

Abstract
Covalent organic frameworks (COFs) have been at the forefront of porous-material research in recent years. With predictable structural compositions and controllable functionalities, the structures and properties of COFs could be controlled to achieve targeted materials. On the other hand, the predesigned structure of COFs allows fruitful postsynthetic modifications to introduce new properties and functions. In this review, the postsynthetic functionalizations of COFs are discussed and their impacts towards structural qualities and performances are comparatively elaborated on. The functionalization involves the formation of specific interactions (covalent or coordination/ionic bonds) and chemical reactions (oxidation/reduction reaction) with pendant groups, skeleton and reactive linkages of COFs. The chemical stability and performance of COFs including catalytic activity, storage, sorption and opto-electronic properties might be enhanced by specific postsynthetic functionalization. The generality of these strategies in terms of chemical reactions and the range of suitable COFs places them as a pivotal role for the development of COF-based smart materials.
Keywords: covalent organic frameworks, crystalline materials, porous materials, postsynthetic functionalization
Introduction
Inspired by the highly complex, yet systematic, organization of porous-nature materials, plentiful efforts have been devoted by researchers to mimicking their properties. Over the past several decades, the regular arrangement of secondary building units of porous inorganic zeolites to establish defined micropores has shown tremendous advantages in high-demand applications [1–4]. On the other hand, with the manageable and controllable porosity under the reticular chemistry principle, metal-organic frameworks (MOFs) have come onto the scene as the second generation of crystalline porous materials [5–7]. In 2005, Yaghi et al. designed a new type of crystalline porous polymer by reticulating predesigned organic building blocks into certain 2D or 3D networks, termed covalent organic frameworks (COFs) [8]. This great discovery tackles the bottleneck of the preconception in which covalent linking of organic molecules into crystalline solids is challenging, since it typically affords amorphous solids or disordered materials.
As an emerging class of highly porous organic polymers, COFs have been researched for over a decade. The rigid backbone and precise arrangement of building blocks in COFs afford highly crystalline materials, making them more robust than amorphous organic polymers [9]. From 2005 until the present, the library of COF compounds has been expanded rapidly with the highly enthusiastic interest of researchers. Since COFs are composed solely of light-organic elements (B, C, O, N and S), they have been remarked as having the lowest density among porous materials [10]. This could promise better performance in several applications, especially in gas storage and other highly demanding applications.
The synthesis of COFs allows the formation of periodic skeleton and defined pores [8]. Under a reversible condensation reaction, the classical crystallization issue found in amorphous organic polymers has been addressed fruitfully in COF synthesis provided by a defect-healing mechanism. In principle, there are two distinct components in COF construction, namely linkers (building blocks) and linkages (bonds connecting the building blocks). The myriad availability of building blocks with different sizes and shapes garners flexibility in designing the porosity and topologies of COFs. Nevertheless, the strong covalent linkages contribute to the high chemical and thermal stabilities of COFs, which are crucial aspects in MOFs and inorganic zeolites. Furthermore, the bottom-up synthesis of COFs allows the designable structural framework that provides the control in the structural–property relationship of the resultant product. This encompasses the pore size, surface physicochemical environment and structural modification. The flexible pore wall decoration (the incorporation of pendant groups and/or active sites) in COFs offers potential exploration in structural functionalization.
Structural functionalization in COFs provides structural qualities and performance enhancements, including gaseous/adsorbate uptake–release, optical/electrical responses and host–guest interactions. Generally, the structural functionalization of COFs can be performed by pre-designing the building blocks (bottom-up approach) and by the postsynthetic functionalization approach. In the bottom-up approach, the building blocks are previously modified with the desired functional groups that are stable during COF synthesis. Meanwhile, in the postsynthetic approach, the functional groups are introduced or attached to the pore surface of COFs while maintaining the structural integrity. Comparatively, the later approach is technically more viable compared to the former one. Indeed, several reported works have shown the structural and performance enhancement of COFs through postsynthetic functionalization over a wide range of applications. In this review, we summarize the postsynthetic functionalization of COFs that encompasses the functionalization of the pendant groups and the linkages, involving the formation of specific bonds (covalent and coordination/ionic bonds), chemical reaction (oxidation/reduction reaction) and host–guest interaction. In addition, we further elaborate comparatively on the contribution of this functionalization to the structural qualities and performances of the resultant COFs.
Structural functionalization approaches in COFs
The design flexibility of COFs through the wide selectivity of building blocks offers opportunities to decorate their backbone with functional moieties. Meanwhile, the adjustable pore size and environment of COFs make them an ideal scaffold for the incorporation of new functional groups. Technically, structural functionalization of COFs can be divided into two approaches: bottom-up and postsynthetic approaches, which are illustrated in Scheme 1. Both approaches produce the desired functionalized COFs with different properties in terms of crystallinity, porosity and stability.
Scheme 1.
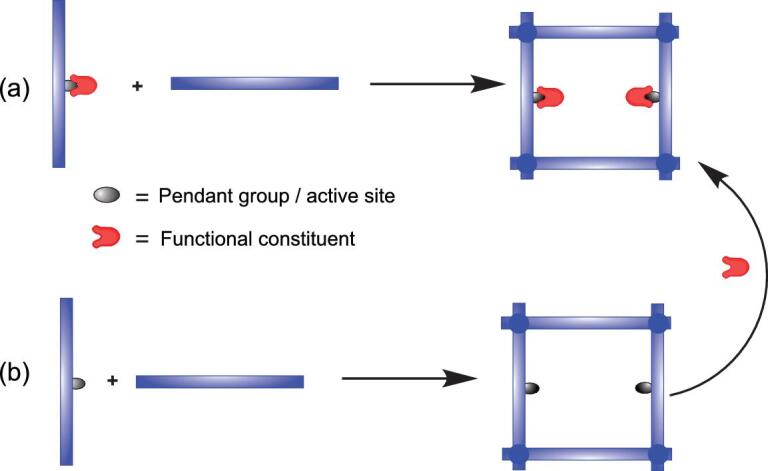
Structural functionalization approaches in COFs. (a) Bottom-up approach and (b) postsynthetic functionalization approach.
Bottom-up functionalization
In the bottom-up functionalization approach, the functional moieties of COFs are originated from the predesigned building blocks (Scheme 1a). One representative piece of work on this approach was reported by Wang et al., in which they synthesized 2D mesoporous COFs with chiral functional groups (COF-LZU-72 and COF-LZU-76) [11]. To produce those COFs, the basic building block (diamino-p–terphenyl) was modified with chiral pyrrolidine moiety previously to afford the desired functionalized unit. However, it should be noted that a certain protective group might be needed during the pre-modification. A similar vision was then adopted to generate several functionalized COFs including thiol-functionalized 2D COF (Thio-COF) and sulfonic acid-functionalized 2D COF (TpPa-SO3H) [12,13].
However, besides the probable protection–deprotection process, there are other crucial disadvantages based on the bottom-up approach, such as the difficulties in synthesizing functionalized building blocks and challenges in the incorporation of large or bulky constituents that may disturb the regularity of the desired COFs. Thus, this approach seems hard for a variety of functional groups.
Postsynthetic functionalization
While some functionalized COFs can be obtained via a bottom-up approach, a postsynthetic functionalization strategy provides a more robust platform (Scheme 1b). Comparatively, it offers a versatile avenue to introduce wider types of functional moieties into COFs without altering the structural regularity. More importantly, it avoids or minimizes the involvement of undesired side reactions during reticulation and functional incorporation. The functionalization of COFs via a postsynthetic approach involves specific bond formation, chemical reactions and host–guest interactions between the pendant groups or the active sites of the established COF and functional constituents. On this point, we classify them into three categories: pendant group and skeleton functionalization, linkage functionalization and host–guest functionalization.
Pendant groups and skeleton functionalization
The specific pendant groups (Table 1) and the skeleton of COFs can be sufficiently functionalized by treating with desired functional constituents. Particularly, the interaction between them includes either the formation of covalent or ionic/coordination bonds or oxidation/reduction reactions, depending on the reaction conditions and types of functional modifiers. In the following sub-section, both interactions are elaborated on, along with their contribution to the stability and performance.
Table 1.
Typical reactive pendant groups appended on COF backbone useful for further functionalization
| Pendant groups | Coupling agents | Bond formed |
|---|---|---|
| -N3 (azide) | -C≡C- (alkyne) | Triazole |
| -OH (hydroxyl) | -C(O)-O-C(O)- (anhydride)-NCS (isothiocyanate)-Br (halide) | EsterThiorcarbamateEther |
| -NH2 (amine) | -C(O)-O-C(O)- (anhydride) | Amide |
| -≡N (nitrile) | -NH2 (amine) | Amidoxime |
| -SH (thiol) | -C=C- (alkene) | Thioether |
Covalent bond
The functionalization of pendant groups of COFs via covalent bonds is performed by linking the incoming functional constituents with the available pendant groups on the pore channel. Practically, typical COFs with reactive pendant groups are designed as the scaffold. Table 1 summarizes the typical pendant groups available in COFs and their corresponding coupling agents that have been demonstrated in functionalizing COFs. The typical bonds of triazole, ester, amide, amidoxime, thioether, thiocarbamate and ether have been adopted so far to postsynthetically functionalize COF as illustrated in Fig. 1.
Figure 1.
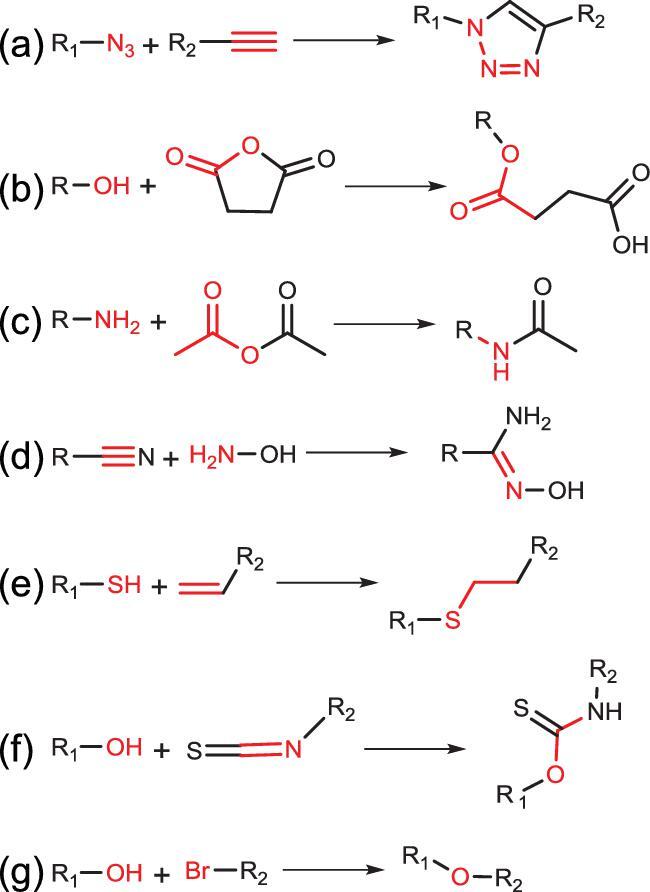
Formation of typical covalent bonds adopted in postsynthetically functionalizing the structural backbone of COFs. (a) Triazole, (b) ester, (c) amide, (d) amidoxime, (e) thioether, (f) thiocarbamate and (g) ether.
The triazole bond that can be prepared under a CuI-assisted click reaction between azide (R-N3) and ethynyl (R≡CH) groups has been widely employed to functionalize COFs (Fig. 1a). For example, treating azide-appended COFs (X%N3-COF-5) with alkynes afforded triazole-functionalized COF-5 (X%RTrz-COF-5) (Fig. 2a) [14]. Notably, this functionalization was compatible for various functional groups attached on the ethynyl groups (R = acetyl (Ac), butyl (Bu), benzyl (Ph), methyl ester (Es) or pyrene (Py)). In addition, it preserved the crystalline nature of the former scaffold and was able to control the density of the triazole groups in the final product. The resultant X%RTrz-COF-5 series possessed different affinities to the different gases and displayed varied adsorption capabilities, which is a potential for
Figure 2.

Postsynthetic functionalization of pendant groups of COFs via various covalent bonds. (a) Functionalization of azide-appended X%N3-COF-5 series into X%RTrz-COF-5 series via triazole bond. (b) Functionalization of hydroxyl-appended 3D-OH-COF into 3D-COOH-COF via ester bond. (c) Functionalization of amine-appended TpBD(NH2)2 COF into TpBD(NHCOCH3)2 via amide bond.
gas separation. Meanwhile, an extended chiral centre and catalytic active site has successfully been endowed within [HC≡C]x-TPB-DMTP-COFs by reacting alkynyl (-C≡CH) pendant groups with azide-containing pyrrolidine (N3-Py) [15]. The resultant [(S)-Py]x-TPB-DMTP-COFs contained chiral and catalytic active pyrrolidine moieties. Thus, they have potential as a chiral organocatalyst. Remarkably, [(S)-Py]x-TPB-DMTP-COFs were crystalline with a significant porosity intake. As a heterogeneous chiral organocatalyst, [(S)-Py]x-TPB-DMTP-COFs could catalyse the organic Michael-addition reaction—a basic C–C bond-formation reaction for synthesizing various important synthetics and natural products [16]. In addition, [(S)-Py]x-TBP-DMTP-COFs were able to promote asymmetric C–C bond formation in water under ambient temperature with prominent activity, enantioselectivity, recyclability and environmental benignity compared to the other metal-based catalysts. For instance, [(S)-Py]0.17-TBP-DMTP-COF proceeded smoothly and cleanly to C–C bond formation with 100% conversion in 12 h (e.e. = 92% and d.r. = 90/10), which largely exceeded the molecular (S)-Py catalyst. Furthermore, several other COFs have been functionalized via triazole linkage, including the transformation from [CH≡C]x-H2P-COF into [Pyr]x-H2P-COF and [CH≡C]0.5-TPB-DMTP-COF into ([uracil]0.5-TPB-DMTP-COF) with preservation of crystallinity and enhanced performance for catalyst and adsorbent applications, respectively [17,18]. Meanwhile, a large molecule of bucky-ball (C60) has also been anchored on the pore surface of predesigned X%[N3]-ZnPc-COF to afford donor–acceptor heterojunction-based [C60]y-ZnPc-COF via covalent triazole linkage [19]. As predicted, the resultant [C60]y-ZnPc-COF exhibited a photoelectric phenomenon with segregated donor–acceptor alignment in which electron-accepting bucky-balls were spatially confined within the nanochannel via triazole units. In addition, it performed photo-induced electron transfer facilitated by an electron-donating COF scaffold and electron-accepting bucky-ball molecules. COFs with high energy-storage performance were achieved by the covalent introduction of redox-active species [20]. In another piece of work, the CO2-capture and -separation properties of COFs were tuned via anchoring diverse functional groups from hydrophobic to hydrophilic and from acid to basic through such approaches [21].
Ester bonds have judiciously been applied to functionalize COFs, which were efficiently obtained under a mild and metal-free ring-opening reaction between anhydride and hydroxyl (-OH) groups (Fig. 1b) [22]. Hydroxyl-decorated square-like 2D porphyrin COFs ([HO]x%-H2P-COFs) were synthesized and employed as scaffolds to demonstrate the structural functionalization of COF via an ester bond [23]. The reaction of these COFs with succinic anhydride generated functionalized COFs with free-carboxyl groups linked by an ester bond ([HO2C]x%-H2P-COFs). This functionalization allows structural rigidity preservation with a well-organized functional constituent within the pore channel and substantial porosity intake. At the end, [HO2C]x%-H2P-COFs were employed as solid sorbents for CO2 storage and separation. Remarkably, the [HO2C]x%-H2P-COF sample demonstrated dramatic CO2-uptake enhancement relative to [HO]x%-H2P-COF. In addition, the ideal absorbed solution theory (IAST) calculation was performed based on their CO2 and N2 sorption isotherms, revealing high CO2-absorption selectivity (Sads) with 323 for [HOOC]100%-H2P-COF (at 0.1 kPa)—much greater than [HO]100%-H2P-COF (only 18). Indeed, this performance was much higher than for other reported adsorbents. On another occasion, a predesigned hydroxyl-appended 3D COF (3D-OH-COF) was prepared and further functionalized via an ester bond (Fig. 2b) [24]. A transformation from hydroxyl pendant groups to ester bonds via the ring-opening reaction with succinic anhydride afforded carboxyl-functionalized 3D COF (3D-COOH-COF). With retained crystallinity and sufficient porosity, the resultant 3D-COOH-COF was employed as a selective adsorbent for Nd3+ over Sr2+ and Fe3+ in aqueous solution. Interestingly, Nd3+ had the highest uptake at low concentrations, as a result of stronger binding with the framework, while Fe3+ possessed the highest uptake at saturation owing to its smaller size. Meanwhile, further IAST calculation showed that 3D-COOH-COF selectively absorbed Nd3+ ion over Sr2+ ion (with IAST selectivity = 27) and Fe3+ ions (IAST selectivity = 18) for a solution containing 5% Nd3+ ion and 95% Sr2+ or Fe3+ ions.
Amide bonds (the strong bonds found in protein) can serve as active sites with secondary amine and carbonyl groups. The incorporation of an amide bond within COFs could trigger the formation of highly porous materials with high chemical stability and abundant active sites. The formation of this bond is typically performed under reaction between primary amine (-NH2) and carbonyl groups (Fig. 1c). However, it seems a challenge to incorporate functional groups such as aldehydes, boronic acids or anhydrides, since they are prone to reacting with the building blocks or likely disturb the formation of the rigid backbone of COFs. A chemically stable amino-functionalized TpBD(NH2)2 was designed by reducing its parent nitro-functionalized COF (TpBD(NO2)2) [25]. The aminolysis reaction [26] between TpBD(NH2)2 and acetic anhydride manifested the amide bond containing COF (TpBD(NHCOCH3)2) (Fig. 2c). Notably, the crystalline was still preserved even after sequential transformation of the COF. Furthermore, the benefit from the high chemical stability in TpBD(NHCOCH3)2 was applied in lactic acid (LA, pH = 2.2, 0.1 M) adsorption. The TpBD(NHCOCH3)2 sample exhibited moderate LA uptake (4.0 wt%) compared to TpBD(NO2)2 and TpBD(NH2)2 (2.5 and 6.6 wt%, respectively). Indeed, these absorption trends were due to the presence of H-bonding and the chemical stability found in amine and amide-functionalized COFs.
In line with the postsynthetic functionalization of COFs mentioned above, another predesigned COF was sequentially functionalized to form a covalent organic nanosheet (CON) with specific functionality [27]. The COF (TpASH) was prepared under a salt-mediated solid-state mixing procedure. Subsequently, TpASH was proceeded into three-sequential postsynthetic functionalization in which its phenolic hydroxyl pendant groups were converted into alkyl-hydroxyl groups under conjugation with glycidol (GIc) in the first step to obtain TpASH-GIc. It was then reacted with 3-aminopropyltriethoxysilane (APTES) to give amine (-NH2)-functionalized CONs (TpASH-APTES). In the last step, the folic acid was incorporated into the pore surface of TpASH-APTES CONs to produce the targeted functionalized CONs (TpASH-FA). Interestingly, under sequential functionalization, stacked layers of COFs were delaminated into a few layers of functionalized CONs with enhanced dispersibility while preserving the structural integrity. In the end, the resultant TpASH-FA was employed as a drug carrier for 5-fluorouracyl (5-FU) molecules. Notably, TpASH-FA could load 12% of the 5-FU and established cancer-specific drug release. Indeed, this report signals the future prospect of CONs for drug-delivery application obtained by postsynthetic functionalization. The electron-rich primary amine and anthracene imine are promising active sites for sorption application. The amidoxime bond particularly possesses primary amine and anthracene imine sites. Functionalization of COFs via amidoxime bonds was performed to incorporate those sites that have potential for the generation of adsorbents for nuclear-waste mitigation (Fig. 1d). Cyan-appended 2D COF (COF-TpDb) was treated with hydroxylamine (NH2-OH) in methanol [28] to obtain amidoxime-functionalized COF (COF-TpDb-AO) (Fig. 3a) [29]. Interestingly, COF-TpDb-AO preserved the structural regularity of the former with remarkable chemical stability and sufficient porosity intake (from 1164 to 826 m2/g). It was further employed as an adsorbent for uranium uptake. Remarkably, COF-TpDb-AO demonstrated superior uranium uptake in terms of saturation adsorption capacity (408 mg/g) compared to the amorphous POP-TpDb-AO sample (355 mg/g). Meanwhile, in terms of contact time, it only needs 10 or 30 min to reach 81 or 95% equilibrium adsorption capacities while POP-TpDb-AO needs 90 min to accomplish 95%. Furthermore, similar postsynthetic functionalization was reported on a newly discovered dioxin-linked 2D COF (COF-316) with remarkable crystallinity intake. Thus, it is a potential material as the solid sorbent for nuclear-waste and other metal-pollutant sequestration [30].
Figure 3.

Postsynthetic functionalization of pendant groups of COFs via various covalent bonds. (a) Functionalization of nitrile-appended COF-TpDb into amidoxime-functionalized COF-TpDb-AO via amidoxime bond. (b) Functionalization vinyl-functionalized COF-V into thioether-functionalized COF-S-SH via thioether bond. (c) Functionalization of hydroxyl-appended T-COF-OH COF into T-COF-OFITC via thiocarbamate bond.
The thioether bond is one of the electron-rich moieties that has been successfully incorporated into COFs under a thiol-ene ‘click’ reaction as depicted in Fig. 1e [31]. A vinyl-functionalized mesoporous 2D COF (COF-V) was prepared and treated with 1,2-ethanedithiol with the assistance of azobis (isobutyronitrile) (AIBN) to afford the thioether-functionalized COF (COF-S-SH) (Fig. 3b) [32]. Sufficient crystallinity and porosity were maintained by COF-S-SH with abundant sulfur species within the channel (20.9 wt%, as thiol and thioether groups). Furthermore, it was accessed as an attractive solid sorbent for heavy-metal (such as Hg) remediation. Practically, COF-S-SH was exposed to Hg2+ aqueous solution (25–700 ppm) and its adsorption isotherm was collected for further analysis. Notably, COF-S-SH managed an Hg2+ uptake capacity as high as 1350 mg/g, which surpassed the performance of all previously reported thiol- and/or thioether-functionalized materials [33–37]. In addition, it remained crystalline after four consecutive adsorption cycles and could effectively remove Hg2+ with high selectivity even in the presence of a high concentration of background metal ions (Ca2+, Zn2+, Mg2+ and Na+). Inspired by this work, the hydrophobic property was successfully integrated into a similar COF scaffold to produce superhydrophobic and water-repellent mesoporous 2D COF (COF-VF) [38]. The COF-VF was obtained by treating COF-V with 1H,1H,2H,2H-perfluorodecanethiol under a common thiol-ene ‘click’ reaction. A test of the water-contact angle revealed that COF-VF was a superhydrophobic material with a static water-contact angle of 167°, while COF-V and alkyl-modified COF-V exhibited contact angles of only 113° and 122°, respectively. In addition, it was used as a hydrophobic coating agent for melamine foam, paper and magnetic liquid in which the resultant coated materials demonstrated hydrophobic properties towards all aqueous liquids including inorganic acidic and basic solutions. Besides a click reaction, the inverse-vulcanization method can also be employed to functionalize COFs via a thioether bond. For instance, the synthesis of electroactive S-COF-V with remarkable electrochemical performance was executed under post-
synthetic functionalization by involving a thioether bond [39]. In addition, a thioether-functionalized 3D COF (COF-102-SPr) has also been constructed under a thiol-ene click reaction with preserved crystallinity relative to its parent COF [40].
The hydroxyl pendant group is one of the versatile functional groups for the functionalization of COFs due to its reactivity to several constituents. For example, a coupling reaction between hydroxyl with isothiocyanate groups affords an extended covalent thiocarbamate bond (Fig. 1f). A fluorescent active COF (T-COF-OFITC) was prepared under a coupling reaction between hydroxyl-appended large-pore T-COF-OH with a fluorescent active fluorescein-isothiocyanate (FITC) molecule via a thiocarbamate bond (Fig. 3c) [41]. Notably, structural preservation was clearly observed under powder X-Ray diffraction (PXRD) analysis and a significant surface-area reduction was found under porosity analysis. In addition, COF-5 was also functionalized under a similar route to obtain COF-5-FITC. As predicted, both functionalized COFs showed strong fluorescent signals, indicating the successful transformation of photo-inactive COFs into photoactive COFs. On another attempt, hydroxyl groups within the backbone of the COF were further condensed with electrophile leaving groups such as halide under Williamson ether reactions to obtain ether-functionalized COFs (Fig. 2g) [42]. Hydroxyl-containing mesoporous 2D imine-linked COF series ([HO]x%-Py-COFs) were prepared and employed as scaffolds for immobilizing catalytically active ionic liquid (IL) molecules to obtain IL-COF systems [43]. The immobilization of (2-bromoethyl)triethylammonium bromide (Et4NBr) on the pore wall of [HO]x%-Py-COFs afforded [Et4NBr]x%-Py-COFs (x = 25–100) (Fig. 4a). Notably, the structural order was seen on [Et4NBr]x%-Py-COFs relative to their pristine scaffold when x = 25 or 50 and crystallinity disturbance occurred once x reached 75 and 100 along with severe porosity reduction. Under CO2-adsorption analysis, [Et4NBr]x%-Py-COFs (x = 25 or 50) exhibited elevated CO2-uptake capacities and higher isosteric heat absorption (Qst) than those [HO]x%-Py-COFs samples and was able to catalyse the transformation of CO2 into value-added chemicals (N-formylation reaction). For instance, [Et4NBr]50%-Py-COF accounted for high CO2-uptake capacity (164.6 mg/g, at 1 bar and 273 K), which was the highest value compared to several reported COFs. Meanwhile, with 5 mol% [Et4NBr]50%-Py-COF, a solely formylated product of N-methylaniline was obtained with an isolated yield of 94% under mild conditions, while [HO]50%-Py-COF could only exhibit 32% conversion under similar conditions. The generality of this postsynthetic functionalization was also demonstrated on other 2D COFs. For example, the functionalization of [OH]x%-TD-COFs into [BE]x%-TD-COFs transformed the neutral skeleton of the COF scaffold into zwitterionic-functionalized COF and the design of ionic-based [SO3−]-DhaTab COF was obtained by reacting DhaTab COF with 1,3-propanesulton in toluene [44,45].
Figure 4.

Postsynthetic functionalization of pendant groups of COFs via covalent bond, cycloaddition reaction and reduction reaction. (a) Functionalization of hydroxyl-containing [HO]x%-Py-COFs into ether-functionalized [Et4NBr]x%-Py-COFs via ether bond. (b) Structural transformation of Ph-An-COF into Ph-AnCD-COF via 4π + 4π cycloaddition reaction. (c) Functionalization of nitrile-appended JUC-505 COF into amine-functionalized JUC-505-NH2 via reduction reaction.
Functionalization of the structural skeleton of COFs via a covalent bond can also be facilitated by a cycloaddition reaction. Meanwhile, anthracene is a typical unit that can be transformed via a (4π + 4π) or (4π + 2π) cycloaddition reaction [46,47]. Thus, COFs with anthracene units could demonstrate this type of functionalization. Structural transformation of planar 2D sheet COF (Ph-An-COF) into a concavo-convex polygon skeleton COF (Ph-AnCD-COF) was performed by irradiating the COF scaffold with Ar (360 nm using a Xenon lamp completed with a band-path filter) (Fig. 4b) [48]. Under this treatment, the Ph-An-COF faced a thermal reversible photo-induced (4π + 4π) cycloaddition reaction that stimulated a drastic conformational change and caused a shortened distance between the central portions of the dimeric molecules. Electronic adsorptions of both materials were essentially different due to the structural transformation while preserving their crystallinities and insignificant porosity change. This phenomenon may promise interesting applications in the field of optics and sensing. Similar structural transformation could also be initiated by using a chemical initiator rather than light irradiation. A predesigned COF scaffold (DaTp) was subjected to a [4π + 2π] Diels-Alder cycloaddition reaction with N-hexylmaleimide molecules, leading to the disturbance of π–π stacking interactions and planarity of the individual layers of DaTp. As a result, layer exfoliation occurred, resulting in thin-layer covalent organic nanosheet formation (DaTp-CON) [49]. Meanwhile, the pendant groups of COFs could also be functionalized or transformed via the reduction or oxidation reaction. For example, highly stable polyarylether COFs (PAE-COFs, JUC-505) bearing nitrile (-CN) pendant groups was successfully reduced to give amine-functionalized COF (JUC-505-NH2) (Fig. 4c) [50]. The reduction reaction was performed by refluxing JUC-505 with LiAlH4 in tetrahydrofuran (THF). Significantly, JUC-505-NH2 could effectively evacuate the tetracycline antibiotics compared to the JUC-505 event at pH = 13. In other sophisticated work, nitro-containing 2D COF (TpBD(NO2)2) was reduced (with SnCl2 under reflux) to produce amine-containing COF (TpBD(NH2)2 and azide-containing COF (X%[N3]-COFs) was also reduced (treated with PPh3 in methanol) to give the corresponding amine-containing COF (X%[NH2]-COFs) [25,51].
Ionic/coordination bond
Unlike the covalent bond that was formed through sharing electrons, coordination-bond formation involves the donor–acceptor electron transfer between the active sites of the components [52].
Meanwhile, ionic interaction occurs on the dipolar region between the two active sites, establishing engagement forces between the components [53,54]. These two concepts were then adopted in functionalizing COFs with metal ions or molecular ions. Based on the components involved, this structural functionalization is categorized into two following sections, namely metal ion/COF hybrid and molecular ion/COF hybrid.
Metal ion/COF hybrid.
COFs with electron-rich s- or p-type orbitals may facilitate the formation of coordination bonds with metals (with electron-less d orbitals). For example, imine linkages found in imine-linked COFs are able to govern coordination bonds with metals or metal ions [55]. More importantly, the electron-rich nitrogen species of imine linkages within 2D COFs are relatively close to each other (approximately 4 Å, the distance from sheet to sheet), thus they promote the efficient establishment of coordination bonds.
The Pd-complex, Pd(OAc)2, was successfully hybridized in between the adjacent layers of a microporous 2D imine-linked COF (COF-LZU1)
under simple overnight stirring in dichloromethane to afford Pd/COF-LZU1 (Fig. 5a) [56]. It was assumed that Pd species were coordinatively bonded on nitrogen atoms between the adjacent sheets. The transmission electron microscopy (TEM) images of Pd/COF-LZU1 exhibited black dots of Pd species, confirming the homogeneous dispersion of Pd species within the skeleton of COF-LZU1 while preserving the crystalline nature of the host. Remarkably, the Pd/COF-LZU1 showed an excellent catalytic activity towards the Suzuki–Miyaura coupling reaction with a broad scope of reactants. This catalytic performance outperformed Pd-containing MOF analogs under similar catalytic conditions [57]. Using a similar approach, Pd(OAc)2 was also hybridized in the channel of a porphyrin-containing 2D COF (H2P-Bph-COF) to generate Pd/H2P-Bph-COF with enhanced catalytic performance towards a typical Suzuki-coupling reaction [58]. Meanwhile, by using Co(OAc)2, the Co-TpBpy was produced by soaking and stirring the bipyridine-containing TpBpy with Co(OAc)2 precursor in methanol [59]. However, the hybridization of Co species in this work is coordinatively bonded in-plane with bipyridine moieties rather than in between the adjacent imine linkages of the COF. Notably, the resultant Co-TpBpy possessed enhanced electrochemical activity (oxygen evolution reaction, OER) compared to the parent COF. In a similar effort, bipyridine-containing COF was used to coordinatively bond a noble metal salt (Re(CO)5Cl) to generate Re-COF [60]. The resultant hybrid was employed as a photocatalyst for CO2 reduction into CO under visible-light illumination with excellent selectivity (98%) and much better activity compared to its homogeneous Re counterpart.
Figure 5.

Postsynthetic functionalization of COFs via coordination bond. (a) Hybridization of Pd(OAc)2 into the channel of COF-LZU1 to obtain Pd/COF-LZU1. (b) Hybridization of Ni(COD)2 into the channel of DBA-3D-COF1 to obtain Ni-DBA-3D-COF. (c) Docking bimetal salts of Mn/Pd into the channel of Py-2,2′-BPyPh to obtain Mn/Pd@Py-2,2′-BPyPh.
Hybridization of metal complex within the 3D COFs comes with a different pathway. 3D COFs possess a 3D pore architecture with a typical longer layer-to-layer distance (approximately 6.6 Å), which is not sufficient to perform coordination spatial cooperation between the adjacent layers. A 3D COF with a high surface area and low density (DBA-3D-COF 1) was prepared and further stirred in toluene containing 10 wt% of Ni(COD)2 at room temperature to afford Ni-DBA-3D-COF (Fig. 5b) [61]. The Ni species (10.1 wt%) were coordinatively bonded exclusively on the cavity of π-conjugated DBA [62] rather than on boronate ester. Interestingly, the structural order was clearly preserved and minimal surface-area reduction was observed. In the end, the Ni-DBA-3D-COF was employed as a solid sorbent for ethane/ethylene separation. On another occasion, Pd(OAc)2 salt was also successfully impregnated into the pore of 3D COF (COF-300) in which the Pd ions were assumed to be coordinatively bonded on solely nitrogen of a particular imine linkage within the skeleton [63]. The X-ray photoelectron spectroscopy (XPS) measurement revealed that Pd species were coordinatively bonded with imine nitrogen atoms at a 1:1 ratio; thus, they largely occupied the pore surface of the COF. The resultant Pd(OAc)2@COF-300 was further employed as a catalyst for a Suzuki–Miyaura coupling reaction. Notably, Pd(OAc)2@COF-300 showed an excellent catalytic activity (barely 99% conversion) toward a phosphine-free Suzuki–Miyaura coupling reaction as well as Heck and Sonogashira reactions. Besides imine and bipyridine moieties, the N,N′-bis [salicylidene] ethylenediamine (Salen) has been considered as an attractive moiety for coordination chemistry due to its ability to hold metal ions [64]. In line with this, a 2D Salen-COF with a large pore size was constructed and impregnated with metal salts (M = Cu, Ni, Zn, Co or Mn) under stirring to afford M/Salen-COFs [65]. This treatment did not sacrifice the crystalline nature of the former COF, since the metal ions occupied most of the Salen pockets. Since these metallosalen-COFs were highly porous and chemically stable, Co/Salen-COF was employed as a heterogeneous catalyst for Henry reactions and exhibited a good performance. Very recently, Fang et al. developed a microporous 3D-Salphen-COF series (JUC-508 and JUC-509) and used them as a perfect scaffold for metalation [66]. The JUC-509 was further metalated via wet chemistry by immersing it in a methanol solution of corresponding metal salts, affording the JUC-509-Y series (Y = Mn, Cu or Eu). They were further employed as heterogeneous catalysts for the catalytic removal of superoxide radical anion (O2.-).
The incorporation of a multi-metal component into the pore of COFs has also been demonstrated. The successful docking of bimetallic salts of Mn/Pd into the pore of a dual-coordinative site containing Py-2,2′-BPyPh was performed via sequential impregnation treatment, where MnCl2 was previously impregnated to give Mn@Py-2,2′-BPyPh and followed by treatment with Pd(OAc)2 to afford bimetal Mn/Pd@Py-2,2′-BPyPh (Fig. 5c) [67]. Based on ICP-OES analysis, the content of the Mn species was 0.8 wt% while the content of the Pd species was 9.3 wt%. The Mn species could only coordinate on the bipyridine moieties and the Pd species docked at the imine sites. Interestingly, the resultant hybrid was able to catalyse a Heck-epoxidation tandem reaction. Notably, Mn/Pd@Py-2,2′-BPyPh performed 94% conversion to achieve the final product with the stepwise reaction giving 95 and 98% conversion for the heck and epoxidation reactions, respectively. By using the similar COF as a scaffold, two metallic salts of Rh and Pd have successfully been docked on the pore surface of Bpy-COF to obtain another bimetallic docking COF hybrid (Rh/Pd@X%Bpy-COF) with the ability to catalyse a one-pot addition–oxidation reaction [68]. Furthermore, another strategy was carried out by preparing dual imine and carboxyl coordinative sites containing COF([HOOC]X-COFs, x = 17, 33, 50 or 100) to hybridize a multi-metal component [69]. Various metal ions (Ca2+, Mn2+ and Sr2+) were then integrated into the [HOOC]17-COF by simply mixing the COF with the chloride salt solution of the respective metals and afforded [MOOC]17-COFs (M = Ca2+, Mn2+ or Sr2+). As predicted, the resultant hybrids exhibited enhanced ammonia uptakes relative to the bare COF.
Similar to the metal ion/COF hybrid, the deposition of metal nanoparticle species within the pore of COFs has also been reported. Typically, the hybridized metal ions within the COF channel are further reduced into either a single nanoparticle or bulky metals to produce M@COF and/or MxOy@COF hybrids. The Au nanoparticles (Au(0)) were deposited within the channel of imine-linked TpPa-1 by reducing (using NaBH4 solution) the deposited Au(III) ions in Au(III)@TpPa-1 to give Au(0)@TpPa-1 (Fig. 6a) [70]. The resultant Au(0)@TpPa-1 was used as a high-performance catalyst for reducing 4-nitrophenol to 4-aminophenol. As predicted, Au(0)@TpPa-1 performed an excellent catalytic activity compared to the pristine COF. For instance, 100% reactant conversion was achieved in only 13 min for Au(0)@TpPa-1 (1.20 wt%), while it required 20 min for the Au salt (HAuCl4·3H2O) and no catalytic activity was observed for the COF-TpPa-1 sample. Meanwhile, the Pd(0)@TpPa-1 has also been obtained under similar procedures and further employed as a catalyst for both C–C Sonogashira/Heck coupling and C–H activation reactions [71–74]. Furthermore, since Pd metal is a highly active catalyst, several Pd(0)@COF hybrids have been produced and utilized as catalysts so far [75,76].
Figure 6.

Postsynthetic functionalization of COFs via coordination bond. (a) Deposition of Au nanoparticle into the channel of imine-linked TpPa-1 to obtain Au(0)@TpPa-1. (b) Deposition of Pt metal cluster into the channel of Thio-COF to obtain PtNPs@COF.
On the other hand, besides imine-linked COFs, other types COFs that have coordinative sites have potential as scaffolds. For example, a hydrazone-linked COF (COF-ASB) was designed and used as a scaffold to accommodate Ru nanoparticles to give Ru@COF-ASB [77]. The deposition of Ru nanoparticles was done by mixing COF-ASB with a CH3CN solution of ruthenium chloride (RuCl3) and further reduced by NaBH4. Notably, the hybrid showed enhanced catalytic performance for a one-pot solvent-free tandem reaction of imine synthesis from alcohols [78] and also in an oxidation reaction relative to the bare COF. Deposition of a metal cluster into the channel of a COF has also been done. An electron-rich thioether-containing COF (Thio-COF) was designed that was able to strongly hold the metal species and control metal nanoparticle nucleation [12,79]. The metalation was performed by mixing Thio-COF suspension with K2PtCl4 solution, which was then reduced with NaBH4 in methanol to afford PtNPs@COF (Fig 6b). Interestingly, under TEM exploration, it was found that the Pt species were obtained as crystalline ultra-small and uniform nanoparticles with an average size of 1.7 nm. The PdNPs@COF analog was also synthesized via a similar route by using K2PdCl4 as the metal precursor. Both hybrids were then employed as catalysts for reducing 4-nitrophenol to 4-aminophenol and Suzuki–Miyaura coupling reactions. Both catalysts presented excellent catalytic performances that outperformed the unsupported Pt nanoparticles (NPs) catalyst. On the other hand, the deposition of metals with two different oxidation states has also been reported, revealing robust postsynthetic metal hybridization in COFs via a coordination bond [80].
Molecular ion/COF hybrid.
The coordinative and ionic sites within the skeleton of COFs can also engage the deposition of molecular ions via ionic interaction. To this end, typical ionic liquids, inorganic/organic salts and other molecular ions have been incorporated within the channel of COFs—for example, the deposition of phosphoric acid (PA, H2PO4−) into the channel of a highly stable 2D COF (TP-Azo) to afford PA@Tp-Azo (Fig. 7a) [81]. The azo moieties within the COF could stabilize H2PO4− once protonated [82]. Notably, PA@Tp-Azo reported remarkable proton conductivity as high as 9.9 × 10−4 S/cm at 332 K and 98% relative humidity (RH)—much higher than PA@Tp-Stb. On another occasion, a molecular ion of PA was again loaded into the channel of two aza-fused COFs (aza-COF-1 and aza-COF-2) to produce aza-COF-1H and aza-COF-2H, respectively [83]. As predicted, both hybrids showed dramatic improvement in proton conductivities relative to their parent COFs. Meanwhile, in another report, the molecular ion of polyoxometalate (POM) was loaded into the channel of a cationic COF (EB-COF:Br, Br as counter ion) [84]. Ion-exchange treatment was performed with PW12O403− (abbreviated as PW12) to give EB-COF:PW12 (Fig. 7b). Remarkably, the ion-exchange treatment did not severely alter the rigidity and crystallinity of the framework, signaling the robustness of the COF scaffold. The porosity of the obtained COF hybrid was almost diminished due to the occupation of the large molecule of POM. For instance, the surface area of EB-COF:Br was 774 m2 g–1, while EB-COF:PW12 showed a surface area of 8 m2 g–1. With POM molecules in the channel of EB-COF, the resultant EB-COF:PW12 demonstrated a proton conductivity 100 times higher than that of the EB-COF:Br sample. Meanwhile, by using an anionic [Mo3S13]2− cluster, Mo3S13@EB-COF has also been fabricated and employed as a photocatalyst material [85,86]. In addition, application in lithium-ion conduction has also met with the fabricated molecular ion/COF hybrid of PEG-Li+@EB-COF-ClO4 [87]. Nevertheless, 3D COF has also been employed to accommodate molecular ions (denoted as PMA@COF-300) with enhanced performance as a catalyst [88].
Figure 7.

Postsynthetic functionalization of COFs via ionic bond. (a) Hybridization of phosphoric acid (PA) into the channel Tp-Azo COF to obtain PA@Tp-Azo. (b) Hybridization of polyoxometalate (PW12) into the channel of cationic EB-COF:Br to obtain EB-COF: PW12.
Linkage functionalization
The linkages in COFs are typically stable but somehow reactive to certain components and under particular conditions. Functionalization or transformation of the linkage of COFs has been performed so far through certain reactions, including oxidation–reduction and ring-fusion reactions. The imine linkage, which is relatively more stable than the boroxine linkage, has been widely transformed. For example, typical 2D imine-linked COF (TPB-TP-COF,1) was treated under oxidation reaction using sodium chlorite (as an oxidant), acetic acid (as a buffer) and 2-methyl-2-butene (Fig. 8a) [89,90]. This oxidation treatment transformed imine linkage into amide linkage, thus a new amide-linked COF (1′) was obtained. Under similar conditions, 4PE-1P-COF (2) was also oxidized into respective amide-linked COF (2′). Notably, the both resultant amide-linked COFs preserved the structural order and remarkable chemical stabilities relative to their pristine COFs, including in 12 M HCl and 1 M NaOH for 24 h. In another occasion, 2D (COF-366-M) and 3D (COF-300) imine-linked COFs were transformed by reducing their linkage into amine-linked COF (366-M-AR and COF-300-AR) [91]. The reduction reaction was performed by quantitatively treating the corresponding COFs with NaBH4. Again, the chemical stability of the resultant amine-linked COFs was somehow enhanced to a high extent relative to their pristine COFs. In addition, both 366-M-AR and COF-300-AR showed improved electrochemical activities compared to the unreduced counterparts.
Figure 8.

Postsynthetic functionalization of linkage of COFs. (a) Linkage transformation of imine-linked TPB-TP-COF(1) into amide-linked COF (1′) under oxidation reaction. (b) Linkage transformation of imine-linked COF-1 into quinone-linked COFs (MF-1a-e) under aza-Diels-Alder cycloaddition reaction. (c) Linkage transformation of imine-linked TTI-COF into thiazole-linked COF (TTT-COF).
Meanwhile, enhancing the structural integrity of an imine-linked COF (termed as COF-1) from moderately stable into ultra-stable quinone-linked COF (denoted as MF-1a-e) has been demonstrated by kinetically fixing its linkage via an aza-Diels-Alder cycloaddition reaction (Fig. 8b) [92]. The reaction involved the efficient Povarov (aza-DA) reaction between aryl imines and arylalkynes by subjecting the COF scaffold to phenylacetylene at 110°C in the presence of BF3.Et2O, chloranil and toluene for 72 h [93]. Drastic chemical stability was observed for all resultant MF-1a-e COFs. For instance, they were stable in strong acid (12 M HCl at 50°C, 8 h), superacid (98% TfOH, 3 days), strong base (14 M NaOH in H2O/MeOH at 60°C, 24 h), strong oxidant (KMnO4 in H2O/CH3CN, 24 h) and reducing agent (NaBH4 in MeOH at 65°C, 24 h). In another report, an imine-linked COF (TTI-COF) was topologically transformed by arresting its crystalline state via a reversible to irreversible ring-fused thiazole-linked COF (TTT-COF) (Fig. 8c) [94,95]. The transformation utilized elemental sulfur reacted with aromatic imine to first oxidize the imine to a thioamide and subsequently oxidatively cyclized the thioamide group to form a thiazole ring [96]. Another reported approach of transforming the linkage of COFs was a linkage-exchange method. An imine-linked COF (ILCOF-1) was treated using either with 4 equiv. 2,5-diaminobenzene-1,4-dithiol dihydrochloride in N,N-dimethylformamide (DMF)/water to
give thiazole-linked COF (COF-921) or with 2,5-diaminohydroquinone dihydrochloride in DMF under oxygen atmosphere to obtain oxazole-linked COF (LZU-192), respectively [97]. These linkage transformations were performed via consecutive steps, initialized by linker exchange, followed by cyclization and end with an oxidation reaction in a one-pot system. As predicted, both COF-921 and LZU-912 were chemically stable relative to ILCOF-1 including in basic (10 M NaOH) and acidic solutions (12.1 M HCl, 18 M H2SO4, 14.8 M H3PO4 and 9 M H2SO4 in dimethyl sulfoxide (DMSO)) for 1 day. Although the linkage transformation discussed in this section mainly utilizes imine linkage, other linkages may also possibly be functionalized via a certain reaction or treatment.
Host–guest functionalization
As highly porous materials, COFs are perfect porous confinement materials acting as hosts to accommodate guest molecules with controlled quantities via the host–guest chemistry approach. This confinement property involves weak forces that have been well studied in general supramolecular chemistry including van der Walls, π–π interaction and hydrophobic/hydrophilic interactions [98].
The photo-responsive bulk-C60 molecules were incorporated into the channel of the highly stable, electronically conjugated 2D phenazine-linked COF (CS-COF) via host–guest chemistry to obtain donor–acceptor-based CS-COF⊃C60 [99]. The loading of bulk-C60 molecules was executed under a thermal sublime diffusion method (Fig. 9a). As high as 25 wt% of bulk-C60 molecules have been successfully loaded into the channel of the host, suggesting a peapod-like encapsulation. The resultant hybrid was then employed as on–off ratio photoswitches and photovoltaic cell. The device made by CS-COF⊃C60 (with optimized polymethyl methacrylate (PMMA) content) showed the best on–off ratio photoswitches (5.9 × 107), photocurrent performance (as high as 2.0 μA) and dark current of 0.029 pA at a bias voltage of 1.5 V. In addition, it allowed multiple rounds of on–off switching without a loss of performance at room temperature that was superior to all other COFs, including hole-conducting-type COFs (8 × 104 for PPy-COF [100] and 150 for NiPc-COF [101]) and electron-conducting type COFs (60 for NiPc-BTBA COF [102], 5 × 104 for ambipolar-conducting ZnP-COF [103] and 1.3 × 104 for 2D D-A COF [104]). In a similar vision, the electron acceptor [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) molecules were loaded into the channel of a boronate ester-linked TT-COF to generate an electron donor–acceptor heterojunction–COF hybrid (TT-COF:PCBM) [105]. The interaction of both components was assumed to involve a van der Waals interaction. Since thieno[2,3-b]thiophene-containing building blocks exhibited high charge-carrier mobilities and efficient photo-induced charge transfer from polymerized thienothiophene derivatives to fullerene acceptor molecules [106,107], the resultant TT-COF:PCBM was an attractive candidate for organic photovoltaic material. Meanwhile, the conductivity of the redox-active DAAQ-TFP COF film was enhanced by electropolymerizing redox-active 3,4-ethylenedioxythiophene (EDOT) monomer into its pores to give PEDOT⊃DAAQ-TFP-COF [108]. As predicted, the hybrid film exhibited dramatically improved current responses in CV experiments when compared with DAAQ-TFP-COF film. Notably, it could accommodate a high charging rate without compromising performance and exhibited both a 10-fold higher current response and stable capacitance over 10 000 cycles relative to the pristine host.
Figure 9.

Postsynthetic host–guest functionalization of COFs. (a) Hybridization of bulk-C60 molecules into the pore channel of phenazine-linked CS-COF to produce CS-COF⊃C60. (b) The loading of proton carriers of triazole (trz) and imidazole (imi) into the pore channel of TPB-DMTP-COF to obtain proton conductors based trz@TPB-DMTP-COF and im@TPB-DMTP-COF.
Another piece of sophisticated work was loading N-heterocyclic proton carriers (triazole (trz) and imidazole (im) molecules) in the channel of hexagonally aligned, mesoporous and highly stable TPB-DMTP-COF to obtain trz@TPB-DMTP-COF and im@TPB-DMTP-COF as proton conductors (Fig. 9b) [109]. The loading of these proton carriers was done via the thermal-vaporization method at 150°C or 120°C overnight, and 180 wt% trz and 155 wt% im could be loaded into the host, respectively. The resultant hybrids were explored for proton conduction experiments in which trz@TPB-DMTP-COF documented a proton conductivity as high as 1.1 × 10−3 S cm–1 at 130°C, while im@TPB-DMTP-COF exhibited a proton conductivity of 4.37 × 10−3 S cm–1, which were pretty much higher compared to their pristine COF. Furthermore, a catalytically active biomolecular has also been engaged into the pore of a predesigned COF as host, exemplifying the generality of host–guest functionalization in COFs [110]. Meanwhile, 3D COF has also been employed as a host to demonstrate the host–guest confinement phenomenon. For example, the organometallic molecules of [Fe(η5-C5H5)2], [Co(η5-C5H5)2] and [Ru(cod)(cot)] were loaded into the pore of COF-102 to obtain (FeCp2)4@COF-102, (CoCp2)4@COF-102 and [Ru(cod)(cot)]@COF-102 [111]. Interestingly, the resultant hybrids showed various confinement phenomena; thus, it provided insights for further research.
Conclusions
COFs materials with tunable structural topologies and high thermal/chemical stabilities provide a versatile postsynthetic functionalization platform to enhance their psychochemical properties and performances toward specific applications. The manageable porosities and functionalities via the bottom-up design of COFs allow judicious incorporation of constituents that can be bonded by strong covalent bonds, ionic/coordination forces and host–guest chemistry interactions. Nevertheless, their linkages can also be postsynthetically functionalized to improve their structural stability. Furthermore, the resultant functionalized COFs with particular hybrid or composite fashions show outstanding performances in the field of catalyst, gaseous uptake–release, optics, energy storage and so on. In the future development of COFs, we assume that there are still many gaps left for further explorations, especially in functionalization of the available linkages that are less studied. In addition, incorporations of functional constituents via host–guest chemistry so far have been mainly developed for electronic and energy-storage applications, thus investigations in wider sophisticated applications are still highly demanded. Last but not least, the postsynthetic functionalization of 3D COFs is still less studied; thus, continuous efforts in the development of 3D COFs may need special concern.
Funding
This work was supported by the National Natural Science Foundation of China (21571079, 21621001, 21390394, 21571076 and 21571078), Overseas Expertise Introduction Project for Discipline Innovation (B07016 and B17020) and the Program for Jilin University Science and Technology Innovative Research Team.
Conflict of interest statement. None declared.
References
- 1. Speybroeck VV, Hemelsoet K, Joos Let al.. Advances in theory and their application within the field of zeolite chemistry. Chem Soc Rev 2015; 44: 7044–111. [DOI] [PubMed] [Google Scholar]
- 2. Tao Y, Kanoh H, Abrams Let al.. Mesopore-modified zeolites: preparation, characterization, and applications. Chem Rev 2006; 106: 896–910. [DOI] [PubMed] [Google Scholar]
- 3. Wang Z, Yu J, Xu R. Needs and trends in rational synthesis of zeolitic materials. Chem Soc Rev 2012; 41: 1729–41. [DOI] [PubMed] [Google Scholar]
- 4. Čejka J, Corma A, Zones S. Zeolites and Catalysis: Synthesis, Reactions and Applications. Mörlenbach: WILEY-VCH Verlag GmbH & Co. KGaA, 2010. [Google Scholar]
- 5. Zhou HC, Long JR, Yaghi OM. Introduction to metal-organic frameworks. Chem Rev 2012; 112: 673–4. [DOI] [PubMed] [Google Scholar]
- 6. Natarajan S, Mahata P. Metal-organic framework structures—how closely are they related to classical inorganic structures. Chem Soc Rev 2009; 38: 2304–18. [DOI] [PubMed] [Google Scholar]
- 7. Guillerm V, Kim D, Eubank JFet al.. A supermolecular building approach for the design and construction of metal-organic frameworks. Chem Soc Rev 2014; 43: 6141–72. [DOI] [PubMed] [Google Scholar]
- 8. Côté AP, Benin AI, Ockwig NW. Porous, crystalline, covalent organic frameworks. Science 2005; 310: 1166–71. [DOI] [PubMed] [Google Scholar]
- 9. Ding S-Y, Wang W. Covalent organic frameworks (COFs): from design to applications. Chem Soc Rev 2013; 42: 548–68. [DOI] [PubMed] [Google Scholar]
- 10. Diercks CS, Yaghi OM. The atom, the molecule, and the covalent organic framework. Science 2017; 355: eaal585. [DOI] [PubMed] [Google Scholar]
- 11. Xu H-S, Ding S-Y, An W-Ket al.. Constructing crystalline covalent organic frameworks from chiral building blocks. J Am Chem Soc 2016; 138: 11489–92. [DOI] [PubMed] [Google Scholar]
- 12. Lu S, Hu Y, Wan Set al.. Synthesis of ultrafine and highly dispersed metal nanoparticles confined in a thioether-containing covalent organic framework and their catalytic applications. J Am Chem Soc 2017; 139: 17082–8. [DOI] [PubMed] [Google Scholar]
- 13. Chandra S, Kundu T, Dey Ket al.. Interplaying intrinsic and extrinsic proton conductivities in covalent organic frameworks. Chem Mater 2016; 28: 1489–94. [Google Scholar]
- 14. Nagai A, Guo Z, Feng Xet al.. Pore surface engineering in covalent organic frameworks. Nat Commun 2011; 2: 536. [DOI] [PubMed] [Google Scholar]
- 15. Xu H, Gao JJiang D. Stable, crystalline, porous, covalent organic frameworks as a platform for chiral organocatalysts. Nat Chem 2015; 7: 905–12. [DOI] [PubMed] [Google Scholar]
- 16. Notz W, Tanaka F, Barbas CF. Enamine-based organocatalysis with proline and diamines: the development of direct catalytic asymmetric aldol, Mannich, Michael, and Diels-Alder reactions. Acc Chem Res 2004; 37: 580–91. [DOI] [PubMed] [Google Scholar]
- 17. Xu H, Chen X, Gao Jet al.. Catalytic covalent organic frameworks via pore surface engineering. Chem Commun 2014; 50: 1292–4. [DOI] [PubMed] [Google Scholar]
- 18. Royuela S, Garcia-Garrido E, Arroyo MMet al.. Uracil grafted imine-based covalent organic framework for nucleobase recognition. Chem Commun 2018; 54: 8729–32. [DOI] [PubMed] [Google Scholar]
- 19. Chen L, Furukawa K, Gao Jet al.. Photoelectric covalent organic frameworks: converting open lattices into ordered donor-acceptor heterojunctions. J Am Chem Soc 2014; 136: 9806–9. [DOI] [PubMed] [Google Scholar]
- 20. Xu F, Xu H, Chen Xet al.. Radical covalent organic frameworks: a general strategy to immobilize open-accessible polyradicals for high-performance capacitive energy storage. Angew Chem Int Ed 2015; 54: 6814–8. [DOI] [PubMed] [Google Scholar]
- 21. Huang N, Krishna R, Jiang D. Tailor-made pore surface engineering in covalent organic frameworks: systematic functionalization for performance screening. J Am Chem Soc 2015; 137: 7079–82. [DOI] [PubMed] [Google Scholar]
- 22. Bartoli G, Bosco M, Carlone Aet al.. Reaction of dicarbonates with carboxylic acids catalyzed by weak Lewis acids: general method for the synthesis of anhydrides and esters. Synthesis 2007; 22: 3489–96. [Google Scholar]
- 23. Huang N, Chen X, Krishna Ret al.. Two-dimensional covalent organic frameworks for carbon dioxide capture through channel-wall functionalization. Angew Chem Int Ed 2015; 54: 2986–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu Q, Ma Y, Li Het al.. Postsynthetic functionalization of three-dimensional covalent organic framework for selective extraction of lanthanide ions. Angew Chem Int Ed 2018; 57: 6042–8. [DOI] [PubMed] [Google Scholar]
- 25. Lohse MS, Stassin T, Naudin Get al.. Sequential pore wall modification in a covalent organic framework for application in lactic acid adsorption. Chem Mater 2016; 28: 626–31. [Google Scholar]
- 26. Hintz H, Wuttke S. Solvent-free and time efficient postsynthetic modification of amino-tagged metal−organic frameworks with carboxylic acid derivatives. Chem Matter 2014; 26: 6722–8. [Google Scholar]
- 27. Mitra S, Sasmal HS, Kundu Tet al.. Targeted drug delivery in covalent organic nanosheets (CONs) via sequential postsynthetic modification. J Am Chem Soc 2017; 139: 4513–20. [DOI] [PubMed] [Google Scholar]
- 28. Vörös A, Baán Z, Mizsey Pet al.. Formation of aromatic amidoximes with hydroxylamine using microreactor technology. Org Process Res Dev 2012; 16: 1717–26. [Google Scholar]
- 29. Sun Q, Aguila B, Earl LDet al.. Covalent organic frameworks as a decorating platform for utilization and affinity enhancement of chelating sites for radionuclide sequestration. Adv Mater 2018; 30: 1705479. [DOI] [PubMed] [Google Scholar]
- 30. Zhang B, Wei M, Mao Het al.. Crystalline dioxin-linked covalent organic frameworks from irreversible reactions. J Am Chem Soc 2018; 140: 12715–9. [DOI] [PubMed] [Google Scholar]
- 31. Ranu BC, Mandal T. Water-promoted highly selective anti-markovniv addition of thiols to unactivated alkenes. SynLett 2007; 6: 925–8. [Google Scholar]
- 32. Sun Q, Aguila B, Perman Jet al.. Postsynthetically modified covalent organic frameworks for efficient and effective mercury removal. J Am Chem Soc 2017; 139: 2786–93. [DOI] [PubMed] [Google Scholar]
- 33. Li B, Zhang Y, Ma Det al.. Removal of mercury (II) from aqueous solution. Nat Commun 2014; 5: 5537. [DOI] [PubMed] [Google Scholar]
- 34. Feng X, Fryxell GE, Wang L-Qet al.. Functionalized monolayers on ordered mesoporous supports. Science 1997; 276: 923–6. [Google Scholar]
- 35. Shin Y, Fryxell GE, Um Wet al.. Sulfur-functionalized mesoporous carbon. Adv Funct Mater 2007; 17: 2897–901. [Google Scholar]
- 36. Yee K-K, Reimer N, Liu Jet al.. Effective mercury sorption by thiol-laced metal-organic frameworks effective mercury sorption by thiol-laced metal-organic frameworks. J Am Chem Soc 2013; 135: 7795–8. [DOI] [PubMed] [Google Scholar]
- 37. Mon M, Lloret F, Ferrando-Soria Jet al.. Mercury selective and efficient removal of mercury from aqueous media with the highly flexible arms of a BioMOF. Angew Chem Int Ed 2016; 55: 11167–72. [DOI] [PubMed] [Google Scholar]
- 38. Sun Q, Aguila B, Perman JAet al.. Integrating superwettability within covalent organic frameworks for functional coating. Chem 2018; 4: 1726–39. [Google Scholar]
- 39. Jiang Q, Li Y, Zhao Xet al.. Inverse-vulcanization of vinylic functionalized covalent organic frameworks as efficient cathode materials for Li-S batteries. J Mater Chem A 2018; 6: 17977–81. [Google Scholar]
- 40. Bunck DN, Dichtel WR. Postsynthetic functionalization of 3D covalent organic frameworks. Chem Commun 2013; 49: 2457–9. [DOI] [PubMed] [Google Scholar]
- 41. Rager S, Dogru M, Werner Vet al.. Pore wall fluorescence labeling of covalent organic frameworks. CrstEngComm 2017; 19: 4886–91. [Google Scholar]
- 42. Williamson A. Theory of etherification. Philos Mag 1850; 37: 350–6. [Google Scholar]
- 43. Dong B, Wang L, Zhao Set al.. Immobilization of ionic liquids to covalent organic frameworks for catalyzing the formylation of amines with CO2 and phenylsilane. Chem Commun 2016; 52: 7082–5. [DOI] [PubMed] [Google Scholar]
- 44. Mu Z-J, Ding X, Chen Z-Yet al.. Zwitterionic covalent organic frameworks as catalysts for hierarchical reduction of CO2 with amine and hydrosilane. ACS Appl Mater Interfaces 2018; 10: 41350–8. [DOI] [PubMed] [Google Scholar]
- 45. Hu H, Yan Q, Wang Met al.. Ionic covalent organic frameworks for highly effective catalysis. Chinese J Catal 2018; 39: 1437–44. [Google Scholar]
- 46. Bouas-Laurent H, Castellan A, Desvergne J-Pet al.. Photodimerization of anthracenes in fluid solution: structural aspects. Chem Soc Rev 2000; 29: 43–55. [Google Scholar]
- 47. Thapaliya ER, Captain B, Rayno FM. Photoactivatable anthracenes. J Org Chem 2014; 79: 3973–81. [DOI] [PubMed] [Google Scholar]
- 48. Huang N, Ding X, Kim Jet al.. A Photoresponsive smart covalent organic framework. Angew Chem Int Ed 2015; 54: 8704–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khayum MA, Kandambeth S, Mitra Set al.. Chemically delaminated free-standing ultrathin covalent organic nanosheets. Angew Chem Int Ed 2016; 55: 15604–8. [DOI] [PubMed] [Google Scholar]
- 50. Guan X, Li H, Ma Yet al.. Chemically stable polyarylether-based covalent organic frameworks. Nat Chem 2019; 11: 587–94. [DOI] [PubMed] [Google Scholar]
- 51. Ji W, Xiao L, Ling Yet al.. Removal of GenX and perfluorinated alkyl substances from water by amine-functionalized covalent organic frameworks. J Am Chem Soc 2018; 140: 12677–81. [DOI] [PubMed] [Google Scholar]
- 52. Liu S. The role of coordination chemistry in the development of target-specific radiopharmaceuticals. Chem Soc Rev 2004; 33: 445–61. [DOI] [PubMed] [Google Scholar]
- 53. Zhang S, Zhang J, Zhang Yet al.. Nanoconfined ionic liquids. Chem Rev 2017; 117: 6755–833. [DOI] [PubMed] [Google Scholar]
- 54. Bideau JL, Viau L, Vioux A. Ionogels, ionic liquid based hybrid materials. Chem Soc Rev 2011; 40: 907–25. [DOI] [PubMed] [Google Scholar]
- 55. Corma A, García H, Xamena FXLet al.. Engineering metal organic frameworks for heterogeneous catalysis. Chem Rev 2010; 110: 4606–55. [DOI] [PubMed] [Google Scholar]
- 56. Ding S-Y, Gao J, Wang Qet al.. Construction of covalent organic framework for catalysis: Pd/COF-LZU1 in Suzuki–Miyaura coupling reaction. J Am Chem Soc 2011; 133: 19816–22. [DOI] [PubMed] [Google Scholar]
- 57. Xamena FXL, Abad A, Corma Aet al.. MOFs as catalysts: activity, reusability and shape-selectivity of a Pd-containing MOF. J Catal 2007; 250: 294–8. [Google Scholar]
- 58. Hou Y, Zhang X, Sun Jet al.. Good Suzuki-coupling reaction performance of Pd immobilized at the metal-free porphyrin-based covalent organic framework. Microporous Mesoporous Mater 2015; 214: 108–14. [Google Scholar]
- 59. Aiyappa HB, Thote J, Shinde DBet al.. Cobalt-modified covalent organic framework as a robust water oxidation electrocatalyst. Chem Mater 2016; 28: 4375–9. [Google Scholar]
- 60. Yang S, Hu W, Zhang Xet al.. 2D covalent organic frameworks as intrinsic photocatalysts for visible light-driven CO2 reduction. J Am Chem Soc 2018; 140: 14614–8. [DOI] [PubMed] [Google Scholar]
- 61. Baldwin LA, Crowe JW, Pyles DAet al.. Metalation of a mesoporous three-dimensional covalent organic framework. J Am Chem Soc 2016; 138: 15134–7. [DOI] [PubMed] [Google Scholar]
- 62. Gao R, Sun W-H, Redshaw C. Nickel complex pre-catalysts in ethylene polymerization: new approaches to elastomeric materials. Cat Sci Technol 2013; 3: 1172–9. [Google Scholar]
- 63. Gonçalves RSB, Oliveira ABV, Sindra HCet al.. Heterogeneous catalysis by covalent organic frameworks (COF): Pd(OAc)2@COF-300 in cross-coupling reactions. Chem Cat Chem 2016; 8: 743–50. [Google Scholar]
- 64. Yoon TP, Jacobsen EN. Privileged chiral catalysts. Science 2003; 299: 1691–3. [DOI] [PubMed] [Google Scholar]
- 65. Li L-H, Feng X-L, Cui X-Het al.. Salen-based covalent organic framework. J Am Chem Soc 2017; 139: 6042–5. [DOI] [PubMed] [Google Scholar]
- 66. Yan S, Guan X, Li Het al.. Three-dimensional salphen-based covalent organic frameworks as catalytic antioxidants. J Am Chem Soc 2019; 141: 2920–4. [DOI] [PubMed] [Google Scholar]
- 67. Leng W, Ge R, Dong Bet al.. Bimetallic docked covalent organic frameworks with high catalytic performance towards tandem reactions. RSC Adv 2016; 6: 37403–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Leng W, Peng Y, Zhang Jet al.. Sophisticated design of covalent organic frameworks with controllable bimetallic docking for a cascade reaction. Chem A Eur J 2016; 22: 9087–91. [DOI] [PubMed] [Google Scholar]
- 69. Yang Y, Faheem M, Wang Let al.. Surface pore engineering of covalent organic frameworks for ammonia capture through synergistic multivariate and open metal site approaches. ACS Cent Sci 2018; 4: 748–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pachfule P, Kandambeth S, Diaz DDet al.. Highly stable covalent organic framework–au nanoparticles hybrids for enhanced activity for nitrophenol reduction. Chem Commun 2014; 50: 3169–72. [DOI] [PubMed] [Google Scholar]
- 71. Pachfule P, Panda MK, Kandambeth Set al.. Multifunctional and robust covalent organic framework-nanoparticle hybrids. J Mater Chem A 2014; 2: 7944–52. [Google Scholar]
- 72. Phan NTS, Sluys MVD, Jones CW. On the nature of the active species in palladium catalyzed Mizoroki–Heck and Suzuki–Miyaura couplings—homogeneous or heterogeneous catalyst, a critical review. Adv Synth Catal 2006; 348: 609–79. [Google Scholar]
- 73. Niu W, Zhang L, Xu G. Shape-controlled synthesis of single-crystalline palladium nanocrystals. ACS Nano 2010; 4: 1987–96. [DOI] [PubMed] [Google Scholar]
- 74. Åkermark B, Eberson L, Jonsson Eet al.. Palladium-promoted cyclization of diphenyl ether, diphenylamine and related compounds. J Org Chem 1975; 40: 1365–7. [Google Scholar]
- 75. Kaleeswaran D, Antony R, Sharma Aet al.. Catalysis and CO2 capture by palladium incorporated covalent organic frameworks. ChemPlusChem 2017; 82: 1253–65. [DOI] [PubMed] [Google Scholar]
- 76. Mullangi D, Nandi S, Shalini Set al.. Pd loaded amphiphilic COF as catalyst for multi-fold heck reactions, C-C couplings and CO oxidation. Sci Rep 2015; 5: 10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen G-J, Li X-B, Zhao C-Cet al.. Ru nanoparticles-loaded covalent organic framework for solvent-free one-pot tandem reactions in air. Inorg Chem 2018; 57: 2678–85. [DOI] [PubMed] [Google Scholar]
- 78. Zahmakıran M, Tonbul Y, Özkar S. Ruthenium(0) nanoclusters stabilized by a nanozeolite framework: isolable, reusable, and green catalyst for the hydrogenation of neat aromatics under mild conditions with the unprecedented catalytic activity and lifetime. J Am Chem Soc 2010; 132: 6541–9. [DOI] [PubMed] [Google Scholar]
- 79. McCaffrey R, Long H, Jin Yet al.. Template synthesis of gold nanoparticles with an organic molecular cage. J Am Chem Soc 2013; 136: 1782–5. [DOI] [PubMed] [Google Scholar]
- 80. Mullangi D, Chakraborty D, Pradeep Aet al.. Highly stable COF-supported co/co (OH)2 nanoparticles heterogeneous catalyst for reduction of nitrile/nitro compounds under mild conditions. Small 2018; 14: 1801233. [DOI] [PubMed] [Google Scholar]
- 81. Chandra S, Kundu T, Kandambeth Set al.. Phosphoric acid loaded azo (−N≡N−) based covalent organic framework for proton conduction. J Am Chem Soc 2014; 136: 6570–3. [DOI] [PubMed] [Google Scholar]
- 82. Asensio JA, Sánchez EM, Gómez-Romero P. Proton-conducting membranes based on benzimidazole polymers for high-temperature PEM fuel cells: a chemical quest. Chem Soc Rev 2010; 39: 3210–39. [DOI] [PubMed] [Google Scholar]
- 83. Meng Z, Aykanat A, Mirica KA. Proton conduction in 2D aza-fused covalent organic frameworks. Chem Mater 2019; 31: 819–25. [Google Scholar]
- 84. Ma H, Liu B, Li Bet al.. Cationic covalent organic frameworks: a simple platform of anionic exchange for porosity tuning and proton conduction. J Am Chem Soc 2016; 138: 5897–903. [DOI] [PubMed] [Google Scholar]
- 85. Cheng Y-J, Wang R, Wang Set al.. Encapsulating [Mo3S13]2− clusters in cationic covalent organic frameworks: enhancing stability and recyclability by converting a homogeneous photocatalyst to a heterogeneous photocatalyst. Chem Commun 2018; 54: 13563–6. [DOI] [PubMed] [Google Scholar]
- 86. Kibsgaard J, Jaramillo TF, Besenbacher F. Building an appropriate active-site motif into a hydrogen-evolution catalyst with thiomolybdate [Mo3S13]2− clusters. Nat Chem 2014; 6: 248–53. [DOI] [PubMed] [Google Scholar]
- 87. Guo Z, Zhang Y, Dong Yet al.. Fast ion transport pathway provided by polyethylene glycol confined in covalent organic frameworks. J Am Chem Soc 2019; 141: 1923–7. [DOI] [PubMed] [Google Scholar]
- 88. Gao W, Sun X, Niu Xet al.. Phosphomolybdic acid functionalized covalent organic frameworks: structure characterization and catalytic properties in olefin epoxidation. Microporous Mesoporous Mater 2015; 213: 59–67. [Google Scholar]
- 89. Waller PJ, Lyle SJ, Popp TMOet al.. Chemical conversion of linkages in covalent organic frameworks. J Am Chem Soc 2016; 138: 15519–22. [DOI] [PubMed] [Google Scholar]
- 90. Mohamed MA, Yamada K, Tomioka K. Accessing the amide functionality by the mild and low-cost oxidation of imine. Tetrahedron Lett 2009; 50: 3436–8. [Google Scholar]
- 91. Liu H, Chu J, Yin Zet al.. Covalent organic frameworks linked by amine bonding for concerted electrochemical reduction of CO2. Chem 2018; 4: 1696–709. [Google Scholar]
- 92. Li X, Zhang C, Cai Set al.. Facile transformation of imine covalent organic frameworks into ultrastable crystalline porous aromatic frameworks. Nat Commun 2018; 9: 2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dibble DJ, Umerani MJ, Mazaheripour Aet al.. An aza-Diels-Alder route to polyquinolines. Macromolecules 2015; 48: 557–61. [Google Scholar]
- 94. Haase F, Gottschling K, Stegbauer Let al.. Tuning the stacking behaviour of a 2D covalent organic framework through non-covalent interactions. Mater Chem Front 2017; 1: 1354–61. [Google Scholar]
- 95. Haase F, Troschke E, Savasci Get al.. Topochemical conversion of an imine- into a thiazole-linked covalent organic framework enabling real structure analysis. Nat Commun 2018; 9: 2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Böttcher VB, Bauer F. The extraction of sulfur from marine base (German). Justus Liebigs Ann Chem 1950; 568: 218–27. [Google Scholar]
- 97. Waller PJ, AlFaraj YS, Diercks CSet al.. Conversion of imine to oxazole and thiazole linkages in covalent organic frameworks. J Am Chem Soc 2018; 140: 9099–103. [DOI] [PubMed] [Google Scholar]
- 98. Steed JW, Atwood JL. Supramolecular Chemistry, 2nd edn. Wiltshire: John-Wiley & Son Ltd, 2006. [Google Scholar]
- 99. Guo J, Xu Y, Jin Set al.. Conjugated organic framework with three-dimensionally ordered stable structure and delocalized π clouds. Nat Commun 2013; 4: 2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wan S, Guo J, Kim Jet al.. A photoconductive covalent organic framework: self-condensed arene cubes composed of eclipsed 2D polypyrene sheets for photocurrent generation. Angew Chem Int Ed 2009; 48: 5439–42. [DOI] [PubMed] [Google Scholar]
- 101. Ding X, Guo J, Feng Xet al.. Synthesis of metallophthalocyanine covalent organic frameworks that exhibit high carrier mobility and photoconductivity. Angew Chem Int Ed 2011; 50: 1289–93. [DOI] [PubMed] [Google Scholar]
- 102. Ding X, Chen L, Honsho Yet al.. An n-channel two-dimensional covalent organic framework. J Am Chem Soc 2011; 133: 14510–3. [DOI] [PubMed] [Google Scholar]
- 103. Feng X, Liu L, Honsho Yet al.. High-rate charge-carrier transport in porphyrin covalent organic frameworks: switching from hole to electron to ambipolar conduction. Angew Chem Int Ed 2012; 51: 2618–22. [DOI] [PubMed] [Google Scholar]
- 104. Feng X, Chen L, Honsho Yet al.. An ambipolar conducting covalent organic framework with self-sorted and periodic electron donor-acceptor ordering. Adv Mater 2012; 24: 3026–31. [DOI] [PubMed] [Google Scholar]
- 105. Dogru M, Handloser M, Auras Fet al.. A photoconductive thienothiophene-based covalent organic framework showing charge transfer towards included fullerene. Angew Chem Int Ed 2013; 52: 2920–4. [DOI] [PubMed] [Google Scholar]
- 106. Savenije TJ, Grzegorczyk WJ, Heeney Met al.. Photoinduced charge carrier generation in blends of poly (thienothiophene) derivatives and [6,6]-phenyl-C61-butyric acid methyl ester: phase segregation versus intercalation. J Phys Chem C 2010; 114: 15116–20. [Google Scholar]
- 107. Heeney M, Bailey C, Genevicius Ket al.. Stable polythiophene semiconductors incorporating thieno[2,3-b]thiophene. J Am Chem Soc 2005; 127: 1078–9. [DOI] [PubMed] [Google Scholar]
- 108. Mulzer CR, Shen L, Bisbey RPet al.. Superior charge storage and power density of a conducting polymer-modified covalent organic framework. ACS Cent Sci 2016; 2: 667–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Xu H, Tao S, Jiang D. Proton conduction in crystalline and porous covalent organic frameworks. Nat Mater 2016; 15: 722–6. [DOI] [PubMed] [Google Scholar]
- 110. Sun Q, Fu C-W, Aguila Bet al.. Pore environment control and enhanced performance of enzymes infiltrated in covalent organic frameworks. J Am Chem Soc 2018; 140: 984–92. [DOI] [PubMed] [Google Scholar]
- 111. Kalidindi SB, Yusenko K, Fischer RA. Metallocenes@COF-102: organometallic host-guest chemistry of porous crystalline organic frameworks. Chem Commun 2011; 47: 8506–8. [DOI] [PubMed] [Google Scholar]