

Abstract
Inorganic arsenic (iAs/As2O32−) is an environmental toxicant found in watersheds around the world including in densely populated areas. iAs is a class I carcinogen known to target the skin, lungs, bladder and digestive organs, but its role as a primary breast carcinogen remains controversial. Here, we examined a different possibility: that exposure to iAs promotes the transition of well-differentiated epithelial breast cancer cells characterized by estrogen and progesterone receptor expression (ER+/PR+), to more basal phenotypes characterized by active proliferation, and propensity to metastasis in vivo. Our results indicate two clear phenotypic responses to low level iAs that depend on the duration of the exposure. Short-term pulses of iAs activate ER signaling, consistent with its reported pseudo-estrogen activity, but longer-term, chronic treatments for over 6 months suppresses both ER and PR expression and signaling. In fact, washout of these chronically exposed cells for up to 1 month failed to fully reverse the transcriptional and phenotypic effects of prolonged treatments, indicating durable changes in cellular physiologic identity. RNA-seq studies found that chronic iAs drives the transition towards more basal phenotypes characterized by impaired hormone receptor signaling despite the conservation of estrogen receptor expression. Because treatments for breast cancer patients are largely designed based on detection of hormone receptor expression, our results suggest greater scrutiny of ER+ cancers in patients exposed to iAs, because these tumors may spawn more aggressive phenotypes than unexposed ER+ tumors, in particular, basal subtypes that tend to develop therapy resistance and metastasis.
Keywords: Breast cancer, estrogen receptor, arsenic, environmental exposures
INTRODUCTION
Inorganic arsenic (iAs) is an established human carcinogen with well-described roles as a skin, lung, digestive tract, kidney, bladder and prostate carcinogen (1–5). Epidemiologic studies of breast cancer, in contrast, have yet to definitively support a role for iAs as a tumor-initiating carcinogen (6–8). Beyond causing malignant transformation, however, it is possible that iAs could drive the evolution of existing tumors toward more aggressive cancer phenotypes. The current study determined if luminal, hormone responsive (ER+/PR+) breast cancers, which are treatable with endocrine therapies such as tamoxifen and aromatase inhibitors, can be converted by exposure to chronic iAs to basal-like subtypes, that are generally more difficult to treat and prone to metastasis. Previous studies provide some support for this concept. Bangladesh, a country with regions facing severe iAs contamination in ground waters, has a disproportionately high incidence of ERlow/ER− breast cancer (9, 10). Additional studies indicated that the incidence of these ERlow/ER− cancer subtypes falls sharply in Bangladeshi women who relocated to areas with lower levels of iAs contamination (11). Epidemiologic analysis, therefore, supports a role for inorganic arsenic in either the development or evolution of breast cancer towards more aggressive, basal ERlow/ER− and PR− phenotypes.
iAs has a well-documented acute effect as a weak pseudo-estrogen. Like estradiol itself, iAs is known to promote the internalization and degradation of the estrogen receptor (12–15). In contrast to these acute effects, the impact of more prolonged exposures to iAs is less clear, despite this being a more likely scenario for environmental exposures through drinking water. In the current study, we sought to determine if prolonged exposure to iAs persistently and/or reversibly changes estrogen receptor expression and function at the molecular and functional levels. Our results demonstrate that chronic iAs is a potent suppressor of ER and PR expression and function, and that even after iAs exposure is suspended, a residual transcriptional ‘memory’ of exposure persists, contributing to more aggressive behavior from luminal breast cancer cells in vivo. We believe that this offers a potential mechanism for the documented link between iAs exposure and breast cancer aggressiveness observed in afflicted women.
MATERIALS AND METHODS
Reagents
17β-estradiol (E2) and progesterone (P4) were purchased from Sigma (#E8875 and #P8783, respectively) and dissolved in ethanol as a 1mM stock solutions. Sodium arsenite (iAs) was also purchased from Sigma (#S7400) and dissolved in ddH2O as a 1mM stock solution. All primers were purchased from Integrated DNA Technologies (IDT). All cell culture reagents were purchased from Gibco/Life Technologies unless otherwise stated. Cell culture ware was purchased from Denville/Thomas Scientific unless otherwise stated.
Cell Culture
MCF-7 cells were purchased from ATCC and were maintained in Minimal Essential Media (MEM) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids (NEAA), 1mM sodium pyruvate, 2 mM L-glutamine, 1% antibiotics penicillin-streptomycin, and 6 ng/ml human recombinant insulin at 37 ˚C in 5% CO2. Before treatment with E2 or P4, cells were cultured in phenol red-free MEM media supplemented with 5% charcoal–stripped fetal bovine serum (i.e. treatment media) for at least 48 hrs. Treatments with E2, P4 and iAs were performed with the doses and for the durations described in the Results section. Unless indicated, E2 or P4 and iAs were added simultaneously. iAs300 MCF-7 cells were continuously exposed to iAs at a concentration of 100nM for up to 10 months, and compared with the unexposed passages of the control cells. Recovery MCF-7 cells were cultured in MCF-7 media without iAs after the 10 month iAs treatment for the indicated amounts of time (typically we used 16 days of recovery).
Proliferation Assay
MCF-7 and MCF-7 iAs300d cells were seeded at a density of 1,000 cells/well of black-walled, 96 well plates (Greiner #655090) and left undisturbed for 24h. After 24h from seeding, an initial cell count reading was made using Celigo Model 200 and the accompanying software for each well, to ensure even seeding. Cells were treated with iAs (100nM) +/− 10 nM E2 every other day for the specified duration of the experiment. The changes in the cell count for each treatment, and changes in proliferation were plotted relative to vehicle treated control cells.
RNA and qPCR
MCF-7, MCF-7 iAs300d and MCF-7 recovery cells were seeded at a density of 150,000 cell/well in 6 well plates in treatment media. After 2 days, the cells were treated with E2 (10 nM), P4 (25 nM) and/or iAs (100 nM) for the duration of the experiments. Total RNA was isolated using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Total RNA (0.5 μg) was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Invitrogen #28025–013). The cDNA product was diluted with 90 μl of ddH20, and then 2 μl was used for the qPCR reaction. The qPCR was performed on an Applied Biosystems Step-One Plus Real-Time PCR system using the Fast SYBR Green qPCR Mastermix (Applied Biosystems). qPCR primers are listed below: FKBP5 forward: CCC CCG CGG CGA CAG GTT CTC TAC; FKBP5 reverse: CCAATCATCGGCGTTTCCTCACCA; GREB1 forward: ACGTGTGGTGACTGGAGTAGC; GREB1 reverse: CCACGCAAGGTAGAAGGTGA; EGR3 forward: TTCTCGTACAGGGTGGCTCC; EGR3 reverse GGCAGAGAGCAACCTTCCC; PS2 forward: GTGTGCAAATAAGGGCTGCTG; PS2 reverse: TGGAGGGACGTCGATGGTA; PR forward: GTCGCCTTAGAAAGTGCTGTCAG; PR reverse: GCTTGGCTTTCATTTGGAACGCC; ER forward: TGCCCTACTACCTGGAGAAC; ER reverse: CCATAGCCATACTTCCCTTGTC; 36B4 forward: GTGTTCGACAATGGCAGCAT; 36B4 reverse: GACACCCTCCAGGAAGCGA; GAPDH forward: GTCTCCTCTGACTTCAACAGCG; GAPDH reverse: ACCACCCTGTTGCTGTAGCCAA; Fold change was calculated using the ΔΔCt method, and 36B4 or GAPDH were used as the internal control. Each qPCR amplification evaluation was performed in triplicate.
Western Blot
MCF-7, MCF-7 iAs300d, and MCF-7 recovery cells were seeded at a density of 150,000 cell/well in 6 well plates. For experiments involving E2 or P4, cells were in treatment media for 2 days prior to treatment. Whole cell extracts were prepared using radioimmunoprecipitation assay buffer (RIPA) containing 1% Protease/ Phosphatase Inhibitor Cocktail (Thermo-Fisher Scientific) and protein content was measured using BCA (Pierce); proteins were denatured and separated by SDS-PAGE using a 4–12% gradient Bis-Tris gel from Invitrogen (NP0321BOX) and then transferred to nitrocellulose membranes (Thermo-Scientific #88018). The membranes were blocked for 1h in Intercept TBS Blocking Buffer (LICOR) and then incubated overnight at 4°C with the appropriate primary antibody. Primary antibodies used include: Vimentin (Abcam #ab16700), E-Cadherin (Cell Signaling #24E10), ER alpha (Abcam #ab18790), PR (Cell Signaling #D8Q2J) and β Actin (Sigma #A5441). The next day the membranes were washed 3x for 5 minutes each wash using TBST 3x and then probed with fluorescent secondary antibodies for 1h prior to the another 3 washes with TBST 3x. Protein bands were detected with the Li-Cor Odyssey Blot Imager (LICOR) and intensity of bands were quantified using Image Studio™ Lite software (LICOR).
Invasion Assay
A transwell invasion assay was performed as previously described (Pan et al, 2015; Morrison et al. 2017). Cells were harvested, disaggregated and re-suspended in cell culture medium with 1% FBS. Cells were then seeded at a 1,000 cells/well density in 12-well cell culture inserts with 8 μm pore size membrane (Falcon). Complete cell culture medium with 10% FBS was added in the lower chamber as chemo-attractant. Cells were incubated at 37°C for 18 h. After this time, cells were fixed with 4% paraformaldehyde (PFA), and non-migrated cells were removed from the inner side of the insert with a cotton swab. All the migrated/invaded cells were gently washed with 1X PBS, then stained with Haemotoxylin & Eosin (H&E) and allowed to dry. The whole insert was photographed with a light microscope (Zeiss) and ImageJ software was used to count the cells.
Mammosphere Formation Assay
Viable cells in culture were re-suspended in organoid culture medium, consisting of: 10 ng/mL epidermal growth factor (EGF) (Corning), 10% MammoCultTM (STEMCELL Technologies), 5% Matrigel (Corning). After resuspension in culture organoid medium, 5,000 cells were dissociated and plated into 6-well ultra-low-attachment plate (Falcon). Cells were fed 200 μL fresh organoid medium every four days, the medium changed every 10 days for up to one month. Mammospheres with ≥ 40 μm diameter were counted and pictures were taken using an inverted microscope as mentioned before.
Fluorescent Immuno-Cytochemical
Immuno-cytochemistry was used to verify specific markers of interest from the protocol previously described (16). Mammospheres were grown up to 21 days, and the matrigel containing the mammospheres was digested with collagenase for 30 min at 37°C, and then collected into a centrifuge tube. After centrifugation, the mammospheres were transferred to a new centrifuge tube, washed with 1X PBS and fixed with 4% PFA (Sigma) for 15 min. For staining, mammospheres were blocked with Bovine Serum Albumin (BSA) for 45 min, washed 3x with 1X TBST for 3 min, incubated with primary antibody (dilution 1:75), CK5 (Abcam, catalog# 17130) and CK8 (Abcam ab9023) overnight at 4°C. Mammospheres were then washed 3x with 1X TBST and incubated with secondary antibody (diluted 1:200) for 1h at room temperature. After incubating with DAPI (Applied Biosystems), mammospheres were placed on slides and mounted with FluoroMount Aqueous Solution (Sigma) and pictures were taken using a Confocal microscope LSM-710META (Zeiss).
RNA-seq
Total RNA from triplicate MCF-7 conditions was submitted to the Genomic Sciences and Precision Medicine Center, Medical College of Wisconsin (Milwaukee, WI) for RNA quality control, library preparation and sequencing. Sample quality was assessed by fragment analysis (Agilent, standard sensitivity RNA catalog #DNF-471) with all samples having RQN > 9.4 and DV200 >90%. Total RNA (1µg) was aliquoted for library preparation utilizing Illumina’s TruSeq Stranded mRNA protocol (catalog #20020594). Briefly, mRNA was captured by poly A magnetic beads, followed by fragmentation, cDNA synthesis, adenylation of fragments and ligation of adaptors for Illumina based sequencing. Final libraries were enriched and purified prior to quantification and paired end 116 base pair sequencing on an Illumina NovaSeq SP flowcell. Raw reads were aligned to the human reference transcriptome GRCh38.78 with at least 33 million mapped read pairs acquired per sample. Sequencing reads were processed through the GSPMC workflow including MapRseq3 (17) and differential expression calculated by EdgeR (18). Differentially expressed genes (DEGs) were filtered based on a false discovery rate (FDR) < 0.05 and an absolute fold change ≥ 2 called between at least one condition and the reference control sample. Hierarchical clustering was performed with reads per kilobase million (RPKM) of DEGs to obtain clusters of gene expression patterns. Pathway analysis of gene clusters was completed with R package RITAN (19) and the Molecular Signatures Database (MSigDB) hallmark gene set collection (20). For generating pathway specific protein-protein interaction (PPI) networks, RITAN was used to query the following PPI databases: STRING (21), CCSB (22) and PID (23). Cytoscape software (24) was used for viewing the PPIs. Results are deposited on a publicly available ArrayExpress database under identifier E-MTAB-9498.
Stem Cell Assays
For the CD44+/CD24− assay, MCF-7 and MCF-7iAs300d cells were labelled as described by Liu et al, 2014. Antibodies for CD44 and CD24 were purchased from BD Pharmingen (# 560890 and # 555428, respectively). SORE6+ assay lentivirus was made from the SORE6 pro-lentiviral plasmids and the control plasmids, obtained from Drs. Binwu Tang and Lalage M. Wakefield at the NIH (25), and cells were subsequently transduced for 72h. Both stem cell assays used the LSRII flow cytometer (Blood Research Institute, MCW). FACS analysis was conducted using FACSDiva to determine changes in the CD44+/CD24− and SORE+ cell populations.
In vivo Metastasis Experiment
Experiments were carried out at the MCW animal facilities and all procedures adhered to guidelines approved by institutional and national policies and committees. For the metastasis studies, MCF-7 and MCF7iAs300d cells were transduced with luciferase virus, and subsequently injected into the lateral tail vein of NOD/SCID immunocompromised mice at a (1×106 cells/animal) concentration. Live animals were imaged for up to 3 months using the IVIS Lumina (Perkin Elmer). RediJect D-Luciferin Bioluminescent Substrate (Perkin Elmer) was injected intraperitoneally (150 mg/kg body weight) and animals were anesthetized in an oxygen chamber with 2.5% isofluorane. The signal was measured as radiance (photons/second/square centimeter/steradian), and analyzed with Living Image software. At the end of the 3 months period, tumors and tissues of interest were excised for histology/pathology analysis.
RESULTS
Chronic iAs exposure increases invasiveness and clonogenicity of MCF-7 luminal A, ERα+ breast carcinoma cells –
Luminal A breast cancers are molecularly characterized by hormone receptor (ER+/PR+) expression and the lack of Her2 activity. Well-differentiated luminal A tumors tend to grow slowly, display epithelial lineage markers and typically have good prognosis. On the contrary, ERlow/−, PR− breast tumors are poorly differentiated, tend to be highly proliferative and often evolve towards poorer outcomes. Epidemiologic studies indicate that these more aggressive ERlow/− tumors occur at a disproportionate frequency in areas where risk for iAs exposure is high (9, 10, 26). In light of this, we sought to develop a system to study the impact of long term, environmentally relevant iAs exposure on phenotypic changes in luminal A breast cancer cells. We therefore exposed MCF-7 cells, a luminal A breast carcinoma line, to 100 nM iAs for 6–10 months in culture. Consistent with previous reports indicating iAs has pseudo-estrogenic effects we observed acceleration in cellular proliferation rates as well as changes in a variety of cellular functional assays. For instance, there was a ca. 2-fold increase in the capacity of iAs exposed cells to invade Matrigel™ when compared to the non-treated MCF-7 controls (Fig. 1 A). Colonies formed by iAs treated cells tended to be smaller in diameter when cells were grown under anchorage-independent conditions, but were more numerous. There was a > 3-fold increase in colony formation (Fig. 1B) by iAs treated cells compared to non-treated MCF-7 controls. Together, these results indicated that iAs induced MCF-7 cells to acquire features compatible with more aggressive breast cancer phenotypes. We also measured proliferation in response to estrogen by performing proliferation assays in the presence of 17β-estradiol (E2), which promotes the proliferation of ER+ breast cancer cells. Indeed, we found that cells exposed to iAs showed a 2-fold increase in the proliferation rate (27). Intriguingly, we saw that E2 stimulation of iAs treated cells exerted a synergistic effect, increasing their proliferation rate by nearly 10-fold compared to the control group (Fig.1C). Combined, these results demonstrate that iAs promotes a less differentiated, more proliferative, and invasive phenotype, which is, nonetheless, exacerbated by E2 treatment.
Figure 1. Chronic exposure to low dose iAs promotes clonogenicity, invasiveness and the proliferation of luminal epithelial breast cancer cells.
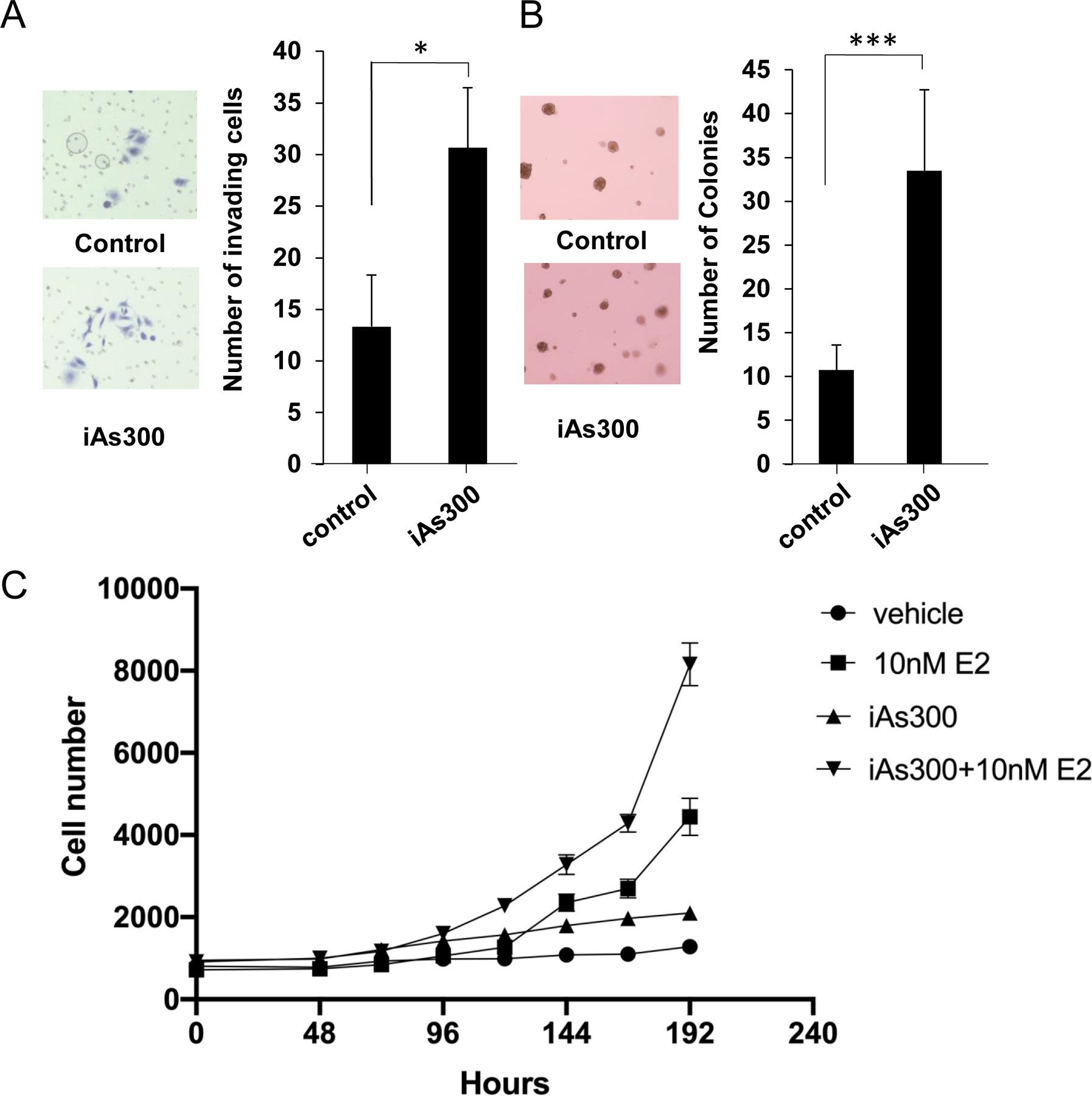
(A) Matrigel™ invasion assay using parental MCF-7 cells (control) and MCF-7 cells cultured in the presence of iAs (100 nM) for 300 days (iAs-300). Cells were platted on the upper chamber of a transwell setup on top of a thin layer of Matrigel™ and allowed to migrate towards the bottom chamber filled with complete media. Cells were stained and counted after 18h. (B) Organoid formation assay using cells platted in Matrigel™ 5% and grown for up to 30 days. Organoids were counted using an inverted microscope. (C) Proliferation curves of MCF-7 and MCF7-iAs300 co-stimulated with E2 (10 nM) for the indicated time points. All experiments represent averages ±SEM of 3 independent biologic replicates. * p < 0.05; *** p<0.001.
Loss of luminal epithelial markers by cells exposed to iAs –
The increased clonogenicity and invasiveness promoted by iAs suggested the possibility of changes at the molecular level. We examined hormone receptor expression and cell lineage (e.g. epithelial and mesenchymal) markers that are used to clinically diagnose breast cancer subtypes. Western blot analyses showed (Fig. 2A–C) that MCF-7 cells exposed to iAs lose epithelial gene expression (E-cadherin) while gaining mesenchymal/basal cell expression (vimentin), suggesting iAs may be an agent promoting epithelial-to-mesenchymal transition. We also detected downregulation of ERα, PRα and PRβ at the protein level (Figs, 2A & 2C) as well as a reduction in PGR mRNA levels (Fig 2 D–E, Suppl. Fig. 1). In fact, iAs reduced expression of the major ER-target gene, PGR, under both basal and E2-stimulated conditions, supporting the hypothesis that long term iAs exposure suppresses ER activity. To compare these long term effects of iAs to more acute exposures, we designed an experiment where cells were treated with iAs for shorter periods starting with one day up to two weeks exposures (Suppl. Fig. 1). Results confirmed that acute iAs exerts pseudo-estrogenic effects, as reflected by an initial upregulation (ca. 3-fold) of PGR gene expression at 24h. Simultaneously, the levels of ERα mRNA were acutely reduced and remained lower than baseline for the duration of the experiment. In contrast, PGR mRNA expression decreased in cells exposed to either iAs or iAs/E2 combination after an acute increase at 24h compared to controls (Suppl. Fig. 1). We also observed that, in response to E2, the expression of PGR became attenuated in cells exposed to iAs for more than 48h and remained significantly lower (ca. 30% compared to either E2 alone or E2 + iAs 24h) after 2 weeks of exposure to iAs (Suppl. Fig. 1) when compared with levels observed for non-iAs exposed cells stimulated with E2 alone. Therefore, the effects of iAs in promoting the loss of ER expression relatively rapidly (< 48 h) set in motion a progressive and persistent reduction in PGR expression early on. Because PGR is an important clinical marker of luminal A tumors, these results suggest that changes to luminal A phenotype occur early during exposure and are accentuated with prolonged treatment regimens.
Figure 2. Molecular markers and hormone receptor status/function of MCF-7 cells treated with iAs.
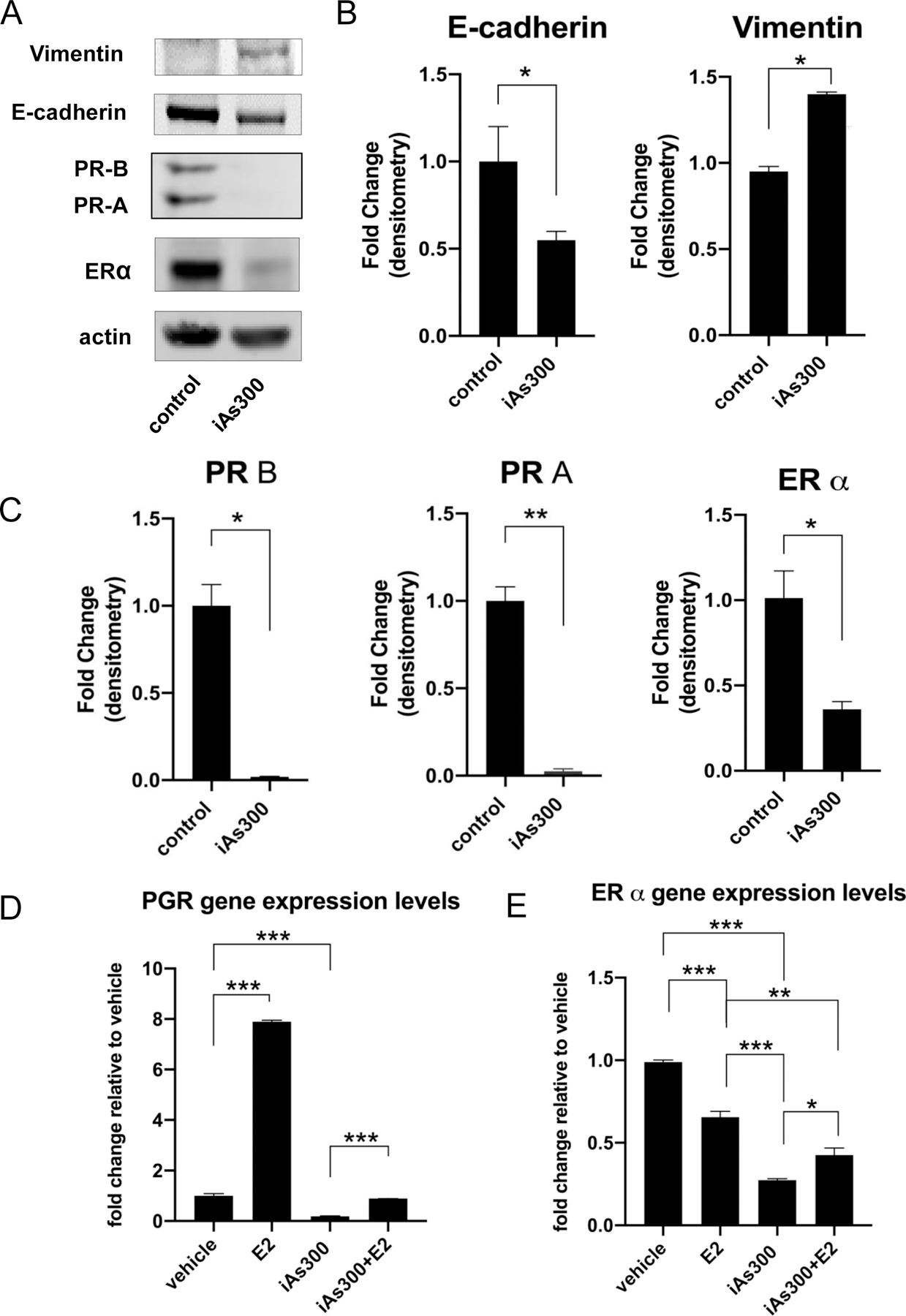
(A) Western blot analysis of epithelial (E-cadherin, CDH1) and mesenchymal (Vimentin) markers as well as hormone receptor status of parental MCF-7 compared to those treated with iAs for 300 days. (B) Quantification of band intensities from E-cadherin and vimentin blots using Image J densitometry (C) Quantification of band intensities from hormone receptor blots using Image J densitometry. (D) RT-QPCR analysis of ER and PR gene expression levels in MCF-7, MCF-7-iAs300 and MCF-7-iAs300 cultured in the absence of iAs for 16 days at baseline and in response to E2. (E) Functional analysis of ER signaling at baseline and in response to E2 using select ER target genes. RT-QPCR was used to quantify ER-target gene expression with data shown as fold-change relative to untreated controls. Quantified data represent averages of at least three independent biological replicates ±SEM. *p < 0.05, **p < 0.01, ***p<0.001.
Prolonged exposure of MCF-7 cells to iAs increases stem cell-like characteristics –
Previous studies indicated that breast cancer cells with characteristics of stem cells are more likely to generate metastases and develop resistance to standard therapies (28, 29). We examined if chronic iAs increased numbers of cells with stem cell-like characteristics by flow cytometry quantification of the membrane markers CD44 and CD24 (30). Fig. 3A shows that iAs treatment increases the subpopulation of CD44+/CD24− cells, with a nearly 2-fold increase in the iAs-treated cells without E2 treatment. We also employed a novel genetically encoded stem cell reporter called SORE6 that measures the activity of the core stem cell transcription factors, Sox2 and Oct4 (25). Exposure to chronic iAs increased SORE6+ stem-like cells by ~2-fold compared to control MCF-7 cells. This increase in stem-like cells is consistent with iAs promoting more basal, less differentiated phenotype that are normally enriched of cancer stem cells.
Figure 3. iAs increases the number of cancer stem cells in MCF-7 cell cultures.
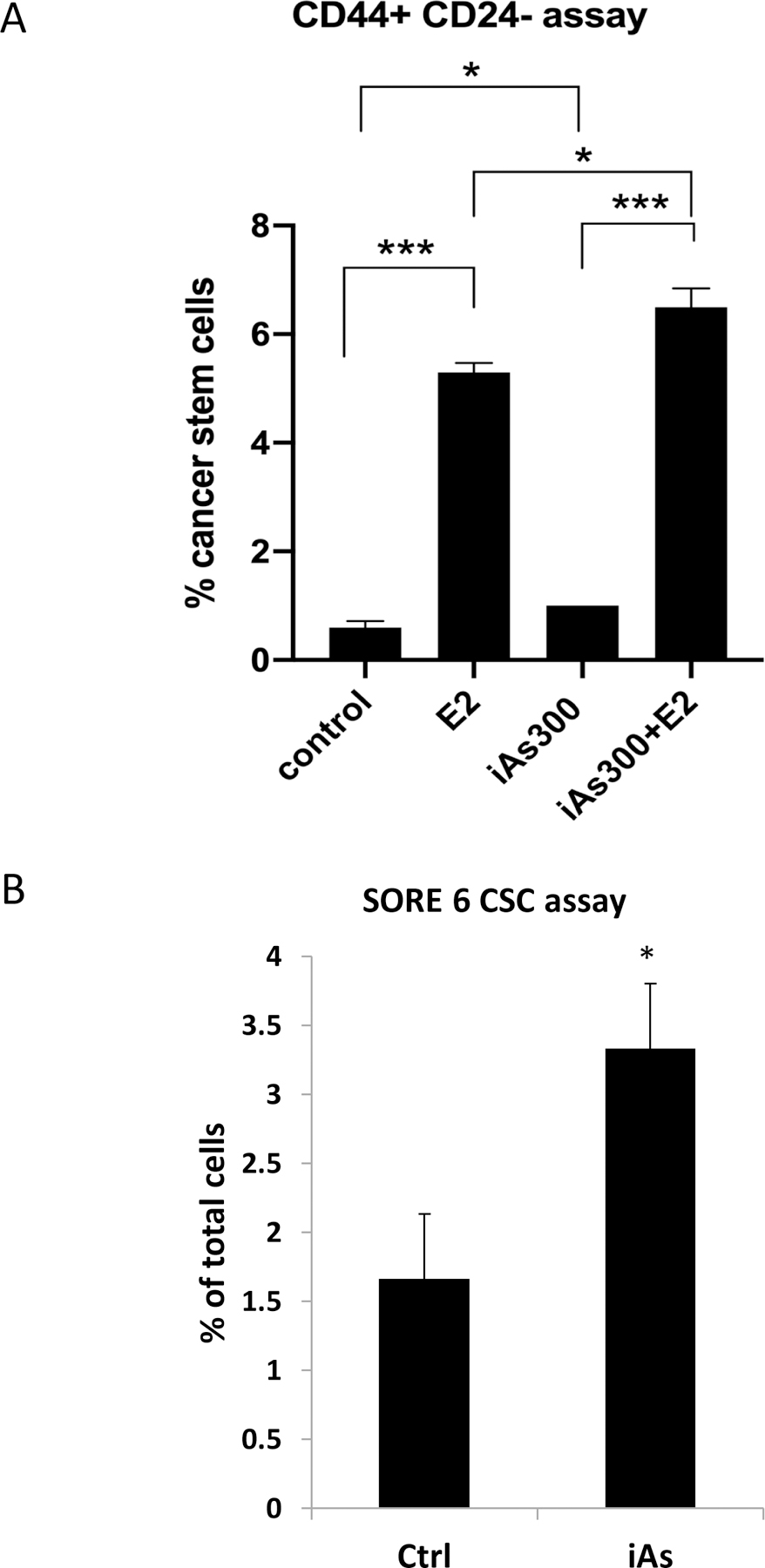
(A) FACS analysis of CD44+/CD24- cancer stem cells. (B) Quantification of cancer stem cell numbers in MCF-7 and iAs-treated MCF-7 cells using the stem cell reporter SORE6. All experiments represent averages ± SEM of 3 independent biologic replicates. * p < 0.05; *** p<0.001.
Exposure to iAs promotes molecular profiles compatible with luminal-to-basal transition in breast cancer cells –
The loss of MCF-7 epithelial marker expression associated with luminal phenotypes suggested the possibility that iAs could promote luminal-basal transition. We, therefore, cultured both untreated and iAs-treated MCF-7 cells in 5% Matrigel™ for 14 days to obtain 3D colonies. These spheroids were subsequently collected, fixed and stained to assess luminal (cytokeratin CK-8) and basal (cytokeratin CK-5) expression. Control cells showed abundant luminal epithelial CK-8 marking with low levels of basal CK-5. In contrast, spheroids grown in iAs containing media reversed this pattern with diminished CK-8 and high levels of CK-5, instead (Fig. 4A). In fact, similarly to our observation using MCF-7 cells cultured as monolayers, iAs increased the number of stem/tumor initiating cells in 3D cultures as well, as indicated by the stem cell reporter SORE6/GFP (Fig. 4B). Compared to 2D cultures where SORE6+ cells increase ~2 fold, we observed >4-fold increases in 3D cultures.
Figure 4. Analysis of luminal (CK8) and basal (CK5) markers as well as cancer stem cells in organoids of MCF-7 cells grown in 3D.
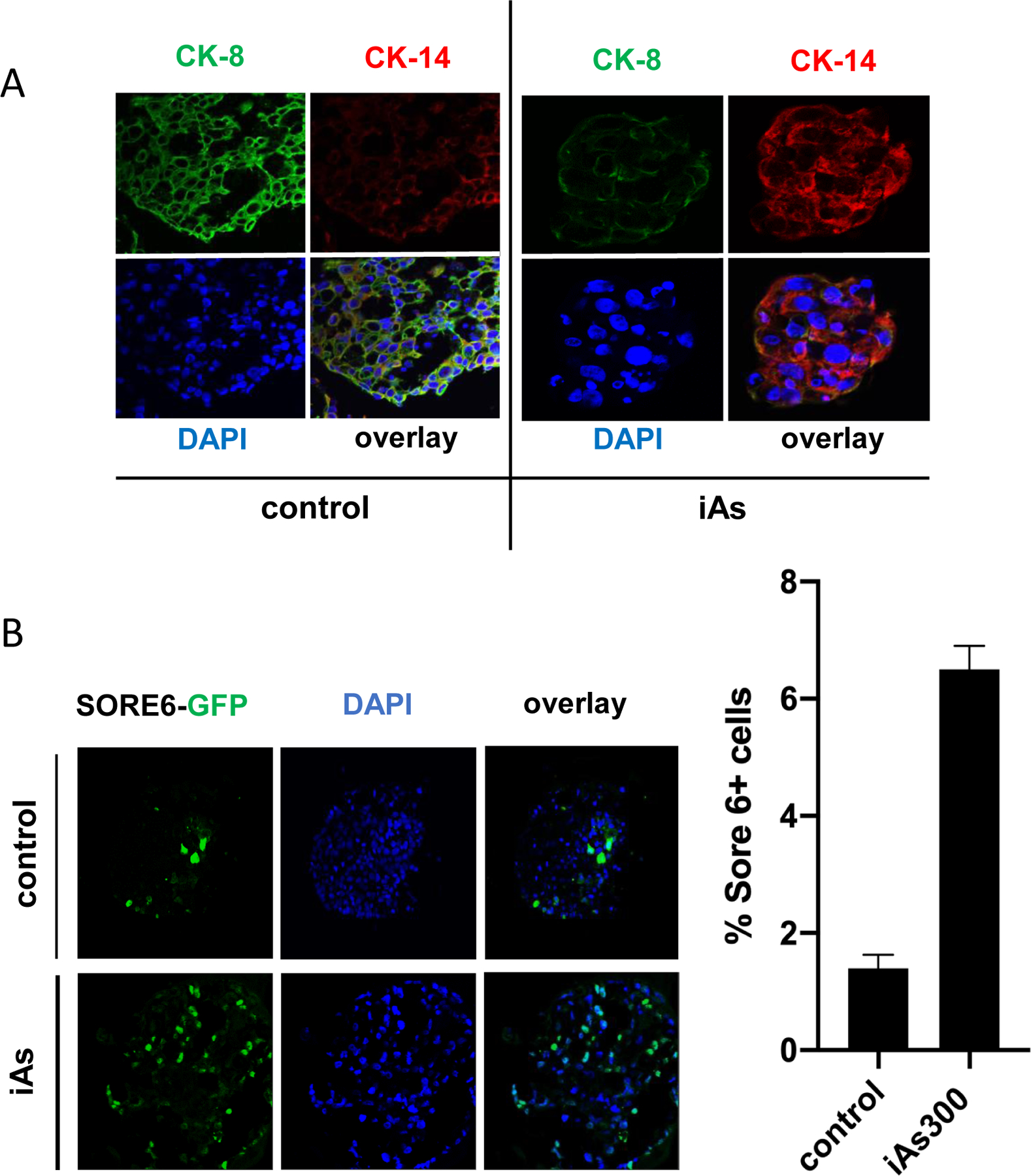
(A) MCF-7 cells (either parental or cells exposed to iAs for 180 days) were grown as organoids in 5% matrigel™. After 21 days, organoids were harvested, fixated with paraformaldehyde and stained for the luminal epithelial cell marker (CK8, shown in green) or basal cell marker (CK5, shown in red). (B) MCF-7 or MCF-7-iAs-180 were transfected with a genetically encoded construct encompassing a Sox2/Oct4 responsive element (SORE6) upstream of GFP. This “stemness” reporter called SORE6-GFP indicated stem cells. Cells were, then platted under low adherence conditions and grown into organoids as in A. Cells displaying GFP positivity were counted using an inverted fluorescence microscope. (C) Quantification of results shown in (B) displayed as averages ± SEM of at least three independent biological replicates. **p < 0.01.
Chronic iAs exposure does not impact ER-target genes equally –
Based on the marked loss of ER protein expression in cells challenged with low level arsenic, we determined if iAs could also change the expression of ER-target genes, either with or without estradiol (E2) treatment. We observed that mRNA levels of the progesterone receptor gene (PGR) were significantly suppressed by chronic iAs, regardless of E2 treatment (Fig. 5). In contrast, levels of another ER-target gene, pS2, were attenuated by iAs in the absence of E2, but stimulated by iAs in the presence of E2. Interestingly, washing iAs out for 16 days (“recovery”) produced a third distinct pattern of gene expression in MCF-7 cells. It caused partial recovery of PGR gene expression independently of E2-stimulation. Cells that underwent the iAs washout treatment showed increases in pS2 expression, both with and without E2. In fact, the washout cells treated with E2 showed levels of pS2 expression nearly 3-fold over cells stimulated with E2 alone. Two additional ER-target genes, GREB1 and EGR3, were attenuated by chronic iAs. While GREB1 expression was unaffected by the 16-day washout period, EGR3 expression was partially recovered. Taken together, these results indicate that iAs differentially affects ER-target genes, suppressing or amplifying mRNA levels in a gene specific manner.
Figure 5. Functional analysis of ER activity assessed by examining target gene expression by RT-QPCR.
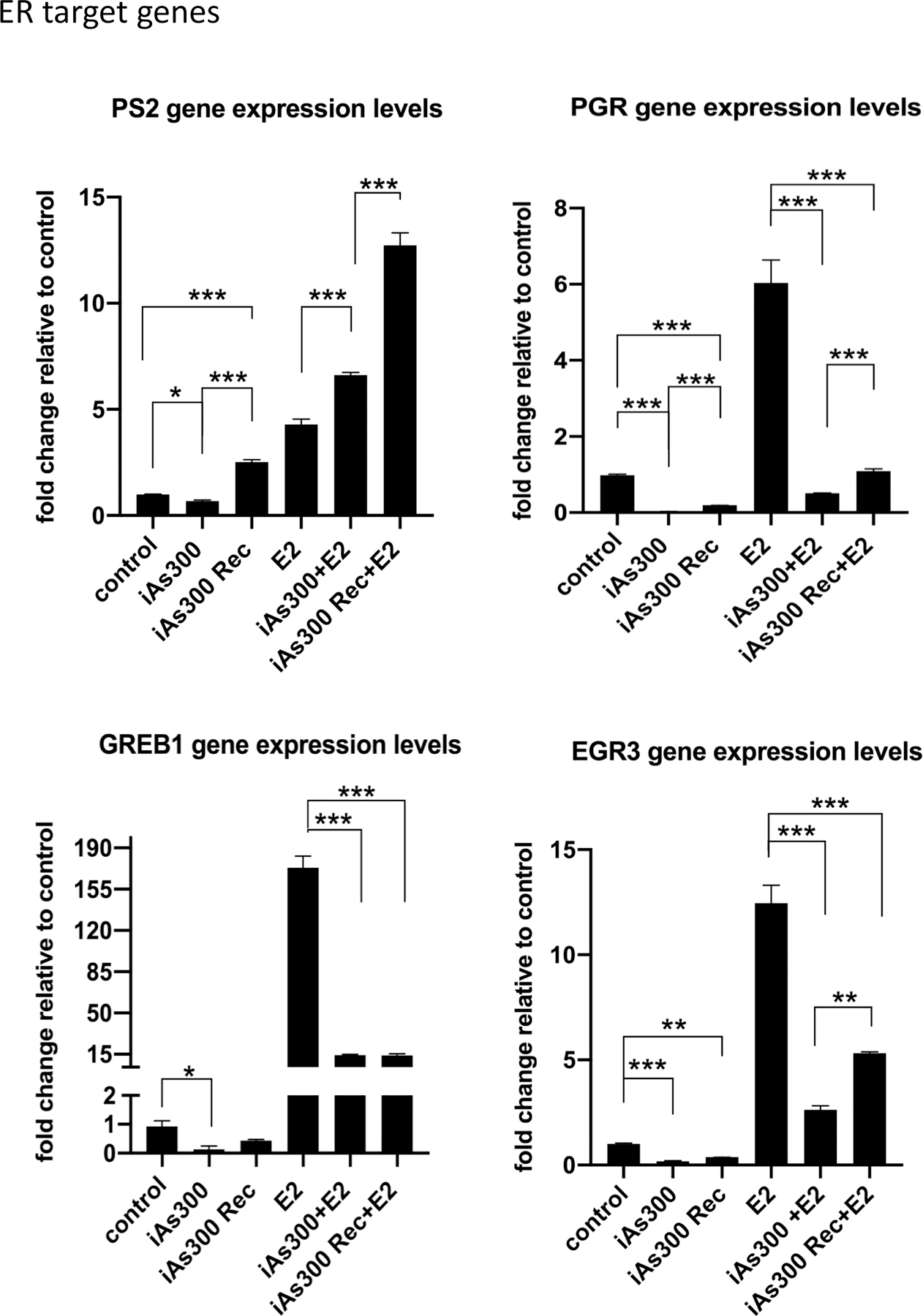
(A-D) RT-QPCR analysis of selected ER-target gene expression in MCF-7 cells as well as MCF-7 exposed to iAs (100 nM) for 300 days prior and after a 16 days recovery period. Analysis were performed at baseline and after stimulation with E2. Results are presented as averages of at least three independent biological replicates ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
iAs suppresses the MCF-7 cell response to progesterone –
The observation that chronic iAs strongly suppressed progesterone receptor (PGR) gene expression suggested that PGR function might be impacted as well, prompting our analysis of PGR-targeted gene expression. Results shown in Fig. 6 demonstrate that iAs suppresses PGR mRNA, and this is not rescued by exogenous progesterone (P4) treatment. Data also indicated that this has functional implications, as well, because the potent suppression of the PGR gene is mirrored by the suppression of the PR-target, FKBP5, whose expression was abolished by iAs treatment. Importantly, FKBP5 levels were not significantly increased by P4 stimulation further indicating PR inactivation. These results indicate that chronic iAs exposure strongly inhibits the cellular response to P4 most likely via suppressing both PGR expression and PR function.
Figure 6. RT-QPCR analysis of progesterone receptor (PGR) as well as PR-target gene FKBP5.
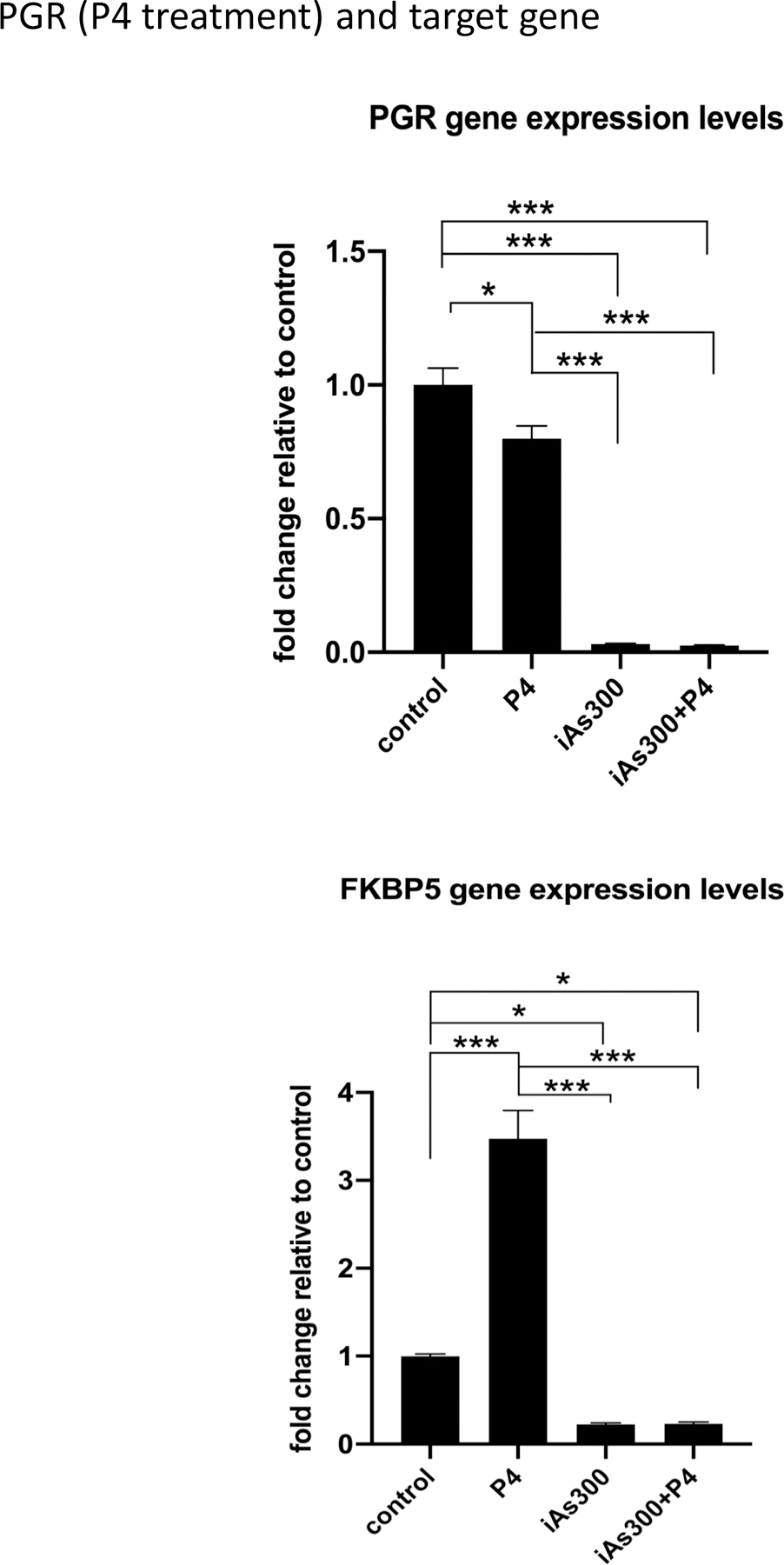
(A) RT-QPCR gene expression analysis of PGR in MCF-7 cells and MCF-7 cells exposed to iAs (100 nM) for 300 days prior and after P4 stimulation. (B) same as (A) but FKBP5 gene expression was analyzed. Results are presented as averages of at least 3 independent biologic replicates ± SEM. *p < 0.05; ***p < 0.001.
RNA-Seq analysis reveals both reversible and persistent transcriptome changes in response to chronic iAs.
We observed profound changes in the transcription of genes regulated by the estrogen receptor pathway with chronically exposed cells and sought to determine how widely iAs treatment affected these cells. RNA-Seq analysis allowed us to ascertain both reversible and persistent effects of iAs exposure on gene networks, particularly those involved in phenotypic transitions. For these analyses, our treatment conditions led to large numbers of differentially expressed genes (DEGs), defined as having mRNA fold changes > 2 and a False Discovery Rate (FDR) < 0.05 compared to controls. Specifically, compared to untreated MCF-7 cells, chronic iAs treatment alone led to 1708 DEGs, iAs treatment with estrogen yielded 3226 DEGs, and iAs treated cells after the 16 day washout recovery period had 3011 DEGs (Fig. 7A). Among these, a total of 855 DEGs changed in all treatment groups, while subsets of DEGs changed only in cells exposed to estrogen alone compared to control cells (542 genes), estrogen and arsenic combinations compared to E2 alone (549 genes), and the washout “recovery” cells compared to iAs treated (341 genes) (Fig. 7A). Plotting the normalized counts for 1708 genes specifically regulated by estrogen revealed the clustering of genes into four major expression patterns (Fig. 7B, color bars). The 427 genes in cluster 1 (green), upregulated by estrogen, becomes downregulated by arsenic and remained at that level even after the recovery period (Fig. 7B). Cluster 2 (purple) contained 323 genes which were upregulated by estrogen and remained upregulated both, upon arsenic treatment or after the washout period (Fig. 7B). Cluster 3 (orange) consisted of 381 genes that are downregulated by estrogen, which however become upregulated in iAs exposed group and remain that way even after recovery. Cluster 4 (red) consisted of 577 genes that were downregulated by estrogen and remained suppressed in cells previously exposed to iAs and after washout (Fig. 7B). These patterns of expression reveal distinct gene networks in breast cancer cells that respond differently to either iAs alone or iAs in combination with E2. In addition, while some of these chronic iAs-induced changes reverted after washing iAs out, some changes persisted even after iAs was removed. Thus, we infer that iAs exposure creates a ‘cellular memory’ of exposure that is detectable in the observed changes to the transcriptome of breast cancer cells. Moreover that these lasting changes are likely to have important functional implications for emerging cancer cell phenotypes.
Figure 7. RNA-seq analysis of MCF-7 cells exposed to iAs followed by a recovery period.
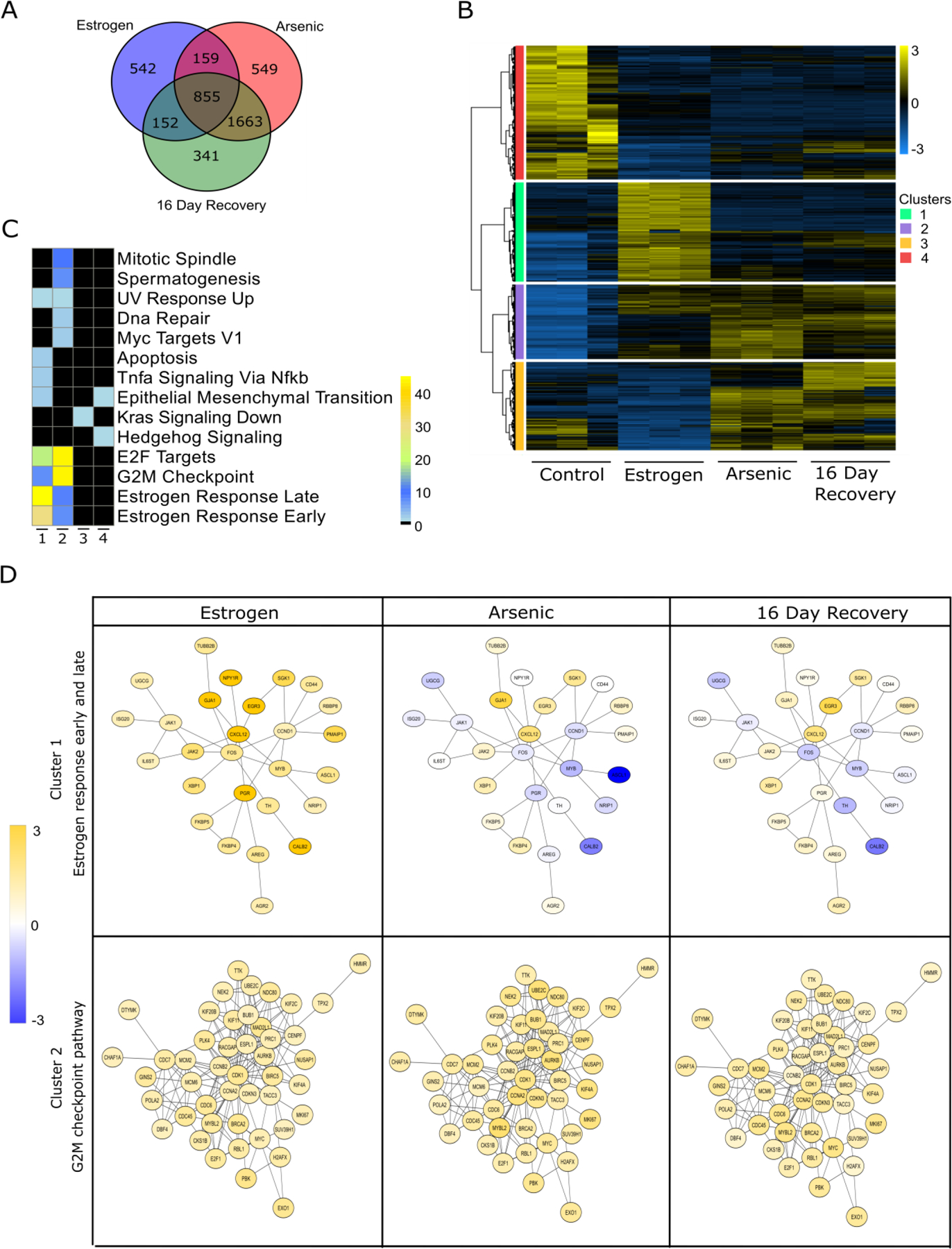
(A) Venn diagram of differentially expressed genes (DEGs) in cells exposed to iAs (100 nM) for 300 days prior to and after a 16 days recovery period. Analysis were performed at baseline and after stimulation with E2. (B) RPKM expression levels of DEGs normalized to the Z scale and plotted for different conditions with respect to control. Hierarchical clustering was performed to obtain clusters of gene expression patterns. (C) DEGs from each cluster in (B) underwent pathway enrichment analysis with RITAN. Color scale represents log10 (FDR) for pathway significance with black indicating no significance. (D) Protein-protein interaction networks of specific pathways present in clusters 1 and 2. Gene fold changes with respect to controls are represented in yellow (upregulation), blue (downregulation) and white (no change).
To support this connection between the transcriptional changes and cellular functional alterations, we investigated the biological roles of the genes found in the different clusters by performing gene enrichment analysis using the R package rapid integration of term annotation and network (RITAN) resources (19) and the molecular signature hallmark database. Intriguingly, cluster 1 is enriched for genes involved in the early and late responses to E2, which is not surprising given their strong induction by it. The loss of this program after iAs treatment is consistent with our observations of attenuation of ER signaling in the cells (Fig. 7C). Cluster 2 had genes that were highly enriched for cellular proliferative pathways, such as the G2M checkpoint and E2F targets (Fig. 7C). Intriguingly, Cluster 3, which showed some of the most potent changes in response iAs treatment, was enriched in genes associated with downregulation of Kras signaling. This suggests these genes may represent attractive prognostic targets for iAs exposed patients. Cluster 4 contained genes that participate in epithelial to mesenchymal transition and hedgehog signaling (Fig. 7C). Suppl. Fig. 3A shows a heatmap analysis of the levels of expression of some of these differentially expressed genes related to processes like epithelial to mesenchymal transition (EMT), metastasis, proliferation and stemness reprogramming in the context of E2 stimulation. We also verified that changes in gene expression correlated with proportional changes in protein levels (Suppl. Fig. 4). We also investigated how genes in cluster 1 and cluster 2 that were enriched in estrogen responses and G2M checkpoint respectively interacted with each other across the conditions. For this purpose, we used RITAN and cystoscope software to build and visualize protein-protein interaction networks that are encoded by genes that form clusters 1 and 2. We observed the E2 upregulates both hormone response and G2M checkpoint networks However, iAs suppressed hormone response networks when combined with E2 and this suppression was maintained after washout. Differently, G2M networks, upregulated by E2, remained upregulated in comparison to baseline in the presence of iAs and after washout. Since our cellular studies indicate that estrogen receptor signaling enforces differentiated luminal epithelial identity, the results shown in Fig. 7 demonstrate that prolonged exposure to environmentally relevant concentrations of iAs suppresses luminal lineage commitment, likely by causing persistent alterations in ER and PR signaling.
iAs persistently changes breast cancer cell phenotypes leading to the emergence of metastatic phenotypes from luminal A cancer cells –
MCF-7 cells are prototypic ER+/PR+ luminal A breast cancer cellular subtype. Cancer cells of this type normally grow tumors in situ in patients and in immunocompromised mice, particularly when E2 is available. According to data presented here, prolonged iAs exposure disrupts the expression of luminal lineage markers as well as promotes the acquisition of less differentiated phenotypes that are display low ER expression and are negative for PR. In humans ERlow/PR- breast cancer phenotypes are fairly aggressive, present many characteristics of more basal carcinomas and are prone to metastatic spreading. To test in vivo whether iAs promotes luminal to basal transition that favors the emergence of aggressive basal subtypes, we injected either control MCF-7 or MCF-7 cells cultured in the presence of iAs for 300 days into the tail vein of female NOD/SCID mice. Cells were transduced with luciferase to allow real time monitoring that was performed for up to 3 months. This experiment showed that MCF-7 cells treated with iAs have increased tumorigenic capacity and spread to distal non-mammary tissues (Fig. 8). Interesting, while injected cells accumulated in the lungs immediately after injection (Suppl. Fig. 5), recurrent tumors were most frequently seen in the lower body/pelvic region. These results provide compelling evidence that exposure to iAs causes persistent changes to ER+/PR+ MCF-7 breast cancer cells that enhance both their tumorigenic as well as metastatic potential compared to parental cells. Thus, we conclude that iAs is a potent stimulant of tumor growth and metastasis in vivo acting primarily via a luminal to basal transition mechanism.
Figure 8. In vivo analysis of systemic tumorigenesis induced by MCF-7 cells injected via tail vein.
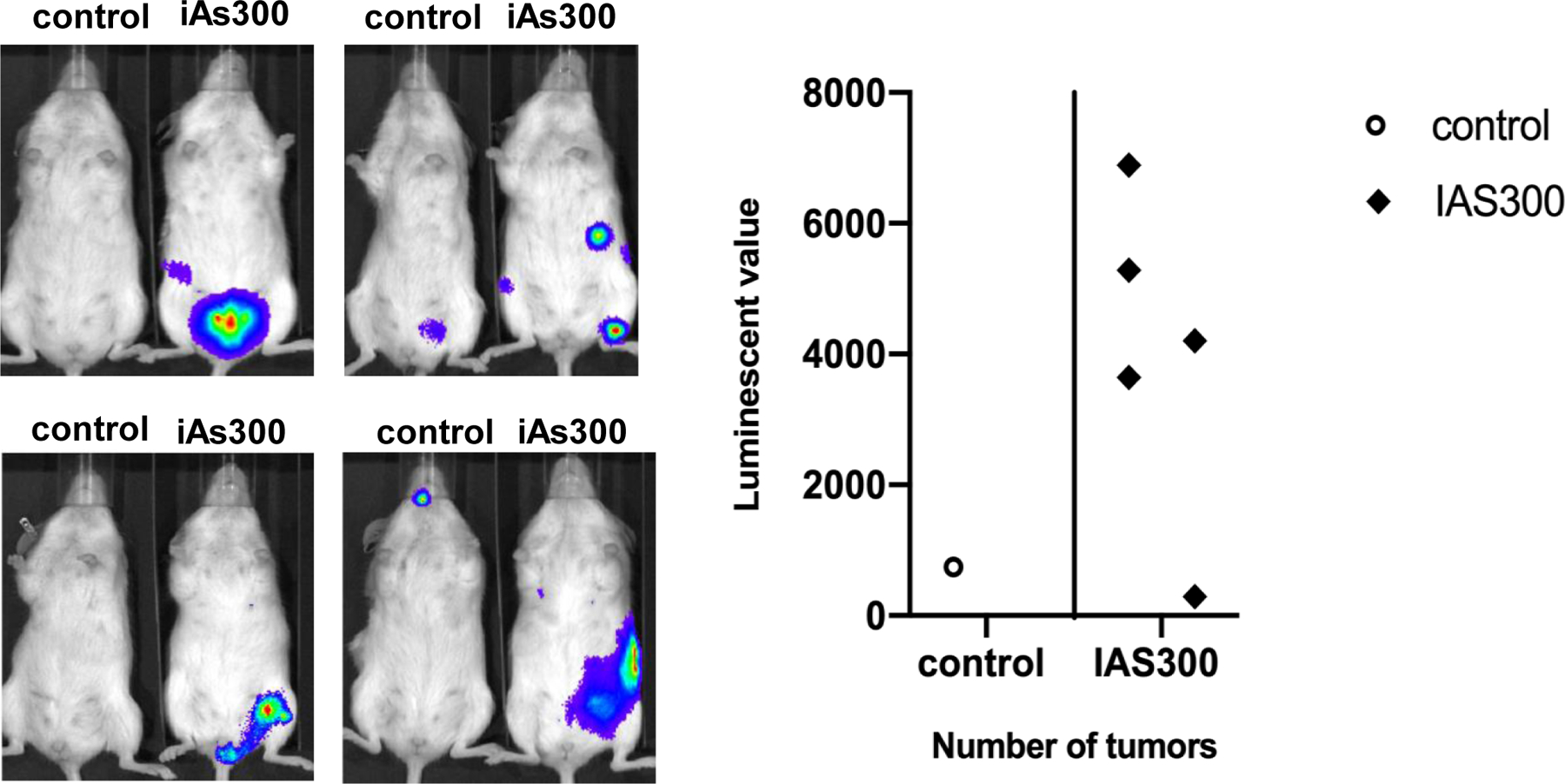
(A) NOD/SCID immunocompromised mice were injected with MCF-7 or MCF-7 exposed to iAs (100 nM) for 300 days (iAs300) via tail vein. Mice were injected with 1×106 luciferase transduced cells per mouse. Tumorigenesis was analyzed after 3 months by injecting firefly luciferin i.p.. Whole body photon count was quantified using an IVIS spectrophotometric system. (B) Quantification of maximum photon counts measured from each individual mouse. Each dot represents a different mouse.
DISCUSSION
Arsenic, in the form of arsenic oxides (iAs), is a major contaminant that occurs naturally in watersheds and soils worldwide, including some densely populated areas. It is an endemic carcinogen to which large numbers of people are chronically exposed around the globe. Although there is debate about the role of iAs as a primary breast carcinogen, we sought to understand the capacity of breast cancer to evolve to more lethal forms of the disease due to exposure to it. To model in vitro a more environmentally relevant exposure we used prolonged treatment (up to 300 days) at a fairly low concentration (up to 100 nM). We determined if these could cause lasting changes to ER+/PR+ cancer cells subsequently leading to reprogramming to more aggressive cancer phenotypes that are more challenging to treat and therefore more lethal. In addition, we sought to determine if eliminating the exposure after prolonged exposures could reverse the effects of iAs. Results presented here showed that iAs promotes changes at the transcriptome level that persist even after iAs is removed leading to phenotypic and functional changes at the cellular level. For instance, increased cellular proliferation with increased numbers of cells that can grow under anchorage independent conditions or invade Matrigel™ were observed indicating the emergence of more aggressive phenotypes.
At the molecular level, prolonged iAs treatment strongly suppressed ER at both mRNA, protein, and functional levels, as shown by the nearly complete suppression of the ER-target progesterone receptor gene (PGR) expression and progesterone receptor (PR) signaling. Though expected from the loss of ER, the near total ablation of PGR/PR expression and function led to a remarkable phenotypic change. Because ER and PR enforce a fully differentiated luminal phenotype, the marked suppression of both hormone receptors by iAs suggested the cells might be transitioning towards more basal phenotypes. These more aggressive phenotypes display increased lineage plasticity and are enriched for stem-cell like subpopulations. We assessed the possibility of an stem-like cell enrichment using a combination of cytokeratin markers (CK8 and CK5, for luminal and basal phenotypes, respectively) as well as a stem cell transcriptional reporter (SORE6). We found that iAs does indeed promote dedifferentiation and leads to the emergence of cells with stem cell characteristics. An important question that arose from these observations was whether these changes caused by iAs could be reversed by suspending iAs treatment. RNA-Seq experiments, therefore, compared naïve MCF-7 cells, chronically exposed and iAs exposed cells after a16-day washout (‘recovery’) period. These transcriptome wide studies indicated that although some of the genes disrupted by iAs are mostly or partially normalized, a significant proportion of the transcriptional program, particularly those associated with luminal identity regulated by hormone receptors, remained altered for weeks after iAs removal. The analysis of selected ER targets even one month after the removal of iAs confirmed that key, clinically relevant ER-targets, like PGR, remained suppressed, while pS2 changes were further accentuated regardless of estradiol treatment. These results suggest that exposure to iAs produces long lasting changes to the transcriptome affecting clinically relevant markers. Hence, experiments were extended to determining if these molecular changes sufficed to produce cancer phenotypes associated with worse prognosis. In this regard, in vivo experiments confirmed that the prolonged exposure to iAs converted the normally non-metastatic MCF-7 line into a more aggressive cancer cell that spawns phenotypes with a high capacity to produce recurrent metastatic lesions without estrogen supplementation (normally required for the growth of MCF7-derived tumors in mice). Our data indicated that changes caused to the transcriptome by chronic iAs exposure are lasting, not reversed weeks after cessation of the exposure suggesting cells conserve a “memory of exposure” that may critically impact breast cancer prognosis. Taken together, our results support the concept that iAs exposure, modeled after the kinds of environmental exposures suffered by people around the world, can promote or accelerate the evolution of ER+/PR+ luminal tumors with typically good prognosis, into more aggressive basal phenotypes with worsened evolution and poorer outcomes. This suggests that patients affected by luminal A breast cancer at risk of co-exposure to arsenic may require specific treatment strategies different than the standard of care for ER+ breast cancer.
Supplementary Material
Acknowledgements:
The authors would like to acknowledge financial support from the National Institute of Environmental Health Sciences (RO1ES028149 to M.G.B.); National Cancer Institute (RO1CA216882 to M.G.B.); National Institute of Allergy and Infectious Diseases (RO1AI131267 to M.G.B.) as well as funding from the Wisconsin Breast Cancer Showhouse to M.G.B. and the Advancing a Healthier Wisconsin Endowment to M.G.B.
ABBREVIATIONS LIST
- iAs/As2O32−
Inorganic arsenic
- MCF7
breast cancer cell line
- iAs300
MCF7 cells treated with inorganic arsenic for 300 days
- ER
Estrogen receptor
- PR
Progesterone receptor, α and β subtypes
- E2
Estradiol
- P4
Progesterone
- PGR
Progesterone receptor gene
- CD24 and CD44
cell membrane markers
- SORE6
Sox2/Oct4 response element six tandem repeats
- Sox2
SRY-Box Transcription Factor 2
- Oct4
Octamer-binding transcription factor 4
- CK-8
cytokeratin 8
- CK-5
cytokeratin 5
- pS2
Trefoil factor 1 gene
- GREB1
Growth Regulating Estrogen Receptor Binding 1 gene
- EGR3
Early Growth Response 3 gene
- FKBP5
FKBP Prolyl Isomerase 5 gene
- G2M
Cell cycle checkpoint
- E2F
E2F transcription factor
- DEG
Differentially expressed genes
- FDR
False discovery rate
- Kras
Kras proto-oncogene
- RITAN
R package rapid integration of term annotation and network
REFERENCES
- 1.Cardoso APF, Al-Eryani L, States JC, Arsenic-Induced Carcinogenesis: The Impact of miRNA Dysregulation. Toxicol Sci 165, 284–290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie H, Huang S, Martin S, Wise JP Sr., Arsenic is cytotoxic and genotoxic to primary human lung cells. Mutat Res Genet Toxicol Environ Mutagen 760, 33–41 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y et al. , Chronic exposure to arsenic and high fat diet induces sex-dependent pathogenic effects on the kidney. Chem Biol Interact 310, 108719 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Eryani L et al. , Differentially Expressed mRNA Targets of Differentially Expressed miRNAs Predict Changes in the TP53 Axis and Carcinogenesis-Related Pathways in Human Keratinocytes Chronically Exposed to Arsenic. Toxicol Sci 162, 645–654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlin DJ et al. , Arsenic and Environmental Health: State of the Science and Future Research Opportunities. Environ Health Perspect 124, 890–899 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khanjani N, Jafarnejad AB, Tavakkoli L, Arsenic and breast cancer: a systematic review of epidemiologic studies. Rev Environ Health 32, 267–277 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Xu Y, Tokar EJ, Waalkes MP, Arsenic-induced cancer cell phenotype in human breast epithelia is estrogen receptor-independent but involves aromatase activation. Arch Toxicol 88, 263–274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Esquinas E et al. , Arsenic exposure and cancer mortality in a US-based prospective cohort: the strong heart study. Cancer Epidemiol Biomarkers Prev 22, 1944–1953 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman M, Ahsan A, Begum F, Rahman K, Epidemiology, Risk Factors and Tumor Profiles of Breast Cancer in Bangladeshi underprivileged women. Gulf J Oncolog 1, 34–42 (2015). [PubMed] [Google Scholar]
- 10.Chatterjee K, Bhaumik G, Chattopadhyay B, Estrogen receptor and progesterone receptor status of breast cancer patients of eastern India: A multi-institutional study. South Asian J Cancer 7, 5–6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Telli ML et al. , Asian ethnicity and breast cancer subtypes: a study from the California Cancer Registry. Breast Cancer Res Treat 127, 471–478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen GC, Guan LS, Hu WL, Wang ZY, Functional repression of estrogen receptor a by arsenic trioxide in human breast cancer cells. Anticancer Res 22, 633–638 (2002). [PubMed] [Google Scholar]
- 13.Selmin OI, Donovan MG, Skovan B, Paine-Murieta GD, Romagnolo DF, Arsenic induced BRCA1 CpG promoter methylation is associated with the downregulation of ERalpha and resistance to tamoxifen in MCF7 breast cancer cells and mouse mammary tumor xenografts. Int J Oncol 54, 869–878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoica A, Pentecost E, Martin MB, Effects of arsenite on estrogen receptor-alpha expression and activity in MCF-7 breast cancer cells. Endocrinology 141, 3595–3602 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Davey JC, Bodwell JE, Gosse JA, Hamilton JW, Arsenic as an endocrine disruptor: effects of arsenic on estrogen receptor-mediated gene expression in vivo and in cell culture. Toxicol Sci 98, 75–86 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Hart PC et al. , Caveolin-1 regulates cancer cell metabolism via scavenging Nrf2 and suppressing MnSOD-driven glycolysis. Oncotarget 7, 308–322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalari KR et al. , MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinformatics 15, 224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarthy DJ, Chen Y, Smyth GK, Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmermann MT, Kabat B, Grill DE, Kennedy RB, Poland GA, RITAN: rapid integration of term annotation and network resources. PeerJ 7, e6994 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liberzon A et al. , The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szklarczyk D et al. , The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39, D561–568 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasad TS, Kandasamy K, Pandey A, Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol Biol 577, 67–79 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Schaefer CF et al. , PID: the Pathway Interaction Database. Nucleic Acids Res 37, D674–679 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shannon P et al. , Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang B et al. , A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Reports 4, 155–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez-Carrillo L, Gamboa-Loira B, Gandolfi AJ, Cebrian ME, Inorganic arsenic methylation capacity and breast cancer by immunohistochemical subtypes in northern Mexican women. Environ Res 184, 109361 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Ruiz-Ramos R, Lopez-Carrillo L, Albores A, Hernandez-Ramirez RU, Cebrian ME, Sodium arsenite alters cell cycle and MTHFR, MT1/2, and c-Myc protein levels in MCF-7 cells. Toxicol Appl Pharmacol 241, 269–274 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Luo M, Brooks M, Wicha MS, Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des 21, 1301–1310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C, Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 24, 65–78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF, Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100, 3983–3988 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.