

Abstract
Motivation
High-throughput sequencing technologies are used increasingly not only in viral genomics research but also in clinical surveillance and diagnostics. These technologies facilitate the assessment of the genetic diversity in intra-host virus populations, which affects transmission, virulence and pathogenesis of viral infections. However, there are two major challenges in analysing viral diversity. First, amplification and sequencing errors confound the identification of true biological variants, and second, the large data volumes represent computational limitations.
Results
To support viral high-throughput sequencing studies, we developed V-pipe, a bioinformatics pipeline combining various state-of-the-art statistical models and computational tools for automated end-to-end analyses of raw sequencing reads. V-pipe supports quality control, read mapping and alignment, low-frequency mutation calling, and inference of viral haplotypes. For generating high-quality read alignments, we developed a novel method, called ngshmmalign, based on profile hidden Markov models and tailored to small and highly diverse viral genomes. V-pipe also includes benchmarking functionality providing a standardized environment for comparative evaluations of different pipeline configurations. We demonstrate this capability by assessing the impact of three different read aligners (Bowtie 2, BWA MEM, ngshmmalign) and two different variant callers (LoFreq, ShoRAH) on the performance of calling single-nucleotide variants in intra-host virus populations. V-pipe supports various pipeline configurations and is implemented in a modular fashion to facilitate adaptations to the continuously changing technology landscape.
Availabilityand implementation
V-pipe is freely available at https://github.com/cbg-ethz/V-pipe.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
RNA viruses are regarded as important models in evolutionary biology, because they exhibit short generation times and higher mutation rates compared to cellular organisms (Duffy et al., 2008). As a result, they evolve rapidly and often exist within their host as a collection of distinct, yet genetically related, viral strains (Lauring and Andino, 2010). This heterogeneity impacts viral transmission, virulence and pathogenesis (Rozera et al., 2014; Tsibris et al., 2009; Vignuzzi et al., 2006), and is of relevance in the context of immune escape (Kuroda et al., 2010; Nowak et al., 1991), vaccine design (Gaschen, 2002) and drug resistance (Mason et al., 2018).
High-throughput sequencing (HTS) technologies have opened up new possibilities for in-depth characterization of the genetic diversity of virus samples (Barzon et al., 2013; Capobianchi et al., 2013). However, analysing viral HTS data is complicated by large volumes of data, short length of the sequencing reads and the high amplification and sequencing error rates relative to the expected intra-host viral diversity (Goodwin et al., 2016). Therefore, statistical and computational challenges remain in disentangling true biological variation from technical errors, especially for low-frequency variants (Beerenwinkel et al., 2012).
Several methods have been proposed for studying genetic diversity in virus populations (Eliseev et al., 2020; Posada-Céspedes et al., 2017), but some of these tools appear no longer actively maintained or limited in terms of robustness and usability. Also, the analysis of HTS data involves additional steps implemented by separate tools, e.g. quality control and read alignment. A prerequisite for incorporating such HTS data into routine diagnostics is to standardize the processing steps end-to-end from raw data input to final output. To this end, several bioinformatics pipelines have been developed for, e.g. virus discovery and metagenomics applications (Ho and Tzanetakis, 2014; Li et al., 2016; Maarala et al., 2018; Naccache et al., 2014; Zhao et al., 2017; Zheng et al., 2017). Other pipelines focus on either (i) the construction of the consensus sequence via reference-guided assembly (Wan et al., 2015; Wymant et al., 2018), (ii) the identification of single nucleotide variants (SNVs) (Howison et al., 2019; Huber et al., 2017; Taylor et al., 2019) or (iii) the reconstruction of viral haplotypes (Jayasundara et al., 2015; Mangul et al., 2014). Often these tools either target specific viruses (Howison et al., 2019; Huber et al., 2017; Taylor et al., 2019; Wymant et al., 2018) or provide limited support for broader applications. For HIV drug resistance testing, Lee et al. (2020) have recently evaluated various bioinformatics pipelines for this specific application.
An important step in inferring viral genetic diversity is the alignment of HTS reads. There are two general strategies for read alignment, namely reference-based approaches and de novo assembly. Shortcomings of the former are the introduction of biases due to low similarity between the reference genome and viral haplotypes (Archer et al., 2010), as well as inaccurate alignment of reads containing insertions or deletions (indels) (Posada-Céspedes et al., 2017). On the other hand, limitations of de novo assembly include increased sensitivity to chimeric reads leading to erroneous contigs, and a segmented coverage of the genome by the assembled contigs (Wymant et al., 2018). To address these limitations, we have developed a read aligner, called ngshmmalign. The aligner borrows ideas from the alignment of protein families to align HTS reads from small and highly diverse genomes. Ngshmmalign models the multiple read alignment as a profile hidden Markov model (HMM) and aligns all reads to this profile. By doing so, ngshmmalign accounts for local heterogeneity, including structural variations.
To support reproducible viral genomics studies for basic research and clinical diagnostics, we have developed V-pipe, a flexible bioinformatics pipeline integrating several tools for analysing viral HTS data. V-pipe allows for assessing viral diversity at the level of SNVs, short variant sequences (or local haplotypes) and long-range haplotypes (or global haplotypes). To provide the flexibility required for adapting to future methodological and technological developments, V-pipe provides a modular and extensible framework which facilitates the introduction of new tools. V-pipe also supports the comparative assessment of different workflows in a standardized environment. To this end, it contains modules to generate synthetic data and to assess the accuracy of the computational inference. We demonstrate the benchmark capabilities of V-pipe by assessing the impact of different read aligners and variant callers on the accuracy of SNV calling. While our focus here is on SNV calling, the performance of various methods for viral haplotype reconstruction has been recently evaluated by Eliseev et al. (2020). We validate V-pipe using sequencing data from a control sample composed of five well-defined HIV-1 strains (Di Giallonardo et al., 2014), and to demonstrate its applicability, we process 92 HIV-1 whole-genome sequencing samples from 11 patients previously reported in Zanini et al. (2015).
2 Materials and methods
We first summarize the core components of V-pipe. We then present the read aligner ngshmmalign and explain the benchmarking functionalities of the pipeline. Lastly, we describe the simulation setup and the control sequencing datasets used to assess the performance of different components of V-pipe.
2.1 Computational pipeline
V-pipe uses the Snakemake workflow management system (Köster and Rahmann, 2012), which enables the well-controlled and scalable execution of the pipeline in local as well as in high-performance computing (HPC) environments. The pipeline integrates various open-source software packages developed for analysing virus samples. For deployment, we provide Conda (https://conda.io) environments to automatically download and install the required tools. The environments include all dependencies and versions which is key for full reproducibility of the analysis settings.
As input, V-pipe requires the raw sequencing data, a reference sequence and a configuration file containing user-defined options. V-pipe supports both single-end and paired-end FASTQ files. In short, the analysis workflow implemented by V-pipe involves the following main steps: (i) quality control, (ii) reference-guided mapping and alignment of sequencing reads and (iii) identification of SNVs and reconstruction of viral haplotypes (Fig. 1).
Fig. 1.

Workflow of V-pipe for the analysis of viral HTS data. As input, the pipeline requires raw sequencing data (FASTQ format) and a reference sequence (FASTA format), which defines the indexing frame for the reporting of variants. (A) For quality control, low-quality bases are removed from both termini. (B) Reads are aligned employing a reference-guided approach using ngshmmalign. (C) For the read alignment, the reference sequence may be provided or it can be built de novo from the read data. (D) Alternatively, reads can be aligned using BWA MEM or Bowtie2. (E) Intermediate results are provided in the form of a consensus sequence per sample, a multiple sequence alignment of all consensus sequences, and the minor allele frequencies (MAFs) for all samples and all loci. (F) Single-nucleotide variants (SNVs) are identified, and (G) local and (H) global haplotypes are reconstructed. Whenever applicable, the pertinent reporting frame (con.: consensus, prof.: profile, ref: reference) is indicated in red. Dotted lines indicate alternative processing steps included in V-pipe
Relatively high error rates are a limitation of HTS technologies. Error sources include the reverse transcription of viral RNA into cDNA, the amplification of the material by multiple cycles of PCR, and the sequencing process itself (Beerenwinkel et al., 2012). To avoid propagating biases to the downstream analysis steps, it is imperative to include quality checks and filtering steps. To this end, we include FastQC (Andrews, 2019) to provide quality control reports and PRINSEQ (Schmieder and Edwards, 2011) for removing low-quality or ambiguous bases from both termini of reads.
For aligning sequencing reads, we developed a novel read aligner called ngshmmalign which is described below. This aligner requires as input a reference sequence to approximately locate reads. The reference sequence can be provided by the user or constructed de novo using the VICUNA software (Yang et al., 2012). We also include two alternative aligners, namely BWA MEM (Li, 2013) and Bowtie2 (Langmead and Salzberg, 2012), to choose from according to particular needs and computational resources. In addition to the alignment file, ngshmmalign outputs two types of consensus sequences constructed from the aligned reads, namely by (i) using a majority vote at each position, and (ii) incorporating ambiguous bases. V-pipe produces these consensus sequences in case an alternative aligner is chosen. We use lowercase characters in the consensus sequences to mark positions with read counts below 50 reads, by default. To report ambiguous bases, every base with a relative frequency greater or equal to a certain threshold, 5% by default, is accounted for, and the corresponding IUPAC ambiguity code is used. V-pipe also reports basic summary statistics, including the number of reads retained after quality control, the number of aligned reads, the region of the genome covered with a minimum number of aligned reads, and the minor allele frequencies per locus for all analysed samples.
We derive SNV calls from local haplotype reconstruction using ShoRAH (Zagordi et al., 2011). By considering co-occurring variants at multiple loci simultaneously, ShoRAH can effectively lower the SNV detection limit, i.e. the minimum frequency at which variants can be called reliably (McElroy et al., 2013). Alternatively, V-pipe also includes the variant caller LoFreq (version 2) (Wilm et al., 2012), which uses a position-wise and hence faster approach. A more complete characterization of the structure of a virus population consists in reconstructing the sequences and relative abundances of all viral haplotypes. For this purpose, we incorporate the quasispecies assembly tools HaploClique (Töpfer et al., 2014) and SAVAGE (Baaijens et al., 2017) to support reference-based and de novo haplotype reconstruction, respectively. Both methods use the overlap between reads to iteratively extend local haplotypes into full-length haplotypes.
As an additional feature, V-pipe includes a module to detect flow-cell cross contamination. To do so, reads are aligned to a panel of reference sequences containing suspected contaminants. We then detect and report the number of reads preferentially mapped to other references. The code developed to support this functionality, as well as various other steps of our pipeline, are maintained as an independent Python package called smallgenomeutilities (Supplementary Section S1.1).
V-pipe allows for constructing, maintaining and using reproducible and traceable data analysis pipelines. In addition to providing a reproducible workflow and pinning dependency versions, V-pipe supports numerical reproducibility by fixing the seeds of all processing steps involving random sampling. Traceability is achieved by providing a well-documented pipeline in a well-defined framework and by ensuring meaningful log output in all processing steps. We have also made additional efforts to improve the reliability of individual tools used as part of our pipeline, such as ShoRAH and HaploClique (Supplementary Section S1.2).
V-pipe is an open source project, the source code is freely available at https://github.com/cbg-ethz/V-pipe. Documentation including user guides can be found on the Wiki (https://github.com/cbg-ethz/V-pipe/wiki) and tutorials for specific viruses are available on our project page (https://cbg-ethz.github.io/V-pipe/). We also promote community involvement by maintaining a mailing list, providing user support and offering workshops.
2.2 ngshmmalign: read aligner
Ngshmmalign performs a three-step alignment (Supplementary Fig. S1). In the first step, reads are mapped to the reference genome to roughly determine their position. This initial mapping is done using a k-mer index of the reference genome. We then determine the mean and standard deviation of the returned location on the genome. If the standard deviation is above a certain threshold, the k-mer index match is considered suboptimal, and we perform a full genome-wide exhaustive alignment (Supplementary Section S1.3.2). After this initial mapping, reads are aligned in a semi-global mode using the Smith-Waterman algorithm. In the second step, the genome is partitioned into overlapping windows, and reads are assigned to windows based on the read-window overlaps. A multiple sequence alignment of the reads is performed independently for each of the windows by employing the L-INS-i iterative refinement approach implemented by MAFFT (Katoh and Standley, 2013) (Supplementary Section S1.3.3). We then infer the parameters of the profile HMM in a supervised manner, by assuming that the multiple read alignment represents a local sample of the profile HMM. In the third step, the final read alignment is obtained by re-aligning all reads to the profile HMM (Supplementary Section S1.3.4). The source code of ngshmmalign is available at https://github.com/cbg-ethz/ngshmmalign.
For every dataset analysed within a single execution of the pipeline, ngshmmalign reports the read alignment using a position numbering relative to the profile. To standardize the position numbering, V-pipe performs a lift-over to report the alignment relative the user-specified reference sequence. To do so, we construct and use a multiple sequence alignment, containing the reference sequence and all consensus sequences from datasets included in the data analysis. This multiple sequence alignment is also an intermediate result of V-pipe, which can be used for other applications, such as phylogenetic analyses.
2.3 Benchmarking
V-pipe provides benchmarking functionality by including two additional modules: (i) simBench can simulate sequencing reads from a mock virus population, and (ii) testBench can evaluate the accuracy of the inference results.
SimBench supports three modes. The first mode can be regarded as a simple quasispecies model (Domingo et al., 2005). A master sequence may be provided or generated by sampling a user-defined number of nucleotides uniformly at random. Simulated haplotype sequences are generated from the master sequence by introducing substitutions, insertions and deletions with user-configurable rates. Additionally, hyper-variable regions are simulated in the form of long deletions of variable length. In the second mode, the simulated haplotype sequences are sampled from a perfect binary tree. Every child node is generated from the parent sequence by introducing substitutions, insertions and deletions with user-configurable rates. Here, we emulate explicitly the hierarchical evolutionary relationships among viral haplotypes. In the third mode, haplotype sequences are defined a priori and given as input to the pipeline, e.g. based on other models of viral evolution or on known viral sequences. The haplotype relative abundances are either (i) set to equal proportions (i.e. where n is the number of haplotypes), (ii) drawn from a Dirichlet distribution with concentration parameters for all haplotypes by default or (iii) obtained using a geometric series with a given common ratio (0.75 by default).
After haplotype sequences have been generated, we use the ART software (Huang et al., 2012) to simulate either single-end or paired-end reads with configurable read length. ART is a read simulator with various built-in, technology-specific read error models and supports common sequencing errors, such as base substitutions, insertions and deletions. We simulate reads from every individual haplotype with read coverage proportional to its relative abundance.
Instead of simulating sequencing reads, a user can also provide FASTQ files as input for the benchmark. This option allows for providing sequences simulated by different means, and it supports the analysis of control sequencing experiments with known haplotypes and relative abundances.
The evaluation module testBench is used to assess the accuracy of the inference results by comparing them to the ground truth. For SNV calling, we obtain a list of true SNVs by constructing a multiple sequence alignment of the underlying haplotypes and the reference sequence. Positions of expected SNVs are reported relative to the reference sequence. We then report the number of true positive, false positive, false negative and true negative SNVs, as well as the inferred versus the expected SNV frequencies, the frequencies of false positives and the number of false negatives per underlying haplotype.
V-pipe supports two modes for carrying out the benchmark, either evaluating a single pipeline arrangement at a time (vpipeBench), or multiple pipeline arrangements simultaneously (vpipeBenchRunner). Although the generation of synthetic datasets is reproducible, the latter mode should be preferred to ensure that other configuration settings are kept constant across different pipeline arrangements (i.e. combinations of processing steps).
2.4 Simulated datasets
We employ simBench to generate simulated reads, varying the number of haplotypes (ranging from 8 to 60), their relative abundances and the total read coverage (either 10 000× or 40 000×, Supplementary Section S1.4). We use equal proportions for the haplotype abundances (denoted as Equal prop.) or sample their frequencies from a Dirichlet distribution. We either use a symmetric Dirichlet distribution with for all i (denoted as Uniform), or choose one haplotype at random and assign to it a greater weight ( and for all ) to emulate a dominant viral strain (denoted as Dirichlet). For all datasets, we simulate paired-end reads with 250 bp read length by using ART with the built-in quality profile for the MiSeq platform.
The datasets are based on HIV-1 or HCV sequences (Supplementary Table S1). For HIV-1, we employ sequences of the subtype B envelope glycoprotein (env) gene obtained for subjects 1051 and BORI0637 in Lee et al. (2009) by single-genome amplification. For HCV, we use sequences from naturally occurring HCV genotype 1 subtype a (1a) E1E2 genes (El-Diwany et al., 2017). Emulating populations using sequences derived from plasma samples of individual patients allows us to mimic the structure of viral populations more faithfully. Moreover, testing different viruses is crucial, as they may display different mechanisms of evolution and, hence, distinct forms of genetic variation.
2.5 Control sample for pipeline validation on real datasets
The control sample consists of an in vitro mixture of five known HIV-1 strains mixed at equal proportions. Four sequencing experiments were carried out starting with approximately 104 (denoted 10 K) and 105 (denoted 100 K) HIV-1 RNA copies. The samples were sequenced by using the Illumina MiSeq platform in paired-end read mode (2 × 250 bp length, v2 kit). The protocol described by Di Giallonardo et al. (2014) was employed for the amplification and sequencing. Primers were designed to cover almost the full HIV genome in five overlapping segments. Two types of sequencing experiments were carried out: one including all five amplicons (denoted A) and another one using only amplicon B (denoted B). This 2 × 2 design gives rise to four datasets referred to as A-10K, A-100K, B-10K and B-100K.
3 Results
We demonstrate the benchmarking capabilities of V-pipe and evaluate the accuracy of SNV detection using different read aligners and mutation callers. In addition, we analyse an in vitro mixture of well-defined viral strains to assess the performance of one particular pipeline configuration on actual sequencing data. We also employ V-pipe to process publicly available longitudinal data from 11 patients (Zanini et al., 2015) to demonstrate the applicability of the pipeline as well as its run time performance.
3.1 Simulation studies
We simulate reads from in silico mixtures of sequences derived from two different viruses, namely HIV-1 and HCV (Section 2.4). We align simulated reads using ngshmmalign and compare it against two widely used read aligners, namely BWA MEM and Bowtie 2. For this comparison, we employ vpipeBenchRunner for the simultaneous execution of all pipeline configurations.
Averaged over all simulated datasets, the evaluated tools align more than 89% of the read pairs concordantly (Supplementary Table S2). Although BWA MEM reports the highest percentage of aligned reads, ngshmmalign aligns a larger portion of sequenced bases resulting in a higher average coverage (Supplementary Fig. S2). To investigate potential read alignment bias due to differences in sequence similarity to the reference sequence, we report the fraction of aligned reads and aligned bases per haplotype. For the HCV-based datasets, sequences exhibit a broad range of divergence from the reference strain (0.005–0.112). BWA MEM aligns most of the reads regardless of the divergence from the reference (Supplementary Fig. S3A), but a large fraction of the bases are soft-clipped, whereas ngshmmalign aligns a higher fraction of the bases for all haplotypes (Supplementary Fig. S3B).
Since the main focus of V-pipe is to infer viral genetic diversity, we evaluate the accuracy of the read aligners based on the score of detecting SNVs using ShoRAH. The score is the harmonic mean of precision and recall. To make the scores comparable, we report the performance metric for the union of all genomic loci covered by at least one of the aligners for each of the evaluated conditions. We evaluate two read coverages (10 000× and 40 000×) and three strategies to generate the underlying haplotype abundances. In most cases, ngshmmalign outperforms BWA MEM and Bowtie 2 in terms of the score (Fig. 2A), and the difference in the scores is statistically significant (corrected P-value < 0.05, Wilcoxon signed-rank test, Supplementary Section S1.5.1). On the other hand, we do not observe substantial differences in the performance of the individual aligners while varying the distribution of haplotype frequencies, the number of haplotypes or the evaluated coverages (Supplementary Figs S4–S6).
Fig. 2.
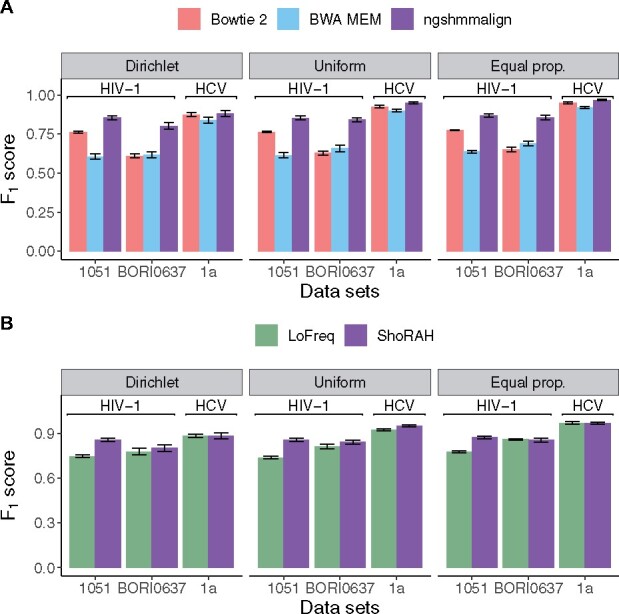
Performance of SNV detection on simulated datasets. (A) We compare ngshmmalign, BWA MEM and Bowtie 2 for read alignment, and fix ShoRAH for mutation calling. (B) We use ngshmmalign for the read alignment, and compare ShoRAH with LoFreq for mutation calling. In both panels, scores are averaged over datasets with various numbers of haplotypes based on HIV-1 subtype B sequences from subjects 1051 and BORI0637, and HCV genotype 1a sequences. Results are shown for a read coverage of 10 000× and for different distributions of haplotype frequencies as described in the Methods Section 2.4 (Dirichlet: . Uniform: . Equal prop.: all haplotype frequencies equal). The error bar corresponds to the standard error
For the evaluations, in addition to single-nucleotide substitutions, we account for position-wise deletions, by including the gap symbol as an alternative base. We further compare the performance in identifying such deletions and find that ngshmmalign outperforms BWA MEM and Bowtie2 for all evaluated conditions (Supplementary Fig. S10A). In addition to the position-wise performance, we also account for the length of individual deletion events and again find ngshmmalign to perform best (Supplementary Fig. S11A).
Next, we focus on mutation calling and compare the accuracy of SNVs obtained by using ShoRAH versus LoFreq, while fixing ngshmmalign for the read alignment. We observe significant differences in the scores for most datasets based on HIV-1 sequences from subject 1051 (corrected P-value < 0.05, Wilcoxon signed-rank test, Supplementary Table S3); otherwise both tools appear to perform equally well (Fig. 2B and Supplementary Fig. S7). Similar results are obtained when comparing performance based on detection of deletions; ShoRAH displays higher scores than LoFreq for most datasets based on HIV-1 sequences from subject 1051 (Supplementary Figs S10B and S11B). Small discrepancies in the scores can be attributed to differences in recall, whereas both tools show almost perfect precision (Supplementary Figs S8 and S9).
Although aligning reads with ngshmmalign and performing mutation calling with ShoRAH resulted in better scores in most cases, we note that there is a trade-off between accuracy and computational resources (Supplementary Section S1.5).
3.2 Validation of V-Pipe on a mixture of five HIV-1 strains
We employ V-pipe using ngshmmalign with a de novo constructed reference sequence, and ShoRAH for SNV calling. In all cases, V-pipe detected more than 86% of the expected SNVs, with almost perfect specificity (Table 1). In datasets for which only amplicon B is sequenced (B-10k and B-100k), V-pipe reports perfect recall, whereas for datasets sequenced with all sets of primers (A-10k and A-100k), V-pipe misses a small fraction of the expected SNVs. The missed variants are predominately located at the genome termini, which correspond to regions of lower coverage. We also observe a decrease in precision for datasets A-100k, B-10k and B-100k. Most of the falsely reported SNVs correspond to single-nucleotide deletions at very low frequencies, whereas such errors are less prominent in sample A-10k. When inspecting the precision of the mutation calls as a function of the variant frequencies, we find that a precision higher than 98% can be attained for SNVs with frequencies at least 0.5% for all the analysed datasets (Fig. 3A). In addition to detecting most of the expected variants, we find the frequencies of unique SNVs per haplotype reported by ShoRAH to be in good agreement with the relative abundances originally reported by Di Giallonardo et al. (2014) (Fig. 3B).
Table 1.
Evaluating mutation calling using V-pipe
| Dataset | Recall | Precision | Specificity |
|---|---|---|---|
| A-10k | 0.860 | 0.944 | 0.992 |
| A-100k | 0.873 | 0.631 | 0.925 |
| B-10k | 1 | 0.770 | 0.977 |
| B-100k | 1 | 0.403 | 0.885 |
Note: Datasets A and B result from using five overlapping amplicons (A) or only the second amplicon covering mainly HIV-1 pol (B), respectively, and the suffix indicates the initial amount of RNA copies.
Fig. 3.
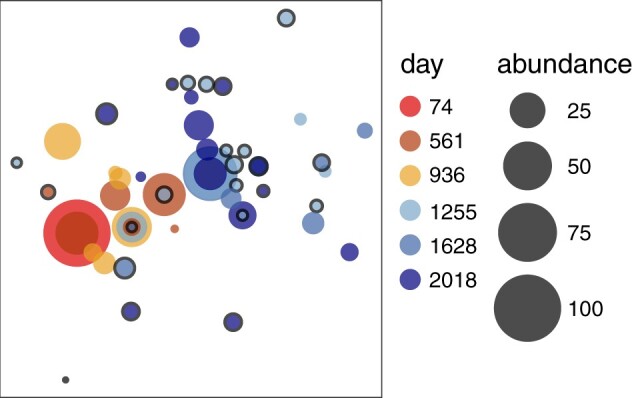
Evaluation of the performance of V-pipe on HTS data derived from the five-virus-mix. (A) Precision of SNV calls as a function of SNV frequency. (B) Distribution of inferred frequencies of unique SNVs for each haplotype in the mix for dataset A-10k. Green diamonds show the corresponding average frequency. The values reported in Di Giallonardo et al. (2014) for the mean and standard deviation of the frequencies estimated from Illumina reads are indicated in red
3.3 Application to clinical samples
To test V-pipe on clinical samples, we analyse publicly available datasets corresponding to longitudinal samples from one of the patients studied by Zanini et al. (2015) (ENA study accession number PRJEB9618). We employ V-pipe to infer local haplotypes on the 6 longitudinal datasets available for patient p2, using ngshmmalign and ShoRAH for local haplotype reconstruction. We compare the inferred haplotypes to the haplotype sequences reported by Zanini et al. (2015) on a region of the p17 gene spanning nucleotides 872–1072 with respect to the HXB2 reference genome. This region has been arbitrarily chosen from regions displaying genetic diversity. After filtering out inferred haplotypes with a posterior probability smaller than 0.9 and an average read count of less than 10 reads, we find a perfect match between haplotype sequences reconstructed using V-pipe and the sequences previously reported by Zanini et al. (2015) using manually curated read alignments. However, V-pipe finds 1, 11, 3 and 8 additional haplotypes in the samples taken 561, 1255, 1628 and 2018 days after the estimated date of infection, respectively. The estimated frequencies of these haplotypes range from 0.08% to 6%. For the matching haplotypes, the absolute error in the estimated abundances is below 4% for all the considered datasets (Supplementary Fig. S12).
We visualise the reconstructed local haplotypes by representing pairwise Hamming distances in a two-dimensional plane (Fig. 4). We observe a drift in sequence space from the initial haplotype (in red, Fig. 4) as time progresses. Sequences of haplotypes recovered after 561 and 1255 days of infection are situated closer to the initial haplotype sequence, whereas sequences corresponding to later time points (after 1628 and 2018 days of infection) are further away.
Fig. 4.
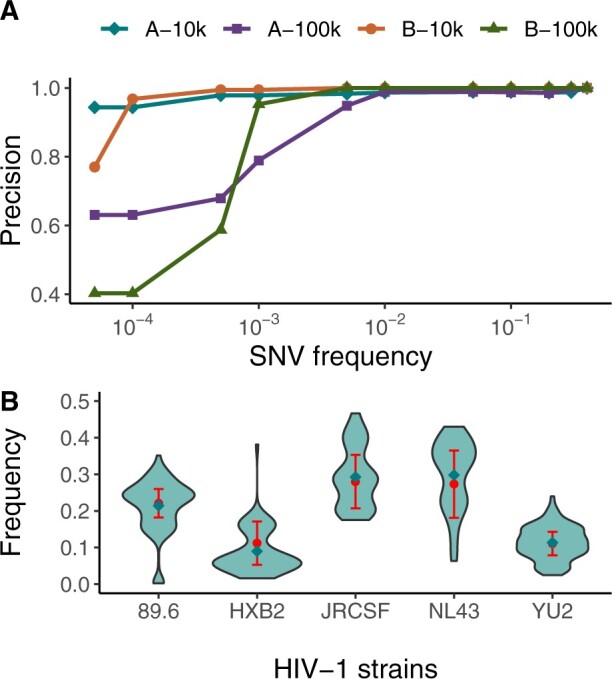
Representation of the reconstructed viral haplotypes in a region of the HIV-1 p17 gene from longitudinal samples of patient p2 of Zanini et al. (2015). Discs represent inferred haplotypes, and their size reflect the relative abundances. Haplotypes are placed in a two-dimensional plane with the aim of preserving all pairwise Hamming distances as much as possible, using multi-dimensional scaling as implemented in scikit-learn (Pedregosa, 2011). The colours indicate the number of days after infection and encircled discs denote haplotypes identified by V-pipe but not by Zanini et al. (2015,)
To illustrate the large-scale applicability of the pipeline, we note that using V-pipe to process all 92 datasets reported in Zanini et al. (2015) yielded an average run time per sample of 16 h on 12-core Intel Xeon E5-2680v3 processors (2.5–3.3 GHz), for an average coverage across samples of . Depending on the computational resources available, the total throughput can be largely independent of the number of samples, because the processing steps for individual datasets can be executed in parallel. Moreover, when computational resources are limited, V-pipe can be executed using alternative aligners, such as BWA MEM. For these particular datasets, BWA MEM aligned reads within 86 s on average, whereas ngshmmalign took on average 4 h 38 min. Similarly, LoFreq can be used for mutation calling as opposed to ShoRAH. In this case, ShoRAH took on average 11 h 7 min using 12 threads, whereas LoFreq took 2 h 42 min executed as a single-threaded program.
4 Discussion and conclusions
Incorporating HTS technologies into viral genomics studies provides new opportunities to characterize intra-host virus populations in unprecedented depth. However, the applicability of these technologies is challenged by the amount and quality of the resulting data. In a clinical diagnostics setting, these large volumes of data need to be analysed accurately and efficiently within a short period of time.
To ensure reproducibility and traceability of the analysis workflows, we developed and implemented the bioinformatics pipeline V-pipe. V-pipe is tailored to studying intra-host diversity from viral HTS data. Although workflows for either the identification of minor variants (Huber et al., 2017) or the reconstruction of viral haplotypes (Jayasundara et al., 2015) have been proposed, to our knowledge, V-pipe is the first pipeline assessing viral diversity at all spatial levels of the viral genome, i.e. SNVs, as well as local and global haplotypes.
We also present a novel read alignment approach tailored to relatively small, yet highly diverse genomes. The aligner prioritizes accuracy over speed and is based on profile hidden Markov models, which allow for capturing features shared among related sequences, such as mutations and indels, through position-specific probabilities.
We evaluated ngshmmalign on simulated reads from mock viral populations that attempt to mimic hyper-variable regions of HIV-1 (env gene) and HCV (E1E2 region). The performance of the aligner was indirectly evaluated based on the detection of SNVs from the aligned reads. In most cases, the F1 score was found to be larger than 0.8, and SNV calls based on alignments obtained using ngshmmalign also reported a higher score compared to alignments by BWA MEM or Bowtie 2. Furthermore, comparing performance outcomes based on HIV-1 datasets, showing variation in the form of indels, with HCV-based datasets, the advantage of using ngshmmalign over conventional aligners became apparent. We further evaluated the performance based solely on detection of deletions, finding that ngshmmalign outperforms the other evaluated aligner. We thus argue that ngshmmalign should be the preferred choice for populations containing indels as sources of variation. Although ngshmmalign performed better under the evaluation conditions, it is possible that the alignments produced by BWA MEM or Bowtie 2 could be further improved by fine-tuning their hyper-parameters. Nevertheless, our results suggest that the SNV detection accuracy is to a larger extent influenced by the upstream alignment step than by the choice of the SNV caller. More importantly, with this simulation study, we demonstrated the benchmarking component implemented in V-pipe, which can be useful for assessing the actual run times of individual tools and adjusting the pipeline to meet specific requirements, e.g. on accuracy of the results or availability of computational resources.
Intrinsic parameters of the analysed sample such as diversity and number of mutations can generally impact the performance of the individual tools integrated in V-pipe. In our simulation study, we analysed synthetic data from populations ranging from 8 to 60 haplotypes, as well as different distributions for the haplotype frequencies, and did not find substantial changes in the F1 score (Supplementary Figs S4 and S7).
Using V-pipe, we were able to reconstruct the genetic diversity present in a five-virus-mix control sample dataset with high recall. While we observed lower precision, this could be improved substantially by filtering out deletion calls with frequencies below 0.5%. Choosing such thresholds for mutation and structural variant calling is highly dependent on the HTS protocols, but detecting SNVs at frequencies as low as 1% is already very close to the Illumina sequencing error rate.
In addition to simulated datasets and control sequencing samples, we demonstrated the applicability of the pipeline by processing data from 92 clinical samples resulting in typical run times of 16 h per sample. Thus, we believe that total turnaround times of a few days are feasible using V-pipe’s default configuration (i.e. with ngshmmalign and ShoRAH) and including sample collection and preparation, sequencing and data analysis. Hence, the accuracy as well as the run times of V-pipe make it suitable for many applications.
Most studies on viral genetic diversity have been limited to the identification of SNVs. Yet, the occurrence of mutations on the same genome might not be independent and the combined effect of co-occurring mutations might not be additive. On the other hand, it is not entirely obvious how to incorporate haplotype information, e.g. into monitoring epidemics, drug resistance surveillance or supporting treatment decisions. There is, however, supporting evidence for using viral haplotypes to infer transmission pairs (Poon et al., 2016). V-pipe can support such research towards the understanding of the missing links in viral haplotyping.
In general, viral haplotype reconstruction is an active research field. Consequently, new computational methods for this task are regularly being proposed. In addition to novel methods, technological improvements are deployed at a high rate. Therefore, any pipeline needs to be actively maintained and constantly adapted to new requirements. We addressed this aspect from two directions. First, additional efforts have been directed into making the individual components of our pipeline, such as ShoRAH and HaploClique, more performant to handle current sequencing throughputs. Second, V-pipe features a modular and extensible architecture, such that the pipeline can be adapted to incorporate new tools.
Furthermore, there is a pressing need to establish standards for data analysis. We thus introduced a benchmark component to support testing of different pipeline configurations. While we mainly focused on performance assessment of mutation calling, the benchmark utilities can be employed in a similar fashion to evaluate haplotype reconstruction, which is subject to future work.
One limitation of the pipeline is that it currently focuses on Illumina data. Other sequencing technologies such as Pacific Biosciences and Oxford Nanopore can produce longer reads which have the potential to reduce the complexity of the haplotype reconstruction problem. However, the higher error rates, compared to Illumina sequencing, can be a limiting factor. Combining data from both short-read and long-read sequencing is a promising direction (Viehweger et al., 2019), and future developments should include extending the pipeline to support Pacific Biosciences and Oxford Nanopore data. Given that long-read technologies often do not provide an output in FASTQ format, the minimum changes required entail input file conversion as well as a customized pipeline configuration, e.g. using BWA-MEM as the aligner. As the need arises, dedicated long-read tools such as minimap2 (Li, 2018) or pbmm2 (https://github.com/PacificBiosciences/pbmm2), read assemblers and even base callers should be added to the pipeline.
Supplementary Material
Acknowledgements
The authors thank Tobias Marschall for outlining the needs of the community, contributing to the initial design of V-pipe and co-organizing the 2017 Basel Computational Biology Conference (BC2) tutorial on ‘Production Pipelines for Virus Sequencing Data’. They thank Maryam Zaheri for testing, refactoring and adding a continuous integration pipeline to the HaploClique software. They also thank Marek Pikulski and Nico Borgsmüller for critical reading.
Funding
This work was supported by the SystemsX.ch [ to K.J.M and N.B.]. V-pipe is supported as a Competitive Resource by the Swiss Institute of Bioinformatics.
Conflict of Interest: none declared.
Data availability
The sequencing data underlying this article are available in Github at https://github.com/cbg-ethz/5-virus-mix and in ENA at https://www.ebi.ac.uk/ena/browser/home and can be accesed with study accession number PRJEB9618.
Contributor Information
Susana Posada-Céspedes, Department of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland; SIB Swiss Institute of Bioinformatics, 4058 Basel, Switzerland.
David Seifert, Department of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland; SIB Swiss Institute of Bioinformatics, 4058 Basel, Switzerland.
Ivan Topolsky, Department of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland; SIB Swiss Institute of Bioinformatics, 4058 Basel, Switzerland.
Kim Philipp Jablonski, Department of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland; SIB Swiss Institute of Bioinformatics, 4058 Basel, Switzerland.
Karin J Metzner, Division of Infectious Diseases and Hospital Epidemiology, University Hospital Zurich, University of Zurich, 8091 Zurich, Switzerland; Institute of Medical Virology, University of Zurich, 8091 Zurich, Switzerland.
Niko Beerenwinkel, Department of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland; SIB Swiss Institute of Bioinformatics, 4058 Basel, Switzerland.
References
- Andrews S. (2019) FastQC a Quality Control Tool for High Throughput Sequence Data. Babraham Institute. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- Archer J. et al. (2010) The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time–an ultra-deep approach. PLoS Comput. Biol., 6, e1001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baaijens J. et al. (2017) De novo assembly of viral quasispecies using overlap graphs. Genome Res., 27, 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzon L. et al. (2013) Next-generation sequencing technologies in diagnostic virology. J. Clin. Virol., 58, 346–350. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N. et al. (2012) Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front Microbiol, 3, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capobianchi M.R. et al. (2013) Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect., 19, 15–22. [DOI] [PubMed] [Google Scholar]
- Di Giallonardo F. et al. (2014) Full-length haplotype reconstruction to infer the structure of heterogeneous virus populations. Nucleic Acids Res., 42, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E. et al. (2005) Quasispecies dynamics and RNA virus extinction. Virus Res., 107, 129–139. [DOI] [PubMed] [Google Scholar]
- Duffy S. et al. (2008) Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet, .9, 267–276. [DOI] [PubMed] [Google Scholar]
- El-Diwany R. et al. (2017) Extra-epitopic hepatitis C virus polymorphisms confer resistance to broadly neutralizing antibodies by modulating binding to scavenger receptor B1. PLoS Pathog., 13, e1006235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliseev A. et al. (2020) Evaluation of haplotype callers for next-generation sequencing of viruses. Infect. Genet. Evol., 82, 104277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschen B. (2002) Diversity considerations in HIV-1 vaccine selection. Science, 296, 2354–2360. [DOI] [PubMed] [Google Scholar]
- Goodwin S. et al. (2016) Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet., 17, 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T., Tzanetakis I.E. (2014) Development of a virus detection and discovery pipeline using next generation sequencing. Virology, 471–473, 54–60. [DOI] [PubMed] [Google Scholar]
- Howison M. et al. (2019) Measurement error and variant-calling in deep Illumina sequencing of HIV. Bioinformatics, 35, 2029–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. et al. (2012) ART: a next-generation sequencing read simulator. Bioinformatics, 28, 593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M. et al. (2017) MinVar: a rapid and versatile tool for HIV-1 drug resistance genotyping by deep sequencing. J. Virol. Methods, 240, 7–13. [DOI] [PubMed] [Google Scholar]
- Jayasundara D. et al. (2015) ViQuaS: an improved reconstruction pipeline for viral quasispecies spectra generated by next-generation sequencing. Bioinformatics, 31, 886–896. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in Performance and Usability. Mol. Biol. Evol., 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster J., Rahmann S. (2012) Snakemake – a scalable bioinformatics workflow engine. Bioinformatics, 28, 2520–2522. [DOI] [PubMed] [Google Scholar]
- Kuroda M. et al. (2010) Characterization of quasispecies of pandemic 2009 influenza A virus (A/H1N1/2009) by de novo sequencing using a next-generation DNA sequencer. PLoS One, 5, e10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring A.S., Andino R. (2010) Quasispecies theory and the behavior of RNA viruses. PLoS Pathog., 6, e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.R. et al. (2020) Performance comparison of next generation sequencing analysis pipelines for HIV-1 drug resistance testing. Sci Rep, 10, 1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.Y. et al. (2009) Modeling sequence evolution in acute HIV-1 infection. J. Theor. Biol., 261, 341–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997. [Google Scholar]
- Li H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2016) VIP: an integrated pipeline for metagenomics of virus identification and discovery. Sci. Rep., 6, 23774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarala A.I. et al. (2018) ViraPipe: scalable parallel pipeline for viral metagenome analysis from next generation sequencing reads. Bioinformatics, 34, 928–935. [DOI] [PubMed] [Google Scholar]
- Mangul S. et al. (2014) Accurate viral population assembly from ultra-deep sequencing data. Bioinformatics, 30, i329–i337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason S. et al. (2018) Comparison of antiviral resistance across acute and chronic viral infections. Antiviral Res., 158, 103–112. [DOI] [PubMed] [Google Scholar]
- McElroy K. et al. (2013) Accurate single nucleotide variant detection in viral populations by combining probabilistic clustering with a statistical test of strand bias. BMC Genomics, 14, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naccache S.N. et al. (2014) A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res., 24, 1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak M.A. et al. (1991) Antigenic diversity thresholds and the development of AIDS. Science, 254, 963–969. [DOI] [PubMed] [Google Scholar]
- Pedregosa F. et al. (2011) Scikit-learn: machine learning in Python. J. Mach. Learn. Res., 12, 2825–2830. [Google Scholar]
- Poon L.L. et al. (2016) Quantifying influenza virus diversity and transmission in humans. Nat. Genet., 48, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada-Céspedes S. et al. (2017) Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Res., 239, 17–32. [DOI] [PubMed] [Google Scholar]
- Rozera G. et al. (2014) Quasispecies tropism and compartmentalization in gut and peripheral blood during early and chronic phases of HIV-1 infection: possible correlation with immune activation markers. Clin. Microbiol. Infect., 20, O157–O166. [DOI] [PubMed] [Google Scholar]
- Schmieder R., Edwards R. (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor T. et al. (2019) A MiSeq-HyDRA platform for enhanced HIV drug resistance genotyping and surveillance. Sci. Rep., 9, 8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Töpfer A. et al. (2014) Viral quasispecies assembly via maximal clique enumeration. PLoS Comput. Biol., 10, e1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsibris A.M.N. et al. (2009) Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One, 4, e5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viehweger,A. et al. (2019) Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Research, 29, 1545–1554. 10.1101/gr.247064.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M. et al. (2006) Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature, 439, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y. et al. (2015) VirAmp: a galaxy-based viral genome assembly pipeline. Gigascience, 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A. et al. (2012) LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res., 40, 11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymant C. et al. ; BEEHIVE Collaboration. (2018) Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol., 4, vey007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. et al. (2012) De novo assembly of highly diverse viral populations. BMC Genomics, 13, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagordi O. et al. (2011) ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics, 12, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanini F. et al. (2015) Population genomics of intrapatient HIV-1 evolution. eLife, 4, e11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G. et al. (2017) VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology, 503, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. et al. (2017) VirusDetect: an automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology, 500, 130–138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data underlying this article are available in Github at https://github.com/cbg-ethz/5-virus-mix and in ENA at https://www.ebi.ac.uk/ena/browser/home and can be accesed with study accession number PRJEB9618.