

Abstract
Pediatric pulmonary hypertension (PPH) is a multifactorial disease with diverse etiologies and presenting features. Pulmonary hypertension (PH), defined as elevated pulmonary artery pressure, is the presenting feature for several pulmonary vascular diseases. It is often a hidden component of other lung diseases, such as cystic fibrosis and bronchopulmonary dysplasia. Alterations in lung development and genetic conditions are an important contributor to pediatric pulmonary hypertensive disease, which is a distinct entity from adult PH. Many of the causes of pediatric PH have prenatal onset with altered lung development due to maternal and fetal conditions. Since lung growth is altered in several conditions that lead to PPH, therapy for PPH includes both pulmonary vasodilators and strategies to restore lung growth. These strategies include optimal alveolar recruitment, maintaining physiologic blood gas tension, nutritional support, and addressing contributing factors, such as airway disease and gastroesophageal reflux. The outcome for infants and children with PH is highly variable and largely dependent on the underlying cause. The best outcomes are for neonates with persistent pulmonary hypertension (PPHN) and reversible lung diseases, while some genetic conditions such as alveolar capillary dysplasia are lethal.
Introduction
Pediatric pulmonary hypertension (PPH) comprises a variety of etiologies spread across the entire age spectrum from newborn to late adolescence. PH is defined as the elevation of pulmonary arterial pressure (PAP) and is commonly diagnosed by echocardiography or cardiac catheterization after it becomes clinically apparent. Use of specific terminology is important to describe PH, which refers to elevated PAP from any cause. Pulmonary arterial hypertension (PAH) refers to precapillary PH with normal or low pulmonary capillary wedge pressure (see below for definition). The major types of PH that occur in the pediatric age group are persistent pulmonary hypertension of the newborn (PPHN), which is classified as 1.7 in current Nice classification, congenital heart disease (CHD) (1.4.4), developmental lung diseases (3.5), and idiopathic pulmonary arterial hypertension (IPAH) (1.1). PPHN has a different etiology, presentation, and clinical course compared to other causes of PPH; a vast majority of affected neonates recover without sequelae. PPH associated with developmental disorders of the lung such as bronchopulmonary dysplasia (BPD) and congenital diaphragmatic hernia (CDH) and PH associated with CHD are important causes of long-term PH in children. It is increasingly being recognized that pediatric PH is different from adult PH, in etiology, clinical presentation, and outcomes. The 6th World Symposium on Pulmonary Hypertension (WSPH) published new definitions and classifications for PH in 2018, which are reflected in this article. Mechanisms of PH usually involve an imbalance between the vasoconstrictor and vasodilator forces in the pulmonary vasculature, which leads to elevated pulmonary vascular resistance (PVR), which in turn leads to increased right ventricular afterload and eventual right ventricular failure. PPH, with or without temporal association with elevated PA pressure, is usually due to disruption of normal development. Therapies to treat PH aim to treat this imbalance and decrease RV afterload and increase cardiac output. PH can occur secondary to three distinct mechanisms: pulmonary vasoconstriction, which is responsive to vasodilator therapy, vascular remodeling with thickening of media and adventitia of affected vessels, and a decrease in angiogenesis with pruning of the vascular tree (Figure 2).
Figure 2.
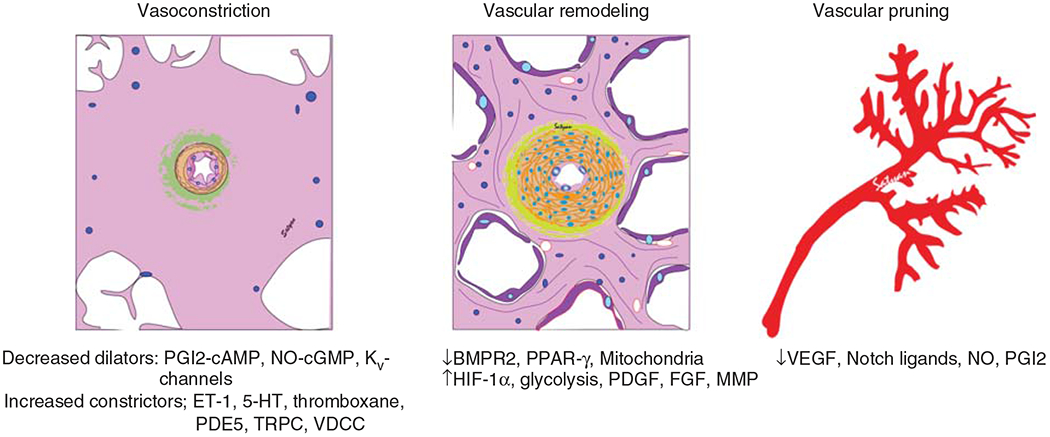
Molecular and structural mechanisms of pulmonary vascular disease. PGI2, prostacyclin; NO, nitric oxide; sGC, soluble guanylate cyclase; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; PDE, phosphodiesterase; Kv channel, voltage-gated potassium channel; ET-1, endothelin-1; 5HT, 5-hydroxytryptamine; TRPC, transient receptor potential cation channel; VDCC, voltage-dependent calcium channel; BMPR2, bone morphogenetic protein receptor-2; PPAR-γ, peroxisome proliferator-activated receptor-γ; PDGF, platelet-derived growth factor; FGF, fibroblast growth factor; MMP, matrix metalloproteinase; HIF-1α, hypoxia inducible factor-1α VEGF, vascular endothelial growth factor.
Definition
The PAP is equal to the systemic pressure in utero and decreases after birth due to a decline in the PVR, reaching adult levels by 2 to 3 months of age. PH has been traditionally defined as a mean pulmonary arterial pressure (mPAP) ≥25 mmHg, while the term PAH refers to elevated PAP with a pulmonary artery wedge pressure ≤15 mmHg in adults, children and term infants >3 months of age at sea level since the 1st WSPH in Geneva in 1973 (4, 38). The criteria for infants and children also include indexed pulmonary vascular resistance (PVRI) as certain classes of pediatric PH cannot be defined with mPAP alone. Children with left-to-right shunts (aortopulmonary or intracardiac shunts) with increased pulmonary blood flow may not have pulmonary hypertensive vascular disease (PHVD) early on, even though they have increased mPAP. Conversely, children without a subpulmonary ventricle might have PHVD even with an mPAP <25 mmHg. Therefore, it was recommended that a PVRI > 3 Wood units (Wu)/M2 be used to define PHVD. The recent 6th WSPH in Nice, France, in 2018 decreased the lower limit for mPAP for adult PH to >20 mmHg to include cases of precapillary PH, as long as PVRI > 3 Wood units (Wu)/M2 based on data showing even mildly elevated mPAP of 21 to 24 mmHg to be an independent predictor of worse outcomes in adult PH and right heart catheterization studies in healthy normal adults demonstrating mPAP of ~14 ± 3.3 mmHg at rest (192, 319, 375, 544). Following this recommendation, the Pediatric Task Force of the 6th WSPH also modified the criteria for diagnosis of pediatric PH to mPAP > 20 mmHg after three months of life, or PVRI ≥ 3 Wu/M2 (506). Table 1 is a comprehensive clinical definition of pediatric PH adapted from the European Pediatric Pulmonary Vascular Disease Network (EPPVDN) modeled on the 6th WSPH definitions.
Table 1.
Definition of Pulmonary Hypertension Adapted from the European Pediatric Pulmonary Vascular Disease Network (EPPVDN) modeled on the 6th WSPH definitions (233–235)
| 1. Pulmonary hypertension |
| a. mPAP > 20 mmHg in children > 3 months at sea level |
| 2. Precapillary PH (e.g., pulmonary arterial hypertension) |
| a. mPAP > 20 mmHg |
| b. PAWP or LVEDP < 15 mmHg |
| c. PVRI ≥ 3 Wu×m2 |
| d. Diastolic TPG ≥ 7 mmHg |
| 3. Isolated postcapillary PH in adults (predominantly LV diastolic dysfunction) |
| a. mPAP > 20 mmHg |
| b. PAWP or LVEDP > 15 mmHg |
| c. PVRI < 3 Wu×m2 |
| d. Diastolic TPG < 7 mmHg |
| 4. Combination of precapillary and postcapillary PH in adults |
| a. mPAP > 20 mmHg |
| b. PAWP or LVEDP > 15 mmHg |
| c. PVRI ≥ 3 Wu×m2 |
| 5. Pulmonary arterial hypertension |
| a. mPAP > 20 mmHg |
| b. PAWP or LVEDP ≤ 15 mmHg |
| c. PVRI ≥ 3 Wu×m2 plus criteria for Group 1 PH |
| 6. Idiopathic PAH (IPAH)–PAH with no underlying disease known to be associated with PAH |
| 7. Heritable PAH (HPAH)–PAH with no known underlying disease but with positive family history or positive genetic testing of the index patient |
| 8. Eisenmenger syndrome–Patient with longstanding pulmonary hypertension, supra-systemic PVR and PAP, and accordingly, right-to-left cardiovascular shunting with systemic hypoxemia (e.g., unrepaired VSD or PDA) |
| 9. Pulmonary hypertensive vascular disease For biventricular circulations: mPAP > 20 mmHg and PVR index ≥ 3 WU×m2 For circulations with cavopulmonary anastomosis (e.g., Fontan physiology): Mean TPG > 6 mmHg (calculate mPAP minus mLAP or PAWP) or PVR index > 3 WU×m2 |
PH, pulmonary hypertension; mPAP, mean pulmonary artery pressure; PAWP, pulmonary arterial wedge pressure; LVEDP, left ventricular end-diastolic pressure; PVRI, pulmonary vascular resistance index; TPG, transpulmonary gradient; PAH, pulmonary arterial hypertension; PVR, pulmonary vascular resistance; VSD, ventricular septal defect; PDA, patent ductus arteriosus; WU, Wood units mLAP, mean left atrial pressure.
Adapted, with permission, from Rosenzweig EB, et al., 2019 (506).
Epidemiology
Comprehensive data on national incidences of PAH in the neonatal and pediatric population are lacking. A recent large-scale insurance claim-based study of pediatric PAH in the United States found an incidence of 4.8 to 8.1 per million children per year and a prevalence of 25.7 to 32.6 per million children (349). The first multinational registry in pediatric pulmonary hypertension (PH) is the Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension (TOPP) registry, which includes data from 31 centers in 19 countries, although they do not report incidence or prevalence data (69). In the TOPP registry, a majority of patients (88%) had PAH, which was primarily IPAH, heritable pulmonary arterial hypertension (HPAH), or PAH associated with congenital heart disease (CHD-PAH); 12% of these patients had PH due to lung disease, with BPD being the most common cause. Another large-scale registry in the United States is the combined adult and pediatric observational cohort, Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). In this cohort, 56% of the children had IPAH/HPAH and 36% had CHD-PAH (58). This is different from the adult data from the same registry where only 12% had CHD-PAH but 24% had PAH associated with connective tissue diseases (CTDs) such as scleroderma (68). 73% of the pediatric cohort had a World Health Organization Functional Class (WHO-FC) I or II at the time of enrolment, whereas 53.7% of the adults were already at WHO-FC III or IV at the same time. Five-year survival for children with PAH was ~75%, with older age being associated with decreased odds of survival. A Netherlands registry-based study reported a yearly incidence of 63.7 cases per million children, with over 80% of these cases being transient PAH (604). The reported incidence and point prevalence of sustained PAH in this Dutch registry were 3 per million children per year and 20 per million children, respectively. A Spanish registry-based study reported an incidence of 4 per million children per year and a prevalence of 20 cases per million children, after excluding transient forms of PH (142).
This data is like the US data, which also excluded transient and early forms of PH such as PPHN and postoperative PH. Data from the UK Service for PH in Children for only IPAH revealed an incidence of 0.48 per million children per year and a prevalence of 2.1 per million children, which are similar to that of other registries (401). BPD, which is the most common morbidity in the preterm infant population, is associated with PH, which increases with increasing BPD severity with numbers of 6%, 12%, and 39% in mild, moderate, and severe BPD reported in a meta-analysis; single-center cohort studies reported the prevalence of BPD-PH to be between 15% and 64% in preterm infants with severe BPD (29, 419, 630). PPHN is the most common cause of transient PAH with an incidence of ~1.9 per 1000 births (613).
Classification of PH with the Most Recent Changes Approved at Nice Conference, 2018
Pulmonary hypertensive diseases were first classified in 1998 at the WSPH in Evian, France (542), and since then have been revised several times. The first Pediatric Task Force of the WSPH met at the 5th WSPH in Nice, France, in 2013 and concluded that a common classification for pediatric and adult PH is preferred as more children with PH are now surviving into adulthood and it is important to share a common language for the purpose of definition and classification (543). The Pediatric Task Force of the 6th WSPH in 2018 proposed some changes that are reflected in the new classification (Table 2) and are more representative of the changing landscape of pediatric PH (506). The four major changes and rationale behind the changes are summarized below:
Table 2.
Revised Clinical Classification of Pediatric Pulmonary Hypertension as Proposed by 2018 World Symposium on Pulmonary Hypertension, Nice, France. Pediatric Task Force
| Group 1. Pulmonary arterial hypertension (PAH) | 1.1 Idiopathic PAH (IPAH) 1.2. Heritable PAH (HPAH) 1.3. Drug and toxin related PAH 1.4. Associated PAH |
|
| 1.4.1. PAH associated with CTD 1.4.2. PAH associated with HIV infection 1.4.3. PAH associated with portal hypertension 1.4.4. Congenital heart disease 1.4.5. Schistosomiasis |
||
| 1.5. PAH long-term responders to CCBs 1.6. PAH with overt features of venous/capillaries (PVOD/PCH) involvement 1.7. Persistent PH of the newborn (PPHN) syndrome |
||
| Group 2. PH due to left heart disease | 2.1. LV systolic dysfunction 2.2. LV diastolic dysfunction 2.3. Valvular disease 2.4. Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathy–pulmonary vein stenosis (isolated or associated with BPD), cor triatriatum, obstructed Total Anomalous Pulmonary Venous Return (TAPVR), Mitral/aortic stenosis (including supra/subvalvular) and coarctation of aorta |
|
| Group 3. PH due to lung disease and/or hypoxia | 3.1. Chronic obstructive pulmonary disease 3.2. Interstitial lung disease 3.3. Other pulmonary diseases with mixed restrictive and obstructive pattern 3.4. Sleep-disordered breathing 3.5. Alveolar hypoventilation syndromes 3.6. Long-term exposure to high altitudes 3.7. Developmental lung diseases |
|
| Bronchopulmonary dysplasia Congenital Diaphragmatic Hernia Down syndrome Alveolar capillary dysplasia with “misalignment of veins” (FOXF1) Lung hypoplasia, acinar dysplasia Surfactant deficiency TTF-1/NKX2-1 TBX4 Pulmonary interstitial glycogenesis Pulmonary alveolar proteinosis Pulmonary lymphangiectasia |
||
| Group 4. PH due to pulmonary artery obstructions | 4.1. Chronic thromboembolic PAH 4.2. Pulmonary artery obstructions either congenital or acquired after cardiac surgery |
|
| Group 5. PH due to unclear/multifactorial mechanisms | 5.1. Hematological disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy 5.2. Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis 5.3. Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid 5.4. Others: tumor obstruction, fibrosing mediastinitis, chronic renal failure, complex CHD–unoperated or operated single ventricle, pulmonary atresia with ventricular septal defect and major aorto-pulmonary collaterals, hemitruncus, absent pulmonary artery and isolated pulmonary artery of ductal origin |
|
CTD, connective tissue disease; HIV, human immunodeficiency virus; CCB, calcium channel blocker; PVOD, pulmonary venous obstructive disease; PCH, pulmonary capillary hemangiomatosis; LV, left ventricle; BPD, bronchopulmonary dysplasia; CHD, congenital heart disease.
Adapted, with permission, from Rosenzweig EB, et al., 2019 (506).
A new class was added to Group 1 PH called PAH long-term responders to calcium channel blockers (CCBs) (Group 1.5), which is similar to adults with PAH who respond positively to an acute vasoreactivity test (AVT). Based on the criteria used (Sitbon vs modified Barst), the percentage of children with PAH who have a positive AVT ranges between 15% and 30% (157, 548).
Dutch registry-based data had shown that among children with nontransient PH, a significant proportion (34%) had PH associated with developmental lung diseases such as BPD, CDH, and congenital pulmonary vascular abnormalities (604). Hence, Group 3.5 was dedicated to developmental lung diseases, which also includes a growing list of genetic developmental lung disorders such as surfactant protein deficiency and alveolar capillary dysplasia (ACD).
Children with single-ventricle physiology may have increased or decreased pulmonary blood flow at various stages and hence do not always fit the classic definition of mPAP > 25mmHg, but they develop PHVD that markedly impairs survival and outcomes. Hence, the 6th WSPH Pediatric Task Force has grouped PHVD in the setting of single-ventricle physiology in Group 5.4.
The Task Force also agreed that Down syndrome-associated PH is variable and does not fit into a single universal classification group and hence will be classified as Group 3 PH unless they have CHD (506).
The contribution of various classes of PH under Nice classification as they apply to pediatric PHVD is described in more detail below. Specific differences between adult and pediatric PH under these categories are discussed. Table 3 discusses the clinical features, hemodynamic findings, and treatment strategies of some of the most common forms of pediatric PHD.
Table 3.
Clinical and Hemodynamic Characteristics of Major Etiologies of Pediatric Pulmonary Hypertension
| Population/risk factors/clinical features | Echocardiography findings | Treatment and prognosis | |
|---|---|---|---|
| Idiopathic and hereditary PAH (IPAH/HPAH) | IPAH – PAH with no disease known to be assocated with it HPAH – Familial history of PAH or known genetic mutation associated with PAH |
mPAP > 20 mmHg, PAWP or LVEDP ≤ 15 mmHg, PVRI ≥ 3Wu×m2 |
Pulmonary vasodilator therapy |
| PAH-CHD | ASD, VSD, PDA, AV canal defects, TGA, Eisenmenger’s syndrome, single ventricle physiology | Elevated PVRI in addition to presence of shunt lesions with affect pulmonary and systemic flow | Catheterization to perform AVT and assess for operablity, selective pulmonary vasodilators, surgical repair |
| BPD-PH | Prematurity, low birthweight, growth restriction, mechanical ventilation | Elevated PVR and TRJV, flattened IVS, RV hypertrophy, suspicion for PVS should trigger CTA or cardiac catheterization | Oxygen, ventilatory management, selective pulmonary vasodilators |
| PPHN | Neonatal population, meconium aspiration, maternal SSRIs, pneumonia, RDS, CDH, pulmonary hypoplasia, renal dysplasias | Suprasystemic PA and RV pressures after birth | Oxygen, acid-base balance, surfactant, mechanical ventilation, iNO, milrinone, inhaled and subcutaneous prostaglandins. ECMO in medically refractory PPHN |
| PH from CDH | Neonates and infants with CDH | Elevated PVR and mPAP, along with possible presence of LV dysfunction | Pulmonary selective vasodilators |
| PH from left heart disease | Older children with CHD, history of repair of coarctation of aorta, VSD repair, heart transplant, HLHS and its variants, cardiomyopathies, LV systolic or diastolic dysfunction | mPAP > 20 mmHg, PAWP or LVEDP > 15 mmHg, PVRI < 3 Wu×m2 | Treatment of left heart disease, surgical repair in children with CHD after assesing operability |
PH, pulmonary hypertension; RA, right atrium; RV, right ventricle; LA, left atrium; LV, left ventricle; PAH, pulmonary arterial hypertension; CHD, congenital heart disease; ASD, atrial septal defect; VSD, ventricular septal defect; PDA, patent ductus arteriosis; HLHS, hypoplastic left heart syndrome; CDH, congenital diaphragmatic hernia; SSRI, selective serotonin reuptake inhbitor; AV, atrioventricular; TGA, transposition of great arteries; BPD, bronchopulmonary dysplasia; PPHN, persistent pulmonary hypertension of the newborn; RDS, respiratory distress syndrome, iNO, inhaled nitric oxide; ECMO, extracorporeal membrane oxygenation, PAWP, pulmonary arterial wedge pressure; LVEDP, left ventricular end-diastolic pressure; PVRI, pulmonary vascular resistance index; mPAP, mean pulmonary artery pressure; TRJV, tricuspid regurgitant jet velocity; IVS, interventricular septum; PVS, pulmonary vein stenosis; CTA, computed tomography with angiography.
Group 1 PH (pulmonary arterial hypertension)
1.1. Idiopathic PAH (IPAH):
IPAH is defined as PAH without any identifiable cause that leads to gradual pulmonary vascular remodeling, which includes adventitial thickening, medial hypertrophy, intimal proliferation, and formation of concentric laminar intimal fibrosis and plexiform lesions. This causes vascular wall thickening and occlusion of small pulmonary arteries, which combined with vasoconstriction, inflammation, and thrombosis increases PVR and pressure. This leads to increased right ventricular afterload and eventual right heart failure and death (262, 277). Estimated incidence rates for IPAH range from 0.47 to 1-2 cases per million children, with estimated prevalence rates varying from 2.1 to 4.4 cases per million children (4). Up to 25% of patients with IPAH have mutations in genes linked to HPAH; these genes are listed under that category. Based on these observations, evaluation of IPAH patients should include genetic screening for known mutations in common genes linked to PAH (233).
1.2. Hereditary PAH (HPAH):
Multiple genetic mutations have been identified in the pediatric PAH population and are implicated in 20% to 30% of sporadic PAH and almost 80% of familial PAH (4). Bone morphogenetic protein receptor type 2 (BMPR2) is the gene most implicated in HPAH, with studies finding ~55% in familial PAH and ~10% in IPAH in both adult and pediatric PAH patients (470, 508, 659). Children and adults with BMPR2 mutations who present with PAH are more likely to have worse disease at diagnosis, present at a younger age, are less likely to respond to AVT, and are at an increased risk of death and/or transplantation (165, 508). Recently, TBX4 gene mutations that cause small-patella syndrome have been implicated in pediatric HPAH (295). Two cohort-based genetic studies found that TBX4 mutations were more enriched in the pediatric PAH population compared to adults (10/130 pediatric vs 0/178 adult onset), and TBX4 gene variant carriers had younger age of disease onset compared to BMPR2 gene variant carriers (347, 659). ACVRL1 mutations have also been implicated in pediatric HPAH, with increased enrichment compared to the adult population (188, 347, 508). Current European Pediatric Pulmonary Vascular Disease Network (PPVDN) and the 6th WSPH Pediatric Task Force recommendations are to offer genetic counseling to all families with children diagnosed with IPAH/HPAH and to evaluate family members of known mutation carriers for PAH if they develop any new cardiorespiratory symptoms (235).
1.3. Drug- and toxin-mediated PAH:
Diazoxide, which is used for the treatment of hyperinsulinemic hypoglycemia in the neonatal population, has been linked to transient PAH that resolves after discontinuation of the drug (385). Neonates on diazoxide should be evaluated for PAH if they develop symptoms of respiratory distress or poor feeding. The illicit use of methamphetamine, a drug used to treat neuropsychiatric disorders, has been linked to PAH—methamphetamine-associated PAH (meth-APAH). Meth-APAH presents with a more severe form of disease, poorer long-term outcomes, and prognosis compared to IPAH (652). Methamphetamine metabolites accumulate within the lung, leading to toxicity and vascular damage (612). PAH patients should be screened for a history of drug use, and, conversely, methamphetamine users should undergo screening for signs and symptoms of PAH (115, 489).
1.4 1.4.1. PAH-CTD:
PAH can be a rare complication of CTD and has mostly been described among patients with systemic sclerosis (SSc), with an estimated prevalence of 5% to 10% (574, 606). It is also a rare manifestation of systemic lupus erythematosus (SLE), mixed connective tissue disease (MCTD), dermatomyositis, polymyositis, Sjogren’s syndrome, and rheumatoid arthritis (89, 236, 280, 323, 487). CTD-associated PAH usually carries a worse prognosis compared to IPAH (517). In general, PH associated with CTD, HIV, and portal hypertension is less common in pediatric compared to adult population (171).
1.4.2. HIV-associated PAH:
As mortality from HIV has decreased, the incidence of cardiovascular diseases due to antiretroviral treatment-associated dyslipidemias and insulin effects as well as HIV-induced chronic endothelial dysfunction, impaired fibrinolysis and chronic inflammation have increased (287). HIV patients are seven times more likely to develop PAH than the rest of the population. The incidence of PAH varies anywhere between 10% and 50% in adult patients with HIV (45, 259, 403), although less common in pediatric population.
1.4.3. PAH associated with portal hypertension:
This can be of two distinct subtypes—hepatopulmonary syndrome (HPS), which is characterized by low PVR and increased pulmonary blood flow, and porto-pulmonary hypertension (POPH), which is characterized by increased pulmonary vascular remodeling and elevated PVR.
1.4.4. PAH-CHD:
This includes all forms of PAH associated with CHD, except complex CHDs (Group 5.4 described later) as well as PAH secondary to Eisenmenger’s syndrome in those with left-to-right shunts. Although adults with PAH and Eisenmenger’s syndrome have better mortality rates than IPAH/HPAH, for children the survival for PAH-CHD and IPAH/HPAH are similar (29% vs 25%) (58). PAH-CHD is associated with a pre- or post-tricuspid shunt lesion with or without pulmonary vascular disease and distinct patterns of right ventricular hypertrophy (RVH). Post-tricuspid lesions are left-to-right shunts (ventricular septal defects for example) that expose pulmonary circulation to systemic pressure and cause LV volume overload, leading to both volume/pressure overload on the pulmonary circulation. Untreated, most of these patients will develop Eisenmenger’s syndrome with a reversal of the shunt direction to right-to-left due to gradual progression of pulmonary pressures to a supra-systemic level (454). Pre-tricuspid lesions like atrial septal defects can be left-to-right or sometimes bidirectional. They are usually slow to progress to florid PAH due to low atrial pressures and rarely develop Eisenmenger physiology. The prognosis of these lesions is excellent if repaired early in life. PAH-CHD not associated with shunt physiology is encountered after cardiac surgery of some cardiac defects such as transposition of great vessels (TGA, transposition of great arteries), truncus arteriosus (TA), Tetralogy of Fallot (TOF), double-outlet LV, pulmonary atresia/intact ventricular septum, and aortopulmonary septal defect. It should be noted that prognosis for PAH-CHD is significantly worse for children with CHD in resource-constrained areas of the world where surgical correction is delayed, perioperative management is variable, and PHD becomes established, leading to a higher mortality risk. PAH can also develop in the setting of single-ventricle physiology. Bidirectional Glenn shunts and Fontan baffles are often used in children whose CHD precludes a direct repair due to hypoplastic ventricle. This leads to systemic venous blood draining directly into the pulmonary arteries, and there is no dedicated subpulmonary ventricle. This can lead to elevated PVR, which affects operability and outcomes of these patients with cavopulmonary anastomoses (200, 362, 397). Children can develop pulmonary arteriovenous fistulae after Glenn procedure, where only the superior vena cava blood flows into the lungs while the inferior vena cava blood bypasses the lungs to enter the systemic circulation directly. Although loss of hepatic venous blood drainage to the lungs has been suspected as being involved in the AV fistula development, cellular mechanism for AV fistula formation in this setting remains unknown. Both bosentan and sildenafil have been used in patients after Fontan repair to improve hemodynamics and oxygen consumption (212, 455).
1.4.5. Schistosomiasis:
It is a rare entity in the developed world and found more commonly in countries with endemic schistosomiasis. Globally, this is one of the most common causes of PAH, with 5% of patients with hepatosplenic schistosomiasis developing PAH.
1.5. PAH long-term responders to CCBs:
A subset of pediatric PAH patients have positive AVT to oxygen and/or inhaled nitric oxide (NO) based on Sitbon or modified Barst criteria and respond to oral calcium channel blockers (CCBs) with decreased pulmonary pressures. These children account for ~ 8% to 15% of all pediatric IPAH patients when using Sitbon criteria (548).
1.6. PAH with overt features of venous/capillaries (PVOD/PCH) involvement:
Pulmonary venous obstructive disease (PVOD) or pulmonary capillary hemangiomatosis (PCH) is rare in children. Biallelic mutations in the EIF2AK4 gene have been implicated in heritable cases of both PVOD and PCH (7, 78, 166). Risk factors for nonidiopathic PVOD include chemotherapy, organic solvent or tobacco exposure, autoimmunity, and inflammatory conditions (233, 405). The incidence of PVOD/PCH is estimated to be ~0.7% to 2% of all PAH cases (506).
1.7. Persistent pulmonary hypertension of the newborn (PPHN):
Estimated at 30.1 cases per million children per year, this is the most common cause of transient PAH. The fetal lung circulation receives 13% to 21% of cardiac output as the placenta is the site for gas exchange (495). After birth there is an eightfold increase in the pulmonary blood flow due to a drop in the PVR mediated by increased oxygen tension, ventilation, shear stress, and increased vasodilatory molecules such as NO and prostacyclin (PGI2) (326). PPHN occurs when one or more of these mechanisms fail to lower the PVR, which leads to extrapulmonary shunting of deoxygenated blood from right-to-left through the patent ductus arteriosus (PDA) and/or patent foramen ovale (PFO) with profound systemic hypoxemia, differential oxygen saturation gradient between the pre- and postductal circulation and increased risk of death or neurodevelopmental impairment in survivors (190, 312, 315). PPHN can be due to (i) increased pulmonary vasoconstriction in the setting of a structurally normal architecture, which is seen in lung parenchymal diseases like meconium aspiration syndrome, respiratory distress syndrome, sepsis, and pneumonia; (ii) pulmonary vascular remodeling and altered vasoreactivity and impaired angiogenesis seen in idiopathic PPHN; and (iii) pulmonary hypoplasia leading to hypoplastic pulmonary vasculature seen in CDH and maternal oligohydramnios (190). The prevalence of PPHN has been historically described as 1.9 per 1000 births; however, with growing recognition of the syndrome especially in preterm infants, the numbers have been rising (613). In addition to the conditions described, other risk factors for developing PPHN include maternal use of selective serotonin reuptake inhibitors (SSRIs) or nonsteroidal anti-inflammatory drugs (NSAIDs), prematurity, male gender, maternal diabetes, asthma, and obesity (143, 245, 423). Mortality for PPHN was >50% prior to extracorporeal membrane oxygenation (ECMO) and use of pulmonary vasodilators like inhaled nitric oxide (iNO). Although mortality rates have decreased to less than 10%, long-term adverse outcomes like cerebral palsy, deafness, and blindness remain high in survivors (317).
Group 2
PH due to left heart disease (LH disease): LH disease is gradually being recognized as an important contributor to pediatric PH. Repair of CHD like coarctation of aorta, VSD, mitral valve replacement, hypoplastic left heart syndrome, and cardiac transplantation can 6lead to left ventricular dysfunction, thereby causing increased back pressure in the pulmonary venous circulation and ultimately postcapillary PH. LV dysfunction is also increasingly being recognized as a cause of BPD-PH (320). Valvular lesions like mitral or aortic stenosis (AS) can also lead to a similar feature of increased pressure in the pulmonary capillary bed. Critical AS or aortic atresia in fetuses and newborns is associated with increased pulmonary vascular muscularization, and pulmonary veins become arterialized in utero, leading to impaired postnatal pulmonary vascular adaptation (241). Outcome for these infants has improved and 5-year survival rates are between 77% and 85% at 5 years (199). Pulmonary vein stenosis (PVS) is associated with very high mortality rates and worse outcomes; medical or surgical therapies are mostly ineffective (609, 649). Like LV dysfunction, this is also becoming an increasing feature in BPD-PH and contributes to increased mortality in this population (320).
Group 3
PH due to lung diseases and/or hypoxemia: Interstitial or parenchymal lung diseases or systemic diseases that affect ventilation of the lung cause chronic hypoxia, which leads to pulmonary vasoconstriction, pulmonary vascular remodeling, and ultimately right heart failure to high afterload. This includes chronic hypoventilation and obstructive sleep apnea (OSA) as well hypobaric hypoxia resulting from high altitudes. OSA in otherwise normal children with enlarged tonsils and adenoids showed almost 20% incidence of RVH by echocardiography and 37% of children with OSA diagnosed on sleep study have decreased RV ejection fraction measured by radionuclide ventriculography (336, 582).
BPD-associated pulmonary hypertension (BPD-PH):
Altered lung development due to growth arrest of alveoli and pulmonary capillaries can lead to the vascular phenotype of BPD-PH (75). Pathogenesis of BPD-PH is multifactorial as shown in Figure 1. Maternal factors such as chorioamnionitis, smoking, preeclampsia, and intrauterine growth restriction, especially if accompanied with reversed or absent end-diastolic flow in the umbilical arteries, are prominent risk factors for BPD-PH in a growing population of extremely preterm infants (75, 114, 387). Small-for-gestational age (SGA) is another risk factor for PH in preterm infants with and without BPD (29). Postnatal lung injury from ventilation and oxygen exposure, infections, inflammatory response, and poor postnatal growth together contribute to alveolar and vascular injury and growth arrest. Two other morbidities associated with prematurity—necrotizing enterocolitis (NEC) and retinopathy of prematurity (ROP)—were also strongly associated with the increased prevalence (29) of BPD-PH in a cohort study from the Children’s Hospital Neonatal Consortium. This study also reported the incidence of PH in preterm infants <32 weeks gestational age with severe BPD to be at 22% (325). During admission, PH was associated with increased mortality and duration of ventilation and after discharge with medical interventions, including tracheostomy, supplemental oxygen use, tube feeds, and increased frequency of readmission through 1 year of life. Presence of PH is strongly associated with increased mortality with reports ranging from 14% to 38% and a meta-analysis reporting 16% mortality before discharge and 40% at two years of life (29, 75). The prevalence of PH increases in step with BPD severity. A meta-analysis and single-center cohort studies reported numbers of 6%, 12%, and 39% in mild, moderate, and severe BPD and the prevalence of BPD-PH to be between 15% and 64% in preterm infants with severe BPD (29, 419, 630). BPD is associated with dysmorphic growth of pulmonary vessels, reduced microcirculation, and altered distribution of vessels. This abnormal vasculature contributes to impaired alveolar-capillary gas exchange causing prolonged hypoxemia, requirement for positive pressure ventilation, and the risk of developing severe PH (2, 18, 139, 588). The pulmonary vasculature shows increased tone and vasoreactivity, decreased growth and increased hypertensive remodeling. This leads to high resting PVR even in the absence of hypoxia and an exaggerated pulmonary vasoconstrictor response to hypoxia. Decreased growth and pruning of vessels lead to severely compromised lung perfusion and right heart strain, especially if significant left-right shunts (8, 79, 416, 417, are, present). Development of pulmonary vascular disease early on in the course of life also strongly correlates with increased severity of BPD, which is an independent risk factor for the development of late BPD-PH (419). Three additional findings on echocardiogram for these preterm infants are being increasingly recognized as contributory and prognostic factors for the development and outcome of BPD-PH—PVS, left ventricular dysfunction, and presence of aortopulmonary collaterals. The prevalence of PVS in a cohort of infants with BPD-PH who underwent cardiac catheterization was 26%, and there have been reports of association with NEC (141, 243, 370, 575). Left ventricular diastolic dysfunction should be suspected in the setting of PH with worsening pulmonary edema or increasing diuretic requirements.
Figure 1.
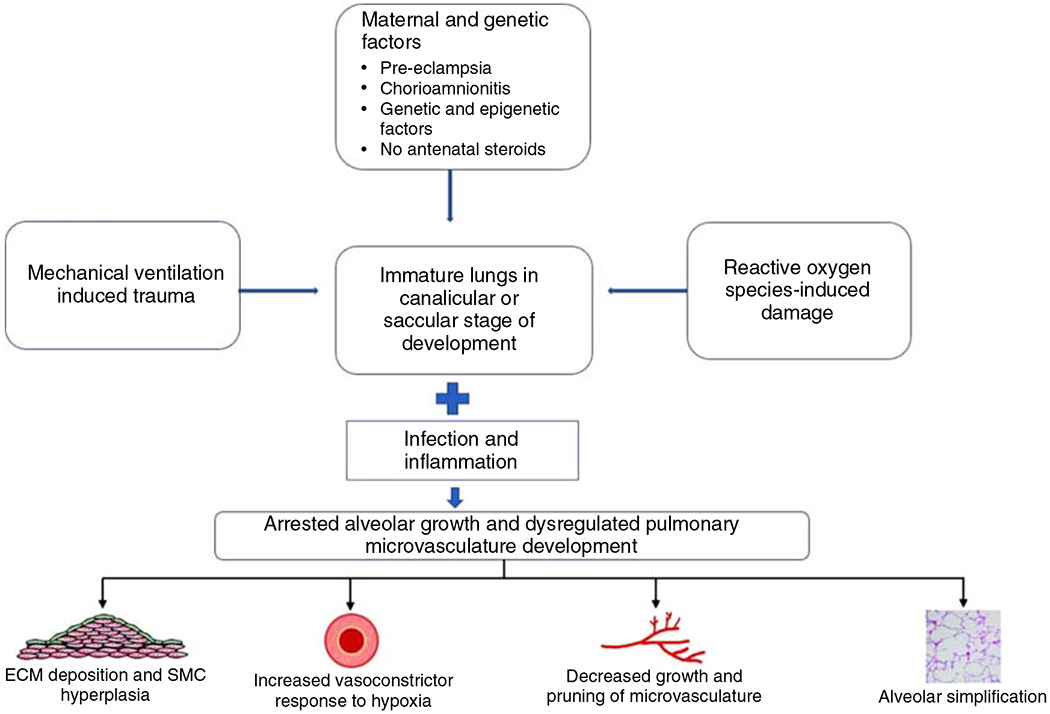
Pathogenesis of bronchopulmonary dysplasia (BPD) associated pulmonary hypertension. The figure highlights the contribution of both prenatal and postnatal factors to the evolution of BPD. ECM, extracellular matrix; SMC, smooth muscle cell.
Congenital diaphragmatic hernia-associated pulmonary hypertension (CDH-PH):
CDH is a birth defect characterized by the herniation of intra-abdominal contents into the thoracic hernia through a diaphragmatic defect in utero. This is a life-threatening condition resulting in death if not medically managed and surgically corrected. With the advent of antenatal diagnosis, better surgical techniques, ventilatory management, and ECMO support, the mortality has decreased to 25% to 30% in the last few decades. The incidence of CDH is ~1 in every 2500 births and PH occurs in nearly 60% to 70% of these infants (266, 556). PH persisting to 1 month of age in CDH is strongly associated with increased mortality (~45%). A multicenter cohort study reported only a 43.9% survival rate when the ratio of RV to systemic pressure at 1 month was greater than 0.67 and 98.6% survival when the ratio was <0.5 (635). The two-hit hypothesis for CDH-PH proposes an early embryonic alteration of the pulmonary vasculature and parenchymal development followed by a later mechanical compression by the herniated abdominal contents leading to pulmonary hypoplasia (292). This leads to a hypoplastic pulmonary vascular bed with decreased arborization and altered vasoreactivity along with increased remodeling with medial and adventitial thickening (239, 400). Left ventricular hypoplasia and dysfunction due to altered mechanics of the thoracic cavity add to this by causing pulmonary venous hypertension (539). LV mass was significantly smaller in nonsurviving infants with CDH, which could be due to compression by the abdominal contents, redistribution of fetal cardiac output from LV to RV in CDH, or less pulmonary venous return to the left side of the heart from the hypoplastic CDH lung (302). This is an important factor contributing to the success or failure of pulmonary vasodilators in the treatment of acute or postoperative PH in the CDH, as they may contribute to worsening wedge pressure and pulmonary edema in the presence of LV dysfunction (359).
ACD with misaligned pulmonary veins is a uniformly fatal disorder characterized by immature lobular development, abnormal air-blood barrier, and an underdeveloped pulmonary capillary bed (81). Mutations in FOXF1 gene are found in 40% to 60% of infants with ACD, particularly in the presence of coexisting anomalies such as anorectal malformations, skeletal defects, and congenital heart defects (576). Most cases of ACD develop severe PAH and die despite maximal PH therapy.
Group 4: PH due to pulmonary artery obstruction
4.1 Chronic thromboembolic PH (CTEPH) occurs in 0.5% to 3.8% of patients with acute or recurrent pulmonary thromboembolism (394). The embolus transforms into a fibrotic residue, resulting in proximal vessel obstruction and distal arteriopathy leading to PH and right ventricular failure (414). CTEPH should be considered in all symptomatic pediatric patients with known hypercoagulable state, history of thromboembolism, or venous catheter placement, especially as the rate of venous thromboembolism in the pediatric population has been rising (488). Pulmonary thromboendarterectomy (PTE), which involves removal of organized thromboembolic material from the vessel intima, is usually well tolerated in these patients with improved hemodynamic and functional status and low perioperative mortality (127, 368).
The illness caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), better known as COVID-19 (coronavirus disease-19), also leads to a coagulopathic state causing venous thromboembolic events. Autopsies of patients who died from COVID-19-induced acute lung injury (ALI) show damage to the pulmonary vascular endothelium and pulmonary capillaries filled with dense fibrin-rich microthrombi (10, 398). Although cases of CTEPH have not been reported in the pediatric population from COVID-19 sequelae, having a follow-up strategy for identifying residual clot burden and potential hemodynamic consequences is important in this group (150). An adult cohort study of right heart catheterization data in mechanically ventilated COVID-19 patients comparing it to patients with acute respiratory distress syndrome (ARDS) from non-COVID-19 causes found that although PVR was lower in COVID-19, there was a high incidence of PH in COVID-19, with a majority being postcapillary PH (101).
Table 4 discusses the key differences between precapillary, postcapillary, and a mixed type of PH in the pediatric population.
Table 4.
Comparison Between Precapillary, Postcapillary and Mixed PH
| Precapillary PH | Postcapillary PH | Mixed pre- and postcapillary PH | |
|---|---|---|---|
| Etiologies | Group 1 and 3 PH predominantly | Group 2 PH (left heart disease including systolic and diastolic LV dysfunction), PVS |
Predominantly left heart disease progressing to increased PVR due to pulmonary vascular remodeling over time |
| Hemodynamic findings and diagnosis | mPAP > 20 mmHg, PAWP or LVEDP ≤ 15 mmHg, PVRI > 3 Wu×m2, Diastolic TPG ≥ 7 mmHg |
mPAP > 20 mmHg, PAWP or LVEDP > 15 mmHg, PVRI < 3 Wu×m2, Diastolic TPG < 7 mmHg |
mPAP > 20 mmHg, PAWP or LVEDP > 15 mmHg, PVRI ≥ 3 Wu×m2, Diastolic TPG ≥ 7 mmHg |
| Treatment | Targeted toward conventional PH therapy |
Targeted toward treating left heart dysfunction | Combination of both approaches of treating pre- and postcapillary PH |
Another clinical classification described in this article is the 2011 Panama classification for pediatric PH (Table 5). This is different from the WSPH classification, which has often been critiqued as more adult PH-oriented. The Panama classification was proposed by the Pulmonary Vascular Research Institute (PVRI) Pediatric Taskforce, which was a group of North and South American pediatric PH experts (328).
Table 5.
Panama Classification for Pediatric Pulmonary Vascular Disease as Proposed by the Pulmonary Vascular Research Institute (PVRI) Pediatric Task Force in 2011
| Category | Description |
|---|---|
| 1 | Prenatal or developmental pulmonary hypertensive vascular disease |
| 2 | Perinatal pulmonary vascular maladaptation |
| 3 | Pediatric cardiovascular disease |
| 4 | Bronchopulmonary dysplasia |
| 5 | Isolated pediatric pulmonary hypertensive vascular disease (isolated pediatric PAH) |
| 6 | Multifactorial pulmonary hypertensive vascular disease in congenital malformation syndromes |
| 7 | Pediatric lung disease |
| 8 | Pediatric thromboembolic disease |
| 9 | Pediatric hypobaric hypoxic exposure |
| 10 | Pediatric pulmonary vascular disease associated with other system disorders |
Modified, with permission, Cerro MJ, et al., 2011 (106). © 2011, SAGE Publications.
Functional classification
Functional classification (FC) of PH is difficult in infants and children due to the practical difficulty of performing exercise tests and the lack of reliable self-reporting of symptoms. The New York Heart Association (NYHA) FC is commonly used by cardiologists to assess clinical status in adults with heart failure. The WHO-FC is a FC for adults with PH, which is modeled on the NYHA FC (26).
WHO-FC:
Class I: Patients with PH but without limitation of physical activity. Ordinary physical activity does not cause undue dyspnea, fatigue, chest pain, or near syncope.
Class II: Patients with PH resulting in slight limitation of physical activity, comfortable at rest. Ordinary physical activity causes undue dyspnea, fatigue, chest pain, or near syncope.
Class III: Patients with PH resulting in marked limitation of physical activity, but comfortable at rest. Less than ordinary physical activity causes undue dyspnea, fatigue, chest pain, or near syncope.
Class IV: Patients with PH resulting in inability to carry out physical activity without symptoms. Symptoms of right heart failure are present, and dyspnea and fatigue are usually present at rest. Syncope or near-syncope may occur.
The Pediatric Task Force of the PVRI proposed a new FC for PH in children in 2011 (Table 6), known as the Panama classification (328). This is stratified into five different classes based on the ages of 0 to 0.5 year, 0.5 to 1 year, 1 to 2 years, 2 to 5 years, and 5 to 16 years. After 16 years, adult FCs can be reliably used. These incorporate weight gain and developmental milestones into the assessment along with increased self-reporting of symptoms as the child grows older.
Table 6.
Panama Functional Classification for Pediatric PH as Proposed by Pulmonary Vascular Research Institute (PVRI) Pediatric Task Force, 2011
| 0 to 0.5 year | 0.5 to 1 year | 1 to 2 years | 2 to 5 years | 5 to 16 years | |
|---|---|---|---|---|---|
| I | Asymptomatic, growing and developing normally, no limitation of physical activity (PA). Gains head control, increases body tone, rolls over, sits without support gradually | Asymptomatic, growing on centiles, no limitation of PA. Mobile, sitting, grasping, crawling, playing | Asymptomatic, growing on centiles, no limitation of PA. Standing, starting to walk, climbing |
Asymptomatic, growing on centiles, no limitation of PA. Attending school normally and playing sports with classmates | Asymptomatic, growing on centiles, no limitation of PA. Attending school normally and playing sports with classmates |
| II | Slight limitation of PA, unduly dyspneic and fatigued, falling behind developmental milestones. Comfortable at rest and gaining weight | Slight limitation of PA, unduly dyspneic when playing. Delayed milestones but normal growth and comfortable at rest | Slight limitation of PA, unduly dyspneic when playing. Delayed milestones but normal growth and comfortable at rest | Slight limitation of PA, unduly dyspneic as compared to classmates. <75% attendance at school. Comfortable at rest and normal weight gain | Slight limitation of PA, unduly dyspneic as compared to classmates. <75% attendance at school. Comfortable at rest and normal weight gain |
| IIIa | Marked limitation of PA, unduly fatigued. Quiet, needs frequent naps, poor feeding, growth, and regression of learned milestones. Comfortable at rest | Marked limitation of PA, unduly fatigued while playing. Quiet, needs frequent naps, poor feeding, growth, and regression of learned milestones. Comfortable at rest | Marked limitation of PA, unduly fatigued while playing. Quiet, needs frequent naps, poor feeding, growth, and regression of learned milestones. Comfortable at rest | Marked limitation of PA, regression of milestones, not climbing stairs, reluctant to play with friends. Less than ordinary activity causes symptoms. <50% attendance at school | Marked limitation of PA, no attempt at sports. Less than ordinary activity causes symptoms. Comfortable at rest. <50% attendance at school |
| IIIb | IIIa plus severely compromised growth and feeding | IIIa plus severely compromised growth and feeding | IIIa plus severely compromised growth and feeding | Unable to attend school, mobile at home, needs wheelchair outside. Compromised growth, poor feeding plus IIIa | Unable to attend school, mobile at home, needs wheelchair outside. Compromised growth, poor feeding plus IIIa |
| IV | Unable to carry out any PA without severe symptoms and is not interacting with family. Right heart failure (RHF) plus syncope | Unable to carry out any PA without severe symptoms and is not interacting with family. RHF plus syncope | Unable to carry out any PA without severe symptoms and is not interacting with family. RHF plus syncope | Unable to carry out any physical activity without severe symptoms, unable to attend school, not interacting with friends, wheelchair dependent, RHF plus syncope | Unable to carry out any physical activity without severe symptoms, unable to attend school, not interacting with friends, wheelchair dependent, RHF plus syncope |
Modified, with permission, from Lammers AE, et al., 2011 (328). Under Creative Commons License.
Cellular and Structural Changes
A variety of cellular and structural changes play a complex role in the pathogenesis of PH (see Figure 2). The primary imbalance between the vasoconstrictor and vasodilator pathways leads to persistent vasoconstriction and pulmonary vascular remodeling, eventually causing right ventricular failure secondary to increased afterload. Alterations in cell biology are presented below for each vascular cell; however, the cell-cell communications are integrated into a complex signaling network that affects the entire vascular wall.
Endothelial cells
The innermost layer of blood vessels is composed of a monolayer of endothelial cells supported by an internal elastic lamina. This endothelium forms a nonthrombogenic, semipermeable barrier between the bloodstream and the extravascular tissues. It regulates vascular tone, hemostasis, growth and differentiation of blood vessels as well as chemotaxis (466). Endothelial cells are the first cells to be exposed to the effects of low oxygen tension in the blood. Chronic hypoxia leads to endothelial cell hypertrophy, as evidenced by the increased DNA synthesis and increased cell number, which are demonstrated by an approximately threefold increase in 3H-thymidine incorporation by endothelial cells early during hypoxia exposure. Endothelial cells undergo disorganized proliferation, which could lead to plexiform lesions or concentric obstructive lesions, both of which lead to obliteration of the pulmonary vascular lumen (596). Plexiform lesions are glomeruloid-like disorganized endothelial cells, which demonstrate markers of angiogenesis such as HIF-1α and VEGF (597). They are most commonly found in IPAH and Group 2 PH. Concentric lesions are onionskin-like proliferative growth of endothelial and/or smooth muscle cells. Rarely, paucicellular lesions can be found in the intima of the pulmonary hypertensive artery, which is characterized by increased extracellular matrix (ECM) and mucopolysaccharides and decreased endothelial cell number (596). The intimal fractional thickness, which is a measure of the contribution of the intima to the overall diameter, shows an almost threefold increase in patients with severe PAH (561). The subendothelial space, which is present between the endothelial cell and its basement membrane, contains increased amounts of collagen, elastin, and microfibrils in autopsy specimens of infants dying from PH. Endothelial cell elastin production, which is suppressed in late fetal and early neonatal life, is upregulated by reexpression of tropoelastin mRNA in endothelial cells in response to hypoxic injury (161, 566). Hypoxia leads to increased expression of neutrophil chemotactic factors on endothelial cells (391). Endothelial cells release mediators that regulate vascular tone and smooth muscle proliferation, and the balance between vasodilatory and vasoconstrictive mediators is lost in PH. The three principal mediators are NO, PGI2, and endothelin (ET-1), which are described in detail in Section 6 in this article.
Smooth muscle cells (SMCs)
SMCs play one of the most important roles in increased pulmonary vascular contractility, increased muscularization of the resistance arteries, medial thickening, abnormal muscularization of the distal nonmuscular pulmonary arteries, and increased ECM production leading to pulmonary vascular remodeling. Pulmonary artery smooth muscle cells (PASMCs), which are in a quiescent state of performing contractile function during the normal physiological state, possess a unique feature unlike other vascular SMCs—they are not terminally differentiated and hence can modulate their phenotype greatly in response to stress and changes in their environment (577). The key processes that change SMC phenotype in response to a PH-causing environment are hypertrophy, proliferation or hyperplasia, resistance to apoptosis, and migration. SMC hypertrophy occurs mainly from increased protein synthesis with decreased breakdown, along with the increased intracellular water content (73). There is increased expression of Na+ ion channels, which are key to the maintenance of increased cell volume. This hypertrophy is also mediated by G-protein-coupled receptor-agonists such as angiotensin II (ANGII), ET-1, thromboxane-A2 (TXA2), and other receptor tyrosine kinases. SMC proliferation, which contributes to the medial thickness and the muscularization of nonmuscular arteries, is regulated by increased Ca2+ levels. There are conflicting reports as to whether hypoxia directly exerts a mitogenic effect on PASMCs, whether hypoxia stimulates the PASMCs to produce an autocrine growth factor, or whether hypoxic stimulation leads to the synthesis of paracrine signals from the neighboring PAECs, which result in PASMC proliferation. PH leads to upregulation of transient receptor potential channel (TRPC) genes and store operated Ca2+ entry (SOCE) channels, which result in increased cytosolic Ca2+ concentration. Ca2+ binds to calmodulin, which activates Ca2+-calmodulin-dependent protein kinases, which in turn phosphorylate transcription factors such as Ca2+/cAMP-response element binding protein (CREB) and Ras, responsible for initiating and maintaining the cell cycle (331). PASMC proliferation has also been linked to the activation of the mTOR pathway, and rapamycin (mTOR inhibitor) normalizes the growth of PASMCs in the monocrotaline (MCT)-induced PH model (258). NO donors were found to inhibit hypoxic PASMC proliferation in vitro in a dose-dependent manner with associated cGMP increases (20). PASMC migration is a phenomenon that occurs during development, vascular injury, and vessel wall remodeling. Growth factors like platelet-derived growth factor (PDGF), epidermal growth factor (EGF), fibroblast growth factor (FGF2), as well as cytokines like IL-6, have all been implicated in increased PASMC migration (202).
Role of fibroblasts and extracellular matrix
Monocytes, macrophages, T lymphocytes, and dendritic cells have all been found in the plexiform and other lesions of PAH-affected human lungs (600). Fibroblasts are the major cell type found in the adventitial layer of the pulmonary vasculature and produce ECM and matricellular proteins (569). They are often the first cells to become activated, proliferate, and differentiate in response to injury (372). In addition, PAH is characterized by endothelial-to-mesenchymal transformation (EndMT) where PAECs lose their cell-to-cell connections due to loss of cell surface markers, detach from the endothelial monolayer, migrate to the medial layer, and dedifferentiate into myofibroblast-like cells with increased expression of α-smooth muscle actin, vimentin, and collagen (567). Inflammation, chronic hypoxia, BMPR2 mutations, increased flow, and shear stress have all been implicated in EndMT (218, 257, 491, 567). Proliferation of the adventitial fibroblasts as well as EndMT leads to changes in the vascular ECM with increased proteolytic enzymes like matrix metalloproteinases (MMPs), metalloproteases, serine elastases, lysyl oxidases and a decrease in the tissue inhibitors of metalloproteinase (TIMPs). This imbalance results in increased collagen deposition, cross-linking of collagen (conversion of soluble to insoluble collagen), elastin deposition and breakdown, and deposition of fibronectin and tenascin. This change in the ECM milieu results in pulmonary vascular remodeling with increased PVR and decreased compliance (77, 253, 345, 589). Animal models with increased expression of MMPs have exaggerated pulmonary vascular remodeling in response to monocrotaline or chronic hypoxia, and, conversely, rats with overexpression of serine elastase inhibitors have an attenuated increase in PAP and pulmonary vascular remodeling when exposed to hypoxia (201, 651). Rodent models of PH (both monocrotaline and chronic hypoxia) have demonstrated that administration of serine elastase inhibitors decreases elastolytic activity, reduces muscularization of nonmuscular distal pulmonary arteries, and lowers PAPs (128, 265, 589, 643).
Pulmonary vasculature
The changes in the pulmonary vasculature differ based on the etiology of PH. BPD is characterized by an arrest in the lung alveolar and vascular development, leading to decreased capillary density and alveolar-capillary area for gas exchange (79). The neonatal rodent hyperoxia model for BPD has demonstrated that the extent of alveolar simplification (less complex interstitial structure with decreased alveolar number and septation) and decreased vessel density depends on the concentration of inspired oxygen and occurs in a dose-dependent manner (615). Angiogenesis, the development of sprouts from existing blood vessels, helps in branching of vascular networks in the developing fetal lung, which then coalesce to permit blood flow. Animal model studies have shown that angiogenic signaling is severely impaired in BPD with disrupted vascular endothelial growth factor (VEGF) signaling, decreased pro-angiogenic factors, and increased vasoconstrictor and inflammatory molecules (39, 40). Hyperoxia-induced damage to the pulmonary microvasculature also causes persistent irreversible pulmonary artery medial thickness and increased EC cytoplasm content (431). Impaired angiogenic signaling leading to decreased microvascular cross-sectional area, impaired vasoreactivity, and increased vascular tone together contribute to increased PVR in BPD-PH. Increased PVR and chronic hypoxic vasoconstriction further lead to pulmonary vascular remodeling with intimal hyperplasia and muscularization of small pulmonary arteries (18, 39, 40, 99, 220). This mechanism of impaired angiogenesis due to the arrest in lung development in BPD is different from that seen in IPAH or HPAH, for example, where the pulmonary vasculature and alveolar growth are usually complete before vascular remodeling happens. The pulmonary vascular remodeling in IPAH/HPAH involves intimal and medial hyperplasia of the muscular pulmonary arteries and distal muscularization of the nonmuscular arteries and precapillary arterioles (536). This is accompanied by proliferation and migration of PASMCs, endothelial-to-mesenchymal transition, and the development of vaso-occlusive lesions comprising PAECs, PASMCs, and migratory and inflammatory cells (596). This fixed obstruction seems to be more dominant in IPAH/HPAH, whereas the dynamic obstruction due to altered vasoreactivity and imbalance between vasodilatory and vasorelaxant mediators is more prominent in PH due to chronic hypoxia, even though pulmonary vascular remodeling is a prominent feature in both. Both conditions eventually reduce the pulmonary arterial cross-sectional area, leading to elevated PVR, which aggravates the remodeling process.
Right ventricular changes
Once PVR is elevated, the right ventricle (RV) must pump the blood against increased afterload, thereby causing increased RV strain. This leads to RV hypertrophy over time with increased protein synthesis and cardiac myocyte size without replication. This is at first a compensatory mechanism, but as the RV assumes a more rounded shape, it compresses the left ventricle (LV) and pushes the IVS leftward. The RV hypertrophy leads to progressive contractile dysfunction compounded by the impaired delivery of oxygen and substrates due to the decreased RV vessel density. This, in turn, leads to gradual decompensation with RV becoming dilated, hypokinetic, and fibrotic, causing RV failure (86, 221). Although increased RV afterload is the initiating event for RV failure, a variety of other mechanisms such as neurohormonal signaling, oxidative stress, inflammation, ischemia, and cell death all contribute to right heart failure (86). The key factors leading to RV failure are (i) limited contractile reserve and adaptability to an elevated transpulmonary gradient, (ii) ischemia due to reduced perfusion pressure of the right coronary artery (RCA) from reduced epicardial systolic flow and/or microvascular rarefaction in the RV, (iii) shift from mitochondrial oxidative phosphorylation to cytosolic aerobic glycolysis, and (iv) downregulation and desensitization of adrenergic receptors in the RV (428, 472, 473, 518). RV failure is the primary cause of death in pediatric and adult PH, and three-dimensional echocardiography of RV function correlates with the severity of pediatric PH (283). In addition to RV systolic failure, PH is also characterized by RV diastolic dysfunction, which is related to RV muscle mass and afterload (196). Decreased RV output leads to impaired LV filling and cardiac output (CO), and decreased LV filling decreases the ability of the LV to assist the failing RV, setting up a feed-forward loop. RV diastolic dysfunction and leftward IVS deviation also impair LV filling and hence diastolic ventricular interaction is as important as systolic interaction in the pathogenesis of heart failure in PH (93, 195).
Molecular Mechanisms
Although many advances have been made in the field of pulmonary vascular biology and molecular mechanisms regulating PVR, much of it is still poorly understood.
NO-sGC-cGMP pathway
NO is synthesized inside endothelial cells by the enzyme endothelial NO synthase (eNOS, NOS3), which cleaves the terminal amino group from the NO precursor, l-arginine, and combines oxygen to generate NO and l-citrulline (461) (see Figure 3). NOS3 gene, which codes for eNOS transcript, is present on chromosome 7. There are two other NO synthases, neuronal and inducible NOS, neither of which are expressed normally in the endothelium. Decreased eNOS expression and function is an important factor in the development of PPHN (251). eNOS uses 5,6,7,8-tetrahydrobiopterin (BH4), nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), and Ca2+ as cofactors. BH4 reduces molecular oxygen to form water, a process coupled to the oxidation of l-arginine to generate NO and l-citrulline. Reductions in BH4 lead to uncoupling of NOS and the reduction of oxygen to superoxide anion instead of water. Superoxide can combine with NO to produce peroxynitrite, which is a potent vasoconstrictor (306). NO works in a paracrine fashion and diffuses out of the endothelial cell and into the smooth muscle cell present in the medial layer of the vessel wall. Here it stimulates soluble guanylate cyclase (sGC), which converts GTP into cyclic 3′,5′-guanosine monophosphate (cGMP), which then activates cGMP-dependent protein kinases, namely, protein kinase G (PKG1) (422). NO-cGMP signaling has been established as one of the key pathways in vascular smooth muscle cell relaxation (410, 451). PKGs, which are responsible for most of the intracellular actions of cGMP, are serine/threonine protein kinases with PKG-I being the predominant isoform in the vascular cells (105). PKG decreases intracellular Ca2+ concentrations by phosphorylation and inactivation of voltage- and receptor-gated Ca2+ channels, which reduce the influx and increase the efflux of Ca2+ (232). cGMP also activates myosin light chain phosphatase, which then decreases vascular tone (343). Acute hypoxia has been shown to decrease PKG activity in fetal pulmonary vascular smooth muscle in animal models of PH, hence indicating decreased NO responsiveness (433). Adult studies have shown decreased levels of eNOS expression in the pulmonary endothelium of the lungs of patients with plexiform PH (206). A dysfunctional NO-sGC-cGMP-PKG pathway is one of the key players in disrupted endothelial cell function and pathogenesis of PH. This results from abnormal eNOS expression, reduced NO production due to eNOS uncoupling, diminished NO bioavailability due to oxidative stress, diminished activities of sGC and PKG, and increased activity of phosphodiesterase-5 (197). Phosphodiesterases (PDEs) are a superfamily of enzymes, from PDE-1 to 11, which can inactivate cAMP and cGMP. The major cGMP-degrading PDE is PDE-5, which is abundantly expressed in the lung tissue. PDE-5 is inhibited by the drugs sildenafil and tadalafil, which are currently used for the treatment of PH (471).
Figure 3.
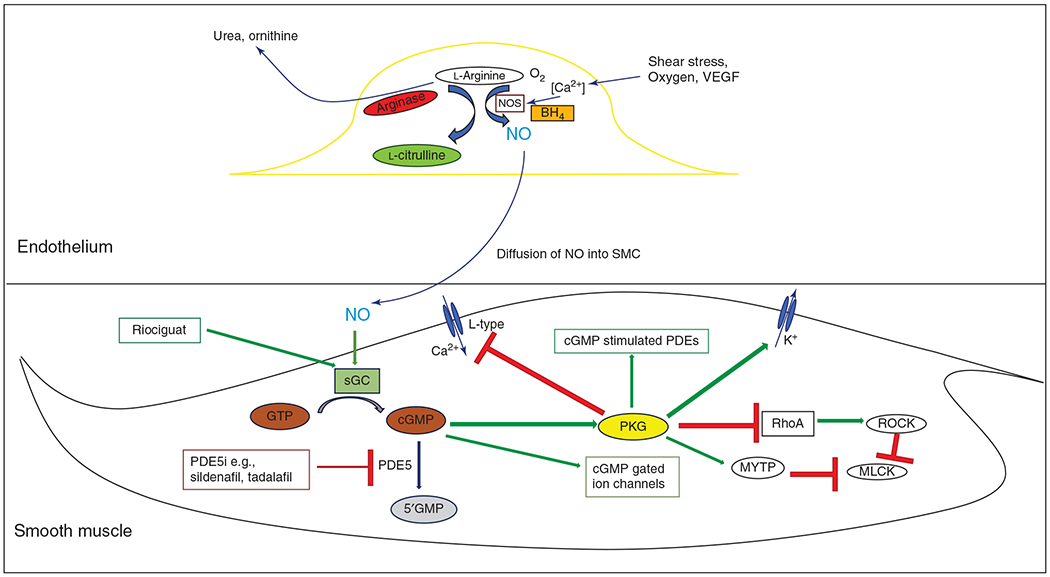
NO-cGMP pathway showing paracrine effect of endothelial NO on vascular smooth muscle cell. NO, nitric oxide; NOS, nitric oxide synthase; BH4, tetrahydrobiopterin; sGC, soluble guanylate cyclase; GTP, guanosine triphosphate; cGMP, cyclic guanosine monophsphate; PDE, phosphodiesterase; PDE5i, PDE5 inhibitor; PKG, protein kinase G; MYTP, myosin phsophatase targeting subunit; MLCK, myosin light chain kinase; ROCK, Rho kinase.
Prostacyclin (PGI2)
PGI2, which is produced by endothelial cells under shear stress, has a variety of functions including inhibition of smooth muscle cell proliferation, vasodilatation, and antiplatelet aggregation. Phospholipase A2 catalyzes the conversion of membrane-bound lipids in endothelial cells to form arachidonic acid (395). Cyclooxygenase-1 (COX2) converts arachidonic acid into intermediate prostaglandins, which serve as precursor molecules to a host of other mediators, including PGI2, which is formed from PGH2 by the action of prostacyclin synthase (PGIS) (396). Both COX-1 and PGIS are abundantly expressed in the endothelium. PGI2 acts via the IP receptor and adenylate cyclase to convert adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP). Increased levels of cAMP mediate increased protein kinase A activity and vascular smooth muscle cell relaxation (Figure 4) (16). Studies in neonatal lambs with PPHN demonstrated that PGIS, COX-1, and COX-2 activity are decreased, contributing to impaired angiogenesis (369).
Figure 4.
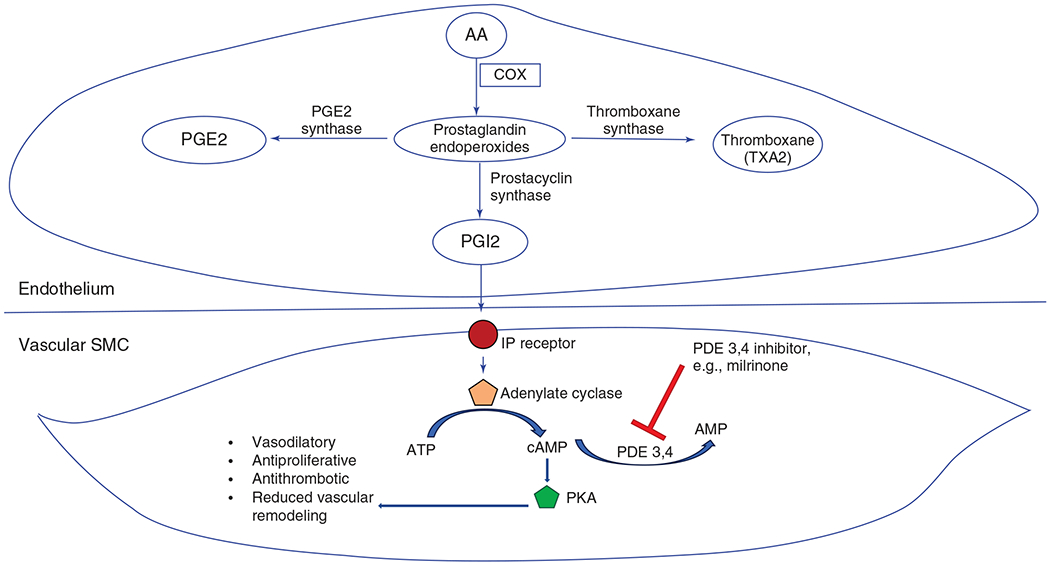
Prostacyclin pathway role in pulmonary hypertension. AA, arachidonic acid; COX, cyclooxygenase; PGE2, prostaglandin E2; PGI2, prostacyclin; IP, inositol phosphate; PDE, phosphodiesterase; ATP, adenosine triphosphate; AMP, adenosine monophosphate; cAMP, cyclic AMP; SMC, smooth muscle cell; PKA, protein kinase A.
In the monocrotaline model of rat PH, COX-2 knockout mice showed increased pulmonary oxidative stress and vasoconstriction. Similarly, the hypoxic mouse model for PH showed that hypoxia induced PH and vascular remodeling was exacerbated in COX-2-deficient pulmonary artery smooth muscle cells (186, 534). Adult lung specimens with severe PH showed a complete lack of PGIS expression in the large pulmonary arteries (598).
Endothelin
ET-1 is a potent endogenous vasoconstrictor and also causes vascular smooth muscle cell proliferation. ET-1 is produced by endothelial cells and acts on the neighboring SMCs in a paracrine fashion (640). Hypoxia, ischemia, and shear stress activate the prepro ET-1 gene promoter, which transcribes the preproET-1 peptide, the precursor molecule of ET-1 (191). NO and PGI2 have been shown to inhibit ET-1 release, resulting in pulmonary vasodilatation (480, 492). ET-1 binds to either ETA receptors found in SMCs and cardiac myocytes or to ETB receptors located in SMCs and endothelial cells. ET-1 binding to ETA on SMCs activates phospholipase C, which increases intracellular Ca2+ concentration through increased inositol triphosphate, leading to vasoconstriction (478). However, activation of endothelial ETB receptors leads to the release of NO and PGI2, increased pulmonary clearance of ET-1 and exerts a mild vasodilator effect (121, 248). ET-1 increases ECM proteins and fibronectin production. ET-1 was shown to enhance the effects of transforming growth factor-beta and platelet-derived growth factor, leading to fibrosis, vascular hypertrophy, and smooth muscle cell proliferation (364). Experimental animal models of hypoxic PH have shown increased ET-1 as well as both ETA and ETB receptors, and newborn models have shown that ETA blockade partly reverses the effects of hypoxic pulmonary vasoconstriction (19, 348, 553). ETA antagonism in a monocrotaline model of PH decreased the RVH and pulmonary vascular thickening significantly, whereas ETB antagonism worsened both endpoints, thereby suggesting the divergent roles of ETA and ETB in PH (442).
Serotonin (5-HT)
5-HT is a potent pulmonary vasoconstrictor and angiogenic agent synthesized from the amino acid l-tryptophan by tryptophan hydroxylase (TPH) and metabolized by monoamine oxidase (MAO). In patients with PAH, TPH expression in PAEC is increased and 5-HT acts in a paracrine fashion on the PASMCs to induce proliferation and contraction and inhibition of voltage-gated K+ channels causing increased vascular tone (162, 339). Serotonin has also been implicated in the activation of mitogen-activated protein kinases through superoxide production and increasing the susceptibility of BMPR2-deficient mice to developing hypoxia-induced PH (344, 361). PAECs and PASMCs isolated from PPHN lambs show increased levels of 5-HT, which contributes to increased PVR through activation of the 5-HT2A receptor, and selective serotonin reuptake inhibitor (SSRI) infusion also increases PVR in the lamb PPHN model (144, 145). 5HT2A and serotonin transporter expression is also increased in the nitrofen CDH model for pulmonary hypoplasia and PH (255). A recent meta-analysis also found that prenatal exposure to SSRIs or serotonin norepinephrine reuptake inhibitors significantly increased the risk of PPHN after birth (OR 1.82, 95% CI = 1.31–2.54) (378).
Reactive oxygen species (ROS)
Multiple studies have shown that increased oxidative stress is a key contributory factor in the pathogenesis of PH as shown in Figure 5 (129, 148, 181, 267, 620). Oxidant stress can disrupt eNOS function by impairing the eNOS chaperone, heat shock protein 90 (Hsp90), depleting BH4, or its many other cofactors (625). In addition, reactive oxygen species (ROS) causes PASMC proliferation, which is attenuated by antioxidants (619, 621). In energy metabolism, oxygen acts as an electron acceptor in the mitochondrial respiratory chain and gets reduced to water eventually. Electron leak in the respiratory complex chain can lead to formation of several ROS such as superoxide anion (O2•−) and hydrogen peroxide (H2O2). Exposure to hyperoxia, uncoupling of eNOS, increased activity of NADPH oxidase, and mitochondrial dysfunction contribute to increased concentrations of ROS (365). Superoxide can combine avidly with endogenous NO to form peroxynitrite (OONO•−), which is a potent vasoconstrictor and also reduces endogenous NO activity by nitration of Hsp90 to decrease its association with eNOS (230). eNOS uncoupling, which can happen due to increased OONO•− levels, itself promotes mitochondrial dysfunction and leads to increased levels of OONO•−, thus causing a feed-forward pathway (573). Superoxide is converted by superoxide dismutase (SOD) into H2O2 under normal conditions, which is further degraded by scavengers such as catalase and glutathione peroxidase. Both superoxide and H2O2 also stimulate PDE5, which degrades cGMP, thereby potentiating vasoconstriction (173, 175, 424). H2O2 also produces hydroxyl free radicals in the presence of iron through the Fenton reaction, which can cause cell damage. PPHN lambs show increased levels of superoxide and H2O2 and NADPH oxidase activity along with decreased sGC activity and impaired angiogenesis (91, 587, 623, 626). PPHN lambs also show decreased levels of mitochondrial DNA copy number and electron transport chain complexes, which thereby lead to accumulation of ROS. This decrease was shown to be dose dependent in relation to oxygen exposure after birth and partly improved by exposure to NO and by reduction in oxygen concentration (11). Other animal models of PH such as the mouse hyperoxia, piglet hypoxia, and the monocrotaline model have all shown increased levels of NADPH oxidase (74, 148, 181, 608). ROS are removed by scavengers including superoxide dismutase (SOD), catalase, and glutathione peroxidase. Overexpression of extracellular SOD ameliorates PH in rats, protects lung development, and attenuates pulmonary vascular remodeling in hypoxic mice (14, 286, 443, 625). Catalase breaks down H2O2, and although mice deficient in catalase develop normally, intratracheal administration of catalase to ventilated PPHN lambs improves oxygenation, increases extracellular SOD activity, decreases superoxide levels, decreases PDE5 activity, and increases cGMP levels in the pulmonary arteries (176, 623).
Figure 5.
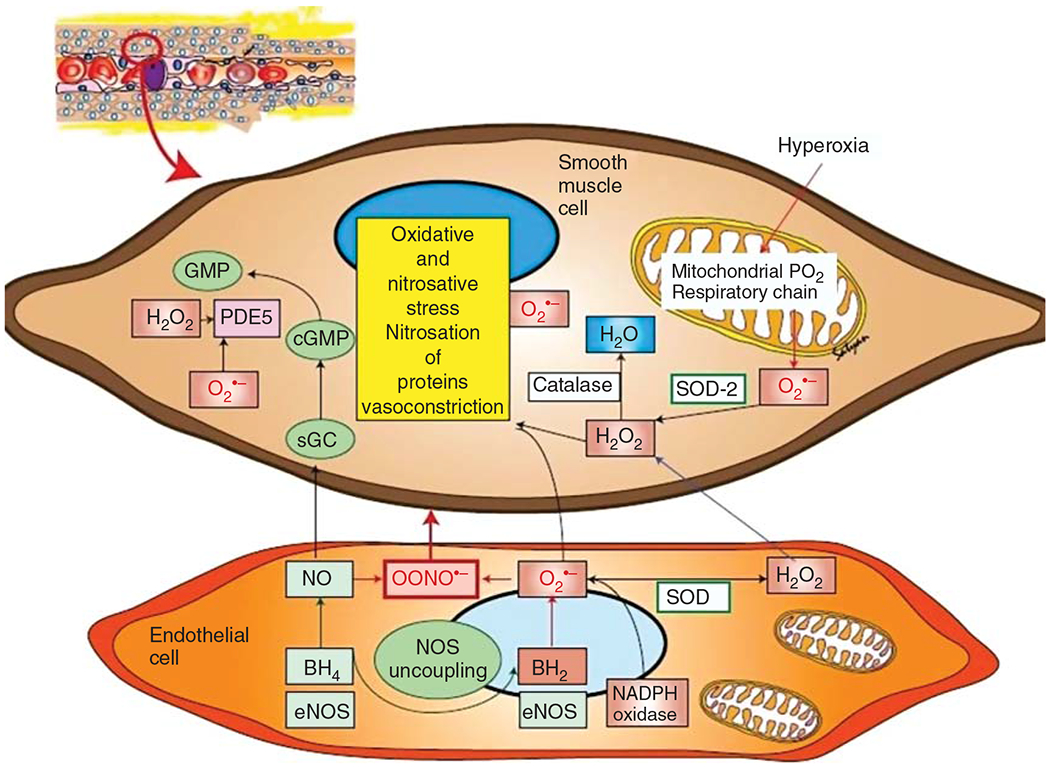
Role of reactive oxygen species in pulmonary hypertension. O2•−, superoxide anion; H2O2, hydrogen peroxide; PDE5, phosphodiesterase-5; GMP, guanosine monophosphate; cGMP, cyclic GMP; GTP, guanosine triphosphate; H2O, water; NO, nitric oxide; OONO•−, peroxynitrite; SOD, superoxide dismutase; BH4, tetrahydrobiopterin; BH2, dihydrobiopterin; NOS, nitric oxide synthase; eNOS, endothelial NOS; NADPH, nicotinamide adenine dinucleotide phosphate dehydrogenase. Reused, with permission, from Apitz C, et al., 2016 (25); Reused, with permission, from Dennis KE, et al., 2009 (148); Reused, with permission, from Fike CD, et al., 2008 (181); Reused, with permission, from Irodova NL, et al., 2002 (267); Reused, with permission, from Wedgwood S and Black SM, 2003 (620).
Potassium (K) channels
Reduced K+ channel expression and activity contribute to depolarization of SMCs in hypoxic PH, and increasing K+ channel expression in PASMCs attenuates changes of PH (96, 647). Oxidative stress has been found to impair the vasodilatory voltage-gated Kv channels in PPHN models, which can be partly restored by superoxide scavengers (313). Depolarization is believed to mediate the increased Ca2+ influx via voltage-gated Ca2+ channels (VGCCs) (537). Administration of dichloroacetate, which inhibits glycolysis, increases the expression of K+ channels and attenuates pulmonary vascular remodeling in both hypoxic and MCT models of rat PH (383, 389).
Calcium (Ca) channels
Increased cytosolic Ca2+ is a major trigger for pulmonary vasoconstriction and PASMC proliferation and migration, leading to remodeling. Increased resting cytosolic levels of Ca2+ as well as increased Ca2+ influx have been noted in PASMCs isolated from PH models. Both VGCCs and TRPC ion channels have been implicated in the Ca2+-mediated pulmonary vascular remodeling and PH pathogenesis. Voltage-gated channels, which are of L and T types, open in response to membrane depolarization and result in Ca2+ influx. Hypoxic mice that developed PH have increased expression of both L-type and T-type Ca2+ channels on vascular SMCs (614). Chronic hypoxia has been shown to upregulate L-type Ca2+ channels in small pulmonary arteries of the neonatal models of PH, and the calcium channel blocker, nifedipine decreased pulmonary pressures in the same model by inhibiting voltage-gated Ca2+ influx (249). Similarly, Rodman et al found an abundance of T-type Ca2+ channels in the medial layer of pulmonary arteries, and siRNA-induced inhibition of these channels decreased PASMC proliferation in vitro (501). TRPCs, which are Ca2+-permeable nonselective cation channels, have been implicated in IPAH and are increasingly recognized as the primary contributors for a sustained increase in cytosolic Ca2+. These, unlike VGCCs, are modulated by phosphorylation, receptor activation, or store depletion. PASMCs from patients with IPAH have increased expression of TRPC3 and TRPC6, and similar increased expression has been found in hypoxic PH models (357, 617). Decreasing the activity of TRPC6 either pharmacologically or by RNA silencing decreased the expression of the TRPCs as well as decreased vascular tone in the pulmonary arteries (321, 357). Data about Ca2+ and PAECs are still conflicting as in vivo models have failed to show elevated intracellular Ca2+ levels in PAEC isolated from hypoxic rats. However, cultured PAECs from these rats show increased Ca2+ levels and increased expression of TRPC4 (170, 459).
Vascular endothelial growth factor (VEGF)
Several different vascular endothelial growth factor (VEGF) splice variants have been identified, of which VEGFA is the most prominent and known for its functions of vascular permeability, angiogenesis, and vascular cell survival (307). VEGFA binds to two different receptor tyrosine kinases (RTKs), VEGFR1 or fms-related tyrosine kinase-1 (Flt1) and VEGFR2 or fetal liver kinase-1 (Flk1). VEGFR1 acts as a negative regulator of VEGF by preventing activation of VEGFR2, which is the functional receptor mediating the mitogenic, proangiogenic, and permeability-enhancing actions of VEGF (307, 463). Most animal studies, including both hypoxic and monocrotaline PH models, have shown increased VEGFA, VEGFR1, and VEGFR2 levels, which have been linked to increased endothelial cell proliferation in PH (118, 119, 599). In contrast, fetal lamb models of in utero generated PH have shown decreased VEGFA levels in endothelial cells, and VEGFA administration improved angiogenesis in vitro (208, 586). These studies highlight the developmental origin of fetal and neonatal PH. The causal relationship of VEGFA in pulmonary arterial remodeling needs further study. However, administration of VEGFA ameliorates the changes of PH in hypoxic animal models, which suggests that the relationship between VEGF, VEGFRs, and PH is complex and context dependent (172, 464). VEGFR inhibition causes apoptotic and emphysematous changes in rat lungs, but when exposed to chronic hypoxia, these rats develop pulmonary vascular angio-proliferative changes leading to severe PH (290, 341, 583). Cord blood levels of VEGFA are decreased in babies with maternal placental hypoperfusion and coexisting BPD-PH, thereby indicating that disrupted angiogenesis starts in utero and contributes to BPD-PH pathogenesis (387). Autopsy specimens have shown increased VEGF and VEGFR1 levels in both BPD-PH and PPHN, likely as a compensatory effect of disrupted endothelial function (332).
Other growth factors
Apart from VEGF, several other growth factors contribute to the pathogenesis of PH. Adult patients with PAH have shown increased expression of basic fibroblast growth factor (bFGF) in plasma and urine (66). Animal models of pediatric PH have shown elevated levels of FGF2 in PASMCs and PAECs (622). FGF-2 has also been shown to be an inducer of VEGF expression in vitro via its primary receptor FGFR-1, both of which are upregulated in PH (533). FGFR-1 knockout mice, when exposed to hypoxia, developed significantly less right ventricular remodeling and had lower RV systolic pressures, and pharmacological inhibition of FGFR-1 using SU5402 nearly reversed a rat model of PH (275). Current knowledge indicates that FGF-2 modulates pulmonary vascular remodeling and PH through the FGFR-1. Hepatocyte growth factor (HGF) levels have been shown to be decreased in hypoxic conditions and in the monocrotaline model (242, 453). HGF gene transfer has also been shown to ameliorate changes of MCT-PH (452).
Platelet-derived growth factor (PDGF) is a mitogen that contributes to vascular remodeling through smooth muscle hyperplasia in chronic PH. PDGF mostly consists of two polypeptide chains, A and B, although later studies have found that C and D chains exist as well. These chains can form dimer isoforms (AA, BB, CC, DD, and AB) and are structurally and functionally analogous to other growth factors like VEGF. They act on two primary tyrosine kinase receptors, PDGF receptor α (PDGFR-α) and PDGF receptor β (PDGFR-β) (24, 187). PDGF-A binds to PDGFR-α, whereas PDGF-B can bind to both receptors. PDGF-A and C are expressed in epithelial cells, whereas PDGF-B is expressed in megakaryocytes and endothelial cells, and PDGF-C and D are expressed in fibroblasts and in vascular SMCs, respectively (22, 24). PDGFR-α is expressed on mesenchymal precursor cells in the lungs, whereas PDGFR-β is expressed in SMCs. Hypoxia is the most common trigger of the PDGF/PDGFR-β pathway leading to a switch in the phenotype of SMCs from the contractile to proliferative phenotype (24, 585, 655). Studies in the ductal ligation lamb model of neonatal PPHN have shown that selective inhibition of PDGF-B decreases RV hypertrophy and pulmonary arterial thickening and increases PDGFR-α and β expression (41). Monocrotaline-induced PH models have shown increased levels of PDGF-B early in the disease process, which decreased to below control levels as the disease progressed (27). Lung specimens from patients undergoing transplants for IPAH have shown increased levels of both PDGF-A and B, as well as PDGFR-α and β in the PASMCs (468). More recently, microRNA-30c, which inhibits PDGFR-β translation, has been implicated in hypoxic PH. Hypoxia leads to decreased levels of microRNA-30c, which causes PDGFR-β overexpression, leading to a switch from the contractile to synthetic type SMCs (637). Both hypoxic and monocrotaline models of PH showed increased expression of PDGF-B and PDGFR-β; inhibition of PDGFR-β has reversed PH in these models (526).
Transforming growth factor (TGF)-β superfamily includes several cytokine growth factors, which play a critical role in regulation of cell growth and differentiation. Bone morphogenetic protein receptor type 2 (BMPR2), ALK1, endoglin, and caveolin-1 are membrane-bound receptors of the TGF-β superfamily, which have been implicated in pediatric PH (188, 238, 648). Caveolin-1 has a cell-specific role in PAH with loss of Cav1 from PAEC and high Cav1 levels in SMC being associated with vascular remodeling, including higher fibroblast proliferation, aberrant Ca2+, and high levels of oxidative stress (377). ALK1 mutation has been associated with younger age at diagnosis and death compared to patients with no mutations and a female predominance, with a female-to-male ratio of 3.5 (211).
Hypoxia inducible factors (HIFs)
HIF-1 and 2 are important transcriptional regulators of the physiological response to hypoxia. HIF-1 has two subunits, α and β, and is a highly conserved transcription factor that regulates the oxygen-dependent expression of hundreds of genes. The β subunit is constitutively expressed, whereas the α subunit only accumulates under hypoxic conditions due to decreased hydroxylation and stabilization from decreased proteasomal degradation (479). HIF-1α then dimerizes with HIF-1β, translocating to the nucleus and activating several genes. Mice heterozygous for HIF1-α null allele when exposed to chronic hypoxia showed significantly less RVH, less RV pressures, and decreased medial thickness compared to wild-type mice (646). At the same time, the heterozygous knockout mice showed an attenuated increase in TRPC expression, cytosolic Ca2+, and Na+/H+ exchanger-isoform 1 and did not show reduced expression of plasma membrane K+ channels in response to chronic hypoxia, demonstrating a protective effect from PH. The HIF-1α downstream targets, which have been implicated in PH, are ET-1, VEGF, and HEK-2. Hypoxia upregulates ET-1 as well as HIF-1α expression in the lungs, and both ET-1 and HIF-1α upregulate each other’s expression, thereby creating a feed-forward loop (350). This effect of ET-1 on HIF-1 expression is only seen in PASMCs and not in aortic SMCs. The fetal ductal ligation lamb model where decreased angiogenesis contributes to the development of PPHN has shown increased HIF-1 expression in PASMCs, and inhibiting HIF-1 expression increased VEGF expression and improved angiogenesis in the PPHN lambs (373, 624). HIF-2 has been shown to activate EPO gene expression, which increases erythropoietin production. Endothelial-specific HIF-2 knockout abolishes PH and right ventricular responses to chronic hypoxia (260).
Rho proteins
RhoA is a member of the Rho family of small GTPases and regulates a variety of cellular responses such as cell contraction, migration, growth, gene expression, and differentiation (164). Activation of RhoA occurs via stimulation of G-protein-coupled receptors by receptor and non-receptor tyrosine kinases; inactivation can occur via protein kinase G, which is activated by the NO-sGC-cGMP pathway. There is also evidence that hypoxia leads to RhoA activation in PAECs and PASMCs (581). RhoA activates its downstream target Rho-kinase (ROCK), which has been widely implicated in PH. In models of neonatal and adult PH, high ROCK levels cause elevated vascular tone, increased myogenic reactivity, and pulmonary vascular remodeling (209, 382, 429, 447). ROCK primarily phosphorylates the myosin-binding subunit of myosin light chain phosphatase (MYPT-1), and thereby increases phosphorylation of myosin light chain and enhances the contraction at any given level of activity of myosin light chain kinase (MLCK) and cytosolic Ca2+ (168, 554). In addition to vascular smooth muscle cell contraction, ROCK affects endogenous NO action by reducing eNOS mRNA stability (335). ET-1, which is a potent vasoconstrictor, has been shown to activate ROCK to cause impaired angiogenesis in fetal lamb PPHN PAECs in vitro (209). ROCK inhibitors, Y-27632 and fasudil, have been shown to inhibit pulmonary artery myogenic responses in hypoxia-exposed adult rats and fetal sheep and to reverse sustained vasoconstriction in response to chronic hypoxia or ET-1 infusion (94, 168, 384, 594, 628). ROCK inhibitors, when systemically administered at the onset of injury in the chronic hypoxia or monocrotaline PH model, prevent changes of PH (1, 168).
Bone morphogenetic protein receptor type 2 (BMPR2)
Mutations of the BMPR2 gene, present on 2q33, have been identified in IPAH and HPAH, and children with BMPR2 mutations are less likely to respond to acute vasodilator testing (13% vs 44%) and are more likely to have severe disease at diagnosis (32, 147, 508). More than 140 distinct BMPR2 mutations have been found and together they are present in 10% to 40% of families with PAH (631). A French cohort found 5 different BMPR2 mutations in children with IPAH/HPAH, along with other mutations in ACVRL1 and TBX4 (347). Presence of a BMPR2 mutation does not guarantee development of the clinical features of PAH, which suggests that there is decreased penetrance. The mechanisms and factors that lead to PAH in some individuals with BMPR2 mutations remain unclear; Figure 6 shows the pathways disrupted in BMPR2 mutations that might lead to PAH. The mechanistic role of BMPR2 mutations in the pathophysiology of PAH is still not clear with most studies indicating upregulation of mitogen-activated protein kinase or reduced activation of transcription factor Smad 1 (515, 642). Several in vitro and in vivo studies have shown that BMPR2 mutations decrease endothelial cell viability and lead to PASMC proliferation, key features of PAH. Several biochemical alterations have been described in cells with decreased BMPR2 function, including decreased mitochondrial function, increased glycolysis, and excess proliferation of PASMCs. Downregulation of BMPR2 has been reported to contribute to pathophysiology of PAH in patients without specific mutations in this gene. BMPR2 has emerged as a major signaling pathway that is altered in a variety of PAH cases, and therapies to promote this signaling are actively being studied as the next frontier in PAH-specific therapies (485, 634).
Figure 6.
A schematic representation of the BMPR2 signaling pathway. ActR, activin Receptor; Akt, protein kinase b; ALK, activin-like receptor; BMP, bone morphogenetic protein; BMPR, bone morphogenetic protein receptor; BRE, BMP response element; c-Src, proto-oncogene tyrosine-protein kinase Src; CLIC4, chloride intracellular channel 4; Dvl, disheveled; Erk, extracellular signal-regulated kinase; FKBP1A, FK binding protein 1A; GSK3-β, glycogen synthase kinase 3-β; ID, inhibitor of differentiation; JNK, c-Jun N-terminal kinase; LIMK, Lin11, Isl-1, and Mec-3 domain kinase; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; PPARγ, peroxisome proliferator-activated receptor gamma; Rac1, Ras-related C3 botulinum toxin substrate 1; RAGE, receptor for advanced glycation end products; RhoA, Ras homolog gene family, member A; SMAD, mothers against decapentaplegic; SMURF, SMAD-specific E3 ubiquitin protein ligase; Tak1, transforming growth factor-β activated kinase 1; VEGFR3, vascular endothelial growth factor receptor 3. Modified, with permission, from Andruska, A et al., 2018 (23). Licensed under CC-BY-4.0.
Other genes that have been implicated in PAH include ALK1, endoglin, CAV1, and KCNK3, which are associated with autosomal dominant diseases, and EIF2AK4, which is associated with the autosomal recessive form of pulmonary veno-occlusive disease (35).
Notch pathway
Notch pathway is a highly conserved canonical pathway important for the determination of cell fate during embryonic development. It consists of the four mammalian Notch receptors—Notch 1 to 4—and five ligands—Delta-like (Dll) 1, 3, 4 and Jagged (Jag) 1 and 2 (392). The interaction of the ligand with the membrane-bound Notch receptor leads to its proteolytic cleavage to release the Notch intracellular domain (NICD), which translocates to the nucleus to activate the C-promoter binding factor 1 (CBF-1). CBF-1 binds to the NICD to form an active transcription factor complex to target the downstream genes belonging to HEY/HES family (318, 638). Notch 1, 3, and 4 and Dll-4, Jag-1 and Jag-2 are present in the human arterial system and are critical to maintaining normal vascular structure, angiogenesis, and vascular remodeling (224, 256, 369). Both hypoxic and MCT models of PH have shown increased expression of Notch 3 and the Notch 3 intracellular domain, and Notch knockout mice do not develop hypoxic PH (352). Chronic hypoxia increases the expression of store-operated Ca2+ channels (SOCE) and activates Notch signaling, and blockade of the TRPC6, a key canonical SOCE, inhibits acute hypoxic pulmonary vasoconstriction and development of PH in chronic hypoxia (552). Notch 3 and its target HES-5 are expressed highly in PASMCs of patients with PAH, and knockdown of HES-5 attenuates the vascular proliferative effects produced by increased expression of Notch 3 in vitro (352). Notch 3 inhibition by itself in hypoxic neonatal rat pups also prevents the changes of chronic PH and decreases PDGFR-β content in the PASMCs (269).
Peroxisome proliferator-activated receptor (PPAR)
PPAR is a member of the nuclear receptor hormone superfamily and is widely expressed in PAECs and PASMCs, where it regulates vascular SMC proliferation (578, 653). Animal models of PPHN have shown decreased levels of PPARγ with increased levels of SOCEs like TRPC1 and TRPC6 (159). PPARγ seems to have a protective effect against PH, as evidenced by the development of spontaneous PH in mice with selective deletion of PPARγ in PASMCs (234). PPAR-γ deletion in vitro also induces PASMC proliferation in cultured human cells, whereas overexpression reduces the same (80). In PPARγ-deficient pulmonary microvascular endothelial cells, migration and angiogenic ability were significantly attenuated via E2F1-mediated gene regulation (607). MCT rat models of PH treated with PPARγ agonists, pioglitazone or troglitazone, were protected against pulmonary vascular remodeling (379, 636). In rats exposed to hypoxia for 3 weeks, rosiglitazone (a PPARγ agonist) attenuated hypoxia-induced RVH, vascular SMC proliferation, pulmonary vascular collagen and elastin deposition, and matrix metalloproteinase activity. Rosiglitazone, however, failed to attenuate hypoxia-induced increases in PAP, which was attributed to the inability of PPARγ ligands to modulate ROCK signaling, a critical mediator of pulmonary vasoconstriction (131, 223). PPARγ ligands decrease hypoxic Nox4 expression, oxidative stress, and PDGF signaling in the lung (441). PPARγ activation also decreases the levels of ET-1 and asymmetric dimethylarginine (an endogenous NO synthase inhibitor), both of which are involved in the pathogenesis of PH (289, 500).
MicroRNA
MicroRNAs (miRs) are short approximately 22 base pair long nucleotide sequences that are conserved across species and produced from transcription of noncoding DNA. miRs can bind to the 3’ untranslated region of mRNA and repress the translational mechanism and hence can modulate several disease processes. Each miR can bind to multiple mRNAs based on the degree of the complementarity between their sequences, and each mRNA has binding sites for multiple miRs. This leads to a complex interplay; hence several miRs have been implicated in PH disease processes. miR-21 is downregulated in the MCT model of rat PH and in human lung tissue and serum of patients with PAH (104). However, hypoxic PH models have shown contrasting results; miR-21 levels were elevated in distal small arteries and downregulating miR-21 expression both in vivo and in vitro decreased PASMC proliferation and pulmonary vascular remodeling (641). miR-21 has several targets, including BMPR2 and dimethylarginine dimethyl-aminohydrolase 1 (DDAH-1), which decreased with increasing levels of miR-21 (264, 484, 641). DDAH-1 is responsible for the metabolism of asymmetric dimethylarginine, which is an endogenous NOS inhibitor. PPAR-γ, which attenuates the effects of hypoxic PH, also decreases miR-21 expression in the presence of hypoxia (222). These data indicate that miR-21 is a likely contributor to pulmonary vascular remodeling due to hypoxia and is therefore a potential therapeutic target for PH. Other miRs like the miR 17 to 92 cluster, miR-145 and miR-210 are also upregulated in experimental models and contribute to PASMC migration and proliferation and PAEC proliferation and resistance to apoptosis. miR-124 has been found to be downregulated in PH and contributes to fibroblast migration, proliferation, and activation in addition to effects on PASMCs (657).
Animal Models of PH
There are multiple preclinical animal models of PH, based on the different pathological states resulting in the final syndrome of elevated PAPs. Understanding the animal models is essential to understand the different molecular mechanisms and treatment strategies targeting PH.
Rat and mouse models have been used to study PH extensively, the most common models being the chronic hypoxia (CH) and the monocrotaline (MCT) models. These models have different strengths and limitations. In general, rat models show more robust vascular remodeling and RVH compared to mouse models and are more widely used for the investigation of PAH pathogenesis. In contrast, mouse models offer a wide array of genetic knockouts and transgenic overexpression to investigate the role of specific signaling pathways in vivo. However, mouse models of hypoxia or monocrotaline fail to develop the full spectrum of vascular remodeling observed in human PH. One of the most commonly used rat models for PAH involves the injection of monocrotaline. Monocrotaline, which is a plant-based alkaloid, is activated in vivo by cytochrome P-450 enzymes to form monocrotaline pyrrole (MCTP), which causes endothelial injury (388). This leads to a cascade of pulmonary vasculitis, PAEC apoptosis, PASMC proliferation, and pulmonary vascular remodeling, leading to obstructive vasculopathy in the pulmonary vasculature. The TGFβ-Smad-BMPR2 signaling pathway is one of the key pathways implicated in the MCT model, along with inflammation and cytokine release playing contributory roles (366). Downregulation of BMPR signaling has been reported in this model in several studies. The MCT model has been criticized as experimental treatments targeting different pathways have shown striking improvements with almost a complete reversal of the PH changes. The rapid response to therapeutic agents with a reversal of PAH changes, which are not found in PAH patients, raised concerns that this model is not reflective of human disease and that the endpoints do not correlate with progression of PH in patients. Chronic hypoxia (CH) model for PH is closely representative of Group 3 PH; however, it has been used for neonatal PPHN studies as well (293). Once altitude and low oxygen tension were found to be causally involved in heart failure in cattle living at >8000 feet in Colorado, several animal CH models have emerged. Animals in these studies are placed in hypobaric or normobaric hypoxia, which induce chronic oxygen deprivation, hypoxic pulmonary vasoconstriction, and pulmonary vascular remodeling. The effects of hypoxia are varied across different species, but most models show extensive vascular remodeling. Rats exhibit consistent increases in mean PAPs, right ventricular mass as well as PASMC hypertrophy and hyperplasia and distal muscularization (486, 568). Mouse CH models, although useful for widely available genetic knockouts, have failed to demonstrate similar changes in PASMC. Mice consistently show less vascular remodeling compared to hypoxic rats or humans, which have less remodeling compared to neonatal calves (60, 458, 568). This might be attributed to differing genetic responses between the two species; CH rats show increased expression of genes involved in EC proliferation and decreased expression of pro-apoptotic genes, whereas CH mice demonstrate decreased expression of genes involved in vascular SMC proliferation (95). Hypoxia-induced inflammatory response also plays a role in the development of PH as evidenced by the presence of early and persistent inflammatory infiltrates (primarily mononuclear cells) along with adhesion molecules and cytokines within the vessel wall (97, 568). One of the prominent issues with the rat CH model is that although pulmonary pressures and RV size are increased, RV failure does not usually occur. Of special note are fawn-hooded rats, which have an inherited deficiency in platelet serotonin uptake, and they develop PH ~4 weeks of life, and this is accentuated by exposure to hypoxia. They also demonstrate decreased alveolarization, lung hypoplasia, and immaturity, which make them suitable for studying BPD-PH (340). Hypoxic vasoconstriction is attenuated in newborns, likely due to enhanced PGI2 release; hence the CH model is not ideal for studying PPHN or neonatal PH.
The combination of Sugen 5416 (SU5416), a VEGF receptor inhibitor and hypoxia, leads to the development of angio-obliterative lesions in rat lungs causing profound PH, more so than hypoxia alone. In mice, it has similar effects with the evolution of a severe PH phenotype compared to hypoxia alone, but it does not produce the same effects of angio-obliterative lesions (610). There have been some critiques of the model related to its almost irreversible and unresponsive PH phenotype, but proponents of the model argue that it more closely resembles the severe type of human PH.
Unique for neonatal and pediatric populations is PH associated with BPD. The alveolar and vascular simplification observed in these infants has been linked to and reproduced by neonatal exposure to hyperoxia. Mice exposed to hyperoxia from postnatal day 1 to 4 showed decreased lung elastance, RVH, reduced distal microvasculature, and reduced expression of BMP receptors and downstream phospho-Smad-1/5/8 (644). Hyperoxia exposure has been shown to increase PDE5 expression in PASMCs and decrease cGMP signaling leading to PH and RVH, which are partly reversed by PDE5 inhibitor, sildenafil (173, 244, 342). Short-term exposure to hyperoxia has been shown to induce mitochondrial matrix ROS, which activate PDE5 in a cGMP-dependent protein kinase-dependent manner in the PASMCs (175). Riociguat, which stimulates sGC, responsible for production of cGMP from GTP, has been shown to prevent lung injury, decrease RV systolic pressure, RVH, and distal pulmonary arteriolar muscularization in hyperoxic neonatal mice (154). Mice with mutations in BMPR2, which itself can lead to PH, when exposed to hyperoxia demonstrated a significantly worse PAH phenotype with increased RV systolic pressure, increased pulmonary vascular occlusion, and decreased cardiac output (179).
PPHN, a unique disorder in which newborns have elevated PAPs at birth, is most closely reproduced by the fetal lamb ductal ligation model (314). The ductus arteriosus, which diverts the blood away from the lung in utero, is mechanically constricted in the fetus to elevate the PVR. Although the pulmonary blood flow increases as well initially, within 2 h it comes back to the baseline, whereas the PVR remains elevated. Fetuses usually survive to delivery and the newborn demonstrates clinical signs of PPHN with elevated PAPs and hypoxemia and morphologic changes of pulmonary vascular remodeling, fibrosis and muscularization of nonmuscular arteries (565). PPHN lambs have been shown to express decreased eNOS gene expression with increased production of ROS from uncoupled eNOS, which contributes to the PPHN phenotype (314, 535). Administration of glucocorticoids and ROS scavengers such as superoxide dismutase attenuate this adaptive response to ROS and restores eNOS function (108, 174). The ductal ligation model shows a clear dose-response relationship of pulmonary vasodilation to NO and was widely used for the preclinical studies, which led to the FDA approval of iNO in PPHN. There have been attempts to create other models of in utero PPHN, either by maternal hypobaric hypoxia or by chronic repeated placental embolization. The first model produced growth retardation but not structural or functional changes in the pulmonary vasculature, and the second was associated with high fetal mortality (565).
Models for CDH: PH is a common feature of CDH and is due to prenatal pulmonary vascular hypoplasia and vascular remodeling. The pulmonary hypoplasia is due to the presence of herniated abdominal contents in the thoracic cavity, limiting lung growth in utero. The models that have been used so far to study CDH are the surgical models to create diaphragmatic defects in rabbit and sheep, the nitrofen exposure model in rats and mice, and the knockout model in mice. The surgical model is based on a surgical intervention creating a diaphragmatic defect in the fetus, which leads to herniation of intra-abdominal contents into the thorax, leading to impaired lung growth (446). Surgical CDH created in late gestation rabbit fetuses showed an increased medial thickness and a decreased internal diameter of the pulmonary arteries. Creation of in utero fetal tracheal occlusion reverses these changes to a certain extent (510, 511). The sheep surgical model has also shown a decreased number of highly proliferative PAECs, and this was hypothesized to be a contributor to the development of pulmonary hypoplasia and impaired angiogenesis, leading to postnatal PH (9). Although this model is valuable for studying in utero interventions for fetal repair of CDH like tracheal occlusion or in utero repair of defects in the diaphragm, the surgical defect is created relatively late in gestation. Creation of this defect late in gestation may miss certain stages of lung development, which are more affected in the human disease (603). This is also a single-hit lung hypoplasia model, whereas CDH is presumed to be a dual-hit model (292). One of the widely studied models is the nitrofen CDH model. Nitrofen when given by gastric lavage to pregnant rats just before fetal lung and diaphragm development begins induces CDH in 70% of the fetuses and pulmonary hypoplasia in 100% (603). Nitrofen model has led to a better understanding of the two-hit hypothesis of CDH, where nitrofen leads to early bilateral lung hypoplasia before the diaphragm is supposed to close (1st hit) and ipsilateral lung hypoplasia and PH from later herniation of bowel and abdominal viscera into the lung cavity from the diaphragmatic defect (2nd hit) (406, 603). The primary mechanism is believed to be via the retinoid signaling pathway as nitrofen is a retinal dehydrogenase 2 inhibitor, and serum levels of both retinol and retinol binding protein were 50% lower in infants with CDH (229). Various other pathways including VEGF, BMP, and Wnt pathways are also implicated in the dysregulated pulmonary endothelium, PAEC dysfunction, and impaired cross talk between PAECs and PASMCs in nitrofen-CDH. Nitrofen-CDH rats also have increased proliferation and resistance to apoptosis in PASMCs as well as increased secretion of ECM proteins and medial wall thickness of the pulmonary vasculature (404). The pulmonary arterioles show blunted oxygen-induced vasodilatation, which further contributes to decreased vasoreactivity and postnatal PH (125). Several genetic models including knockout models of genes like Wilms Tumor 1 (Wt1), Sonic Hedgehog (Shh), Slit 3, Fog 2, COUP-TFII, Gata 4, and Gata 6 have also been proposed, but a well-defined knockout model is still lacking. Knockouts of retinoic acid receptor genes have resulted in CDH, which is aligned with the altered vitamin A and retinoid signaling pathway hypothesis of CDH (117).
CHD and shunt lesions leading to PHVD is an unique entity that has been studied in lamb and piglet models. The lamb model is created by insertion of a polytetrafluoroethylene graft between the ascending aorta and the pulmonary artery in late gestation, which leads to the elevated pulmonary-systemic blood flow ratio and mPAP, although the PVR is not significantly increased. The model closely simulates infants with left-to-right shunts with relatively normal Qp/Qs at birth but a progressive increase in Qp during first six weeks of life as the PVR declines postnatally. These lambs develop increased pulmonary vasoconstrictor response to hypoxia and thromboxane A2 (497). One piglet model of CHD-PAH involves shunts between the left pulmonary artery and the subclavian artery, leading to flow-induced PAH and vascular remodeling. These piglets with shunts develop RV failure even with mild PAH, likely due to the decreased levels of VEGF and increased pro-inflammatory cytokines such as interleukin-1a and TNF-β seen in this model (502, 503). Pigs with chronic systemic-to-pulmonary shunting also have increased expression of the pulmonary ET-1 system, which is prevented by the ET receptor antagonists bosentan and sitaxsentan (502, 504).
Diagnostic Evaluation
Pediatric PH has multiple etiologies and a wide spectrum in presentation. Hence, a comprehensive, systematic approach to the diagnosis will aid in the correct classification and choice of treatment. Recent studies have shown that most children do not undergo the full evaluation necessary for an accurate diagnosis of PH (61, 270). Several diagnostic algorithms have been suggested and were modified over the years with more available evidence about the full spectrum of pediatric PH and with changing classifications. The most recent algorithm from the Pediatric Task Force of the 6th WSPH in 2018 is shown in Figure 7. Based on history, biomarkers, echocardiographic, cardiac catheterization, and MRI findings, children with PAH can be stratified into high-risk or low-risk categories (Table 7), which determines treatment arms. Table 8 discusses the recommendations for use of different diagnostic tools in the pediatric PHD.
Figure 7.
Diagnostic algorithm for a child or young adult with suspected pulmonary hypertension. PH, pulmonary hypertension; ECG, electrocardiogram; CHD, congenital heart disease; PFT, pulmonary function test; CT, computed tomography; DLCO, diffusing capacity for carbon monoxide; RV, right ventricle; CTEPH, chronic thromboembolic pulmonary hypertension; CTA, CT angiogram; PA, pulmonary artery; PEA, pulmonary endarterectomy; AVT, acute vasoreactivity testing; mPAP, mean pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PVRI, pulmonary vascular resistance index; WU, Wood units; 6MWT, 6-minute walk test; MRI, magnetic resonance imaging; CPET, cardiopulmonary exercise testing; CTD, connective tissue disease; HIV, human immunodeficiency virus; PVOD, pulmonary veno-occlusive disease; PCH, pulmonary capillary hemangiomatosis; IPAH, idiopathic pulmonary arterial hypertension; FPAH, familial pulmonary arterial hypertension. Adapted, with permission, from Rosenzweig EB, et al., 2019 (506). © 2019, The European Respiratory Society.
Table 7.
Determinants of Risk in Pediatric Pulmonary Hypertension
| Lower risk | Determinants of risk | Higher risk |
|---|---|---|
| No | Clinical evidence of RV failure | Yes |
| No | Progression of symptoms | Yes |
| No | Syncope | Yes |
| Normal height and BMI | Growth | Failure to thrive |
| I and II | WHO FC | III and IV |
| Minimally elevated for age or not elevated | Serum NT-proBNP | Greatly elevated for age > 1200 pg/mL (>1 year old) Rising NT-proBNP level |
| Minimal RA/RV enlargement | Echocardiography, CMRI | Severe RA/RV enlargement |
| No RV systolic dysfunction | RV systolic dysfunction | |
| RV/LV e.s. ratio < 1 | RV/LV e.s. ratio > 1.5 | |
| TAPSE normal (z > −2) | TAPSE abnormal (z < −3) | |
| S/D ratio < 1 (TR jet) | S/D ratio > 1.4 (TR jet) | |
| PAAT > 100 ms (> 1 year old) | PAAT < 70 ms (> 1 year old) Pericardial effusion |
|
| CI > 3.0L/min/m2 | Invasive hemodynamics | CI < 2.5 L/min/m2 |
| mRAP < 10 mmHg | mRAP > 15 mmHg | |
| mPAP/mSAP < 0.5 | mPAP/mSAP > 0.75 | |
| Acute vasoreactivity positive | PVRI > 15 Wu×m2 |
BMI, body mass index; BNP, brain natriuretic peptide; CI, cardiac index; CMR, cardiovascular magnetic resonance imaging; e.s., end-systolic; IPAH, idiopathic pulmonary arterial hypertension; mPAP, mean pulmonary artery pressure; mRAP, mean right atrial pressure; mSAP, mean systemic artery pressure; NT-proBNP, N-terminal proBNP; PAAT, pulmonary artery acceleration time by transthoracic Doppler echocardiography; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; PHVD, pulmonary hypertensive vascular disease; PVR, pulmonary vascular resistance; PVRi, pulmonary vascular resistance index; RA, right atrium; RV, right ventricle; S/D ratio, systolic/diastolic duration ratio by Doppler echocardiography; SVR, systemic vascular resistance; SVRi, systemic vascular resistance index; TAPSE, tricuspid annular plane systolic excursion; TR, tricuspid regurgitation; Vo2max, maximum rate of oxygen consumption; WHO, World Health Organization; WU, Wood units.
Adapted, with permission, from Rosenzweig EB, et al., 2019 (506). © 2019, The European Respiratory Society.
Table 8.
Recommendations for Different Diagnostic Strategies for Pediatric PH
| Diagnostic tool for PH | Recommendations for use at time of diagnosis of PH | Recommendations for use during follow-up |
|---|---|---|
| Chest X-ray | Recommended but not necessary to perform at baseline | No need at follow-up visits, unless there is clinical reason |
| NT-proBNP | Part of initial workup | Serial measurements important to follow up disease severity, progression and response to treatment |
| Transthoracic echocardiography | Necessary part; need to include mPAP, end-diastolic PAP, RV longitudinal systolic function, RV strain, RV size and function, RV base/apex ratio, RV systolic-to-diastolic duration ratio, tissue Doppler velocities, RVOT size enlargement, RA and RV size enlargement, RA function, end-systolic LV eccentricity index, end-systolic RV/LV diameter ratio and indicators of diastolic LV dysfunction | Should be performed every 3 to 6 months, sooner if there is clinical worsening |
| Cardiac catheterization | Indicated in all patients to confirm diagnosis, determine severity and anytime when PH-specific therapy considered; exceptions are PPHN and BPD-PH, weight <2 to 5 kg who might be unstable and in critically ill patients; need to include AVT, PVR/SVR ratio, PVRI, oximetry, pressure measurements of RA, RV, PA, LA, PA wedge and systemic artery, VO2 | Repeated at clinical discretion based on clinical worsening, failure to reach treatment goals with multidrug therapy, listing for heart or heart-lung transplant, and every 12 to 24 months in stable patients after full non-invasive evaluation has been performed |
| CT angiography | High-resolution chest CT with angiography at initial evaluation; evaluate MPA/AO ratio, lung parenchyma, PA pruning and pulmonary veins | Not recommended for repeat unless presence of PVS or before considering transplant |
| Cardiac MRI | Gold standard for evaluating RV size, mass, and function. Ventricular EDV, RVEF, and stroke volume predict morbidity and mortality in adults | Based on clinical discretion |
| Genetic testing | Recommended for all children diagnosed with IPAH/HPAH in addition to counseling and for first-degree family members of patients with PAH and a known mutation | Not indicated |
| Polysomnography | Recommended in patients diagosed with PH at risk for sleep-disordered breathing including trisomy 21, patients with small upper airways, daytime sleepiness | Not indicated unless abnormal during initial evaluation |
PH, pulmonary hypertension; PAH, pulmonary arterial hypertension; IPAH, idiopathic PAH; HPAH, hereditary PAH; CT, computed tomography; MRI, magnetic resonance imaging; NT-proBNP, N-terminal pro-brain natriuretic peptide; RV, right ventricle; LV, left ventricle; RVOT, RV outflow tract; RA, right atrium; LA, left atrium; PA, pulmonary artery; mPAP, mean pulmonary arterial pressure; RVEF, RV ejection fraction; EDV, end diastolic volume; MPA, main PA; AO, aorta; BPD, bronchopulmonary dysplasia; PPHN, persistent pulmonary hypertension of the newborn; PVS, pulmonary vein stenosis.
Chest X-ray
PAH is clinically silent in early stages and not visible on X-ray imaging. Chest X-rays may show features of RA and RV enlargement in later stages. Dilation of central pulmonary arteries and diminished peripheral lung vasculature based on decreased distal pulmonary blood flow may also become apparent with disease progression. Increasing PVR is represented as worsening oligemia in lung fields, particularly in specific disorders, such as primary PPHN. Increasing pulmonary edema or signs of pulmonary venous congestion on X-ray should raise concern for other etiologies like PVS or left ventricular dysfunction leading to back pressure. In specific syndromes such as PPHN or BPD-PH, X-rays may be useful to track the parenchymal lung disease. PPHN due to meconium aspiration syndrome shows hyperexpanded lung fields mixed with scattered areas of atelectasis due to the obstructive nature of meconium in air passages (430). BPD will show signs of chronic lung disease such as increased lung texture and hypolucency (92, 351).
Electrocardiogram (ECG)
ECG changes in pediatric pulmonary hypertension include right atrial enlargement, right axis deviation (RAD), and RVH with secondary T-wave changes (507). A study of electrocardiogram-echocardiogram pairs for known cases of pediatric PH showed a 69% sensitivity and 67% positive predictive value of ECG in predicting PH when echocardiogram was used as the diagnostic gold standard (481). When ECG was compared to cardiac catheterization for known pediatric PH, the sensitivity, specificity, positive and negative predictive values of RAD and RVH on baseline ECG for disease progression were 92%, 48%, 33%, and 95%, respectively (334). ECG-based screening when applied to a school-based population of children in Japan helped in early recognition and treatment of pediatric PH patients, associated with severe PH and preserved right heart function (525).
Echocardiography
Conventional 2-D transthoracic echocardiography remains one of the most widely used tools to diagnose pediatric PH. The advantages of echo are its noninvasive nature, wide availability, safety and feasibility in the pediatric population. A meta-analysis in adult PH found a pooled sensitivity of 74% and a pooled specificity of 85% in diagnosing PH for echo (437). PH leads to poor right ventricular compliance and RV diastolic dysfunction, leading to progressive right atrial dilatation. Imaging of the RA in the apical four-chamber view to measure the RA major and minor axes and planimetry of the RA area in end-systole will determine the presence of RA dilatation (282). Progression of PH might also lead to inferior vena cava (IVC) dilatation, and estimating the IVC diameter and the presence of inspiratory collapse can help in indirect estimation of the RA pressures. The usual RV:LV area ratio is less than 0.6, and both acute and chronic increases in PA pressures affect this number. Acute PH leading to RV distension and chronic PH leading to ventricular remodeling to maintain cardiac output will increase the RV:LV area ratio assessed by echocardiography.
Estimation of pulmonary arterial pressure (PAP)
The peak tricuspid regurgitation (TR) gradient is the most commonly used estimator of RV systolic pressures, which reflects the systolic PA pressure (sPAP), provided there is no obstruction of blood flow between the RV and PA. The TR gradient is measured as the continuous wave (CW) Doppler velocity across the tricuspid valve in line with the regurgitation flow and is then converted into a pressure gradient using the modified Bernoulli equation [4×(TR velocity)] (276). This is the pressure gradient across the tricuspid valve and hence is the difference between systolic pressures of RA and RV. Adding the RA pressure (usually 5-10 mmHg in pediatric patients) to this number will give the RV systolic pressure, which gives us the estimate of the sPAP. This is not always practical as the TR jet is not present in all patients (418). In the cases of RV outflow tract obstruction, PA stenosis, or ventricular septal defects, the accuracy of estimation of sPAP based on TR jet diminishes (310). Using CW Doppler to measure pulmonary regurgitation (PR), the mean PAP and the diastolic PAP can also be estimated from the early-diastolic and end-diastolic PR velocities, respectively, and then applying the modified Bernoulli’s equation. The mPAP can also be calculated from the sPAP as mPAP = 0.61 × sPAP + 2 mmHg (308). Other measures of RV and PA systolic pressures are the measurement of pressure gradient in the presence of a VSD or a PDA and subtracting this from the systemic pressures at that time. PA acceleration time (PAAT), which is the interval in milliseconds from the onset of ejection to the peak flow velocity, is another measure of PVR and a PAAT < 120 ms in children is indicative of PH (107). PVR can also be estimated in children from the echocardiographic estimation by a simple ratio of peak TR velocity to the velocity time integral of the right ventricular outflow tract (VTI-RVOT), with a value of >38 providing a 100% specificity for a PVR of >8 WU (462, 611). Echocardiographic estimates of PVR are not always accurate, especially when the PVR is very high, and in such situations cardiac catheterization is the next preferred step.
Estimation of RV function
Assessing the RV longitudinal systolic function is an important part of the echocardiographic PH diagnosis, as this RV dysfunction places the patient at a higher risk of complications and mortality and thus potentially changing management strategies. The tricuspid annular plane systolic excursion (TAPSE) is the longitudinal excursion of the tricuspid annulus toward the apex as measured by M-mode, and varies based on gestational age and postnatal age, and there are studies establishing reference values for the same (311, 322). TAPSE is a surrogate for RV function and a decreased TAPSE implies diminished RV function, with a strong correlation (R = 0.86) to RV function estimated by cardiac MRI, considered to be the gold standard (322, 524). The TR jet/TAPSE has also been found to correlate positively and strongly with the NYHA FC, thus implying future use in PH diagnosis and prognostication (309). TAPSE, however, does not document radial systolic RV function, which contributes to the RV ejection in the setting of RV hypertrophy.
RV strain and strain rate are also important measures of RV function, which determine regional wall motion abnormalities, which can be abnormal in PH. Strain is the measure of the myocardial length change from the baseline and strain rate is the velocity of this change (324). The severity of PH in adult patients has a strong correlation with the decreased peak systolic strain, which decreases with increasing RV afterload, and serial measurements of RV systolic strain have been found to be useful in predicting long-term prognosis in adult patients (237, 353). This has not been well studied in pediatric PH, although one study found RV strain to be an early predictor of RV dysfunction in children with IPAH (448). Recently, the RV-arterial coupling ratio using stroke volume to end-systolic volume has been found to be an independent predictor of adverse clinical outcomes in pediatric PH (284). Both TAPSE and RV global longitudinal peak strains have been found to be associated with progression to death or extracorporeal membrane oxygenation (ECMO) in infants with PPHN (374).
Estimation of LV function
Identifying whether there is any left heart disease is important as this can change the management approach of PH completely. LV dysfunction leads to increased LV filling pressures, which in turn can lead to increased back pressure in the pulmonary vascular bed. LV systolic function is measured by the shortening fraction and ejection fraction (EF), with a normal EF being 56% to 78% (254). LV diastolic function is measured by assessing the mitral inflow by Doppler, with biphasic waves that peak during early diastolic filling (E waves) and atrial contraction (A waves). E waves are greater in velocity than A waves, and a reversed relationship between these two implies impaired ventricular filling as a result of decreased relaxation (305). A recent consensus statement from the PPHNet mentioned the importance of measuring LV systolic and diastolic function in babies with BPD-PH as this contributes to worsening pulmonary edema in these patients and needs a different approach (320). A recent study also found some degree of LV diastolic dysfunction through echocardiographic indices in nearly all pediatric patients with PH, thus further stressing the point of assessing LV function during PH diagnosis and evaluation (98). Infants with PPHN who have diminished LV size and function have increased mortality risk and are more likely to need advanced therapies (469, 570). Another specific patient population is infants with CDH as they have high rates of LV dysfunction even at birth, which impacts gas exchange and systemic perfusion in addition to the existing PH, and studies have shown that infants with CDH who have intact LV function might be more likely to respond to pulmonary vasodilators like iNO (207, 338).
RV-to-LV diameter ratio
While the RV is more compliant, the LV is better suited to handling a pressure overload. In PH, as RV pressures increase, the interventricular septum (IVS) starts flattening during systole, resulting in a “D-shaped” LV in the parasternal short-axis view (see Figure 8). In severe PH, where RV pressures are supra-systemic, the septum bulges out into the LV cavity at end-systole (308). This has been used to measure the RV/LV end-systolic diameter ratio, which has been shown to be significantly higher in children with PH compared to controls and correlates positively with adverse outcomes in pediatric PH (281). Flattening of the IVS offers indirect evidence of elevated pulmonary pressures in the absence of a TR jet, and end-systolic flattening of the IVS is a sensitive marker of RV systolic hypertension in pediatric PH (297). Severe septal flattening or bulging can also impact LV diastolic filling and in some situations lead to decreased cardiac output.
Figure 8.
Two-dimensional echocardiogram of a patient with severe PAH in the parasternal short-axis view showing D-shaped left ventricle (yellow line) and severe right ventricular dilatation (red line), along with biventricular remodelling.
Systolic-to-diastolic duration ratio
The Doppler derived ratio of systolic to diastolic duration (S/D ratio) has been shown to be an indicator of RV dysfunction and is independent of heart size, hence valuable in the assessment of pediatric PH. Children with significant PH have marked decreases in their diastolic duration resulting in an increased S/D ratio, which progressively worsens with increasing heart rate. S/D ratio greater than 1.4 inversely correlated with survival in pediatric PH in one study (17).
Tissue Doppler velocities and three-dimensional (3-D) echocardiography
Tissue Doppler imaging (TDI) measures myocardial velocities, which is an estimator of RV systolic function. RV TDI correlates well with cardiac catheterization measurements in pediatric CHD patients with PH and has been used for follow-up of children with IPAH (281). TDI has been shown to accurately document reduced systolic and early diastolic RV velocities in infants with CDH and PH (465). TDI varies with age and heart rate, and hence normal values for adults cannot be applied to children. Also, adult studies have shown poor sensitivity of 33% but a 100% specificity in identifying precapillary PH (231). 3-D echo eliminates the need for geometric assessments and hence gives more accurate estimates of RV function and volume, which correlate with cardiac MRI estimated volumes in the pediatric population (363).
Evaluation of cardiac anatomy
During diagnosis and follow-up for PH, a careful evaluation of the entire cardiac anatomy and extracardiac structures including shunts, aortic coarctation, and pulmonary veins (PV) is important. Pulmonary vein stenosis (PVS) has been recently identified to be a risk factor for developing severe PH in infants with BPD and is associated with a significant increase in mortality in these patients (141, 370, 575). What complicates this picture further is that PVS might develop over time or even after discharge from the NICU and is not evident often during the first imaging; one study showed that infants with BPD-PH received a median of five echocardiograms before they were diagnosed with PVS (370). Pulsed Doppler imaging of all pulmonary veins to look for PVS and either continuous, nonphasic flow, or absence of late diastolic flow reversal in the presence of nonphasic flow suggests PVS.
When echocardiograms of premature neonates were analyzed by blinded pediatric cardiologists who followed a standardized reading protocol, there was greater than 80% concordance on the diagnosis of PH, suggesting that the presence of a standardized protocol leads to a more consistent and accurate diagnosis of PH (381).
Cardiac catheterization
Cardiac catheterization remains the gold standard for diagnosis and monitoring of treatment response of pediatric PH. Catheterization helps to accurately confirm the diagnosis and severity of PH and to assess the response to pulmonary vasodilators (AVT). Catheterization is also needed to evaluate the response to vasodilator therapy, evaluate other diagnoses, and identify intra- or extracardiac conditions that affect prognosis such as left ventricular dysfunction or PVS, and to determine suitability for transplant. Ideally, every patient with an echocardiographic diagnosis of PH would be evaluated by cardiac catheterization at least once before starting therapy. However, this is not always feasible since infants and children undergoing catheterization require conscious sedation or general anesthesia, which increases the risk of adverse effects significantly. General anesthesia, which is preferred in infants and children younger than 12 years, provides a secure airway, a steady level of sedation, and control over gas exchange. However, it predisposes children to increased episodes of systemic hypotension, alters the pulmonary vascular hemodynamics, which might not be a representative of the true values during the awake state, and uses positive pressure ventilation, which might impair RV function (102). Although conscious sedation might avoid these side effects, it poses risks of developing hypoxia or hypercarbia, especially in the setting of lung disease or airway obstruction. Both induction and emergence from anesthesia have been identified as time points of increased risk, as well as any episodes of systemic hypotension or acute hypoxia as the RV is exposed to supra-systemic pressures. The risk of adverse events associated with catheterization range from 1.4% to 3.5% in pediatric PH, and mortality ranges from 0% to 1.4% (84,444, 445). This risk increases almost threefold in infants and children under the age of 2 (140). Risk factors for adverse events after catheterization include patient characteristics like prematurity, lower systemic arterial saturations or inotrope or systemic vasodilator treatment prior to catheterization, hemodialysis, and higher pulmonary vascular resistance and PAPs (444, 445). Centers with high volume of pediatric PH patients and catheterization numbers have lower rates of adverse events (445). Cardiac catheterization should be avoided in classic PPHN and should be postponed or even omitted in infants and children <2 to 5 kg who are at a higher risk of complications. It should also be avoided in acute presentation of PH or critically ill patients requiring immediate initiation of therapy (235). Having a pediatric pulmonary hypertension expert and a pediatric anesthesiologist and performing the procedure in a center capable of postprocedural care of this vulnerable population in an intensive care setting are important for having better outcomes during and after the procedure.
The measurements obtained during cardiac catheterization include oxygen saturations, pressures in different chambers and vessels, systemic and pulmonary vascular blood flow, and AVT.
Oximetry
Blood sampling to determine oxygen saturation for calculation of flow should be performed both proximal and distal to the presence of shunts, if there are any. Occasionally, there are multiple sources of pulmonary blood flow (post-Fontan operation, for example); the true mixed PA saturation can be difficult to estimate and using cardiac MRI to quantify pulmonary and systemic blood flow is a better option in such cases (225). Obtaining the hemoglobin concentration during the time of the procedure is also important, as it affects the pulmonary vascular resistance and it is integral to the Fick equation (252).
Calculation of pressure, flow, and resistance
Systolic, mean and diastolic systemic arterial, RA, RV systolic and end-diastolic, systolic, mean and diastolic pulmonary arterial and bilateral pulmonary arterial wedge pressures are usually measured (25). Pulmonary arterial wedge pressure (PAWP) is indicative of LA pressures and LV dysfunction. Inability to obtain PAWP should prompt measurement of LA pressure and LV end-diastolic pressure. Ideal time point for obtaining measurements is end expiration in a spontaneously breathing patient and end inspiration in a mechanically ventilated patient. The Fick principle states that blood flow is proportional to the oxygen consumption (Vo2) divided by the extraction of oxygen across the same vascular bed (169). Calculation of Vo2 in real time and in real-life clinical settings is hard, especially in intubated patients. The breath-by-breath method for measuring Vo2 correlated well with mass spectrometry measurements in pediatric cardiac catheterization but has not been validated in infants less than 3.5 kg (228). Systemic and pulmonary blood flows can be estimated using the Fick principle, and they can be used to calculate the vascular resistances. Both flood flow and resistance are usually then indexed to the body surface area. The Fick principle decreases in accuracy at high blood flows as the arteriovenous differences decrease. The thermodilution catheter method can also be used to estimate pulmonary blood flow if there are no intra or extracardiac shunts present.
Acute vasoreactivity testing (AVT)
AVT evaluates the response of the pulmonary vasculature to pulmonary vasodilators. It has two goals—(i) to assess prognosis and indication of PH-specific therapy and (ii) to assess the operability for PAH-CHD (4, 25, 355). True PAH acute responders (IPAH), when treated with calcium channel blockers, have an excellent prognosis with a 95% five-year survival rate (650). However, the long-term impact of defect closure in patients with CHD in the setting of PAH is unknown. Neonates with BPD-PH who underwent AVT with either 100% O2 alone or in combination with iNO and had a positive response had better long-term outcomes compared to nonresponders (185). Hemodynamic and oxygen transport mechanisms are measured at the patient’s baseline and then AVT is performed using iNO (20–80 ppm). In addition to iNO, 100% oxygen alone or in combination with iNO, aerosolized or intravenous PGI2 analogs, intravenous adenosine, and intravenous sildenafil have also been used (33, 49, 354, 531). The use of intravenous epoprostenol or adenosine is not recommended in the pediatric population as data regarding optimal dosing are not well defined. In children with PH and elevated PVR, more acute responders were identified with iNO/O2 combination than with O2 alone, and although there was no difference in the responder rate between iNO alone and iNO/O2 group, the latter showed improved pulmonary hemodynamics, which may warrant some caution during interpretation of results (49). Use of 100% O2 alone should be avoided in patients with PAH-CHD, as this would lead to increased oxygen in the pulmonary venous blood and, consequently, the fraction bound to hemoglobin. This would lead to overestimation of pulmonary blood flow and hence an underestimation of the pulmonary resistance (25).
There are considerable differences between centers in identifying responders to AVT and selecting patients for treatment (157). The European Pediatric Pulmonary Vascular Disease Network suggests the use of the modified Barst criteria for AVT, whereas the recent 6th WSPH Pediatric Task Force recommends the use of the Sitbon criteria (4, 25, 235, 506).
Modified Barst criteria for AVT:
In patients with IPAH/HPAH: For patients without a shunt, a positive response to AVT is considered as a 20% decrease in mPAP and indexed PVR(PVRi)/indexed SVR(SVRi) ratio without a decrease in cardiac output.
In patients with PAH-CHD and shunts: The hemodynamic response defined as a positive response and operability in shunt defects (Qp:Qs > 1.5:1) is a >2% fall in the PVRi and PVRi/SVRi with respective final values <6 indexed WU (iWU) and <0.3.
Sitbon criteria: This is defined as the decrease in mPAP by at least 10 mmHg to a value of <40 mmHg with sustained cardiac output. If the mPAP is less than 40 mmHg to begin with, a drop by at least 10 mmHg without decrease in the cardiac output is defined as a positive AVT.
The Sitbon criteria were found to identify AVT responders who had better outcomes when treated with long-term CCBs (157, 548) and has been recommended by the latest WSPH Pediatric Task Force. It should be noted, however, that no fall in PAP does not necessarily mean no fall in PVR. Response to pulmonary vasodilators with a decrease in PVR and increase in Qp is possible without changes in the PAP, and hence hemodynamic indicators such as PVR/SVR and PVRi are considered better markers of AVT response.
Computed tomography (CT) scan
High-resolution computed tomography (HRCT) scan of the lung parenchyma along with computed tomography angiography (CTA) to evaluate the pulmonary, bronchial, and systemic thoracic vasculature is a commonly used tool in the diagnosis of neonatal and pediatric PH. Chest CT has significant utility in staging of pulmonary interstitial disease and in Group 3 PH due to lung hypoxia. CT with contrast and CTA can be used to rule out chronic thromboembolic pulmonary hypertensive disease. CT-measured ratio of main PA to ascending aorta of ≥1.3 raises the index of suspicion of PAH in children (235). Chest CT with CTA is also useful in identifying obstructive pulmonary vascular disease like peripheral pulmonary stenosis or pulmonary venous stenosis, which worsens mortality and morbidity in BPD-PH. Lymph node enlargement, centrilobular ground-glass opacities, and septal thickening with pulmonary artery enlargement all point toward venous obstructive disease, whereas smooth interlobular septal thickening, diffuse multifocal ground-glass opacifications, and enlarged central pulmonary arteries are more indicative of pulmonary capillary hemangiomatosis (4). CTA can also identify systemic pulmonary collaterals that are present in up to 30% of infants with BPD-associated PH (141). These collaterals contribute to increased PAP, and closure of the collateral by interventional cardiologists may be needed to alleviate PH in select cases. It is also recommended that every patient undergoing evaluation for lung transplantation should receive a chest CT (329, 595).
Cardiac magnetic resonance imaging (CMRI)
Cardiac magnetic resonance imaging (CMRI) is recommended both as a part of the initial diagnostic workup and as a part of follow-up to assess ventricular function (333). CMRI is the gold standard to which all echocardiographic measures for evaluation of RV volume and function are compared (524, 616). CMRI is usually performed in infants and children with either some degree of sedation or under general anesthesia, which again pose similar risks as stated above during cardiac catheterization (584). CMRI helps in reliable assessment of the RV and LV size and volume indices as well as the RV ejection fraction (RVEF). RVEF and LV stroke volume were found to be most strong predictors of death or need for heart transplant in pediatric PH (2.6- and 2.5-fold increase in mortality for every 1-SD decrease, respectively) out of all variables measured by CMRI (402). It is recommended that all pediatric CMRI should include cine CMRI, which is the gold standard for assessment of biventricular volumes, muscle mass, and global pump function (226, 291). In addition, selective blood flow measurements in pulmonary and systemic circulation and quantification of shunt flow can be performed with increased accuracy. CMRI when performed in preterm neonates found that when controlled for BPD severity, birthweight and gestational age, MRI LV eccentricity index and PA/aorta ratio correlated positively with the need for PH therapy either during hospitalization or after discharge (130). Other parameters for which CMRI is used are visualization of myocardial fibrosis using late gadolinium enhancement, strain and strain rate, septal curvature, pulmonary artery stiffness, and RV-PA coupling (67, 90, 333). MR angiography is also useful for evaluation of any pulmonary venous thromboembolism.
Ventilation/perfusion scan (VP scan)
Ventilation perfusion mismatch in a patient with known or suspected PH raises concern for thromboembolic disease of the pulmonary vasculature. This is especially important in IPAH as well as in known Eisenmenger’s syndrome due to the increased incidence of thromboembolic disease in these patients (76). Chronic thromboembolic PH shows areas of ventilation-perfusion mismatch in VP scans, usually one area, sometimes two or more (177, 415).
Biomarkers
Brain natriuretic peptide (BNP) and its precursor, NT-proBNP, are the most studied biomarkers in both adult and pediatric PH (83). A recent meta-analysis of pediatric PH biomarkers found that low levels of NT-proBNP are strong predictors of survival and children who stay at NT-proBNP levels below 1200 ng/L during treatment have significantly better survival rates, a statistic that has been shown to be true in adult PH as well (438, 475, 477). BNP has a shorter half-life than NT-pro-BNP; hence the latter is more commonly used, although it is more susceptible to changes in renal function. Reference values of NT-proBNP have been established for the pediatric population and increase in the first few days after life and then fall drastically after the first week, and then gradually throughout childhood (440). Recently, urinary NT-pro-BNP has also been studied as a screening tool for PH in preterm infants (425). A proteomic analysis of early serum angiogenic proteins showed that early increases in bone morphogenetic protein 10 (BMP10) are strongly associated with late increases in BPD and PH (28). A decrease in cord blood angiogenic factors associated with placental maternal vascular under-perfusion has been also associated with an increased risk of BPD-PH in preterm neonates (387). MicroRNAs, which have recently acquired a great deal of attention as biomarkers for diseases in which angiogenesis is impaired, have shown some promise in predicting pediatric PH (296). Circulating endothelial cells (CECs) and endothelial cell progenitors (ECPs) have been shown to be present in blood of PH patients (124, 360, 549). Measurement of CECs in children with IPAH and CHD-PAH before and after treatment showed that rising levels of CECs preceded clinical deterioration (346). Furthermore, elevated CEC levels were associated with irreversibility in CHD-PAH (550). Several other biomarkers such as uric acid, atrial natriuretic peptide, and Troponin T have also been shown to be associated with worse outcomes in both adult and pediatric PH (4).
Genetic testing
Genetic mutations are increasingly being identified in children with IPAH/HPAH, with BMPR2 being the most common (almost 70% of HPAH and 10%–40% of IPAH cases) (15, 347). With the emerging recognition of the need for genetic testing, other genes have been found to be involved in pediatric PAH, including, but not limited to, ALK1, ABCC8, ENG, CAV1, KCKN3, EIF2AK4, and TBX4 (7, 35, 188, 347, 367, 411). Genetic testing for these commonly found genes is currently recommended for families of all children diagnosed with IPAH/HPAH. Gene testing for less implicated genes in PAH such as NOTCH3, SMAD9, GDF2, AQP1, SMAD8, SOX17, and ATP13A3 can be performed as a second-tier test in children with PAH of unknown cause with a negative test for the previously mentioned genes (235). Next-generation sequencing should be preferably performed to maximize the depth of coverage for the affected genes. Most PAH-associated mutations are inherited in an autosomal dominant fashion with incomplete penetrance. Hence, first-degree relatives of all PAH patients with a genetic mutation known to be implicated in PAH should at least undergo genetic counseling. Children who are found to have PAH-associated mutations and are asymptomatic should undergo screening echocardiograms every 1 to 3 years to detect elevated RV pressure, and asymptomatic first-degree relatives of patients with PAH-associated mutations should undergo PH screening if they develop new cardiorespiratory symptoms (4, 235, 506).
Six-minute walk test (6MWT)
The six-minute walking distance (6MWD) is considered a useful tool for follow-up and as a therapeutic endpoint for treatment goals in pediatric PH. In adult PH, the 6MWD correlates well with other parameters of disease severity like the WHO-FC, and the magnitude of oxygen desaturations during the test and the heart rate recovery after it have been used for prognostication of adult PH (393, 456, 490). Studies have shown that it is feasible to perform the 6MWT in children and that it reflects disease severity and clinically relevant exercise tolerance (156, 198, 330). The 6MWD is higher in children than in adults, and reference values have been established for the pediatric population (330). Shorter 6MWD combined with lower transcutaneous oxygen saturations during the 6MWT correlated with higher WHO-FC and NT-proBNP levels and worse transplant-free outcomes in pediatric PH patients in one study (156).
Cardiopulmonary exercise testing (CPET)
CPET is performed to both evaluate and follow up patients with PH. Adult studies have shown that low peak oxygen uptake and low systolic blood pressure at peak of exercise in patients with PH undergoing CPET correlate with impaired survival (632). CPET has been shown to be feasible and safe to perform in children with decreased peak oxygen uptake and decreased baseline oxygen saturation at peak exercise compared to healthy controls (8, 551). The peak oxygen uptake has also been shown to strongly correlate with invasive measures of disease severity, including the pulmonary vascular resistance index. The type of exercise, treadmill versus cycle ergometer, or the specific exercise protocol is not important to the success of the test if the protocol has been standardized and is performed in a controlled environment. Changes in oxygen consumption, CO2 production, minute ventilation, heart rate, and blood pressure should be obtained at rest, during exercise and during recovery. Subtle changes in exercise tolerance may suggest deterioration prior to clinical manifestations, which might prompt earlier reevaluation including cardiac catheterization (198).
Treatment
The 6th WSPH Pediatric Task Force has proposed a treatment algorithm based on expert consensus opinion and is mostly relevant for the treatment of pediatric IPAH/HPAH (Figure 9). Similar algorithms exist for the treatment of BPD-PH (Figure 10), which are based on expert consensus opinion. There is a lack of randomized clinical trials evaluating therapies in the pediatric PH population, and most data are based off extrapolation from adult trials or case series from off-label use.
Figure 9.
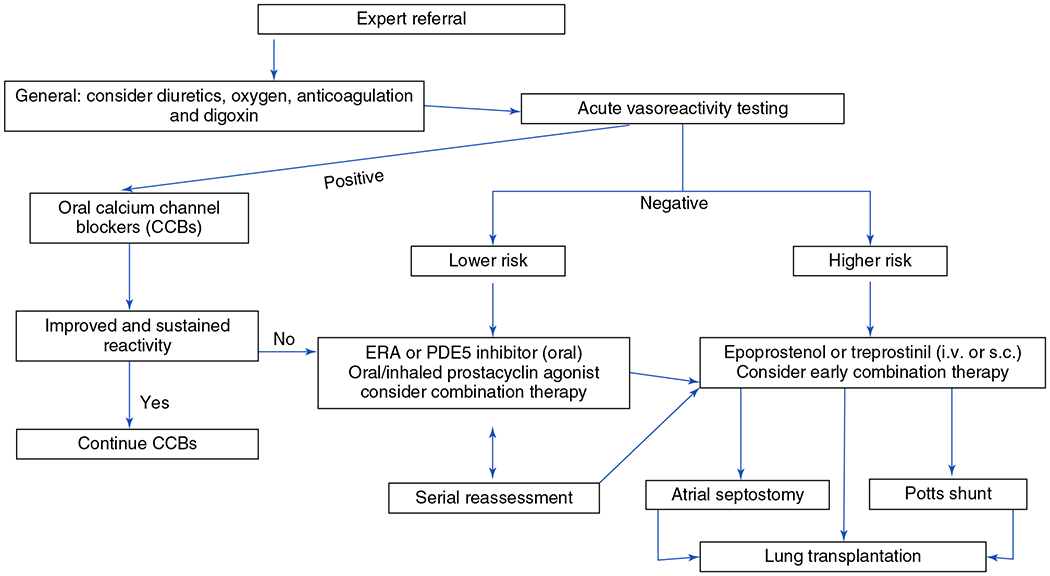
Treatment algorithm for pediatric IPAH/HPAH as recommended by the 6th WSPH Pediatric Task Force. ERA, endothelin receptor antagonist; PDE5, phosphodiesterase 5; i.v., intravenous; s.c, subcutaneous. Adapted, with permission, from Rosenzweig EB, et al., 2019 (506). © 2019, The European Respiratory Society.
Figure 10.
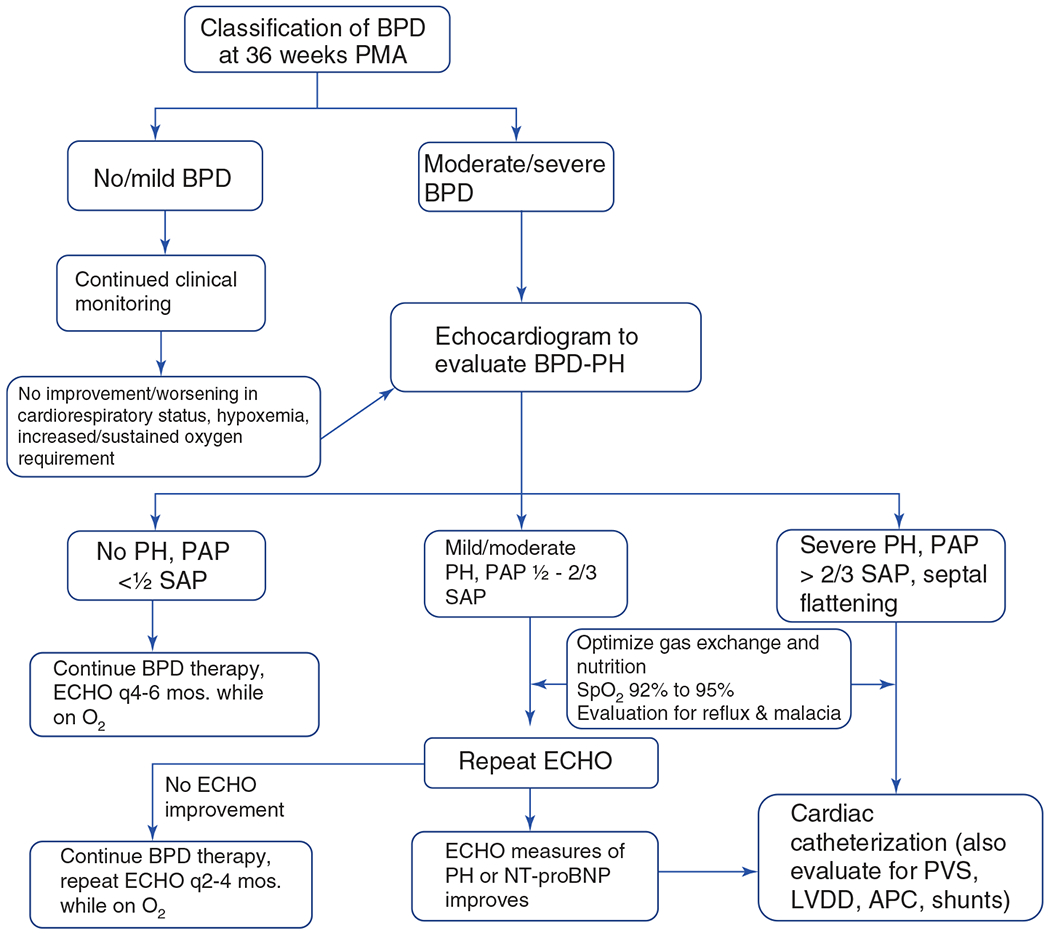
Algorithm for the diagnosis and treatment of pulmonary hypertension in infants with bronchopulmonary dysplasia based on recommendations from PPHNet. PPHNet, pediatric pulmonary hypertension network; BPD, bronchopulmonary dysplasia; PH, pulmonary hypertension; ECHO, echocardiogram; PAP, pulmonary arterial pressure; SAP, systemic arterial pressure; BNP, brain-type natriuretic peptide; NT-proBNP, N-terminal proBNP; PVS, pulmonary vein stenosis; LVDD, left ventricular diastolic dysfunction; APC, aortopulmonary collateral. Modified, with permission, from Krishnan U, et al., 2017 (320).
Oxygen therapy
Maintaining adequate oxygenation is key to preventing the vicious cycle of hypoxic pulmonary vasoconstriction and the VP mismatch and hypoxemia that ensues (629). The effect of oxygen on pulmonary vasodilatation increases with increasing gestational age (110, 494). Extremely preterm neonates have diminished pulmonary blood flow with a poor vasodilatory response to oxygen. During resuscitation of these infants, 100% oxygen decreases the PVR rapidly; however, this effect is not sustained and might lead to blunted responsiveness to iNO later and increased oxygen toxicity from free radicals (109, 110). To answer the question of the optimal target oxygen saturation for extremely premature infants, several randomized clinical trials were conducted, and these were recently studied in a meta-analysis called the Neonatal Oxygenation Prospective Meta-analysis (NeoPROM) (36, 134, 436, 528, 529). These studies did not measure PH as an outcome measure but did document a higher mortality risk when preterm infants were randomized to lower oxygen saturations (30). Observational cohort studies have shown that changing the oxygen saturation parameters for preterm neonates from lower (tolerating a lower limit of up to 85%) to higher (at least above 90%) targets decreased the incidence of elevated PVR and PH at 36 weeks postmenstrual age in these infants (288, 327). This comes as a trade-off since the incidence of BPD was found to be higher in the Neonatal Research Network units after the saturation target parameters were changed to higher levels (182). In preterm infants with BPD-PH, minor episodes of desaturations increase pulmonary pressures and should be avoided (3, 6, 417). A recent consensus from the PPHNet recommends maintaining oxygen saturations between 92% and 95% in these infants and use of chronic supplemental oxygen before starting pharmacological vasodilators (320). The European PPVDN recommends target oxygen saturations of >93% in preterm neonates and >95% for neonates with an echocardiographic diagnosis of BPD-PH (246). In PPHN, although increased oxygen is usually required to reverse the pulmonary vasoconstriction and hypoxemia, acute hyperoxia should also be avoided as it increases oxidant stress, alters pulmonary vasoreactivity, and augments pulmonary vascular dysfunction associated with lung disease (564). Exposure to prolonged hyperoxia and high oxygenation indices prior to start of iNO was associated with a higher incidence of ECMO and/or death in several clinical trials (109). Therefore, maintaining a strategy to minimize oxygen exposure with lung recruitment, surfactant administration and iNO are essential to reduce the toxic effects of free radical damage in PPHN (564).
For the treatment of PAH in the intensive care unit as well as at home in PAH or CHD-PAH population, it is advisable to use continuous supplemental oxygen to keep oxygen saturations >95% or the arterial pO2 above 60 mmHg (227). These parameters change during shunt physiology. For patients with CHD-PAH and significant left-to-right shunt, oxygen therapy might lead to pulmonary overcirculation, which may worsen the right heart function without lowering the mPAP in the long term. For patients with right-to-left shunts, maintaining the shunt flow may be critical in maintaining adequate systemic oxygen delivery. Hence, oxygen is not indicated unless there is parenchymal lung disease or there is profound cyanosis (75%-85% are acceptable saturation parameters in these children) (285).
Diuretics and fluid balance
Fluid and volume status need delicate balancing in PH. In neonates and children with severe PH, the RV is preload dependent and volume depletion can lead to acute worsening and PH crises. However, severe PH by itself leads to RV failure, volume overload, increased central venous pressure, hepatic congestion, ascites, and peripheral edema. There have been no randomized trials to study the effect of diuretic use on PH outcomes, either in adults or in children. Hence, current recommendations are to limit the use of diuretics to loop diuretics and aldosterone antagonists in patients with the signs of systemic venous congestion or severe left-to-right shunting causing pulmonary overcirculation (439). Aldosterone antagonists such as spironolactone and eplerenone block mineralocorticoid receptor action and have been shown to improve RV and LV function in adults, but pediatric data are lacking (85, 138). It is important to monitor electrolyte levels while on diuretics, and to carefully monitor fluid status as the RV is preload dependent in such situations. Infants with BPD-PH are often on chronic diuretics to reduce pulmonary vascular congestion from the sequelae of BPD. This, however, leads to a chronic low-volume state and might mask left ventricular dysfunction, which can be a cause of PH in these patients. These patients might be given a small fluid bolus during cardiac catheterization to evaluate the left ventricular function when subjected to an increased afterload (320).
Digoxin
Digoxin or digitalis has been shown to acutely improve cardiac output in adult IPAH patients and slow the ventricular rate down in PAH patients with tachyarrhythmias (499). There are no studies on the long-term effects of digoxin on the right ventricular function, and as such it is not a drug commonly recommended in pediatric PH.
Acid-base balance
Acidosis increases PVR and causes pulmonary vasoconstriction and may impede the action of inotropes (111). Therefore, acidosis should be avoided, and attempts should be made to normalize the arterial pH. Alkalization is effective for the treatment of acute PH crises in the intensive care unit (111, 285, 412). In the era before the use of iNO, alkalization was induced in newborns with PPHN with serum bicarbonate infusions. Although alkalization brings about a transient decrease in PVR and improvement in oxygenation, animal models have shown an exaggerated pulmonary vasoconstrictive response to hypoxia after prolonged alkalosis (516). Alkalosis also causes cerebral vasoconstriction and diminished cerebral blood flow and has been shown to worsen neurodevelopmental outcomes and hearing deficits in newborns and thus should be strongly avoided (376).
Anticoagulation
Children with PH are often on anticoagulants or antiplatelet agents. There are no long-term data on the benefit of children on chronic anticoagulation, but the current consensus is that it may benefit certain classes of pediatric PH such as progressive IPAH/HPAH, chronic thromboembolic PH, hypercoagulable states, and patients in low cardiac output states, which predispose to blood stasis and increased coagulability (235). The target international normalized ratio in IPAH/HPAH is between 1.5 and 2.0; however, this is an empirical target (4). Anticoagulant and antiplatelet therapy should be avoided in patients with hereditary hemorrhagic telangiectasia (HHT) and porto-pulmonary hypertension and should be critically reviewed in smaller children who are at a higher risk for hemorrhagic complications and congenital or acquired von Willebrand disease. A class of pediatric PH that is at a higher risk for pulmonary vascular thromboses are children with Eisenmenger’s syndrome, but they are also at a higher risk of severe pulmonary hemorrhage and hence the use of anticoagulation in them warrants caution. A retrospective study of adults with Eisenmenger’s syndrome on anticoagulants showed no impact of anticoagulant use on long-term survival (523).
Inhaled nitric oxide (iNO)
iNO is one of the most used therapies to treat PH in the acute setting of PPHN, PH in CDH, acute postoperative PH or PH crises. It has been approved by the FDA since 1999 as a pulmonary vasodilator therapy for the treatment of PPHN in term and near-term infants, based on two landmark multicenter placebo-controlled randomized controlled trials that showed a significant decrease in the need for extracorporeal membrane oxygenation (ECMO) in the iNO group (120, 435). It is delivered as an inhaled gas blended with air or oxygen and simulates the action of endogenous NO to activate sGC in the pulmonary arterial smooth muscle cells, leading to increased cGMP levels and SMC relaxation. NO can cross the alveolar-capillary membrane to enter the smooth muscles of the precapillary pulmonary arterioles, causing selective vasodilation and attenuation of vascular remodeling (306). iNO has a relatively short half-life of 15 to 30 s and is rapidly metabolized by Hb in the RBC in the pulmonary circulation, preventing its systemic effects (51). Chronic use of iNO is associated with methemoglobinemia, and hence methemoglobin levels should be monitored in these patients (496, 590). iNO is usually started at a dose of 20 parts per million (ppm) regardless of the etiology. Higher doses do not improve oxygenation and contribute to increased risks of methemoglobinemia and NO2 exposure. Once oxygenation improves, iNO dose can be rapidly weaned off in steps to 5ppm and then gradually weaned in 1 ppm decrements to 1 ppm before discontinuation. There are reports of life-threatening rebound PH after discontinuation, and this can usually be prevented by slow weaning from 5 to 1 ppm and waiting for a few hours for clinical stability before turning it off (136).
The use of iNO in term and near-term infants for the treatment of PPHN is well studied and documented through several double-blinded placebo-controlled trials (44, 135, 137, 180, 303, 358, 435, 520, 633). There have also been several trials in term and near-term infants with PPHN who have been randomized to either iNO or placebo and compared infants based on their severity of respiratory failure as determined by the increased oxygenation index or alveolar-arterial oxygen gradient (44, 126, 217, 316). A randomized trial also found that starting iNO at an earlier oxygenation index (15–25 vs >25) does not decrease mortality or the need for ECMO (316). A 2016 Cochrane review for the use of iNO in term and near-term infants with hypoxic respiratory failure studied 17 randomized controlled trials and found that iNO improved outcomes in hypoxic term and near-term infants by reducing the combined endpoint of death or need for ECMO, which was primarily due to the reduction in the use of ECMO (47). iNO also showed an improved oxygenation index within 30 to 60 min of start of the drug and improved arterial oxygen saturation, and these improvements are not limited to those who show echocardiographic signs of decrease in PAPs (47).
A unique population with PPHN is the CDH population, where in addition to altered pulmonary vasoreactivity, pulmonary hypoplasia and associated left ventricular dysfunction often complicate the presentation and management. The only two randomized trials studying the effects of iNO in infants with hypoxic respiratory failure due to CDH and PH were the NINOS 1997 trial and the diaphragmatic hernia subpopulation of the Clark 2000 trial (120, 467). Both these trials found that death or need for ECMO did not change either separately or as a composite outcome in infants with CDH who were randomized to either placebo or iNO. Two other large-scale database studies from the Pediatric Health Information System (PHIS) and from the CDH Study Group (CDHSG) were conducted to evaluate the use of iNO in CDH (100, 483). The PHIS data showed that out of 1713 neonates with CDH in the United States, 57% of the infants received iNO and that only half of these infants showed an improvement in oxygenation. However, there was no change in mortality or need for ECMO for these infants (100). The CDHSG data, which included over 3300 infants from 13 different countries, showed that 74% of the infants who received an echocardiographic diagnosis of PH in the first week of life were started on iNO. The infants who were diagnosed with PH were also more likely to need ECMO, but iNO did not change the need for ECMO or the mortality for this cohort (483). A recent single-center retrospective review of infants with CDH who were started on iNO either due to clinical hypoxemia or an echocardiographic diagnosis of PH found a subset of patients who responded to iNO (338). Responders were less likely to have left ventricular systolic dysfunction and were less likely to need ECMO. Current recommendations from the AHA allow the use of iNO in infants with CDH and normal left ventricular function but advise against continuation for more than 24 h if no clinical benefit is seen (4).
iNO has also been studied for preterm infants with hypoxemic respiratory failure as an initial rescue therapy, as a routine adjunct to conventional ventilatory support, or as a later treatment in infants at risk for BPD (4, 42, 133, 240, 298, 304, 386, 530, 560, 571, 572, 605, 627). These studies were heterogeneous for the birthweight and gestational age of infants recruited as well as the eligibility criteria. There was no effect of iNO on death before 36 weeks postmenstrual age or death before discharge or on BPD at 36 weeks postmenstrual age (46). However, there is a subpopulation of infants with oligohydramnios and/or preterm premature prolonged rupture of membranes who have shown improved oxygenation and pulmonary hemodynamics, and a recent expert panel recommended use of iNO in this cohort (298, 299 301).
iNO is also used in acute postoperative PH and in PH crises in acute care settings as well as during cardiopulmonary bypass for congenital heart surgery (63, 113). Currently, a multicenter randomized trial to evaluate the benefits of iNO during the entire duration of cardiopulmonary bypass in CHD surgery is underway and results are awaited (527).
Phosphodiesterase-5 (PDE-5) inhibitors
Sildenafil and tadalafil are the PDE-5 inhibitors that have been used in the pediatric population. PDE-5 degrades cGMP, which is responsible for pulmonary vascular smooth muscle relaxation. Sildenafil was initially studied as a candidate drug for the treatment of angina pectoris in the 1980s. Urologic studies at the same time showed that cGMP was also responsible for smooth muscle relaxation and vasodilatation leading to penile erection; hence use of sildenafil started in the treatment of erectile dysfunction (ED) and was approved by the FDA for ED in 1998 (205, 215, 408). As the role of PDE-5 in the lung vasculature became more evident in the 1990s, sildenafil was studied for adult PH and the first intravenous placebo control study was performed in late 1990s, which showed that sildenafil selectively reduced pulmonary pressure and pulmonary vascular resistance in patients with PAH, pulmonary venous hypertension, and hypoxic pulmonary hypertension (205). This led to the approval for the use of oral sildenafil in adult Group 1 PH in 2005 by the US-FDA, and later in 2009 the intravenous formulation was also approved.
Sildenafil is a water-soluble compound that has a similar half-life in the pediatric population compared to adults, although the volume of distribution and the peak concentration reached are higher in children (449). An open-label trial of intravenous sildenafil in term neonates with PPHN found similar results with fourfold higher volume of distribution; however, this population also had significantly longer plasma half-life. This decreased with increasing clearance and reached adult values by one week of life, attributed to the postnatal maturation of N-demethylation mechanism in neonates (420). Oral bioavailability of sildenafil is ~40% and it undergoes first-pass metabolism in the liver by the hepatic cytochrome P450 pathway (261). For children weighing above 20 kg, the recommended dose is 20 mg three times daily (TID) and for 8–20 kg it is 10 mg TID as per European guidelines (149). The common practice for infants and children weighing below 8 kg is to start at 0.5 mg/kg every 8 h and escalate to 1 mg/kg every 8 h, which is continued as the maintenance dosing for oral sildenafil at most PH centers (123). There have been multiple case series and small studies on the use of both oral and intravenous sildenafil in pediatric PH, which included CHD-PAH, PPHN, postoperative PH, BPD, and CDH (43, 183, 210, 212, 247, 263, 407, 434, 460, 546, 563, 601, 602). Sildenafil has been shown to be useful in prevention and treatment of postoperative PH in children after CHD surgery, either as an adjunct to iNO or to help weaning from it (34, 432). A recent meta-analysis of perioperative PH in children with CHD found that sildenafil decreased ICU stay significantly, although it did not decrease total length of stay or mortality before discharge (278). For PPHN, intravenous sildenafil was associated with immediate and sustained improvements in oxygenation in those infants who received higher infusion doses (563). A recent Cochrane review that analyzed 5 trials where sildenafil was used for the treatment of PPHN found a significant decrease in mortality when sildenafil was compared to placebo; however, the differences ceased to exist when compared to iNO or when iNO was used in both groups (294). Recently, a multicenter international trial has started recruiting patients with CDH and PH to be randomized to intravenous sildenafil infusion or iNO; the outcomes are absence of PH by day of life 14 or death at day of life 28 (122). The first randomized, double-blinded, placebo-controlled trial of oral sildenafil monotherapy in children with PAH was the STARTS-1 trial (Sildenafil in Treatment-Naïve Children, Aged 1 to 17 years, with PAH), which randomized children >8 kg to either low-, medium-, or high-dose sildenafil or placebo for 16 weeks and peak oxygen consumption (PVO2) was measured during CPET. Although this study found that PVO2 was only marginally changed in the sildenafil groups combined together, when medium- and high-dose groups were combined, they showed efficacy in PVO2, WHO-FC, and hemodynamic parameters (55). When these groups were followed in the long term as a part of STARTS-2 trial, they showed increased mortality with higher doses of sildenafil for unexplained reasons (50). This led to the issuance of a US-FDA warning in 2012 against the use of sildenafil in the treatment of pediatric PH, which requires closer monitoring and surveillance of patients on sildenafil (5, 380). A multivariate analysis of the STARTS-2 trial data had shown that the increased mortality was primarily associated with HPAH, high PVRI, and high RA pressures, and adjusting for these factors decreased the hazard ratio for high-dose versus low-dose sildenafil. The latest FDA recommendations for sildenafil use in pediatric PH issued in 2014 do not recommend against the routine use of sildenafil and recommend closer monitoring of children on long-term sildenafil, especially on higher doses (149).
Tadalafil is another selective PDE-5 inhibitor that has once daily dosing and a longer half-life than sildenafil and has been shown to improve exercise capacity and quality of life measures in adults with PH. It was studied in children with PH either as an initial drug or as a transition from sildenafil and was shown to improve mPAP and PVRI in both cases (579). An Iranian pediatric PH cohort also reported similar findings after transitioning to tadalafil from sildenafil with no worsening in the NYHA FC or pulmonary hemodynamics (519). Postmarketing surveillance in Japan has shown that tadalafil is safe and effective as monotherapy in pediatric PH (639).
Udenafil is a newer selective PDE-5 inhibitor having a longer duration of action and was found to improve myocardial performance in pediatric patients with Fontan physiology in a phase I/II clinical trial conducted by the Pediatric Heart Network (213). Patients with Fontan physiology have a circulation that is dependent on low pulmonary vascular resistance to maintain adequate cardiac output; hence the patients enrolled in this trial did not have traditional parameters of PAH. This study was subsequently extended to a multicenter international trial (Fontan Udenafil Exercise Longitudinal trial) and found no difference in the myocardial performance index. Udenafil use was not associated with improvements in peak oxygen consumption during exercise, but it was associated with improvements in several measures of exercise performance at the ventilatory anaerobic threshold (214).
Calcium channel blockers (CCBs)
CCBs are used infrequently in children with PAH as first-line therapy; however, it is efficacious in children who are AVT responders (592). Based on the Sitbon criteria, those children with IPAH or HPAH who respond to NO or 100% oxygen during AVT (around 8%-15% of children with IPAH), it is prudent to offer CCBs as first-line monotherapy (593). AVT establishes a relative contribution of reversible vasoconstriction versus fixed stenosis in children with PAH (593). Those who have a negative AVT are unlikely to benefit from CCBs, and additionally may have deleterious adverse reactions (498, 547). CCBs are not meant to be used in pediatric PAH without a prior documented positive response to AVT as they can cause systemic hypotension, worsen right heart failure, and potentially lead to death (53, 194). The reasons for such effects range from depression in the myocardial contractility and negative inotropic effects to the activation of renin-angiotensin system and hypotension, leading to decreased coronary perfusion and myocardial dysfunction from ischemia (457, 593). Those children who respond to CCBs as initial monotherapy can be continued on them with close follow-up, keeping in mind that they can become unresponsive and deteriorate later, necessitating further evaluation and addition of other drugs (235, 650). It should also be kept in mind that children with PAH and a significant intracardiac left-to-right shunt or those with Eisenmenger’s syndrome most likely will not benefit from CCB therapy regardless of the AVT and hence CCBs should not be used in this setting (235). CCBs are also not indicated in infants less than 1 year of age as the negative inotropic effects are pronounced in this age group. The CCBs used in pediatric PAH are nifedipine (2-5 mg/kg/day), diltiazem (3-5 mg/kg/day), and amlodipine (2.5-10 mg/day) (37). Diltiazem lowers heart rates more prominently than the other ones affecting cardiac output and systemic blood pressure; hence it is preferred in children who have higher resting heart rates. Verapamil is also contraindicated in PAH due to its tendency to cause bradycardia without significant pulmonary vasodilatory properties (4).
Prostacyclin analogs
PGI2 analogs, which fall in the larger group of prostanoids, mimic endogenous PGI2 and stimulate G-protein-coupled receptors on the surface of endothelial and smooth muscle cells to increase intracellular cAMP levels, which result in pulmonary vasodilatation and decrease in PVR. There is an imbalance in the favor of vasoconstrictive thromboxane A2 instead of vasodilatory PGI2 in PAH. PGI2s are FDA-approved in adults with PAH and are used off-label in the pediatric PH population as monotherapy in those who are in high-risk PH group and fail AVT or those who do not show any improvement on CCBs after a positive AVT. They are also used as combined therapy in pediatric PH patients who are in low-risk PH group but fail to improve on monotherapy of PDE-5 inhibitor or ERAs (235). The three PGI2s used in the pediatric population are epoprostenol, iloprost, and treprostinil.
Epoprostenol is the first prostanoid to be FDA approved and is still the gold standard of treatment for severe PH. It has a very rapid onset of action and a short half-life; hence it is preferably given as a continuous intravenous infusion. In acute postoperative PH as well as in neonates with severe PH from BPD, CDH, or PPHN, intravenous epoprostenol can be used as an alternative if iNO is unavailable. Multiple small studies and retrospective data have shown improved survival and quality of life in adult and pediatric PAH treated with intravenous epoprostenol (56, 272, 540, 650, 660). A cohort of 77 children with IPAH on epoprostenol who were followed through the 1990s to early 2000s showed survival of 94%, 81% and 61% at 1, 5, and 10 years, respectively (650). In neonates with PPHN refractory to iNO, a subpopulation responds to intravenous epoprostenol with a decrease in the oxygenation index and need for ECMO (13). There is a need to study the use of prostanoids in the treatment of PPHN using randomized trials as was pointed out by a recent meta-analysis (538). The side effects of epoprostenol include headache, gastrointestinal disturbances, jaw pain, bradycardia, hypotension, and thrombocytopenia. Epoprostenol when given to patients with parenchymal lung disease such as BPD and interstitial lung disease may lead to worsening of ventilation perfusion matching. In patients with veno-occlusive disease and PVS, epoprostenol can lead to worsening of pulmonary edema. It can also affect platelet counts and lead to an increased risk of bleeding (167). Inhaled epoprostenol has been used recently in acute care settings for PPHN where iNO might be unavailable or infants are unresponsive to iNO and has been shown to improve oxygenation and echocardiographic parameters of PH significantly (72).
Iloprost is a synthetic PGI2 analogue approved by the FDA for adult PAH in 2004. It has a short half-life of 20 to 25 min, longer than epoprostenol. The benefit of aerosolized iloprost over other prostanoids is that it lowers PVR but does not affect systemic blood pressure. Like epoprostenol, iloprost has also been used as an adjunct or in place of iNO in acute postoperative PH and in PPHN, where it lowers mean pulmonary pressure and improves oxygenation (356). A retrospective study of the use of inhaled iloprost in IPAH and CHD-PAH in the pediatric population showed that it was effective and well tolerated in this population (409). Similarly, another retrospective study of inhaled iloprost in preterm neonates with severe respiratory distress syndrome and PPHN showed benefits with improved oxygenation and with no systemic hypotension (645). There are other smaller studies where iloprost has been used in conjunction with oral sildenafil or bosentan for the treatment of pediatric PH (132, 421, 545). In both IPAH and CHD-PAH, inhaled iloprost has been shown to improve the functional status when studied in the long term (273). One major drawback of the use of inhaled iloprost in the pediatric population is that it needs to be administered using nebulization every 6 to 8 h and requires patient compliance, which might be difficult in a population already facing a lot of quality of life challenges (273, 450). There have also been reports of worsening reactive airway disease on inhaled iloprost (273).
Treprostinil is another PGI2 analogue approved for use as oral, inhaled, intravenous, and subcutaneous forms. This has a longer half-life (steady state in 10 h) compared to other prostanoids and is stable at neutral pH at room temperature and hence can be given as continuous infusion. Subcutaneous mode of delivery avoids problems associated with central lines; however, it causes pain and reactions at the infusion site. Adults with PAH on long-term intravenous or subcutaneous treprostinil have displayed good long-term results (52, 216). There have been reports of pediatric patients on intravenous epoprostenol who were transitioned over to intravenous treprostinil due to the longer half-life of the latter, and these patients had no change in exercise capacity, WHO-FC, hemodynamics, and echocardiographic determination of right ventricular systolic pressure. The side effects associated with epoprostenol of headache, rash, diarrhea, and jaw pain have decreased on treprostinil (271). Intravenous treprostinil is also associated with catheter-associated infections, but these can be decreased by protecting catheter connections, avoiding water on any connection and a more basic buffer (155, 184). Subcutaneous treprostinil has also been used in pediatric and neonatal PH and has been well tolerated and efficacious (178). Inhaled treprostinil is available as well; however, it achieves lower plasma concentrations than the subcutaneous or intravenous forms and hence should not be used in patients who are not responding to the maximal doses of parenteral treprostinil (167). Inhaled treprostinil reaches peak levels in 5 to 10 min and needs to be administered every 4 to 6 h. Oral treprostinil, although approved in adults, has not been studied in the pediatric population much, primarily since the tablet cannot be crushed and there is no oral suspension available (167). Treprostinil clearance is decreased in patients with liver disease, and coadministration with anticoagulants or other vasodilators may increase the risk of bleeding and systemic hypotension.
Beraprost is an oral PGI2 analog that has not been approved in the United States or Europe and has not been well studied in children. A double-blinded, placebo-controlled, randomized trial of beraprost in adult PAH showed improved hemodynamics initially; however, this was not sustained over a long period (57).
Endothelin antagonists
Endothelin receptor antagonists (ERAs) are now considered first-line oral pharmacotherapy in pediatric IPAH/HPAH patients who either have a negative AVT and are low risk based on risk stratification or those who did not show sustained and improved reactivity on oral CCBs after a positive AVT (4). As described in a separate section, ET-1 acts on both ETA and ETB, which are G-protein-coupled receptors present on smooth muscle cells and endothelial cells. ETA binding leads to increased intracellular Ca2+ causing vasoconstriction, whereas ETB stimulation leads to the release of NO and PGI2, increased ET-1 clearance and a minor effect on pulmonary vasodilatation, and reduced pulmonary vascular remodeling (65, 160). The ERAs used in clinical medicine for PH include bosentan, ambrisentan, and macitentan.
Bosentan is a nonselective ERA and inhibits binding of ET-1 to both ETA and ETB receptors and has been shown to improve exercise capacity and pulmonary vascular hemodynamics in adults with PAH (112). Pharmacokinetics of bosentan in pediatric PAH and adult patients are similar with a ~50% oral bioavailability and half-life of 5.4 h (54). It is metabolized by the liver isoenzymes, CYP3A4 and 2C9, and is a potent CYP3A4 inducer. Thus, other drugs that are metabolized by CYP3A4 like sildenafil need to be dose adjusted when bosentan is coadministered (618). Bosentan also elevated liver enzymes and has been shown to cause cirrhosis with chronic use. Hence, monthly monitoring of liver enzymes is important while on bosentan (618). It has been listed by the FDA as an indication for children aged three years and older with IPAH/HPAH at a dose of 2 mg/kg twice a day (618). Small prospective cohort studies and retrospective reviews have found bosentan to improve the 6MWD, decrease mPAP and PVR in pediatric patients with IPAH (250, 371, 401, 493). Bosentan, in conjunction with or independent of other PAH-specific therapies, showed improved survival in children with IPAH at 1, 2, 3, and 4 years of 98%, 88%, 82%, and 82%, respectively (274). FUTURE-1 (pediatric formulation of bosentan in PAH), which enrolled 36 patients and followed 33 of them to the FUTURE-2 trial, showed that the pediatric bosentan formulation was well tolerated and its safety profile was comparable to that of the adult formulation when used in children (70). The FUTURE-1 trial had shown that dosing of 2 mg/kg twice a day versus 4 mg/kg twice a day yielded similar concentrations of bosentan in the plasma (64). A third pharmacokinetic study looked at 2 mg/kg three times daily versus twice daily dosing of bosentan in pediatric PAH and found no clinically relevant difference in exposure to bosentan or safety profile between the two, and hence current recommendations are to use 2 mg/kg twice daily (71). For older children, the dosing recommendations for bosentan are based on the BREATHE-3 trial: 31.25, 62.5, and 125 twice daily for 10 to 20, 20 to 40, and >40 kg, respectively (37, 54, 103). Bosentan was found to significantly improve the oxygenation index and decrease PAPs in newborns with PPHN compared to placebo without noticeable side effects in a single-center study done in a setting where iNO was not available (399). However, when bosentan was used as an adjunct in newborns with PPHN on iNO (FUTURE-4 trial), it did not improve oxygenation or other outcomes compared to placebo, and there was no difference in time to weaning from iNO or mechanical ventilation from both groups (562).
Ambrisentan is a selective ETA receptor antagonist, requires once-daily dosing, and has a half-life of around 9 h. Ambrisentan also does not affect liver enzymes; hence they do not need to be monitored as in bosentan therapy. Ambrisentan has been approved by the FDA for the treatment of adult PAH and in two multicenter adult trials (ARIES-1 and ARIES-2) was found to improve the 6MWD and delay clinical worsening (193). A retrospective study of ambrisentan use in children with PAH as an add-on therapy to or as a transition from bosentan found improved mPAP and WHO-FC in the cohort, with 13% of patients discontinuing ambrisentan due to severe headache, lack of efficacy, or near-syncopal events (580).
Macitentan is another nonselective ERA; however, it has greater affinity for ETA receptors. Long-term macitentan therapy in adult patients with PAH was associated with significant reductions in morbidity and mortality compared to placebo (SERAPHIN trial) (482, 555). A current multicenter, open-label, phase III trial to study the pharmacokinetics and long-term effects of macitentan in pediatric PAH is underway (167).
Soluble guanylate cyclase stimulators
This class of drugs acts along the NO-cGMP pathway and increases the intracellular concentration of cGMP in the smooth muscle cells, leading to the downstream cascade of smooth muscle relaxation. Adult patients with chronic thromboembolic PH when treated with riociguat showed improvements in exercise capacity and PVR (203, 541). Adults with PAH who were either treatment-naïve or were pretreated with ERAs or prostanoids when started on riociguat showed improvements in several clinically relevant endpoints, including WHO-FC and exercise capacity (204, 514). When a subpopulation of these adult PAH patients with CHD were analyzed, they were also found to display similar improvements in exercise capacity and WHO-FC, which were sustained at the two-year follow-up (505). A case report of a child with severe PAH with supra-systemic PVR who had failed treatment with amlodipine, bosentan, and sildenafil showed sustained improvement in PVR and RV function when switched to a bosentan/riociguat combination for off-label use (559). There are no other reports or human studies of the use of riociguat in the pediatric PH population.
Novel therapies
FK506
Germline mutations causing loss of BMPR2 function are present in >80% of HPAH and ~20% of IPAH patients (adult data), and the presence of BMPR2 mutations is associated with worse pulmonary vascular remodeling (147, 413, 591). In addition, patients with IPAH without a BMPR2 mutation or with PAH associated with other conditions have reduced expression of BMPR2 in pulmonary arteries (558). Low-dose FK506 (tacrolimus) has been identified as a potent activator of BMPR2 and was shown to reverse pulmonary arterial occlusive changes in animal models. In PAECs isolated from patients with IPAH, low-dose tacrolimus reversed dysfunctional BMPR2 signaling (558). A randomized, placebo-controlled trial of tacrolimus showed improvements in WHO-FC, hemodynamics, and increased BMPR2 expression in peripheral mononuclear cells (557). The improvements noted in this trial were not significant and were only observed in a subset of patients with PAH (558).
Fasudil
Rho-kinase (ROCK) activity has been associated with several animal models of PAH and was found to be increased in expression in lung and pulmonary arteries from patients with severe PH (153, 532). Fasudil is an intravenous ROCK inhibitor that competes with ATP for the ATP-binding site on ROCK, thereby blocking ROCK activity and myosin light chain phosphorylation, which leads to ultimate vasodilation (268, 512). It has been studied in China and Japan for the treatment of PAH, PAH-CHD as well as PAH due to left ventricular dysfunction in adult patients and has been shown to improve hemodynamics (189, 279, 513, 654). There have been no studies of fasudil in the pediatric PH population.
Endothelial progenitor cells
Bone-marrow-derived endothelial progenitor cells (EPCs) have been shown to regenerate pulmonary vascular endothelium and reverse the changes of PAH in animal models (656). A recent meta-analysis concluded that stem cells are useful in the treatment of PAH in preclinical models and further human studies need to be performed (152). A small pilot study performed in China demonstrated that autologous EPCs transfused into children with PAH led to significant improvements in PVR, mPAP, 6MWD, and cardiac output with no adverse events (658). Intrapulmonary artery injection of stem cells has also been shown to improve persistent PAH after surgical correction of cardiac defects in three patients (21). Larger studies are needed to delineate the potential benefit of this therapy in patients refractory to established treatment protocols.
Surgical interventions
Pediatric PAH patients with supra-systemic PVR, multiple syncopal episodes, poor WHO-FC who are on maximal combined pharmacological therapy are candidates for surgical interventions either as a therapeutic intervention or as a palliative bridge to lung transplantation. The two procedures performed are balloon atrial septostomy (BAS) and reversed Potts shunt, both of which convert the physiology from that of PAH with supra-systemic PVR and increased RV afterload to the one in Eisenmenger’s syndrome. Eisenmenger’s syndrome is seen in longstanding left-to-right shunting lesions in congenital or acquired cardiac disease, where there is gradual development of PAH and ultimately the shunt reverses to a right-to-left one (521). The long-term outcomes for children with severe PAH are very poor with five-year survival rates ranging between 57% and 75%, with lung transplantation being the only option for severe PAH refractory to combined pharmacological treatment. In comparison, patients with Eisenmenger’s syndrome have been reported to have superior long-term survival and transplant-free survival outcomes, and thus provided the concept of BAS and Potts shunt (151).
Atrial septostomy (AS)
AS is a percutaneous procedure by which an atrial communication is created via balloon dilation of the atrial septum and has been shown to improve symptoms and hemodynamics in patients refractory to vasodilator therapy (48, 337, 390). The atrial communication creates a right-to-left shunt to allow for decompression of the right heart with increased left ventricular preload and cardiac output with increased cyanosis, thus simulating Eisenmenger physiology (48). AS is considered either a palliative bridge to lung transplant in IPAH to increase survival while waiting for a donor organ or in patients with severe PH, WHO-FC III or IV and with recurrent syncope on combined medical therapy (235, 509). In resource-poor countries with limited access to PH drugs, it might be considered a therapeutic intervention but the long-term benefit of AS in the absence of an end-goal of lung transplant is unclear and should be weighed against the significant risks the procedure poses in pediatric patients with severe PAH. Data for outcomes of BAS mainly come from retrospective single-center studies with no randomized trials performed. One US center reported lung-transplantation free and repeat BAS-free survival at 30 days, 1 year, and 5 years to be 87%, 61%, and 32%, respectively (116). This data included both pediatric and adult patients (1-56 years) with a median age of 23 years and did not find any difference in serum biomarkers or hemodynamic findings pre-BAS and at 1 year or later follow-up. This finding was different from another group that reported improvements in hemodynamic parameters after BAS, but a majority of the patients in the second study were not on pharmacological PH treatment (522). Another US center reported their data on event-free survival at 1, 2, and 3 years of 84%, 77%, and 69%, respectively, with significant improvements in symptoms and hemodynamic parameters, in patients who survived beyond 30 days post intervention (337). One reason for such high survival rates from this study is because they did not include patients who died within the first 30 days, which was 22% of their initial cohort. The increased postprocedural mortality from BAS stems from the sudden severe right-to-left shunting, which might lead to life-threating hypoxemia and subsequent hypoxic pulmonary vasoconstriction and impaired cardiac output. The sizing of the defect in BAS is critical since too much right-to-left shunt at the atrial level could be immediately life threatening because of insufficient pulmonary blood flow as well as severe desaturation in the brain and in the coronary circulation, and too small of a shunt may require repeated procedures because of spontaneous closure of the defect (390). The current recommendations from the European PPVDN and the 6th WSPH Pediatric Task Force are to avoid BAS in the following group of patients: (i) mean right atrial pressure >20 mmHg, (ii) resting arterial oxygen saturation <90%, (iii) severe RV failure, and (iv) patients with impending death (235, 506).
Reversed Potts shunt
Reversed Potts shunt is like BAS in that it creates a right-to-left shunt pathway in patients with severe PH and thus converting them into Eisenmenger physiology. This is performed by a direct side-by-side anastomosis from the left pulmonary artery (LPA) to the descending aorta and was first described in the pediatric population by Blanc et al (59, 82). This helps to decrease the RV afterload and act as a palliative bridge to lung transplant in severe PH patients who are on maximal combined pharmacological therapy with poor WHO-FC class, similar to BAS. Data about effectiveness of the Potts shunt in improving pulmonary hemodynamics and transplant-free survival are limited and mostly from case series. In one series of pediatric PAH patients who underwent elective Potts shunt placement, 8 out of 12 patients survived for a median of 27 months post shunt with significant improvements in pulmonary hemodynamics and WHO-FC at follow-up (12). Another similar small series with a median age of 13.5 months at the time of shunt placement were followed for a median of 17 months post shunt. Among those who survived the initial period after the shunt, there was improvement in their WHO-FC as well as the clinical symptomatology of RV failure (219). The largest case series of pediatric PH patients who received a Potts shunt included 24 patients with a median age of 7.7 years. They all had drug-refractory PAH with supra-systemic PVR, except for one patient who was operated on due to multiple central-line associated infections while on intravenous epoprostenol. After a median follow-up of 2.1 years, this cohort showed significant improvements in their WHO-FC, 6MWD, serum BNP/NT-proBNP levels, syncopal events, and ability to wean pulmonary vasodilator therapy. One child in this series progressed to lung transplantation (59). There have also been reports on transcatheter creation of Potts shunt by stenting the patent ductus arteriosus (PDA) in pediatric IPAH patients, with similar outcomes as the surgical procedure (87, 88, 163). The major benefit of the Potts shunt over BAS is that the right-to-left shunt is created after the coronary and cerebral circulation are supplied by the oxygenated left ventricular output, thus avoiding myocardial and cerebral ischemia, with only the lower part of the body being cyanotic. The other benefit is that this directly offloads the RV both in systole and diastole, hence shifting the interventricular septum toward the RV and improving LV filling and cardiac output (12, 59, 163). This data is mostly anecdotal and has been challenged in the recent times using the CircAdapt model, which showed that the Potts shunt successfully transferred the supra-systemic PAH to an Eisenmenger physiology, but failed to decompress and offload the RV (146). There are no clinical trials comparing BAS to Potts shunt, and whether one is superior over the other is still unknown. Centers with higher volume and experience performing the Potts shunt and with extracorporeal life support backup for handling severe postoperative hypoxemia and low cardiac output states have better outcomes, especially as the initial postoperative period is associated with higher mortality.
Treatment Goals and Prognostic Tools for Monitoring and Follow Up
The identification of treatment goals is important as the US-FDA requires inclusion of a clinical endpoint for determining treatment efficacy of any drug or combination therapy. Important goals include death, transplantation and hospitalization, and the quality of the child’s life. Other goals like weight gain, serum biomarkers, echocardiographic signs, invasive hemodynamics, CMRI, and exercise testing can also be studied. Exercise tests and 6MWTs are difficult to perform in the pediatric population, and invasive hemodynamic data from cardiac catheterization solely for the purpose of follow-up are not pursued due to the risks associated with the procedure in the pediatric population (62). A small cohort study of pediatric IPAH/HPAH and PAH-CHD patients found pulmonary stroke volume, mean systemic arterial pressure, and heart rate were the strongest predictors of survival (158). Observational studies have shown that echocardiographic parameters correlate with meaningful outcomes in the pediatric PH population. Right and left ventricular dimensions, TAPSE, and right-to-left ventricular dimension ratios correlate with WHO-FC, hemodynamics, and survival (476). A meta-analysis in 2015 reported that WHO-FC, NT-proBNP, mean RA pressure, PVRi, cardiac index, and AVT have been consistently reported as prognostic factors for outcomes in pediatric PH (477). Composite clinical worsening has been used as an endpoint for adult PAH and was recently studied in a Dutch national cohort as well as by the TOPP registry. Two-year outcomes from the REVEAL registry showed that the soft clinical worsening endpoints were highly predictive of subsequent mortality (474). The Dutch cohort reported occurrences of hospitalization, initiation of intravenous prostanoids, or functional deterioration (defined as WHO-FC deterioration, >15% decrease in 6MWD or both) were individually predictive of death or lung transplantation, and a composite outcome of the three components was suggested as an endpoint for further study (474). Another Dutch cohort study for pediatric PAH reported WHO-FC, TAPSE, and NT-proBNP were predictors of transplant-free survival, and improvements in these variables were associated with improved survival. The TOPP registry investigators reported composite clinical worsening (cCW) outcomes comprising PAH-related hospitalization, atrial septostomy, WHO-FC deterioration, intravenous/subcutaneous prostanoid initiation, syncope and occurrence/worsening of ≥PAH symptoms were associated with a higher risk of transplantation/death (62). They created three different cCW models, all of which were associated with an increased risk of death or lung transplant for all PAH subtypes combined. However, when patients were separated based on etiology, for the PAH-CHD category none of the models or the individual components were associated with death and/or transplant.
Conclusion and Future Directions
In the last two decades, pediatric PH has been increasingly recognized as a separate entity with a different etiology and pathophysiology from adult PH. With the development of the Pediatric Task Force of the WSPH, TOPP registry, and PPH-Net, there has been increasing attention drawn to the pediatric-specific etiologies such as BPD-PH, PPHN, CDH-PH, and CHD-PAH. There are also an increasing number of off-label studies of drug regimens for pediatric PH. However, iNO is still the only drug approved by FDA for pediatric PH use. This is primarily due to the lack of randomized trials in the pediatric population and a lack of long-term safety data. The future goals of pediatric PH research should be focused on novel therapies for conditions unique to this age group and the conduct of well-designed, multicenter pediatric clinical trials of the drugs already being used in the adult patients. Identifying and validating composite clinical outcomes that can be reproduced across centers and different ages is key to developing clinical trials for monitoring treatment outcomes and defining goals and endpoints.
Didactic Synopsis.
Major Teaching Points
Pulmonary hypertension (PH) is often a hidden component, occurring by itself or in association with lung diseases, and requires echocardiography and/or cardiac catheterization for diagnosis.
Epidemiology and classification of pediatric pulmonary hypertension (PPH) has changed over time. Premature birth and bronchopulmonary dysplasia (BPD) are emerging as important causes of PPH; the risk is influenced by both prenatal and postnatal factors that adversely affect pulmonary vascular development.
Several developmental disorders of the lung contribute to pediatric PH and are unique to this age group. They include lung hypoplasia secondary to congenital diaphragmatic hernia and genetic conditions such as alveolar capillary dysplasia.
Congenital heart disease (CHD) is a major contributor to PPH; its presentation and clinical course are highly variable based on the type of CHD.
Understanding the molecular mechanisms and changes at cellular and structural level in PH is the key to developing future drug targets for PPH.
Developing diagnostic and treatment algorithms specific to PPH will help in identifying and properly classifying this component of PH.
Inhaled nitric oxide is the only approved drug for the treatment of neonatal PPHN, and bosentan is the only approved drug for the treatment of pediatric PAH in the United States. Other drugs that target nitric oxide-cyclic GMP pathway, endothelin receptors, and prostacyclin pathways are also effective in PPH and are currently being used off-label.
Future direction of PPH research needs to focus on developing novel drugs, other approaches besides vasodilation and on designing randomized clinical trials specifically in the pediatric population.
Acknowledgements
This work was supported by grant funding from the National Institutes of Health—RO1HL 136597-01, Pilot Innovative Research Grant from Children’s Research Institute, Milwaukee, WI, and Muma Endowed Chair in Neonatology (Girija Ganesh Konduri).
References
- 1.Abe K, Shimokawa H, Morikawa K, Uwatoku T, Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K, Takeshit A. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res 94: 385–393, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Abman SH. Bronchopulmonary dysplasia: “A vascular hypothesis”. Am J Respir Crit Care Med 164: 1755–1756, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Abman SH, Accurso FJ, Koops BL. Experience with home oxygen in the management of infants with bronchopulmonary dysplasia. Clin Pediatr (Phila) 23: 471–476, 1984. [DOI] [PubMed] [Google Scholar]
- 4.Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, Hanna BD, Rosenzweig EB, Raj JU, Cornfield D, Stenmark KR, Steinhorn R, Thebaud B, Fineman JR, Kuehne T, Feinstein JA, Friedberg MK, Earing M, Barst RJ, Keller RL, Kinsella JP, Mullen M, Deterding R, Kulik T, Mallory G, Humpl T, Wessel DL, American Heart Association Council on Cardiopulmonary CCP, Resuscitation, Council on Clinical C, Council on Cardiovascular Disease in the Y, Council on Cardiovascular R, Intervention, Council on Cardiovascular S, Anesthesia, and the American Thoracic S. Pediatric pulmonary hypertension: Guidelines from the American heart association and American thoracic society. Circulation 132: 2037–2099, 2015. [DOI] [PubMed] [Google Scholar]
- 5.Abman SH, Kinsella JP, Rosenzweig EB, Krishnan U, Kulik T, Mullen M, Wessel DL, Steinhorn R, Adatia I, Hanna B, Feinstein J, Fineman J, Raj U, Humpl T, Pediatric Pulmonary Hypertension N. Implications of the U.S. Food and Drug Administration warning against the use of sildenafil for the treatment of pediatric pulmonary hypertension. Am J Respir Crit Care Med 187: 572–575, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Abman SH, Wolfe RR, Accurso FJ, Koops BL, Bowman CM, Wiggins JW Jr. Pulmonary vascular response to oxygen in infants with severe bronchopulmonary dysplasia. Pediatrics 75: 80–84, 1985. [PubMed] [Google Scholar]
- 7.Abou Hassan OK, Haidar W, Arabi M, Skouri H, Bitar F, Nemer G, Akl IB. Novel EIF2AK4 mutations in histologically proven pulmonary capillary hemangiomatosis and hereditary pulmonary arterial hypertension. BMC Med Genet 20: 176, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abumehdi MR, Wardle AJ, Nazzal R, Charalampopoulos A, Schulze-Neick I, Derrick G, Moledina S, Giardini A. Feasibility and safety of cardiopulmonary exercise testing in children with pulmonary hypertension. Cardiol Young 26: 1144–1150, 2016. [DOI] [PubMed] [Google Scholar]
- 9.Acker SN, Seedorf GJ, Abman SH, Nozik-Grayck E, Partrick DA, Gien J. Pulmonary artery endothelial cell dysfunction and decreased populations of highly proliferative endothelial cells in experimental congenital diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 305: L943–L952, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, Li WW, Li VW, Mentzer SJ, Jonigk D. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 383: 120–128, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Afolayan AJ, Eis A, Alexander M, Michalkiewicz T, Teng RJ, Lakshminrusimha S, Konduri GG. Decreased endothelial nitric oxide synthase expression and function contribute to impaired mitochondrial biogenesis and oxidative stress in fetal lambs with persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 310: L40–L49, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aggarwal M, Grady RM, Choudhry S, Anwar S, Eghtesady P, Singh GK. Potts shunt improves right ventricular function and coupling with pulmonary circulation in children with suprasystemic pulmonary arterial hypertension. Circ Cardiovasc Imaging 11: e007964, 2018. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad KA, Banales J, Henderson CL, Ramos SE, Brandt KM, Powers GC. Intravenous epoprostenol improves oxygenation index in patients with persistent pulmonary hypertension of the newborn refractory to nitric oxide. J Perinatol 38: 1212–1219, 2018. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed MN, Suliman HB, Folz RJ, Nozik-Grayck E, Golson ML, Mason SN, Auten RL. Extracellular superoxide dismutase protects lung development in hyperoxia-exposed newborn mice. Am J Respir Crit Care Med 167: 400–405, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G, Galie N, Manes A, Corris P, Simonneau G, Humbert M, Morrell NW, Trembath RC. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat 27: 212–213, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Alfranca A, Iniguez MA, Fresno M, Redondo JM. Prostanoid signal transduction and gene expression in the endothelium: Role in cardiovascular diseases. Cardiovasc Res 70: 446–456, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Alkon J, Humpl T, Manlhiot C, McCrindle BW, Reyes JT, Friedberg MK. Usefulness of the right ventricular systolic to diastolic duration ratio to predict functional capacity and survival in children with pulmonary arterial hypertension. Am J Cardiol 106: 430–436, 2010. [DOI] [PubMed] [Google Scholar]
- 18.Alvira CM. Aberrant pulmonary vascular growth and remodeling in bronchopulmonary dysplasia. Front Med (Lausanne) 3: 21, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ambalavanan N, Bulger A, Murphy-Ullrich J, Oparil S, Chen YF. Endothelin-A receptor blockade prevents and partially reverses neonatal hypoxic pulmonary vascular remodeling. Pediatr Res 57: 631–636, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ambalavanan N, Mariani G, Bulger A, Philips IJ. Role of nitric oxide in regulating neonatal porcine pulmonary artery smooth muscle cell proliferation. Biol Neonate 76: 291–300, 1999. [DOI] [PubMed] [Google Scholar]
- 21.Amoozgar H, Banafi P, Mohammadi H, Edraki MR, Mehdizadegan N, Ajami G, Borzouee M, Keshaarz K, Moradi P, Dehghani E. Management of persistent pulmonary hypertension after correction of congenital heart defect with autologous marrow-derived mononuclear stem cell injection into the pulmonary artery: A pilot study. Pediatr Cardiol 41: 398–406, 2020. [DOI] [PubMed] [Google Scholar]
- 22.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev 22: 1276–1312, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andruska A, Spiekerkoetter E. Consequences of BMPR2 deficiency in the pulmonary vasculature and beyond: Contributions to pulmonary arterial hypertension. Int J Mol Sci 19: 2499, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.SA Antoniu. Targeting PDGF pathway in pulmonary arterial hypertension. Expert Opin Ther Targets 16: 1055–1063, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Apitz C, Hansmann G, Schranz D. Hemodynamic assessment and acute pulmonary vasoreactivity testing in the evaluation of children with pulmonary vascular disease. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 102 (Suppl 2): ii23–ii29, 2016. [DOI] [PubMed] [Google Scholar]
- 26.Archer S, Rich S. Primary pulmonary hypertension: A vascular biology and translational research “Work in progress”. Circulation 102: 2781–2791, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Arcot SS, Lipke DW, Gillespie MN, Olson JW. Alterations of growth factor transcripts in rat lungs during development of monocrotaline-induced pulmonary hypertension. Biochem Pharmacol 46: 1086–1091, 1993. [DOI] [PubMed] [Google Scholar]
- 28.Arjaans S, Wagner BD, Mourani PM, Mandell EW, Poindexter BB, Berger RMF, Abman SH. Early angiogenic proteins associated with high risk for bronchopulmonary dysplasia and pulmonary hypertension in preterm infants. Am J Physiol Lung Cell Mol Physiol 318: L644–L654, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arjaans S, Zwart EAH, Ploegstra MJ, Bos AF, Kooi EMW, Hillege HL, Berger RMF. Identification of gaps in the current knowledge on pulmonary hypertension in extremely preterm infants: A systematic review and meta-analysis. Paediatr Perinat Epidemiol 32: 258–267, 2018. [DOI] [PubMed] [Google Scholar]
- 30.Askie LM, Darlow BA, Finer N, Schmidt B, Stenson B, Tarnow-Mordi W, Davis PG, Carlo WA, Brocklehurst P, Davies LC, Das A, Rich W, Gantz MG, Roberts RS, Whyte RK, Costantini L, Poets C, Asztalos E, Battin M, Halliday HL, Marlow N, Tin W, King A, Juszczak E, Morley CJ, Doyle LW, Gebski V, Hunter KE, Simes RJ, Neonatal Oxygenation Prospective Meta-analysis C. Association between oxygen saturation targeting and death or disability in extremely preterm infants in the neonatal oxygenation prospective meta-analysis collaboration. JAMA 319: 2190–2201, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Assad TR, Hemnes AR, Larkin EK, Glazer AM, Xu M, Wells QS, Farber-Eger EH, Sheng Q, Shyr Y, Harrell FE, Newman JH, Brittain EL. Clinical and biological insights Into combined post- and precapillary pulmonary hypertension. J Am Coll Cardiol 68: 2525–2536, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 105: 1672–1678, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Atz AM, Adatia I, Lock JE, Wessel DL. Combined effects of nitric oxide and oxygen during acute pulmonary vasodilator testing. J Am Coll Cardiol 33: 813–819, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Atz AM, Lefler AK, Fairbrother DL, Uber WE, Bradley SM. Sildenafil augments the effect of inhaled nitric oxide for postoperative pulmonary hypertensive crises. J Thorac Cardiovasc Surg 124: 628–629, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res 115: 189–202, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Australia B-I, >United Kingdom Collaborative G, Tarnow-Mordi W, Stenson B, Kirby A, Juszczak E, Donoghoe M, Deshpande S, Morley C, King A, Doyle LW, Fleck BW, Davis PG, Halliday HL, Hague W, Cairns P, Darlow BA, Fielder AR, Gebski V, Marlow N, Simmer K, Tin W, Ghadge A, Williams C, Keech A, Wardle SP, Kecskes Z, Kluckow M, Gole G, Evans N, Malcolm G, Luig M, Wright I, Stack J, Tan K, Pritchard M, Gray PH, Morris S, Headley B, Dargaville P, Simes RJ, Brocklehurst P. Outcomes of two trials of oxygen-saturation targets in preterm infants. N Engl J Med 374: 749–760, 2016. [DOI] [PubMed] [Google Scholar]
- 37.Avitabile CM, Vorhies EE, Ivy DD. Drug treatment of pulmonary hypertension in children. Paediatr Drugs 22: 123–147, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, McGoon M, Naeije R, Olschewski H, Oudiz RJ, Torbicki A. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 54: S55–S66, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Baker CD, Abman SH. Impaired pulmonary vascular development in bronchopulmonary dysplasia. Neonatology 107: 344–351, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker CD, Abman SH, Mourani PM. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Pediatr Allergy Immunol Pulmonol 27: 8–16, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balasubramaniam V, Le Cras TD, Ivy DD, Grover TR, Kinsella JP, Abman SH. Role of platelet-derived growth factor in vascular remodeling during pulmonary hypertension in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 284: L826–L833, 2003. [DOI] [PubMed] [Google Scholar]
- 42.Ballard RA, Truog WE, Cnaan A, Martin RJ, Ballard PL, Merrill JD, Walsh MC, Durand DJ, Mayock DE, Eichenwald EC, Null DR, Hudak ML, Puri AR, Golombek SG, Courtney SE, Stewart DL, Welty SE, Phibbs RH, Hibbs AM, Luan X, Wadlinger SR, Asselin JM, Coburn CE, Group NCS. Inhaled nitric oxide in preterm infants undergoing mechanical ventilation. N Engl J Med 355: 343–353, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Baquero H, Soliz A, Neira F, Venegas ME, Sola A. Oral sildenafil in infants with persistent pulmonary hypertension of the newborn: A pilot randomized blinded study. Pediatrics 117: 1077–1083, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Barefield ES, Karle VA, Phillips JB 3rd, Carlo WA. Inhaled nitric oxide in term infants with hypoxemic respiratory failure. J Pediatr 129: 279–286, 1996. [DOI] [PubMed] [Google Scholar]
- 45.Barnett CF, Hsue PY, Machado RF. Pulmonary hypertension: An increasingly recognized complication of hereditary hemolytic anemias and HIV infection. JAMA 299: 324–331, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Barrington KJ, Finer N, Pennaforte T. Inhaled nitric oxide for respiratory failure in preterm infants. Cochrane Database Syst Rev 1: CD000509, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barrington KJ, Finer N, Pennaforte T, Altit G. Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst Rev 1: CD000399, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barst RJ. Role of atrial septostomy in the treatment of pulmonary vascular disease. Thorax 55: 95–96, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barst RJ, Agnoletti G, Fraisse A, Baldassarre J, Wessel DL, Group NODS. Vasodilator testing with nitric oxide and/or oxygen in pediatric pulmonary hypertension. Pediatr Cardiol 31: 598–606, 2010. [DOI] [PubMed] [Google Scholar]
- 50.Barst RJ, Beghetti M, Pulido T, Layton G, Konourina I, Zhang M, Ivy DD, Investigators S. STARTS-2: Long-term survival with oral sildenafil monotherapy in treatment-naive pediatric pulmonary arterial hypertension. Circulation 129: 1914–1923, 2014. [DOI] [PubMed] [Google Scholar]
- 51.Barst RJ, Channick R, Ivy D, Goldstein B. Clinical perspectives with long-term pulsed inhaled nitric oxide for the treatment of pulmonary arterial hypertension. Pulm Circ 2: 139–147, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barst RJ, Galie N, Naeije R, Simonneau G, Jeffs R, Arneson C, Rubin LJ. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J 28: 1195–1203, 2006. [DOI] [PubMed] [Google Scholar]
- 53.Barst RJ, Gibbs JS, Ghofrani HA, Hoeper MM, McLaughlin VV, Rubin LJ, Sitbon O, Tapson VF, Galie N. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 54: S78–S84, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barst RJ, Ivy D, Dingemanse J, Widlitz A, Schmitt K, Doran A, Bingaman D, Nguyen N, Gaitonde M, van Giersbergen PL. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther 73: 372–382, 2003. [DOI] [PubMed] [Google Scholar]
- 55.Barst RJ, Ivy DD, Gaitan G, Szatmari A, Rudzinski A, Garcia AE, Sastry BK, Pulido T, Layton GR, Serdarevic-Pehar M, Wessel DL. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation 125: 324–334, 2012. [DOI] [PubMed] [Google Scholar]
- 56.Barst RJ, Maislin G, Fishman AP. Vasodilator therapy for primary pulmonary hypertension in children. Circulation 99: 1197–1208, 1999. [DOI] [PubMed] [Google Scholar]
- 57.Barst RJ, McGoon M, McLaughlin V, Tapson V, Rich S, Rubin L, Wasserman K, Oudiz R, Shapiro S, Robbins IM, Channick R, Badesch D, Rayburn BK, Flinchbaugh R, Sigman J, Arneson C, Jeffs R, Beraprost Study G. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol 41: 2119–2125, 2003. [DOI] [PubMed] [Google Scholar]
- 58.Barst RJ, McGoon MD, Elliott CG, Foreman AJ, Miller DP, Ivy DD. Survival in childhood pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 125: 113–122, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Baruteau AE, Belli E, Boudjemline Y, Laux D, Levy M, Simonneau G, Carotti A, Humbert M, Bonnet D. Palliative Potts shunt for the treatment of children with drug-refractory pulmonary arterial hypertension: Updated data from the first 24 patients. Eur J Cardiothorac Surg 47: e105–e110, 2015. [DOI] [PubMed] [Google Scholar]
- 60.Bauer NR, Moore TM, McMurtry IF. Rodent models of PAH: Are we there yet? Am J Physiol Lung Cell Mol Physiol 293: L580–L582, 2007. [DOI] [PubMed] [Google Scholar]
- 61.Beghetti M, Berger RM, Schulze-Neick I, Day RW, Pulido T, Feinstein J, Barst RJ, Humpl T, Investigators TR. Diagnostic evaluation of paediatric pulmonary hypertension in current clinical practice. Eur Respir J 42: 689–700, 2013. [DOI] [PubMed] [Google Scholar]
- 62.Beghetti M, Brand M, Berger RMF, Humpl T, Wheeler JG, Ivy DD, Bonnet D, investigators T. Meaningful and feasible composite clinical worsening definitions in paediatric pulmonary arterial hypertension: An analysis of the TOPP registry. Int J Cardiol 289: 110–115, 2019. [DOI] [PubMed] [Google Scholar]
- 63.Beghetti M, Habre W, Friedli B, Berner M. Continuous low dose inhaled nitric oxide for treatment of severe pulmonary hypertension after cardiac surgery in paediatric patients. Br Heart J 73: 65–68, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beghetti M, Haworth SG, Bonnet D, Barst RJ, Acar P, Fraisse A, Ivy DD, Jais X, Schulze-Neick I, Galie N, Morganti A, Dingemanse J, Kusic-Pajic A, Berger RM. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: The FUTURE-1 study. Br J Clin Pharmacol 68: 948–955, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benigni A, Remuzzi G. Endothelin antagonists. Lancet 353: 133–138, 1999. [DOI] [PubMed] [Google Scholar]
- 66.Benisty JI, McLaughlin VV, Landzberg MJ, Rich JD, Newburger JW, Rich S, Folkman J. Elevated basic fibroblast growth factor levels in patients with pulmonary arterial hypertension. Chest 126: 1255–1261, 2004. [DOI] [PubMed] [Google Scholar]
- 67.Benza R, Biederman R, Murali S, Gupta H. Role of cardiac magnetic resonance imaging in the management of patients with pulmonary arterial hypertension. J Am Coll Cardiol 52: 1683–1692, 2008. [DOI] [PubMed] [Google Scholar]
- 68.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 122: 164–172, 2010. [DOI] [PubMed] [Google Scholar]
- 69.Berger RM, Beghetti M, Humpl T, Raskob GE, Ivy DD, Jing ZC, Bonnet D, Schulze-Neick I, Barst RJ. Clinical features of paediatric pulmonary hypertension: A registry study. Lancet 379: 537–546, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berger RM, Haworth SG, Bonnet D, Dulac Y, Fraisse A, Galie N, Ivy DD, Jais X, Miera O, Rosenzweig EB, Efficace M, Kusic-Pajic A, Beghetti M. FUTURE-2: Results from an open-label, long-term safety and tolerability extension study using the pediatric FormUlation of bosenTan in pUlmonary arterial hypeRtEnsion. Int J Cardiol 202: 52–58, 2016. [DOI] [PubMed] [Google Scholar]
- 71.Berger RMF, Gehin M, Beghetti M, Ivy D, Kusic-Pajic A, Cornelisse P, Grill S, Bonnet D, investigators F. A bosentan pharmacokinetic study to investigate dosing regimens in paediatric patients with pulmonary arterial hypertension: FUTURE-3. Br J Clin Pharmacol 83: 1734–1744, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berger-Caron F, Piedboeuf B, Morissette G, Simonyan D, Chetaille P, Pellerin A, Hebert A. Inhaled epoprostenol for pulmonary hypertension treatment in neonates: A 12-year experience. Am J Perinatol 36: 1142–1149, 2019. [DOI] [PubMed] [Google Scholar]
- 73.Berk BC. Vascular smooth muscle growth: Autocrine growth mechanisms. Physiol Rev 81: 999–1030, 2001. [DOI] [PubMed] [Google Scholar]
- 74.Berkelhamer SK, Kim GA, Radder JE, Wedgwood S, Czech L, Steinhorn RH, Schumacker PT. Developmental differences in hyperoxia-induced oxidative stress and cellular responses in the murine lung. Free Radic Biol Med 61: 51–60, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berkelhamer SK, Mestan KK, Steinhorn R. An update on the diagnosis and management of bronchopulmonary dysplasia (BPD)-associated pulmonary hypertension. Semin Perinatol 42: 432–443, 2018. [DOI] [PubMed] [Google Scholar]
- 76.Berman EB, Barst RJ. Eisenmenger’s syndrome: Current management. Prog Cardiovasc Dis 45: 129–138, 2002. [DOI] [PubMed] [Google Scholar]
- 77.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang YY, Rehman S, Sugahara M, Qi Z, Gorcsan J 3rd, Vargas SO, Saggar R, Saggar R, Wallace WD, Ross DJ, Haley KJ, Waxman AB, Parikh VN, De Marco T, Hsue PY, Morris A, Simon MA, Norris KA, Gaggioli C, Loscalzo J, Fessel J, Chan SY. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 126: 3313–3335, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Best DH, Sumner KL, Austin ED, Chung WK, Brown LM, Borczuk AC, Rosenzweig EB, Bayrak-Toydemir P, Mao R, Cahill BC, Tazelaar HD, Leslie KO, Hemnes AR, Robbins IM, Elliott CG. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 145: 231–236, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med 164: 1971–1980, 2001. [DOI] [PubMed] [Google Scholar]
- 80.Bijli KM, Kleinhenz JM, Murphy TC, Kang BY, Adesina SE, Sutliff RL, Hart CM. Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation. Free Radic Biol Med 80: 111–120, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bishop NB, Stankiewicz P, Steinhorn RH. Alveolar capillary dysplasia. Am J Respir Crit Care Med 184: 172–179, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blanc J, Vouhe P, Bonnet D. Potts shunt in patients with pulmonary hypertension. N Engl J Med 350: 623, 2004. [DOI] [PubMed] [Google Scholar]
- 83.Blyth KG, Groenning BA, Mark PB, Martin TN, Foster JE, Steedman T, Morton JJ, Dargie HJ, Peacock AJ. NT-proBNP can be used to detect right ventricular systolic dysfunction in pulmonary hypertension. Eur Respir J 29: 737–744, 2007. [DOI] [PubMed] [Google Scholar]
- 84.Bobhate P, Guo L, Jain S, Haugen R, Coe JY, Cave D, Rutledge J, Adatia I. Cardiac catheterization in children with pulmonary hypertensive vascular disease. Pediatr Cardiol 36: 873–879, 2015. [DOI] [PubMed] [Google Scholar]
- 85.Boehm M, Arnold N, Braithwaite A, Pickworth J, Lu C, Novoyatleva T, Kiely DG, Grimminger F, Ghofrani HA, Weissmann N, Seeger W, Lawrie A, Schermuly RT, Kojonazarov B. Eplerenone attenuates pathological pulmonary vascular rather than right ventricular remodeling in pulmonary arterial hypertension. BMC Pulm Med 18:41, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 135: 794–804, 2009. [DOI] [PubMed] [Google Scholar]
- 87.Boudjemline Y, Patel M, Malekzadeh-Milani S, Szezepanski I, Levy M, Bonnet D. Patent ductus arteriosus stenting (transcatheter Potts shunt) for palliation of suprasystemic pulmonary arterial hypertension: A case series. Circ Cardiovasc Interv 6: e18–e20, 2013. [DOI] [PubMed] [Google Scholar]
- 88.Boudjemline Y, Sizarov A, Malekzadeh-Milani S, Mirabile C, Lenoir M, Khraiche D, Levy M, Bonnet D. Safety and feasibility of the transcatheter approach to create a reverse potts shunt in children with idiopathic pulmonary arterial hypertension. Can J Cardiol 33: 1188–1196, 2017. [DOI] [PubMed] [Google Scholar]
- 89.Bournia VK, Tsangaris I, Rallidis L, Konstantonis D, Frantzeskaki F, Anthi A, Orfanos SE, Demerouti E, Karyofillis P, Voudris V, Laskari K, Panopoulos S, Vlachoyiannopoulos PG, Sfikakis PP. Cardiac catheterization versus echocardiography for monitoring pulmonary pressure: A prospective study in patients with connective tissue disease-associated pulmonary arterial hypertension. Diagnostics (Basel) 10: 49, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bradlow WM, Assomull R, Kilner PJ, Gibbs JS, Sheppard MN, Mohiaddin RH. Understanding late gadolinium enhancement in pulmonary hypertension. Circ Cardiovasc Imaging 3: 501–503, 2010. [DOI] [PubMed] [Google Scholar]
- 91.Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: A role for NADPH oxidase. Circ Res 92: 683–691, 2003. [DOI] [PubMed] [Google Scholar]
- 92.Breysem L, Smet MH, Van Lierde S, Devlieger H, De Boeck K. Bronchopulmonary dysplasia: Correlation of radiographic and clinical findings. Pediatr Radiol 27: 642–646, 1997. [DOI] [PubMed] [Google Scholar]
- 93.Bronicki RA, Baden HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med 11: S15–S22, 2010. [DOI] [PubMed] [Google Scholar]
- 94.Broughton BR, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 294: L797–L806, 2008. [DOI] [PubMed] [Google Scholar]
- 95.Bull TM, Coldren CD, Geraci MW, Voelkel NF. Gene expression profiling in pulmonary hypertension. Proc Am Thorac Soc 4: 117–120, 2007. [DOI] [PubMed] [Google Scholar]
- 96.Burg ED, Remillard CV, Yuan JX. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br J Pharmacol 153 (Suppl 1): S99–S111, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burke DL, Frid MG, Kunrath CL, Karoor V, Anwar A, Wagner BD, Strassheim D, Stenmark KR. Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol 297: L238–L250, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burkett DA, Slorach C, Patel SS, Redington AN, Ivy DD, Mertens L, Younoszai AK, Friedberg MK. Impact of pulmonary hemodynamics and ventricular interdependence on left ventricular diastolic function in children with pulmonary hypertension. Circ Cardiovasc Imaging 9, 2016. DOI: 10.1161/aRCIMAGING.116.004612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bush A, Busst CM, Knight WB, Hislop AA, Haworth SG, Shinebourne EA. Changes in pulmonary circulation in severe bronchopulmonary dysplasia. Arch Dis Child 65: 739–745, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Campbell BT, Herbst KW, Briden KE, Neff S, Ruscher KA, Hagadorn JI. Inhaled nitric oxide use in neonates with congenital diaphragmatic hernia. Pediatrics 134: e420–e426, 2014. [DOI] [PubMed] [Google Scholar]
- 101.Caravita S, Baratto C, Di Marco F, Calabrese A, Balestrieri G, Russo F, Faini A, Soranna D, Perego GB, Badano LP, Grazioli L, Lorini FL, Parati G, Senni M. Hemodynamic characteristics of COVID-19 patients with acute respiratory distress syndrome requiring mechanical ventilation. An invasive assessment using right heart catheterization. Eur J Heart Fail 22 (12): 2228–2237, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carmosino MJ, Friesen RH, Doran A, Ivy DD. Perioperative complications in children with pulmonary hypertension undergoing noncardiac surgery or cardiac catheterization. Anesth Analg 104: 521–527, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carter NJ, Keating GM. Bosentan: In pediatric patients with pulmonary arterial hypertension. Paediatr Drugs 12: 63–73, 2010. [DOI] [PubMed] [Google Scholar]
- 104.Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R, Denby L, Dempsie Y, Long L, Morrell NW, Baker AH. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol 30: 716–723, 2010. [DOI] [PubMed] [Google Scholar]
- 105.Carvajal JA, Germain AM, Huidobro-Toro JP, Weiner CP. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J Cell Physiol 184: 409–420, 2000. [DOI] [PubMed] [Google Scholar]
- 106.Cerro MJ, Abman S, Diaz G, Freudenthal AH, Freudenthal F, Harikrishnan S, Haworth SG, Ivy D, Lopes AA, Raj JU, Sandoval J, Stenmark K, Adatia I. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 1: 286–298, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cevik A, Kula S, Olgunturk R, Tunaoglu FS, Oguz AD, Saylan B, Cilsal E, Sanli C. Assessment of pulmonary arterial hypertension and vascular resistance by measurements of the pulmonary arterial flow velocity curve in the absence of a measurable tricuspid regurgitant velocity in childhood congenital heart disease. Pediatr Cardiol 34: 646–655, 2013. [DOI] [PubMed] [Google Scholar]
- 108.Chandrasekar I, Eis A, Konduri GG. Betamethasone attenuates oxidant stress in endothelial cells from fetal lambs with persistent pulmonary hypertension. Pediatr Res 63: 67–72, 2008. [DOI] [PubMed] [Google Scholar]
- 109.Chandrasekharan P, Lakshminrusimha S. Oxygen therapy in preterm infants with pulmonary hypertension. Semin Fetal Neonatal Med 25: 101070, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chandrasekharan P, Rawat M, Gugino SF, Koenigsknecht C, Helman J, Nair J, Vali P, Lakshminrusimha S. Effect of various inspired oxygen concentrations on pulmonary and systemic hemodynamics and oxygenation during resuscitation in a transitioning preterm model. Pediatr Res 84: 743–750, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chang AC, Zucker HA, Hickey PR, Wessel DL. Pulmonary vascular resistance in infants after cardiac surgery: Role of carbon dioxide and hydrogen ion. Crit Care Med 23: 568–574, 1995. [DOI] [PubMed] [Google Scholar]
- 112.Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, Badesch DB, Roux S, Rainisio M, Bodin F, Rubin LJ. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: A randomised placebo-controlled study. Lancet 358: 1119–1123, 2001. [DOI] [PubMed] [Google Scholar]
- 113.Checchia PA, Bronicki RA, Muenzer JT, Dixon D, Raithel S, Gandhi SK, Huddleston CB. Nitric oxide delivery during cardiopulmonary bypass reduces postoperative morbidity in children—a randomized trial. J Thorac Cardiovasc Surg 146: 530–536, 2013. [DOI] [PubMed] [Google Scholar]
- 114.Check J, Gotteiner N, Liu X, Su E, Porta N, Steinhorn R, Mestan KK. Fetal growth restriction and pulmonary hypertension in premature infants with bronchopulmonary dysplasia. J Perinatol 33: 553–557, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheng Y, Tung CK, Chung AKK, Liu WW, Huang D, Chan PH, Lam M, Chan WC, Siu CW, Hai JJ. Screening of pulmonary hypertension in methamphetamine abusers (SOPHMA): Rationale and design of a multicentre, cross-sectional study. BMJ Open 9: e027193, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chiu JS, Zuckerman WA, Turner ME, Richmond ME, Kerstein D, Krishnan U, Torres A, Vincent JA, Rosenzweig EB. Balloon atrial septostomy in pulmonary arterial hypertension: Effect on survival and associated outcomes. J Heart Lung Transplant 34: 376–380, 2015. [DOI] [PubMed] [Google Scholar]
- 117.Chiu PP. New insights into congenital diaphragmatic hernia—A surgeon’s introduction to CDH animal models. Front Pediatr 2: 36, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cho YJ, Han JY, Lee SG, Jeon BT, Choi WS, Hwang YS, Roh GS, Lee JD. Temporal changes of angiopoietins and Tie2 expression in rat lungs after monocrotaline-induced pulmonary hypertension. Comp Med 59: 350–356, 2009. [PMC free article] [PubMed] [Google Scholar]
- 119.Christou H, Yoshida A, Arthur V, Morita T, Kourembanas S. Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 18: 768–776, 1998. [DOI] [PubMed] [Google Scholar]
- 120.Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, Roy BJ, Keszler M, Kinsella JP. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. N Engl J Med 342: 469–474, 2000. [DOI] [PubMed] [Google Scholar]
- 121.Clozel M, Breu V, Gray GA, Loffler BM. In vivo pharmacology of Ro 46-2005, the first synthetic nonpeptide endothelin receptor antagonist: Implications for endothelin physiology. J Cardiovasc Pharmacol 22 (Suppl 8): S377–S379, 1993. [DOI] [PubMed] [Google Scholar]
- 122.Cochius-den Otter S, Schaible T, Greenough A, van Heijst A, Patel N, Allegaert K, van Rosmalen J, Tibboel D, Consortium CE. The CoDiNOS trial protocol: An international randomised controlled trial of intravenous sildenafil versus inhaled nitric oxide for the treatment of pulmonary hypertension in neonates with congenital diaphragmatic hernia. BMJ Open 9: e032122, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cohen JL, Nees SN, Valencia GA, Rosenzweig EB, Krishnan US. Sildenafil use in children with pulmonary hypertension. J Pediatr 205: 29–34 e21, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Colvin kL, Dufva MJ, Delaney RP, Ivy DD, Stenmark KR, Yeager ME. Biomarkers for pediatric pulmonary arterial hypertension - A call to collaborate. Front Pediatr 2: 7, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Coppola CP, Gosche JR. Oxygen-induced vasodilation is blunted in pulmonary arterioles from fetal rats with nitrofen-induced congenital diaphragmatic hernia. J Pediatr Surg 36: 593–597, 2001. [DOI] [PubMed] [Google Scholar]
- 126.Cornfield DN, Maynard RC, deRegnier RA, Guiang SF 3rd, Barbato JE, Milla CE. Randomized, controlled trial of low-dose inhaled nitric oxide in the treatment of term and near-term infants with respiratory failure and pulmonary hypertension. Pediatrics 104: 1089–1094, 1999. [DOI] [PubMed] [Google Scholar]
- 127.Corsico AG, D’Armini AM, Cerveri I, Klersy C, Ansaldo E, Niniano R, Gatto E, Monterosso C, Morsolini M, Nicolardi S, Tramontin C, Pozzi E, Vigano M. Long-term outcome after pulmonary endarterectomy. Am J Respir Crit Care Med 178: 419–424, 2008. [DOI] [PubMed] [Google Scholar]
- 128.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med 6: 698–702, 2000. [DOI] [PubMed] [Google Scholar]
- 129.Cracowski JL, Cracowski C, Bessard G, Pepin JL, Bessard J, Schwebel C, Stanke-Labesque F, Pison C. Increased lipid peroxidation in patients with pulmonary hypertension. Am J Respir Crit Care Med 164: 1038–1042, 2001. [DOI] [PubMed] [Google Scholar]
- 130.Critser PJ, Higano NS, Tkach JA, Olson ES, Spielberg DR, Kingma PS, Fleck RJ, Lang SM, Moore RA, Taylor MD, Woods JC. Cardiac magnetic resonance imaging evaluation of neonatal bronchopulmonary dysplasia-associated pulmonary hypertension. Am J Respir Crit Care Med 201: 73–82, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Crossno JT Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol 292: L885–L897, 2007. [DOI] [PubMed] [Google Scholar]
- 132.Daftari B, Alejos JC, Perens G. Initial experience with sildenafil, bosentan, and nitric oxide for pediatric cardiomyopathy patients with elevated pulmonary vascular resistance before and after orthotopic heart transplantation. J Transplant 2010: 656984, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dani C, Bertini G, Pezzati M, Filippi L, Cecchi A, Rubaltelli FF. Inhaled nitric oxide in very preterm infants with severe respiratory distress syndrome. Acta Paediatr 95: 1116–1123, 2006. [DOI] [PubMed] [Google Scholar]
- 134.Darlow BA, Marschner SL, Donoghoe M, Battin MR, Broadbent RS, Elder MJ, Hewson MP, Meyer MP, Ghadge A, Graham P, McNeill NJ, Kuschel CA, Tarnow-Mordi WO, Benefits of Oxygen Saturation Targeting-New Zealand Collaborative Group. Randomized controlled trial of oxygen saturation targets in very preterm infants: Two year outcomes. J Pediatr 165: 30–35 e32, 2014. [DOI] [PubMed] [Google Scholar]
- 135.Davidson D, Barefield ES, Kattwinkel J, Dudell G, Damask M, Straube R, Rhines J, Chang CT. Inhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: A randomized, double-masked, placebo-controlled, dose-response, multicenter study. The I-NO/PPHN Study Group. Pediatrics 101: 325–334, 1998. [DOI] [PubMed] [Google Scholar]
- 136.Davidson D, Barefield ES, Kattwinkel J, Dudell G, Damask M, Straube R, Rhines J, Chang CT. Safety of withdrawing inhaled nitric oxide therapy in persistent pulmonary hypertension of the newborn. Pediatrics 104: 231–236, 1999. [DOI] [PubMed] [Google Scholar]
- 137.Day RW, Lynch JM, White KS, Ward RM. Acute response to inhaled nitric oxide in newborns with respiratory failure and pulmonary hypertension. Pediatrics 98: 698–705, 1996. [PubMed] [Google Scholar]
- 138.de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, Francois C, Schalij I, Dorfmuller P, Simonneau G, Fadel E, Perros F, Boonstra A, Postmus PE, van der Velden J, Vonk-Noordegraaf A, Humbert M, Eddahibi S, Guignabert C. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 780–789, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.De Paepe ME, Mao Q, Powell J, Rubin SE, DeKoninck P, Appel N, Dixon M, Gundogan F. Growth of pulmonary microvasculature in ventilated preterm infants. Am J Respir Crit Care Med 173: 204–211, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Del Cerro MJ, Moledina S, Haworth SG, Ivy D, Al Dabbagh M, Banjar H, Diaz G, Heath-Freudenthal A, Galal AN, Humpl T, Kulkarni S, Lopes A, Mocumbi AO, Puri GD, Rossouw B, Harikrishnan S, Saxena A, Udo P, Caicedo L, Tamimi O, Adatia I. Cardiac catheterization in children with pulmonary hypertensive vascular disease: Consensus statement from the Pulmonary Vascular Research Institute. Pediatric and Congenital Heart Disease Task Forces. Pulm Circ 6: 118–125, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.del Cerro MJ, Sabate Rotes A, Carton A, Deiros L, Bret M, Cordeiro M, Verdu C, Barrios MI, Albajara L, Gutierrez-Larraya F. Pulmonary hypertension in bronchopulmonary dysplasia: Clinical findings, cardiovascular anomalies and outcomes. Pediatr Pulmonol 49: 49–59, 2014. [DOI] [PubMed] [Google Scholar]
- 142.del Cerro Marin MJ, Sabate Rotes A, Rodriguez Ogando A, Mendoza Soto A, Quero Jimenez M, Gavilan Camacho JL, Raposo Sonnenfeld I, Moya Bonora A, Albert Brotons DC, Moreno Galdo A, Investigators R. Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish registry. Am J Respir Crit Care Med 190: 1421–1429, 2014. [DOI] [PubMed] [Google Scholar]
- 143.Delaney C, Cornfield DN. Risk factors for persistent pulmonary hypertension of the newborn. Pulm Circ 2: 15–20, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Delaney C, Gien J, Grover TR, Roe G, Abman SH. Pulmonary vascular effects of serotonin and selective serotonin reuptake inhibitors in the late-gestation ovine fetus. Am J Physiol Lung Cell Mol Physiol 301: L937–L944, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Delaney C, Gien J, Roe G, Isenberg N, Kailey J, Abman SH. Serotonin contributes to high pulmonary vascular tone in a sheep model of persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 304: L894–L901, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Delhaas T, Koeken Y, Latus H, Apitz C, Schranz D. Potts shunt to be preferred above atrial septostomy in pediatric pulmonary arterial hypertension patients: A modeling study. Front Physiol 9: 1252, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67: 737–744, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L596–L607, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dhariwal AK, Bavdekar SB. Sildenafil in pediatric pulmonary arterial hypertension. J Postgrad Med 61: 181–192, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dhawan RT, Gopalan D, Howard L, Vicente A, Park M, Manalan K, Wallner I, Marsden P, Dave S, Branley H, Russell G, Dharmarajah N, Kon OM. Beyond the clot: Perfusion imaging of the pulmonary vasculature after COVID-19. Lancet Respir Med, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Diller GP, Dimopoulos K, Broberg CS, Kaya MG, Naghotra US, Uebing A, Harries C, Goktekin O, Gibbs JS, Gatzoulis MA. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: A combined retrospective and case-control study. Eur Heart J 27: 1737–1742, 2006. [DOI] [PubMed] [Google Scholar]
- 152.Ding XF, Liang HY, Yuan B, Li LF, Wang T, Kan QC, Wang LX, Sun TW. Efficacy of stem cell therapy for pulmonary arterial hypertension: A systematic review and meta-analysis of preclinical studies. Stem Cell Res Ther 10: 55, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Do e Z, Fukumoto Y, Takaki A, Tawara S, Ohashi J, Nakano M, Tada T, Saji K, Sugimura K, Fujita H, Hoshikawa Y, Nawata J, Kondo T, Shimokawa H. Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ J 73: 1731–1739, 2009. [DOI] [PubMed] [Google Scholar]
- 154.Donda K, Zambrano R, Moon Y, Percival J, Vaidya R, Dapaah-Siakwan F, Luo S, Duncan MR, Bao Y, Wang L, Qin L, Benny M, Young K, Wu S. Riociguat prevents hyperoxia-induced lung injury and pulmonary hypertension in neonatal rats without effects on long bone growth. PLoS One 13: e0199927, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Doran AK, Ivy DD, Barst RJ, Hill N, Murali S, Benza RL, Scientific Leadership Council of the Pulmonary Hypertension A. Guidelines for the prevention of central venous catheter-related blood stream infections with prostanoid therapy for pulmonary arterial hypertension. Int J Clin Pract Suppl 5–9, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Douwes JM, Hegeman AK, van der Krieke MB, Roofthooft MT, Hillege HL, Berger RM. Six-minute walking distance and decrease in oxygen saturation during the six-minute walk test in pediatric pulmonary arterial hypertension. Int J Cardiol 202: 34–39, 2016. [DOI] [PubMed] [Google Scholar]
- 157.Douwes JM, Humpl T, Bonnet D, Beghetti M, Ivy DD, Berger RM, Investigators T. Acute vasodilator response in pediatric pulmonary arterial hypertension: current clinical practice fromthe TOPP registry. J Am Coll Cardiol 67: 1312–1323, 2016. [DOI] [PubMed] [Google Scholar]
- 158.Douwes JM, Roofthooft MT, Bartelds B, Talsma MD, Hillege HL, Berger RM. Pulsatile haemodynamic parameters are predictors of survival in paediatric pulmonary arterial hypertension. Int J Cardiol 168: 1370–1377, 2013. [DOI] [PubMed] [Google Scholar]
- 159.Du Y, Fu J, Yao L, Qiao L, Liu N, Xing Y, Xue X. Altered expression of PPARgamma and TRPC in neonatal rats with persistent pulmonary hypertension. Mol Med Rep 16: 1117–1124, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dupuis J Endothelin-receptor antagonists in pulmonary hypertension. Lancet 358: 1113–1114, 2001. [DOI] [PubMed] [Google Scholar]
- 161.Durmowicz AG, Stenmark KR. Mechanisms of structural remodeling in chronic pulmonary hypertension. Pediatr Rev 20: e91–e102, 1999. [PubMed] [Google Scholar]
- 162.Eddahibi S, Guignabert C, Barlier-Mur AM, Dewachter L, Fadel E, Dartevelle P, Humbert M, Simonneau G, Hanoun N, Saurini F, Hamon M, Adnot S. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: Critical role for serotonin-induced smooth muscle hyperplasia. Circulation 113: 1857–1864, 2006. [DOI] [PubMed] [Google Scholar]
- 163.Esch JJ, Shah PB, Cockrill BA, Farber HW, Landzberg MJ, Mehra MR, Mullen MP, Opotowsky AR, Waxman AB, Lock JE, Marshall AC. Transcatheter Potts shunt creation in patients with severe pulmonary arterial hypertension: Initial clinical experience. J Heart Lung Transplant 32: 381–387, 2013. [DOI] [PubMed] [Google Scholar]
- 164.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 420: 629–635, 2002. [DOI] [PubMed] [Google Scholar]
- 165.Evans JD, Girerd B, Montani D, Wang XJ, Galie N, Austin ED, Elliott G, Asano K, Grunig E, Yan Y, Jing ZC, Manes A, Palazzini M, Wheeler LA, Nakayama I, Satoh T, Eichstaedt C, Hinderhofer K, Wolf M, Rosenzweig EB, Chung WK, Soubrier F, Simonneau G, Sitbon O, Graf S, Kaptoge S, Di Angelantonio E, Humbert M, Morrell NW. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir Med 4: 129–137, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Eyries M, Montani D, Girerd B, Perret C, Leroy A, Lonjou C, Chelghoum N, Coulet F, Bonnet D, Dorfmuller P, Fadel E, Sitbon O, Simonneau G, Tregouet DA, Humbert M, Soubrier F. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 46: 65–69, 2014. [DOI] [PubMed] [Google Scholar]
- 167.Ezekian JE, Hill KD. Management of pulmonary arterial hypertension in the pediatric patient. Curr Cardiol Rep 21: 162, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L656–L664, 2004. [DOI] [PubMed] [Google Scholar]
- 169.Fagard R, Conway J. Measurement of cardiac output: Fick principle using catheterization. Eur Heart J 11 (Suppl I): 1–5, 1990. [DOI] [PubMed] [Google Scholar]
- 170.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 285: L1233–L1245, 2003. [DOI] [PubMed] [Google Scholar]
- 171.Farhat N, Lador F, Beghetti M. Diagnosis and treatment of pediatric pulmonary arterial hypertension. Expert Rev Cardiovasc Ther 17: 161–175, 2019. [DOI] [PubMed] [Google Scholar]
- 172.Farkas L, Farkas D, Ask K, Moller A, Gauldie J, Margetts P, Inman M, Kolb M. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest 119: 1298–1311, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Farrow KN, Groh BS, Schumacker PT, Lakshminrusimha S, Czech L, Gugino SF, Russell JA, Steinhorn RH. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ Res 102: 226–233, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Farrow KN, Lakshminrusimha S, Reda WJ, Wedgwood S, Czech L, Gugino SF, Davis JM, Russell JA, Steinhorn RH. Superoxide dismutase restores eNOS expression and function in resistance pulmonary arteries from neonatal lambs with persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L979–L987, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Farrow KN, Lee KJ, Perez M, Schriewer JM, Wedgwood S, Lakshminrusimha S, Smith CL, Steinhorn RH, Schumacker PT. Brief hyperoxia increases mitochondrial oxidation and increases phosphodiesterase 5 activity in fetal pulmonary artery smooth muscle cells. Antioxid Redox Signal 17: 460–470, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Farrow KN, Wedgwood S, Lee KJ, Czech L, Gugino SF, Lakshminrusimha S, Schumacker PT, Steinhorn RH. Mitochondrial oxidant stress increases PDE5 activity in persistent pulmonary hypertension of the newborn. Respir Physiol Neurobiol 174: 272–281, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fedullo PF, Auger WR, Channick RN, Kerr KM, Rubin LJ. Chronic thromboembolic pulmonary hypertension. Clin Chest Med 22: 561–581, 2001. [DOI] [PubMed] [Google Scholar]
- 178.Ferdman DJ, Rosenzweig EB, Zuckerman WA, Krishnan U. Subcutaneous treprostinil for pulmonary hypertension in chronic lung disease of infancy. Pediatrics 134: e274–e278, 2014. [DOI] [PubMed] [Google Scholar]
- 179.Fessel JP, Flynn CR, Robinson LJ, Penner NL, Gladson S, Kang CJ, Wasserman DH, Hemnes AR, West JD. Hyperoxia synergizes with mutant bone morphogenic protein receptor 2 to cause metabolic stress, oxidant injury, and pulmonary hypertension. Am J Respir Cell Mol Biol 49: 778–787, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Field D, Elbourne D, Hardy P, Fenton AC, Ahluwalia J, Halliday HL, Subhedar N, Heinonen K, Aikio O, Grieve R, Truesdale A, Tomlin K, Normand C, Stocks J, Group ITC. Neonatal ventilation with inhaled nitric oxide vs. ventilatory support without inhaled nitric oxide for infants with severe respiratory failure born at or near term: The INNOVO multicentre randomised controlled trial. Neonatology 91: 73–82, 2007. [DOI] [PubMed] [Google Scholar]
- 181.Fike CD, Slaughter JC, Kaplowitz MR, Zhang Y, Aschner JL. Reactive oxygen species from NADPH oxidase contribute to altered pulmonary vascular responses in piglets with chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L881–L888, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Foglia EE, Carper B, Gantz M, DeMauro SB, Lakshminrusimha S, Walsh M, Schmidt B, Eunice Kennedy Shriver National Institute of Child H, and Human Development Neonatal Research N. Association between policy changes for oxygen saturation alarm settings and neonatal morbidity and mortality in infants born very preterm. J Pediatr 209: 17–22. e12, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fraisse A, Butrous G, Taylor MB, Oakes M, Dilleen M, Wessel DL. Intravenous sildenafil for postoperative pulmonary hypertension in children with congenital heart disease. Intensive Care Med 37: 502–509, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Frank BS, Ivy DD. Diagnosis, evaluation and treatment of pulmonary arterial hypertension in children. Children (Basel) 5: 44, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Frank BS, Schafer M, Grenolds A, Ivy DD, Abman SH, Darst JR. Acute vasoreactivity testing during cardiac catheterization of neonates with bronchopulmonary dysplasia-associated pulmonary hypertension. J Pediatr 208: 127–133, 2019. [DOI] [PubMed] [Google Scholar]
- 186.Fredenburgh LE, Liang OD, Macias AA, Polte TR, Liu X, Riascos DF, Chung SW, Schissel SL, Ingber DE, Mitsialis SA, Kourembanas S, Perrella MA. Absence of cyclooxygenase-2 exacerbates hypoxia-induced pulmonary hypertension and enhances contractility of vascular smooth muscle cells. Circulation 117: 2114–2122, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fredriksson L, Li H, Eriksson U. The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev 15: 197–204, 2004. [DOI] [PubMed] [Google Scholar]
- 188.Fujiwara M, Yagi H, Matsuoka R, Akimoto K, Furutani M, Imamura S, Uehara R, Nakayama T, Takao A, Nakazawa M, Saji T. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J 72: 127–133, 2008. [DOI] [PubMed] [Google Scholar]
- 189.Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, Kihara Y, Kawano M, Watanabe H, Takeda Y, Adachi T, Osanai S, Tanabe N, Inoue T, Kubo A, Ota Y, Fukuda K, Nakano T, Shimokawa H. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J 77: 2619–2625, 2013. [DOI] [PubMed] [Google Scholar]
- 190.Fuloria M, Aschner JL. Persistent pulmonary hypertension of the newborn. Semin Fetal Neonatal Med 22: 220–226, 2017. [DOI] [PubMed] [Google Scholar]
- 191.Galie N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res 61: 227–237, 2004. [DOI] [PubMed] [Google Scholar]
- 192.Galie N, McLaughlin VV, Rubin LJ, Simonneau G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J 53: 1802148, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Galie N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, Badesch DB, McGoon MD, McLaughlin VV, Roecker EB, Gerber MJ, Dufton C, Wiens BL, Rubin LJ, Ambrisentan in Pulmonary Arterial Hypertension RD-BP-CMESG. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 117: 3010–3019, 2008. [DOI] [PubMed] [Google Scholar]
- 194.Galie N, Torbicki A, Barst R, Dartevelle P, Haworth S, Higenbottam T, Olschewski H, Peacock A, Pietra G, Rubin LJ, Simonneau G, Priori SG, Garcia MA, Blanc JJ, Budaj A, Cowie M, Dean V, Deckers J, Burgos EF, Lekakis J, Lindahl B, Mazzotta G, McGregor K, Morais J, Oto A, Smiseth OA, Barbera JA, Gibbs S, Hoeper M, Humbert M, Naeije R, Pepke-Zaba J, Task F. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The task force on diagnosis and treatment of pulmonary arterial hypertension of the European Society of Cardiology. Eur Heart J 25: 2243, 2004–2278. [DOI] [PubMed] [Google Scholar]
- 195.Gan C, Lankhaar JW, Marcus JT, Westerhof N, Marques KM, Bronzwaer JG, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Impaired left ventricular filling due to right-to-left ventricular interaction in patients with pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 290: H1528–H1533, 2006. [DOI] [PubMed] [Google Scholar]
- 196.Gan CT, Holverda S, Marcus JT, Paulus WJ, Marques KM, Bronzwaer JG, Twisk JW, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right ventricular diastolic dysfunction and the acute effects of sildenafil in pulmonary hypertension patients. Chest 132: 11–17, 2007. [DOI] [PubMed] [Google Scholar]
- 197.Gao Y, Chen T, Raj JU. Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. Am J Respir Cell Mol Biol 54: 451–460, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Garofano RP, Barst RJ. Exercise testing in children with primary pulmonary hypertension. Pediatr Cardiol 20: 61–64; discussion 65, 1999. [DOI] [PubMed] [Google Scholar]
- 199.Gaynor JW, Bull C, Sullivan ID, Armstrong BE, Deanfield JE, Taylor JF, Rees PG, Ungerleider RM, de Leval MR, Stark J, et al. Late outcome of survivors of intervention for neonatal aortic valve stenosis. Ann Thorac Surg 60: 122–125; discussion 125-126, 1995. [PubMed] [Google Scholar]
- 200.Gentles TL, Gauvreau K, Mayer JE Jr, Fishberger SB, Burnett J, Colan SD, Newburger JW, Wernovsky G. Functional outcome after the Fontan operation: Factors influencing late morbidity. J Thorac Cardiovasc Surg 114: 392–403; discussion 404-395, 1997. [DOI] [PubMed] [Google Scholar]
- 201.George J, D’Armiento J. Transgenic expression of human matrix metalloproteinase-9 augments monocrotaline-induced pulmonary arterial hypertension in mice. J Hypertens 29: 299–308, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res 100: 607–621, 2007. [DOI] [PubMed] [Google Scholar]
- 203.Ghofrani HA, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, Mayer E, Simonneau G, Wilkins MR, Fritsch A, Neuser D, Weimann G, Wang C, Group C-S. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 369: 319–329, 2013. [DOI] [PubMed] [Google Scholar]
- 204.Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, Keogh AM, Langleben D, Kilama MO, Fritsch A, Neuser D, Rubin LJ, Group P-S. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369: 330–340, 2013. [DOI] [PubMed] [Google Scholar]
- 205.Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov 5: 689–702, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333: 214–221, 1995. [DOI] [PubMed] [Google Scholar]
- 207.Gien J, Kinsella JP. Management of pulmonary hypertension in infants with congenital diaphragmatic hernia. J Perinatol 36 (Suppl 2): S28–S31, 2016. [DOI] [PubMed] [Google Scholar]
- 208.Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH. Intrauterine pulmonary hypertension impairs angiogenesis in vitro: Role of vascular endothelial growth factor nitric oxide signaling. Am J Respir Crit Care Med 176: 1146–1153, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gien J, Tseng N, Seedorf G, Roe G, Abman SH. Endothelin-1 impairs angiogenesis in vitro through Rho-kinase activation after chronic intrauterine pulmonary hypertension in fetal sheep. Pediatr Res 73: 252–262, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Giordano R, Palma G, Poli V, Palumbo S, Russolillo V, Cioffi S, Mucerino M, Mannacio VA, Vosa C. First experience with sildenafil after Fontan operation: Short-term outcomes. J Cardiovasc Med (Hagerstown) 16: 552–555, 2015. [DOI] [PubMed] [Google Scholar]
- 211.Girerd B, Montani D, Coulet F, Sztrymf B, Yaici A, Jais X, Tregouet D, Reis A, Drouin-Garraud V, Fraisse A, Sitbon O, O’Callaghan DS, Simonneau G, Soubrier F, Humbert M. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med 181: 851–861, 2010. [DOI] [PubMed] [Google Scholar]
- 212.Goldberg DJ, French B, McBride MG, Marino BS, Mirarchi N, Hanna BD, Wernovsky G, Paridon SM, Rychik J. Impact of oral sildenafil on exercise performance in children and young adults after the fontan operation: A randomized, double-blind, placebo-controlled, crossover trial. Circulation 123: 1185–1193, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Goldberg DJ, Zak V, Goldstein BH, Chen S, Hamstra MS, Radojewski EA, Maunsell E, Mital S, Menon SC, Schumacher KR, Payne RM, Stylianou M, Kaltman JR, deVries TM, Yeager JL, Paridon SM, Pediatric Heart Network I. Results of a phase I/II multi-center investigation of udenafil in adolescents after fontan palliation. Am Heart J 188: 42–52, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Goldberg DJ, Zak V, Goldstein BH, Schumacher KR, Rhodes J, Penny DJ, Petit CJ, Ginde S, Menon SC, Kim SH, Kim GB, Nowlen TT, DiMaria MV, Frischhertz BP, Wagner JB, McHugh KE, McCrindle BW, Shillingford AJ, Sabati AA, Yetman AT, John AS, Richmond ME, Files MD, Payne RM, Mackie AS, Davis CK, Shahanavaz S, Hill KD, Garg R, Jacobs JP, Hamstra MS, Woyciechowski S, Rathge KA, MG MB, Frommelt PC, Russell MW, Urbina EM, Yeager JL, Pemberton VL, Stylianou MP, Pearson GD, Paridon SM, Pediatric Heart Network I. Results of the FUEL trial. Circulation 141: 641–651, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Goldstein I, Lue TF, Padma-Nathan H, Rosen RC, Steers WD, Wicker PA. Oral sildenafil in the treatment of erectile dysfunction. Sildenafil Study Group. N Engl J Med 338: 1397–1404, 1998. [DOI] [PubMed] [Google Scholar]
- 216.Gomberg-Maitland M, Tapson VF, Benza RL, McLaughlin VV, Krichman A, Widlitz AC, Barst RJ. Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am J Respir Crit Care Med 172: 1586–1589, 2005. [DOI] [PubMed] [Google Scholar]
- 217.Gonzalez A, Fabres J, D’Apremont I, Urcelay G, Avaca M, Gandolfi C, Kattan J. Randomized controlled trial of early compared with delayed use of inhaled nitric oxide in newborns with a moderate respiratory failure and pulmonary hypertension. J Perinatol 30: 420–424, 2010. [DOI] [PubMed] [Google Scholar]
- 218.Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, Holmes AM. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol 185: 1850–1858, 2015. [DOI] [PubMed] [Google Scholar]
- 219.Gorbachevsky SV, Shmalts AA, Barishnikova IY, Zaets SB. Potts shunt in children with pulmonary arterial hypertension: Institutional experience. Interact Cardiovasc Thorac Surg 25: 595–599, 2017. [DOI] [PubMed] [Google Scholar]
- 220.Gorenflo M, Vogel M, Obladen M. Pulmonary vascular changes in bronchopulmonary dysplasia: A clinicopathologic correlation in short-and long-term survivors. Pediatr Pathol 11: 851–866, 1991. [DOI] [PubMed] [Google Scholar]
- 221.Graham BB, Kumar R, Mickael C, Kassa B, Koyanagi D, Sanders L, Zhang L, Perez M, Hernandez-Saavedra D, Valencia C, Dixon K, Harral J, Loomis Z, Irwin D, Nemkov T, D’Alessandro A, Stenmark KR, Tuder RM. Vascular adaptation of the right ventricle in experimental pulmonary hypertension. Am J Respir Cell Mol Biol 59: 479–489, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Green DE, Murphy TC, Kang BY, Searles CD, Hart CM. PPARgamma ligands attenuate hypoxia-induced proliferation in human pulmonary artery smooth muscle cells through modulation of MicroRNA-21. PLoS One 10: e0133391, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Green DE, Sutliff RL, Hart CM. Is peroxisome proliferator-activated receptor gamma (PPARgamma) a therapeutic target for the treatment of pulmonary hypertension? Pulm Circ 1: 33–47, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Gridley T Notch signaling in vascular development and physiology. Development 134: 2709–2718, 2007. [DOI] [PubMed] [Google Scholar]
- 225.Grosse-Wortmann L, Al-Otay A, Yoo SJ. Aortopulmonary collaterals after bidirectional cavopulmonary connection or Fontan completion: Quantification with MRI. Circ Cardiovasc Imaging 2: 219–225, 2009. [DOI] [PubMed] [Google Scholar]
- 226.Grothues F, Moon JC, Bellenger NG, Smith GS, Klein HU, Pennell DJ. Interstudy reproducibility of right ventricular volumes, function, and mass with cardiovascular magnetic resonance. Am Heart J 147: 218–223, 2004. [DOI] [PubMed] [Google Scholar]
- 227.Grunig E, Benjamin N, Kruger U, Kaemmerer H, Harutyunova S, Olsson KM, Ulrich S, Gerhardt F, Neurohr C, Sablotzki A, Halank M, Marra AM, Kabitz HJ, Thimm G, Fliegel KG, Klose H. General measures and supportive therapy for pulmonary arterial hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int J Cardiol 272S: 30–36, 2018. [DOI] [PubMed] [Google Scholar]
- 228.Guo L, Cui Y, Pharis S, Walsh M, Atallah J, Tan MW, Rutledge J, Coe JY, Adatia I. Measurement of oxygen consumption in children undergoing cardiac catheterization: Comparison between mass spectrometry and the breath-by-breath method. Pediatr Cardiol 35: 798–802, 2014. [DOI] [PubMed] [Google Scholar]
- 229.Gupta VS, Harting MT. Congenital diaphragmatic hernia-associated pulmonary hypertension. Semin Perinatol 44: 151167, 2020. [DOI] [PubMed] [Google Scholar]
- 230.Haddad IY, Ischiropoulos H, Holm BA, Beckman JS, Baker JR, Matalon S. Mechanisms of peroxynitrite-induced injury to pulmonary surfactants. Am J Physiol 265: L555–L564, 1993. [DOI] [PubMed] [Google Scholar]
- 231.Hammerstingl C, Schueler R, Bors L, Momcilovic D, Pabst S, Nickenig G, Skowasch D. Diagnostic value of echocardiography in the diagnosis of pulmonary hypertension. PLoS One 7: e38519, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Hampl V, Herget J. Role of nitric oxide in the pathogenesis of chronic pulmonary hypertension. Physiol Rev 80: 1337–1372, 2000. [DOI] [PubMed] [Google Scholar]
- 233.Hansmann G Pulmonary hypertension in infants, children, and young adults. J Am Coll Cardiol 69: 2551–2569, 2017. [DOI] [PubMed] [Google Scholar]
- 234.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest 118: 1846–1857, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hansmann G, Koestenberger M, Alastalo TP, Apitz C, Austin ED, Bonnet D, Budts W, D’Alto M, Gatzoulis MA, Hasan BS, Kozlik-Feldmann R, Kumar RK, Lammers AE, Latus H, Michel-Behnke I, Miera O, Morrell NW, Pieles G, Quandt D, Sallmon H, Schranz D, Tran-Lundmark K, Tulloh RMR, Warnecke G, Wahlander H, Weber SC, Zartner P. 2019 updated consensus statement on the diagnosis and treatment of pediatric pulmonary hypertension: The European Pediatric Pulmonary Vascular Disease Network (EPPVDN), endorsed by AEPC, ESPR and ISHLT. J Heart Lung Transplant 38: 879–901, 2019. [DOI] [PubMed] [Google Scholar]
- 236.Hao YJ, Jiang X, Zhou W, Wang Y, Gao L, Wang Y, Li GT, Hong T, Huo Y, Jing ZC, Zhang ZL. Connective tissue disease-associated pulmonary arterial hypertension in Chinese patients. Eur Respir J 44: 963–972, 2014. [DOI] [PubMed] [Google Scholar]
- 237.Hardegree EL, Sachdev A, Villarraga HR, Frantz RP, McGoon MD, Kushwaha SS, Hsiao JF, McCully RB, Oh JK, Pellikka PA, Kane GC. Role of serial quantitative assessment of right ventricular function by strain in pulmonary arterial hypertension. Am J Cardiol 111: 143–148, 2013. [DOI] [PubMed] [Google Scholar]
- 238.Harrison RE, Berger R, Haworth SG, Tulloh R, Mache CJ, Morrell NW, Aldred MA, Trembath RC. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 111: 435–441, 2005. [DOI] [PubMed] [Google Scholar]
- 239.Harting MT. Congenital diaphragmatic hernia-associated pulmonary hypertension. Semin Pediatr Surg 26: 147–153, 2017. [DOI] [PubMed] [Google Scholar]
- 240.Hascoet JM, Fresson J, Claris O, Hamon I, Lombet J, Liska A, Cantagrel S, Al Hosri J, Thiriez G, Valdes V, Vittu G, Egreteau L, Henrot A, Buchweiller MC, Onody P. The safety and efficacy of nitric oxide therapy in premature infants. J Pediatr 146: 318–323, 2005. [DOI] [PubMed] [Google Scholar]
- 241.Haworth SG, Reid L. Quantitative structural study of pulmonary circulation in the newborn with aortic atresia, stenosis, or coarctation. Thorax 32: 121–128, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hayashi S, Morishita R, Nakamura S, Yamamoto K, Moriguchi A, Nagano T, Taiji M, Noguchi H, Matsumoto K, Nakamura T, Higaki J, Ogihara T. Potential role of hepatocyte growth factor, a novel angiogenic growth factor, in peripheral arterial disease: Downregulation of HGF in response to hypoxia in vascular cells. Circulation 100: II301–II308, 1999. [DOI] [PubMed] [Google Scholar]
- 243.Heching HJ, Turner M, Farkouh-Karoleski C, Krishnan U. Pulmonary vein stenosis and necrotising enterocolitis: Is there a possible link with necrotising enterocolitis? Arch Dis Child Fetal Neonatal Ed 99: F282–F285, 2014. [DOI] [PubMed] [Google Scholar]
- 244.Heilman RP, Lagoski MB, Lee KJ, Taylor JM, Kim GA, Berkelhamer SK, Steinhorn RH, Farrow KN. Right ventricular cyclic nucleotide signaling is decreased in hyperoxia-induced pulmonary hypertension in neonatal mice. Am J Physiol Heart Circ Physiol 308: H1575–H1582, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hernandez-Diaz S, Van Marter LJ, Werler MM, Louik C, Mitchell AA. Risk factors for persistent pulmonary hypertension of the newborn. Pediatrics 120: e272–e282, 2007. [DOI] [PubMed] [Google Scholar]
- 246.Hilgendorff A, Apitz C, Bonnet D, Hansmann G. Pulmonary hypertension associated with acute or chronic lung diseases in the preterm and term neonate and infant. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 102 (Suppl 2): ii49–ii56, 2016. [DOI] [PubMed] [Google Scholar]
- 247.Hill KD, Tunks RD, Barker PC, Benjamin DK Jr, Cohen-Wolkowiez M, Fleming GA, Laughon M, Li JS. Sildenafil exposure and hemodynamic effect after stage II single-ventricle surgery. Pediatr Crit Care Med 14: 593–600, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Hirata Y, Emori T, Eguchi S, Kanno K, Imai T, Ohta K, Marumo F. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J Clin Invest 91: 1367–1373, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hirenallur SD, Haworth ST, Leming JT, Chang J, Hernandez G, Gordon JB, Rusch NJ. Upregulation of vascular calcium channels in neonatal piglets with hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L915–L924, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Hislop AA, Moledina S, Foster H, Schulze-Neick I, Haworth SG. Long-term efficacy of bosentan in treatment of pulmonary arterial hypertension in children. Eur Respir J 38: 70–77, 2011. [DOI] [PubMed] [Google Scholar]
- 251.Ho JJ, Man HS, Marsden PA. Nitric oxide signaling in hypoxia. J Mol Med (Berl) 90: 217–231, 2012. [DOI] [PubMed] [Google Scholar]
- 252.Hoffman JI. Pulmonary vascular resistance and viscosity: The forgotten factor. Pediatr Cardiol 32: 557–561, 2011. [DOI] [PubMed] [Google Scholar]
- 253.Hoffmann J, Marsh LM, Pieper M, Stacher E, Ghanim B, Kovacs G, Konig P, Wilkens H, Haitchi HM, Hoefler G, Klepetko W, Olschewski H, Olschewski A, Kwapiszewska G. Compartment-specific expression of collagens and their processing enzymes in intrapulmonary arteries of IPAH patients. Am J Physiol Lung Cell Mol Physiol 308: L1002–L1013, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hoffmann R, von Bardeleben S, ten Cate F, Borges AC, Kasprzak J, Firschke C, Lafitte S, Al-Saadi N, Kuntz-Hehner S, Engelhardt M, Becher H, Vanoverschelde JL. Assessment of systolic left ventricular function: A multi-centre comparison of cineventriculography, cardiac magnetic resonance imaging, unenhanced and contrast-enhanced echocardiography. Eur Heart J 26: 607–616, 2005. [DOI] [PubMed] [Google Scholar]
- 255.Hofmann AD, Friedmacher F, Hunziker M, Takahashi H, Duess JW, Gosemann JH, Puri P. Upregulation of serotonin-receptor-2a and serotonin transporter expression in the pulmonary vasculature of nitrofen-induced congenital diaphragmatic hernia. J Pediatr Surg 49: 871–874; discussion 874-875, 2014. [DOI] [PubMed] [Google Scholar]
- 256.Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels: Who is talking to whom about what? Circ Res 100: 1556–1568, 2007. [DOI] [PubMed] [Google Scholar]
- 257.Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 133: 1783–1794, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, Marcos E, Tissot CM, Dubois-Rande JL, Amsellem V, Adnot S. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol 48: 568–577, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hsue PY, Deeks SG, Farah HH, Palav S, Ahmed SY, Schnell A, Ellman AB, Huang L, Dollard SC, Martin JN. Role of HIV and human herpesvirus-8 infection in pulmonary arterial hypertension. AIDS 22: 825–833, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hu CJ, Poth JM, Zhang H, Flockton A, Laux A, Kumar S, McKeon B, Mouradian G, Li M, Riddle S, Pugliese SC, Brown RD, Wallace EM, Graham BB, Frid MG, Stenmark KR. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur Respir J 54: 1900378,2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Huddleston AJ, Knoderer CA, Morris JL, Ebenroth ES. Sildenafil for the treatment of pulmonary hypertension in pediatric patients. Pediatr Cardiol 30: 871–882, 2009. [DOI] [PubMed] [Google Scholar]
- 262.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43: 13S–24S, 2004. [DOI] [PubMed] [Google Scholar]
- 263.Humpl T, Reyes JT, Erickson S, Armano R, Holtby H, Adatia I. Sildenafil therapy for neonatal and childhood pulmonary hypertensive vascular disease. Cardiol Young 21: 187–193, 2011. [DOI] [PubMed] [Google Scholar]
- 264.Iannone L, Zhao L, Dubois O, Duluc L, Rhodes CJ, Wharton J, Wilkins MR, Leiper J, Wojciak-Stothard B. miR-21/DDAH1 pathway regulates pulmonary vascular responses to hypoxia. Biochem J 462: 103–112, 2014. [DOI] [PubMed] [Google Scholar]
- 265.Ilkiw R, Todorovich-Hunter L, Maruyama K, Shin J, Rabinovitch M. SC-39026, a serine elastase inhibitor, prevents muscularization of peripheral arteries, suggesting a mechanism of monocrotaline-induced pulmonary hypertension in rats. Circ Res 64: 814–825, 1989. [DOI] [PubMed] [Google Scholar]
- 266.Iocono JA, Cilley RE, Mauger DT, Krummel TM, Dillon PW. Postnatal pulmonary hypertension after repair of congenital diaphragmatic hernia: Predicting risk and outcome. J Pediatr Surg 34: 349–353, 1999. [DOI] [PubMed] [Google Scholar]
- 267.Irodova NL, Lankin VZ, Konovalova GK, Kochetov AG, Chazova IE. Oxidative stress in patients with primary pulmonary hypertension. Bull Exp Biol Med 133: 580–582, 2002. [DOI] [PubMed] [Google Scholar]
- 268.Ishikura K, Yamada N, Ito M, Ota S, Nakamura M, Isaka N, Nakano T. Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ J 70: 174–178, 2006. [DOI] [PubMed] [Google Scholar]
- 269.Ivanovska J, Shah S, Wong MJ, Kantores C,Jain A, Post M, Yeganeh B, Jankov RP. mTOR-Notch3 signaling mediates pulmonary hypertension in hypoxia-exposed neonatal rats independent of changes in autophagy. Pediatr Pulmonol 52: 1443–1454, 2017. [DOI] [PubMed] [Google Scholar]
- 270.Ivy DD, Abman SH, Barst RJ, Berger RM, Bonnet D, Fleming TR, Haworth SG, Raj JU, Rosenzweig EB, Schulze Neick I, Steinhorn RH, Beghetti M. Pediatric pulmonary hypertension. J Am Coll Cardiol 62: D117–D126, 2013. [DOI] [PubMed] [Google Scholar]
- 271.Ivy DD, Claussen L, Doran A. Transition of stable pediatric patients with pulmonary arterial hypertension from intravenous epoprostenol to intravenous treprostinil. Am J Cardiol 99: 696–698, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ivy DD, Doran A, Claussen L, Bingaman D, Yetman A. Weaning and discontinuation of epoprostenol in children with idiopathic pulmonary arterial hypertension receiving concomitant bosentan. Am J Cardiol 93: 943–946, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ivy DD, Doran AK, Smith KJ, Mallory GB Jr, Beghetti M, Barst RJ, Brady D, Law Y, Parker D, Claussen L, Abman SH. Short- and long-term effects of inhaled iloprost therapy in children with pulmonary arterial hypertension. J Am Coll Cardiol 51: 161–169, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Ivy DD, Rosenzweig EB, Lemarie JC, Brand M, Rosenberg D, Barst RJ. Long-term outcomes in children with pulmonary arterial hypertension treated with bosentan in real-world clinical settings. Am J Cardiol 106: 1332–1338,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B, Raffestin B, Eddahibi S. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest 119: 512–523, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Janda S, Shahidi N, Gin K, Swiston J. Diagnostic accuracy of echocardiography for pulmonary hypertension: A systematic review and meta-analysis. Heart 97: 612–622, 2011. [DOI] [PubMed] [Google Scholar]
- 277.Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis 45: 173–202, 2002. [DOI] [PubMed] [Google Scholar]
- 278.Jiang L, Sun W, Zhang K, Zhou B, Kong X. Perioperative sildenafil therapy in pediatric congenital cardiac disease patients. Int Heart J 59: 1333–1339, 2018. [DOI] [PubMed] [Google Scholar]
- 279.Jiang X, Wang YF, Zhao QH, Jiang R, Wu Y, Peng FH, Xu XQ, Wang L, He J, Jing ZC. Acute hemodynamic response of infused fasudil in patients with pulmonary arterial hypertension: A randomized, controlled, crossover study. Int J Cardiol 177: 61–65, 2014. [DOI] [PubMed] [Google Scholar]
- 280.Johnson SR, Granton JT. Pulmonary hypertension in systemic sclerosis and systemic lupus erythematosus. Eur Respir Rev 20: 277–286, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Jone PN, Hinzman J, Wagner BD, Ivy DD, Younoszai A. Right ventricular to left ventricular diameter ratio at end-systole in evaluating outcomes in children with pulmonary hypertension. J Am Soc Echocardiogr 27: 172–178, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Jone PN, Ivy DD. Echocardiography in pediatric pulmonary hypertension. Front Pediatr 2: 124, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Jone PN, Patel SS, Cassidy C, Ivy DD. Three-dimensional echocardiography of right ventricular function correlates with severity of pediatric pulmonary hypertension. Congenit Heart Dis 11: 562–569, 2016. [DOI] [PubMed] [Google Scholar]
- 284.Jone PN, Schafer M, Pan Z, Ivy DD. Right ventricular-arterial coupling ratio derived from 3-dimensional echocardiography predicts outcomes in pediatric pulmonary hypertension. Circ Cardiovasc Imaging 12: e008176, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kaestner M, Schranz D, Warnecke G, Apitz C, Hansmann G, Miera O. Pulmonary hypertension in the intensive care unit. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 102 (Suppl 2): ii57–ii66, 2016. [DOI] [PubMed] [Google Scholar]
- 286.Kamezaki F, Tasaki H, Yamashita K, Tsutsui M, Koide S, Nakata S, Tanimoto A, Okazaki M, Sasaguri Y, Adachi T, Otsuji Y. Gene transfer of extracellular superoxide dismutase ameliorates pulmonary hypertension in rats. Am J Respir Crit Care Med 177: 219–226, 2008. [DOI] [PubMed] [Google Scholar]
- 287.Kamin DS, Grinspoon SK. Cardiovascular disease in HIV-positive patients. AIDS 19: 641–652, 2005. [DOI] [PubMed] [Google Scholar]
- 288.Kanaan U, Srivatsa B, Huckaby J, Kelleman M. Association of unitwide oxygen saturation target on incidence of pulmonary hypertension in very low birthweight premature infants. J Perinatol 38: 148–153, 2018. [DOI] [PubMed] [Google Scholar]
- 289.Kang BY, Kleinhenz JM, Murphy TC, Hart CM. The PPARgamma ligand rosiglitazone attenuates hypoxia-induced endothelin signaling in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol 301: L881–L891, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106: 1311–1319, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Katz J, Whang J, Boxt LM, Barst RJ. Estimation of right ventricular mass in normal subjects and in patients with primary pulmonary hypertension by nuclear magnetic resonance imaging. J Am Coll Cardiol 21: 1475–1481, 1993. [DOI] [PubMed] [Google Scholar]
- 292.Keijzer R, Liu J, Deimling J, Tibboel D, Post M. Dual-hit hypothesis explains pulmonary hypoplasia in the nitrofen model of congenital diaphragmatic hernia. Am J Pathol 156: 1299–1306, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Keith IM, Tjen ALS, Kraiczi H, Ekman R. Three-week neonatal hypoxia reduces blood CGRP and causes persistent pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol 279: H1571–H1578, 2000. [DOI] [PubMed] [Google Scholar]
- 294.Kelly LE, Ohlsson A, Shah PS. Sildenafil for pulmonary hypertension in neonates. Cochrane Database Syst Rev 8: CD005494, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Kerstjens-Frederikse WS, Bongers EM, Roofthooft MT, Leter EM, Douwes JM, Van Dijk A, Vonk-Noordegraaf A, Dijk-Bos KK, Hoefsloot LH, Hoendermis ES, Gille JJ, Sikkema-Raddatz B, Hofstra RM, Berger RM. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet 50: 500–506, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Kheyfets VO, Sucharov CC, Truong U, Dunning J, Hunter K, Ivy D, Miyamoto S, Shandas R. Circulating miRNAs in pediatric pulmonary hypertension show promise as biomarkers of vascular function. Oxid Med Cell Longev 2017: 4957147, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.King ME, Braun H, Goldblatt A, Liberthson R, Weyman AE. Interventricular septal configuration as a predictor of right ventricular systolic hypertension in children: A cross-sectional echocardiographic study. Circulation 68: 68–75, 1983. [DOI] [PubMed] [Google Scholar]
- 298.Kinsella JP. Inhaled nitric oxide therapy in premature newborns. Curr Opin Pediatr 18: 107–111, 2006. [DOI] [PubMed] [Google Scholar]
- 299.Kinsella JP, Cutter GR, Steinhorn RH, Nelin LD, Walsh WF, Finer NN, Abman SH. Noninvasive inhaled nitric oxide does not prevent bronchopulmonary dysplasia in premature newborns. J Pediatr 165: 1104–1108. e1101, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Kinsella JP, Cutter GR, Walsh WF, Gerstmann DR, Bose CL, Hart C, Sekar KC, Auten RL, Bhutani VK, Gerdes JS, George TN, Southgate WM, Carriedo H, Couser RJ, Mammel MC, Hall DC, Pappagallo M, Sardesai S, Strain JD, Baier M, Abman SH. Early inhaled nitric oxide therapy in premature newborns with respiratory failure. N Engl J Med 355: 354–364, 2006. [DOI] [PubMed] [Google Scholar]
- 301.Kinsella JP, Steinhorn RH, Krishnan US, Feinstein JA, Adatia I, Austin ED, Rosenzweig EB, Everett AD, Fineman JR, Hanna BD, Hopper RK, Humpl T, Ivy DD, Keller RL, Mullen MP, Raj JU, Wessel DL, Abman SH. Recommendations for the use of inhaled nitric oxide therapy in premature newborns with severe pulmonary hypertension. J Pediatr 170: 312–314, 2016. [DOI] [PubMed] [Google Scholar]
- 302.Kinsella JP, Steinhorn RH, Mullen MP, Hopper RK, Keller RL, Ivy DD, Austin ED, Krishnan US, Rosenzweig EB, Fineman JR, Everett AD, Hanna BD, Humpl T, Raj JU, Abman SH, Pediatric Pulmonary Hypertension N. The left ventricle in congenital diaphragmatic hernia: implications for the management of pulmonary hypertension. J Pediatr 197: 17–22, 2018. [DOI] [PubMed] [Google Scholar]
- 303.Kinsella JP, Truog WE, Walsh WF, Goldberg RN, Bancalari E, Mayock DE, Redding GJ, deLemos RA, Sardesai S, McCurnin DC, Moreland SG, Cutter GR, Abman SH. Randomized, multicenter trial of inhaled nitric oxide and high-frequency oscillatory ventilation in severe, persistent pulmonary hypertension of the newborn. J Pediatr 131: 55–62, 1997. [DOI] [PubMed] [Google Scholar]
- 304.Kinsella JP, Walsh WF, Bose CL, Gerstmann DR, Labella JJ, Sardesai S, Walsh-Sukys MC, McCaffrey Mj, Cornfield DN, Bhutani VK, Cutter GR, Baier M, Abman SH. Inhaled nitric oxide in premature neonates with severe hypoxaemic respiratory failure: A randomised controlled trial. Lancet 354: 1061–1065, 1999. [DOI] [PubMed] [Google Scholar]
- 305.Kirkpatrick EC. Echocardiography in pediatric pulmonary hypertension. Paediatr Respir Rev 14: 157–164, 2013. [DOI] [PubMed] [Google Scholar]
- 306.Klinger JR, Abman SH, Gladwin MT. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 188: 639–646, 2013. [DOI] [PubMed] [Google Scholar]
- 307.Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2: a006502, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Koestenberger M, Apitz C, Abdul-Khaliq H, Hansmann G. Transthoracic echocardiography for the evaluation of children and adolescents with suspected or confirmed pulmonary hypertension. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by iShLT and D6PK. Heart 102 (Suppl 2): ii14–ii22, 2016. [DOI] [PubMed] [Google Scholar]
- 309.Koestenberger M, Avian A, Cantinotti M, Meinel K, Hansmann G. A novel echocardiographic approach indicates disease severity in pediatric pulmonary hypertension. Pediatr Int, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Koestenberger M, Friedberg MK, Nestaas E, Michel-Behnke I, Hansmann G. Transthoracic echocardiography in the evaluation of pediatric pulmonary hypertension and ventricular dysfunction. Pulm Circ 6: 1529, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Koestenberger M, Nagel B, Ravekes W, Urlesberger B, Raith W, Avian A, Halb V, Cvirn G, Fritsch P, Gamillscheg A. Systolic right ventricular function in preterm and term neonates: Reference values of the tricuspid annular plane systolic excursion (TAPSE) in 258 patients and calculation of Z-score values. Neonatology 100: 85–92, 2011. [DOI] [PubMed] [Google Scholar]
- 312.Konduri GG. New approaches for persistent pulmonary hypertension of newborn. Clin Perinatol 31: 591–611, 2004. [DOI] [PubMed] [Google Scholar]
- 313.Konduri GG, Bakhutashvili I, Eis A, Gauthier KM. Impaired voltage gated potassium channel responses in a fetal lamb model of persistent pulmonary hypertension of the newborn. Pediatr Res 66: 289–294, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Konduri GG, Bakhutashvili I, Eis A, Pritchard K Jr. Oxidant stress from uncoupled nitric oxide synthase impairs vasodilation in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 292: H1812–H1820, 2007. [DOI] [PubMed] [Google Scholar]
- 315.Konduri GG, Kim UO. Advances in the diagnosis and management of persistent pulmonary hypertension of the newborn. Pediatr Clin North Am 56: 579–600, Table of Contents, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Konduri GG, Solimano A, Sokol GM, Singer J, Ehrenkranz RA, Singhal N, Wright LL, Van Meurs K, Stork E, Kirpalani H, Peliowski A, Neonatal Inhaled Nitric Oxide Study G. A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infants with hypoxic respiratory failure. Pediatrics 113: 559–564, 2004. [DOI] [PubMed] [Google Scholar]
- 317.Konduri GG, Vohr B, Robertson C, Sokol GM, Solimano A, Singer J, Ehrenkranz RA, Singhal N, Wright LL, Van Meurs K, Stork E, Kirpalani H, Peliowski A, Johnson Y, Neonatal Inhaled Nitric Oxide Study G. Early inhaled nitric oxide therapy for term and near-term newborn infants with hypoxic respiratory failure: Neurodevelopmental follow-up. J Pediatr 150: 235–240 e231, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kopan R, Ilagan MX. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 137: 216–233, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: A systematic review. Eur Respir J 34: 888–894, 2009. [DOI] [PubMed] [Google Scholar]
- 320.Krishnan U, Feinstein JA, Adatia I, Austin ED, Mullen MP, Hopper RK, Hanna B, Romer L, Keller RL, Fineman J, Steinhorn R, Kinsella JP, Ivy DD, Rosenzweig EB, Raj U, Humpl T, Abman SH, Pediatric Pulmonary Hypertension N. Evaluation and management of pulmonary hypertension in children with bronchopulmonary dysplasia. J Pediatr 188: 24–34. e21, 2017. [DOI] [PubMed] [Google Scholar]
- 321.Kunichika N, Landsberg JW, Yu Y, Kunichika H, Thistlethwaite PA, Rubin LJ, Yuan JX. Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am J Respir Crit Care Med 170: 1101–1107, 2004. [DOI] [PubMed] [Google Scholar]
- 322.Kurath-Koller S, Avian A, Cantinotti M, Burmas A, Grangl G, Schweintzger S, Gamillscheg A, Koestenberger M. Normal pediatric values of the subcostal tricuspid annular plane systolic excursion (S-TAPSE) and its value in pediatric pulmonary hypertension. Can J Cardiol 35:899–906, 2019. [DOI] [PubMed] [Google Scholar]
- 323.Kuzuya K, Tsuji S, Matsushita M, Ohshima S, Saeki Y. Systemic sclerosis and systemic lupus erythematosus overlap syndrome with pulmonary arterial hypertension successfully treated with immunosuppressive therapy and riociguat. Cureus 11: e4327, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.La Gerche A, Jurcut R, Voigt JU. Right ventricular function by strain echocardiography. Curr Opin Cardiol 25: 430–436, 2010. [DOI] [PubMed] [Google Scholar]
- 325.Lagatta JM, Hysinger EB, Zaniletti I, Wymore EM, Vyas-Read S, Yallapragada S, Nelin LD, Truog WE, Padula MA, Porta NFM, Savani RC, Potoka KP, Kawut SM, DiGeronimo R, Natarajan G, Zhang H, Grover TR, Engle WA, Murthy K, Children’s Hospital Neonatal Consortium Severe BPDFG. The impact of pulmonary hypertension in preterm infants with severe bronchopulmonary dysplasia through 1 year. J Pediatr 203: 218–224. e213, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Lakshminrusimha S, Steinhorn RH. Pulmonary vascular biology during neonatal transition. Clin Perinatol 26: 601–619, 1999. [PubMed] [Google Scholar]
- 327.Laliberte C, Hanna Y, Ben Fadel N, Lemyre B, Bijelic V, Barrowman N, Hoey L, Thebaud B, Katz SL. Target oxygen saturation and development of pulmonary hypertension and increased pulmonary vascular resistance in preterm infants. Pediatr Pulmonol 54: 73–81, 2019. [DOI] [PubMed] [Google Scholar]
- 328.Lammers AE, Adatia I, Cerro MJ, Diaz G, Freudenthal AH, Freudenthal F, Harikrishnan S, Ivy D, Lopes AA, Raj JU, Sandoval J, Stenmark K, Haworth SG. Functional classification of pulmonary hypertension in children: Report from the PVRI pediatric taskforce, Panama 2011. Pulm Circ 1: 280–285, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Lammers AE, Apitz C, Zartner P, Hager A, Dubowy KO, Hansmann G. Diagnostics, monitoring and outpatient care in children with suspected pulmonary hypertension/paediatric pulmonary hypertensive vascular disease. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 102 (Suppl 2): ii1–ii13, 2016. [DOI] [PubMed] [Google Scholar]
- 330.Lammers AE, Hislop AA, Flynn Y, Haworth SG. The 6-minute walk test: Normal values for children of 4-11 years of age. Arch Dis Child 93: 464–468, 2008. [DOI] [PubMed] [Google Scholar]
- 331.Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci 19: 44–50, 2004. [DOI] [PubMed] [Google Scholar]
- 332.Lassus P, Turanlahti M, Heikkila P, Andersson LC, Nupponen I, Sarnesto A, Andersson S. Pulmonary vascular endothelial growth factor and Flt-1 in fetuses, in acute and chronic lung disease, and in persistent pulmonary hypertension of the newborn. Am J Respir Crit Care Med 164: 1981–1987, 2001. [DOI] [PubMed] [Google Scholar]
- 333.Latus H, Kuehne T, Beerbaum P, Apitz C, Hansmann G, Muthurangu V, Moledina S. Cardiac MR and CT imaging in children with suspected or confirmed pulmonary hypertension/pulmonary hypertensive vascular disease. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 102 (Suppl 2): ii30–ii35, 2016. [DOI] [PubMed] [Google Scholar]
- 334.Lau KC, Frank DB, Hanna BD, Patel AR. Utility of electrocardiogram in the assessment and monitoring of pulmonary hypertension (idiopathic or secondary to pulmonary developmental abnormalities) in patients ≤18 years of age. Am J Cardiol 114: 294–299, 2014. [DOI] [PubMed] [Google Scholar]
- 335.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 273: 24266–24271, 1998. [DOI] [PubMed] [Google Scholar]
- 336.Laurikainen E, Aitasalo K, Erkinjuntti M, Wanne O. Sleep apnea syndrome in children-secondary to adenotonsillar hypertrophy? Acta Otolaryngol Suppl 492: 38–41, 1992. [PubMed] [Google Scholar]
- 337.Law MA, Grifka RG, Mullins CE, Nihill MR. Atrial septostomy improves survival in select patients with pulmonary hypertension. Am Heart J 153: 779–784, 2007. [DOI] [PubMed] [Google Scholar]
- 338.Lawrence KM, Monos S, Adams S, Herkert L, Peranteau WH, Munson DA, Hopper RK, Avitabile CM, Rintoul NE, Hedrick HL. Inhaled nitric oxide is associated with improved oxygenation in a subpopulation of infants with congenital diaphragmatic hernia and pulmonary hypertension. J Pediatr 219: 167–172, 2020. [DOI] [PubMed] [Google Scholar]
- 339.Lawrie A, Spiekerkoetter E, Martinez EC, Ambartsumian N, Sheward WJ, MacLean MR, Harmar AJ, Schmidt AM, Lukanidin E, Rabinovitch M. Interdependent serotonin transporter and receptor pathways regulate S100A4/Mts1, a gene associated with pulmonary vascular disease. Circ Res 97: 227–235, 2005. [DOI] [PubMed] [Google Scholar]
- 340.Le Cras TD, Kim DH, Gebb S, Markham NE, Shannon JM, Tuder RM, Abman SH. Abnormal lung growth and the development of pulmonary hypertension in the Fawn-Hooded rat. Am J Physiol 277: L709–L718, 1999. [DOI] [PubMed] [Google Scholar]
- 341.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol 283: L555–L562, 2002. [DOI] [PubMed] [Google Scholar]
- 342.Lee KJ, Berkelhamer SK, Kim GA, Taylor JM, O’Shea KM, Steinhorn RH, Farrow KN. Disrupted pulmonary artery cyclic guanosine monophosphate signaling in mice with hyperoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 50: 369–378, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Lee MR, Li L, Kitazawa T. Cyclic GMP causes Ca2+ desensitization in vascular smooth muscle by activating the myosin light chain phosphatase. J Biol Chem 272: 5063–5068, 1997. [DOI] [PubMed] [Google Scholar]
- 344.Lee SL, Wang WW, Finlay GA, Fanburg BL. Serotonin stimulates mitogen-activated protein kinase activity through the formation of superoxide anion. Am J Physiol 277: L282–L291, 1999. [DOI] [PubMed] [Google Scholar]
- 345.Lepetit H, Eddahibi S, Fadel E, Frisdal E, Munaut C,Noel A, Humbert M, Adnot S, D’Ortho MP, Lafuma C. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur Respir J 25: 834–842, 2005.15863640 [Google Scholar]
- 346.Levy M, Bonnet D, Mauge L, Celermajer DS, Gaussem P, Smadja DM. Circulating endothelial cells in refractory pulmonary hypertension in children: Markers of treatment efficacy and clinical worsening. PLoS One 8: e65114,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Levy M, Eyries M, Szezepanski I, Ladouceur M, Nadaud S, Bonnet D, Soubrier F. Genetic analyses in a cohort of children with pulmonary hypertension. Eur Respir J 48: 1118–1126,2016. [DOI] [PubMed] [Google Scholar]
- 348.Li H, Chen SJ, Chen YF, Meng QC, Durand J, Oparil S, Elton TS. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J Appl Physiol (1985) 77: 1451–1459, 1994. [DOI] [PubMed] [Google Scholar]
- 349.Li L, Jick S, Breitenstein S, Hernandez G, Michel A, Vizcaya D. Pulmonary arterial hypertension in the USA: An epidemiological study in a large insured pediatric population. Pulm Circ 7: 126–136, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Li M, Liu Y, Jin F, Sun X, Li Z, Liu Y, Fang P, Shi H, Jiang X. Endothelin-1 induces hypoxia inducible factor 1alpha expression in pulmonary artery smooth muscle cells. FEBS Lett 586: 3888–3893, 2012. [DOI] [PubMed] [Google Scholar]
- 351.Li R, Zhang J. Diagnostic value of chest CT combined with X-ray for premature infants with bronchopulmonary dysplasia. Medicine (Baltimore) 97: e9723, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JX, Jamieson SW, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med 15: 1289–1297, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Li Y, Xie M, Wang X, Lu Q, Fu M. Right ventricular regional and global systolic function is diminished in patients with pulmonary arterial hypertension: A 2-dimensional ultrasound speckle tracking echocardiography study. Int J Cardiovasc Imaging 29: 545–551, 2013. [DOI] [PubMed] [Google Scholar]
- 354.Limsuwan A, Khosithseth A, Wanichkul S, Khowsathit P. Aerosolized iloprost for pulmonary vasoreactivity testing in children with longstanding pulmonary hypertension related to congenital heart disease. Catheter Cardiovasc Interv 73: 98–104, 2009. [DOI] [PubMed] [Google Scholar]
- 355.Limsuwan A, Khowsathit P. Assessment of pulmonary vasoreactivity in children with pulmonary hypertension. Curr Opin Pediatr 21: 594–599, 2009. [DOI] [PubMed] [Google Scholar]
- 356.Limsuwan A, Wanitkul S, Khosithset A, Attanavanich S, Samankatiwat P. Aerosolized iloprost for postoperative pulmonary hypertensive crisis in children with congenital heart disease. Int J Cardiol 129: 333–338, 2008. [DOI] [PubMed] [Google Scholar]
- 357.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: A novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. [DOI] [PubMed] [Google Scholar]
- 358.Liu CQ, Ma L, Tang LM, He XJ, Wei SF, Wang SX, Zhang GY. A randomized controlled study on the efficacy of inhaled nitric oxide in treatment of neonates with meconium aspiration syndrome. Zhonghua Er Ke Za Zhi 46: 224–228, 2008. [PubMed] [Google Scholar]
- 359.Loh E, Stamler JS, Hare JM, Loscalzo J, Colucci WS. Cardiovascular effects of inhaled nitric oxide in patients with left ventricular dysfunction. Circulation 90: 2780–2785, 1994. [DOI] [PubMed] [Google Scholar]
- 360.Lohani O, Colvin KL, Yeager ME. Biomarkers for pediatric pulmonary arterial hypertension: Challenges and recommendations. Paediatr Respir Rev 16: 225–231, 2015. [DOI] [PubMed] [Google Scholar]
- 361.Long L, MacLean MR, Jeffery TK, Morecroft I, Yang X, Rudarakanchana N, Southwood M, James V, Trembath RC, Morrell NW. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res 98: 818–827, 2006. [DOI] [PubMed] [Google Scholar]
- 362.Lopes A, and Barst R. A proposed algorithm for management of patients with congenital cardiac defects associated with pulmonary hypertension. https://pvrinstitute.org [Google Scholar]
- 363.Lu X, Nadvoretskiy V, Bu L, Stolpen A, Ayres N, Pignatelli RH, Kovalchin JP, Grenier M, Klas B, Ge S. Accuracy and reproducibility of real-time three-dimensional echocardiography for assessment of right ventricular volumes and ejection fraction in children. J Am Soc Echocardiogr 21: 84–89, 2008. [DOI] [PubMed] [Google Scholar]
- 364.Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: Therapeutic considerations for a novel class of cardiovascular drugs. Circulation 102: 2434–2440, 2000. [DOI] [PubMed] [Google Scholar]
- 365.Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 21: 269–280, 2006. [DOI] [PubMed] [Google Scholar]
- 366.Maarman G, Lecour S, Butrous G, Thienemann F, Sliwa K. A comprehensive review: The evolution of animal models in pulmonary hypertension research; are we there yet? Pulm Circ 3: 739–756, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Machado RD, Eickelberg O, Elliott CG, Geraci MW, Hanaoka M, Loyd JE, Newman JH, Phillips JA 3rd, Soubrier F, Trembath RC, Chung WK. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 54: S32–S42, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Madani MM, Wittine LM, Auger WR, Fedullo PF, Kerr KM, Kim NH, Test VJ, Kriett JM, Jamieson SW. Chronic thromboembolic pulmonary hypertension in pediatric patients. J Thorac Cardiovasc Surg 141: 624–630, 2011. [DOI] [PubMed] [Google Scholar]
- 369.Mahajan CN, Afolayan AJ, Eis A, Teng RJ, Konduri GG. Altered prostanoid metabolism contributes to impaired angiogenesis in persistent pulmonary hypertension in a fetal lamb model. Pediatr Res 77: 455–462, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Mahgoub L, Kaddoura T, Kameny AR, Lopez Ortego P, Vanderlaan RD, Kakadekar A, Dicke F, Rebeyka I, Calderone CA, Redington A, Del Cerro MJ, Fineman J, Adatia I. Pulmonary vein stenosis of ex-premature infants with pulmonary hypertension and bronchopulmonary dysplasia, epidemiology, and survival from a multicenter cohort. Pediatr Pulmonol 52: 1063–1070, 2017. [DOI] [PubMed] [Google Scholar]
- 371.Maiya S, Hislop AA, Flynn Y, Haworth SG. Response to bosentan in children with pulmonary hypertension. Heart 92: 664–670, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Majesky MW. Adventitia and perivascular cells. Arterioscler Thromb Vasc Biol 35: e31–e35, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Makker K, Afolayan AJ, Teng RJ, Konduri GG. Altered hypoxia-inducible factor-1alpha (HIF-1alpha) signaling contributes to impaired angiogenesis in fetal lambs with persistent pulmonary hypertension of the newborn (PPHN). Physiol Rep 7: e13986, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Malowitz JR, Forsha DE, Smith PB, Cotten CM, Barker PC, Tatum GH. Right ventricular echocardiographic indices predict poor outcomes in infants with persistent pulmonary hypertension of the newborn. Eur Heart J Cardiovasc Imaging 16: 1224–1231, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Maron BA, Brittain EL, Choudhary G, Gladwin MT. Redefining pulmonary hypertension. Lancet Respir Med 6: 168–170, 2018. [DOI] [PubMed] [Google Scholar]
- 376.Marron MJ, Crisafi MA, Driscoll JM Jr, Wung JT, Driscoll YT, Fay TH, James LS. Hearing and neurodevelopmental outcome in survivors of persistent pulmonary hypertension of the newborn. Pediatrics 90: 392–396, 1992. [PubMed] [Google Scholar]
- 377.Marsboom G, Chen Z, Yuan Y, Zhang Y, Tiruppathi C, Loyd JE, Austin ED, Machado RF, Minshall RD, Rehman J, Malik AB. Aberrant caveolin-1-mediated Smad signaling and proliferation identified by analysis of adenine 474 deletion mutation (c.474delA) in patient fibroblasts: A new perspective on the mechanism of pulmonary hypertension. Mol Biol Cell 28: 1177–1185, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Masarwa R, Bar-Oz B, Gorelik E, Reif S, Perlman A, Matok I. Prenatal exposure to selective serotonin reuptake inhibitors and serotonin norepinephrine reuptake inhibitors and risk for persistent pulmonary hypertension of the newborn: A systematic review, meta-analysis, and network meta-analysis. Am J Obstet Gynecol 220: 57 e1–57 e13, 2019. [DOI] [PubMed] [Google Scholar]
- 379.Matsuda Y, Hoshikawa Y, Ameshima S, Suzuki S, Okada Y, Tabata T, Sugawara T, Matsumura Y, Kondo T. Effects of peroxisome proliferator-activated receptor gamma ligands on monocrotaline-induced pulmonary hypertension in rats. Nihon Kokyuki Gakkai Zasshi 43: 283–288, 2005. [PubMed] [Google Scholar]
- 380.Maxey DM, Ivy DD, Ogawa MT, Feinstein JA. Food and Drug Administration (FDA) postmarket reported side effects and adverse events associated with pulmonary hypertension therapy in pediatric patients. Pediatr Cardiol 34: 1628–1636, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.McCrary AW, Barker PCA, Torok RD, Spears TG, Li JS, Hornik CP, Laughon MM. Agreement of an echocardiogram-based diagnosis of pulmonary hypertension in infants at risk for bronchopulmonary dysplasia among masked reviewers. J Perinatol 39: 248–255, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.McMurtry IF, Bauer NR, Fagan KA, Nagaoka T, Gebb SA, Oka M. Hypoxia and Rho/Rho-kinase signaling. Lung development versus hypoxic pulmonary hypertension. Adv Exp Med Biol 543: 127–137, 2003. [PubMed] [Google Scholar]
- 383.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 95: 830–840, 2004. [DOI] [PubMed] [Google Scholar]
- 384.McNamara PJ, Murthy P, Kantores C, Teixeira L, Engelberts D, van Vliet T, Kavanagh BP, Jankov RP. Acute vasodilator effects of Rho-kinase inhibitors in neonatal rats with pulmonary hypertension unresponsive to nitric oxide. Am J Physiol Lung Cell Mol Physiol 294: L205–L213, 2008. [DOI] [PubMed] [Google Scholar]
- 385.Menendez Suso JJ, Del Cerro Marin MJ, Dorao Martinez-Romillo P, Labrandero deLera C, Fernandez Garcia-Moya L, Rodriguezgonzalez JI. Nonketotic hyperglycinemia presenting as pulmonary hypertensive vascular disease and fatal pulmonary edema in response to pulmonary vasodilator therapy. J Pediatr 161: 557–559, 2012. [DOI] [PubMed] [Google Scholar]
- 386.Mercier JC, Hummler H, Durrmeyer X, Sanchez-Luna M, Carnielli V, Field D, Greenough A, Van Overmeire B, Jonsson B, Hallman M, Baldassarre J, Group ES. Inhaled nitric oxide for prevention of bronchopulmonary dysplasia in premature babies (EUNO): A randomised controlled trial. Lancet 376: 346–354, 2010. [DOI] [PubMed] [Google Scholar]
- 387.Mestan KK, Gotteiner N, Porta N, Grobman W, Su EJ, Ernst LM. Cord blood biomarkers of placental maternal vascular underperfusion predict bronchopulmonary dysplasia-associated pulmonary hypertension. J Pediatr 185: 33–41, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Meyrick B, Gamble W, Reid L. Development of crotalaria pulmonary hypertension: Hemodynamic and structural study. Am J Physiol 239: H692–H702, 1980. [DOI] [PubMed] [Google Scholar]
- 389.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 105: 244–250, 2002. [DOI] [PubMed] [Google Scholar]
- 390.Micheletti A, Hislop AA, Lammers A, Bonhoeffer P, Derrick G, Rees P, Haworth SG. Role of atrial septostomy in the treatment of children with pulmonary arterial hypertension. Heart 92: 969–972, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Michiels C, Arnould T, Remacle J. Endothelial cell responses to hypoxia: Initiation of a cascade of cellular interactions. Biochim Biophys Acta 1497: 1–10, 2000. [DOI] [PubMed] [Google Scholar]
- 392.Miele L, Golde T, Osborne B. Notch signaling in cancer. Curr Mol Med 6: 905–918, 2006. [DOI] [PubMed] [Google Scholar]
- 393.Minai OA, Gudavalli R, Mummadi S, Liu X, McCarthy K, Dweik RA. Heart rate recovery predicts clinical worsening in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 185:400–408, 2012. [DOI] [PubMed] [Google Scholar]
- 394.Miniati M, Monti S, Bottai M, Scoscia E, Bauleo C, Tonelli L, Dainelli A, Giuntini C. Survival and restoration of pulmonary perfusion in a long-term follow-up of patients after acute pulmonary embolism. Medicine (Baltimore) 85: 253–262, 2006. [DOI] [PubMed] [Google Scholar]
- 395.Mitchell JA, Warner TD. Cyclo-oxygenase-2: Pharmacology, physiology, biochemistry and relevance to NSAID therapy. Br J Pharmacol 128: 1121–1132, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Mitchell JA, Warner TD. COX isoforms in the cardiovascular system: Understanding the activities of non-steroidal anti-inflammatory drugs. Nat Rev Drug Discov 5: 75–86, 2006. [DOI] [PubMed] [Google Scholar]
- 397.Mitchell MB, Campbell DN, Ivy D, Boucek MM, Sondheimer HM, Pietra B, Das BB, Coll JR. Evidence of pulmonary vascular disease after heart transplantation for Fontan circulation failure. J Thorac Cardiovasc Surg 128: 693–702, 2004. [DOI] [PubMed] [Google Scholar]
- 398.Mitchell WB. Thromboinflammation in COVID-19 acute lung injury. Paediatr Respir Rev 35: 20–24, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Mohamed WA, Ismail M. A randomized, double-blind, placebo-controlled, prospective study of bosentan for the treatment of persistent pulmonary hypertension of the newborn. J Perinatol 32: 608–613, 2012. [DOI] [PubMed] [Google Scholar]
- 400.Mohseni-Bod H, Bohn D. Pulmonary hypertension in congenital diaphragmatic hernia. Semin Pediatr Surg 16: 126–133, 2007. [DOI] [PubMed] [Google Scholar]
- 401.Moledina S, Hislop AA, Foster H, Schulze-Neick I, Haworth SG. Childhood idiopathic pulmonary arterial hypertension: A national cohort study. Heart 96: 1401–1406, 2010. [DOI] [PubMed] [Google Scholar]
- 402.Moledina S, Pandya B, Bartsota M, Mortensen KH, McMillan M, Quyam S, Taylor AM, Haworth SG, Schulze-Neick I, Muthurangu V. Prognostic significance of cardiac magnetic resonance imaging in children with pulmonary hypertension. Circ Cardiovasc Imaging 6: 407–414, 2013. [DOI] [PubMed] [Google Scholar]
- 403.Mondy KE, Gottdiener J, Overton ET, Henry K, Bush T, Conley L, Hammer J, Carpenter CC, Kojic E, Patel P, Brooks JT, Investigators SUNS. High prevalence of echocardiographic abnormalities among HIV-infected persons in the era of highly active antiretroviral therapy. Clin Infect Dis 52: 378–386, 2011. [DOI] [PubMed] [Google Scholar]
- 404.Montalva L, Antounians L, Zani A. Pulmonary hypertension secondary to congenital diaphragmatic hernia: Factors and pathways involved in pulmonary vascular remodeling. Pediatr Res 85: 754–768, 2019. [DOI] [PubMed] [Google Scholar]
- 405.Montani D, Girerd B, Jais X, Levy M, Amar D, Savale L, Dorfmuller P, Seferian A, Lau EM, Eyries M, Le Pavec J, Parent F, Bonnet D, Soubrier F, Fadel E, Sitbon O, Simonneau G, Humbert M. Clinical phenotypes and outcomes of heritable and sporadic pulmonary veno-occlusive disease: A population-based study. Lancet Respir Med 5: 125–134, 2017. [DOI] [PubMed] [Google Scholar]
- 406.Montedonico S, Nakazawa N, Puri P. Retinoic acid rescues lung hypoplasia in nitrofen-induced hypoplastic foetal rat lung explants. Pediatr Surg Int 22: 2–8, 2006. [DOI] [PubMed] [Google Scholar]
- 407.Morchi GS, Ivy DD, Duster MC, Claussen L, Chan KC, Kay J. Sildenafil increases systemic saturation and reduces pulmonary artery pressure in patients with failing Fontan physiology. Congenit Heart Dis 4: 107–111,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Moreland RB, Goldstein I, Traish A. Sildenafil, a novel inhibitor of phosphodiesterase type 5 in human corpus cavernosum smooth muscle cells. Life Sci 62: PL 309–PL 318, 1998. [DOI] [PubMed] [Google Scholar]
- 409.Moreno-Galdo A, Torrent-Vernetta A, de Mir Messa I, Rovira Amigo S, Gran Pina F, Gartner S, Albert Brotons B. Use of inhaled iloprost in children with pulmonary hypertension. Pediatr Pulmonol 50: 370–379, 2015. [DOI] [PubMed] [Google Scholar]
- 410.Moro MA, Russel RJ, Cellek S, Lizasoain I, Su Y, Darley-Usmar VM, Radomski MW, Moncada S. cGMP mediates the vascular and platelet actions of nitric oxide: Confirmation using an inhibitor of the soluble guanylyl cyclase. Proc Natl Acad Sci USA 93: 1480–1485, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J 53, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Morris K, Beghetti M, Petros A, Adatia I, Bohn D. Comparison of hyperventilation and inhaled nitric oxide for pulmonary hypertension after repair of congenital heart disease. Crit Care Med 28: 2974–2978, 2000. [DOI] [PubMed] [Google Scholar]
- 413.Morty RE, Nejman B, Kwapiszewska G, Hecker M, Zakrzewicz A, Kouri FM, Peters DM, Dumitrascu R, Seeger W, Knaus P, Schermuly RT, Eickelberg O. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 27: 1072–1078, 2007. [DOI] [PubMed] [Google Scholar]
- 414.Moser KM, Bloor CM. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest 103: 685–692, 1993. [DOI] [PubMed] [Google Scholar]
- 415.Moser KM, Page GT, Ashburn WL, Fedullo PF. Perfusion lung scans provide a guide to which patients with apparent primary pulmonary hypertension merit angiography. West J Med 148: 167–170, 1988. [PMC free article] [PubMed] [Google Scholar]
- 416.Mourani PM, Abman SH. Pulmonary vascular disease in bronchopulmonary dysplasia: Pulmonary hypertension and beyond. Curr Opin Pediatr 25: 329–337, 2013. [DOI] [PubMed] [Google Scholar]
- 417.Mourani PM, Ivy DD, Gao D, Abman SH. Pulmonary vascular effects of inhaled nitric oxide and oxygen tension in bronchopulmonary dysplasia. Am J Respir Crit Care Med 170: 1006–1013, 2004. [DOI] [PubMed] [Google Scholar]
- 418.Mourani PM, Sontag MK, Younoszai A, Ivy DD, Abman SH. Clinical utility of echocardiography for the diagnosis and management of pulmonary vascular disease in young children with chronic lung disease. Pediatrics 121: 317–325, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Mourani PM, Sontag MK, Younoszai A, Miller JI, Kinsella JP, Baker CD, Poindexter BB, Ingram DA, Abman SH. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am J Respir Crit Care Med 191: 87–95, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Mukherjee A, Dombi T, Wittke B, Lalonde R. Population pharmacokinetics of sildenafil in term neonates: Evidence of rapid maturation of metabolic clearance in the early postnatal period. Clin Pharmacol Ther 85: 56–63, 2009. [DOI] [PubMed] [Google Scholar]
- 421.Mulligan C, Beghetti M. Inhaled iloprost for the control of acute pulmonary hypertension in children: A systematic review. Pediatr Crit Care Med 13: 472–480, 2012. [DOI] [PubMed] [Google Scholar]
- 422.Munzel T, Feil R, Mulsch A, Lohmann SM, Hofmann F, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3’,5’-cyclic monophosphate-dependent protein kinase [corrected]. Circulation 108: 2172–2183, 2003. [DOI] [PubMed] [Google Scholar]
- 423.Muraskas JK, Juretschke LJ, Weiss MG, Bhola M, Besinger RE. Neonatal-perinatal risk factors for the development of persistent pulmonary hypertension of the newborn in preterm newborns. Am J Perinatol 18: 87–91, 2001. [DOI] [PubMed] [Google Scholar]
- 424.Muzaffar S, Shukla N, Bond M, Sala-Newby GB, Newby AC, Angelini GD, Jeremy JY. Superoxide from NADPH oxidase upregulates type 5 phosphodiesterase in human vascular smooth muscle cells: Inhibition with iloprost and NONOate. Br J Pharmacol 155: 847–856, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Naeem B, Ayub A, Aly AM, Malloy MH, Okorodudu AO, Jain SK. Urinary NT-proBNP as a potential noninvasive biomarker for screening of pulmonary hypertension in preterm infants: A pilot study. J Perinatol 40: 628–632, 2020. [DOI] [PubMed] [Google Scholar]
- 426.Naeije R, Chin K. Differentiating precapillary from postcapillary pulmonary hypertension. Circulation 140: 712–714, 2019. [DOI] [PubMed] [Google Scholar]
- 427.Naeije R, Gerges M, Vachiery JL, Caravita S, Gerges C, Lang IM. Hemodynamic phenotyping of pulmonary hypertension in left heart failure. Circ Heart Fail 10: e004082, 2017. [DOI] [PubMed] [Google Scholar]
- 428.Naeije R, Vachiery JL, Yerly P, Vanderpool R. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J 41: 217–223, 2013. [DOI] [PubMed] [Google Scholar]
- 429.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665–L672, 2004. [DOI] [PubMed] [Google Scholar]
- 430.Nair J, Lakshminrusimha S. Update on PPHN: Mechanisms and treatment. Semin Perinatol 38: 78–91, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Nakanishi H, Morikawa S, Kitahara S, Yoshii A, Uchiyama A, Kusuda S, Ezaki T. Morphological characterization of pulmonary microvascular disease in bronchopulmonary dysplasia caused by hyperoxia in newborn mice. Med Mol Morphol 51: 166–175, 2018. [DOI] [PubMed] [Google Scholar]
- 432.Namachivayam P, Theilen U, Butt WW, Cooper SM, Penny DJ, Shekerdemian LS. Sildenafil prevents rebound pulmonary hypertension after withdrawal of nitric oxide in children. Am J Respir Crit Care Med 174: 1042–1047, 2006. [DOI] [PubMed] [Google Scholar]
- 433.Negash S, Narasimhan SR, Zhou W, Liu J, Wei FL, Tian J, Raj JU. Role of cGMP-dependent protein kinase in regulation of pulmonary vascular smooth muscle cell adhesion and migration: Effect of hypoxia. Am J Physiol Heart Circ Physiol 297: H304–H312, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Nemoto S, Sasaki T, Ozawa H, Katsumata T, Kishi K, Okumura K, Mori Y, Umegaki O. Oral sildenafil for persistent pulmonary hypertension early after congenital cardiac surgery in children. Eur J Cardiothorac Surg 38: 71–77, 2010. [DOI] [PubMed] [Google Scholar]
- 435.Neonatal Inhaled Nitric Oxide Study G. Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure. N Engl J Med 336: 597–604, 1997. [DOI] [PubMed] [Google Scholar]
- 436.Network SSGotEKSNNR, Carlo WA, Finer NN, Walsh MC, Rich W, Gantz MG, Laptook AR, Yoder BA, Faix RG, Das A, Poole WK, Schibler K, Newman NS, Ambalavanan N, Frantz ID 3rd, Piazza AJ, Sanchez PJ, Morris BH, Laroia N, Phelps DL, Poindexter BB, Cotten CM, Van Meurs KP, Duara S, Narendran V, Sood BG, O’Shea TM, Bell EF, Ehrenkranz RA, Watterberg KL, Higgins RD. Target ranges of oxygen saturation in extremely preterm infants. N Engl J Med 362: 1959–1969, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Ni JR, Yan PJ, Liu SD, Hu Y, Yang KH, Song B, Lei JQ. Diagnostic accuracy of transthoracic echocardiography for pulmonary hypertension: A systematic review and meta-analysis. BMJ Open 9: e033084, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Nickel N, Golpon H, Greer M, Knudsen L, Olsson K, Westerkamp V, Welte T, Hoeper MM. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 39: 589–596, 2012. [DOI] [PubMed] [Google Scholar]
- 439.Nicolarsen J, Ivy D. Progress in the diagnosis and management of pulmonary hypertension in children. Curr Opin Pediatr 26: 527–535, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Nir A, Lindinger A, Rauh M, Bar-Oz B, Laer S, Schwachtgen L, Koch A, Falkenberg J, Mir TS. NT-pro-B-type natriuretic peptide in infants and children: Reference values based on combined data from four studies. Pediatr Cardiol 30: 3–8, 2009. [DOI] [PubMed] [Google Scholar]
- 441.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol 42: 482–490, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Nishida M, Eshiro K, Okada Y, Takaoka M, Matsumura Y. Roles of endothelin ETA and ETB receptors in the pathogenesis of monocrotaline-induced pulmonary hypertension. J Cardiovasc Pharmacol 44: 187–191, 2004. [DOI] [PubMed] [Google Scholar]
- 443.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 295: L422–L430, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.O’Byrne ML, Glatz AC, Hanna BD, Shinohara RT, Gillespie MJ, Dori Y, Rome JJ, Kawut SM. Predictors of catastrophic adverse outcomes in children with pulmonary hypertension undergoing cardiac catheterization: A multi-institutional analysis from the pediatric health information systems database. J Am Coll Cardiol 66: 1261–1269, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.O’Byrne ML, Kennedy KF, Kanter JP, Berger JT, Glatz AC. Risk factors for major early adverse events related to cardiac catheterization in children and young adults with pulmonary hypertension: An analysis of data from the IMPACT (improving adult and congenital treatment) registry. J Am Heart Assoc 7, 2018. DOI: 10.1161/JAHA.117.008142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Ohi R, Suzuki H, Kato T, Kasai M. Development of the lung in fetal rabbits with experimental diaphragmatic hernia. J Pediatr Surg 11:955–959, 1976. [DOI] [PubMed] [Google Scholar]
- 447.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. [DOI] [PubMed] [Google Scholar]
- 448.Okumura K, Humpl T, Dragulescu A, Mertens L, Friedberg MK. Longitudinal assessment of right ventricular myocardial strain in relation to transplant-free survival in children with idiopathic pulmonary hypertension. J Am Soc Echocardiogr 27: 1344–1351, 2014. [DOI] [PubMed] [Google Scholar]
- 449.Olguin HJ, Martinez HO, Perez CF, Mendiola BR, Espinosa LR, Pacheco JLC, Perez JF, Magana IM. Pharmacokinetics of sildenafil in children with pulmonary arterial hypertension. World J Pediatr 13: 588–592, 2017. [DOI] [PubMed] [Google Scholar]
- 450.Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, Nikkho S, Speich R, Hoeper MM, Behr J, Winkler J, Sitbon O, Popov W, Ghofrani HA, Manes A, Kiely DG, Ewert R, Meyer A, Corris PA, Delcroix M, Gomez-Sanchez M, Siedentop H, Seeger W, Aerosolized Iloprost Randomized Study G. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 347: 322–329, 2002. [DOI] [PubMed] [Google Scholar]
- 451.Olson LJ, Knych ET Jr, Herzig TC, Drewett JG. Selective guanylyl cyclase inhibitor reverses nitric oxide-induced vasorelaxation. Hypertension 29: 254–261, 1997. [DOI] [PubMed] [Google Scholar]
- 452.Ono M, Sawa Y, Fukushima N, Suhara H, Nakamura T, Yokoyama C, Tanabe T, Matsuda H. Gene transfer of hepatocyte growth factor with prostacyclin synthase in severe pulmonary hypertension of rats. Eur J Cardiothorac Surg 26: 1092–1097, 2004. [DOI] [PubMed] [Google Scholar]
- 453.Ono M, Sawa Y, Mizuno S, Fukushima N, Ichikawa H, Bessho K, Nakamura T, Matsuda H. Hepatocyte growth factor suppresses vascular medial hyperplasia and matrix accumulation in advanced pulmonary hypertension of rats. Circulation 110: 2896–2902, 2004. [DOI] [PubMed] [Google Scholar]
- 454.Opotowsky AR. Clinical evaluation and management of pulmonary hypertension in the adult with congenital heart disease. Circulation 131: 200–210, 2015. [DOI] [PubMed] [Google Scholar]
- 455.Ovaert C, Thijs D, Dewolf D, Ottenkamp J, Dessy H, Moons P, Gewillig M, Mertens L. The effect of bosentan in patients with a failing Fontan circulation. Cardiol Young 19: 331–339, 2009. [DOI] [PubMed] [Google Scholar]
- 456.Paciocco G, Martinez FJ, Bossone E, Pielsticker E, Gillespie B, Rubenfire M. Oxygen desaturation on the six-minute walk test and mortality in untreated primary pulmonary hypertension. Eur Respir J 17: 647–652, 2001. [DOI] [PubMed] [Google Scholar]
- 457.Packer M Pathophysiological mechanisms underlying the adverse effects of calcium channel-blocking drugs in patients with chronic heart failure. Circulation 80: IV59–IV67, 1989. [PubMed] [Google Scholar]
- 458.Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns HH, Kummer W, Braun-Dullaeus RC. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res 8: 15, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Paffett ML, Naik JS, Resta TC, Walker BR. Reduced store-operated Ca2+ entry in pulmonary endothelial cells from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 293: L1135–L1142, 2007. [DOI] [PubMed] [Google Scholar]
- 460.Palma G, Giordano R, Russolillo V, Cioffi S, Palumbo S, Mucerino M, Poli V, Vosa C. Sildenafil therapy for pulmonary hypertension before and after pediatric congenital heart surgery. Tex Heart Inst J 38: 238–242, 2011. [PMC free article] [PubMed] [Google Scholar]
- 461.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524–526, 1987. [DOI] [PubMed] [Google Scholar]
- 462.Pande A, Sarkar A, Ahmed I, Naveen Chandra G, Patil SK, Kundu CK, Arora R, Samanta A. Non-invasive estimation of pulmonary vascular resistance in patients of pulmonary hypertension in congenital heart disease with unobstructed pulmonary flow. Ann Pediatr Cardiol 7: 92–97, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem 269: 25646–25654, 1994. [PubMed] [Google Scholar]
- 464.Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, Lemarchand P, Eddahibi S. Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol 23: 762–771, 2000. [DOI] [PubMed] [Google Scholar]
- 465.Patel N, Mills JF, Cheung MM. Assessment of right ventricular function using tissue Doppler imaging in infants with pulmonary hypertension. Neonatology 96: 193–199; discussion 200–192, 2009. [DOI] [PubMed] [Google Scholar]
- 466.Pearson JD. Normal endothelial cell function. Lupus 9: 183–188, 2000. [DOI] [PubMed] [Google Scholar]
- 467.The Neonatal Inhaled Nitric Oxide Study Group (NINOS). Inhaled nitric oxide and hypoxic respiratory failure in infants with congenital diaphragmatic hernia. Pediatrics 99: 838–845, 1997. [DOI] [PubMed] [Google Scholar]
- 468.Perros F, Montani D, Dorfmuller P, Durand-Gasselin I, Tcherakian C, Le Pavec J, Mazmanian M, Fadel E, Mussot S, Mercier O, Herve P, Emilie D, Eddahibi S, Simonneau G, Souza R, Humbert M. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 178: 81–88, 2008. [DOI] [PubMed] [Google Scholar]
- 469.Peterson AL, Deatsman S, Frommelt MA, Mussatto K, Frommelt PC. Correlation of echocardiographic markers and therapy in persistent pulmonary hypertension of the newborn. Pediatr Cardiol 30: 160–165, 2009. [DOI] [PubMed] [Google Scholar]
- 470.Pfarr N, Fischer C, Ehlken N, Becker-Grunig T, Lopez-Gonzalez V, Gorenflo M, Hager A, Hinderhofer K, Miera O, Nagel C, Schranz D, Grunig E. Hemodynamic and genetic analysis in children with idiopathic, heritable, and congenital heart disease associated pulmonary arterial hypertension. Respir Res 14: 3, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Phosphodiesterase Type 5 (PDE5) Inhibitors. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases, 2012. [Google Scholar]
- 472.Piao L, Fang YH, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: A maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med (Berl) 91:1185–1197, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Piao L, Fang YH, Parikh KS, Ryan JJ, D’Souza KM, Theccanat T, Toth PT, Pogoriler J, Paul J, Blaxall BC, Akhter SA, Archer SL. GRK2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: Therapeutic implications in pulmonary hypertension. Circulation 126: 2859–2869, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Ploegstra MJ, Arjaans S, Zijlstra WMH, Douwes JM, Vissia-Kazemier TR, Roofthooft MTR, Hillege HL, Berger RMF. Clinical worsening as composite study end point in pediatric pulmonary arterial hypertension. Chest 148: 655–666, 2015. [DOI] [PubMed] [Google Scholar]
- 475.Ploegstra MJ, Douwes JM, Roofthooft MT, Zijlstra WM, Hillege HL, Berger RM. Identification of treatment goals in paediatric pulmonary arterial hypertension. Eur Respir J 44: 1616–1626, 2014. [DOI] [PubMed] [Google Scholar]
- 476.Ploegstra MJ, Roofthooft MT, Douwes JM, Bartelds B, Elzenga NJ, van de Weerd D, Hillege HL, Berger RM. Echocardiography in pediatric pulmonary arterial hypertension: Early study on assessing disease severity and predicting outcome. Circ Cardiovasc Imaging 8: e000878, 2015. [DOI] [PubMed] [Google Scholar]
- 477.Ploegstra MJ, Zijlstra WM, Douwes JM, Hillege HL, Berger RM. Prognostic factors in pediatric pulmonary arterial hypertension: A systematic review and meta-analysis. Int J Cardiol 184: 198–207, 2015. [DOI] [PubMed] [Google Scholar]
- 478.Pollock DM, Keith TL, Highsmith RF. Endothelin receptors and calcium signaling. FASEB J 9: 1196–1204, 1995. [DOI] [PubMed] [Google Scholar]
- 479.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 92: 967–1003, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Prins BA, Hu RM, Nazario B, Pedram A, Frank HJ, Weber MA, Levin ER. Prostaglandin E2 and prostacyclin inhibit the production and secretion of endothelin from cultured endothelial cells. J Biol Chem 269: 11938–11944, 1994. [PubMed] [Google Scholar]
- 481.Puchalski mD, Lozier JS, Bradley DJ, Minich LL, Tani LY. Electrocardiography in the diagnosis of right ventricular hypertrophy in children. Pediatrics 118: 1052–1055, 2006. [DOI] [PubMed] [Google Scholar]
- 482.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, Jansa P, Jing ZC, Le Brun FO, Mehta S, Mittelholzer CM, Perchenet L, Sastry BK, Sitbon O, Souza R, Torbicki A, Zeng X, Rubin LJ, Simonneau G, Investigators S. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 369: 809–818, 2013. [DOI] [PubMed] [Google Scholar]
- 483.Putnam LR, Tsao K, Morini F, Lally PA, Miller CC, Lally KP, Harting MT, Congenital Diaphragmatic Hernia Study G. Evaluation of variability in inhaled nitric oxide use and pulmonary hypertension in patients with congenital diaphragmatic hernia. JAMA Pediatr 170: 1188–1194, 2016. [DOI] [PubMed] [Google Scholar]
- 484.Qin W, Zhao B, Shi Y, Yao C, Jin L, Jin Y. BMPRII is a direct target of miR-21. Acta Biochim Biophys Sin (Shanghai) 41: 618–623, 2009. [DOI] [PubMed] [Google Scholar]
- 485.Rabinovitch M Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306–4313, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: Hemodynamic and structural features. Am J Physiol 236: H818–H827, 1979. [DOI] [PubMed] [Google Scholar]
- 487.Rachdi I, Tougorti M, Daoud F, Aydi Z, Zoubeidi H, Ben Dhaou B, Boussema F. Pulmonary hypertension on systemic sclerosis-lupus erythematosus overlap syndrome. Ann Cardiol Angeiol (Paris) 68: 221225, 2019. [DOI] [PubMed] [Google Scholar]
- 488.Raffini L, Huang YS, Witmer C, Feudtner C. Dramatic increase in venous thromboembolism in children’s hospitals in the United States from 2001 to 2007. Pediatrics 124: 1001–1008, 2009. [DOI] [PubMed] [Google Scholar]
- 489.Ramirez RL 3rd, Perez VJ, Zamanian RT. Methamphetamine and the risk of pulmonary arterial hypertension. Curr Opin Pulm Med 24: 416–424, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Ramos RP, Arakaki JS, Barbosa P, Treptow E, Valois FM, Ferreira EV, Nery LE, Neder JA. Heart rate recovery in pulmonary arterial hypertension: Relationship with exercise capacity and prognosis. Am Heart J 163: 580–588, 2012. [DOI] [PubMed] [Google Scholar]
- 491.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 131: 1006–1018, 2015. [DOI] [PubMed] [Google Scholar]
- 492.Rapoport RM. Nitric oxide inhibition of endothelin-1 release in the vasculature: In vivo relevance of in vitro findings. Hypertension 64: 908–914,2014. [DOI] [PubMed] [Google Scholar]
- 493.Raposo-Sonnenfeld I, Otero-Gonzalez I, Blanco-Aparicio M, Ferrer-Barba A, Medrano-Lopez C. Treatment with sildenafil, bosentan, or both in children and young people with idiopathic pulmonary arterial hypertension and Eisenmenger’s syndrome. Rev Esp Cardiol 60: 366–372, 2007. [PubMed] [Google Scholar]
- 494.Rasanen J, Wood DC, Debbs RH, Cohen J, Weiner S, Huhta JC. Reactivity of the human fetal pulmonary circulation to maternal hyperoxygenation increases during the second half of pregnancy: A randomized study. Circulation 97: 257–262, 1998. [DOI] [PubMed] [Google Scholar]
- 495.Rasanen J, Wood DC, Weiner S, Ludomirski A, Huhta JC. Role of the pulmonary circulation in the distribution of human fetal cardiac output during the second half of pregnancy. Circulation 94: 1068–1073, 1996. [DOI] [PubMed] [Google Scholar]
- 496.Raut MS, Maheshwari A. Inhaled nitric oxide, methemoglobinemia, and route of delivery. Saudi J Anaesth 11: 364, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Reddy VM, Meyrick B, Wong J, Khoor A, Liddicoat JR, Hanley FL, Fineman JR. In utero placement of aortopulmonary shunts. A model of postnatal pulmonary hypertension with increased pulmonary blood flow in lambs. Circulation 92: 606–613, 1995. [DOI] [PubMed] [Google Scholar]
- 498.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 327:76–81, 1992. [DOI] [PubMed] [Google Scholar]
- 499.Rich S, Seidlitz M, Dodin E, Osimani D, Judd D, Genthner D, McLaughlin V, Francis G. The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest 114: 787–792, 1998. [DOI] [PubMed] [Google Scholar]
- 500.Richir MC, Ellger B, Teerlink T, Siroen MP, Visser M, Spreeuwenberg M, Girbes AR, van der Hoven B, van den Berghe G, Wilhelm AJ, de Vries TP, van Leeuwen PA. The effect of rosiglitazone on asymmetric dimethylarginine (ADMA) in critically ill patients. Pharmacol Res 60: 519–524, 2009. [DOI] [PubMed] [Google Scholar]
- 501.Rodman DM, Reese K, Harral J, Fouty B, Wu S, West J, Hoedt-Miller M, Tada Y, Li KX, Cool C, Fagan K, Cribbs L. Low-voltage-activated (T-type) calcium channels control proliferation of human pulmonary artery myocytes. Circ Res 96: 864–872, 2005. [DOI] [PubMed] [Google Scholar]
- 502.Rondelet B, Kerbaul F, Motte S, van Beneden R, Remmelink M, Brimioulle S, McEntee K, Wauthy P, Salmon I, Ketelslegers JM, Naeije R. Bosentan for the prevention of overcirculation-induced experimental pulmonary arterial hypertension. Circulation 107: 1329–1335, 2003. [DOI] [PubMed] [Google Scholar]
- 503.Rondelet B, Kerbaul F, Van Beneden R, Motte S, Fesler P, Hubloue I, Remmelink M, Brimioulle S, Salmon I, Ketelslegers JM, Naeije R. Signaling molecules in overcirculation-induced pulmonary hypertension in piglets: Effects of sildenafil therapy. Circulation 110: 2220–2225, 2004. [DOI] [PubMed] [Google Scholar]
- 504.Rondelet B, Kerbaul F, Vivian GF, Hubloue I, Huez S, Fesler P, Remmelink M, Brimiouille S, Salmon I, Naeije R. Sitaxsentan for the prevention of experimental shunt-induced pulmonary hypertension. Pediatr Res 61: 284–288, 2007. [DOI] [PubMed] [Google Scholar]
- 505.Rosenkranz S, Ghofrani HA, Beghetti M, Ivy D, Frey R, Fritsch A, Weimann G, Saleh S, Apitz C. Riociguat for pulmonary arterial hypertension associated with congenital heart disease. Heart 101: 1792–1799, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, Ivy DD, Berger RMF. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur Respir J 53: 1801916, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Rosenzweig EB, Feinstein JA, Humpl T, Ivy DD. Pulmonary arterial hypertension in children: Diagnostic work-up and challenges. Prog Pediatr Cardiol 27: 4–11, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Rosenzweig EB, Morse JH, Knowles JA, Chada KK, Khan AM, Roberts KE, McElroy JJ, Juskiw NK, Mallory NC, Rich S, Diamond B, Barst RJ. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant 27: 668–674, 2008. [DOI] [PubMed] [Google Scholar]
- 509.Rothman A, Sklansky MS, Lucas VW, Kashani IA, Shaughnessy RD, Channick RN, Auger WR, Fedullo PF, Smith CM, Kriett JM, Jamieson SW. Atrial septostomy as a bridge to lung transplantation in patients with severe pulmonary hypertension. Am J Cardiol 84: 682–686, 1999. [DOI] [PubMed] [Google Scholar]
- 510.Roubliova X, Verbeken E, Wu J, Yamamoto H, Lernt T, Tibboel D, Deprest J. Pulmonary vascular morphology in a fetal rabbit model for congenital diaphragmatic hernia. J Pediatr Surg 39: 1066–1072, 2004. [DOI] [PubMed] [Google Scholar]
- 511.Roubliova XI, Verbeken EK, Wu J, Vaast P, Jani J, Deprest JA. Effect of tracheal occlusion on peripheric pulmonary vessel muscularization in a fetal rabbit model for congenital diaphragmatic hernia. Am J Obstet Gynecol 191: 830–836, 2004. [DOI] [PubMed] [Google Scholar]
- 512.Ruan H, Zhang Y, Liu R, Yang X. The acute effects of 30 mg vs 60 mg of intravenous Fasudil on patients with congenital heart defects and severe pulmonary arterial hypertension. Congenit Heart Dis 14: 645–650, 2019. [DOI] [PubMed] [Google Scholar]
- 513.Ruan HY, Zhang YG, Liu R. Acute effects of intravenous fasudil with different dosage on patients with congenital heart defects and severe pulmonary arterial hypertension. Zhonghua Yi Xue Za Zhi 98: 678–681, 2018. [DOI] [PubMed] [Google Scholar]
- 514.Rubin LJ, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, Keogh A, Langleben D, Fritsch A, Menezes F, Davie N, Ghofrani HA. Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur Respir J 45: 1303–1313, 2015. [DOI] [PubMed] [Google Scholar]
- 515.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet 11: 1517–1525, 2002. [DOI] [PubMed] [Google Scholar]
- 516.Rudolph AM, Yuan S. Response of the pulmonary vasculature to hypoxia and H+ ion concentration changes. J Clin Invest 45: 399–411, 1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Ruiz-Cano MJ, Escribano P, Alonso R, Delgado J, Carreira P, Velazquez T, Sanchez MA, Saenz de la Calzada C. Comparison of baseline characteristics and survival between patients with idiopathic and connective tissue disease-related pulmonary arterial hypertension. J Heart Lung Transplant 28: 621–627, 2009. [DOI] [PubMed] [Google Scholar]
- 518.Ryan JJ, Huston J, Kutty S, Hatton ND, Bowman L, Tian L, Herr JE, Johri AM, Archer SL. Right ventricular adaptation and failure in pulmonary arterial hypertension. Can J Cardiol 31: 391–406, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Mr Sabri, Beheshtian E. Comparison of the therapeutic and side effects of tadalafil and sildenafil in children and adolescents with pulmonary arterial hypertension. Pediatr Cardiol 35: 699–704, 2014. [DOI] [PubMed] [Google Scholar]
- 520.Sadiq HF, Mantych G, Benawra RS, Devaskar UP, Hocker JR. Inhaled nitric oxide in the treatment of moderate persistent pulmonary hypertension of the newborn: A randomized controlled, multicenter trial. J Perinatol 23: 98–103, 2003. [DOI] [PubMed] [Google Scholar]
- 521.Said S, Porres-Aguilar M, Porres-Munoz M, Mukherjee D. Eisenmenger syndrome: Recent advances in pharmacotherapy. Cardiovasc Hematol Agents Med Chem 11: 289–296, 2013. [DOI] [PubMed] [Google Scholar]
- 522.Sandoval J, Gaspar J, Pena H, Santos LE, Cordova J, del Valle K, Rodriguez A, Pulido T. Effect of atrial septostomy on the survival of patients with severe pulmonary arterial hypertension. Eur Respir J 38: 1343–1348, 2011. [DOI] [PubMed] [Google Scholar]
- 523.Sandoval J, Santos LE, Cordova J, Pulido T, Gutierrez G, Bautista E, Martinez Guerra ML, Pena H, Broberg CS. Does anticoagulation in Eisenmenger syndrome impact long-term survival? Congenit Heart Dis 7: 268–276, 2012. [DOI] [PubMed] [Google Scholar]
- 524.Sato T, Tsujino I, Ohira H, Oyama-Manabe N, Yamada A, Ito YM, Goto C, Watanabe T, Sakaue S, Nishimura M. Validation study on the accuracy of echocardiographic measurements of right ventricular systolic function in pulmonary hypertension. J Am Soc Echocardiogr 25: 280–286, 2012. [DOI] [PubMed] [Google Scholar]
- 525.Sawada H, Mitani Y, Nakayama T, Fukushima H, Kogaki S, Igarashi T, Ichida F, Ono Y, Nakanishi T, Doi S, Ishikawa S, Matsushima M, Yamada O, Saji T. Detection of pediatric pulmonary arterial hypertension by school electrocardiography mass screening. Am J Respir Crit Care Med 199: 1397–1406, 2019. [DOI] [PubMed] [Google Scholar]
- 526.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 115: 2811–2821, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Schlapbach LJ, Horton SB, Long DA, Beca J, Erickson S, Festa M, d’Udekem Y, Alphonso N, Winlaw D, Johnson K, Delzoppo C, van Loon K, Gannon B, Fooken J, Blumenthal A, Young P, Jones M, Butt W, Schibler A, Nitric Study Group tA, New Zealand Intensive Care Society Clinical Trials Group tPCCRg, and the APSG. Study protocol: NITric oxide during cardiopulmonary bypass to improve Recovery in Infants with Congenital heart defects (NITRIC trial): A randomised controlled trial. BMJ Open 9: e026664, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Schmidt B, Whyte RK. Oxygen saturation target ranges and alarm settings in the NICU: What have we learnt from the neonatal oxygenation prospective meta-analysis (NeOProM)? Semin Fetal Neonatal Med 25: 101080, 2020. [DOI] [PubMed] [Google Scholar]
- 529.Schmidt B, Whyte RK, Asztalos EV, Moddemann D, Poets C, Rabi Y, Solimano A, Roberts RS, Canadian Oxygen Trial G. Effects of targeting higher vs lower arterial oxygen saturations on death or disability in extremely preterm infants: A randomized clinical trial. JAMA 309: 2111–2120, 2013. [DOI] [PubMed] [Google Scholar]
- 530.Schreiber MD, Gin-Mestan K, Marks JD, Huo D, Lee G, Srisuparp P. Inhaled nitric oxide in premature infants with the respiratory distress syndrome. N Engl J Med 349: 2099–2107, 2003. [DOI] [PubMed] [Google Scholar]
- 531.Schulze-Neick I, Hartenstein P, Li J, Stiller B, Nagdyman N, Hubler M, Butrous G, Petros A, Lange P, Redington AN. Intravenous sildenafil is a potent pulmonary vasodilator in children with congenital heart disease. Circulation 108 (Suppl 1): II167–II173, 2003. [DOI] [PubMed] [Google Scholar]
- 532.Schwenke DO, Pearson JT, Sonobe T, Ishibashi-Ueda H, Shimouchi A, Kangawa K, Umetani K, Shirai M. Role of Rho-kinase signaling and endothelial dysfunction in modulating blood flow distribution in pulmonary hypertension. J Appl Physiol (1985) 110: 901–908, 2011. [DOI] [PubMed] [Google Scholar]
- 533.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: An autocrine mechanism contributing to angiogenesis. J Cell Biol 141: 1659–1673, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Seta F, Rahmani M, Turner PV, Funk CD. Pulmonary oxidative stress is increased in cyclooxygenase-2 knockdown mice with mild pulmonary hypertension induced by monocrotaline. PLoS One 6: e23439, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Shaul PW, Yuhanna IS, German Z, Chen Z, Steinhorn RH, Morin FC 3rd. Pulmonary endothelial NO synthase gene expression is decreased in fetal lambs with pulmonary hypertension. Am J Physiol 272: L1005–L1012, 1997. [DOI] [PubMed] [Google Scholar]
- 536.Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med (Berl) 91: 297–309, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca(2+) channels, resting [Ca(2+)](i), and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol 279: L884–L894, 2000. [DOI] [PubMed] [Google Scholar]
- 538.Shivanna B, Gowda S, Welty SE, Barrington KJ, Pammi M. Prostanoids and their analogues for the treatment of pulmonary hypertension in neonates. Cochrane Database Syst Rev 10: CD012963, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Siebert JR, Haas JE, Beckwith JB. Left ventricular hypoplasia in congenital diaphragmatic hernia. J Pediatr Surg 19: 567–571, 1984. [DOI] [PubMed] [Google Scholar]
- 540.Siehr SL, Ivy DD, Miller-Reed K, Ogawa M, Rosenthal DN, Feinstein JA. Children with pulmonary arterial hypertension and prostanoid therapy: Long-term hemodynamics. J Heart Lung Transplant 32: 546–552, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Simonneau G, D’Armini AM, Ghofrani HA, Grimminger F, Hoeper MM, Jansa P, Kim NH, Wang C, Wilkins MR, Fritsch A, Davie N, Colorado P, Mayer E. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: A long-term extension study (CHEST-2). Eur Respir J 45: 1293–1302, 2015. [DOI] [PubMed] [Google Scholar]
- 542.Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, Rich S, Fishman A. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 43: 5S–12S, 2004. [DOI] [PubMed] [Google Scholar]
- 543.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62: D34–D41, 2013. [DOI] [PubMed] [Google Scholar]
- 544.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 53: 1801913, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Simonneau G, Rubin LJ, Galie N, Barst RJ, Fleming TR, Frost A, Engel P, Kramer MR, Serdarevic-Pehar M, Layton GR, Sitbon O, Badesch DB, Group PS. Long-term sildenafil added to intravenous epoprostenol in patients with pulmonary arterial hypertension. J Heart Lung Transplant 33: 689–697, 2014. [DOI] [PubMed] [Google Scholar]
- 546.Singh RK, Richmond ME, Zuckerman WA, Lee TM, Giblin TB, Rodriguez R, Chen JM, Addonizio LJ. The use of oral sildenafil for management of right ventricular dysfunction after pediatric heart transplantation. Am J Transplant 14: 453–458, 2014. [DOI] [PubMed] [Google Scholar]
- 547.Sitbon O, Humbert M, Jagot JL, Taravella O, Fartoukh M, Parent F, Herve P, Simonneau G. Inhaled nitric oxide as a screening agent for safely identifying responders to oral calcium-channel blockers in primary pulmonary hypertension. Eur Respir J 12: 265–270, 1998. [DOI] [PubMed] [Google Scholar]
- 548.Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Herve P, Simonneau G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 111: 3105–3111, 2005. [DOI] [PubMed] [Google Scholar]
- 549.Smadja DM, Gaussem P, Mauge L, Israel-Biet D, Dignat-George F, Peyrard S, Agnoletti G, Vouhe PR, Bonnet D, Levy M. Circulating endothelial cells: A new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart disease. Circulation 119: 374–381, 2009. [DOI] [PubMed] [Google Scholar]
- 550.Smadja DM, Gaussem P, Mauge L, Lacroix R, Gandrille S, Remones V, Peyrard S, Sabatier F, Bonnet D, Levy M. Comparison of endothelial biomarkers according to reversibility of pulmonary hypertension secondary to congenital heart disease. Pediatr Cardiol 31: 657–662, 2010. [DOI] [PubMed] [Google Scholar]
- 551.Smith G, Reyes JT, Russell JL, Humpl T. Safety of maximal cardiopulmonary exercise testing in pediatric patients with pulmonary hypertension. Chest 135: 1209–1214, 2009. [DOI] [PubMed] [Google Scholar]
- 552.Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D, Yuan JX. Notch activation of Ca(2+) signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol 53: 355–367, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Soma S, Takahashi H, Muramatsu M, Oka M, Fukuchi Y. Localization and distribution of endothelin receptor subtypes in pulmonary vasculature of normal and hypoxia-exposed rats. Am J Respir Cell Mol Biol 20: 620–630, 1999. [DOI] [PubMed] [Google Scholar]
- 554.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325–1358, 2003. [DOI] [PubMed] [Google Scholar]
- 555.Souza R, Channick RN, Delcroix M, Galie N, Ghofrani HA, Jansa P, Le Brun FO, Mehta S, Perchenet L, Pulido T, Sastry BKS, Sitbon O, Torbicki A, Rubin LJ, Simonneau G. Association between six-minute walk distance and long-term outcomes in patients with pulmonary arterial hypertension: Data from the randomized SERAPHIN trial. PLoS One 13:e0193226, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Spaggiari E, Stirnemann JJ, Sonigo P, Khen-Dunlop N, De Saint BL, Ville Y. Prenatal prediction of pulmonary arterial hypertension in congenital diaphragmatic hernia. Ultrasound Obstet Gynecol 45: 572–577, 2015. [DOI] [PubMed] [Google Scholar]
- 557.Spiekerkoetter E, Sung YK, Sudheendra D, Scott V, Del Rosario P, Bill M, Haddad F, Long-Boyle J, Hedlin H, Zamanian RT. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J 50: 1602449, 2017. [DOI] [PubMed] [Google Scholar]
- 558.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, Haghighat L, de Jesus PV, Wang L, Reddy S, Zhao M, Bernstein D, Solow-Cordero DE, Beachy PA, Wandless TJ, Ten Dijke P, Rabinovitch M. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 123: 3600–3613, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Spreemann T, Bertram H, Happel CM, Kozlik-Feldmann R, Hansmann G. First-in-child use of the oral soluble guanylate cyclase stimulator riociguat in pulmonary arterial hypertension. Pulm Circ 8: 2045893217743123, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Srisuparp P, Heitschmidt M, Schreiber MD. Inhaled nitric oxide therapy in premature infants with mild to moderate respiratory distress syndrome. J Med Assoc Thai 85 (Suppl 2): S469–S478, 2002. [PubMed] [Google Scholar]
- 561.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, Tuder RM. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 261–272, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Steinhorn RH, Fineman J, Kusic-Pajic A, Cornelisse P, Gehin M, Nowbakht P, Pierce CM, Beghetti M, investigators F-s. Bosentan as adjunctive therapy for persistent pulmonary hypertension of the newborn: Results of the randomized multicenter placebo-controlled exploratory trial. J Pediatr 177:90–96 e93,2016. [DOI] [PubMed] [Google Scholar]
- 563.Steinhorn RH, Kinsella JP, Pierce C, Butrous G, Dilleen M, Oakes M, Wessel DL. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr 155: 841–847 e841, 2009. [DOI] [PubMed] [Google Scholar]
- 564.Steinhorn RH, Lakshminrusimha S. Oxygen and pulmonary vasodilation: The role of oxidative and nitrosative stress. Semin Fetal Neonatal Med 25: 101083, 2020. [DOI] [PubMed] [Google Scholar]
- 565.Steinhorn RH, Morin FC 3rd, Fineman JR. Models of persistent pulmonary hypertension of the newborn (PPHN) and the role of cyclic guanosine monophosphate (GMP) in pulmonary vasorelaxation. Semin Perinatol 21: 393–408, 1997. [DOI] [PubMed] [Google Scholar]
- 566.Stenmark KR, Durmowicz AG, Roby JD, Mecham RP, Parks WC. Persistence of the fetal pattern of tropoelastin gene expression in severe neonatal bovine pulmonary hypertension. J Clin Invest 93: 1234–1242, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Stenmark KR, Frid M, Perros F. Endothelial-to-mesenchymal transition: An evolving paradigm and a promising therapeutic target in PAH. Circulation 133: 1734–1737, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 569.Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, Riddle SR, Frid MG. The adventitia: Essential regulator of vascular wall structure and function. Annu Rev Physiol 75: 23–47, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Su BH, Peng CT, Tsai CH. Persistent pulmonary hypertension of the newborn: Echocardiographic assessment. Acta Paediatr Taiwan 42: 218–223, 2001. [PubMed] [Google Scholar]
- 571.Su PH, Chen JY. Inhaled nitric oxide in the management of preterm infants with severe respiratory failure. J Perinatol 28: 112–116, 2008. [DOI] [PubMed] [Google Scholar]
- 572.Subhedar NV, Ryan SW, Shaw NJ. Open randomised controlled trial of inhaled nitric oxide and early dexamethasone in high risk preterm infants. Arch Dis Child Fetal Neonatal Ed 77: F185–F190, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Sud N, Wells SM, Sharma S, Wiseman DA, Wilham J, Black SM. Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: Role of mitochondrial dysfunction. Am J Physiol Cell Physiol 294: C1407–C1418, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Sundaram SM, Chung L. An update on systemic sclerosis-associated pulmonary arterial hypertension: A review of the current literature. Curr Rheumatol Rep 20: 10, 2018. [DOI] [PubMed] [Google Scholar]
- 575.Swier NL, Richards B, Cua CL, Lynch SK, Yin H, Nelin LD, Smith CV, Backes CH. Pulmonary vein stenosis in neonates with severe bronchopulmonary dysplasia. Am J Perinatol 33: 671–677, 2016. [DOI] [PubMed] [Google Scholar]
- 576.Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, Schuette J, Godiwala N, Yatsenko SA, Sebastian J, Madan-Khetarpal S, Surti U, Abellar RG, Bateman DA, Wilson AL, Markham MH, Slamon J, Santos-Simarro F, Palomares M, Nevado J, Lapunzina P, Chung BH, Wong WL, Chu YWY, Mok GTK, Kerem E, Reiter J, Ambalavanan N, Anderson SA, Kelly DR, Shieh J, Rosenthal TC, Scheible K, Steiner L, Iqbal MA, McKinnon ML, Hamilton SJ, Schlade-Bartusiak K, English D, Hendson G, Roeder ER, DeNapoli TS, Littlejohn RO, Wolff DJ, Wagner CL, Yeung A, Francis D, Fiorino EK, Edelman M, Fox J, Hayes DA, Janssens S, De Baere E, Menten B, Loccufier A, Vanwalleghem L, Moerman P, Sznajer Y, Lay AS, Kussmann JL, Chawla J, Payton DJ, Phillips GE, Brosens E, Tibboel D, de Klein A, Maystadt I, Fisher R, Sebire N, Male A, Chopra M, Pinner J, Malcolm G, Peters G, Arbuckle S, Lees M, Mead Z, Quarrell O, Sayers R, Owens M, Shaw-Smith C, Lioy J, McKay E, de Leeuw N, Feenstra I, Spruijt L, Elmslie F, Thiruchelvam T, Bacino CA, Langston C, Lupski JR, Sen P, Popek E, Stankiewicz P. Pathogenetics of alveolar capillary dysplasia with misalignment of pulmonary veins. Hum Genet 135: 569–586, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Tajsic T, Morrell NW. Smooth muscle cell hypertrophy, proliferation, migration and apoptosis in pulmonary hypertension. Compr Physiol 1: 295–317,2011. [DOI] [PubMed] [Google Scholar]
- 578.Takano H, Komuro I. Peroxisome proliferator-activated receptor gamma and cardiovascular diseases. Circ J 73: 214–220, 2009. [DOI] [PubMed] [Google Scholar]
- 579.Takatsuki S, Calderbank M, Ivy DD. Initial experience with tadalafil in pediatric pulmonary arterial hypertension. Pediatr Cardiol 33:683–688, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Takatsuki S, Rosenzweig EB, Zuckerman W, Brady D, Calderbank M, Ivy DD. Clinical safety, pharmacokinetics, and efficacy of ambrisentan therapy in children with pulmonary arterial hypertension. Pediatr Pulmonol 48: 27–34, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation 106: 57–62, 2002. [DOI] [PubMed] [Google Scholar]
- 582.Tal A, Leiberman A, Margulis G, Sofer S. Ventricular dysfunction in children with obstructive sleep apnea: Radionuclide assessment. Pediatr Pulmonol 4: 139–143, 1988. [DOI] [PubMed] [Google Scholar]
- 583.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. [DOI] [PubMed] [Google Scholar]
- 584.Taylor CJ, Derrick G, McEwan A, Haworth SG, Sury MR. Risk of cardiac catheterization under anaesthesia in children with pulmonary hypertension. Br J Anaesth 98: 657–661, 2007. [DOI] [PubMed] [Google Scholar]
- 585.ten Freyhaus H, Dagnell M, Leuchs M, Vantler M, Berghausen EM, Caglayan E, Weissmann N, Dahal BK, Schermuly RT, Ostman A, Kappert K, Rosenkranz S. Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am J Respir Crit Care Med 183: 1092–1102, 2011. [DOI] [PubMed] [Google Scholar]
- 586.Teng RJ, Du J, Afolayan AJ, Eis A, Shi Y, Konduri GG. AMP kinase activation improves angiogenesis in pulmonary artery endothelial cells with in utero pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 304: L29–L42, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Teng RJ, Eis A, Bakhutashvili I, Arul N, Konduri GG. Increased superoxide production contributes to the impaired angiogenesis of fetal pulmonary arteries with in utero pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 297: L184–L195, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Thebaud B, Abman SH. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med 175: 978–985, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Thenappan T, Chan SY, Weir EK. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 315: H1322–H1331, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Thomas CA, Valentine K. Utility of routine methemoglobin laboratory assays in critically ill pediatric subjects receiving inhaled nitric oxide. J Crit Care 48: 63–65, 2018. [DOI] [PubMed] [Google Scholar]
- 591.Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, Higenbottam T, Gibbs JS, Egan J, Crozier A, Peacock A, Allcock R, Corris P, Loyd JE, Trembath RC, Nichols WC. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet 37: 741–745, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Tissot C, Ivy DD, Beghetti M. Medical therapy for pediatric pulmonary arterial hypertension. J Pediatr 157: 528–532, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Tonelli AR, Alnuaimat H, Mubarak K. Pulmonary vasodilator testing and use of calcium channel blockers in pulmonary arterial hypertension. Respir Med 104: 481–496, 2010. [DOI] [PubMed] [Google Scholar]
- 594.Tourneux P, Chester M, Grover T, Abman SH. Fasudil inhibits the myogenic response in the fetal pulmonary circulation. Am J Physiol Heart Circ Physiol 295: H1505–H1513, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Tsai IC, Tsai WL, Wang KY, Chen MC, Liang KW, Tsai HY, Liao WC. Comprehensive MDCT evaluation of patients with pulmonary hypertension: Diagnosing underlying causes with the updated Dana Point 2008 classification. AJR Am J Roentgenol 197: W471–W481, 2011. [DOI] [PubMed] [Google Scholar]
- 596.Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res 367: 643–649, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J Pathol 195: 367–374, 2001. [DOI] [PubMed] [Google Scholar]
- 598.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch D, Voelkel NF. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 159: 1925–1932, 1999. [DOI] [PubMed] [Google Scholar]
- 599.Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest 95: 1798–1807, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 144: 275–285, 1994. [PMC free article] [PubMed] [Google Scholar]
- 601.Tunks RD, Barker PC, Benjamin DK Jr, Cohen-Wolkowiez M, Fleming GA, Laughon M, Li JS, Hill KD. Sildenafil exposure and hemodynamic effect after Fontan surgery. Pediatr Crit Care Med 15: 28–34, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Uslu S, Kumtepe S, Bulbul A, Comert S, Bolat F, Nuhoglu A. A comparison of magnesium sulphate and sildenafil in the treatment of the newborns with persistent pulmonary hypertension: A randomized controlled trial. J Trop Pediatr 57: 245–250, 2011. [DOI] [PubMed] [Google Scholar]
- 603.van Loenhout RB, Tibboel D, Post M, Keijzer R. Congenital diaphragmatic hernia: Comparison of animal models and relevance to the human situation. Neonatology 96: 137–149, 2009. [DOI] [PubMed] [Google Scholar]
- 604.van Loon RL, Roofthooft MT, Hillege HL, ten Harkel AD, van Osch-Gevers M, Delhaas T, Kapusta L, Strengers JL, Rammeloo L, Clur SA, Mulder BJ, Berger RM. Pediatric pulmonary hypertension in the Netherlands: Epidemiology and characterization during the period 1991 to 2005. Circulation 124: 1755–1764, 2011. [DOI] [PubMed] [Google Scholar]
- 605.Van Meurs KP, Wright LL, Ehrenkranz RA, Lemons JA, Ball MB, Poole WK, Perritt R, Higgins RD, Oh W, Hudak ML, Laptook AR, Shankaran S, Finer NN, Carlo WA, Kennedy KA, Fridriksson JH, Steinhorn RH, Sokol GM, Konduri gG, Aschner JL, Stoll BJ, D’Angio CT, Stevenson DK. Preemie Inhaled Nitric Oxide Study. Inhaled nitric oxide for premature infants with severe respiratory failure. N Engl J Med 353: 13–22, 2005. [DOI] [PubMed] [Google Scholar]
- 606.Vandecasteele E, Drieghe B, Melsens K, Thevissen K, De Pauw M, Deschepper E, Decuman S, Bonroy C, Piette Y, De Keyser F, Brusselle G, Smith V. Screening for pulmonary arterial hypertension in an unselected prospective systemic sclerosis cohort. Eur Respir J 49: 1602275, 2017. [DOI] [PubMed] [Google Scholar]
- 607.Vattulainen-Collanus S, Akinrinade O, Li M, Koskenvuo M, Li CG, Rao SP, de Jesus Perez V, Yuan K, Sawada H, Koskenvuo JW, Alvira C, Rabinovitch M, Alastalo TP. Loss of PPARgamma in endothelial cells leads to impaired angiogenesis. J Cell Sci 129: 693–705, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608.Veit F, Pak O, Egemnazarov B, Roth M, Kosanovic D, Seimetz M, Sommer N, Ghofrani HA, Seeger W, Grimminger F, Brandes RP, Schermuly RT, Weissmann N. Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid Redox Signal 19: 2213–2231, 2013. [DOI] [PubMed] [Google Scholar]
- 609.Viola N, Alghamdi AA, Perrin DG, Wilson GJ, Coles JG, Caldarone CA. Primary pulmonary vein stenosis: The impact of sutureless repair on survival. J Thorac Cardiovasc Surg 142: 344–350, 2011. [DOI] [PubMed] [Google Scholar]
- 610.Vitali SH, Hansmann G, Rose C, Fernandez-Gonzalez A, Scheid A, Mitsialis SA, Kourembanas S. The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: Long-term follow-up. Pulm Circ 4: 619–629, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Vlahos AP, Feinstein JA, Schiller NB, Silverman NH. Extension of Doppler-derived echocardiographic measures of pulmonary vascular resistance to patients with moderate or severe pulmonary vascular disease. J Am Soc Echocardiogr 21: 711–714, 2008. [DOI] [PubMed] [Google Scholar]
- 612.Volkow ND, Fowler JS, Wang GJ, Shumay E, Telang F, Thanos PK, Alexoff D. Distribution and pharmacokinetics of methamphetamine in the human body: Clinical implications. PLoS One 5: e15269, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Walsh-Sukys MC, Tyson JE, Wright LL, Bauer CR, Korones SB, Stevenson DK, Verter J, Stoll BJ, Lemons JA, Papile LA, Shankaran S, Donovan EF, Oh W, Ehrenkranz RA, Fanaroff AA. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: Practice variation and outcomes. Pediatrics 105: 14–20, 2000. [DOI] [PubMed] [Google Scholar]
- 614.Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, Choudhury MS, Ko EA, Wang J, Wang C, Makino A, Yuan JX. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am J Physiol Lung Cell Mol Physiol 305: L154–L164, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Wang H, Jafri A, Martin RJ, Nnanabu J, Farver C, Prakash YS, Mac-Farlane PM. Severity of neonatal hyperoxia determines structural and functional changes in developing mouse airway. Am J Physiol Lung Cell Mol Physiol 307: L295–L301, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Wang J, Prakasa K, Bomma C, Tandri H, Dalal D, James C, Tichnell C, Corretti M, Bluemke D, Calkins H, Abraham TP. Comparison of novel echocardiographic parameters of right ventricular function with ejection fraction by cardiac magnetic resonance. J Am Soc Echocardiogr 20: 1058–1064, 2007. [DOI] [PubMed] [Google Scholar]
- 617.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. [DOI] [PubMed] [Google Scholar]
- 618.Wang Y, Chen S, Du J. Bosentan for treatment of pediatric idiopathic pulmonary arterial hypertension: State-of-the-art. Front Pediatr 7: 302, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Wedgwood S, Black SM. Induction of apoptosis in fetal pulmonary arterial smooth muscle cells by a combined superoxide dismutase/catalase mimetic. Am J Physiol Lung Cell Mol Physiol 285: L305–L312, 2003. [DOI] [PubMed] [Google Scholar]
- 620.Wedgwood S, Black SM. Role of reactive oxygen species in vascular remodeling associated with pulmonary hypertension. Antioxid Redox Signal 5: 759–769, 2003. [DOI] [PubMed] [Google Scholar]
- 621.Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 281: L1058–L1067, 2001. [DOI] [PubMed] [Google Scholar]
- 622.Wedgwood S, Devol JM, Grobe A, Benavidez E, Azakie A, Fineman JR, Black SM. Fibroblast growth factor-2 expression is altered in lambs with increased pulmonary blood flow and pulmonary hypertension. Pediatr Res 61: 32–36, 2007. [DOI] [PubMed] [Google Scholar]
- 623.Wedgwood S, Lakshminrusimha S, Fukai T, Russell JA, Schumacker PT, Steinhorn RH. Hydrogen peroxide regulates extracellular superoxide dismutase activity and expression in neonatal pulmonary hypertension. Antioxid Redox Signal 15: 1497–1506, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Wedgwood S, Lakshminrusimha S, Schumacker PT, Steinhorn RH. Hypoxia inducible factor signaling and experimental persistent pulmonary hypertension of the newborn. Front Pharmacol 6: 47, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Wedgwood S, Steinhorn RH. Role of reactive oxygen species in neonatal pulmonary vascular disease. Antioxid Redox Signal 21: 1926–1942, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Wedgwood S, Steinhorn RH, Bunderson M, Wilham J, Lakshminrusimha S, Brennan LA, Black SM. Increased hydrogen peroxide downregulates soluble guanylate cyclase in the lungs of lambs with persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 289: L660–L666, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Wei QF, Pan XN, Li Y, Feng L, Yao LP, Liu GL, Meng DH, Xu J, Guo XF, Liu XZ. Efficacy of inhaled nitric oxide in premature infants with hypoxic respiratory failure. Zhongguo Dang Dai Er Ke Za Zhi 16: 805–809, 2014. [PubMed] [Google Scholar]
- 628.Weigand L, Sylvester JT, Shimoda LA. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 290: L284–L290, 2006. [DOI] [PubMed] [Google Scholar]
- 629.Weir EK, Archer SL. The mechanism of acute hypoxic pulmonary vasoconstriction: The tale of two channels. FASEB J 9: 183–189, 1995. [DOI] [PubMed] [Google Scholar]
- 630.Weismann CG, Asnes JD, Bazzy-Asaad A, Tolomeo C, Ehrenkranz RA, Bizzarro MJ. Pulmonary hypertension in preterm infants: Results of a prospective screening program. J Perinatol 37: 572–577, 2017. [DOI] [PubMed] [Google Scholar]
- 631.Welch cL Austin ED, Chung WK. Genes that drive the pathobiology of pediatric pulmonary arterial hypertension. Pediatr Pulmonol, 2020. DOI: 10.1002/ppul.24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Wensel R, Opitz CF, Anker SD, Winkler J, Hoffken G, Kleber FX, Sharma R, Hummel M, Hetzer R, Ewert R. Assessment of survival in patients with primary pulmonary hypertension: Importance of cardiopulmonary exercise testing. Circulation 106: 319–324, 2002. [DOI] [PubMed] [Google Scholar]
- 633.Wessel DL, Adatia I, Van Marter LJ, Thompson JE, Kane JW, Stark AR, Kourembanas S. Improved oxygenation in a randomized trial of inhaled nitric oxide for persistent pulmonary hypertension of the newborn. Pediatrics 100: E7, 1997. [DOI] [PubMed] [Google Scholar]
- 634.West J, Austin E, Fessel JP, Loyd J, Hamid R. Rescuing the BMPR2 signaling axis in pulmonary arterial hypertension. Drug Discov Today 19: 1241–1245, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Wynn J, Krishnan U, Aspelund G, Zhang Y, Duong J, Stolar CJ, Hahn E, Pietsch J, Chung D, Moore D, Austin E, Mychaliska G, Gajarski R, Foong YL, Michelfelder E, Potolka D, Bucher B, Warner B, Grady M, Azarow K, Fletcher SE, Kutty S, Delaney J, Crombleholme T, Rosenzweig E, Chung W, Arkovitz MS. Outcomes of congenital diaphragmatic hernia in the modern era of management. J Pediatr 163: 114–119 e111, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Xie X, Wang G, Zhang D, Zhang Y, Zhu Y, Li F, Li S, Li M. Activation of peroxisome proliferator-activated receptor gamma ameliorates monocrotaline-induced pulmonary arterial hypertension in rats. Biomed Rep 3: 537–542, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Xing Y, Zheng X, Li G, Liao L, Cao W, Xing H, Shen T, Sun L, Yang B, Zhu D. MicroRNA-30c contributes to the development of hypoxia pulmonary hypertension by inhibiting platelet-derived growth factor receptor beta expression. Int J Biochem Cell Biol 64: 155–166, 2015. [DOI] [PubMed] [Google Scholar]
- 638.Yamamura H, Yamamura A, Ko EA, Pohl NM, Smith KA, Zeifman A, Powell FL, Thistlethwaite PA, Yuan JX. Activation of Notch signaling by short-term treatment with Jagged-1 enhances store-operated Ca(2+) entry in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 306: C871–C878, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Yamazaki H, Kobayashi N, Taketsuna M, Tajima K, Suzuki N, Murakami M. Safety and effectiveness of tadalafil in pediatric patients with pulmonary arterial hypertension: A sub-group analysis based on Japan post-marketing surveillance. Curr Med Res Opin 33: 2241–2249, 2017. [DOI] [PubMed] [Google Scholar]
- 640.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332: 411–415, 1988. [DOI] [PubMed] [Google Scholar]
- 641.Yang S, Banerjee S, Freitas A, Cui H, Xie N, Abraham E, Liu G. miR-21 regulates chronic hypoxia-induced pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 302: L521–L529, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res 96: 1053–1063, 2005. [DOI] [PubMed] [Google Scholar]
- 643.Ye CL, Rabinovitch M. Inhibition of elastolysis by SC-37698 reduces development and progression of monocrotaline pulmonary hypertension. Am J Physiol 261: H1255–H1267, 1991. [DOI] [PubMed] [Google Scholar]
- 644.Yee M, White RJ, Awad HA, Bates WA, McGrath-Morrow SA, O’Reilly MA. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol 178: 2601–2610, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Yilmaz O, Kahveci H, Zeybek C, Ciftel M, Kilic O. Inhaled iloprost in preterm infants with severe respiratory distress syndrome and pulmonary hypertension. Am J Perinatol 31: 321–326, 2014. [DOI] [PubMed] [Google Scholar]
- 646.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest 103: 691–696, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98: 1400–1406, 1998. [DOI] [PubMed] [Google Scholar]
- 648.Yuan SM. Pulmonary artery hypertension in childhood: The transforming growth factor-beta superfamily-related genes. Pediatr Neonatol 59: 112–119,2018. [DOI] [PubMed] [Google Scholar]
- 649.Yun TJ, Coles JG, Konstantinov IE, Al-Radi OO, Wald RM, Guerra V, de Oliveira NC, Van Arsdell GS, Williams WG, Smallhorn J, Caldarone CA. Conventional and sutureless techniques for management of the pulmonary veins: Evolution of indications from postrepair pulmonary vein stenosis to primary pulmonary vein anomalies. J Thorac Cardiovasc Surg 129: 167–174, 2005. [DOI] [PubMed] [Google Scholar]
- 650.Yung D, Widlitz AC, Rosenzweig EB, Kerstein D, Maislin G, Barst RJ. Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation 110: 660–665, 2004. [DOI] [PubMed] [Google Scholar]
- 651.Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation 105: 516–521, 2002. [DOI] [PubMed] [Google Scholar]
- 652.Zamanian RT, Hedlin H, Greuenwald P, Wilson DM, Segal JI, Jorden M, Kudelko K, Liu J, Hsi A, Rupp A, Sweatt AJ, Tuder R, Berry GJ, Rabinovitch M, Doyle RL, de Jesus Perez V, Kawut SM. Features and outcomes of methamphetamine-associated pulmonary arterial hypertension. Am J Respir Crit Care Med 197: 788–800, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Zhang D, Wang G, Han D, Zhang Y, Xu J, Lu J, Li S, Xie X, Liu L, Dong L, Li M. Activation of PPAR-gamma ameliorates pulmonary arterial hypertension via inducing heme oxygenase-1 and p21(WAF1): An in vivo study in rats. Life Sci 98: 39–43, 2014. [DOI] [PubMed] [Google Scholar]
- 654.Zhang X, Zhang X, Wang S, Luo J, Zhao Z, Zheng C, Shen J. Effects of fasudil on patients with pulmonary hypertension associated with left ventricular heart failure with preserved ejection fraction: A prospective intervention study. Can Respir J 2018: 3148259, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Zhao Y, Biswas SK, McNulty PH, Kozak M, Jun JY, Segar L. PDGF-induced vascular smooth muscle cell proliferation is associated with dysregulation of insulin receptor substrates. Am J Physiol Cell Physiol 300: C1375–C1385, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: Efficacy of combined cell and eNOS gene therapy in established disease. Circ Res 96: 442–450, 2005. [DOI] [PubMed] [Google Scholar]
- 657.Zhou G, Chen T, Raj JU. MicroRNAs in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 52: 139–151, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Zhu JH, Wang XX, Zhang FR, Shang YP, Tao QM, Zhu JH, Chen JZ. Safety and efficacy of autologous endothelial progenitor cells transplantation in children with idiopathic pulmonary arterial hypertension: Open-label pilot study. Pediatr Transplant 12: 650–655, 2008. [DOI] [PubMed] [Google Scholar]
- 659.Zhu N, Gonzaga-Jauregui C, Welch CL, Ma L, Qi H, King AK, Krishnan U, Rosenzweig EB, Ivy DD, Austin ED, Hamid R, Nichols WC, Pauciulo MW, Lutz KA, Sawle A, Reid JG, Overton JD, Baras A, Dewey F, Shen Y, Chung WK. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genom Precis Med 11: e001887, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Zijlstra WMH, Douwes JM, Rosenzweig EB, Schokker S, Krishnan U, Roofthooft MTR, Miller-Reed K, Hillege HL, Ivy DD, Berger RMF. Survival differences in pediatric pulmonary arterial hypertension: Clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol 63: 2159–2169, 2014. [DOI] [PubMed] [Google Scholar]