigens out of Spike‐f, S1 or NC‐C, or reactivity towards two out of the three antigens, based on the analysis of 331 positive and 2090 negative controls
| Antigen | Host | Positive controls | Negative controls | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sensitivity [%] | 95% CI | Positive result | Negative result | Specificity [%] | 95% CI | Positive result | Negative result | ||
|
Spike‐f NC‐C |
HEK E. coli |
99.7 | 98.3–100 | 330 | 1 | 100 | 99.8–100 | 0 | 2090 |
|
S1 NC‐C |
CHO E. coli |
99.1 | 97.4–99.8 | 328 | 3 | 99.9 | 99.7–100 | 2 | 2088 |
|
Spike‐f S1 |
HEK CHO |
99.1 | 97.4–99.8 | 328 | 3 | 99.5 | 99.1–99.8 | 10 | 2080 |
|
Spike‐f |
HEK |
||||||||
|
S1 |
CHO |
99.7 | 98.3–100 | 330 | 1 | 99.4 | 99.0–99.7 | 12 | 2078 |
|
NC‐C |
E. coli |
||||||||
Discussion
Accurate serological tests are of utmost importance to map the progress of the ongoing SARS‐CoV‐2 pandemic but also to understand how and when the immune system responds to the virus and to vaccines as well as the duration of the response. Here, we present the development and validation of a robust high‐throughput serological method that takes advantage of a panel of antigens for an accurate serological evaluation.
As for other coronaviruses, SARS‐CoV‐2 infection has been reported to induce antibody responses towards different viral antigens. 22 The spike glycoprotein (full‐length protein, as well as S1 and S2 subunits and also RBD on its own) and the nucleocapsid protein are the targets that most antibody detection assays are based on, as they are recognised as the most immunogenic targets. Most of the available SARS‐CoV‐2 antibody assays are based on single antigens and detection of antibody of the IgG subclass. Our assay, as well as other similar published efforts, 23 , 24 , 25 demonstrates that a multiplex approach offers a more accurate classification, which has also the potential of remaining more stable over time as well as to provide a more detailed evaluation of the antibody response dynamics in COVID‐19.
Compared to the other multiplex serological assays for SARS‐CoV‐2, 23 , 24 , 25 the uniqueness in the development of our assay resides in the high number (N = 111) and different types of tested antigens which, to our knowledge, is the broadest described. We included antigens produced in different cellular systems, either eukaryotic (HEK and CHO cell lines) or prokaryotic (mainly E. coli), and each antigen was evaluated by testing its ability to classify SARS‐CoV‐2‐positive cases and negative samples collected pre‐pandemic. To guarantee a high performance in a serological test, it is key to select a validation sample set representing the serological spectrum in the general population. 26 Therefore, we included pre‐pandemic serum and plasma samples collected from healthy individuals, as well as individuals infected by other coronaviruses or other viruses (i.e. EBV and CMV), and individuals affected by autoimmune diseases (i.e. SLE, MS and RA) that could induce a perturbation of the immune system resulting in possible false‐positive reactions to SARS‐CoV‐2 antigens. 27 Based on this selection, we could evaluate the background distribution across a wide and comprehensive sample set and thereafter select the 12 negative controls that best represent such background distribution to be used as reference in each assay run for the evaluation of the antigen‐specific cut‐offs (Supplementary figure 2). These 12 negative controls allowed us to evaluate the inter‐assay variability and thereby provide high precision. Moreover, to evaluate the assay sensitivity it is of utmost importance that the test is validated by using not only COVID‐19 severe cases but also cases with milder symptoms. For this reason, we included in our validation process positive cases from hospitalised patients as well as plasma and serum samples from convalescents who reported mild and moderate symptoms. The sensitivity of our assay was also evaluated over time for a subgroup of confirmed SARS‐CoV‐2 cases sampled at 1 or up to 53 days after symptom onset or a positive PCR test, to identify the earliest day at which the majority of the cases had a detectable antibody production. Several antigens showed an increase in the prevalence of reactivity from day 1, reaching a plateau of seropositivity for samples collected at least 17 days after symptoms onset or a positive SARS‐CoV‐2 PCR test. This allowed us to define this as the earliest time point at which our assay performs the best in discriminating positive from negative samples, in agreement with the CDC guidelines for serologic testing indicating that the majority of PCR‐confirmed SARS‐CoV‐2 cases will start developing antibodies between 7 and 14 days after infection onset. 28 This parameter was therefore set as criteria for the definition of positive controls in our specific setting.
The classification performance that we observed varied substantially between the different antigens. As expected, predicted B‐cell epitopes produced as peptides provided low sensitivity. This is in agreement with what is reported in literature, where the reactivity to short peptides was demonstrated to be highly variable among patients. 29 Therefore, the use of full‐length proteins or protein domains, characterised by a higher similarity to the native conformation of the original antigen, as well as by the parallel presence of several linear and conformational epitopes, may allow to better capture the predicted polyclonal humoral response to SARS‐CoV‐2. 30 In agreement to this, the first‐stage investigation of our study resulted in the selection of 12 candidate antigens, of which eight include different full‐length and protein domain versions of the spike protein, produced in HEK or CHO cells, and three versions of the NC, produced in E. coli. Following additional verifications in a large number of positive and negative samples, all versions of the spike protein showed very good classification performance. The NTD from the spike protein was shown to give high specificity but suffer from low sensitivity. Different versions of NC have earlier been shown to have high immunogenicity and are therefore used in a number of commercial serological tests. 15 , 16 Here, we could see that the selectivity and sensitivity were dependent on which part of NC that was used. The N terminus (NC‐N) gave the lowest sensitivity while the C‐terminal part (NC‐C) gave the highest sensitivity. Further, the NC‐N provided the highest specificity (Table 1).
Five proteins were finally included in the latter stage of the validation, of which three were derived from the spike protein and two from the NC. As expected, a good correlation was detected for the intensity signals towards the different spike‐based antigens (Figure 5b) and between the two NC‐based antigens (Figure 5d). When comparing the signal intensities originating from the NC‐ and the spike‐based antigens, a lower correlation could be seen as individuals with high levels of antibodies against the NC do not always show high levels against the spike protein and vice versa (Figure 5c). Since only antibodies directed to the spike protein mediate virus neutralisation, these findings have implications for the usefulness of the NC‐based assays.
While sensitivity is determined by the least performing antigen, specificity can be increased by combining more than one antigen. With the attempt to increase the specificity of our test without losing sensitivity, we combined the binary data from two of the antigens providing the highest sensitivity. The highest specificity was achieved by combining antigens from the two different viral proteins. Hence, through combining one spike‐based antigen and one NC‐based antigen, we were able to keep the high sensitivity (99.7%) and still achieve a specificity as high as 100% (Table 3). If two out of three positive responses were demanded, of which two of the three antigens were based on spike, a sensitivity of 99.7% and a specificity of 99.4% (Spike‐f, S1 and NC‐C) were achieved (Table 3). These findings strengthen the importance of using a multiplex serology test to be able to maximise the assay sensitivity and specificity, and still provide more stable sample classification.
Another uniqueness of our assay development is related to the evaluation of the performance of antigens produced in different cellular systems. The data provided by the spike antigens (full‐length, RBD and S1) produced in HEK and CHO eukaryotic cell systems showed a high concordance with minor differences in the sample classification. This may suggest that possible post‐translational modifications that are different in human and hamster cells may not influence dramatically the reactivity to these antigens.
A limitation of our assay development is related to the absence of verified PCR‐confirmed SARS‐CoV‐2 cases from asymptomatic individuals which may have further improved the assay sensitivity. Another limitation is related to the absence of antigen representing other coronaviruses, at this stage of the validation. However, none of the samples from individuals previously infected by common coronaviruses showed reactivity to any of the SARS‐CoV‐2 representing antigens tested in the presented work, excluding cross‐reactivity of these antibodies to SARS‐CoV‐2 antigens. The assay is here focused on the detection of IgG antibodies, thereby excluding the aspects of COVID‐19 serology associated with IgA and IgM, 31 , 32 even though it is certainly also applicable to the detection of those immunoglobulin classes. All these biases can have an influence on both the false‐positive and false‐negative results and should be kept in consideration. 33
Conclusions
Because of the currently ongoing COVID‐19 pandemic in combination with the heterogeneous symptomatology of the disease and prevalence across different areas of the world, there is a high demand on sensitive and specific methods to identify individuals who have already been infected and thereby possibly developed an immune response. Further, the current global vaccination campaign emphasises the importance to have such methods available. Here, we describe a multi‐step validation of a serology assay based on a multiplex bead‐based SARS‐CoV‐2 array. By combining the analysis of antibodies reacting to different SARS‐CoV‐2 antigens, we designed a highly sensitive and stringent serological assay providing a robust assay performance of 99.7% sensitivity and specificity of up to 100% when utilising a combination of at least two seropositive antigens. Analysing multiple antigens simultaneously can generate data that can be used for diagnostic purposes as well as to further understand the immune system response to SARS‐CoV‐2. Furthermore, the multifaceted data that our method delivers allow for an adaptation of the outcome to the individual analysis, providing accurate and granular data on the reactivity pattern to different SARS‐CoV‐2 antigens at early stage after infection onset.
Methods
Study design
The assay was developed in three stages. First, we designed, constructed, produced and collected a wide range of representations of SARS‐CoV‐2 antigens, including full‐length proteins, protein domains and fragments as well as peptides, and evaluated their utility in antibody detection using a limited set of positive and negative control samples (Figure 1). The antigens to which antibodies could be detected and that initially revealed a clear difference between positive (n = 9) and negative (n = 40) control samples were further included in the second stage, wherein 227 positive and 462 negative control samples were utilised to determine sensitivity and specificity. At this stage, also the performance of antigens produced in bacterial and different mammalian cells was compared. Lastly, the antigens providing the highest sensitivity and specificity were further assessed using an extended sample set including in total 331 positive and 2090 negative control samples. The performance of the selected antigens was evaluated one by one and in different combinations.
Protein design and production
The genes related to the complete genome sequence available at GenBank (severe acute respiratory syndrome coronavirus 2 isolate Wuhan‐Hu‐1, NCBI Reference Sequence: NC_045512.2) were used as base for the design of protein variants representing the SARS‐CoV‐2 virus (Supplementary table 1). Genes and their corresponding proteins were analysed and used to design constructs encoding either full‐length versions, domains, folded chains or fragments of the four structural proteins (Spike, NC, E and M) as well as 19 non‐structural/other parts (Figure 1a). Structures available at PDB (6VYB, 6LVN, 6M3M, 6WZO) were used to select boundaries for domains and folded chains. Fragments of approximately 100 residues were also designed, for both structural and other proteins. Full‐length versions, domains and selected folded chains were cloned for expression in both mammalian cells (human—HEK and hamster—CHO, see below) and bacteria (E. coli), while fragments and most of the folded chains were cloned only for expression in E. coli. In order to facilitate trimerisation of the full‐length spike protein, a C‐terminal T4 fibritin trimerisation motif (foldon) was included in the version denoted Spike‐f. 9 Proteins that did not express well or did not result in a reasonable amount of pure sample were omitted from the study. Previously predicted B‐cell epitopes 34 expected to be exposed in the protein structure (6VYB, 6M3M, 6WZO) were designed as 20 residues long peptides with an N‐terminal biotin (Sigma‐Aldrich, St. Louis, MO, USA). In the initial stage, relevant proteins, available from commercial actors as well as through collaborators, were also included in the assay (Supplementary table 1).
Bacterial expression system, Escherichia coli
For protein production in Escherichia coli (E. coli), BL21 (DE3) in combination with the T7 promoter was used as described by Tegel et al. 35
Human cells, Expi293‐F (HEK)
Expi293‐F cells (Thermo Fisher Scientific, Waltham, MA, USA) were cultivated in Expi293 Expression medium (A1435101, Gibco, Waltham, MA, USA) at 37 °C, 8% CO2 and 85% humidity. For transient production, cells were split the day before transfection to 2 million mL−1, ≥ 98% viability. 1.5 µg plasmid DNA mL−1 transfection culture was used. Transfection reagent, PEI MAX (Polysciences, Inc, Warrington, PA, USA) (1 mg mL−1), was used in a ratio of 1:4 (plasmid:PEI MAX). The transfection reagent and plasmid DNA were separately diluted in water prior to complex formation. The complex was incubated for 15 min before addition to the cells. Twenty‐four hours after transfection, cultures were diluted (1:2) with expression medium. Harvest was performed by centrifugation (4000 g, 30 min) 4 days after transfection, and PMSF stock solution (100 mm phenylmethanesulfonyl fluoride [P7626‐5G, Sigma‐Aldrich] in isopropyl alcohol) was added to the supernatant (5 µL mL−1). The harvested supernatant was finally filtered, using a 0.2‐µm filter, prior to purification. For proteins produced with strep‐tag, 18.1 µL BioLock Biotin blocking solution (2‐0205‐050, IBA Lifesciences, Göttingen, Germany) was added per mL harvest supernatant before filtration.
Hamster cells, ExpiCHO‐S (CHO)
ExpiCHO‐S cells (Thermo Fisher Scientific) were cultivated in ExpiCHO Expression Medium (Gibco, A2910001) at 37 °C, 8% CO2 and 85% humidity. For transient production, cells were split the day before transfection to 4 million mL−1, ≥ 98% viability. Transient transfection was performed with ExpiFectamine™ CHO Transfection Kit (Gibco, A29129) according to the manufacturer's user guide, and the high‐titre protocol was followed. Harvest was performed by centrifugation (4000 g, 30 min) between 5 and 13 days post‐transfection depending on cell concentration and viability > 75%. PMSF stock solution (100 mm phenylmethanesulfonyl fluoride [Sigma‐Aldrich, P7626‐5G]) in isopropyl alcohol was added to the supernatant (5 µL mL−1). The harvested supernatant was finally filtered, using a 0.2‐µm filter, prior purification. For proteins produced with strep‐tag, 25.7 µL BioLock Biotin blocking solution (IBA Lifesciences, 2‐0205‐050) was added per mL harvest supernatant before filtration.
Protein purification and analysis
For the proteins produced in E. coli, a His6‐tag was included for purification by utilising immobilised metal ion affinity chromatography (IMAC) on an ASPEC GX‐274 four‐probe SPE system as described by Tegel et al. 35 All purifications of proteins from mammalian sources were performed on ÄKTAxpress chromatography systems (Cytiva, Uppsala, Sweden). Protein C‐tagged proteins were purified on HPC4 columns according to Kanje et al. 36 Strep‐tagged proteins were purified on StrepTrap columns (Cytiva) according to the manufacturer's instructions. Proteins fused to a mouse IgG2 Fc fragment were purified on MabSelect PrismA or MabSelect SuRe columns (Cytiva) according to the manufacturer's instructions. Protein concentration was determined using absorbance at 280 nm, and analysis of protein purity was performed by SDS–PAGE and western blot. All proteins were finally identified using MS/MS.
Samples
Evaluation of antibody development was performed in samples from 301 individuals with mild‐to‐severe disease collected at 1–53 days after symptom onset or a positive PCR test. A subset of these were longitudinal samples from patients admitted at Danderyd Hospital, Stockholm, Sweden, who were sampled every 2 days during their admittance. For the longitudinal samples, the sample closest to and passing the day criteria was included at each given day in the evaluation (Figure 2). For validation of the method, serum and plasma samples from 331 positive and 2090 negative controls were analysed. The positive control samples used here were defined based on the described antibody development evaluation as collected at least 17 days after positive PCR or confirmed disease onset, and included convalescent blood donors (n = 142), early confirmed cases (n = 143) and admitted COVID‐19 patients (n = 46, last sample in longitudinal sampling used). The negative control samples were collected in 2019 and earlier from healthy controls (n = 1198), individuals with other coronavirus infections (n = 45) as well as patients with other diseases including multiple sclerosis, rheumatoid arthritis and systemic lupus erythematosus (n = 847). See Supplementary table 2 for more detailed sample information.
The study was approved by the Swedish Ethical Review Authority (EPN decision numbers: 2020‐01479, 2020‐01620, 2020‐01649, 2020‐01653, 2020‐01744, 2020‐01881, 2020‐02724, 2020‐02811 and 03‐556).
Generating the multiplex antigen bead array
The viral peptides and proteins were immobilised on the surface of colour‐coded magnetic beads (MagPlex, Luminex Corp., Austin, TX, USA) as previously described. 37 Briefly, each antigen was diluted to a final concentration of 80 µg mL−1 in 100 mm 2‐(N‐morpholino) ethanesulfonic acid buffer, pH 4.5 (Sigma‐Aldrich) and coupled to one bead identity (colour code). The carboxylated surface of 1 × 106 colour‐coded magnetic beads per bead identity was activated by using 100 µL phosphate buffer complemented with 0.5 mg 1‐(3‐dimethylaminopropyl)‐3‐ethylcarbodiimide hydrochloride (ProteoChem, Inc., Hurricane, UT, USA) and 0.5 mg N‐hydroxysulfosuccinimide (Thermo Fisher Scientific). Activated beads were incubated for 2 h with the diluted antigen, followed by overnight incubation in blocking buffer (Blocking Reagent for ELISA, Roche, supplemented with 0.1% (v/v) ProClin, Sigma‐Aldrich). The bead identities were then pooled to form the antigen‐bead array. Besides the viral antigens, two bead identities were coupled with anti‐human IgG (309‐005‐082, Jackson Immunoresearch, West Grove, PA, USA) and the EBNA1 protein (ab138345, Abcam, Cambridge, UK) from the common Epstein–Barr virus and used as sample loading controls.
Antibody profiling on bead arrays
Plasma and serum samples from positive and negative controls were tested with the antigen bead array as previously published. 37 Briefly, samples were thawed at 4°C and diluted 1:50 in assay buffer composed of 3% (w/v) bovine serum albumin (Saveen‐Werner, Limhamn, Sweden) and 5% (w/v) non‐fat milk (Sigma‐Aldrich) in 1×PBS supplemented with 0.05% (v/v) Tween‐20 (Thermo Fisher Scientific) (PBS‐T) using a liquid handling system (EVO150, TECAN, Männedorf, Switzerland). After dilution, the samples were distributed into a 384‐well microtitre plate (Greiner BioOne, Kremsmunster, Austria) containing the antigen bead array and incubated for 1 h at room temperature. The beads were then washed with PBS‐T, and antibody–antigen immunocomplexes, cross‐linked by applying 0.2% paraformaldehyde for 10 min at room temperature. After further washing, beads were incubated with R‐phycoerythrin‐conjugated anti‐human IgG (H10104, Invitrogen, Carlsbad, CA, USA) diluted to 0.4 µg mL−1 in 1×PBS‐T for 30 min at room temperature. Finally, signal detection was performed using a FlexMap3D instrument and Luminex xPONENT software (Luminex Corp.). In total, 15 assays were run to cover all included samples. In each assay, 4 positive and 12 negative reference samples were included. The negative reference samples were thoroughly selected among the pre‐pandemic samples in stage 2 (n = 462) to represent the wide range of possible background signals of the tested antigens (Supplementary figure 2) and used to calculate the seropositivity cut‐off (see section Data analysis for bead array data). The positive reference samples were selected from post‐infection cases to represent clearly seropositive samples and were included as a technical control in each assay run to confirm that the desired separation of true‐negative and true‐positive samples was achieved. All samples were analysed once at a dilution of 1:50, utilising a dynamic range of approximately 10e4.
Data analysis for bead array data
Statistics and data visualisations were performed using R (version 3.6.1) with RStudio (1.2.1335) and the additional packages tidyverse (1.3.0), pheatmap (1.0.10), viridis (0.5.1), ComplexUpset (0.8.1), patchwork (1.0.1), ggbeeswarm (0.6.0), ggpubr (0.2.5), gtable (0.2.0), WriteXLS (5.0.0), xlsx (0.6.1), reshape2 (1.4.3), plotROC (2.2.1) and in‐house developed functions for instrument file import and quality control.
The data are based on relative quantification and acquired as median fluorescent intensity (MFI) per sample and bead identity and presented here as signal intensity (arbitrary unit, AU). Unprocessed data are used for all visualisations when using continuous data, except for the heatmap of peptides (Supplementary figure 1b). The peptide data were normalised per sample to compensate for sample specific backgrounds using the algorithm (MFI − sample median)/sample MAD, where MAD is the median absolute deviation. The resulting value is number of MADs around the sample median. In each assay, a cut‐off for seropositivity was calculated per antigen as the mean + 6 × SD, rounded up to the nearest integer, of the 12 negative reference samples included in each run. The cut‐off values are therefore antigen and assay specific, to account for inter‐assay variation. The 95% confidence interval of the test performances was calculated using an exact binomial test (binom.test, stats). Correlations were evaluated using the Pearson correlation coefficient (cor.test, stats). The inter‐assay variability was evaluated as % CV for the antigens in stage 3 across the positive reference samples included in each assay, totalling to 12 unique samples present in 17–85 assays each and 129 assays in total. The receiver operating characteristic (ROC) curves and resulting area under the curve (AUC) were generated using unprocessed signal intensities (melt_roc, geom_roc and calc_auc, plotROC).
Conflict of interest
The authors declare no conflict of interest.
Author Contribution
Sophia Hober: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Cecilia Hellström: Data curation; Formal analysis; Methodology; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Jennie Olofsson: Formal analysis. Eni Andersson: Formal analysis. Sofia Bergstrom: Formal analysis. August Jernbom Falk: Formal analysis. Shaghayegh Bayati: Formal analysis. Sara Mravinacova: Formal analysis. Ronald Sjoberg: Resources; Software. Jamil Yousef: Formal analysis. Lovisa Skoglund: Formal analysis. Sara Kanje: Investigation; Methodology. Anna Berling: Investigation; Methodology. Anne‐Sophie Svensson : Investigation; Methodology. Gabriella Jensen: Investigation; Methodology. Henric Enstedt: Investigation; Methodology. Delaram Afshari: Investigation; Methodology. Lan Lan Xu: Investigation; Methodology. Martin Zwahlen: Software. Kalle von Feilitzen: Software. Leo Hanke: Investigation; Writing‐review & editing. Ben Murrell: Investigation; Writing‐review & editing. Gerald McInerney: Investigation; Writing‐review & editing. Gunilla B Karlsson Hedestam: Investigation; Writing‐review & editing. Christofer Lendel: Investigation. Robert G Roth: Investigation. Ingmar Skoog: Resources; Writing‐review & editing. Elisabet Svenungsson: Resources. Tomas Olsson: Resources; Writing‐review & editing. Anna Fogdell‐Hahn: Resources; Writing‐review & editing. Ylva Lindroth: Resources. Maria Lundgren: Resources. Kimia Maleki: Resources. Nina Lagerqvist: Resources. Jonas Klingström: Methodology; Resources; Writing‐review & editing. Rui Da Silva Rodrigues: Resources. Sandra Muschiol: Resources. Gordana Bogdanovic: Resources. Laila Sara Arroyo Mühr: Resources. Carina Eklund: Resources. Camilla Lagheden: Resources. Joakim Dillner: Resources; Writing‐review & editing. Åsa Sivertsson: Investigation; Methodology; Software; Writing‐review & editing. Sebastian Havervall: Investigation; Resources; Writing‐review & editing. Charlotte Thålin: Funding acquisition; Investigation; Resources; Writing‐review & editing. Hanna Tegel: Investigation; Methodology; Resources; Writing‐review & editing. Elisa Pin: Conceptualization; Investigation; Methodology; Writing‐original draft; Writing‐review & editing. Anna Månberg: Conceptualization; Investigation; Methodology; Writing‐original draft; Writing‐review & editing. My Hedhammar: Investigation; Methodology; Resources; Supervision; Writing‐review & editing. Peter Nilsson: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Supporting information
Acknowledgments
This study was funded by Region Stockholm, Region Skåne, the Knut and Alice Wallenberg Foundation, the Science for Life Laboratory (SciLifeLab), the Erling Persson family foundation, Christian and Jennifer Dahlberg, the Atlas Copco and an EU grant (CoroNab). We express our gratitude to Malin Westin, Malte Nygren, Issra Ahmed Dawi Ali, Faranak Bidad, David Just, Julia Remnestål, Mimmi Olofsson, Cornelia Westerberg, Emmie Pohjanen, MariaJesus Iglesias, Anna Mattsson, Nastya Kharlamova for technical assistance and all members of the Schwenk laboratory for great support. Maria Stenvall is acknowledged for help with the illustrations. The AstraZeneca Protein Reagent team is acknowledged for contributing protein reagents.
References
- 1. Zhou P, Yang XL, Wang XG et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; 579: 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dong E, Du H, Gardner L. An interactive web‐based dashboard to track COVID‐19 in real time. Lancet Infect Dis 2020; 20: 533–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rudberg AS, Havervall S, Månberg A et al. SARS‐CoV‐2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. Nat Commun 2020; 11: 5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pascarella G, Strumia A, Piliego C et al. COVID‐19 diagnosis and management: a comprehensive review. J Intern Med 2020; 288: 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan JF, Yip CC, To KK et al. Improved molecular diagnosis of COVID‐19 by the novel, highly sensitive and specific COVID‐19‐RdRp/Hel real‐time reverse transcription‐PCR assay validated. J Clin Microbiol 2020; 58: e00310‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neuman BW, Adair BD, Yoshioka C et al. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J Virol 2006; 80: 7918–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 2015; 1282: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. Proteolytic activation of the SARS‐coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res 2013; 100: 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wrapp D, Wang N, Corbett KS et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science 2020; 367: 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daly JL, Simonetti B, Klein K et al. Neuropilin‐1 is a host factor for SARS‐CoV‐2 infection. Science 2020; 370: 861–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang CK, Hou MH, Chang CF, Hsiao CD, Huang TH. The SARS coronavirus nucleocapsid protein–forms and functions. Antiviral Res 2014; 103: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Premkumar L, Segovia‐Chumbez B, Jadi R et al. The receptor binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS‐CoV‐2 patients. Sci Immunol 2020; 5: eabc8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ravi N, Cortade DL, Ng E, Wang SX. Diagnostics for SARS‐CoV‐2 detection: a comprehensive review of the FDA‐EUA COVID‐19 testing landscape. Biosens Bioelectron 2020; 165: 112454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. COVID‐19 In Vitro Diagnostic Medical Devices. Available from: https://covid‐19‐diagnostics.jrc.ec.europa.eu/devices [last accessed November 2020].
- 15. Lisboa Bastos M, Tavaziva G, Abidi SK et al. Diagnostic accuracy of serological tests for covid‐19: systematic review and meta‐analysis. BMJ 2020; 370: m2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Espejo AP, Akgun Y, Al Mana AF et al. Review of current advances in serologic testing for COVID‐19. Am J Clin Pathol 2020; 154: 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ayouba A, Thaurignac G, Morquin D et al. Multiplex detection and dynamics of IgG antibodies to SARS‐CoV2 and the highly pathogenic human coronaviruses SARS‐CoV and MERS‐CoV. J Clin Virol 2020; 129: 104521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pisanic N, Randad PR, Kruczynski K et al. COVID‐19 serology at population scale: SARS‐CoV‐2‐specific antibody responses in saliva. J Clin Microbiol 2020; 59: e02204‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tregoning JS, Brown ES, Cheeseman HM et al. Vaccines for COVID‐19. Clin Exp Immunol 2020; 202: 162–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Elslande J, Oyaert M, Ailliet S et al. Longitudinal follow‐up of IgG anti‐nucleocapsid antibodies in SARS‐CoV‐2 infected patients up to eight months after infection. J Clin Virol 2021; 136: 104765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stromer A, Rose R, Grobe O et al. Kinetics of Nucleo‐ and Spike Protein‐Specific Immunoglobulin G and of Virus‐Neutralizing Antibodies after SARS‐CoV‐2 Infection. Microorganisms 2020; 8: 1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shah VK, Firmal P, Alam A, Ganguly D, Chattopadhyay S. Overview of immune response during SARS‐CoV‐2 infection: lessons from the past. Front Immunol 2020; 11: 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grossberg AN, Koza LA, Ledreux A et al. A multiplex chemiluminescent immunoassay for serological profiling of COVID‐19‐positive symptomatic and asymptomatic patients. Nat Commun 2021; 12: 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosado J, Pelleau S, Cockram C et al. Multiplex assays for the identification of serological signatures of SARS‐CoV‐2 infection: an antibody‐based diagnostic and machine learning study. Lancet Microbe 2021; 2: e60–e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bray RA, Lee JH, Brescia P et al. Development and validation of a multiplex, bead‐based assay to detect antibodies directed against SARS‐CoV‐2 proteins. Transplantation 2021; 105: 79–89. [DOI] [PubMed] [Google Scholar]
- 26. Takahashi S, Greenhouse B, Rodríguez‐Barraquer I. Are seroprevalence estimates for severe acute respiratory syndrome coronavirus 2 biased? J Infect Dis 2020; 222: 1772–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tzouvelekis A, Karampitsakos T, Krompa A, Markozannes E, Bouros D. False positive COVID‐19 antibody test in a case of granulomatosis with polyangiitis. Front Med 2020; 7: 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Center for Disease Control and Prevention. Available from: https://www.cdc.gov/coronavirus/2019‐ncov/lab/resources/antibody‐tests‐guidelines.html (accessed 7 Feb 2021).
- 29. Li Y, Ma ML, Lei Q et al. Linear epitope landscape of the SARS‐CoV‐2 Spike protein constructed from 1,051 COVID‐19 patients. Cell Rep 2021; 34: 108915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wheatley AK, Juno JA, Wang JJ et al. Evolution of immune responses to SARS‐CoV‐2 in mild‐moderate COVID‐19. Nat Commun 2021; 12: 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Semmler G, Traugott MT, Graninger M et al. Assessment of S1‐, S2‐, and NCP‐specific IgM, IgA, and IgG antibody kinetics in acute SARS‐CoV‐2 infection by a microarray and 12 other immunoassays. J Clin Microbiol 2021; 59: e02890‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sterlin D, Mathian A, Miyara M et al. IgA dominates the early neutralizing antibody response to SARS‐CoV‐2. Sci Transl Med 2021; 13: eabd2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu G, Rusling JF. COVID‐19 antibody tests and their limitations. ACS Sens 2021; 6: 593–612. [DOI] [PubMed] [Google Scholar]
- 34. Ahmed SF, Quadeer AA, McKay MR. COVIDep: a web‐based platform for real‐time reporting of vaccine target recommendations for SARS‐CoV‐2. Nat Protoc 2020; 15: 2141–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tegel H, Steen J, Konrad A et al. High‐throughput protein production–lessons from scaling up from 10 to 288 recombinant proteins per week. Biotechnol J 2009; 4: 51–57. [DOI] [PubMed] [Google Scholar]
- 36. Kanje S, Enstedt H, Dannemeyer M, Uhlén M, Hober S, Tegel H. Improvements of a high‐throughput protein purification process using a calcium‐dependent setup. Protein Expr Purif 2020; 175: 105698. [DOI] [PubMed] [Google Scholar]
- 37. Pin E, Sjöberg R, Andersson E et al. Array‐based profiling of proteins and autoantibody repertoires in CSF. Methods Mol Biol 2019; 2044: 303–318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials