

Abstract
Azoxy-, azo- and amino-aromatics are among the most widely used building blocks in materials science pharmaceuticals and synthetic chemistry, but their controllable and green synthesis has not yet been well established. Herein, a facile potential-tuned electrosynthesis of azoxy-, azo- and amino-aromatics via aqueous selective reduction of nitroarene feedstocks over a CoP nanosheet cathode is developed. A series of azoxy-, azo- and amino-compounds with excellent selectivity, good functional group tolerance and high yields are produced by applying different bias input. The synthetically significant and challenging asymmetric azoxy-aromatics can be controllably synthesized in moderate to good yields. The use of water as the hydrogen source makes this strategy remarkably fascinating and promising. In addition, deuterated aromatic amines with a high deuterium content can be readily obtained by using D2O. By pairing with anodic oxidation of aliphatic amines to nitriles, synthetically useful building blocks can be simultaneously produced in a CoP||Ni2P two-electrode electrolyzer. Only 1.25 V is required to achieve a current density of 20 mA cm−2, which is much lower than that of overall water splitting (1.70 V). The paired oxidation and reduction reactions can also be driven using a 1.5 V battery to synthesize nitrile and azoxybenzene with good yields and selectivity, further emphasizing the flexibility and controllability of our method. This work paves the way for a promising approach to the green synthesis of valuable chemicals through potential-controlled electrosynthesis.
Keywords: azoxy, azo- and amino-aromatics, electrosynthesis, metal phosphides, nitroarenes, selectivity
INTRODUCTION
Azoxy-, azo- and amino-compounds are important organic molecules that have attracted much interest within the chemistry community due to their unique properties arising from the N−N−O and N=N moieties in azoxy- and azo-molecules and the vital applications of amines for the industrial production of dyestuffs, pharmaceuticals, and other chemicals [1,2]. A substantial effort has been devoted to their synthesis in a profitable and efficient manner, where selective reduction of nitro feedstocks outperforms other strategies due to their wide commercial availability and ease of implementation [3–5]. However, the nitro reduction proceeds via multiple electron-proton coupled steps [6–8]. This poses practical difficulties in controlling the degree of reduction against the production of thermodynamically favorable, over-hydrogenated amines [9–11]. Thus, the synthesis of highly valuable azoxy and azo compounds remains a critical challenge. Several strategies have been proposed for selective synthesis of azoxy and azo compounds by changing the adsorption/desorption energy of some reaction intermediates (such as nitrosobenzene and phenylhydroxylamine) and/or the reactive hydrogen (*H) to the catalysts. However, most of these strategies have been focused on the modifications of catalysts [12–15] or use of light with different wavelengths [16] to realize different product selectivity, inevitably suffering from tedious modifications or requirements for the use of specific light sources. High pressure H2 and other expensive and hard-to-control donors (such as NH2NH2·H2O, NaBH4) remain the primary hydrogen sources, posing serious safety risk and environmental concerns and leading to the poor functional group compatibility of the current methods. Thus, it is highly desirable to develop a facile, efficient and sustainable method to achieve the synthesis of azoxy-, azo- and amino-compounds through the reduction of nitro feedstocks in a controllable manner.
Recently, electrochemical transformation has offered a tunable and efficient alternative in synthetic chemistry due to its precise control by the external potential or current [17–25]. Electrons can act as green reductants to drive chemical reactions. By designing appropriate catalytic materials, the chemoselectivity can be well tuned by adjusting the potential or current input, altering the concentration and types of reactive intermediates at the electrode surface and finally determining the product distributions. For example, in the electrochemical CO2 reduction reactions (CO2RR) on Cu electrodes, generally, 2e− products (H2, CO and HCOOH) are preferred at lower overpotentials, whereas higher e− products (CH4: 8e− and C2H4: 6e−) are formed at higher overpotentials [26–28]. Inspired by the fundamental understanding of CO2RR and the electrochemical hydrogen evolution reaction (HER), it is intriguing to develop an electrochemical pathway to reduce nitro substrates specifically to azoxy-, azo- and amino-products in aqueous solution at different electrochemical conditions by balancing the nitro reduction with the competitive HER. In this case, the use of water as the sole hydrogen source will provide not only a safer, cleaner and more sustainable way to traditional nitro reductions using H2, metal hydrides, hydrazine hydrate, silane, and other reagents [3–15] but also the synthesis of value-added deuterated amines (–ND2) with potential utilization for deuterated drugs with enhanced therapeutic effect and LC/MS analysis with increased sensitivity, ionization efficiency, and chromatographic performance [29–31]. The latter can be readily realized by using D2O to replace H2O, in sharp contrast to previous –ND2 synthesis using expensive and hard-to-obtain deuterated D2, LiAlD4 and DCl [32,33]. Currently, the electrochemical reduction of nitro compounds relies on the use of noble metal electrodes and suffers from low conversion, narrow substrate scopes and poor selectivity [34,35]. Furthermore, the operational conditions in acidic solutions (such as HClO4) make it impractical and challenging to synthesize –ND2 with a high deuteration content [36,37].
Herein, we reported a highly efficient aqueous electrosynthesis of azoxy-, azo- and amino-aromatics by selective reduction of nitro substrates over a cobalt phosphide (CoP) nanosheet cathode (Fig. 1). The product selectivity can be flexibly controlled by adjusting the applied potentials to modulate the competition between HER and nitro reduction. The use of CoP as cathode and water as solvent avoids the excessive reduction of nitro aromatics to cyclohexylamine [38]. Deuterated amino compounds (–ND2) can be efficiently fabricated by using D2O as the hydrogen source.
Figure 1.
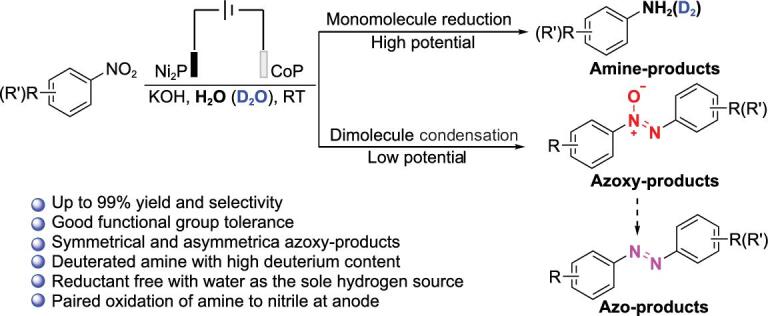
Highly selective electrochemical reduction of nitroarenes to azoxy-, azo- and amino-aromatics over a CoP cathode.
RESULTS AND DISCUSSION
CoP nanosheet electrode
Co-related catalysts are widely used in catalysis due to their low cost, abundance and high selectivity [39–41]. The moderate Co-*H formation energy makes it highly suitable for use in hydrogenation and HER through the H2 dissociation and H-H combination. CoP nanosheets have shown electrocatalytic performance comparable to that of Pt-based materials due to their abundance of exposed catalytically active sites, high catalytic activity and excellent stability toward electrochemical HER [42–45]. Thus, CoP nanosheets were chosen as the model electrocatalyst to evaluate our conceptual strategy on potential-tuned selective electrosynthesis of azoxy-, azo- and amino-aromatics. The CoP nanosheet electrode was prepared by direct phosphating of cobalt hydroxide precursors [42]. Scanning electron microscopy (SEM) images show that the nanosheet arrays are anchored on nickel foam (Fig. 2a). The transmission electron microscopy (TEM) image confirms the nanosheet morphology (Fig. 2b). The high-resolution TEM (HRTEM) image shows a lattice spacing of 0.25 nm indexed to the (111) plane of orthorhombic CoP (Fig. 2c). All diffraction peaks in the X-ray diffraction (XRD) pattern can be indexed to orthorhombic CoP (JCPDS No. 29–0497) (Supplementary Fig. 1a). X-ray photoelectron spectroscopy (XPS) spectra also show the characteristic peaks of CoP (Supplementary Fig. 1b-d), as observed in [42]. Additionally, the energy-dispersive X-ray spectroscopy (EDS) elemental mapping images indicate that the Co and P elements are uniformly distributed (Fig. 2d). These results suggest the successful synthesis of the CoP nanosheet electrode.
Figure 2.

(a) SEM (inset: enlargement), (b) TEM, (c) HRTEM, and (d) EDS elemental mapping images of CoP nanosheets.
Selective electrochemical reduction of nitroarenes to azoxy-, azo- and amino-aromatics
The electrochemical reduction of nitro substrate was first applied to the selective synthesis of azoxy-aromatic compounds, which is the most challenging synthetic task among azoxy-, azo- and amino-aromatics using reported methods [4,10–12,16]. The reaction was implemented in a 1.0 M KOH aqueous solution using a standard three-compartment electrochemical cell. Not that, all the electrochemical measurements were carried out under ambient atmosphere. Nitrobenzene (1a) was chosen as the model substrate. Different potentials were first applied ranging from −0.6 V to −1.2 V (vs. Ag/AgCl) to investigate the product distributions through long-term chronoamperometry with 1.0 mmol of 1a (Fig. 3a and Supplementary Table 1). As expected, similar to CO2RR, azoxybenzene (2a) with 99% selectivity and full conversion of 1a was realized at less negative potentials, whereas aniline (4a) (6e−) dominated at high reductive voltages. Although we were unable to directly synthesize azobenzene (3a) by tuning the potential, post-transformation of 2a at higher reductive voltages led to the formation of 3a with high yield and selectivity, as will be discussed below. Figure 3b shows the linear sweep voltammetry (LSV) curves of nitro reduction and HER over a CoP cathode with and without 1.0 mmol of 1a, respectively. For HER, the onset potential is approximately −1.10 V (vs. Ag/AgCl), and H2 bubbles can be clearly observed when the potential reaches −1.12 V. Upon the addition of 1a, the current density starts to increase from −0.6 V and the H2 bubbles clearly appear until the potential reaches −1.12 V. These results show that the reduction of 1a occurs preferentially at more positive potentials than HER due to its more electron-deficient property. Time-depended transformations reveal that the reaction can be finished within 6 h at −0.8 V (Fig. 3d). Using Ni2P or FeP as the cathodes, inferior yield and selectivity were observed, which may be ascribed to the moderate adsorption/desorption of highly active *H on the CoP surface [46]. Pure Ni foam (NF) was almost useless. All of the control experiments indicated the remarkable superiority of CoP in the selective conversion of 1a into 2a (Supplementary Fig. 2). In addition, no detectable chemical transformation occurred without electricity, suggesting the reduction reaction is mediated by electro-catalysis. The use of CH3CN/H2O (25 mL/40 μL) and CH3OH as the solvents under otherwise identical conditions led to poor yields and selectivity (Supplementary Fig. 3a), revealing the important role of water in controlling and promoting the selective nitro reduction. Finally, an undivided cell was adopted for more convenient production. However, 2a is delivered in lower yield due to the re-oxidation of the intermediates or azoxy products at the anode (Supplementary Fig. 3b).
Figure 3.

(a) Cathodic products from the electrochemical reduction of 1a over CoP obtained at different potentials. (b) LSV curves of CoP at a scan rate of 5 mV s−1 in 1.0 M KOH with and without 0.5 mmol of 1a. (c) Cycle-dependent selectivity and FEs of 2a over CoP at −0.8 V vs. Ag/AgCl. (d-f) Time-dependent conversion plots for the electrochemical reduction of 1a into 2a, 3a and 4a over a CoP cathode, respectively, in 1.0 M KOH at different potentials.
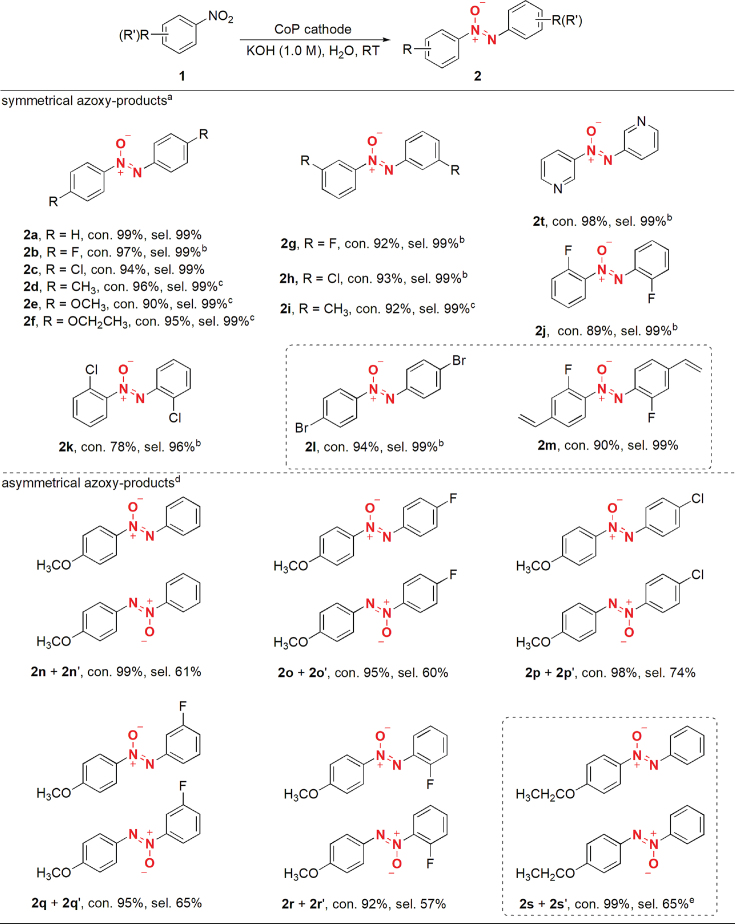
To validate the universality of this electrochemical synthesis of azoxy aromatic compounds, a variety of nitroarenes were examined (Supplementary Fig. 4). The results in Table 1 demonstrate that this strategy has excellent functional-group tolerance. The nitro substrates with the electron-donating or -withdrawing groups on the aryl ring all work well to give the corresponding azoxy-aromatic products with high conversion yields and excellent selectivity. No obvious decreases in reaction efficiency are observed when installing the -F and -Cl groups at the ortho-position of the nitro group (Table 1, 2j and 2k), indicating an inessential steric hindrance. Due to the mild reaction conditions, the quite fragile C-Br and C=C bonds are retained well, which is challenging in the traditional hydrogenation methods (Table 1, 2l-m), providing good opportunities for the subsequent synthesis of more complex molecules. Furthermore, this method can be applied to gram-scale synthesis with no obvious decrease in the reaction yields (Supplementary Fig. 5). All of these results prove that our established strategy is versatile and efficient for the synthesis of azoxy-aromatic compounds.
Table 1.
Electrochemical reduction of nitroarenes to symmetrical and asymmetrical azoxy-aromatics over a CoP cathode.
|
aNitro substrates (0.5 mmol), CoP (working area: 1 cm2), 1.0 M KOH (25 mL), room temperature, −0.75 V vs. Ag/AgCl. Con. is the abbreviation for conversion and sel. for selectivity. The con. and sel. are determined by gas chromatography. b−0.7 V vs. Ag/AgCl was used. c−0.8 V vs. Ag/AgCl was used. dp-methoxynitrobenzene (0.5 mmol), other nitro substrates (1.5 mmol). ep-ethoxynitrobenzene (0.5 mmol), 1a (1.5 mmol), −0.8 V vs. Ag/AgCl.
Importantly, asymmetrically substituted azoxy-aromatic compounds that are difficult to be synthesized by the present methods can be constructed using this controllable electrochemical method under the optimized reaction conditions. Note that, p-methoxynitrobenzene is used as 1.0 equivalent, while the other nitro substrates are in excess for enhanced yields of the cross-coupling products due to the faster kinetics of the electron-withdrawing aryl substituents (Table 1, 2o + o’ to 2s + s’). Nearly complete conversion of p-methoxynitrobenzene with moderate yields of the asymmetric products was obtained. The slightly low yield for each case is ascribed to the competitive role of the homocoupling reaction, but its yield is still much higher than the reported yield values [47]. Notably, our method can provide a convenient approach to the efficient synthesis of p-azoxyphenetole (2s + s’), which is an important nematic liquid crystal [48].
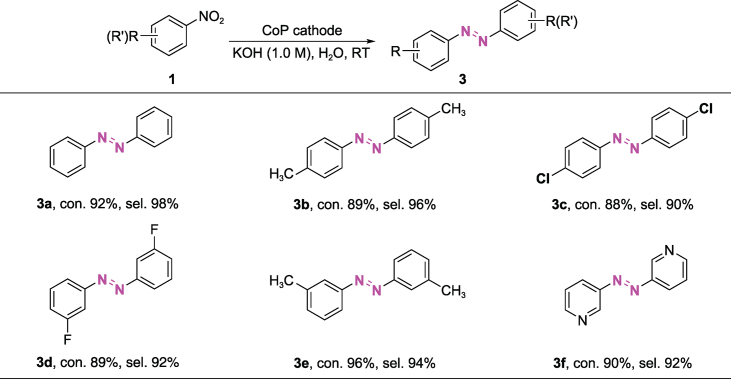
Azobenzenes are regarded as one of the largest and most commonly used organic dyes, and their selective synthesis has been a long-standing challenge [2,5,13–16,49]. By using our electrochemical method, azobenzene can be efficiently synthesized through a one-pot two-step procedure involving the first formation of azoxy products at a less negative bias (−0.8 V vs. Ag/AgCl) and their subsequent deep reduction to azo at the more negative potentials (−1.1 V vs. Ag/AgCl) (Fig. 3e and Supplementary Fig. 6). This one-pot two-step production of azo-aromatics from nitro reduction further demonstrates the good controllability of this potential-tuned strategy. We note that the amino product could not be detected during this procedure. This proves that the formation of amine via mono-molecule reduction is not involved in the formation of an azo-intermediate (Supplementary Table 1 and Supplementary Fig. 7). Some representative examples with high yields and selectivity are displayed in Table 2, providing a new alternative to the current synthesis methods of azo-compounds.
Table 2.
Electrochemical reduction of nitroarenes to azo-aromatics over a CoP cathodea.
|
aNitro substrates (0.5 mmol), CoP (working area: 1 cm2), 1.0 M KOH (25 mL), room temperature, −0.75 V and then turned to −1.1 V vs. Ag/AgCl. Con. is the abbreviation for conversion and sel. for selectivity. The con. and sel. are determined by gas chromatography.
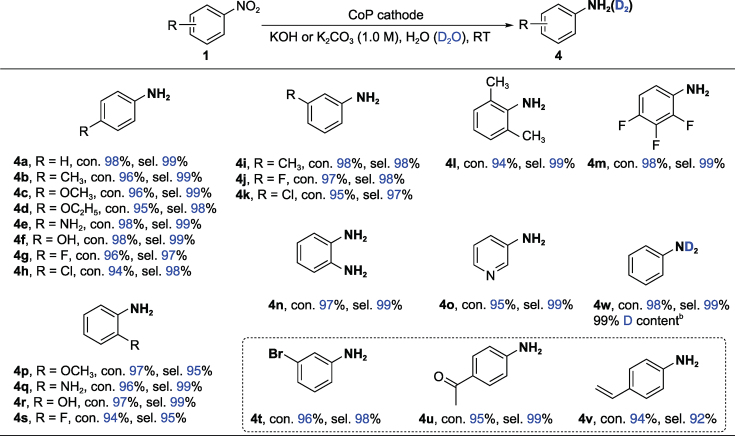
Amino aromatic compounds are a class of essential and versatile building blocks for drug synthesis, material preparations and catalyst designs/modifications [1,3,39,40]. Our method was also quite applicable to the rapid synthesis of amino aromatic compounds within 2.5 h (Fig. 3f). As observed from the data presented in Table 3, a variety of nitro substrates with electro-donating and electron-withdrawing groups on the ortho, meta or para positions on the aryl ring were selectively transformed into the corresponding aromatic amines with yields in the 94–98% range (4a-k and 4p-s). The multi-substituted and sterically constrained substrates worked smoothly to provide 4l and 4m in good yields. No decreases in the yields were observed for o-dinitrobenzene and 3-nitropyridine (4n-o). We note that the highly fragile -C–Br, -C=O and -C=C groups were well retained to the products without their simultaneous hydrogenation and without the detection of coupling or condensation products between -C–Br or -C=O and -NH2; these features are always challenging to obtain by other methods [50]. These unexpected chemoselective properties may be ascribed to both the electron-deficient effect and the preferred adsorption of the -NO2 group on the surface of CoP compared to the other groups [51,52]. Benefiting from this electrocatalytic method, deuterated aromatic amines with high deuterium content are easily synthesized (4w), demonstrating that this electroreductive strategy has remarkable advantages relative to the traditional approaches in terms of the operation, cost and sustainability.
Table 3.
Electrochemical reduction of nitroarenes to amino-aromatics over a CoP cathodea.
|
aNitro substrates (0.5 mmol), CoP (working area: 1 cm2), 1.0 M KOH (25 mL), room temperature, −0.9 V ∼ −1.2 V vs. Ag/AgCl. Con. is the abbreviation for conversion and sel. for selectivity. The con. and sel. are determined by gas chromatography. b1.0 M K2CO3 was used as the electrolyte.
Stability and reusability are essential properties for evaluating the potential utility of a catalyst. At a constant potential of −0.8 V vs. Ag/AgCl, one piece of catalyst was reused to reduce 2.0 mmol of nitrobenzene for six times. High Faradaic efficiencies (FEs) and 99% yield of 2a can be maintained for at least six cycle use, revealing good durability of the CoP cathode for such electrochemical reduction process (Fig. 3c). No obvious changes in the used CoP cathode were observed by SEM and XPS, suggesting the good stability of the cathode (Supplementary Figs. 8 and 10). The diffraction peaks located at 31.774, 36.413, 46.282, 48.363, 56.489 and 76.081 of used CoP could be indexed to the (011), (111), (112), (211), (301) and (222) plane of orthorhombic CoP (JCPDS No. 29–0497), which is consistent well with the fresh one (Supplementary Fig. 9).
Finally, to enhance the practicability of our method, a two-electrode configuration using CoP as the cathode and Ni2P as the anode was assembled in a separated CoP||Ni2P cell. To reduce the total energy input, oxidation of the thermodynamically more favorable organic compounds (e.g. alcohols, amines) was used to replace the kinetically sluggish oxygen evolution reaction at the anode [53–58]. Here, cathodic electrochemical reduction of 1a and anodic conversion of octylamine were selected as the model cases (Fig. 4a). As depicted in Fig. 4b and c, the required cell voltage for achieving the current density of 20 mA cm−2 is only 1.25 V (vs. counter electrode) after adding 1.0 mmol of 1a and 1.0 mmol of octylamine in the cathode and anode chambers, respectively. This cell voltage is much lower than that required for overall water splitting (1.70 V). Furthermore, a long-term electrolysis at −1.2 V over a CoP||Ni2P electrolyzer was performed to quantify the products of the cathode and anode chambers. Both 2a and octylnitrile with nearly 100% selectivity were obtained (Supplementary Fig. 11). Impressively, this two-electrode configuration can be used at the gram scale (∼2 g) under standard conditions with comparable conversion and selectivity, indicating the potential utility of this two-electrode electrolyzer for both the nitro reduction and amine oxidation that cannot be achieved by traditional methods. Finally, as revealed in Fig. 3a and Supplementary Fig. 12, 2a can be obtained with ∼99% yield and FEs at the potentials ranging from −0.65 to −0.95 V, leading to the possibility of distributed synthesis of azoxy using either a photo-voltaic-integrated solar panel or a 1.5 V battery (Fig. 4d and e). Comparable yields and selectivities for both 2a and nitrile products were ultimately obtained using the two-electrode electrolyzer, demonstrating the remarkable feasibility and practicability of our method.
Figure 4.

(a) Illustration for coupling cathodic electro-reductive synthesis of 2a with anodic conversion of octylamine into octylnitrile in a CoP||Ni2P electrolyzer. (b) LSV curves and (c) comparison of potential for achieving benchmark current densities in a CoP||Ni2P electrolyzer at a scan rate of 5 mV s−1 in 1.0 M KOH before and after adding 1.0 mmol of 1a to cathode chamber and 1.0 mmol of octylamine to anode chamber. (d-f) Time-dependent photos of a CoP||Ni2P electrolyzer setup driven by 1.5 V battery for both anodic oxidation of octylamine and cathodic reduction of 1a to 2a.
As mentioned above, the nitro reduction proceeds via multiple electron-proton coupled steps. Catalyst engineering to alter the adsorption/desorption energy of some reaction intermediates to the catalysts is a favorable method for tuning the product selectivity [12–15]. For our electrochemical method, potential tuning can be used to control the intermediates. It is well-accepted that the formation of azoxy- and azo-products involves the condensation of two intermediates, namely, nitrosobenzene and phenylhydroxylamine [8]. Additionally, 1.0 M KOH is essential because it will suppress the unwanted hydrogenation of the two intermediates due to the trace amount of protons at low potentials. At a higher reductive potential, more active *H will be generated at the surface of CoP via water splitting, trapping phenylhydroxylamine to form aniline and inhibiting the condensation of nitrosobenzene and phenylhydroxylamine (Supplementary Fig. 7). Cobalt in CoP is known to have a partial positive charge (δ+) [59], favoring adsorption of nitrobenzene and facilitating the reduction reactions.
CONCLUSIONS
In summary, we demonstrated a potential-tuned strategy for aqueous selective reduction of nitro substrates to azoxy-, azo- and amino-aromatics over a CoP nanosheet cathode. A variety of products bearing different functional groups were efficiently synthesized. In particular, the highly fragile -C-Br, -C=O and -C=C bonds were retained well for the synthesis of aromatic amines under our conditions. This good chemoselectivity is attributed to the electron-deficient effect and the preferred adsorption of the -NO2 group on CoP. Additionally, our protocol can also achieve the synthesis of unique asymmetric azoxy-aromatic compounds that are challenging for the traditional methods. Compared to the methods that rely on catalyst modifications to realize product selectivity, our potential-tuned strategy is more convenient to operate to tune the products selectivity at a gram scale for practical applications in industrial production. The adoption of water as the sole hydrogen source also provides a convenient method for producing deuterated amino aromatics with high deuterated efficiency. Furthermore, pairing with anodic oxidation of aliphatic amines, gram-scale production of azoxybenzene and octylnitrile can be simultaneously achieved with high efficiency in a two-electrode electrolyzer. Impressively, the paired reaction can be operated using a 1.5 V battery with good yield and selectivity, paving the way for low-cost and efficient production of both azoxy-aromatic and aliphatic nitrile. This potential-tuned strategy by using water as the hydrogenation source with good product selectivity can find wide applications in other types of electrochemical reduction reactions for controllable and green synthesis.
METHODS
Synthesis of CoP nanosheet electrode
Firstly, a piece of nickel foam (NF) (3 cm × 1 cm × 0.1 cm) was ultrasonicated with acetone, water and 3.0 M HCl aqueous solution for 5 min, respectively. Then, the NF was rinsed with distilled water (DIW) and anhydrous alcohol. Lastly, the NF was quickly dried under ambient conditions. The CoP electrode was prepared through modifying the reported method [42]. Typically, a α-Co(OH)2 precursor was electrodeposited onto the fresh-treated NF (α-Co(OH)2/NF) at −1.0 V vs. SCE (saturated calomel electrode) in 0.025 M Co(NO3)2 solution. Then, a piece of α-Co(OH)2/NF and NaH2PO2 (0.1 g) were put at two separate position in a porcelain boat with NaH2PO2 at the upstream side of the furnace. Subsequently, the sample was heated at 300°C for 2 h in static Ar atmosphere, and then naturally cooled to ambient temperature under Ar atmosphere.
Selective electrochemical reduction of nitroarenes to azoxy-aromatics
In 25 mL aqueous electrolyte (1.0 M KOH) containing 0.5 mmol of nitroarenes, the linear sweep voltammetry (LSV) curves tests with the scan rate of 5 mV s−1 ranging from −0.6 to −1.3 V vs. Ag/AgCl were implemented to study the onset reductive potentials of nitroarenes. To achieve azoxy-aromatics products, chronoamperometry was carried out at a given constant potential ranging from −0.7 to −0.8 V vs. Ag/AgCl in 1.0 M KOH solution.
Selective electrochemical reduction of nitroarenes to azo-aromatics
To achieve azo aromatics products, chronoamperometry was firstly carried out at a low potential ranging from −0.7 to −0.8 V vs. Ag/AgCl in 25 mL aqueous electrolyte (1.0 M KOH) containing 0.5 mmol of nitroarenes to produce azoxy-aromatics first. After nitroarenes were completely transferred to azoxy-aromatics, the constant potential is quickly swift to a high potential (e.g. −1.1 V vs. Ag/AgCl for azobenzene) to synthesize azo-aromatics.
Selective electrochemical reduction of nitroarenes to amino-aromatics
To achieve amino aromatics products, firstly long-term chronoamperometry was carried out at more negative constant potentials ranging from −0.9 to −1.2 V vs. Ag/AgCl in 25 mL aqueous electrolyte (1.0 M KOH) containing 0.5 mmol of nitroarenes.
Characterization
The scanning electron microscopy (SEM) images were taken with a Hitachi S-4800 scanning electron microscope (3 kV). The transmission electron microscopy (TEM) images, the energy-dispersive X-ray spectroscopy (EDS) elemental mapping images and higher-resolution transmission electron microscopy (HRTEM) images were obtained with JEOL-2100F system equipped with EDAX Genesis XM2. The X-ray diffraction (XRD) patterns of the products were recorded with Bruker D8 Focus Diffraction System using a Cu Kα source (λ = 0.15406 nm). X-ray photoelectron spectroscopy (XPS) measurements were performed on a photoelectron spectrometer using Al Kα radiation as the excitation source (PHI 5000 VersaProbe). All the peaks were calibrated with C 1 s spectrum at binding energy of 284.8 eV. The NMR spectra were recorded on Varian Mercury Plus 400 instruments at 400 MHz (1H NMR) and 101 MHz (13C NMR). Chemical shifts were reported in parts per million (ppm) down field from internal tetramethysilane. Multiplicity was indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), mm (multiplet), dd (doublet of doublet), br (broad). Coupling constants were reported in hertz (Hz). The gas chromatograph-mass spectrometer (GC-MS) was carried out with TRACE DSQ. The gas chromatograph (GC) was measured on Aglient 7890A with thermal conductivity (TCD) and flame ionization detector (FID). The injection temperature was set at 300°C. Nitrogen was used as the carrier gas at 1.5 mL min−1. All reported data are averages of experiments performed at least thrice.
Electrochemical measurements
Electrochemical measurements were carried out in a standard three-compartment electrochemical cell consisting of a working electrode, a Pt plate counter electrode, and a Ag/AgCl (1.0 M KCl) reference electrode, and all the potentials in this work are referred to Ag/AgCl unless otherwise stated. Linear sweep voltammetry (LSV), chronoamperometry was performed using an electrochemical workstation (CHI 660D, Chenhua, Shanghai). The solution of 1.0 M KOH (pH = 13.6) was employed as electrolyte. The NF with catalyst samples directly grown on the surface was used as the working electrode with exposed surface area of 1.0 cm2. The electrochemical hydrogen evolution reaction and electrocatalytic reduction reactions of nitroarenes experiments were conducted in 25 mL of 1.0 M KOH solution with and without 1.0 mmol of nitroarenes. The electrochemical OER experiment was also conducted in 25 mL of 1.0 M KOH solution. For two-electrode electrolysis, CoP and Ni2P were employed as cathode and anode, respectively. The scan rates of LSV curves were 5 mV s−1.
The stability tests of CoP electrode for 1a reduction were evaluated by chronoamperometry at −0.8 V in 25 mL of 1.0 M KOH solution using 2.0 mmol of 1a. All experiments were carried out at room temperature (RT, 25 ± 1°C) under ambient atmosphere.
Quantitative analysis of reduction and oxidative products
To analyze the products of nitrobenzene reduction and calculate the corresponding FEs, 25 mL of electrolyte solution was extracted with acetic ether after chronoamperometry testing with ∼289 C of total passing charge. Simultaneously, in the two-electrode system, the generated octylnitrile at anode was also collected. The extracted products were confirmed by the comparisons of their GC retention time and mass spectra. Yields were determined by GC analysis. Here, we take azoxy-compounds and octylnitrile as the examples to show the calculated equations of the cathodic and anodic products, respectively. For azo- and amino-products, the calculated equations are similar to those of azoxy-compounds.
The conversion (%) and selectivity (%) of the formed octylnitrile were calculated using equations (1) and (2), respectively:
 |
(1) |
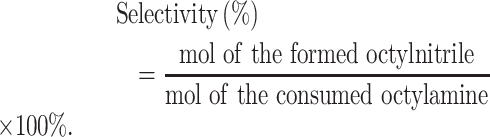 |
(2) |
The FEs for the formation of azoxy-aromatic compounds on CoP cathode and octylnitrile on Ni2P anode were calculated using the equation (3) and (4), respectively:
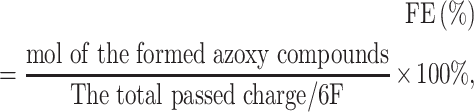 |
(3) |
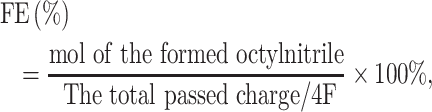 |
(4) |
where F is the Faraday constant (96 485 C mol−1).
A theoretical charge for the complete reduction of nitroarenes (1.0 mmol) to azoxy aromatic compounds (0.5 mmol): (1.0 × 10−3 mol) ×3 × (6.02 × 1023 mol−1) × (1.6 × 10−19 C) =∼289 C.
A theoretical charge for the complete oxidation of octylamine (1.0 mmol) to octylnitrile (1.0 mmol): (1.0 × 10−3 mol) × 4 × (6.02 × 1023 mol−1) ×(1.6 × 10−19 C) = ∼385 C.
Supplementary Material
Acknowledgements
We acknowledge the characterization support from the Analysis & Testing Center at Tianjin University.
FUNDING
This work was supported by the National Natural Science Foundation of China (21871206 and 21422104) and the Natural Science Foundation of Tianjin City (17JCJQJC44700).
Conflict of interest statement. None declared.
REFERENCES
- 1. Formenti D, Ferretti F, Scharnagl FKet al. Reduction of nitro compounds using 3d-non-noble metal catalysts. Chem Rev 2019; 119: 2611–80. [DOI] [PubMed] [Google Scholar]
- 2. Belowicha ME, Stoddart JF. Dynamic imine chemistry. Chem Soc Rev 2012; 41: 2003–24. [DOI] [PubMed] [Google Scholar]
- 3. Jagadeesh RV, Murugesan K, Alshammari ASet al. MOF-derived cobalt nanoparticles catalyze a general synthesis of amines. Science 2017; 358: 326–32. [DOI] [PubMed] [Google Scholar]
- 4. Li J, Song S, Long Yet al. Investigating the hybrid-structure-effect of CeO2-encapsulated Au nanostructures on the transfer coupling of nitrobenzene. Adv Mater 2018; 30: 1704416. [DOI] [PubMed] [Google Scholar]
- 5. Grirrane A, Corma A, García H. Gold-catalyzed synthesis of aromatic azo compounds from anilines and nitroaromatics. Science 2008; 322: 1661–4. [DOI] [PubMed] [Google Scholar]
- 6. Jensen SC, Homan SB, Weiss EA. Photocatalytic conversion of nitrobenzene to aniline through sequential proton-coupled one-electron transfers from a cadmium sulfide quantum dot. J Am Chem Soc 2016; 138: 1591–600. [DOI] [PubMed] [Google Scholar]
- 7. Serna P, Corma A. Transforming nano metal nonselective particulates into chemoselective catalysts for hydrogenation of substituted nitrobenzenes. ACS Catal 2015; 5: 7114–21. [Google Scholar]
- 8. Song JJ, Huang ZF, Pan Let al. Review on selective hydrogenation of nitroarene by catalytic, photocatalytic and electrocatalytic reactions. Appl Catal B Environ 2018; 227: 386–408. [Google Scholar]
- 9. Shiraishi Y, Togawa Y, Tsukamoto Det al. Highly efficient and selective hydrogenation of nitroaromatics on photoactivated rutile titanium dioxide. ACS Catal 2012; 2: 2475–81. [Google Scholar]
- 10. Pahalagedara MN, Pahalagedara LR, He Jet al. Room temperature selective reduction of nitrobenzene to azoxybenzene over magnetically separable urchin-like Ni/graphene nanocomposites. J Catal 2016; 336: 41–8. [Google Scholar]
- 11. Wang Y, Yu Y, Jia Ret al. Electrochemical synthesis of nitric acid from air and ammonia through waste utilization. Natl Sci Rev 2019; 6: 730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan H, Zhao X, Guo Net al. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat Common 2018; 9: 3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu X, Ye S, Li Het al. Selective and switchable transfer reduction of nitroarenes catalyzed by supported gold nanoparticles. Cat Sci Technol 2013; 3: 3200–6. [Google Scholar]
- 14. Zhu H, Ke X, Yang Xet al. Reduction of nitroaromatic compounds on supported gold nanoparticles by visible and ultraviolet light. Angew Chem Int Ed 2010; 49: 9657–61. [DOI] [PubMed] [Google Scholar]
- 15. Naya S, Niwa T, Kume Tet al. Visible-light-induced electron transport from small to large nanoparticles in bimodal gold nanoparticle-loaded titanium (IV) oxide. Angew Chem Int Ed 2014; 53: 7305–9. [DOI] [PubMed] [Google Scholar]
- 16. Dai Y, Li C, Shen Yet al. Light-tuned selective photosynthesis of azo- and azoxy-aromatics using graphitic C3N4. Nat Common 2018; 9: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yan M, Kawamata Y, Baran PS. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem Rev 2017; 117: 13230–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pletcher D, Green RA, Brown RCD. Flow electrolysis cells for the synthetic organic chemistry laboratory. Chem Rev 2018; 118: 4573–91. [DOI] [PubMed] [Google Scholar]
- 19. Liu K, Tang S, Wu Tet al. Electrooxidative Para-selective C–H/N–H cross-coupling with hydrogen evolution to synthesize triarylamine derivatives. Nat Common 2019; 10: 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peters BK, Rodriguez KX, Reisberg SHet al. Scalable and safe synthetic organic electroreduction inspired by Li-ion battery chemistry. Science 2019; 363: 838–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu N, Sauer GS, Saha Aet al. Metal-catalyzed electrochemical diazidation of alkenes. Science 2017; 357: 575–9. [DOI] [PubMed] [Google Scholar]
- 22. Asefa T. Metal-free and noble metal-free heteroatom-doped nanostructured carbons as prospective sustainable electrocatalysts. Acc Chem Res 2016; 499: 1873–83. [DOI] [PubMed] [Google Scholar]
- 23. Hou ZW, Mao ZY, Melcamu YYet al. Electrochemical synthesis of Imidazo-fused n-heteroaromatic compounds through a C-N bond-forming radical cascade. Angew Chem Int Ed 2018; 57: 1636–9. [DOI] [PubMed] [Google Scholar]
- 24. Yang QL, Wang XY, Lu JYet al. Copper-catalyzed electrochemical C–H amination of arenes with secondary amines. J Am Chem Soc 2018; 140: 11487–94. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Z, Zhang L, Cao Yet al. Mn-mediated electrochemical trifluoromethylation/C (sp2)–H functionalization cascade for the synthesis of azaheterocycles. Org Lett 2019; 21: 762–6. [DOI] [PubMed] [Google Scholar]
- 26. Vasileff A, Xu CC, Jiao Yet al. Surface and interface engineering in copper-based bimetallic materials for selective CO2 electroreduction. Chem 2018; 4: 1809–31. [Google Scholar]
- 27. Ren D, Fong JH, Yeo BS. The effects of currents and potentials on the selectivities of copper toward carbon dioxide electroreduction. Nat Common 2018; 9: 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang Y, Long R, Xiong Y. Regulating C–C coupling in thermocatalytic and electrocatalytic COx conversion based on surface science. Chem Sci 2019; 10: 7310–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Atzrodt J, Derdau V, Kerr WJet al. Deuterium- and tritium-labelled compounds: applications in the life sciences. Angew Chem Int Ed 2018; 57: 1758–84. [DOI] [PubMed] [Google Scholar]
- 30. Chatterjee B, Krishnakumar V, Gunanathan C. Selective α-deuteration of amines and amino acids using D2O. Org Lett 2016; 18: 5892–5. [DOI] [PubMed] [Google Scholar]
- 31. Qiu C, Xu Y, Fan Xet al. Highly crystalline K-intercalated polymeric carbon nitride for visible-light photocatalytic alkenes and alkynes deuterations. Adv Sci 2019; 6: 1801403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang YM, Liu Z, Gao Get al. Visible light-driven selective hydrogenation of unsaturated aromatics in an aqueous solution by direct photocatalysis of Au nanoparticles. Cat Sci Technol 2018; 8: 726–34. [Google Scholar]
- 33. Wiltshire HR, Prior KJ, Dhesi Jet al. The synthesis of labelled forms of Saquinavir. J Labelled Cpd Radiopharm 1998; 41: 1103–26. [Google Scholar]
- 34. Sheng X, Wouters B, Breugelmans Tet al. Pure and alloyed copper-based nanoparticles supported on activated carbon: synthesis and electrocatalytic application in the reduction of nitrobenzene. ChemElectroChem 2014; 1: 1198–210. [Google Scholar]
- 35. Zhang P, Sheng X, Chen Xet al. Paired electrocatalytic oxygenation and hydrogenation of organic substrates with water as the oxygen and hydrogen source. Angew Chem Int Ed 2019; 58: 9155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Daemsa N, Wouters J, Goethem CVet al. Selective reduction of nitrobenzene to aniline over electrocatalysts based on nitrogen-doped carbons containing non-noble metals. Appl Catal Environ 2018; 226: 509–22. [Google Scholar]
- 37. Cyr A, Huot P, Marcoux JFet al. The electrochemical reduction of nitrobenzene and azoxybenzene in neutral and basic aqueous methanolic solutions at polycrystalline copper and nickel electrodes. Electrochim Acta 1989; 34: 439–45. [Google Scholar]
- 38. Yuan XZ, Ma ZF, Jiang QZet al. Cogeneration of cyclohexylamine and electrical power using PEM fuel cell reactor. Electrochem Commun 2001; 3: 599–602. [Google Scholar]
- 39. Ai WY, Zhong R, Liu XFet al. Hydride transfer reactions catalyzed by cobalt complexes. Chem Rev 2019; 119: 2876–953. [DOI] [PubMed] [Google Scholar]
- 40. Crossley SWM, Obradors C, Martinez RMet al. Mn-, Fe-, and Co-catalyzed radical hydrofunctionalizations of olefins. Chem Rev 2016; 116: 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun J, Deng L. Cobalt complex-catalyzed hydrosilylation of alkenes and alkynes. ACS Catal 2016; 6: 290–300. [Google Scholar]
- 42. Tian J, Liu Q, Asiri AMet al. Self-supported nanoporous cobalt phosphide nanowire arrays: an efficient 3D hydrogen-evolving cathode over the wide range of pH 0–14. J Am Chem Soc 2014; 136: 7587–90. [DOI] [PubMed] [Google Scholar]
- 43. Zhang C, Huang Y, Yu Yet al. Sub-1.1 nm ultrathin porous CoP nanosheets with dominant reactive {200} facets: a high mass activity and efficient electrocatalyst for the hydrogen evolution reaction. Chem Sci 2017; 8: 2769–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xue ZH, Su H, Yu QYet al. Janus Co/CoP nanoparticles as efficient Mott–Schottky electrocatalysts for overall water splitting in wide pH range. Adv Energy Mater 2017; 7: 1602355. [Google Scholar]
- 45. Jiang N, You B, Sheng Met al. Electrodeposited cobalt-phosphorous-derived films as competent bifunctional catalysts for overall water splitting. Angew Chem Int Ed 2015; 54: 6251–4. [DOI] [PubMed] [Google Scholar]
- 46. Kibsgaard J, Tsai C, Chan Ket al. Designing an improved transition metal phosphide catalyst for hydrogen evolution using experimental and theoretical trends. Energ Environ Sci 2015; 8: 3022–9. [Google Scholar]
- 47. Chen YF, Chen J, Lin LJet al. Synthesis of azoxybenzenes by reductive dimerization of nitrosobenzene. J Org Chem 2017; 82: 11626–30. [DOI] [PubMed] [Google Scholar]
- 48. Phaovibul O, Sungsittayakorn P, Limcharoen Pet al. Optical studies on binary mesophase mixtures containing p-azoxyphenetole (PAP). Mol Cryst Liq Cryst 1981; 73: 81–93. [Google Scholar]
- 49. Zhang YF, Mellah M. Convenient electrocatalytic synthesis of azobenzenes from nitroaromatic derivatives using SmI2. ACS Catal 2017; 7: 8480–6. [Google Scholar]
- 50. Zhou BW, Song JL, Zhou HCet al. Using the hydrogen and oxygen in water directly for hydrogenation reactions and glucose oxidation by photocatalysis. Chem Sci 2016; 7: 463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boronat M, Concepcion P, Corma Aet al. A molecular mechanism for the chemoselective hydrogenation of substituted nitroaromatics with nanoparticles of gold on TiO2 catalysts: a cooperative effect between gold and the support. J Am Chem Soc 2007; 129: 16230–7. [DOI] [PubMed] [Google Scholar]
- 52. Blaser HU, Steiner H, Studer M. Selective catalytic hydrogenation of functionalized nitroarenes: an update. ChemCatChem 2009; 1: 210–21. [Google Scholar]
- 53. Yu Y, Shi Y, Zhang B. Synergetic transformation of solid inorganic-organic hybrids into advanced nanomaterials for catalytic water splitting. Acc Chem Res 2018; 51: 1711–21. [DOI] [PubMed] [Google Scholar]
- 54. Xu Y, Zhang B. Recent advances in electrochemical hydrogen production from water assisted by alternative oxidation reactions. ChemElectroChem 2019; 6: 3214–26. [Google Scholar]
- 55. You B, Sun Y. Innovative strategies for electrocatalytic water splitting. Acc Chem Res 2018; 51: 1571–80. [DOI] [PubMed] [Google Scholar]
- 56. Zhang J, Wang H, Tian Yet al. Anodic hydrazine oxidation assists energy-efficient hydrogen evolution over a bifunctional cobalt perselenide nanosheet electrode. Angew Chem Int Ed 2018; 57: 7649–53. [DOI] [PubMed] [Google Scholar]
- 57. Huang Y, Chong X, Liu Cet al. Boosting hydrogen production by anodic oxidation of primary amines over a NiSe nanorod electrode. Angew Chem Int Ed 2018; 57: 13163–6. [DOI] [PubMed] [Google Scholar]
- 58. Tang C, Zhang R, Lu Wet al. Energy-saving electrolytic hydrogen generation: Ni2P nanoarray as a high-performance non-noble-metal electrocatalyst. Angew Chem Int Ed 2017; 56: 842–6. [DOI] [PubMed] [Google Scholar]
- 59. Liu Q, Tian J, Cui Wet al. Carbon nanotubes decorated with CoP nanocrystals: a highly active non-noble-metal nanohybrid electrocatalyst for hydrogen evolution. Angew Chem Int Ed 2014; 53: 6710–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.