

Abstract
Pangolins are among the most critically endangered animals due to heavy poaching and worldwide trafficking. However, their demographic histories and the genomic consequences of their recent population declines remain unknown. We generated high-quality de novo reference genomes for critically endangered Malayan (Manis javanica, MJ) and Chinese (M. pentadactyla, MP) pangolins and re-sequencing population genomic data from 74 MJs and 23 MPs. We recovered the population identities of illegally traded pangolins and previously unrecognized genetic populations that should be protected as evolutionarily distinct conservation units. Demographic reconstruction suggested environmental changes have resulted in a population size fluctuation of pangolins. Additionally, recent population size declines due to human activities have resulted in an increase in inbreeding and genetic load. Deleterious mutations were enriched in genes related to cancer/diseases and cholesterol homeostasis, which may have increased their susceptibility to diseases and decreased their survival potential to adapt to environmental changes and high-cholesterol diets. This comprehensive study provides not only high-quality pangolin reference genomes, but also valuable information concerning the driving factors of long-term population size fluctuations and the genomic impact of recent population size declines due to human activities, which is essential for pangolin conservation management and global action planning.
Keywords: pangolins, population genomics, population declines, genetic diversity, inbreeding, genetic load, demographic history
INTRODUCTION
Pangolins (monotypic order Pholidota and family Manidae) are believed to be the most heavily poached and trafficked mammals in the world, accounting for as much as 20% of all illegal transnational wildlife trade [1–4]. Overexploitation driven by escalating demand for their meat as luxury food consumption and scales for traditional medicines has driven pangolins to the edge of extinction [5–10]. More than 1 million pangolins had been poached in the decade prior to 2014 [11]. All eight pangolin species (four Asian pangolins: Malayan pangolin Manis javanica, Chinese pangolin M. pentadactyla, Indian pangolin M. crassicaudata and Philippine pangolin M. culionensis; four African pangolins: Tree pangolin Phataginus tricuspis, Long-tailed pangolin P. tetradactyla, Temminck's ground pangolin Smutsia temminckii and Giant pangolin S. gigantean) [12,13] have been listed in ‘Appendix I’ of the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) [14]. Notably, two Asian pangolins, Malayan and Chinese pangolins, are the most threatened pangolin species with the highest extinction risk. The Malayan pangolin is mainly found in Southeast Asia [9,14,15] and the Chinese pangolin mainly inhabits the south of China and some of northern Southeast Asia [16–18]. Both species have been recognized as the most-traded mammals in Asia [2,5,7,14] and classified as Critically Endangered on the International Union for Conservation of Nature (IUCN) red list [19]. Although a zero-trade quota for wild-caught individuals of them has been exerted since 2000 [20], wildlife poaching and international illegal trades have been still reported and the population decline continues [2,5,9,14].
The two critically endangered pangolins have attracted much attention. Earlier studies have focused on the confiscated species identification [6,9,13,21] and the phylogeny [12,22,23] by using mitochondrial (mt) genes and microsatellites. Recent genomic studies have investigated the illegal trade routes [15] and the adaptive evolution of pangolin scales and immunity [16], while several transcriptomic studies have explored the adaptive evolution of their myrmecophagous feeding [24,25]. However, very limited knowledge was available about their demographic histories and the genomic consequences associated with their recent population declines, which is essential for pangolin conservation management and global action planning.
Therefore, in this study, we have generated a data set comprising two high-quality de novo assembled genomes of Malayan and Chinese pangolins, which are significantly improved compared with the published pangolin assemblies [16,26], and re-sequenced the genomes of 74 Malayan and 23 Chinese pangolins. The population genomic analyses provide a comprehensive evaluation of their survival potential, the driving factors of long-term population size fluctuations and the genomic impact of recent population size declines due to human activities.
RESULTS AND DISCUSSION
Generation of two high-quality pangolin reference genomes
The two newly assembled pangolin genomes were generated with a very-high-coverage whole-genome shotgun and 10X genomic sequencing strategy. We generated a total of 980 Gb of sequence data (411.8-fold genome coverage) from a Malayan pangolin (M. javanica, referred to hereafter as MJ) and 831 Gb of sequence data (289.6-fold genome coverage) from a Chinese pangolin (M. pentadactyla, referred to hereafter as MP) (Supplementary Table 1).
The genome sizes of the de novo assembly for MJ and MP are 2.45 and 2.40 Gb, with scaffold N50 of 13.85 and 7.77 Mb, respectively (Supplementary Table 2). The base error rates for the reference genomes are 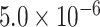 and
and 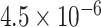 for MJ and MP, respectively, which are both <
for MJ and MP, respectively, which are both <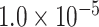 . Both of them are significantly improved compared with the published pangolin assemblies (genome size: 2.55 and 2.21 Gb, scaffold N50: 0.2 and 0.16 Mb in Choo et al. [16]; genome size: 2.85 and 2.91 Gb, scaffold N50: 1.97 and 1.88 Mb in Huang et al. [26]). For the MJ genome, 94.93% of the short reads were mapped onto the 99.98% assembled genome and, for the MP genome, 98.20% of the short reads were mapped onto the 99.96% assembled genome, indicating high reliability of both pangolin genome assemblies.
. Both of them are significantly improved compared with the published pangolin assemblies (genome size: 2.55 and 2.21 Gb, scaffold N50: 0.2 and 0.16 Mb in Choo et al. [16]; genome size: 2.85 and 2.91 Gb, scaffold N50: 1.97 and 1.88 Mb in Huang et al. [26]). For the MJ genome, 94.93% of the short reads were mapped onto the 99.98% assembled genome and, for the MP genome, 98.20% of the short reads were mapped onto the 99.96% assembled genome, indicating high reliability of both pangolin genome assemblies.
To annotate the genomes, we combined three approaches to predict genes including ab initio gene prediction, RNA-seq data and homology-based gene prediction. We identified 21 785 and 23 865 protein-coding genes in the Malayan and Chinese pangolin genomes, respectively. The numbers of identified protein-coding genes are similar to those in the previous study (23 446 and 20 298 for the two pangolins) [16]. Core Eukaryotic Genes Mapping Approach (CEGMA) [27] evaluation of MJ and MP genome assemblies indicated that 91.13% and 89.52% of 248 core eukaryotic genes are complete. However, in the published pangolin assemblies [16], only 58% and 55% of 248 core eukaryotic genes are complete in the CEGMA evaluation. Benchmarking Universal Single-Copy Orthologs (BUSCO) [28] assessment of the two new pangolin assemblies also showed that 94.10% and 94.50% of 4104 single-copy mammalian orthologous genes are complete (Supplementary Table 2). Thus, the new genome assemblies provide better coverage for protein-coding genes than the previous ones [16].
In summary, the two newly assembled pangolin genomes were both significantly improved compared with the previous pangolin genome assemblies [16,26] (Supplementary Table S2) and the high-quality pangolin reference genomes are not only important for the population genomic analyses of the pangolin, but also essential for mammalian study.
Generation of population genomic data and variant discovery
Individual genome re-sequencing of 74 MJs at an average 15.15-fold coverage (ranging between 12.12 and 21.16 for different individuals) and 23 MPs at an average of 38.64-fold coverage (ranging between 31.92 and 45.51 for different individuals) were generated for population genomic analyses (Supplementary Table 3). The average alignment rate to the reference genomes are 96.75% (88.71%–98.42%) and 98.80% (97.98%–99.44%) for Malayan and Chinese pangolins, respectively (Supplementary Table S3). The high alignment rate ensures the accurate identification of genetic variation. The single-nucleotide polymorphism (SNP) calling and filtering produced a total of 20.02 and 21.82 million high-quality autosomal SNPs for MJs and MPs, respectively.
Revealing population identity for illegally traded pangolins
We traced the origin locations of illegally traded pangolins in this study, which has been collected from different times and different places, via genetic clustering with pangolins of known origin. The most available molecular data of pangolins are mt DNA data. In this study, deep coverage of the mt genome allowed us to obtain the accurate mt sequences for all pangolin samples. The mt data of these Malayan pangolins were combined together with the publicly available mt data of Malayan pangolins from multiple Southeast Asian countries, including Indonesia (Borneo, Java and Sumatra), Singapore, Thailand and Malaysia [12,15,23]. The haplotype network analyses revealed two clusters of Malayan pangolins (Fig. 1a), named MJAmt and MJBmt populations. These two clusters were also distinguished by the nuclear genome tree of the Malayan pangolin, named MJA and MJB populations (Fig. 2a). The MJAmt and MJA populations contained the individuals sampled from the mainland (MJ09, MJ16 and MJ17 from Yunnan and MJ26 from Myanmar) that were divergent from other samples, corroborating the MJA’s non-overlapping geographic locality that stands for the mainland population. In comparison, the MJBmt and MJB populations contained the individuals sampled from the Southeast Asian islands, except for Java (Fig. 1a). The relatively deep splits among MJB lineages (MJB1–3; Fig. 2a) are also expected because of the isolation effect of islands.
Figure 1.

Haplotype network analyses based on the extracted mt-gene sequences of our samples and those of previously available samples with known origin. The newly defined MJAmt and MPBmt populations in this study are framed in a red circle. (a) The concatenated CYTB and COI gene sequences from 74 Malayan pangolin individuals (MJ01–74) in this study and those of 60 individuals in previous studies. Purple : Java [15], blue : Borneo [15], green : Singapore/Sumatra [15], black : Indonesia [15], orange : Thailand [12,23], pink : Malaysia (NCBI GenBank), yellow : MJAmt (this study; all are framed in a red circle; samples of known geographic locations are labeled with location names after the sample code names), red : MJBmt (this study; samples of known geographic locations are labeled with location names after the sample code names). (b) The COI gene sequences from 23 Chinese pangolin individuals (MP01–23) in this study and those of 10 individuals in previous studies [9,12]. (c) The CYTB gene sequences from 23 Chinese pangolin individuals (MP01–23) in this study and those of 15 individuals in previous studies [6,12]. Blue : published data; orange : MPAmt (this study), purple : MPBmt (this study; all are framed in a red circle). Samples of known geographic locations are labeled with location names after the sample code names.
Figure 2.

Phylogenetic analyses, PCA and admixture analyses based on nuclear genome sequences. (a) NJ phylogeny of 74 Malayan pangolins (MJ) (5 samples with known geographic sources; * Yunnan, 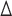 Myanmar, + Malaysia). ML phylogeny produced similar results and is shown as a small version to the right of the figure as a sub-figure. Two populations (MJA and MJB) and subpopulations within the two populations (MJA1-2 and MJB1-3) are labeled. MJA1 and MJA2 are also shown as a magnified version in the top-left of the figure. The values around the black point represent bootstrap support. (b) NJ phylogeny of 23 Chinese pangolins (MP) (9 samples with known geographic sources; * Yunnan, o Taiwan island). ML phylogeny produced similar results and is shown as a small version to the right of the figure as a sub-figure. Two populations (MPA and MPB) are labeled. The values around the black point represent bootstrap support. (c) Principal component analysis (PCA) for 74 MJ individuals. PCA for the MJA population is also shown. (d) PCA for 23 MP individuals. (e) Admixture analyses for 74 MJ individuals. The postulated number of ancestral clusters (K) was set from 2 to 5. (f) Admixture analyses for 23 MP individuals. The postulated number of ancestral clusters (K) was set from 2 to 5.
Myanmar, + Malaysia). ML phylogeny produced similar results and is shown as a small version to the right of the figure as a sub-figure. Two populations (MJA and MJB) and subpopulations within the two populations (MJA1-2 and MJB1-3) are labeled. MJA1 and MJA2 are also shown as a magnified version in the top-left of the figure. The values around the black point represent bootstrap support. (b) NJ phylogeny of 23 Chinese pangolins (MP) (9 samples with known geographic sources; * Yunnan, o Taiwan island). ML phylogeny produced similar results and is shown as a small version to the right of the figure as a sub-figure. Two populations (MPA and MPB) are labeled. The values around the black point represent bootstrap support. (c) Principal component analysis (PCA) for 74 MJ individuals. PCA for the MJA population is also shown. (d) PCA for 23 MP individuals. (e) Admixture analyses for 74 MJ individuals. The postulated number of ancestral clusters (K) was set from 2 to 5. (f) Admixture analyses for 23 MP individuals. The postulated number of ancestral clusters (K) was set from 2 to 5.
The results of principal component analysis (PCA) (Fig. 2c) and the admixture analysis (Fig. 2e) from the nuclear genomes recapitulated the phylogenetic and network analysis results in Malayan pangolins. The first PCA component (PC1) separates MJA and MJB (Tracy-Widom, P = 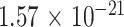 ) and the second component (PC2) divides MJB into subpopulations (MJB1–3; P = 7.28
) and the second component (PC2) divides MJB into subpopulations (MJB1–3; P = 7.28  10−4) (Fig. 2c). Moreover, the PCA also revealed two subpopulations within MJA (MJA1 and MJA2; P = 3.15
10−4) (Fig. 2c). Moreover, the PCA also revealed two subpopulations within MJA (MJA1 and MJA2; P = 3.15 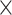 10−6) (Fig. 2c). The admixture analysis (Fig. 2e) showed the lowest cross-validation error when K = 2 (Supplementary Fig. 1) and supported two genetically distinct populations in the Malayan pangolins. When K = 5, MJs were separated into five clusters (Fig. 2e), corresponding to five subpopulations identified in the phylogenetic analyses and PCA.
10−6) (Fig. 2c). The admixture analysis (Fig. 2e) showed the lowest cross-validation error when K = 2 (Supplementary Fig. 1) and supported two genetically distinct populations in the Malayan pangolins. When K = 5, MJs were separated into five clusters (Fig. 2e), corresponding to five subpopulations identified in the phylogenetic analyses and PCA.
Based on mt DNA, the haplotype network analyses revealed two clusters of Chinese pangolins (Figs. 1b and c), named MPAmt and MPBmt populations. The MPAmt population was composed of the samples that originated from Yunnan, Guangdong, Hunan, Hainan, Taiwan in China and Thailand [6,9,12]. Interestingly, the MPBmt population appeared as an independent cluster, which is highly diverged from the MPAmt population and has short intra-population branches. All of the MPB individuals were seized from at least four illegal trades on the Sino–Burmese border between 2016 and 2017 (Supplementary Table 3). Therefore, it is very likely that MPB individuals originate from Myanmar. We suggest that the MPB population needs conservation attention for their unique evolutionary status. The same two populations (MPA and MPB) were inferred from the nuclear genome tree (Fig. 2b). Similarly to the network inferred from the mt DNA, the intra-population branches of the MPB population in the nuclear genome tree are short. Interestingly, within the MPA population, the individual from Taiwan [17] (number MP07; the representative of the Taiwan subspecies M. pentadactyla pentadactyla in the previous study [16], referred to hereafter as MPP) demonstrated a significantly long branch compared with the other MPA individuals (Wilcox-test; P < 0.05 in neighbor-joining (NJ) and maximum-likelihood (ML) trees). In the PCA results, PC1 separated MPA and MPB (P = 3.46 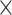 10−4) (Fig. 2d). PC2 divided Taiwan pangolins from other MPA pangolins, although it was marginally significant (P = 0.055). The admixture analysis (Fig. 2e) showed the lowest cross-validation error when K = 2 (Supplementary Fig. 1) and supported two genetically distinct populations in the Chinese pangolins. For the Taiwan pangolin individual, it showed an MPA genetic component from K = 2 to 4 (Fig. 2f). At K = 5, a component specific to the Taiwan individual then appeared.
10−4) (Fig. 2d). PC2 divided Taiwan pangolins from other MPA pangolins, although it was marginally significant (P = 0.055). The admixture analysis (Fig. 2e) showed the lowest cross-validation error when K = 2 (Supplementary Fig. 1) and supported two genetically distinct populations in the Chinese pangolins. For the Taiwan pangolin individual, it showed an MPA genetic component from K = 2 to 4 (Fig. 2f). At K = 5, a component specific to the Taiwan individual then appeared.
Intriguingly, for both pangolin species, some individuals demonstrated the signal of admixture between two populations at K = 2 (Fig. 2e and f). Particularly, all individuals of the MJB3 subgroup were primarily formed by admixture of the ancestral MJA and MJB populations (Fig. 2e). The admixture between the populations within MJs and MPs was also reflected by the finding that these admixed individuals varied between the mt haplotype network and the nuclear genome tree due to migration (see Figs 1 and 2).
As a summary, we combined the mt and nuclear genome data together to trace the population identity of the illegally traded pangolins. MJA pangolins are continentally distributed, while MJB pangolins are from the Southeast Asian islands, except
Java. The MPA pangolins are composed of the samples that originated from southern China and Thailand, while the MPB pangolins are most likely to originate from Myanmar.
Genetic diversity of pangolins
The mean heterozygosity (He) of MP (0.127%) is significantly higher than that of MJ (0.085%) (Wilcox-test, P = 4.79 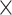 10−3; Fig. 3a). Among the four main populations, the mean heterozygosity of MJA (
10−3; Fig. 3a). Among the four main populations, the mean heterozygosity of MJA ( 0.043%) is the lowest compared with the other three main populations (MJB, MPA, MPB) (
0.043%) is the lowest compared with the other three main populations (MJB, MPA, MPB) ( 0.141%, 0.130% and 0.123%, respectively; P = 2.34
0.141%, 0.130% and 0.123%, respectively; P = 2.34 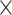 10−13, 8.20
10−13, 8.20 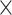 10−7 and 3.18
10−7 and 3.18 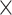 10−6; Fig. 3a). These observations suggest that the significant difference in
10−6; Fig. 3a). These observations suggest that the significant difference in 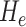 between MJ and MP is mainly due to the severely reduced genetic diversity of MJA.
between MJ and MP is mainly due to the severely reduced genetic diversity of MJA.
Figure 3.
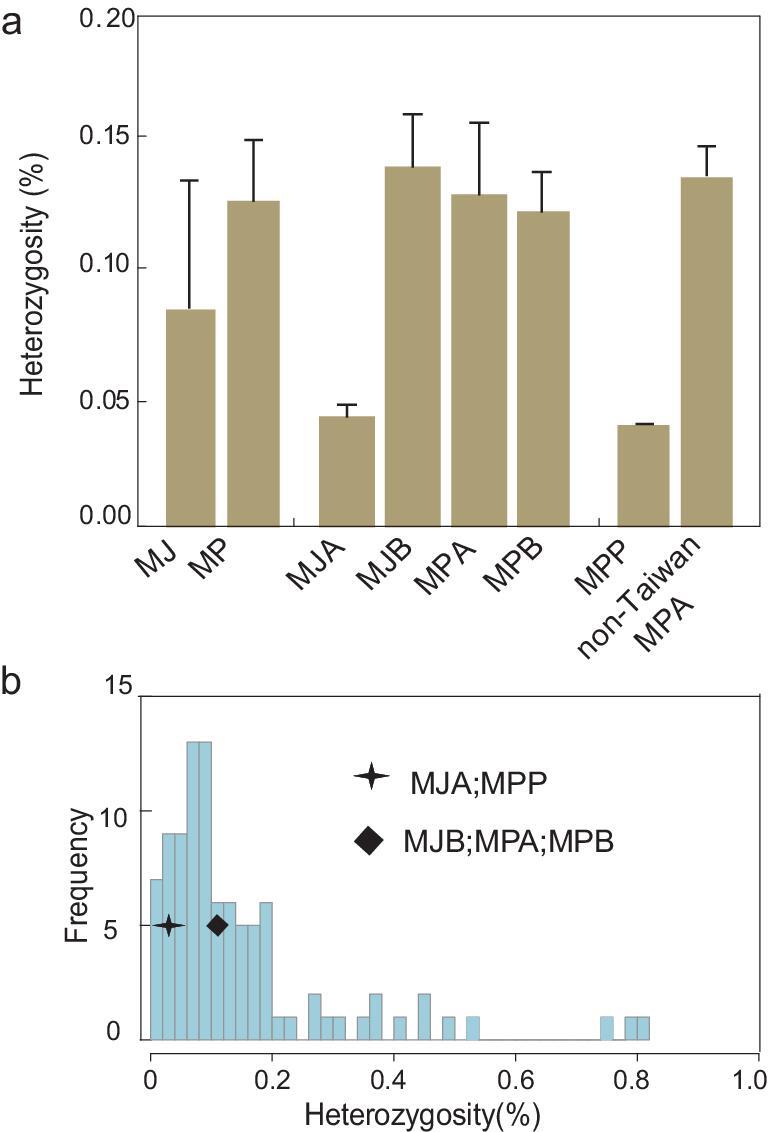
Genetic diversity of pangolins. Error bars represent SD. (a) Heterozygosity of MJ, MP, MJA, MJB, MPA, MPB, MPP and non-Taiwan MPA. (b) The frequency of the distribution of the genome-wide heterozygosity in 94 published mammals (Supplementary Table 4) and pangolins.
The 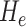 of the Taiwan pangolin (MPP) (0.04%) is notably lower than the
of the Taiwan pangolin (MPP) (0.04%) is notably lower than the 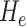 of the other MPA pangolins (0.125%–0.162%; P =
of the other MPA pangolins (0.125%–0.162%; P =  ), MPB pangolins (0.107%–0.155%; P =
), MPB pangolins (0.107%–0.155%; P = 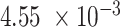 ) and MJB pangolins (0.11%–0.17%; P =
) and MJB pangolins (0.11%–0.17%; P = 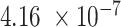 ), but similar to the extremely low
), but similar to the extremely low 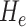 of MJA pangolins (0.036%–0.062%) as described above (Fig. 3a). The low genetic diversity in MPP is most likely due to the small founding population on the island. Moreover, both phylogenetic and PCA analyses (Fig. 2b and d) indicate that MPP is indeed a differentiated and isolated population.
of MJA pangolins (0.036%–0.062%) as described above (Fig. 3a). The low genetic diversity in MPP is most likely due to the small founding population on the island. Moreover, both phylogenetic and PCA analyses (Fig. 2b and d) indicate that MPP is indeed a differentiated and isolated population.
Next, we compared the 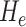 of pangolins with that of other species (Fig. 3b and Supplementary Table 4). The
of pangolins with that of other species (Fig. 3b and Supplementary Table 4). The 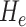 of MJA is similar to those of the endangered golden snub-nosed monkeys (0.042%) [29] and carnivores, including the Bengal tiger, White lion, Amur tiger and San Nicolis Island fox (0.040%–0.049%) [30–32], whereas those of MJB, MPA and MPB were comparable to those in the endangered Siamang (0.13%–0.15%) [33], giant panda (0.132%) [34], critically endangered Sumatran orangutan (0.12%) [35], Sumatran Rhinoceros (0.13%) [36], Western lowland gorilla (0.144%) [37] and the extinct Oimyakon woolly mammoth (0.125%) [38]. Such low level of genetic diversity
of MJA is similar to those of the endangered golden snub-nosed monkeys (0.042%) [29] and carnivores, including the Bengal tiger, White lion, Amur tiger and San Nicolis Island fox (0.040%–0.049%) [30–32], whereas those of MJB, MPA and MPB were comparable to those in the endangered Siamang (0.13%–0.15%) [33], giant panda (0.132%) [34], critically endangered Sumatran orangutan (0.12%) [35], Sumatran Rhinoceros (0.13%) [36], Western lowland gorilla (0.144%) [37] and the extinct Oimyakon woolly mammoth (0.125%) [38]. Such low level of genetic diversity
suggests that pangolins likely have decreased adaptive potential.
Demographic history of pangolins
The ancient demography of pangolins is very important for understanding the driving factors, i.e. the environmental changes, behind the population fluctuation of pangolins in the past. The joint site-frequency spectrum (joint SFS) is one of the optimal summary statistics for addressing these issues and the parameters can be reliably inferred by coalescent-based simulations [39]. Thus, we assessed various demographic scenarios of MJ and MP species by coalescent-based simulations (Supplementary Figs 2 and 3 and Supplementary Tables 5–7). Based on the best-fitting model, the parameters, including divergence time, current effective population size and migration rate, were estimated (Supplementary Tables 8–10). All these pangolin populations were subject to multiple population size changes in their evolutionary history (Fig. 4).
Figure 4.

Demographic histories of MJ (a) and MP (b) inferred from the demographic model simulations. The arrows indicate migration events and those with stronger migration events in pairwise comparisons are indicated with bold arrows ((a) and (b)). The blue shadows represent the marine isotope stage (MIS) during the Pleistocene [40] (a). The red and orange shadows represent the last interglacial period and last glacial period (LGP) during the Pleistocene, respectively (a). The green shadows represent the Holocene era ((a) and (b)). The surface temperature (temp.) over the past 1 million years based on δ18O data [42] is indicated (b).
Two Malayan-pangolin populations (MJA and MJB) split 300 thousand years ago (Kya), corresponding to the beginning of marine isotope stage (MIS) 8 [40]—a period during which the sea level fell precipitously, potentially resulting in land-habitat expansion and climate changes, and contributing to the population differentiation. Interestingly, the MJA population showed a small population size fluctuation until a severe population size decline at 3.2 Kya (from about 106 000 to 2000 individuals; Supplementary Table 8). The MJB population mainly lives on islands and demonstrates numerous changes in population size, potentially as a result of sea-level changes (Fig. 4a). The subsequent MIS periods associated with alternating episodes of sea-level fall (i.e. MIS8, MIS6 and MIS2) and rise (i.e. MIS7 and MIS5e) [40–42] partially explained the population expansions and bottlenecks (Fig. 4a), respectively. The close conjunction of population size fluctuation and the sea-level change accords with known and potential locality sources of MJB samples from Southeast Asian islands (Fig. 1a).
Two Chinese pangolin populations (MPA and MPB) split at 130 Kya when the last interglacial period began [43] and the most recent and intense uplift event of the Tibetan plateau occurred (i.e. the Gonghe movement) [44]. These two factors were known to have led to the speciation of a large number of organisms in southern China [45]. Therefore, this indicates that the climate warming and the Tibetan-plateau uplift may also have contributed to the split of MP populations that mainly live in southern China [12,16,18]. After the split, MPA and MPB experienced an abrupt population decline around the last glacial period (LGP, ∼10–120 Kya) (70% reduction for MPA, from about 2300 to 12 000 individuals; corresponding to PA1 in Fig. 4b) or the last glacial maximum (LGM; ∼25–13 Kya [46]; 97% reduction for MPB, from about 110 000 to 2900 individuals; corresponding to PB2 in Fig. 4b) (Supplementary Table 8). Therefore, the impacts of LGP and LGM on the population-size decline were revealed for MP species. The similar phenomenon was also observed in the giant panda [47] and the golden snub-nosed monkeys [29,48], which geographically largely overlap with MP species.
As a sub-species of Chinese pangolins, the Taiwan pangolin is of specific interest as an island-endemic pangolin. Based on phylogenetic and admixture analysis, the Taiwan pangolin was phylogenetically mixed with other MPA individuals and showed 100% MPA ancestry component (Fig. 2b and f), indicating that the Taiwan pangolin originated from the mainland counterpart MPA. Divergence time estimation (Fig. 5a) and demographic history reconstruction (Fig. 5b) showed that the Taiwan pangolin began to separate from its ancestral MPA group about 10 Kya, consistently with the fossil records documented for the immigration of numerous mammals, including the elephant and rhinoceros, from the mainland to Taiwan through a land bridge during the late Pleistocene (26–11 Kya) [49,50]. Afterwards, the Taiwan pangolin population went through a continuous and steep population decline.
Figure 5.

Demographic history of the Taiwan pangolin. (a) Divergence time of the Taiwan pangolin (MP07) from its ancestral MPA group (MP01–MP14). (b) Population size dynamics of the Taiwan pangolin (MP07) and non-Taiwan MPA. Two non-Taiwan MPA individuals (MP05 and MP08) that have the highest sequence coverage were used to represent the non-Taiwan MPA populations.
Effects of recent population decline on the inbreeding level and genetic load
The recent population decline due to human activities has been documented widely in pangolins [1,5,11,19]. We then studied the effects of the recent population decline on inbreeding levels by using the runs of homozygosity (ROH). A long ROH is an indicator of inbreeding arising within tens of generations [51,52], as reported in wolfs [51] and gorillas [52]. The long ROHs between MJ and MP indicate that the recent inbreeding occurred in pangolins <20 generations/years ago (Supplementary Fig. 4).
Additionally, MJ pangolins harbored more ROHs >100 kb (974.26 Mb in total, 42.23% of the genome) than MP genomes (294.31 Mb in total, 11.99% of the genome) (Fig. 6a) and had significantly more long ROHs above 1 Mb (19.54 Mb in total, 8.8% of the genome) than those of MPs (3.54 Mb in total, 1.4% of the genome) (Wilcox-test, P = 1.34  10–8). Further comparisons among populations showed that MJA had more ROHs >100 kb (1559 Mb in total, 62.29% of the genome) than the other three populations (MJB, 390 Mb in total, 15.9% of the genome; MPA, 320 Mb in total, 12.79% of the genome; MPB, 269 Mb in total, 10.76% of the genome; Fig. 6a). MJA also had 8- to 12-fold more long ROHs above 1 Mb (34.91 Mb in total, 14.25% of the genome) than the other three populations (4.17, 2.89 and 4.18 Mb in total; 1.59%, 1.16% and 1.67% of the genome for MJB, MPA and MPB, respectively). In addition, consistently with the ROH results above, we found a longer extent of linkage disequilibrium (LD) in MJA than in the other three populations (Fig. 6b), which showed that LD decayed to r2 < 0.2 within 10 kb in MJB, MPA and MPB, but declined much more slowly for MJA (>300 kb). Overall, these results suggested the substantial increase of recent inbreeding in the MJ species, especially in the MJA population, possibly due to the recent human impact in recent decades.
10–8). Further comparisons among populations showed that MJA had more ROHs >100 kb (1559 Mb in total, 62.29% of the genome) than the other three populations (MJB, 390 Mb in total, 15.9% of the genome; MPA, 320 Mb in total, 12.79% of the genome; MPB, 269 Mb in total, 10.76% of the genome; Fig. 6a). MJA also had 8- to 12-fold more long ROHs above 1 Mb (34.91 Mb in total, 14.25% of the genome) than the other three populations (4.17, 2.89 and 4.18 Mb in total; 1.59%, 1.16% and 1.67% of the genome for MJB, MPA and MPB, respectively). In addition, consistently with the ROH results above, we found a longer extent of linkage disequilibrium (LD) in MJA than in the other three populations (Fig. 6b), which showed that LD decayed to r2 < 0.2 within 10 kb in MJB, MPA and MPB, but declined much more slowly for MJA (>300 kb). Overall, these results suggested the substantial increase of recent inbreeding in the MJ species, especially in the MJA population, possibly due to the recent human impact in recent decades.
Figure 6.
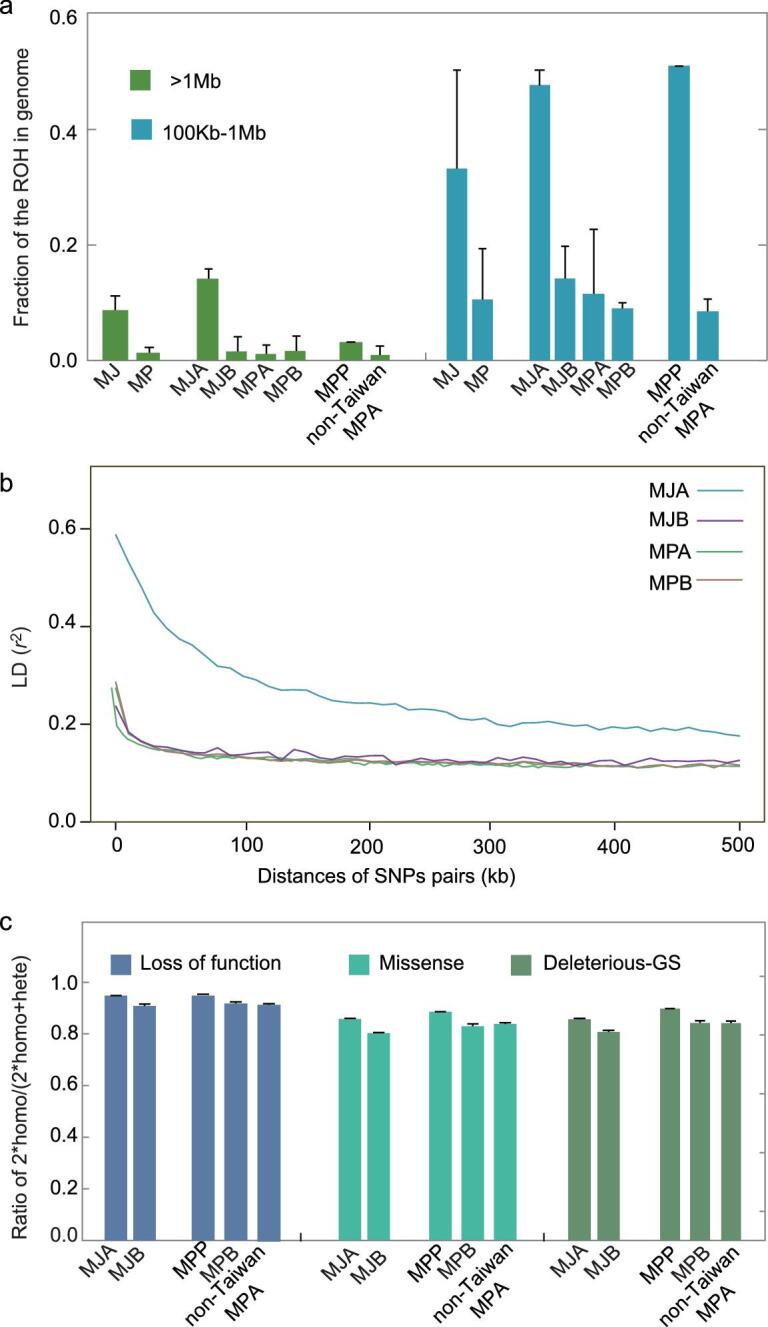
Inbreeding levels and genetic load of pangolins. Error bars represent SD. (a) Fraction of the runs of homozygosity (ROH) in the genome, including the medium (100 kb–1 Mb) and long (>1 Mb) ROH, of MJ, MP, MJA, MJB, MPA, MPB, MPP and non-Taiwan MPA. (b) Linkage disequilibrium (LD) of MJA, MJB, MPA and MPB. (c) Genetic load of MJA, MJB, MPP, non-Taiwan MPA and MPB.
We also assessed the inbreeding extent in the Taiwan pangolin. ROH analyses showed that the Taiwan pangolin population had more ROHs >100 kb (1366 Mb in total, 54.65% of the genome) than its mainland counterpart MPA (239 Mb in total, 9.57% of the genome) and MPB populations (269 Mb in total, 10.76% of the genome) (Fig. 6a). It also had more long ROHs above 1 Mb (8.04 Mb in total, 3.22% of the genome) than the latter two (2.49 Mb in total, 1.00% of the genome; 4.18 Mb in total, 1.67% of the genome). These results demonstrated that the Taiwan population also experienced an increase in recent inbreeding.
Next, we assessed the genetic load in the MJA and MPP populations by identifying putative deleterious loss-of-function (LOF) and missense variants (Fig. 6c). The mutation of homozygous-derived alleles under evolutionary constraints is measured as an indicator of genetic load and predicted as approximate fitness consequences [37,52–54]. The results showed that the ratio of the number of homozygous sites to the homozygous and heterozygous sites in the MJA population was significantly higher than in the MJB population (0.948 vs. 0.908 for LOF; 0.855 vs. 0.800 for missense mutations; Wilcox-test, P < 0.001) (Fig. 6c). A similar difference was observed when comparing the Taiwan pangolin with other MPA individuals (0.947 vs. 0.917 for LOF; 0.883 vs. 0.838 for missense mutations; P < 0.001) and the MPB population (0.947 vs. 0.917 for LOF; 0.883 vs. 0.827 for missense mutations; P < 0.001) (Fig. 6c). In addition, the derived deleterious missense variants were predicted by Grantham Score (≥150) [55] that measured the physical and chemical properties of amino-acid changes. We also found an excess of homozygous deleterious-derived mutations in MJA and MPP (Fig. 6c). Therefore, the genetic load varied among populations, with a significantly higher genetic load in the MJA and MPP populations than in other populations. Interestingly, we found that the number of genes with homozygous deleterious mutations in ROHs was significantly higher in the MJA population than those in the MJB population (P < 0.001; Fig. 7). A similar difference was observed when comparing the Taiwan pangolin with other MPA individuals and the MPB population (P < 0.01; Fig. 7). Therefore, the increased genetic load was likely due to the inbreeding. Although MPP was the only Chinese pangolin population that has increased over the past two decades as a result of successful conservation efforts [56], our result indicated that the genetic diversity in this population was very low and the risk of inbreeding will need to be addressed in the future. We suggest that future conservation efforts could consider the possibility of rescuing this population by introducing mainland pangolins (e.g. from the MPA population) to increase the genetic diversity and adaptive potential of Taiwan pangolins.
Figure 7.
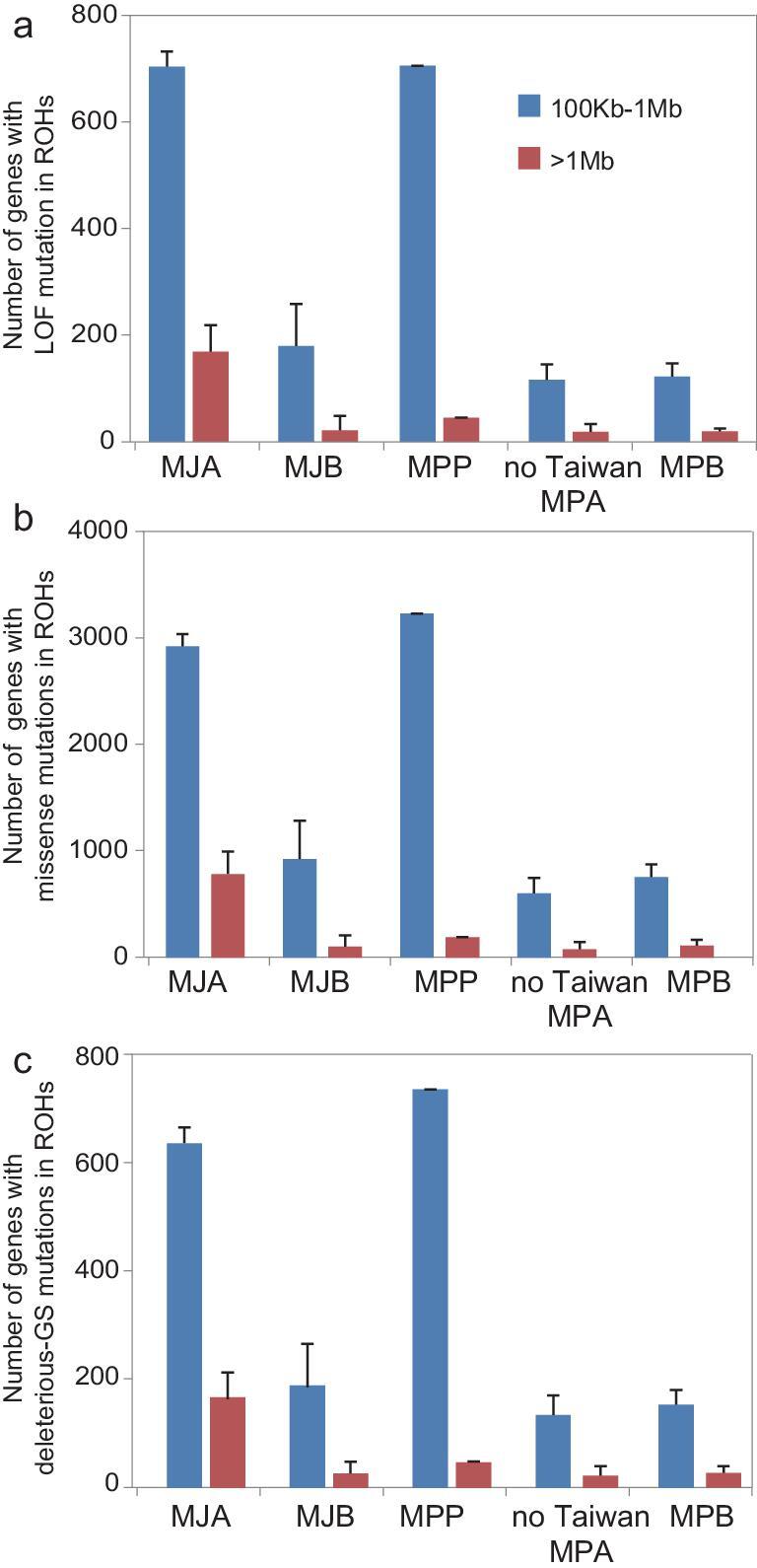
The number of genes with homozygous deleterious mutations in ROHs, including LOF mutations (a), missense mutations (b) and deleterious-GS mutations (c), of MJA, MJB, MPP, non-Taiwan MPA and MPB. Error bars represent SD.
We analyzed the genes affected by the homozygous putative deleterious LOF mutations [52] in MJA and MPP, and found that a number of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) categories were involved in processes relating to cancer, disease and metabolism (Supplementary Table 11). For example, LOF mutations were found to enrich in the cholesterol homeostasis (Gene Ontology term, GO0042632) in MJA (Supplementary Table 11). The insufficient or excessive cholesterol has serious cellular consequences and leads to metabolic syndrome and cardiovascular diseases [57]. Pangolins live on a unique diet of high-cholesterol ants and termites [17,24,25]. The LOF mutations in genes related to cholesterol homeostasis may confer increased susceptibility to diseases due to abnormal cholesterol levels, which could be caused by a decrease in the utilization of food, leading to malnutrition and loss of competitiveness in the community.
CONCLUSION
In this study, we recovered the population identities of illegally traded pangolins and revealed the previously unrecognized genetic lineages, including a Malayan pangolin population that might be continentally distributed (MJA in Fig. 2) and a Chinese pangolin population that might originate from Myanmar (MPB in Fig. 2) that should be protected and managed as evolutionarily distinct conservation units. The ancient demographic history analyses showed the effects of long-term environmental changes, including climate and sea-level changes. The ROHs and genetic load analyses demonstrated the increased inbreeding and genetic load effect of recent population decline due to recent human activities. Considering the short generation time and the small population size of pangolins, it would be interesting to investigate museum pangolin specimens, which could further quantify the impacts of human activities. In the future, it would be essential to establish population-specific genetic fingerprints of pangolins across the distribution of pangolins. The population-specific genetic fingerprints would be very useful to trace the origin of illegally traded pangolins, which will ultimately support long-term pangolin conservation efforts.
A note added after acceptance
By coincidence, the acceptance of this study coincides with the COVID-19 outbreak in China. It has been widely reported that the causative coronavirus (SARS-CoV-2) may have been passed from the primary host (bats) to humans via pangolins as the secondary host. We have recently obtained the RNA-seq data from several tissues of a Malayan pangolin individual and made them available to the public (deposited under project in NCBI: PRJNA531293/NGDC: PRJCA002221). Our preliminary analysis of the five tissues (large intestine, small intestine, stomach, lung and cerebrum) of this pangolin individual did not show any evidence of the SARS-CoV-2 virus. While a comprehensive survey will be carried out by adding more tissues from more pangolin individuals (as well as other wild animals), this preliminary analysis suggests that SARS-CoV-2 viruses should not be expected to exist in all pangolins, if they indeed do.
METHODS
Sample collection
All necessary research permits and ethics approvals were granted in this study. In total, 73 Malayan pangolin samples between 2000 and 2017 and 22 Chinese pangolin samples between 1990 and 2017 were collected by the Animal Branch of the Germplasm Bank of Wild Species of Chinese Academy of Sciences, Guangzhou Wildlife Rescue Center and Nujiang Forest Public Security Bureau. Eleven pangolins were sampled from Yunnan Province, China and one from Kachin, Myanmar. Others were from seizures of illegal wildlife trade with unknown sampling locations. Detailed sample information is provided in Supplementary Table 3.
In addition, the short-read library reads from a Malayan pangolin individual (from Malaysia) and a Chinese pangolin individual (from Taiwan) [16] were included in our analyses.
Species identification
Genomic DNA was extracted from the collected specimens using a QIAamp DNA Tissure Mini Kit (Qiagen, USA). Pangolin species identification of the specimens was confirmed by using a 658-bp region of mt cytochrome c oxidase subunit I (COI) gene with two pairs of mixed primer sets LepF1/LepR1 and VF1d/VR1d [58]. Polymerase chain reaction (PCR) amplification conditions were initial denaturation (98°C) for 2 min, followed by 35 cycles of denaturation (10 s at 98°C), annealing (15 s at 58°C) and extension (1 min at 72°C) and a final extension at 72°C (5 min). The PCR products were sequenced using LepF1 and LepR1. The generated COI sequences were checked carefully and queried in BLAST searches of GenBank to identify the pangolin species. COI sequences for species identification have been deposited in GenBank with accession numbers MK810451–MK810545.
De novo genome sequencing and assembly
DNA samples from a Malayan pangolin (MJ74) and a Chinese pangolin (MP20) that was confiscated in Yunnan Province were used for de novo sequencing (Supplementary Tables 1–3). A whole-genome shotgun sequencing strategy was applied. Eleven libraries consisting of 4 short paired-end inserts (250 and 500 bp) and 8 long mate-paired inserts (2k∼15k) for a Chinese pangolin and 36 libraries consisting of 4 short paired-end inserts (220 and 500 bp) and 32 mate-paired inserts (3k∼18k) for a Malayan pangolin were constructed (Supplementary Table 1). In addition, 10X Genomes libraries for both pangolins were constructed (Supplementary Table 1). All libraries were sequenced on the Illumina HiSeq platform.
All the Illumina paired-end reads were trimmed for adaptor sequence and the reads containing >10% ambiguous nucleotides and >20% low-quality nucleotides (Q ≤ 5) were discarded. The average sequence error rate for each base was 0.04% (see Supplementary Table 1). The filtered Illumina paired-end reads were assembled into scaffolds using ALLpaths-LG [59] and the gaps were then filled using GapCloser v.1.12 [60]. Finally, the 10X linked-reads were used to link the scaffolds using fragScaff [61].
To assess the quality of the genome assembly, the short insert library reads were mapped onto the assembled genome using a Burrows-Wheeler Aligner (BWA) v.0.7.12 [62] to calculate the mapping ratio and assess the assembly integrity. In addition, CEGMA [27] and BUSCO [28] pipelines were used to validate the completeness of the assembly. Two genome assemblies have been deposited in NCBI under projects PRJNA529512 and PRJNA529513.
Repeat identification, gene prediction and annotation
Repeats of the Malayan- and Chinese-pangolin genomes were identified using RepeatMsker [63], the Repbase library [63] and RepeatProteinMask [63]. The de novo repeat annotation was performed using LTR_FINDER [64], RepeatScout [65], RepeatModeler [66] and TRF [67].
Using the repeat-masked genome, we predicted genes by combining three approaches. First, AUGUSTUS [68], Glimmer-HMM [69], SNAP [70], Geneid [71] and Genscan [72] were used for ab initio prediction. Second, we assembled the RNA-seq reads from the tissues of a Malayan pangolin individual into transcripts using Tophat [73] and Cufflinks [74] for gene-structure annotation. Thirteen tissues (large intestine, small intestine, stomach, liver, pancreas, lung, ovary, cerebrum, tongue, kidney, heart, eyes and muscle) of the Malayan pangolin, which unfortunately died in the process of rescue, were obtained from Guangzhou Wildlife Rescue Center. Total RNA was extracted from each tissue using the TRIzol kit (Life Technologies). Libraries were constructed and sequenced according to the Illumina protocol and the average size of sequencing raw data was 6.98 Gb (deposited under project in NCBI: PRJNA531293/NGDC: PRJCA002221). Third, we used BLAST (E-value ≤ ) [75] and Genewise [76] to align the protein sequences from Homo sapiens, Mus musculus, Canis familiaris, Felis catus, Capra hircus, Equus caballus and Myotis brandtii to the Malayan pangolin and Chinese pangolin genomes for homology-based gene prediction. The gene sets predicted by the three approaches were integrated using EvidenceModeler [77] to produce consensus gene models, which were then updated by PASA [78].
) [75] and Genewise [76] to align the protein sequences from Homo sapiens, Mus musculus, Canis familiaris, Felis catus, Capra hircus, Equus caballus and Myotis brandtii to the Malayan pangolin and Chinese pangolin genomes for homology-based gene prediction. The gene sets predicted by the three approaches were integrated using EvidenceModeler [77] to produce consensus gene models, which were then updated by PASA [78].
Genome re-sequencing of Malayan and Chinese pangolin populations
Libraries with an insert size of 500 bp were constructed to re-sequence Malayan and Chinese pangolin individuals according to the Illumina protocol. All libraries were sequenced on the Illumina HiSeq platform. All the Illumina paired-end reads were trimmed for adaptor sequence and the reads containing >10% ambiguous nucleotides and >20% low-quality nucleotides (Q ≤ 5) were discarded; 95.79% of the nucleotide sites in the filtered reads were of high quality, with a phred score 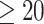 , and 91.73% of the sites had a phred score
, and 91.73% of the sites had a phred score 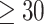 . The average sequence error rate for each base was 0.03% (see Supplementary Table 3).
. The average sequence error rate for each base was 0.03% (see Supplementary Table 3).
In addition, the short-read library reads were also collected from a previously published de novo Malayan pangolin individual (from Malaysia; NCBI, SRA, SRR3949728) and a Chinese pangolin individual (from Taiwan; NCBI, SRA, SRR770330-SRR770587) [16] as well as those from the two newly sequenced de novo Malayan and Chinese pangolin individuals in this study. In total, 74 Malayan and 23 Chinese pangolins were used for population genomic analyses (Supplementary Table 3). Re-sequencing data are available at NCBI Read Archive under project PRJNA529540.
Re-sequencing data processing and SNP calling
We used BWA v.0.7.12 [62] to align the trimmed paired-end reads from the Malayan pangolin individual and the Chinese pangolin individual to our de novo assembled Malayan and Chinese pangolin reference genomes, respectively. Binary sequence alignment files were generated using SAMtools v.1.3 [79]. PCR duplicates were removed using PICARD (http://picard.sourceforge.net). Insert/InDel (insert and deletions) realignment was performed using the IndelRealigner algorithm implemented in the Genome Analysis Toolkit (GATK) v.3.5.0 [80].
Before conducting the SNP-calling analyses, the candidate sex-chromosome scaffolds from the assembled genomes that show >60% sequence similarity with sex chromosomes of Canis lupus familiaris (Chr X from CanFam3.1 [81]; Chr Y from KP081776.1 [82]) and those of Homo sapiens (GRCh38.p12) [83] were excluded by LASTZ (http://www.bx.psu.edu/∼rsharris/lastz/). The autosomal scaffolds were used in the subsequent analyses. In addition, the scaffolds with length <100 kb were excluded.
We identified autosomal SNPs using SAMtools mpileup [79]. SNPs with an allele frequency <20% of each individual and with a depth distribution of all sites <2.5% or >97.5% were filtered using a custom script to obtain high-quality SNPs. Moreover, low-quality SNPs were filtered out when the base-quality and mapping-quality score was <20.
Haplotype network analyses based on mt DNA
More than 1 million pangolins have been poached in recent years. It is important to trace the original locations of illegally traded pangolins. The most available molecular data of pangolins are from mt DNA. Previous studies have reported concatenated mt COI and CYTB analyses (1575 bp) of Malayan pangolins with known and assigned geographic sources from Southeast Asia [12,15,23], so we extracted the COI and CYTB sequences from our genomic sequences. The COI and CYTB sequences of Malayan pangolins in this study were determined using BWA [62] by mapping the short reads to the reference mt sequences of Malayan pangolins (GenBank accession number KT445979). The consensus COI and CYTB sequences between the mapped short reads and the reference mt sequence were extracted using SAMtools [79] and an in-house Perl script. The concatenated COI and CYTB sequences of 74 Malayan pangolins in this study and those of another 60 individuals in previous studies [12,15,23] were generated.
The same strategy was also applied to Chinese pangolins. We downloaded the sequences of the COI gene (600 bp) from 10 individuals sampled from Guangdong, Hunan, Hainan and Taiwan in China and Thailand [6,9,12] and the CYTB gene (782 bp) from 15 individuals sampled from Taiwan in China and Thailand [6,12]. The reference mt sequence (GenBank accession number MG196307) was used. In total, the COI data set from 33 Chinese pangolins and the CYTB data set from 38 Chinese pangolins were obtained.
The resulting data sets were aligned using ClustalW [84] and the ambiguous sites were removed using Gblocks (gap = all) [85]. We generated a haplotype network using the Median-Joining method in PopART [86].
Phylogenetic tree reconstruction based on nuclear genomes
To build a genome tree based on autosomal SNPs data sets, we first converted VCF files to FASTA files and then constructed an NJ tree using MEGA X [87] with the Kimura's two-parameter model. To validate the converting process, we also built the genome NJ tree from the VCF file using the eGPS software [88] with the same substitution model. We also constructed an ML tree using SNPhylo [89]. One thousand bootstraps were performed for both trees. SNPhylo is a newly developed pipeline for constructing phylogenetic trees based on large SNP data sets and has steps for removing low-quality data and considering LD to increase the reliability of a tree. We first used SNPhylo with parameter -p 25 to generate sequences from the extracted representative SNPs and then reconstructed an ML tree by running the RAxML v.8.2.4 program [90] with the GTRGAMMA model.
Population structure and admixture analyses
The autosomal SNPs were thinned by LD values using PLINK v.1.1 [91], resulting in a set of ∼1.13- and ∼0.42-Mb SNPs for the population-structure and admixture analyses of MJ and MP, respectively. PCA was carried out using the smartPCA program from the Eigensoft package [92] and the Tracy-Widom test. Admixture analysis was performed using Frappe v.1.1 [93]. The postulated number of ancestral clusters (K) was set from 2 to 5 and the maximum number of expectation-maximization iterations was set to 10 000. The best K value was identified using the cross-validation procedure by ADMIXTURE [94].
Demographic history analyses
We used the maximum-composite-likelihood approach based on the joint SFS implemented in fastsimcoal2 [95] to assess the fit of various demographic models and to infer the final optimal demographic scenario of the Malayan and Chinese pangolins. The scripts for the demographic modeling are available upon the request.
First, the joint SFS of different populations was inferred by assuming the allele state of the out-group as the ancestral allele state and removing the heterozygote allele sites in the out-group genome. The two newly sequenced de novo Malayan and Chinese pangolin genomes were used as the out-group genomes for each other's data sets. A total of 27 735 083 SNPs and 36 739 658 SNPs were used for the Malayan pangolin data set and Chinese pangolin data set analyses, respectively. The mutation rate per generation was set as 1.47 × 10−8 [16] and the generation time as 1 year [96,97].
In each parameter-estimation procedure, 100 000 coalescent simulations and ≥20 expectation-conditional maximization cycles, up to a maximum of 40, were used. To get a reliable global maximum estimation for each scenario and avoid a local maximum, we ran between 200 and 300 replicates. The AIC [98] was used to compare the fit of different models. The preferred point estimations of all parameters, including the migration rate, divergence time and effective population size, were chosen by the ML method and their confidence interval (CI) was calculated by simulating 100 independent site-frequency spectra conditional on the preferred demographic scenario. Re-estimation procedures were implemented for each simulated data set at least twice. We further validated our results using the SFS analyses (Supplementary Fig. 2) and the genetic-diversity comparison between the simulation and observed data set (Supplementary Fig. 3 and Supplementary Table 10).
In addition, we also reconstructed the demographic history of the Taiwan pangolin, which is of specific interest in this study as an island-endemic pangolin compared to their continental relatives among the Chinese pangolins. We used the multiple-sequential Markovian coalescent model [99] based on the phased haplotypes to estimate the divergence time and demographic history of the Taiwan pangolin. The haplotypes were phased using BEAGLE v.5.0 [100]. The mutation rate (μ) and the generation time were set as the same as those described above.
Genetic diversity, ROH and LD analyses
The heterozygosity, which was used to assess the genetic diversity, was calculated based on autosomal SNPs with a 50-kb non-overlapping sliding-window size using VCFtools [101]. To test whether the resulting higher heterozygosity was caused by the higher sequence coverage, we subsampled the sequence reads from the MP de novo genome to the mean depth in MJ (15 X) and found a higher heterozygosity of MP (0.128%) than that of MJ (0.085%). Therefore, the higher heterozygosity of MP was not caused by the higher sequence coverage of MP.
PLINK [91] was used to identify the ROH in each pangolin following the method described before [31]. Only filtered ROHs that are >10 kb and contain <20 SNPs were taken into consideration. The extreme 5% ROH differences between MJ and MP were longer than 2.5 Mb (Supplementary Fig. 4). Using the physical length of the ROH as approximation for the genetic length [52], the ROH of a 2.5-Mb trace (g = 100/2 * ROHlength, where g is the number of generations and ROHlength is the ROH length in centimorgans) was estimated to occur <20 generations ago.
The LD was calculated using Haploview [102]. We calculated the r square statistic (r2), which is the correlation coefficient between two focal loci of interest.
Deleterious mutation patterns
Deleterious mutations are expected to disrupt gene function or reduce an individual's viability [32]. We used SnpEff v.4.3t [103] to categorize the derived allele mutations of the coding regions of each individual into missense and LOF mutations. Similarly to Feng et al.’s strategy [53], the genotypes of major homozygous alleles in each pangolin species (>50% of the individuals) and also the same homozygous alleles in the other pangolin species (as the out-group) were used to represent the ancestral state. We built the databases from the annotations and the reference-genome sequences of the two pangolin species. The input files in VCF format were used to annotate SNPs and we assigned a mutation category to the input SNPs per individual. LOF mutations included premature stop codons (nonsense) and splice-site-disrupting single-nucleotide variants. The ratio of the number of homozygous sites (two per site) to both homozygous and heterozygous sites (two per homozygous site and one per heterozygous site) for each category was evaluated for estimating the deleterious load [32,53].
The deleteriousness of the missense mutations was also diagnosed using the Grantham Score (GS) [55]—a measure of the physical/chemical properties of amino-acid changes. Grantham scores ≥150 were designated as deleterious [104].
For all identified homozygous LOF mutations (with an absolute allele frequency difference between populations >0.2) [52], we obtained the associated genes based on the human-genome annotation using BLASTP (E-value 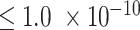 ) [75]. The GO and KEGG functional enrichment of genes affected by those LOF mutations was assessed based on the DAVID database [105]. To avoid the influence of assembly and annotation artifacts on the LOF results, we also confirmed these results by comparing them with the two genomes independently published previously [16].
) [75]. The GO and KEGG functional enrichment of genes affected by those LOF mutations was assessed based on the DAVID database [105]. To avoid the influence of assembly and annotation artifacts on the LOF results, we also confirmed these results by comparing them with the two genomes independently published previously [16].
Supplementary Material
Acknowledgements
We are grateful to the Animal Branch of the Germplasm Bank of Wild Species of Chinese Academy of Sciences (Large Research Infrastructure Funding), Nujiang Forest Public Security Bureau and the Guangzhou Wildlife Rescue Center for providing the specimens.
FUNDING
This work was supported by a grant from the National Natural Science Foundation of China to L.Y. (31925006) and the Joint Funding of the Yunnan Provincial Science and Technology Department and Yunnan University to L.Y. (2018FY001-009), a grant from the National Natural Science Foundation of China to X.L. (31760620) and the National Youth Talent Support Program to L.Y. (W8133006).
AUTHOR CONTRIBUTIONS
L.Y. conceived and designed the study; J.-Y.H., Z.-Q.H., H.L., H.W. and W.-M.K. carried out data analyses; S.-F.W., Y.-F.J., W.C. and Y.-P.Z. provided specimens; J.-Y.H., L.Y., H.L. and Z.-Q.H. wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest statement . None declared.
REFERENCES
- 1. Challender DWS, Harrop SR, Macmillan DC. Understanding markets to conserve trade-threatened species in CITES. Biol Conserv 2015; 187: 249–59. [Google Scholar]
- 2. Cheng W, Xing S, Bonebrake TC. Recent pangolin seizures in China reveal priority areas for intervention. Conserv Lett 2017; 10: 757–64. [Google Scholar]
- 3. Nijman V, Zhang MX, Shepherd CR. Pangolin trade in the Mong La wildlife market and the role of Myanmar in the smuggling of pangolins into China. Glob Ecol Conserv 2016; 5: 118–26. [Google Scholar]
- 4. Wu SB, Ma GZ. The status and conservation of pangolins in China. Traffic East Asia News 2007; 4: 1–5. [Google Scholar]
- 5. Challender DWS. Asian pangolins: increasing affluence driving hunting pressure. Traffic Bull 2011; 23: 92–3. [Google Scholar]
- 6. Hsieh HM, Lee JC, Wu JHet al. Establishing the pangolin mitochondrial D-loop sequences from the confiscated scales. Forensic Sci Int Genet 2011; 5: 303–7. [DOI] [PubMed] [Google Scholar]
- 7. Liu Y, Weng Q. Fauna in decline: plight of the pangolin. Science 2014; 345: 884. [DOI] [PubMed] [Google Scholar]
- 8. Luczon AU, Ong PS, Quilang JPet al. Determining species identity from confiscated pangolin remains using DNA barcoding. Mitochondrial DNA Part B 2016; 1: 763–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang H, Miller MP, Feng Yet al. Molecular tracing of confiscated pangolin scales for conservation and illegal trade monitoring in Southeast Asia. Glob Ecol Conserv 2015; 4: 414–22. [Google Scholar]
- 10. Zhou ZM, Zhou Y, Newman Cet al. Scaling up pangolin protection in China. Front Ecol Environ 2014; 12: 97–8. [Google Scholar]
- 11. IUCN . The IUCN Red List of Threatened Species. Version2014.
- 12. Gaubert P, Antunes A, Meng Het al. The complete phylogeny of pangolins: scaling up resources for the molecular tracing of the most trafficked mammals on earth. J Hered 2018; 109: 347–59. [DOI] [PubMed] [Google Scholar]
- 13. Gaudin TJ, Emry RJ, Wible JR. The phylogeny of living and extinct pangolins (Mammalia, Pholidota) and associated taxa: a morphology based analysis. J Mammal Evol 2009; 16: 235–305. [Google Scholar]
- 14. Heinrich S, Wittmann TA, Prowse TAAet al. Where did all the pangolins go? International CITES trade in pangolin species. Glob Ecol Conserv 2016; 8: 241–53. [Google Scholar]
- 15. Nash HC, Wirdateti, Low GWet al. Conservation genomics reveals possible illegal trade routes and admixture across pangolin lineages in Southeast Asia. Conserv Genet 2018; 19: 1083–95. [Google Scholar]
- 16. Choo SW, Rayko M, Tan TKet al. Pangolin genomes and the evolution of mammalian scales and immunity. Genome Res 2016; 26: 1312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heath ME. Manis pentadactyla. Mammal Species 1992; 414: 1–6. [Google Scholar]
- 18. Yang L, Chen M, Challender DWSet al. Historical data for conservation: reconstructing range changes of Chinese pangolin (Manis pentadactyla) in eastern China (1970-2016) Dryad Digital Repository. P RoySoc B: Biol Sci 2018; 285: 20181084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. IUCN WCC Resolution 015-Greater protection needed for all pangolin species . IUCN World Conservation Congress, Hawaii: (United States; ). 2016. [Google Scholar]
- 20. CITES . Amendments to Appendices I and II of the Convention adopted by the Conference of the Parties at its 11th meeting in Gigiri, Kenya, from 10 to 20 April 2000. CITES Secretariat. 2000.
- 21. Luo S, Cai Q, David VAet al. Isolation and characterization of microsatellite markers in pangolins (Mammalia, Pholidota, Manis spp.). Mol Ecol Resour 2007; 7: 269–72. [Google Scholar]
- 22. Du Toit Z, Grobler JP, Kotzé Aet al. The complete mitochondrial genome of Temminck's ground pangolin (Smutsia temminckii; Smuts, 1832) and phylogenetic position of the Pholidota (Weber, 1904). Gene 2014; 551: 49–54. [DOI] [PubMed] [Google Scholar]
- 23. Hassanin A, Hugot JP, van Vuuren BJ. Comparison of mitochondrial genome sequences of pangolins (Mammalia, Pholidota). C R Biol 2015; 338: 260–5. [DOI] [PubMed] [Google Scholar]
- 24. Ma JE, Li LM, Jiang HYet al. Transcriptomic analysis identifies genes and pathways related to myrmecophagy in the Malayan pangolin (Manis javanica). PeerJ 2017; 5: e4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yusoff MA, Tan TK, Hari Ret al. De novo sequencing, assembly and analysis of eight different transcriptomes from the Malayan pangolin. Sci Rep 2016; 6: 28199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang Z, Xu J, Xiao Set al. Comparative optical genome analysis of two pangolin species: Manis pentadactyla and Manis javanica. Gigascience 2016; 5: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parra G, Korf I, Bradnam K. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007; 23: 1061–7. [DOI] [PubMed] [Google Scholar]
- 28. Simao FA, Waterhouse RM, Panagiotis Iet al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015; 31: 3210–2. [DOI] [PubMed] [Google Scholar]
- 29. Zhou X, Meng X, Liu Zet al. Population genomics reveals low genetic diversity and adaptation to hypoxia in snub-nosed monkeys. Mol Biol Evol 2016; 33: 2670–81. [DOI] [PubMed] [Google Scholar]
- 30. Cho YS, Hu L, Hou Het al. The tiger genome and comparative analysis with lion and snow leopard genomes. Nat Commun 2013; 4: 2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dobrynin P, Liu S, Tamazianet al. Genomic legacy of the African cheetah, Acinonyx jubatus. Genome Biol 2015; 16: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson J, OrtegaDel Vecchyo D, Fan Zet al. Genomic flatlining in the endangered island fox. Curr Biol 2016; 26: 1183–9. [DOI] [PubMed] [Google Scholar]
- 33. Carbone L, Harris RA, Gnerre Set al. Gibbon genome and the fast karyotype evolution of small apes. Nature 2014; 513: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li R, Fan W, Tian Get al. The sequence and de novo assembly of the giant panda genome. Nature 2010; 463: 311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Locke DP, Hillier LDW, Warren WCet al. Comparative and demographic analysis of orang-utan genomes. Nature 2011; 469: 529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mays J, Herman L, Hung CMet al. Genomic analysis of demographic history and ecological niche modeling in the endangered sumatran rhinoceros Dicerorhinus sumatrensis. Curr Biol 2018; 28: 70–76.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xue Y, Prado Martinez J, Sudmant PHet al. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 2015; 348: 242–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Palkopoulou E, Mallick S, Skoglund Pet al. Complete genomes reveal signatures of demographic and genetic declines in the woolly mammoth. Curr Biol 2015; 25: 1395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H, Stephan W. Inferring the demographic history and rate of adaptive substitution in Drosophila. PLoS Genet 2006; 2: e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rohling EJ, Grant K, Bolshaw Met al. Antarctic temperature and global sea level closely coupled over the past five glacial cycles. Geoscience 2009; 2: 500–4. [Google Scholar]
- 41. Rovere A, Raymo M, Vacchi Met al. The analysis of last interglacial (MIS 5e) relative sea-level indicators: reconstructing sea-level in a warmer world. Earth Sci Rev 2016; 159: 404–27. [Google Scholar]
- 42. Kawamura K, Parrenin F, Lisiecki Let al. Northern Hemisphere forcing of climatic cycles in Antarctica over the past 360,000 years. Nature 2007; 448: 912–6. [DOI] [PubMed] [Google Scholar]
- 43. Otto-Bliesner BL, Marshall SJ, Overpeck JTet al. Simulating arctic climate warmth and icefield retreat in the last interglaciation. Science 2006; 311: 1751–3. [DOI] [PubMed] [Google Scholar]
- 44. Han W, Fang X, Ye Cet al. Tibet forcing Quaternary stepwise enhancement of westerly jet and central Asian aridification: carbonate isotope records from deep drilling in the Qaidam salt playa, NE Tibet. Glob Planet Change 2014; 116: 68–75. [Google Scholar]
- 45. He K, Jiang XL. Sky islands of southwest China. I: an overview of phylogeographic patterns. Chin Sci Bull 2014; 59: 585–97. [Google Scholar]
- 46. Hoffecker JF. Desolate Landscapes: Ice-Age Settlement in Eastern Europe. New Brunswick: Rutgers University Press, 2002. [Google Scholar]
- 47. Zhao S, Zheng P, Dong Set al. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet 2013; 45: 67–71. [DOI] [PubMed] [Google Scholar]
- 48. Kuang WM, Ming C, Li HPet al. The origin and population history of the endangered golden snub-nosed monkey (Rhinopithecus roxellana). Mol Biol Evol 2019; 36: 487–99. [DOI] [PubMed] [Google Scholar]
- 49. Cai B. The later Pleistocene fossil mammals and the palaeogeographical environment of the Taiwan Strait. J Xiamen Univ 1999; 4: 29–33. [Google Scholar]
- 50. Kawamura A, Chang CH, Kawamura Y. Middle Pleistocene to Holocene mammal faunas of the Ryukyu Islands and Taiwan: an updated review incorporating results of recent research. Quat Int 2016; 397: 117–35. [Google Scholar]
- 51. Kardos M, Akesson M, Fountain Tet al. Genomic consequences of intensive inbreeding in an isolated wolf population. Nat Ecol Evol. 2018; 2: 124–31. [DOI] [PubMed] [Google Scholar]
- 52. Van der Valk T, Diez-Del-Molino D, Marques-Bonet Tet al. Historical genomes reveal the genomic consequences of recent population decline in eastern gorillas. Curr Biol 2019; 29: 165–70.e6. [DOI] [PubMed] [Google Scholar]
- 53. Feng S, Fang Q, Barnett Ret al. The genomic footprints of the fall and recovery of the crested ibis. Curr Biol 2019; 29: 340–49.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marsden CD, Ortega-Del Vecchyo D, O’Brien DPet al. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc Natl Acad Sci USA 2016; 113: 152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grantham R. Amino acid difference formula to help explain protein evolution. Science 1974; 185: 862–4. [DOI] [PubMed] [Google Scholar]
- 56. Sun NCM, Arora B, Lin JSet al. Mortality and morbidity in wild Taiwanese pangolin (Manis pentadactyla pentadactyla). PLoS One 2019; 14: e0198230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goedeke L, Fernández-Hernando C. Regulation of cholesterol homeostasis. Cell Mol Life Sci 2012; 69: 915–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ivanova NS, Zemlak T, Hanner Ret al. Universal primer cocktails for fish DNA barcoding. Mol Ecol Res 2007; 7: 544–8. [Google Scholar]
- 59. Gnerre S, Maccallum I, Przybylski Det al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA 2011; 108: 1513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li R, Zhu HJ, Qian Wet al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 2010; 20: 265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mostovoy Y, Levy-Sakin M, Lam Jet al. A hybrid approach for de novo human genome sequence assembly and phasing. Nat Methods 2016; 13: 587–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Currt Protoc Bioinform 2009; 4: 11–4. [DOI] [PubMed] [Google Scholar]
- 64. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 2007; 35: W265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics 2005; 21: i351–8. [DOI] [PubMed] [Google Scholar]
- 66. Smith AFA, Hubley R. RepeatModeler open-1.0.5 [Internet]. 2012. Available online at: http://www.repeatmasker.org. [Google Scholar]
- 67. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999; 27: 573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003; 19: ii215–25. [DOI] [PubMed] [Google Scholar]
- 69. Majoros W, Pertea MS. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 2004; 20: 2878–9. [DOI] [PubMed] [Google Scholar]
- 70. Korf I. Gene finding in novel genomes. BMC Bioinformatics 2004; 5: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr Protoc Bioinformatics 2007; 18: Unit 4.3. [DOI] [PubMed] [Google Scholar]
- 72. Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res 2000; 10: 516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Trapnell C, Pachter L, Lsalzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009; 25: 1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Trapnell C, Williams BA, Pertea Get al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biol 2010; 28: 511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Altschul SF, Madden TL, SchFfer AAet al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Birney E, Clamp M, Durbin R. GeneWise and GenomeWise. Genome Res 2004; 14: 988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Haas BJ, Salzberg SL, Zhu Wet al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 2008; 9: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Haas BJ, Delcher AL, Mount SMet al. Improving the arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 2003; 31: 5654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Li H, Handsaker B, Wysoker Aet al. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McKenna A, Hanna M, Banks Eet al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hoeppner MP, Lundquist A, Pirun Met al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS One 2014; 9: e91172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li G, Davis BW, Raudsepp Tet al. Comparative analysis of mammalian Y chromosomes illuminates ancestral structure and lineage-specific evolution. Genome Res 2013; 23: 1486–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schneider VA, Graves-Lindsay T, Howe Ket al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 2017; 27: 849–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics 2002; Chapter 2: Unit 2.3. [DOI] [PubMed] [Google Scholar]
- 85. Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17: 540–52. [DOI] [PubMed] [Google Scholar]
- 86. Leigh JW, Bryant D. PopART: full-feature software for haplotype network construction. Methods Ecol Evol 2015; 6: 1110–6. [Google Scholar]
- 87. Kumar S, Stecher Gand Li Met al. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018; 35: 1547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yu D, Dong L, Yan Fet al. eGPS 1.0: comprehensive software for multi-omic and evolutionary analyses. Natl Sci Rev 2019; 6: 867–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lee TH, Guo H, Wang Xet al. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 2014; 15: 162–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014; 30: 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Purcell S, Neale B, Todd-Brown Ket al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006; 2: e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tang H, Peng J, Wang Pet al. Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol 2005; 28: 289–301. [DOI] [PubMed] [Google Scholar]
- 94. Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19:1655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Excoffier L, Dupanloup I, Huerta-Sanchez Eet al. Robust demographic inference from genomic and SNP data. PLoS Genet 2013; 9: e1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang F, Wu S, Li Yet al. Reproductive parameters of the Sunda pangolin, Manis javanica. Folia Zoologica 2015; 64: 129–35. [Google Scholar]
- 97. Zhang F, Wu S, Zou Cet al. A note on captive breeding and reproductive parameters of the Chinese pangolin, Manis pentadactyla Linnaeus. Zookeys 2016; 1758: 129–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr 1974; 19: 716–23. [Google Scholar]
- 99. Schiffels S, Durbin R. Inferring human population size and separation history from multiple genome sequences. Nat Genet 2014; 46: 919–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Browning BL, Zhou Y, Browning SR. A one-penny imputed genome from next-generation reference panels. Am J Hum Genet 2018; 103: 338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Danecek P, Auton A, Abecasis Get al. The variant call format and VCFtools. Bioinformatics 2011; 27: 2156–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Barrett JC, Fry B, Maller Jet al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–5. [DOI] [PubMed] [Google Scholar]
- 103. Cingolani P, Platts A, Wangle Let al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012; 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Li WH, Wu CI, Luo CC. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J Mol Evol 1984; 21: 58–71. [DOI] [PubMed] [Google Scholar]
- 105. Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.