

Abstract
Aside from the first week postnatal, murine heart regeneration is restricted and responses to damage follow classic fibrotic remodeling. Recent transcriptomic analyses have suggested that significant cross talk with the sterile immune response could maintain a more embryonic-like signaling network that promotes acute, transient responses. However, with age, this response—likely mediated by neonatal yolk sac macrophages—then transitions to classical macrophage-mediated, cardiac fibroblast (CF)-based remodeling of the extracellular matrix (ECM) after myocardial infarction (MI). The molecular mechanisms that govern the change with age and drive fibrosis via inflammation are poorly understood. Using multiple ribonucleic acid sequencing (RNA-Seq) datasets, we attempt to resolve the relative contributions of CFs and macrophages in the bulk-healing response of regenerative (postnatal day 1) and nonregenerative hearts (postnatal day 8+). We performed an analysis of bulk RNA-Seq datasets from myocardium and cardiac fibroblasts as well as a single-cell RNA-Seq dataset from cardiac macrophages. MI-specific pathway differences revealed that nonregenerative hearts generated more ECM and had larger matricellular responses correlating with inflammation, produced greater chemotactic gradients to recruit macrophages, and expressed receptors for danger-associated molecular patterns at higher levels than neonates. These changes could result in elevated stress-response pathways compared with neonates, converging at NF-κB and activator protein-1 (AP-1) signaling. Profibrotic gene programs, which greatly diverge on day 3 post MI, lay the foundation for chronic fibrosis, and thus postnatal hearts older than 7 days typically exhibit significantly less regeneration. Our analyses suggest that the macrophage ontogenetic shift in the heart postnatally could result in detrimental stress signaling that suppresses regeneration.
NEW & NOTEWORTHY Immediately postnatal mammalian hearts are able to regenerate after infarction, but the cells, pathways, and molecules that regulate this behavior are unclear. By comparing RNA-Seq datasets from regenerative mouse hearts and older, nonregenerative hearts, we are able to identify biological processes that are hallmarks of regeneration. We find that sterile inflammatory processes are upregulated in nonregenerative hearts, initiating profibrotic gene programs 3 days after myocardial infarction that can cause myocardial disease.
Listen to this article’s corresponding podcast at https://ajpheart.podbean.com/e/macrophage-regulation-of-matrix-remodeling/.
Keywords: fibrosis, gene expression and regulation, inflammation, myocardial infarction, myocardial regeneration
INTRODUCTION
The heart wall is often mistakenly viewed as being enriched in contractile cells, but cardiomyocytes only compose ∼25% of the myocardium; endothelial cells (∼60%), and cardiac fibroblasts (∼15%) make up the majority of the tissue along with other smaller cell populations (1). During myocardial infarction (MI), coronary artery occlusion results in ischemic injury to cardiac tissue, which recruits several white blood cell populations (2). The resulting sterile inflammatory cascade begins with neutrophils, mast cells, and macrophages sensing damage-associated molecular patterns (DAMPS) or hypoxia (3, 4). Responding cells pick up molecular cues from the microenvironment, which then dictate their inflammatory status (5, 6). Once educated, these cells are able to directly or indirectly steer tissue resorption, growth, and extracellular matrix (ECM) deposition, as well as recruit regulatory T-lymphocytes to quench the inflammatory process (7). What naturally results from excessive matrix production is a noncontractile, rigid scar that dramatically reduces heart ejection fraction.
Although the steps in this process are well known, contributions by individual resident myocardial cell types, the specific molecular pathways they utilize, and how they change with age are not completely clear. For example, epicardially derived cardiac fibroblasts (CFs) are the primary ECM producers in the heart and secrete a wide variety of scaffolding proteins for parenchymal cells (8). They also sense and respond to many structural and secreted cues, e.g., proinflammatory signals to increase their contractility and ECM assembly (9) as they become “myofibroblasts.” In addition to traditional cues such as transforming growth factor-β (TGF-β), fibroblasts can be activated via Toll-like receptors (TLRs), such as TLR2 and TLR4 (10). Both receptors are promiscuous and bind to many DAMPS, including lipopolysaccharide (LPS), low-molecular-weight hyaluronic acid (LMW HA), the chromatin binding protein HMGB1, and others (11). The end result of this signaling is increased collagen I, collagen III, and fibronectin synthesis, matrix cross-linking, and secretion of ECM-binding proteins (12–17) that create a stiff scar and can induce myofibroblast transdifferentiation (18). Scar formation is also balanced by ECM degradation rate; fibroblast-secreted matrix metalloproteinases, metallopeptidases, calpain, cathepsins, and caspases enzymatically digest and help recycle matrix (19), whereas tissue inhibitors of metalloproteases (TIMPs) skew the equilibrium toward matrix deposition. Chronically, age-associated heart stiffening is both a symptom as well as an agonist of disease (20).
Alongside resident CFs, macrophages are also present early in heart development and arise from yolk sac (YS) progenitors that migrate between developing organs before differentiating into tissue-specific, resident macrophages (21, 22). Later in development, definitive hematopoiesis generates marrow-derived monocytes (23), which are also recruited to the myocardium and then become macrophages. YS and bone marrow-derived macrophages (BMDMs) appear functionally distinct in many organs, including responding differently to pathological cues (24, 25); for example, lymphatic vessel endothelial receptor 1 (LYVE1) and T-cell immunoglobulin and mucin domain containing 4 (TIMD4) are restricted to the YS-lineage (4) and facilitate hyaluronan binding (26) and phagocytosis (27), respectively, to help “cloak” proinflammatory signals (28). In contrast, more BMDMs are recruited after infarction (4) via well-documented chemokine cascades, e.g., CCL2-CCR2, that create acute inflammation (29) detrimental to the repair process (30). Yet, once these BMDMs have integrated with the destination tissue, they become transcriptionally similar to cardiac YS macrophages (4) but without the regenerative capacity. Thus, YS macrophages may be the only proregenerative subpopulation in the heart.
Differences in developmental linage, as well as tissue priming and response to pathogens can lead to a diverse set of macrophage phenotypes. Although many groups have relied on the M1/M2 dichotomy that was introduced in the late 1990s to describe macrophages impacted by a Th1 or Th2 response (31), this system fails to encompass the plurality of characteristics that macrophages exhibit (32). Therefore, additional discussion of macrophage phenotype and marker expression will be primarily described by functional attributes for the remainder of this study.
The complex signaling networks introduced above imply that matrix expression and healing are not simply composed of “on” or “off” cues. Identifying clusters of genes that are coordinated by conserved regulatory mechanisms and are involved in CF-macrophage cross talk may more easily identify mechanism(s) and reveal better targets for therapy. Thus, we analyzed immediately postnatal (P1) and 1+ week old (P8) ribonucleic acid sequencing (RNA-Seq) datasets from bulk ventricular tissue (33) (GSE123868), sorted cardiac fibroblasts (34) (GSE49906), and single-cell macrophages (4) (GSE119355) with the goal of elucidating age and MI-dependent programmatic changes. Although the bulk ventricular data set contains all experimental groups (postnatal day 1 and day 8 and MI/sham), it lacks the cellular resolution to understand macrophage and cardiac fibroblast signaling differences. Therefore, we employ the latter two datasets to attribute tissue-level changes to either cell type. Together, the literature and our analyses suggest that the window of opportunity for successful regeneration, likely mediated by CF cross talk with YS macrophages rather than with BMDMs, is restricted to 3 days post MI.
METHODS
Bulk RNA-Seq Processing
Sequencing files were obtained from the Gene Expression Omnibus database under accessions GSE123868, GSE49906, and GSE119355. Bulk FASTQ files were aligned to the mm10 genome using STAR with the following settings: –readFilesCommand zcat –genomeLoad LoadAndRemove –outFilterType BySJout –outFilterMultimapNmax 10 –alignSJoverhangMin 8 –alignSJDBoverhangMin 1 –outFilterMismatchNmax 4 –alignIntronMin 20 –alignIntronMax 1000000 –alignMatesGapMax 100000. BAM files were sorted and indexed using samtools. Raw and transcripts per kilobase million (TPM) normalized tag directories were generated using HOMER command makeTagDirectory and analyzeRepeats scripts. Statistical significance for Giudice et al. (35) and Wang et al. (33) raw counts were determined using EdgeR (36) and DESEQ2 (37) in the getDiffExpression HOMER script, respectively, based on replicate numbers (no replicates justified EdgeR, replicate of 3 justified DESEQ2). Biological process and molecular function gene ontologies were generated using PANTHER (38) overrepresentation with a Bonferroni test. Graphs of TPM-normalized values were generated using R and ggplot and pheatmap packages. Principal component analysis (PCA) plots were generated using ClustVis (39).
Single-Cell RNA-Seq Processing
Single-cell macrophage data (4) was read into the Seurat (40) package of R (v. 3.1) using provided matrix and TSV files. Data were filtered by selecting only cells with 1,000 to 5,800 features (referring to unique genes), where those features were detected in at least three cells. Cells with >18% mitochondrial reads were removed from analysis due to indicating apoptosis. Default log normalization was performed across separate Seurat objects for control, infarcted, and combined datasets. Differential gene expression was determined using default FindVariableFeatures parameters, and then the data were scaled to regress mitochondrial counts. Dimensionality of each data set was determined using Elbow and JackStraw Plots for each Seurat object. A chemokine-receptor-specific Seurat object was subset from the original combined object, selecting only cells with expression of CCR2 > 1, CXCR4 > 1, and Timd4 > 0.5, encompassing 60% of the original cells. Values were experimentally determined based on yielding a significant sample size and specific marker expression of the cells that best binned into the three categories while minimizing noise. This object was used to generate Spp1 and Ccl24 plots from Fig. 1.
Figure 1.
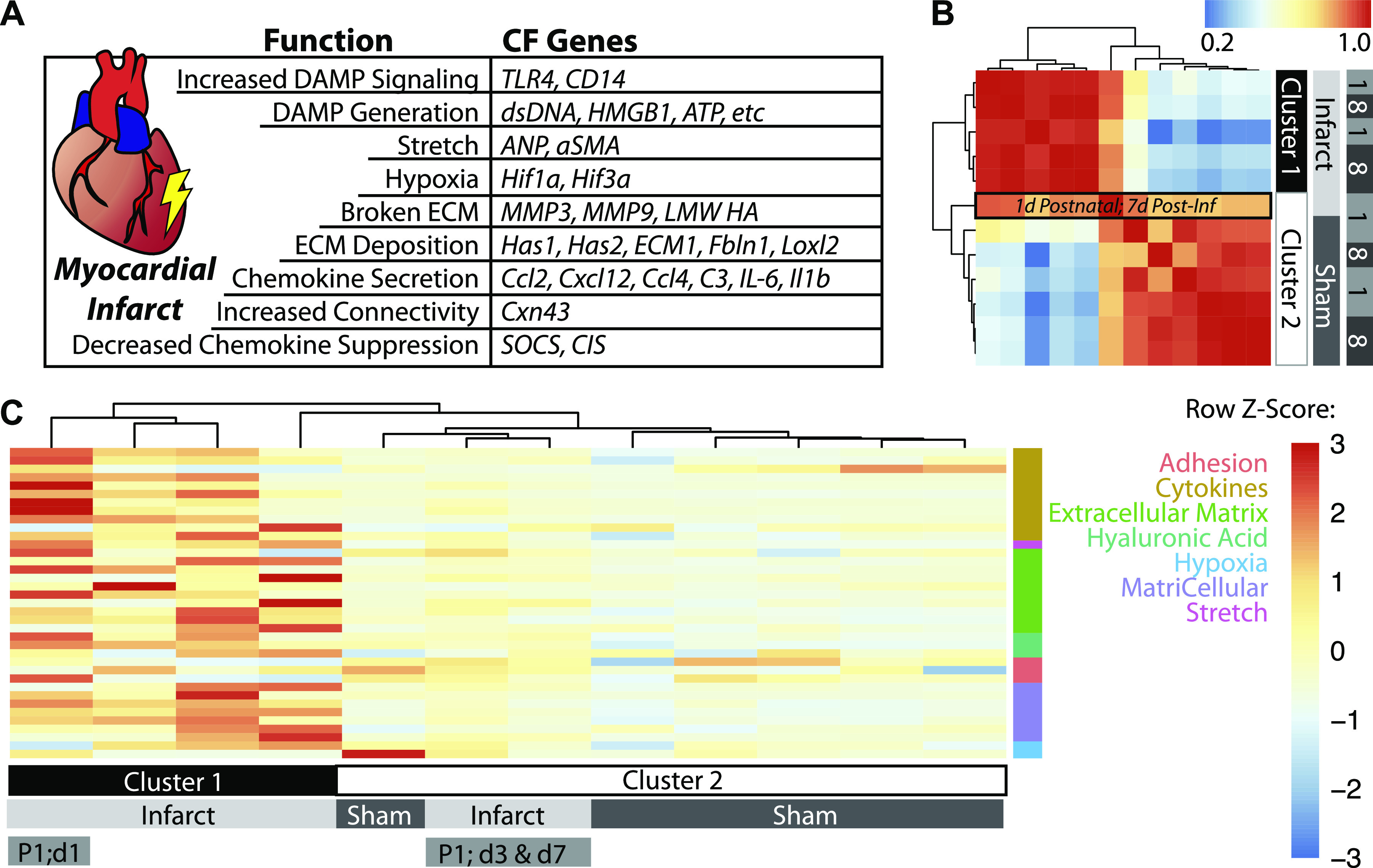
Transcriptomic analyses of infarct and sham bulk highlight changes in specific remodeling pathways. A: table of gene groupings and corresponding genes that literature suggest are differentially expressed with myocardial infarction. B: heatmap of bulk RNA-Seq data (averaged across three mice per group) showing hierarchical clustering of myocardia based on infarction. The only MI group that clustered with sham controls is indicated in the black box and is the 1 day postnatal MI group after 7 days of healing. C: heatmap of the TPM Z-scores of the 37 genes, with rows grouped by functional process and columns clustered using K-means. Cluster 1 denotes all the infarct groups from day 8 postnatal mice (independent of days postinfarct) and the first time point after infarction for day 1 mice. Days 3 and 7 postinfarct groups of postnatal day 1 infarcted hearts clustered with sham samples, indicating their return to baseline in as little as 3 days. MI, myocardial infarction; RNA-Seq, ribonucleic acid sequencing; TPM, transcripts per kilobase million.
Statistical Analyses
Statistical significance was determined using default parameters of HOMER’s DESEQ2 for bulk myocardium and EdgeR for cardiac fibroblast datasets, respectively. For DESEQ2 (v. 1.22.1), samples were grouped by age, time point, and infarction status, with each permutation representing a treatment in the design matrix. Counts were generated using the DESEQDataSetFromMatrix command and compared using a Wald test and corrected using the Benjamini–Hochberg procedure. P values, log2 fold change, and adjusted P values were generated for each gene and filtered manually using R, selecting only genes with an adjusted P value of 0.05, minimum fold change of ±2, and minimum 32 tags in one data set per gene. For EdgeR (v. 3.26.0), counts were read in using DGEList, whereas library sizes and normalization factors were calculated from Tag Directory sizes. Reads were counted using DGEList, with each sample constituting a treatment in the design matrix. Common dispersion was estimated at 0.05 as recommended. P values were generated using an Exact Test and corrected using the Benjamini–Hochberg method. As before, only genes with an adjusted P value of 0.05, minimum fold change of ±2, and minimum 32 tags in one data set per gene. For single-cell data, FindMarkers was used to identify gene landmarks of clusters. Summarily, a log(variance) and log(mean) relationship was determined using local polynomial regression and variance was calculated using standardized values after clipping to a maximum. Statistical significance was calculated by only comparing positive markers with a minimum log2 fold change of 0.25 and minimum percentage positive of 0.25 using a Wilcoxon test.
RESULTS AND DISCUSSION
The onset of a myocardial infarction produces many biological, chemical, and physical signals that activate the microenvironment: ECM degradation, excessive wall stretch, necrotizing cells that release damage-associated molecular patterns (DAMPS) and disrupt cell-cell communication, and hypoxia. These signals result in an increase in ECM deposition, upregulation of cell adhesion molecules, an increase in DAMP sensitivity, chemokine production, and reduction of cytokine suppressors. Each of these signals may impact cell types antagonistically or synergistically, so analyses here focus on key myocardial cell types and the genes that regulate their behavior. More specifically, we analyzed gene transcription in critical functional categories mentioned above and found 37 that were statistically significant and differentially regulated with age and infarction in the bulk ventricular data set (Fig. 1A).
Hierarchical clustering of these genes in infarcted and sham myocardia from postnatal day 1 and day 8 (P1/P8) hearts showed clustering based on injury and not postnatal age, except for the P1 samples 1 wk after infarction (Fig. 1B). At this time point, prior analyses indicated functional recovery of the tissue (33). Since the clustering of our 37 genes mirror that of the whole transcriptome (i.e., Fig. 1B in Ref. 33), these genes are likely to play a key role in regeneration, or at least be representative of processes that drive global transcriptional changes. K-means clustering of the per-gene heatmap revealed activation of a distinct transcriptional program in P1 hearts immediately after infarction, in which every process except hypoxia was upregulated (Fig. 1C, Supplemental Table S1; see https://doi.org/10.6084/m9.figshare.14249012). To further highlight these genes and their functional groupings, we examined their change in expression over time postinjury. We found that regenerative hearts resembled sham more quickly after infarction (i.e., there were fewer differentially expressed genes); extracellular matrix genes largely returned to baseline after 7 days (Fig. 2A). In contrast, hearts infarcted at day 8 upregulated ECM and matricellular genes through day 7 (Supplemental Fig. S1), and had prolonged differential expression of cytokine genes. These data indicate that the acute response to MI of postnatal day 8 hearts activates gene programs associated with chronic fibrosis and are detectable as early as 3–7 days postinfarction.
Figure 2.

MI induces the largest transcriptomic changes initially in younger mice but older mice maintain significant transcriptional differences. A: MA plots show the relationship between the MI/sham gene ratio (i.e., fold change) and the transcript per million reads for murine myocardia infarcted 1 day postnatal and chased for up to 7 days post-MI as indicated in each panel. Gene functional groupings listed are annotated in the figure by color and with large data points for visualization (when individual gene is statistically significant by Wald test with Benjamini–Hochberg correction; q < 0.05). Gray data points are coded by size for significance but are not the 37 literature-identified genes used in the rest of the analysis. B: box-and-whisker plot indicate the changes, broken down by category, in gene ratio (MI/sham) between transcriptome sampled over time as indicated at bottom. Data for mice infarcted 1 day and 8 days postnatal are separated by a dashed line. C: stacked bar plot annotates the log2 of the fold change for genes of the indicated ontologies/functions (genes with ratios less than one result in a negative number). MI, myocardial infarction.
Expression differences over time indicate differential gene program acceleration/deceleration between time points; ECM, matricellular, cytokine, and hyaluronic acid (HA) signaling processes were among the most volatile after infarction, but day 1 hearts largely stabilized 3 days after infarction as their fold-change differences are markedly reduced at day 7. In contrast, processes such as hypoxia, stretch, and adhesion and growth underwent modest fold-change differences, but remained persistent in their expression over time (Fig. 2, B and C). These data are suggestive of two key changes in cardiac fibroblasts and macrophages that result from these transcriptional differences, namely, that inflammatory cascades (29, 41) cause cardiac fibroblasts to activate and resemble contractile myofibroblasts (9) (Fig. 3A), and that macrophages transition to a BMDM origin through activation of CCL2/7 and CXCL12 (Fig. 3B). To better understand how post-MI processes are affected by aging, we created a signaling model from our RNA-Seq meta-analysis and fibroblast literature; in this model in Fig. 3C, we map important inputs (red), which our meta-analysis shows are most differentially expressed, intermediaries (yellow), and resulting outcomes that are linked to poor patient prognosis (orange). The summary outcome is that nonregenerative day 8 hearts exaggerate hypoxic response, chemoattraction, and DAMP generation and sensitivity while losing embryonic-restricted growth signals through macrophages and fibroblasts. The culmination of these processes results in increased nuclear factor κB (NF-κB) and activator protein-1 (AP-1) signaling (yellow noted in Fig. 3C) that spurs matricellular and extracellular matrix protein production. Compositional differences between day 1 and day 8 hearts activate divergent responses, which either return to baseline or activate the processes highlighted here that ultimately become pathogenic. Each post-MI process in this model is explained in greater detail below, with background provided before analysis in each category.
Figure 3.

Model for cellular and molecular changes with age and infarction. A: age-related transcriptional differences in fibroblasts. As fibroblasts progress through development, they upregulate TLR2 expression and downregulate STAT3 and Igf2bp3. B: macrophage composition of the heart shifts from CCL24-producing YS lineage cells to two ontogenies of BMDMs: CXCR4+ and CCR2+. C: proposed molecular mechanism for nonregenerative cardiac fibroblasts. MI in postnatal day 8 hearts generate inflammatory ligands (red) to a greater extent than postnatal day 1 hearts, which are sensed to a greater extent by TLR2, and ultimately result in NF-κB and AP-1 activation (yellow). Lastly, the signal propagation results in excess matricellular protein and ECM deposition (orange). Signaling from receptors and continuing to the right is believed to occur in fibroblasts. AP-1, activator protein-1; BMDM, bone marrow-derived macrophages; ECM, extracellular matrix; MI, myocardial infarction; NF, necrosis factor; TLR, Toll-like receptors.
Chemokines, Cytokines, Suppressors, and Interferon Responses
Chemokines, a chemotactic subset of cytokines, are responsible for immune recruitment to the site of injury, and many of these proteins are expressed in response to infarction. When cross-referenced with a whole heart data set (33), several chemokines are differentially expressed between regenerative and nonregenerative hearts initially but decrease by day 3, e.g., Cxcl2 and Ccl3/4 (Supplemental Fig. S2A). In the context of the regenerative phenotype, Ccl3 binds Ccr1, which is expressed on YS macrophages, as well as some interferon-responsive and recruited macrophages that are unique to injury. Ccl3/4 also bind to Ccr5 (42), which is expressed across all populations (Supplemental Fig. S2C); these ligands likely serve as generic macrophage recruitment ligands. Cxcl2 recruits neutrophils by binding to Cxcr2 in the bone marrow, though neutrophils can also be recruited by Ccr2 and Ccr5 (42). This demonstrates that a wide variety of neutrophils and macrophages are recruited by regenerative hearts 24-h postinfarct and that their contribution to healing is less likely ontogenically based (since Ccl3/4 and Cxcl2 are promiscuous and do not recruit specific subsets) but rather dictated by the microenvironment.
In contrast to nonspecific and lowly expressed chemokines, Ccl2/7 have similar levels at day 1 but peak at day 3 in nonregenerative P8 hearts, whereas the expression is quenched in regenerative postnatal regenerative day 1 hearts (Fig. 4A, Supplemental Fig. S2A). In the context of the signaling network, Ccl7 is secreted by IFN-β stimulated monocytes and B cells to attract classical monocytes and neutrophils, which scavenge dead cells (2, 37, 38). IFN-β is secreted by nonregenerative cardiac fibroblasts at steady-state after myocardial remodeling (Supplemental Fig. S2B) and by macrophages that have phagocytosed dead cardiomyocytes (2). Blockade of either Ccl7 or IFN-β signaling increases fractional shortening, decreases infarct size, and improves survival after MI (2, 43). Similarities between Ccl2/7 and common receptor targets suggests redundancy in recruiting Ccr2+ monocytes. As a third expression pattern, we found that the second highest expressed chemokine, Cxcl12, is uniquely upregulated at day 7 in nonregenerative hearts (Fig. 4A); this ligand binds the receptor CXCR4 and could explain the appearance of this third subset of macrophages before harvest on postnatal day 11. Chemokine signatures of nonregenerative (postnatal day 8) hearts, e.g., Cxcl12 and Ccl2/7, are expressed orders of magnitude higher than less specific chemokines, and this very clearly differentiates ontogenies of macrophages post MI. These observations reinforce the concept that subset-specific chemokines likely ascribe function, while less specific chemokines broadly recruit cells that are informed by local environmental cues to reinforce regeneration.
Figure 4.
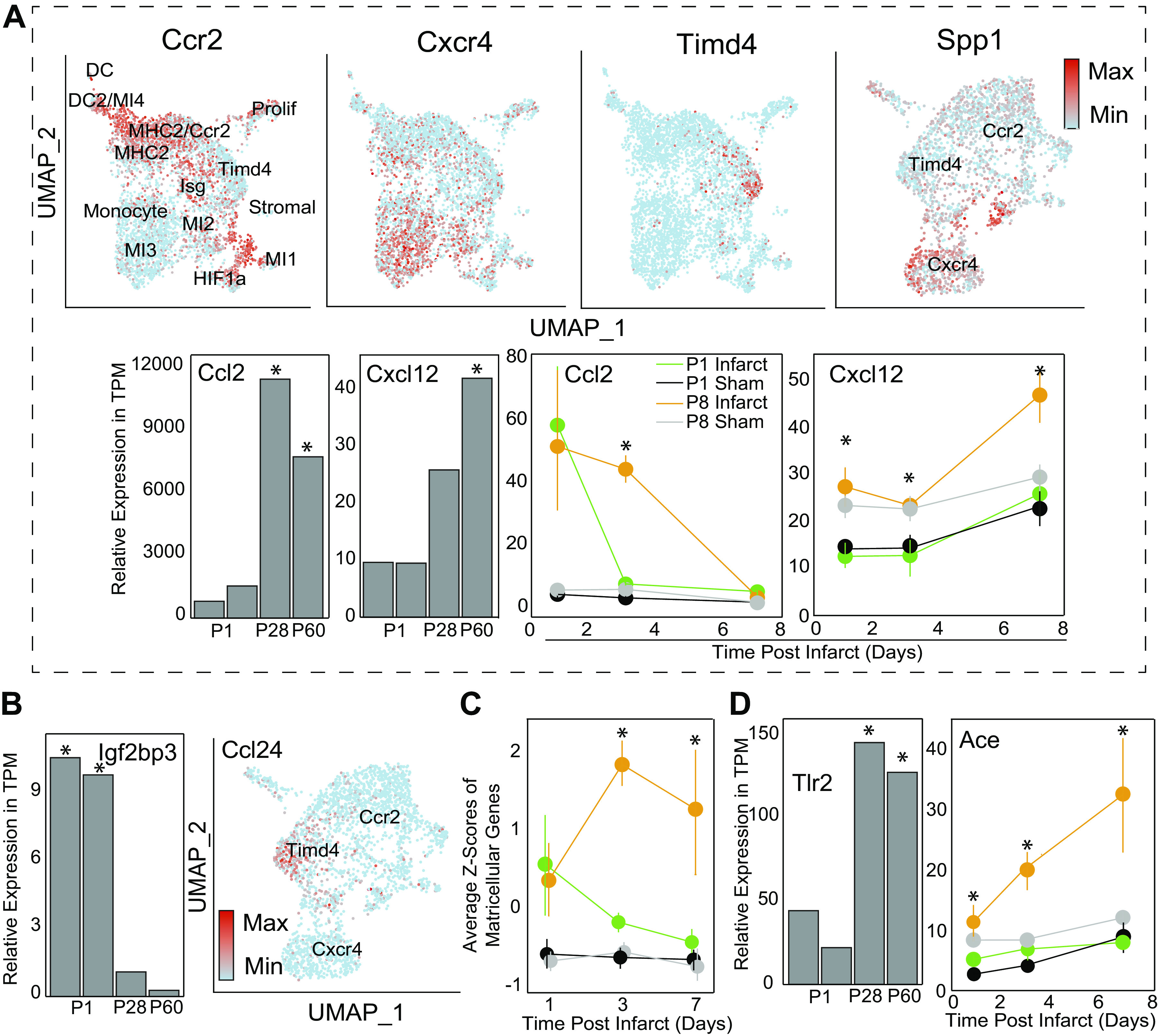
P8 cytokines recruit BMDMs deficient in growth proteins to an increasingly sensitive inflammatory microenvironment. A: post-MI Macrophages were clustered according to original (4) study’s markers. Macrophages either express high levels of CCR2 (classical monocytes), CXCR4 (late-phase monocytes) or Timd4 (YS macrophages). Spp1 graph was generated by examining a subset of cells based on expression of previous three genes and then re-clustering and demonstrates high Spp1 expression by Cxcr4+ macrophages. Bar graphs are from fibroblast dataset, line graphs are from bulk tissue, and UMAPs are from single-cell macrophages. B: growth factors identified from original bulk analysis identified in neonatal CF and Mac populations, respectively. Ccl24 was from same Seurat object that generated Spp1 plot. C: average Z-scores of matricellular genes demonstrating similar trends between genes by group and timepoint. Individual genes and functional groupings are listed in Supplemental Table 1. D: TLR2 expression in fibroblasts on postnatal days 1, 28, and 60 (34) is plotted here. Significance is indicated as *P < 0.05 as determined by exact test with Benjamini–Hochberg correction. Conversely, ACE expression is plotted from the bulk heart dataset (33), with significance determined by Wald test with Benjamini–Hochberg correction, P < 0.05. ACE, angiotensin converting enzyme; BMDM, bone marrow-derived macrophages; CF, cardiac fibroblasts; ECM, extracellular matrix; MI, myocardial infarction; NF, necrosis factor; P8, postnatal day 8; TLR, Toll-like receptors; YS, yolk sac.
While understanding white blood cell recruitment helps describe phenotype at the time of injury, cellular effects are achieved primarily through their in situ phenotype during the remodeling process. Classical cytokines associated with wound healing and fibrosis include IL-1β, TNFα, IL-6, and TGF-β, though only the latter two cytokines were differently expressed between regenerative and nonregenerative hearts (i.e., upregulated for nonregenerative hearts). IL-1β, IL-6, and TNFα are all regulated by NF-κB and less classically signal transducer and activator of transcription 3 (STAT3) and AP-1 (44–46). In macrophages, NF-κB is also stimulates secretion of Ccl2 (47), which attracts pathogenic CCR2+ monocytes and is upregulated in day 8 hearts 3 days post-MI (Fig. 4A). Along with CCR2+ macrophages, the microenvironment is populated with inflammatory proteins linked to poor prognoses. For instance, when fibroblasts bind TGF-β, they become activated, triggering the secretion of IL-6 (35) on day 3 post infarction in older hearts and correlating with the TGF-β spike (Supplemental Fig. S2B). These spikes cause hypertrophy and decrease cardiomyocyte contractility (48), suggesting that signaling immediately after infarction ultimately leads to the divergent chronic outcomes, i.e., regeneration or pathogenic remodeling as outlined in Fig. 3C. This signaling is often transcribed via JAK/STAT pathways, which encode SOCS genes as a negative feedback mechanism to prevent cytokines storms. STAT3 is typically inhibited by SOCS2 and SOCS3, the latter of which results in a downregulation of IL-6 (49). We found that STAT3 is upregulated on day 1 by regenerative hearts, but quickly returns to baseline, correlating with the observed transcription of IL-6. Though SOCS3 is widely expressed across macrophage populations and in steady-state adult cardiac fibroblasts (Supplemental Fig. S2, and D), SOCS2 was not highly expressed in either macrophage or fibroblast datasets, suggesting that the primary source of the protein is another cardiac cell type. In summary, STAT3 signaling occurs earlier in day 1 hearts, leading to an earlier resolution of TGF-β and IL-6 production versus day 8 hearts. The delayed cytokine signaling in these day 8 hearts is then more likely to affect a greater number of leukocytes and amplify inflammatory processes.
Along with cytokine diffusion, regenerative hearts produced a number of growth factors. Igfbp3, which enhances IGF-2 translation, was originally (33) associated with day 1 hearts and we were able to identify neonatal CFs as a cellular source (Fig. 4B, Supplemental Fig. S3A). IGF-2 has been shown to induce cardiomyocyte proliferation and aid in heart regeneration (33). An additional neonatal-restricted growth factor was identified as CCL24, which also induces cardiomyocyte cell cycle reentry (33). When referenced with the macrophage single-cell data set, YS macrophages were found to be the primary transcribers of this protein (Fig. 4B). Thus, we have identified the cellular contributions of both neonatal growth factors, though it is possible that YS macrophages are required for the CF production of CCL24.
Cellular Connectivity after Infarction
Cytokines may diffuse over significant distance, but for cardiac cells, additional cell-cell communication is possible through intercellular structures such as gap junctions, i.e., homotypic gated intercellular connections. In the heart, the primary gap junctions—connexin 43 and 45—propagate not only ion currents between cells, but also DAMPS and secondary messengers (50). In response to infarction, day 1 regenerative mouse hearts increased connexin expression after 3 days, while day 8 hearts downregulated production of these junctions (Supplemental Fig. S3A). An increase in intercellular permeability may help disperse DAMPS around the infarct area and reduce local concentrations; this, in turn, lowers the concentration of danger signals received by individual cells and reduces their inflammatory response. Moreover, DAMP dispersion allows a greater number of cells to bind and degrade the ligands, reducing the duration of danger signals.
For extracellular adhesions, no significant differential changes were observed between regenerative and nonregenerative hearts for common leukocyte adhesion molecules in the bulk data set, but nonregenerative heart fibroblasts expressed more adhesion molecules (Supplemental Fig. S3A). In addition to having a higher affinity for leukocytes, fibroblasts from nonregenerative hearts become larger, suggesting that these day 8 hearts are “stickier” to white blood cells (51) and thus more effective in inducing inflammation versus regenerative hearts. Once adhered to the myocardium, platelets, and neutrophils secrete TGF-β and PDGF into the infarct zone (52), binding to TGFβR and activating fibroblasts to produce the long isoforms of the large glycosaminoglycan called hyaluronic acid (HA) via HAS1 and HAS2 (53). HA is then cleaved in nonregenerative hearts into lower molecular weights, which are then able to bind many receptors and initiate detrimental functions as highlighted in the schematic in Fig. 3C. For example, high molecular weight HA sterically hinders Toll-like receptor (TLR) signaling and induces IL-4 producing macrophages in vitro (54). In contrast, low molecular weight HA is able to bind many receptors, e.g., CD44, receptor for HA-mediated motility (RHAMM), TLR2, TLR4, and PDGFR-β, some of which complex together (55, 56), resulting in SMAD2/3, FAK/ERK, and p38 and PI3K/AKT signaling. Since HA is able to bind many receptors with diverging downstream signaling, preventing this proinflammatory signaling by limiting low molecular weight HA production is likely the best approach. Hypertrophied hearts contain greater concentration of HA, especially lower molecular weight oligomers (57); HA degradation from high to low molecular weight is typically mediated by hyaluronidases (58), hence HYAL1 transcript was elevated in nonregenerative mice (7) (Supplemental Fig. S3B). This HA size conversion has also been a therapeutic target; when RHAMM, but not CD44 or TLR2/4, was blocked by a peptide receptor mimic, macrophage influx was prevented and TGF-β production decreased (55), preventing dermal scar formation in rats. Conversely, NF-κB is a central regulator of RHAMM that activates CCL2 production in a variety of cell types (47, 59) and this cytokine is overexpressed by nonregenerative hearts on day 3 (Fig. 4A). Thus, we conclude that HA conversion is a critical node in converting hearts into a nonregenerative mode and is mediated by upstream signals from inflammatory cells, e.g., platelets and neutrophils.
Beyond HA, several other matrix constituents undergo significant remodeling; matrix naturally turns over slowly with time in a tightly regulated process. However, in nonregenerative hearts, expression of several matrix components, e.g., Fbln1, Col1a2, Fn1, and Col3a1, is noticeably increased. Loxl2, a gene in the family of collagen cross-linking enzymes, is also highly expressed in nonregenerative hearts, which along with matrix overproduction could suggest why infarct scars are hard (60). Components that process and remodel matrix, primary matrix metalloproteases (MMPs) 2, 3, 9, and 14 (61–64), are also differentially expressed over time between P1 and P8 mice. Similarly, the duration of high TIMP1 expression, the inhibitor of MMPs, was longer for nonregenerative P8 mice (Supplemental Fig. S3C), suggesting that MMP activity may be inhibited in nonregenerative hearts to enable further accumulation of matrix. This additional matrix (including low-molecular-weight HA) present in the infarct could become an extracellular adhesion substrate for myofibroblast transdifferentiation (18) and disease progression.
In addition to proteins that form the ECM network, many other smaller matricellular proteins modify the properties of this network, e.g., Ccn3, periostin, osteopontin, and tenascin C among many others; these matricellular proteins are critical in the balance between healing and fibrosis. Of all the gene genres analyzed in these datasets, matricellular proteins show the most striking differences between regenerative and nonregenerative hearts (Fig. 4C, Supplemental Fig. S4A). These proteins are secreted by fibroblasts as well as activate them (65), forming a positive feedback loop, though they each have unique functions; for example, thrombospsondin-1 cleaves the latent form of TGF-β to activate it, whereas osteopontin and periostin increase fibroblast activation in response to TGF-β (66–68). Osteopontin (e.g., Spp1) was primarily detected in CXCR4+ recruited macrophages, and in negligible amounts by Timd4+ macrophages, suggesting that the later wave of recruited macrophages could contribute to fibrosis. TnC is overexpressed in nonregenerative hearts (Supplemental Fig. S4A) and together with TGF-β, induces the production of each other in fibroblasts, along with collagen 1 and smooth muscle actin (69). Another upregulated protein, SPARC (i.e., osteonectin) helps process and assemble collagen fibrils (70) and propagates mechanotransductive signals. SPARC knockout increases cardiac rupture risk after MI as well as decreases SMAD2/3 signaling (71). Finally, Thrombospondins (Thbs1 and Thbs2) are calcium-binding glycoproteins that bind collagens, fibrinogen, and integrins (72). Thbs1 is inducible via angiotensin II, whereas Thbs2 is regulated through reactive oxygen species (73, 74). Both stimulate TGF-β signaling through NF-κB (75) but Thrombospondin 1 also binds to TLR4, further propagating damage-associated signaling. Taken together, these matricellular proteins are necessary to prevent cardiac rupture, but upregulation is associated with poor outcomes via increased cardiac fibroblast activation and secretion and assembly of fibronectin, fibulin, and several collagens.
Cardiac Stress That Are Enhanced in Nonregenerative Hearts
Working in concert with the emergence of many biological inflammatory signals after MI, the establishment of an acute hypoxic microenvironment is equally important in spurring fibroblast activation and matrix deposition. Responses to hypoxia were more severe in nonregenerative mice, resulting in peak expression of hypoxia-inducible factor (HIF)1α, positive regulation of hypoxic response, and a decrease in HIF3α, negative regulation of hypoxic response (76) (Supplemental Fig. S4B). Hif1α regulates NF-κΒ signaling (77), which binds the periostin promoter during fibrosis (16), and induces BMDM recruitment through CXCL12 (78) further causing activation. Thus, hypoxia is able to activate cardiac fibroblasts to divide and secrete matrix (79).
Another hypoxia-related impact of MI is stretch response. When cardiomyocytes in the infarct region are deprived of oxygen, they stop contracting, although peri-infarct myocytes continue to beat. This creates a region of high tension around the infarct, spurring atrial-natriuretic peptide (ANP, whose gene is Nppa) production and the activation of fibroblasts (18). Although Herum et al. (15) were able to decouple the biological effects of stretch and stiffening on cardiac fibroblasts, biological activation of fibroblasts is usually accompanied by an increase in matrix production. Interestingly, an increase in Acta2 expression (which encodes smooth muscle actin, a marker of fibroblast to myofibroblast conversion), is not accompanied by a spike in collagen 1 mRNA production in P1 mice (Supplemental Fig. S4C). An increase in Acta2 expression suggests that P1 hearts are more sensitive to stretch stimuli, and this is corroborated by an increase in ANP transcription. ANP has been shown to inhibit fibroblast proliferation and matrix deposition (80), and the P1 spike in ANP correlates with decreased collagen production. This trend is not conserved in nonregenerative P8 hearts, suggesting that additional signaling overrides ANP-associated matrix suppression.
When cells apoptose or necrose such as with excessive stretch, they release their intracellular content into the interstitium where they can then be sensed by membrane-bound TLRs. Sensing of nuclei acids, histones, and other nuclear components by TLRs 2 and 4 result in NF-κB induced CXCL12-mediated monocyte recruitment to the heart (78) and an inflammatory response. TLRs 2 and 4 are the primary TLRs expressed on cardiac fibroblasts, though only TLR2 expression increases from postnatal day 1 to day 60 mice (Fig. 4D, Supplemental Table S2) (33, 34). TLR2 heterodimerizes with TLRs 1 and 6, and can recognize the chromatin binding protein HMBG1, hyaluronan, heparin sulfate, fibrinogen, and angiotensin II (55, 56). These native proteins are generated by cell lysis, through the clotting cascade, or in the case of angiotensin II, through the renin-angiotensin-aldosterone system, which is engaged by MI-induced hypotension. This causes systemic release of angiotensin I after conversion to its active form via angiotensin converting enzyme (ACE). Angiotensin can then bind to TLR2, induce downstream NK-κB activation, macrophage recruitment, and ultimately fibrosis (10). ACE expression is upregulated in nonregenerative day 8 hearts after MI, but not regenerative day 1 samples (Fig. 4D), suggesting that ACE or Ang II inhibitors may help reduce mortality via TLR inhibition; thus, this treatment strategy is modeled after neonatal-like healing response.
Limitations of Analysis
The observations in this analysis are based on mRNA expression across several cardiac populations in mice (4, 33, 34). It is important to note that while next generation RNA sequencing has provided an in-depth tool for mRNA quantification, it has several limitations. Particularly in single-cell analysis, low-level transcripts are difficult to detect at current sequencing depths, making it more difficult to separate true and false negatives. For that reason, this analysis focused on positive data or population differences in nonzero comparisons of differentially expressed genes. Moreover, mRNA does not necessarily scale to protein production and especially not to biological function or protein half-life; they also do not account for the effects of any posttranslational modifications. Therefore, the conclusions from these studies were compared against existing protein-level or in vivo studies examining the function of the resultant proteins. Finally, this analysis is based on datasets with limited time course. Significant follow-up could strengthen the conclusions drawn here. Conversely, other attempts at longitudinal assessment exist but are restricted to older heart (81), further motivating the need for longer observations post-MI of postnatal day 1 regenerative hearts. The strengths of this approach include many instances of compounding evidence across several datasets, researchers, and models of mice. Common regulation of MI-response pathways bodes well for evolutionarily preserved mechanisms that are likely similar in humans. Although additional studies will need to be conducted to compare human and mouse differences in stress responses, we hope that this study helps to parcel critical pathways and compare them against a regenerative positive control model for subsequent analyses in other platforms.
SUMMARY
Differential regulation of nonregenerative versus regenerative hearts seems restricted to key pathways: NF-κB, AP-1, hypoxia, stretch, and STAT3. On day 3, many proteins are upregulated by NF-κB, including but not limited to TnC, Ccl2, Ang II, thrombospondins, HAS, and MMP2/9. Although NF-κB is likely induced immediately after infarction in both regenerative and nonregenerative hearts, only regenerative hearts quench the signaling cascade (Supplemental Fig. S4D). This could be due to stronger NF-κΒ induction in nonregenerative hearts via greater TLR2 and 4 expression and ligand availability, particularly low molecular weight HA. Moreover, NF-κB induction and the macrophage recruitment steadily increases TGF-β and IL-6 postinfarction. Combined with the sudden emergence of synergistic profibrotic matricellular proteins and increased TGF-β sensitivity, fibroblasts are more likely to be activated and secrete an overabundance of matrix, resulting in a myocardial scar as outlined in Fig. 3C. In contrast, regenerative hearts have an acute induction of STAT3 signaling on day 1, which activates SOCS3 as a negative feedback regulator, reducing TLR sensitivity, inhibiting IL-6, and likely reducing the induction of NF-κB. Although many of the detrimental effects of nonregenerative P8 signaling can be attributed to NF-κB, improper dosing could be fatal. Instead, our analysis suggests that pharmacological inhibition of TLR/TGFβR/RHAMM ligands such as low-molecular-weight HA, Angiotensin II, stretch signaling, and monocyte recruitment could provide more promise for clinical translation.
GRANTS
This work was supported by the National Institute on Aging Grant R01AG045428 (to A.J.E.), the National Science Foundation Graduate Research Fellowship Program (to A.J.W.), and the Achievement Rewards for College Scientists Foundation (to A.J.W.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.J.W. conceived and designed research; A.J.W. analyzed data; A.J.W. and A.J.E. interpreted results of experiments; A.J.W. and A.J.E. prepared figures; A.J.W. and A.J.E. drafted manuscript; A.J.W. and A.J.E. edited and revised manuscript; A.J.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Kevin R. King (University of California, San Diego) for helpful discussions.
REFERENCES
- 1.Perbellini F, Watson SA, Bardi I, Terracciano CM. Heterocellularity and cellular cross-talk in the cardiovascular system. Front Cardiovasc Med 5: 143, 2018. doi: 10.3389/fcvm.2018.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP, Kohler RH, Arlauckas SP, Yoshiko V, Savo A, Sadreyev RI, Kelly M, Fitzgibbons TP, Fitzgerald KA, Mitchison T, Libby P, Nahrendorf M, Weissleder R. IRF3 and type i interferons fuel a fatal response to myocardial infarction. Nat Med 23: 1481–1487, 2017. doi: 10.1038/nm.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction. Circ Res 119: 91–112, 2016. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, Robbins CS, Epalman S. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20: 29–39, 2019. doi: 10.1038/s41590-018-0272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res 191: 15–28, 2018. doi: 10.1016/j.trsl.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res 110: 51–61, 2016. doi: 10.1093/cvr/cvw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Dembowsky K, Chevalier E, Stüve P, Korf-Klingebiel M, Lochner M, Napp LC, Frank H, Brinkmann E, Kanwischer A, Bauersachs J, Gyöngyösi M, Sparwasser T, Wollert KC. C-X-C motif chemokine receptor 4 blockade promotes tissue repair after myocardial infarction by enhancing regulatory T cell mobilization and immune-regulatory function. Circulation 139: 1798–1812, 2019. doi: 10.1161/CIRCULATIONAHA.118.036053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang M, Xiang FL, Braitsch CM, Yutzey KE. Epicardium-derived fibroblasts in heart development and disease. J Mol Cell Cardiol 91: 23–27, 2016. doi: 10.1016/j.yjmcc.2015.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest 124: 2921–2934, 2014. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Li YL, Zhang CC, Cui W, Wang X, Xia Y, Du J, Li HH. Inhibition of toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc Res 101: 383–392, 2014. doi: 10.1093/cvr/cvt258. [DOI] [PubMed] [Google Scholar]
- 11.Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol 87: 989–999, 2010. doi: 10.1189/jlb.1209775. [DOI] [PubMed] [Google Scholar]
- 12.Wang KX, Denhardt DT. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev 19: 333–345, 2008. doi: 10.1016/j.cytogfr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Fan D, Wang C, Wang JY, Cui XB, Wu D, Zhou Y, Wu LL. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac fibroblasts. Cardiovasc Res 91: 80–89, 2011. doi: 10.1093/cvr/cvr067. [DOI] [PubMed] [Google Scholar]
- 14.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol 180: 2625–2633, 2008. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 15.Hao J, Ju H, Zhao S, Junaid A, Scammell-La Fleur T, Dixon IMC. Elevation of expression of Smads 2, 3, and 4, decorin and TGF-β in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol 31: 667–678, 1999. doi: 10.1006/jmcc.1998.0902. [DOI] [PubMed] [Google Scholar]
- 16.Prakoura N, Kavvadas P, Kormann R, Dussaule JC, Chadjichristos CE, Chatziantoniou C. NF κ B induced periostin activates integrin-β3 signaling to promote renal injury in GN. J Am Soc Nephrol 28: 1475–1490, 2017. doi: 10.1681/ASN.2016070709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghatak S, Misra S, Norris RA, Moreno-Rodriguez RA, Hoffman S, Levine RA, Hascall VC, Markwald RR. Periostin induces intracellular cross-talk between kinases and hyaluronan in atrioventricular valvulogenesis. J Biol Chem 289: 8545–8561, 2014. doi: 10.1074/jbc.M113.539882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol Biol Cell 28: 1871–1882, 2017. doi: 10.1091/mbc.E17-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Müller AL, Dhalla NS. Role of various proteases in cardiac remodeling and progression of heart failure. Heart Fail Rev 17: 395–409, 2012. doi: 10.1007/s10741-011-9269-8. [DOI] [PubMed] [Google Scholar]
- 20.Ho CY, López B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, González A, Colan SD, Seidman JG, Díez J, Seidman CE. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med 363: 552–563, 2010. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity 41: 21–35, 2014. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takata K, Kozaki T, Lee CZW, Thion MS, Otsuka M, Lim S, Utami KH, Fidan K, Park DS, Malleret B, Chakarov S, See P, Low D, Low G, Garcia-Miralles M, , et al. Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity 47: 183–198.e6, 2017. doi: 10.1016/j.immuni.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Lin WY, Xu D, Austin CD, Caplazi P, Senger K, Sun Y, Jeet S, Young J, Delarosa D, Suto E, Huang Z, Zhang J, Yan D, Corzo C, Barck K, , et al. Function of CSF1 and IL34 in macrophage homeostasis, inflammation, and cancer. Front Immunol 10: 2019, 2019. doi: 10.3389/fimmu.2019.02019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 13: 432–438, 2007. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 25.Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, Varol C. Infiltrating monocyte-derived macrophages and resident Kupffer cells display different ontogeny and functions in acute liver injury. J Immunol 193: 344–353, 2014. doi: 10.4049/jimmunol.1400574. [DOI] [PubMed] [Google Scholar]
- 26.Lawrance W, Banerji S, Day AJ, Bhattacharjee S, Jackson DG. Binding of hyaluronan to the native lymphatic vessel endothelial receptor LYVE-1 is critically dependent on receptor clustering and hyaluronan organization. J Biol Chem 291: 8014–8030, 2016. doi: 10.1074/jbc.M115.708305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature 450: 435–439, 2007. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 28.Uderhardt S, Martins AJ, Tsang JS, Lämmermann T, Germain RN. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell 177: 541–555.e17, 2019. doi: 10.1016/j.cell.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical monocytes in health and disease. Annu Rev Immunol 37: 439–456, 2019. doi: 10.1146/annurev-immunol-042617-053119. [DOI] [PubMed] [Google Scholar]
- 30.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D, Lavine KJ. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res 124: 263–278, 2019. doi: 10.1161/CIRCRESAHA.118.314028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med 176: 287–292, 1992. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nahrendorf M, Swirski FK. Abandoning M1/M2 for a network model of macrophage function. Circ Res 119: 414–417, 2016. doi: 10.1161/CIRCRESAHA.116.309194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Cui M, Shah AM, Ye W, Tan W, Min YL, Botten GA, Shelton JM, Liu N, Bassel-Duby R, Olson EN. Mechanistic basis of neonatal heart regeneration revealed by transcriptome and histone modification profiling. Proc Natl Acad Sci USA 116: 18455–18465, 2019. doi: 10.1073/pnas.1905824116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giudice J, Xia Z, Wang ET, Scavuzzo MA, Ward AJ, Kalsotra A, Wang W, Wehrens XHT, Burge CB, Li W, Cooper TA. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat Commun 5: 3603, 2014. doi: 10.1038/ncomms4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eickelberg O, Pansky A, Mussmann R, Bihl M, Tamm M, Hildebrand P, Perruchoud AP, Roth M. Transforming growth factor-β1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J Biol Chem 274: 12933–12938, 1999. doi: 10.1074/jbc.274.18.12933. [DOI] [PubMed] [Google Scholar]
- 36.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140, 2010. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13: 2129–2141, 2003. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43: W566–W570, 2015. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive integration of single-cell data. Cell 177: 1888–1902.e21, 2019. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willenborg S, Lucas T, Van Loo G, Knipper JA, Krieg T, Haase I, Brachvogel B, Hammerschmidt M, Nagy A, Ferrara N, Pasparakis M, Eming SA. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 120: 613–625, 2012. doi: 10.1182/blood-2012-01-403386. [DOI] [PubMed] [Google Scholar]
- 42.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7: 678–689, 2007. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 43.Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guérin C, Vilar J, Caligiuri G, Tsiantoulas D, Laurans L, Dumeau E, Kotti S, Bruneval P, Charo IF, Binder CJ, Danchin N, Tedgui A, Tedder TF, Silvestre J-S, Mallat Z. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med 19: 1273–1280, 2013. doi: 10.1038/nm.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 56: 225–231, 2010. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haghikia A, Ricke-Hoch M, Stapel B, Gorst I, Hilfiker-Kleiner D. STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc Res 102: 281–289, 2014. doi: 10.1093/cvr/cvu034. [DOI] [PubMed] [Google Scholar]
- 46.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res 108: 1122–1132, 2011. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 47.Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T. NF-kappa B and Sp1 regulate transcription of the human moncyte chemoattractant protein-1 gene. J Immunol 153: 2052–2063, 1994. [PubMed] [Google Scholar]
- 48.Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA 92: 4862–4866, 1995. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Förster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 4: 540–545, 2003. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 50.Wang P, Yang H, Chen W, Ochani M, Qiang X, Al-Abed Y, Zhu S, D’Angelo J, Wang S, He M, Wang H, Tracey KJ, Ma G, Bao G, Li W. Connexin 43 hemichannel as a novel mediator of sterile and infectious inflammatory diseases. Sci Rep 8: 166, 2018. doi: 10.1038/s41598-017-18452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jonsson MKB, Hartman RJG, Ackers-Johnson M, Tan WLW, Lim B, van Veen TAB, Foo RS. A transcriptomic and epigenomic comparison of fetal and adult human cardiac fibroblasts reveals novel key transcription factors in adult cardiac fibroblasts. JACC Basic Transl Sci 1: 590–602, 2016. doi: 10.1016/j.jacbts.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frangogiannis NG. The role of transforming growth factor (TGF)-β in the infarcted myocardium. J Thorac Dis 9: S52–S63, 2017. doi: 10.21037/jtd.2016.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stuhlmeier KM, Pollaschek C. Differential effect of transforming growth factor β (TGF-β) on the genes encoding hyaluronan synthases and utilization of the p38 MAPK pathway in TGF-β-induced hyaluronan synthase 1 activation. J Biol Chem 279: 8753–8760, 2004. doi: 10.1074/jbc.M303945200. [DOI] [PubMed] [Google Scholar]
- 54.Rayahin JE, Buhrman JS, Zhang Y, Koh TJ, Gemeinhart RA. High and low molecular weight hyaluronic acid differentially influence macrophage activation. ACS Biomater Sci Eng 1: 481–493, 2015. doi: 10.1021/acsbiomaterials.5b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tolg C, Hamilton SR, Zalinska E, McCulloch L, Amin R, Akentieva N, Winnik F, Savani R, Bagli DJ, Luyt LG, Cowman MK, McCarthy JB, Turley EA. A RHAMM mimetic peptide blocks hyaluronan signaling and reduces inflammation and fibrogenesis in excisional skin wounds. Am J Pathol 181: 1250–1270, 2012. doi: 10.1016/j.ajpath.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Porsch H, Mehić M, Olofsson B, Heldin P, Heldin CH. Platelet-derived growth factor β-receptor, transforming growth factor β type I receptor, and CD44 protein modulate each other’s signaling and stability. J Biol Chem 289: 19747–19757, 2014. doi: 10.1074/jbc.M114.547273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lorén C, Dahl C, Do L, Almaas V, Geiran O, Mörner S, Hellman U. Low molecular mass myocardial hyaluronan in human hypertrophic cardiomyopathy. Cells 8: 97, 2019. doi: 10.3390/cells8020097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fallacara A, Baldini E, Manfredini S, Vertuani S. Hyaluronic acid in the third millennium. Polymers (Basel) 10: 701, 2018. doi: 10.3390/polym10070701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol 10: 1498–1506, 1990. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, Morine KJ, Gardner TJ, Discher DE, Sweeney HL. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol 290: H2196–H2203, 2006. doi: 10.1152/ajpheart.01017.2005. [DOI] [PubMed] [Google Scholar]
- 61.Kaplan RC, Smith NL, Zucker S, Heckbert SR, Rice K, Psaty BM. Matrix metalloproteinase-3 (MMP3) and MMP9 genes and risk of myocardial infarction, ischemic stroke, and hemorrhagic stroke. Atherosclerosis 201: 130–137, 2008. doi: 10.1016/j.atherosclerosis.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Squire IB, Evans J, Ng LL, Loftus IM, Thompson MM. Plasma MMP-9 and MMP-2 following acute myocardial infarction in man: Correlation with echocardiographic and neurohumoral parameters of left ventricular dysfunction. J Card Fail 10: 328–333, 2004. doi: 10.1016/j.cardfail.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 63.Donnini S, Morbidelli L, Taraboletti G, Ziche M. ERK1-2 and p38 MAPK regulate MMP/TIMP balance and function in response to thrombospondin-1 fragments in the microvascular endothelium. Life Sci 74: 2975–2985, 2004. doi: 10.1016/j.lfs.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 64.Vanhoutte D, Schellings M, Pinto Y, Heymans S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res 69: 604–613, 2006. doi: 10.1016/j.cardiores.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 65.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180: 1340–1355, 2012. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murphy-Ullrich JE, Schultz-Cherry S, Höök M. Transforming growth factor-β complexes with thrombospondin. Mol Biol Cell 3: 181–188, 1992. doi: 10.1091/mbc.3.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R. Osteopontin expression is required for myofibroblast differentiation. Circ Res 102: 319–327, 2008. doi: 10.1161/CIRCRESAHA.107.160408. [DOI] [PubMed] [Google Scholar]
- 68.Ashley SL, Wilke CA, Kim KK, Moore BB. Periostin regulates fibrocyte function to promote myofibroblast differentiation and lung fibrosis. Mucosal Immunol 10: 341–351. 2017. doi: 10.1038/mi.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, Lafyatis R, Lee J, Hinchcliff M, Feghali-Bostwick C, Lakota K, Budinger GRS, Raparia K, Tamaki Z, Varga J. Tenascin-C drives persistence of organ fibrosis. Nat Commun 7: 11703–11714, 2016. doi: 10.1038/ncomms11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harris BS, Zhang Y, Card L, Rivera LB, Brekken RA, Bradshaw AD. SPARC regulates collagen interaction with cardiac fibroblast cell surfaces. Am J Physiol Heart Circ Physiol 301: H841–H847, 2011. doi: 10.1152/ajpheart.01247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schellings MWM, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, Van Leeuwen REW, D’Hooge J, Van Werf FD, Carmeliet P, Pinto YM, Sage EH, Heymans S. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med 206: 113–123, 2009. doi: 10.1084/jem.20081244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol 3: a009712, 2011. doi: 10.1101/cshperspect.a009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Y, Poczatek MH, Berecek KH, Murphy-Ullrich JE. Thrombospondin 1 mediates angiotensin II induction of TGF-β activation by cardiac and renal cells under both high and low glucose conditions. Biochem Biophys Res Commun 339: 633–641, 2006. doi: 10.1016/j.bbrc.2005.11.060. [DOI] [PubMed] [Google Scholar]
- 74.Lopes N, Gregg D, Vasudevan S, Hassanain H, Goldschmidt-Clermont P, Kovacic H. Thrombospondin 2 regulates cell proliferation induced by Rac1 redox-dependent signaling. Mol Cell Biol 23: 5401–5408, 2003. doi: 10.1128/mcb.23.15.5401-5408.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 18: 6853–6866, 1999. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 76.Serocki M, Bartoszewska S, Janaszak-Jasiecka A, Ochocka RJ, Collawn JF, Bartoszewski R. miRNAs regulate the HIF switch during hypoxia: a novel therapeutic target. Angiogenesis 21: 183–202, 2018. doi: 10.1007/s10456-018-9600-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.D'Ignazio L, Bandarra D, Rocha S. NF-κB and HIF crosstalk in immune responses. FEBS J 283: 413–424, 2016. doi: 10.1111/febs.13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kew RR, Penzo M, Habiel DM, Marcu KB. The IKKα-dependent NF-κB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J Immunol 188: 2380–2386, 2012. doi: 10.4049/jimmunol.1102454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pillai MS, Sapna S, Shivakumar K. P38 MAPK regulates G1-S transition in hypoxic cardiac fibroblasts. Int J Biochem Cell Biol 43: 919–927, 2011. doi: 10.1016/j.biocel.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 80.Moubarak M, Magaud C, Saliba Y, Chatelier A, Bois P, Faivre JF, Farès N. Effects of atrial natriuretic peptide on rat ventricular fibroblasts during differentiation into myofibroblasts. Physiol Res 64: 495–503, 2015. doi: 10.33549/physiolres.932839. [DOI] [PubMed] [Google Scholar]
- 81.Deleon-Pennell KY, Iyer RP, Ma Y, Yabluchanskiy A, Zamilpa R, Chiao YA, Cannon PL, Kaplan A, Cates CA, Flynn ER, Halade GV, de Castro Brás LE, Lindsey ML. The mouse heart attack research tool 1.0 database. Am J Physiol Heart Circ Physiol 315: H522–H530, 2018. doi: 10.1152/ajpheart.00172.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]