

Abstract
Nonstructural protein 1 (NS1) plays a crucial function in the replication, spread, and pathogenesis of influenza virus by inhibiting the host innate immune response. Here we report the discovery and optimization of novel pyrazolopyridine NS1 antagonists that can potently inhibit influenza A/PR/8/34 replication in MDCK cells, rescue MDCK cells from cytopathic effects of seasonal influenza A strains, reverse NS1-dependent inhibition of IFN-β gene expression, and suppress the slow growth phenotype in NS1-expressing yeast. These pyrazolopyridines will enable researchers to investigate NS1 function during infection and how antagonists can be utilized in the next generation of treatments for influenza infection.
Keywords: Influenza, NS1, Antagonist, Pyrazolopyridine, Antiviral
Influenza is an acute respiratory disease caused by members of the orthomyxoviridae family of RNA viruses. Recent analysis by the Centers for Disease Control and Prevention (CDC) estimates that influenza has resulted in, on average, about 600,000 hospitalization and 40,000 deaths annually in the US.1, 2 Coupled with the morbidity and mortality is the substantial financial impact on medical cost, loss of work, and hospitalizations. The “Spanish flu” of 1918, which caused an estimated 20-50 million deaths worldwide, is a stark reminder of a flu pandemic’s potential for devastation. The keystones of current public policy remain surveillance and prevention via administration of the seasonal vaccine containing the most commonly circulating viral strains. However, mutations within the viral genome lead to antigenic drift, eventually rendering the flu shots ineffective.3 Additionally, novel viruses may be formed by antigenic shift when segments of human viral strains reassort with those from other animal reservoirs (e.g. birds and pigs).4-6 The danger of antigenically shifted pathogens is exemplified by the millions of deaths in the 1957 and 1968 influenza pandemics caused by viruses derived from avian sources for which humans were immunologically unprepared.6 The dangerous introduction of an antigenically novel virus for which humans have faint immunological memory was felt again in 2009 when a version of the 1918 H1N1 virus, after circulating in pigs for nearly 100 years, led to the emergence of a pandemic strain from Mexico and California.7 This outbreak exposed shortcomings in global preparedness, as it was clear that it would take at least 6 months to generate a vaccine supply which would be useful, a timeline which could easily be superseded by a rapidly spreading virus.
An effective response to pandemic strains will require a wider variety of antiviral drugs than those that are currently available. Adamantane antivirals, which target the viral M2 protein required for viral uncoating within the host cell, are no longer recommended for use due to widespread resistance. Inhibitors of the neuraminidase protein (i.e. oseltamivir and zanamivir), which block the release of new virions from the host cells, are the most commonly prescribed treatments today.8 However, given the emergence of oseltamivir-resistant strains8 and the rapid spread of the highly pathogenic H5N1 strain among birds, it is vital to continue the pursuit of new treatment modalities.9-10 Recent FDA approval of baloxavir marboxil, which targets the viral polymerase, highlights the need for additional viral targets to be exploited therapeutically.11
A proposed antiviral mechanism targets the viral nonstructural protein 1 (NS1), which plays a key role in virus replication by repressing the innate host immune system.12-10 NS1 is a well conserved, 230-237 amino acid multifunctional protein that contains an RNA binding domain and an effector domain that are connected by a variable linker,17 and it is highly expressed during infection.18 NS1 is the centerpiece of the viral response to the host interferon (IFN) system, functioning as a key component in the temporal regulation of viral RNA synthesis, its splicing, and translation. Specifically, NS1: i) suppresses the host IFN-β response to viral infection,19-23 ii) inhibits the function of 2’-5’- oligoadenylate synthase and protein kinase R, iii) inhibits the maturation of host pre-mRNAs,24-25 iv) interferes with the host RNAi pathway, adaptive immune response, and the apoptotic response,26 and v) binds to the human PAF1 transcription elongation complex (hPAF1C), a crucial player in transcription of antiviral genes.27 In doing so, NS1 promotes and enhances viral replication, making it a compelling target for influenza treatment. As such, a number of chemical series that can disrupt NS1 function have been reported.14, 28-33 Here we report a structurally distinct class of pyrazolopyridine antagonists of NS1 function, discovered using a yeast-based screening strategy,29 their structure-activity relationship (SAR), and their ability to reduce virus replication, rescue infected cells from virus-induced cytotoxicity and restore the NS1-inhibited expression of IFN mRNA.
Expanding on a previous approach,29, 34 which uses a NS1-pYES yeast strain that exhibits a pronounced slow-growth phenotype upon expression of NS1 protein, we have performed a quantitative high-throughput screen (qHTS) against a library of 269,572 compounds from the NIH Molecular Libraries-Small Molecule Repository (MLSMR) collection for activities that specifically reversed the NS1-induced slow-growth phenotype. The published protocol29 was adapted for 1536-well plate format at 4 doses (100 nM-57 μM). The screening effort and subsequent cherry-picking experiments identified 200 hits, representing 0.08% of the chemical library.35 These hits were then evaluated for their antiviral activity and cytotoxicity. Their antiviral activity was examined by testing the compounds ability to slow replication of the A/PR/8/34 strain of influenza A virus (H1N1) in MDCK cells infected at a multiplicity of infection (MOI) of 0.1 for 48 hours. The reduction of viral titer was measured by a hemagglutination assay of the supernatant and standard TCID50 analysis. Cytotoxicity was tested in MDCK cells using the CellTiter Glo ATP cell viability assay. Twenty-three compounds belonging to two structural clusters, and seven singletons showed antiviral activity. The pyrazolopyridine hit 1 (Figure 1) demonstrated the most robust antiviral activity without toxicity towards the host MDCK cells. Based on this activity and its chemical tractability, hit 1 was chosen for further optimization. 36
Figure 1.

Pyrazolopyridine hit from qHTS screen.
In total, 40 pyrazolopyridines were evaluated in the aforementioned influenza virus replication and cytotoxicity assays. The non-commercially available analogs were synthesized via the route shown in Scheme 1.
Scheme 1.

Synthetic route to pyrazolopyridine analogs for SAR evaluation.
Most of the compounds were non-cytotoxic, and several compounds reduced viral titer > 100-fold at 10 μM. Table 1 summarizes the fold reductions in viral titer after incubating the compounds at 10 μM for 48 hours. The fold decrease in viral replication was determined by calculating the inverse log of the difference between the logTCID50 value for each compound and the DMSO control. The SAR summary of the series is illustrated in Figure 2.
Table 1.
SAR around hit 1
| Cmpd | R1 | R2 | R3 | R4 | R5 | R6 | Viral fold reduction at 10 μMa |
|
|---|---|---|---|---|---|---|---|---|
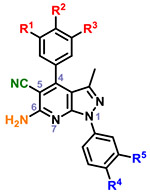 |
2 | OH | OH | H | H | H | 32 | |
| 3 | OH | OH | H | Me | H | 64 | ||
| 4 | OH | OH | H | Me | Me | 256 | ||
| 5 | OH | OH | H | F | H | 16 | ||
| 6 | OH | OH | H | Cl | H | 256 | ||
| 7 | OMe | OH | H | H | H | 32 | ||
| 8 | OMe | OH | H | Me | H | 4 | ||
| 9 | OMe | OH | H | Me | Me | 4 | ||
| 10 | OMe | OH | H | F | H | 256 | ||
| 11 | OMe | OH | H | Cl | H | 32 | ||
| 12 | OMe | OMe | H | H | H | 2 | ||
| 13 | OMe | OMe | H | Me | H | 0 | ||
| 14 | OMe | OMe | H | F | H | 2 | ||
| 15 | OMe | OMe | H | Cl | H | 0 | ||
| 16 | OMe | OMe | OMe | H | H | 16 | ||
| 17 | OMe | OMe | OMe | Me | H | 32 | ||
| 18 | OMe | OMe | OMe | Me | Me | 0 | ||
| 19 | OMe | OMe | OMe | F | H | 0 | ||
| 20 | OMe | OMe | OMe | Cl | H | 0 | ||
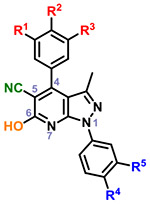 |
21 | OMe | OH | H | H | H | 16 | |
| 22 | OMe | OMe | H | H | H | 2 | ||
| 23 | OMe | OMe | OMe | H | H | 16 | ||
| 24 | OMe | OMe | H | F | H | 0 | ||
|
1 (hit) |
OEt | OH | H | F | H | 2 | ||
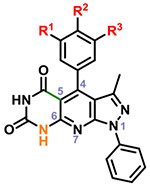 |
25 | OMe | OH | H | 2 | |||
| 26 | OMe | OMe | H | 4 | ||||
| 27 | OMe | OH | OMe | 4 | ||||
| 28 | OH | OMe | H | 2 | ||||
| 29 | -OCH2O- | H | 4 | |||||
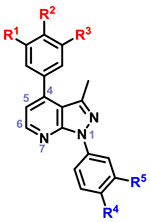 |
30 | OMe | OH | H | H | F | 0 | |
| 31 | OMe | OH | H | H | OM | 0 | ||
| 32 | OMe | OH | H | CF3 | H | 128 | ||
| 33 | OMe | OH | H | H | CF3 | 256 | ||
| 34 | OMe | OH | H | H | H | 2 | ||
| 35 | OMe | OH | H | OMe | H | 32 | ||
| 36 | OMe | OH | H | F | H | 2 | ||
| 37 | OMe | H | H | F | H | 4 | ||
| 38 | OMe | OMe | H | F | H | 2 | ||
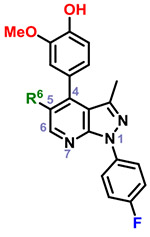 |
39 | CO2H | 4 | |||||
| 40 | CO2Et | 16 | ||||||
MDCK cell lines infected at a MOI of 0.1 for 48 h (time) (with or without compounds) were used to determine virus titer using TCID50 analysis.
Figure 2.

Structure activity relationship summary for chemical series.
Within the context of the 5-cyano-6-amino pyrazolopyridine core with a catechol-like substituent in the C4 position (i.e. 2–6), the presence of a lipophilic methyl or chloro group at R4 and/or R5 seemed to cause potent antiviral activity, with 4 and 6 showing 256-fold reduction in virus production. Capping one of the phenols (R1) with a methyl group (i.e. 7-11) led to compounds that did not strictly recapitulate this trend, but still produced 10 with similar potency. The antiviral activity of analogs with a m,p-dimethoxy phenyl (i.e. 12-15) or m,p,m-trimethoxy phenyl (i.e. 16-20) at C4 was attenuated in general. Analogs with a 6-hydroxy core (i.e. 21-24) was evaluated briefly as it was present in hit 1, and modest 16-fold reductions were observed with compounds 21 and 23. A limited examination of a pyridinedione tricyclic core (i.e. 25-29) with an unsubstituted phenyl ring at N1 revealed modest antiviral activity. The necessity of substitutions at C5 and C6 of the core was also interrogated using synthesized analogs 30-38. Significant reductions in viral titer were observed with 32 and 33, reiterating the ideal presence of a small lipophilic group at R3 or R4. This potent activity was obtained with a m-OMe-p-OH phenyl substituent at C4, implying that the presence of a catechol-like substituent, like that in analogs 2-6, is not necessary for potent activity. Modest activity was also observed with a carboxylic acid 39 or ester 40 at C5 in the context of a 4-F phenyl group at N1.
Our efforts at exploring the SAR of the screening hit 1 had therefore resulted in several compounds with potent antiviral activity. It was also noteworthy that, besides compound 37, no other analogs exhibited cytotoxicity towards the host MDCK cells. Key potent compounds with ≥128 fold reduction in viral titer (i.e. 4, 6, 10, 32, and 33) were analyzed for dose response in the TCID50 assay, and they successfully decreased viral titer with increasing concentration. As an illustrative example, analog 32 had an IC90 of 155 nM (Figure 3), and since it did not affect cell viability at 5, 10, 50 and 100 μM, it had a greater than 100-fold selectivity index (SI).
Figure 3.

The activity of 32 on viral titer. Triplicate cultures were infected with A/PR/8/34 at a MOI of 0.1 and treated with the indicated concentrations of compound. After 48 h (time), the supernatants were collected and analyzed to determine the reduction in virus by assaying the supernatant infectivity. The data are presented (left) as a percent decrease in titer back-calculated from TCID50S where IC90 is 155 nM and (right) the actual TCID50s where 3 log units of viral suppression are observed at 1.6 μM concentration.
Because the A/PR/8/34 is a mouse adapted laboratory strain, we wanted to evaluate these compounds in other viral strains that are more relevant to human infection. To that end, we utilized resources available at the National Institute of Allergy and Infectious Diseases (NIAID) and evaluated key compounds (1, 4, 6, 10, 19, 32 and 33) in an assay that measured their ability to protect host cells from cytopathic effects of various viral strains. The compounds were tested against 3 additional human influenza viruses: H1N1 (strain: A/California/7/2009), H3N2 (strain: Influenza A/Brisbane/10/20070, and H5N1 (strain: a recombinant A/PR/8/34 strain that encoded NS1 from avian A/Vietnam/1203/2004), and the results are presented in Table 2. The best activity was observed against a seasonal A/H1N1 strain, with most compounds (i.e. 1, 4, 10, 33) able to rescue cytopathic effects caused by the H1N1 virus at single digit micromolar concentrations. Compound 32 was most potent with a sub-micromolar EC50 value. The antiviral activity of these compounds was in general attenuated towards H3N2 and H5N1, and all the compounds were inactive against a common Influenza B strain, B/Brisbane/60/2008 (data not shown). Notably, compound 10 showed broad-spectrum antiviral activity against all three strains of influenza A viruses, and compound 33 showed potent activity against H1N1 and H3N2 with high selectivity indices. Curiously, we noticed that the sensitivity of this NIAID assay was reduced compared to our primary assay.
Table 2.
Selected compounds’ ability to rescue virus-mediated cytopathy of MDCK cells
| Virus Screened | Influenza A virus H1N1 | Influenza A virus H3N2 | Influenza A virus H5N1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Virus Strain | Influenza A/California/7/2009 | Influenza A/Brisbane/10/2007 |
Influenza A/Vietnam/1203/2004
× A/PR/8/34 (recombinant H5N1) |
|||||||||
| cmpd | EC50a | EC90a | CC50a | SI50a | EC50 | EC90 | CC50 | SI50 | EC50 | EC90 | CC50 | SI50 |
| 1 | 1.7 | 4.9 | >50 | >29 | 47.6 | >50 | >50 | >1 | 28.5 | >50 | >50 | >1 |
| 4 | 1.2 | 3.4 | 39.3 | 34 | >50 | >50 | 39.0 | <1 | >50 | >50 | 24.5 | <1 |
| 6 | >50 | >50 | 31.9 | <1 | >50 | >50 | 20.4 | <1 | >50 | >50 | 24.5 | <1 |
| 10 | 2.1 | 4.6 | 48.3 | 23 | 10.1 | >50 | 45.8 | 5 | 3.6 | 9.9 | 24.7 | 7 |
| 19 | 18.3 | 42.4 | 5.6 | <1 | >50 | >50 | 6.0 | <1 | >50 | >50 | 0.87 | <1 |
| 32 | 0.7 | >50 | 25.3 | 38 | 17.0 | >50 | 27.8 | 2 | >50 | >50 | 19.8 | <1 |
| 33 | 2.4 | 5.7 | 49.0 | 21 | 2.4 | >50 | 48.6 | 20 | >50 | >50 | 24.5 | <1 |
| Ribavirin | 2.2 | 3.5 | >100 | >44 | 28.8 | 34.9 | >100 | >3 | 32.2 | 68.6 | >100 | >3 |
The EC50, EC90 (Effective compound Concentration that reduces viral mediated cell death by 50% and 90% respectively) and CC50 (compound concentration that reduces cell viability by 50%) are in μM and derived after analysis at four concentrations (0.8, 4, 20, 100 μM). The selectivity index SI50 is CC50/EC50.
The most promising compounds (i.e. 4, 6, 10, 32, 33) were then cross-examined for their ability to inhibit NS1 function in the yeast-based assay that discovered the screening hit, which demonstrated compound-dependent restoration of yeast growth. The data for the lead molecule 32 is presented in Figure 4 (compounds 4, 6, 10 & 33 show the same behavior and are not shown). Examination at three time intervals (22, 42 & 60 h) clearly indicated a reversal of the slow growth phenotype that was observed with the NS1-pYES strain. Thus, 32 antagonized NS1 function in yeast, confirming that structural changes to the hit had not compromised the functional activity. Moreover, restoration of yeast growth indicates that the compound does not display acute toxicity in this assay.
Figure 4.

Compound-dependent restoration of yeast growth: Growth of the control yeast strain pYES (without NS1) and the NS1-expressing stain NS1-pYES measured as OD600nm at 22, 42 and 60 h. pYES shows growth in the presence of DMSO and 32 (50 μM), while the NS1-pYES strain exhibits a slow growth phenotype, which is reversed by the presence of 32 (50 μM).
One of the ways that NS1 enhances viral replication is by blocking the cellular IFN response.12, 15, 26 Consequently, antagonists of NS1 function might be expected to restore IFN-β mRNA levels in infected cells. As such, compounds 4, 6, 10 and 32 were further evaluated for their ability to restore IFN-β mRNA levels in MDCK cells. MDCK cells were infected with A/PR/8/34, at a MOI of 2, for 6 hours (enough time to allow an initial round of viral replication in a majority of cells), and then harvested to detect IFN-β mRNA via RT-PCR analysis. The four representative pyrazolopyridines significantly restored IFN-β mRNA levels in infected cells (Figure 5A). Compounds 6 and 32 were the best, with 6 able to restore IFN-β level similar to the known NS1 antagonist JJ329731, 37 (lane 4) or the known IFN-β inducer poly(I:C)38 (lane 2). Importantly, the compounds did not induce IFN-β in the absence of A/PR/8/34 infection (Figure 5B), demonstrating that they were antagonizing specific functions of response. Also, as shown in Figure 5C, none of the compounds directly affected the steady-state level of NS1 protein in infected cells, which indicates that the inhibitors act functionally, and not at the level of NS1 production or stability.
Figure 5.

A) MDCK cells were infected with A/PR/8/34 at a MOI of 2 and incubated in the presence of 20 μM concentration of indicated compounds for 6 h (time). Cells were harvested for RT-PCR analysis of IFN-β and β-actin mRNA. Second lane showing induction of IFN-β in the presence of poly(I:C) represents a positive control for the RT-PCR and induction of IFN- β mRNA; B) Cells were uninfected, except for the third lane, where cells were infected with A/PR/8/34. E)rug treatment and RT-PCR were as described for panel (A); C) Cells were uninfected (first lane) or infected with A/PR/8/34 in the presence of DMSO or 20 μM of the indicated compounds. Western blots were performed for NS1 and α-tubulin as loading control.
We then set out to demonstrate that the ability for the pyrazolopyridines to restore IFN-β mRNA levels in MDCK cells is dependent on NS1. Poly(I:C) is a strong inducer of IFN-β mRNA, and NS1 is known to specifically block this induction. We recapitulated this activity by co-transfection of an NS1 expression plasmid and an IFN-β luciferase reporter plasmid, where poly(I:C)-induced luciferase activity was abrogated in the presence, but not in the absence, of NS1 (Figure 6A). Treatment with NS1-inhibitory compounds in the presence of both the plasmids efficiently restored poly(I:C)-induced luciferase activity, demonstrating that that they block NS1 function. Importantly, in the absence of NS1 transfection and poly(I:C) treatment (Figure 6B), the compounds had no effect on luciferase activity, indicating that they do not induce the IFN-β reporter on their own and that their action strictly requires NS1 expression. Thus, the compounds are able to exert their effect only in the presence of the NS1 protein.
Figure 6.

A) 293 cells were co-transfected with a firefly luciferase reporter driven by the human IFN-β promoter and NS1-encoding expression constructs. At 16 h after transfection, cells were treated with 50 μg of poly(I:C)/mL and the indicated compounds (20 μM), and incubated for an additional 24 h. The cells were harvested, and their luciferase activity was measured using a Glo-max luminometer (Promega). “con” indicates the luciferase activity of the untransfected cells; “DMSO” indicates treatment with 1% DMSO; B) 293 cells were transfected with the IFN-β reporter and treated with poly(I:C), 1% DMSO, or 20 μM of the indicated compounds in the absence of poly(I:C).
Together, these experiments strongly suggest that the key pyrazolopyridines interfere with the function of NS1, specifically its ability to ameliorate the host innate immune response. Because our goal is to provide the research community with tool compounds that can be used in therapeutically relevant, advanced in vivo, proof-of-concept studies, and because no previous NS1 antagonists have been evaluated in vivo in the literature, we carried out preliminary examination of the key analogs’ in vitro ADME properties. To that end, most compounds showed good stability in mouse liver microsomes (MLM), moderate solubility, and moderate permeability (Table 3). The compounds also showed low efflux in a Caco-2 monolayer, though the absolute permeability numbers in the Caco-2 assay were also low, which is characteristic of compounds with low solubility. In order to position this series towards in vivo evaluation, we selected 32 and 33 as two representative members for a pharmacokinetics (PK) study.
Table 3.
In vitro ADME data for key analogs.
| cmpd | Aqueous
Kinetic Solubility |
Mouse Liver
Microsomal Stability (% remaining) |
Caco-2 Permeability (10−6 cm/s after 2 h) |
Efflux Ratio | ||||
|---|---|---|---|---|---|---|---|---|
| (μM) | 0 min | 15 min | 30 min | Papp A to B | Papp B to A | |||
| 32 | 3.1 | 100 | 66 | 45 | 0.10 | 0.23 | 2.29a | |
| 3% BSA | 0.22 | 0.27 | 1.20b | |||||
| 33 | 1.7 | 100 | 25 | 16 | 0.13 | 0.38 | 2.88 | |
| 6 | 1.9 | 100 | 93 | 90 | 0.34 | 2.64 | 7.78 | |
| 4 | 11.6 | 100 | 78 | 64 | ||||
| 10 | 3.0 | 100 | 74 | 60 | ||||
The mass recovery was 5 and 9%, respectively
The mass recovery was 49 and 81%, respectively, with 3%BSA.
A single intraperitoneal (IP) dose of 30 mg/kg of 32 and 33 in male C57BL/6 mice led to an AUCINF of 53894 and 49100 h*ng/mL, respectively, and >1 μM concentration in the lungs, the primary site of infection, for over 12 hours (Table 4, Figure 7). The overall exposures in the liver and lungs were ~10-20 and ~7-8 times that in plasma. The half-life was the same (Ελ ~ 4-5 h) in all organs for both compounds in plasma, liver and lung, with no signs of drug accumulation or toxicity. There were also no adverse effects observed at this single dose. The reasonable PK profile of these pyrazolopyridines should allow future in vivo experiments to examine NS1 antagonism.
Table 4.
Pharmacokinetics parameters for 32 and 33.
| PK parameters | Unit | Plasma | Liver | Lung | Plasma | Liver | Lung |
|---|---|---|---|---|---|---|---|
| Compound 32 | Compound 33 | ||||||
| Tmax | h (time) | 0.250 | 0.250 | 0.250 | 0.083 | 0.083 | 0.083 |
| Cmax | ng/mL | 2437 | 49600 | 18567 | 4470 | 64000 | 36200 |
| Terminal t1/2 | h | 4.6 | 4.7 | 4.5 | 5.2 | 5.3 | 5.0 |
| AUC (AUClast) | h* ng/mL | 7655 | 80218 | 53854 | 5960 | 119000 | 49100 |
| AUCINF | h* ng/mL | 53894 | 80282 | 53894 | 5970 | 120000 | 49100 |
| AUCorgan/AUCplasma | % | 1048 | 704 | 2010 | 823 | ||
Figure 7.

Mean plasma, liver, and lung concentrations-time profiles for compounds 32 and 33 after IP dose of 30 mpk in male C57BL/6 mice (N=3).
In summary, following a qHTS campaign for NS1 antagonists, we discovered a novel class of pyrazolopyridines that inhibits NS1 function. SAR evaluation produced several compounds that can potently reduce virus replication, cytoprotect against seasonal influenza A H1N1, and restore IFN-β mRNA levels in infected MDCK cells. Two of these analogs, 32 and 33, have acceptable pharmacokinetic profiles, making them amenable for in vivo experiments. Compound 32 has previously been disclosed as the probe ML303 in a report deposited in the public domain.39 We believe that this pyrazolopyridine series should represent useful tools that will allow future researchers to gauge NS1 antagonism in mouse models of influenza, and determine if NS1 antagonism can either reduce viral load or rescue animals from toxic effects of the influenza virus to validate NS1 as a therapeutic target.
Supplementary Material
Novel pyrazolopyridine NS1-antagonists for influenza A.
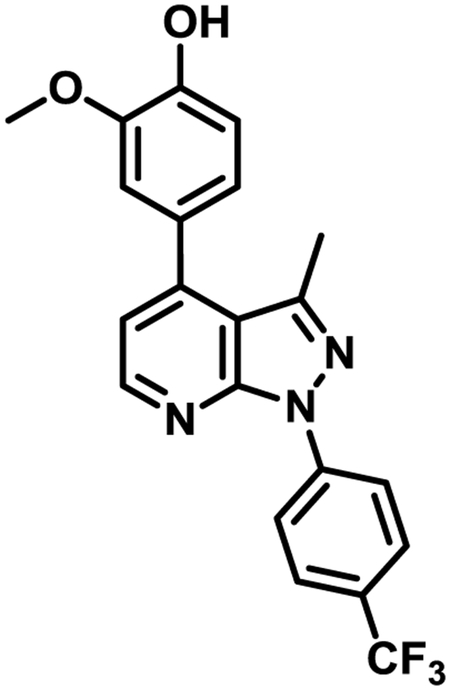
Reduces viral titer by 128-fold at 10 μM
Inhibits cytopathic effects of a seasonal influenza A strain (EC50 = 660 nM)
Restores IFN-β mRNA levels in virus-infected MDCK cells
Pharmacokinetics: > 1μM concentration in lungs for over 12 h after 30mpk IP single dose in mouse
Acknowledgments
This work was supported by NIH grant R03MH085680 to DAE and R44AI084244 to D.B. This work was also supported by contract HHSN272201100019I from the Respiratory Diseases Branch, Division of Microbiology and Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA.
Footnotes
Competing Interests Statement
The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Rolfes MA; Foppa IM; Garg S; Flannery B; Brammer L; Singleton JA; Burns E; Jernigan D; Reed C; Olsen SJ; Bresee J Estimated influenza illnesses, medical visits, hospitalizations, and deaths averted by vaccination in the united states, https://www.cdc.gov/flu/about/disease/2015-16.htm (accessed 01/31/2019). [Google Scholar]
- 2.Estimated influenza illnesses, medical visits, and hospitalizations averted by vaccination in the united states. https://www.cdc.gov/flu/about/disease/2016-17.htm (accessed 01/31/2019). [Google Scholar]
- 3.Taubenberger JK; Morens DM, The pathology of influenza virus infections. Annu Rev Pathol 2008, 3, 499–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown IH, The epidemiology and evolution of influenza viruses in pigs. Veterinary microbiology 2000, 74 (1-2), 29–46. [DOI] [PubMed] [Google Scholar]
- 5.Claas EC, Pandemic influenza is a zoonosis, as it requires introduction of avian-like gene segments in the human population. Veterinary microbiology 2000, 74(1–2), 133–9. [DOI] [PubMed] [Google Scholar]
- 6.Harder TC; Werner O, Influenza report. Ch. 2. Avian influenza. (http://www.Influenzareport.Com/ir/ai.Htm). Kamps BS; Hoffmann C; Preiser W, Eds. Flying publishers; 2006. [Google Scholar]
- 7.Bavagnoli L; Maga G, The 2009 influenza pandemic: Promising lessons for antiviral therapy for future outbreaks. Current medicinal chemistry 2011, 18 (35), 5466–75. [DOI] [PubMed] [Google Scholar]
- 8.Moscona A, Neuraminidase inhibitors for influenza. The New England journal of medicine 2005, 353 (13), 1363–73. [DOI] [PubMed] [Google Scholar]
- 9.Trampuz A; Prabhu RM; Smith TF; Baddour LM, Avian influenza: A new pandemic threat? Mayo Clinic proceedings 2004, 79 (4), 523–30; quiz 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webster RG; Govorkova EA, H5n1 influenza — continuing evolution and spread. New England Journal of Medicine 2006, 355 (21), 2174–2177. [DOI] [PubMed] [Google Scholar]
- 11.Hayden FG; Sugaya N; Hirotsu N; Lee N; de Jong MD; Hurt AC; Ishida T; Sekino H; Yamada K; Portsmouth S; Kawaguchi K; Shishido T; Arai M; Tsuchiya K; Uehara T; Watanabe A, Baloxavir marboxil for uncomplicated influenza in adults and adolescents. New England Journal of Medicine 2018, 379 (10), 913–923. [DOI] [PubMed] [Google Scholar]
- 12.Ayllon J; Garcia-Sastre A, The ns1 protein: A multitasking virulence factor. Current topics in microbiology and immunology 2015, 386, 73–107. [DOI] [PubMed] [Google Scholar]
- 13.Dash P; Thomas PG, Host detection and the stealthy phenotype in influenza virus infection. Current topics in microbiology and immunology 2015, 386, 121–47. [DOI] [PubMed] [Google Scholar]
- 14.Engel DA, The influenza virus ns1 protein as a therapeutic target. Antiviral research 2013, 99 (3), 409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krug RM, Functions of the influenza a virus ns1 protein in antiviral defense. Current opinion in virology 2015, 12, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marc D, Influenza virus non-structural protein ns1: Interferon antagonism and beyond. The Journal of general virology 2014, 95 (Pt 12), 2594–611. [DOI] [PubMed] [Google Scholar]
- 17.Bornholdt AA; Carrilli B; Venkataraman Prasad BV, Structural insights into the function of the influenza ns1. In Influenza: Molecular virology, Wang Q; Tao YJ, Eds. Caister Academic PressQinghua Wang and Yizhi Jane Tao: 2010. [Google Scholar]
- 18.Palese P; Shaw ML, Orthomyxoviridae: The viruses and their replication. In Fields virology, 5th Edition ed.; Knipe DM; Howley PM, Eds. Lippincott Williams & Wilkins: Philadelphia, 2007; pp 1647–1689. [Google Scholar]
- 19.Ludwig S; Wang X; Ehrhardt C; Zheng H; Donelan N; Planz O; Pleschka S; Garcia-Sastre A; Heins G; Wolff T, The influenza a virus ns1 protein inhibits activation of jun n-terminal kinase and ap-1 transcription factors. Journal of virology 2002, 76 (21), 11166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X; Li M; Zheng H; Muster T; Palese P; Beg AA; Garcia-Sastre A, Influenza a virus ns1 protein prevents activation of nf-kappab and induction of alpha/beta interferon. Journal of virology 2000, 74 (24), 11566–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez-Aparicio MT; Ayllon J; Leo-Macias A; Wolff T; Garcia-Sastre A, Subcellular localizations of rig-i, trim25, and mavs complexes. Journal of virology 2017, 91 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fortes P; Beloso A; Ortin J, Influenza virus ns1 protein inhibits pre-mrna splicing and blocks mrna nucleocytoplasmic transport. The EMBO journal 1994, 13 (3), 704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu Y; Krug RM, The influenza virus ns1 protein is a poly(a)-binding protein that inhibits nuclear export of mrnas containing poly(a). Journal of virology 1994, 68 (4), 2425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z; Li Y; Krug RM, Influenza a virus ns1 protein targets poly(a)-binding protein ii of the cellular 3'-end processing machinery. The EMBO journal 1999, 18 (8), 2273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nemeroff ME; Barabino SM; Li Y; Keller W; Krug RM, Influenza virus ns1 protein interacts with the cellular 30 kda subunit of cpsf and inhibits 3'end formation of cellular pre-mrnas. Molecular cell 1998, 1 (7), 991–1000. [DOI] [PubMed] [Google Scholar]
- 26.Hale BG; Randall RE; Ortin J; Jackson D, The multifunctional ns1 protein of influenza a viruses. The Journal of general virology 2008, 89 (Pt 10), 2359–76. [DOI] [PubMed] [Google Scholar]
- 27.Marazzi I; Ho JS; Kim J; Manicassamy B; Dewell S; Albrecht RA; Seibert CW; Schaefer U; Jeffrey KL; Prinjha RK; Lee K; Garcia-Sastre A; Roeder RG; Tarakhovsky A, Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483 (7390), 428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barman S; You L; Chen R; Codrea V; Kago G; Edupuganti R; Robertus J; Krug RM; Anslyn EV, Exploring naphthyl-carbohydrazides as inhibitors of influenza a viruses. European journal of medicinal chemistry 2014, 71, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu D; Walkiewicz MP; Frieman M; Baric RS; Auble DT; Engel DA, Novel influenza virus ns1 antagonists block replication and restore innate immune function. Journal of virology 2009, 83 (4), 1881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jablonski JJ; Basu D; Engel DA; Geysen HM, Design, synthesis, and evaluation of novel small molecule inhibitors of the influenza virus protein ns1. Bioorganic & medicinal chemistry 2012, 20 (1), 487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walkiewicz MP; Basu D; Jablonski JJ; Geysen HM; Engel DA, Novel inhibitor of influenza non-structural protein 1 blocks multi-cycle replication in an rnase l-dependent manner. The Journal of general virology 2011, 92 (Pt 1), 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho EJ; Xia S; Ma LC; Robertus J; Krug RM; Anslyn V; Montelione GT; Ellington AD, Identification of influenza virus inhibitors targeting ns1a utilizing fluorescence polarization-based high-throughput assay. Journal of biomolecular screening 2012, 17 (4), 448–59. [DOI] [PubMed] [Google Scholar]
- 33.Nayak MK; Agrawal AS; Bose S; Naskar S; Bhowmick R; Chakrabarti S; Sarkar S; Chawla-Sarkar M, Antiviral activity of baicalin against influenza virus h1n1-pdm09 is due to modulation of ns1-mediated cellular innate immune responses. The Journal of antimicrobial chemotherapy 2014, 69 (5), 1298–310. [DOI] [PubMed] [Google Scholar]
- 34.Ward AC; Azad AA; Macreadie IG, Expression and characterisation of the influenza a virus non-structural protein ns1 in yeast. Archives of virology 1994, 138 (3-4), 299–314. [DOI] [PubMed] [Google Scholar]
- 35.The results of this screening effort were deposited in pubchem: "Assay for inhibitors of influenza ns1 protein function" https://pubchem.Ncbi.Nlm.Nih.Gov/bioassay/2326
- 36.The details of the other hits' structure and activity in the secondary antiviral assay was deposited in pubchem: "Assay for inhibitors of influenza ns1 protein function: Influenza virus replication assay in mdck cells infected with virus a/pr/8/34" https://pubchem.Ncbi.Nlm.Nih.Gov/assay/assaydata.Html?Aid=504647&act=act.
- 37.Kleinpeter AB; Jureka AS; Falahat SM; Green TJ; Petit CM, Structural analyses reveal the mechanism of inhibition of influenza virus ns1 by two antiviral compounds. Journal of Biological Chemistry 2018, 293 (38), 14659–14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magee WE; Griffith MJ, The liver as a site for interferon production in response to poly i: Poly c. Life Sciences 1972, 11 (22, Part 2), 1081–1086. [DOI] [PubMed] [Google Scholar]
- 39.Patnaik S; Basu D; Dehdashti S; Zheng W; Ferrer M; Southall N; Taylor M; Engel DA; Marugan JJ, Discovery of small molecule influenza virus ns1 antagonist. In Probe reports from the nih molecular libraries program National Center for Biotechnology Information (US): Bethesda, MD, 2013; https://www.ncbi.nlm.nih.gov/books/NBK169451/. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.