

Abstract
Abnormallyactivated FGFR1 has been validated as a therapeutic target for differentcancers. Although a variety of FGFR inhibitors have shown benefit in manyclinical patients with FGFR1 aberration, FGFR1 mutant resistance such as V561Mmutation, has been reported. To date however, no FGFR inhibitors have beenapproved to treat patients with FGFR mutant resistance. Herein, we report that GZD824, athird generation ABL inhibitor (Phase II, China), overcomes FGFR1‐V561F/M mutant resistance in vitro and in vivo. GZD824potently suppresses FGFR1/2/3 with an IC50 value of 4.14 ± 0.96, 2.77 ± 0.082, and 8.10 ± 0.15 nmol/L. It effectively overcomes FGFR1‐V561F/M and other mutantresistance in Ba/F3 stable cells (IC50:8.1–55.0 nM), and effectively inhibits the growth of Ba/F3‐FGFR1‐V561F/M mutantxenograft tumors in vivo (TGI=73.4%, 49.8% at20mg/kg, p.o, q2d). GZD824may be considered to be an effective drug to treat patients with FGFR1 abnormalactivation or mutant resistance in clinical trials.
Keywords: FGFR1, GZD824, resistance, V561F
FGFR1‐V561F/M mutation is resistant to erdafitinib, pemigatinib, BGJ398, and TAS120 but sensitive to GZD824. GZD824 strongly suppresses FGFR1‐3 and effectively overcomes FGFR1‐V561F, V561M, and other mutant resistance in vitro (IC50:8.1–55.0 nM) and in vivo (TGI=73.4%, 49.8% at 20mg/kg, p.o, q2d). GZD824 as a type II FGFR1 inhibitor overcomes FGFR1‐V561F/M mutant resistance.

1. INTRODUCTION
The fibroblast growth factor receptor (FGFR) family consists of four isoforms FGFR1–FGFR4, whose natural ligands include a family of 22 fibroblast growth factors (FGF). The binding of FGFs and FGFRs induces receptor dimerization, recruits the fibroblast growth factor receptor substrate 2 (FRS2) adaptor proteins, 1 then phosphorylates and activates the downstream signaling cascades via MAPK and PI3K/AKT. 2 FGFR1 abnormalities have been found in a variety of cancers and play an important role in the growth, angiogenesis, migration, and resistance to cancer therapies. 3 FGFR amplification has been frequently reported in lung squamous cell carcinoma (SqCC, 5.6%–24.8%), small cell lung cancers (SCLC, 5.6%–6%), estrogen receptor‐positive breast cancers (10%), and ovary (~5%) and bladder (<2%) cancers. FGFR1 fusions have been described in lung squamous cell carcinoma, breast cancer, glioblastoma, stem cell leukemia/lymphoma syndrome (SCLL), and myeloproliferative syndrome. 4 , 5 Less common activating mutations of FGFR1, such as N546K and K656E, have been identified in dysembryoplastic neuroepithelial tumor (DNET), 6 small cell lung cancer (SCLC), 7 Ewing sarcoma, 8 and in other cancers. 9
Two non‐selective FGFR1–3 inhibitors, erdafitinib (JNJ‐42756493) and pemigatinib (INCB054828) have been approved and are being marketed to treat bladder cancer and cholangiocarcinoma in patients with FGFR2 or FGFR3 rearrangements. 10 , 11 Several selective FGFR1–3 inhibitors, including infigratinib (NVP‐BGJ398), 12 erdafitinib (JNJ42756493), 13 AZD4547, 14 and ICP‐192, have been developed and are being studied in different phase clinical trials. Other multiple kinase inhibitors, including ponatinib, 15 dovitinib, 16 lucitanib (E‐3810), 17 and nintedanib (BIBF 1120), 18 also have been shown to possess inhibitory activity against FGFRs. Currently, multiple clinical studies are in progress in patients with FGFR1 aberrantly activated cancers, including SCLL, breast, prostate, and lung cancer. 4 , 11
Although these FGFR inhibitors have shown benefits in many clinical patients with FGFR1 aberration, occurrence of FGFR1 mutant resistance has been reported. FGFR1 gatekeeper mutations such as V561M and N546K are the common mechanism of resistance to FGFR1–3 inhibitors such as BGJ398, AZD4547, 19 lucitanib (E‐3810), and FIIN‐1 in cells or patients. 20 , 21 Some FGFRs inhibitors such as FIIN‐2, 22 TAS‐120, 23 , 24 and LY2874455 25 have been shown to overcome FGFRs mutant resistance but they are insensitive to FGFR2‐V564 M/F or FGFR3‐V555 M/F excluding LY2874455. In addition, it is unclear if FGFRs inhibitors overcome the FGFR1‐V561F mutation. There is, therefore, an unmet need to evaluate novel FGFR inhibitors which could overcome FGFR1‐V561F/M mutant resistance.
GZD824 (HQP1351, 3‐methyl‐4‐((5‐methyl‐7,8‐dihydropyrido[2,3‐d]pyridazin‐3‐yl)ethynyl)‐N‐(4‐((4‐methylpiperazin‐1‐yl)methyl)‐3‐(trifluoromethyl)phenyl)benzamide 26 is a third generation ABL inhibitor designed and synthesized by our group. It has completed Phase II clinical trials for chronic myeloid leukemia (CML) patients harboring Abl mutant resistant and is included in the priority review for new drug applications (NDA) by NMPA, China. Through a KINOMEscan screening of 442 kinases, we have reported that GZD824 possess binding activity with FGFR1–3, FLT3, and PDGFRα, but its activity against FGFR1 mutant resistance has not been disclosed.
Herein, we report the activities and mechanisms by which GZD824 can overcome FGFR1‐V561F/M mutant resistance in vitro and in vivo, and explore the potential applications of GZD824 in patients with FGFR aberrant activation and mutant resistance. We report that FGFR1‐V561F mutation is resistant to erdafitinib, pemigatinib, BGJ398, and TAS120 but is sensitive to GZD824. GZD824 strongly suppresses FGFR1–3 and effectively overcomes FGFR1‐V561F/M and other mutant resistance in vitro and in vivo, and may serve as a novel drug in clinical trials of cancer therapy.
2. MATERIALS AND METHODS
2.1. Agents
GZD824 was designed and synthesized by our laboratory. NVP‐BGJ398, TAS120, pemigatinib, and erdafitinib were purchased from the Selleckchem Company (Houston, TX, USA). These compounds were dissolved in DMSO (10 mM) and stored at −20℃. Primary antibodies against FRS2α (sc‐17841) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary antibodies against FGFR1(9740S), FGFR2(23328S), FGFR3(4574S), FGFR4(8562S), phosphor‐FGFR (3471S), phosphor‐FRS2α (3861S), FRS2α (sc‐17841), ERK1/2 (4695S), phosphor‐ERK1/2 (4370S), AKT(4685S), phosphor‐AKT (4060S), caspase‐3 (9662S), cleaved caspase‐3 (9664S), caspase‐9 (9502S), cleaved caspase‐9 (9505S), cyclin E1(20808S), cyclin D2 (3741S), cyclin B1 (12231S), CDK2 (2546S), CDK4 (12790S), GAPDH (2118), and secondary antibodies were purchased from Cell Signaling Technology (Boston, MA, USA).
2.2. In vitro kinase assays
FGFR1 and the Z′‐Lyte kinase assay kit were purchased from Invitrogen (Waltham, MA, USA) and the assays were performed according to the manufacturer's instructions as described previously. 27
2.3. Cell cultures
FGFR sensitive cell lines, H1581, A204, KATOIII, NCI‐H716, SNU‐16, RT112, and MDA‐MB‐453 and other negative control cell lines MKN‐1, A549, and MDA‐MB‐231, were purchased from Shanghai Cell Bank (Type Culture Collection, China) or the American Type Culture Collection (ATCC). The base medium for MDA‐MB‐453 cell line is DMEM medium (CORNING, USA). All other cell lines were cultured in RPMI‐1640 medium supplemented with 10% FBS, 100 U/ml of penicillin, and 50 mg/ml of streptomycin in 5% CO2 at 37℃. The cells in the logarithmic growth phase were used for the experiments.
2.4. Construction of Ba/F3‐FGFR1 stable cells
The Ba/F3 cell lines stably expressing FGFR1, FGFR1‐V561M, FGFR1‐V561F, FGFR1‐K656N, and FGFR1‐K656E were self‐established through electroporation pCDNA3.1‐TEL plasmids using the Amaxa Cell Line Nucleofector Kit V (Lonza, Cologne, Germany). Stable cell lines were selected by G418 (Merck, USA) and then withdrawal of IL‐3 (R&D). All Ba/F3 stable cell lines were verified by positive drugs activities, DNA sequencing, and WB analysis. Parental Ba/F3 cells were maintained in RPMI 1640 supplemented with 10% FBS, 100 U/ml of penicillin, 50 mg/ml of streptomycin, and 10 ng/ml of IL‐3, while all Ba/F3 stable cell lines were cultured in the same medium without IL‐3.
2.5. In vitro cell proliferation Assay
Cells were seeded in 96‐well plate in complete medium (1000–6000 cells/well) and cultured for overnight. Then the cells were exposed to gradient concentrations (0.0005–10 μM) of compounds. Cell proliferation was analyzed by Cell Counting Kit 8 (CK04, Dojindo Laboratories, Japan) after co‐incubation for a further 72 h. IC50 values were calculated by fitting of concentration–response curves using GraphPad Prism 8.01 software. Each assay was repeated at least three times. Each IC50 value is displayed as mean ±SD of at least three independent experiments.
2.6. Cell cycle analysis
Cells were treated with indicated concentrations of GZD824 or DMSO for 24 h. About 6 × 105 cells were re‐suspended in 150 μl of BD Cytofix/Cytoperm buffer solution I for 10 min at room temperature. The cell suspensions were added with BD Cytofix/Cytoperm buffer solution II and incubated with another 10 min at room temperature, and then incubated with 200 μl of propidium iodide buffer (0.1 mg/ml) in the dark for 10 min at 4℃. Assays were performed with a Guava easyCyte flow cytometer (Merck, USA).
2.7. Apoptosis assay
Cells were treated with indicated concentrations of GZD824 or DMSO for 48 h at 37℃. After incubation, cells were collected and washed twice with pre‐cold PBS. About 6 × 105 cells were resuspended in 100 μl of 1× BD binding buffer solution (#556454, BD), and then stained with 7‐ADD (#559925, BD) and Annexin V‐PE (#556422, BD) in the dark for 15 min. Finally, 400 μl of 1×BD binding buffer solution were added to stop dyeing. The cells were then measured on a Guava easyCyte flow cytometer (Merck, USA).
2.8. Western blot analysis (WB)
Cells were treated with different concentrations of compound or DMSO for indicated time. Then, cells were lysed with 1 × SDS sample lysis buffer (recommended by CST). After ultra‐sonication and boiling, the cell lysates were loaded and then electrophoresed to 10%–12% SDS‐PAGE gel, and then the separated proteins were transferred to PVDF membranes. After blocking with 5% BSA in TBST solution (0.5% Tween‐20) at room temperature for 4 hrs, the membranes were incubated with the corresponding primary antibody (1:2000–1:200) at 4℃ overnight. After washing with TBST for 3–5 times, the HRP‐conjugated secondary antibodies were incubated for 2 hrs. The protein signals were detected with Amersham Imager 600 system (GE, Boston, MA, USA) using an ECL western blotting detection kit (Thermo Fisher Scientific, Waltham, MA, USA).
2.9. Induced fit docking studies
Ligands were built and prepared using LigPrep (Maestro Version 11.9, Schrodinger LLC, New York, NY, USA) with the default settings and the OPLS3e force field. The crystal structure of FGFR1 in complex with ponatinib (PDB code: 4V04) was extracted from the Protein Data Bank (http://www.pdb.org). The protein was prepared using the Protein Preparation Wizard workflow in Maestro to assign bond orders and add hydrogens. Crystal water molecules and all heteroatom (het) residues except those in ponatinib were removed. Docking studies were performed using the induced fit docking (IFD) protocol of Schrödinger (Schrödinger LLC, New York, NY, USA) software. The centroid of the cocrystal structure ligand ponatinib was used as the center of the grid box. Residues within 5 Å of the ligand poses were refined. The default protocols were used to perform induced fit docking.
2.10. Mouse xenograft tumor models
All animal studies were approved by the Institutional Animal Care and Use Committee. Male CB17‐SCID mice were purchased from Vital River Laboratory Animal Technology Inc. (Beijing, China). The right flanks of SCID mice were subcutaneously injected with 2 x 106 Ba/F3‐FGFR1, Ba/F3‐FGFR1‐V561F, or Ba/F3‐FGFR1‐V561M cells, respectively. When the average tumor volume reached 100–200 mm3, mice bearing tumor were randomly divided into groups and treated with compounds or vehicle. All the compounds were suspended in 0.5% CMC‐Na aqueous solution. GZD824 (10 or 20 mg / kg, q2d), BGJ398 (qd), or vehicle (q2d) were administrated by gavage for 12–20 days. The body weight and tumor volume of animals were measured once every 2 days. Tumor volumes were calculated as L×W×W/2, where L and W are the length and width of the tumor, respectively. At the experiment terminal, tumor tissues were dissected, paraffin embedded, and sectioned for hematoxylin and eosin (H&E), immunohistochemistry (IHC), and TUNEL analysis.
2.11. Cellular thermal shift assay (CETSA)
Engagement between FGFR1 protein and GZD824 was assessed by CETSA. For each group, 2 × 107 cells were collected and resuspended in PBS after treatment with GZD824 (1 μM for H1581, 100 nM for Ba/F3‐FGFR1‐V561F) or DMSO 4 h, then the cell suspensions of each group were divided into eight equal parts. Pairs consisting of one experimental aliquot and one DMSO aliquot were heated from 38.0 to 57.8℃ for 3 min. Then, the samples were exposed to liquid nitrogen for 10 min, and then placed in a water bath at 37℃ for 10 min. After three freeze–thaw cycles, cell lysates were centrifuged at 12000 rpm at 4℃ for 10 min, and then boiled for 15 min. The soluble supernatant was used for WB analysis.
2.12. Statistical analysis
Data are expressed as the mean ±SD of three independent experiments. Two‐tailed Student's t‐test was used for comparison between the two groups, and one‐way ANOVA was used for comparison among the multiple groups. Differences with p < 0.01 and p < 0.05 were considered very significant or significant, and marked as ** and *.
3. RESULTS
3.1. GZD824 strongly inhibits FGFR1–3 activities in vitro
We have developed GZD824 (Figure 1A) as an ABL inhibitor, and has also displayed its potential off‐target kinases to be FGFR1/2/3/4. To further explore the clinical applications of GZD824, the kinase inhibitory activities against FGFRs of GZD824 were evaluated using the FRET‐based Z′‐Lyte assay. Under the experimental conditions, GZD824 effectively inhibits FGFR1/2/3/4 with IC50 values of 4.14 ± 0.96, 2.77 ± 0.082, 8.10 ± 0.15, and 30.27 ± 21.66 nM, respectively (Table 1).
FIGURE 1.

GZD824 is a pan‐FGFR inhibitor. (A) Chemical structure of GZD824; (B) GZD824 inhibits FGFR1/2/3/4 kinase activities in vitro by FRET‐based Z′‐Lyte assay; (C) GZD824 possess binding activities with FGFR1 in H1581 lung cancer cells by CETSA; (D) GZD824 possess binding activities with FGFR1‐V561F in Ba/F3‐FGFR1‐V561F cells by CETSA. Cells were treated with DMSO or GZD824 (1000 nM for H1581, 100 nM for Ba/F3‐FGFR1‐V561F) for 3 h, respectively. Drug‐treated cells were heated at gradient temperature from 38℃ to 57.8℃ for 3 min and then lysed, centrifuged to separate the soluble fractions from precipitates. The soluble fractions were analyzed by immunoblotting. Relative band intensities were plotted as a function of temperature followed a sigmoidal trend. The corresponding sigmoidal curve is on the right. GZD824 protected FGFR1 against thermal degradation, suggesting that the compound interacts with the receptor
TABLE 1.
IC50 and Kd values of GZD824 against FGFR1–4
| FGFRs | Kd (nM) | IC50 (nM) |
|---|---|---|
| FGFR1 | 7.3 ± 0.3 | 4.14 ± 0.96 |
| FGFR2 | 15.5 ± 3.5 | 2.77 ± 0.082 |
| FGFR3 | 17.0 ± 1.4 | 8.10 ± 0.15 |
| FGFR4 | 19.0 ± 1.4 | 30.27 ± 21.66 |
Binding activities were performed by KINOMEscan™ assay. Kinase inhibitory was performed by Z′‐Lyte method‐based FRET principle.
A commercial binding affinity assay, KINOMEscan™ was used to evaluate the potential activities of GZD824 against FGFR mutant resistance. It was shown that GZD824 possesses high binding affinities with FGFR1/2/3/4, with Kd values of 7.3 ± 0.3, 15.5 ± 3.5, 17.0 ± 1.4, and 19.0 ± 1.4 nM (Table 1), respectively, and also displays strong binding affinities to mutants FGFR2‐N550K, FGFR3‐G697C, FGFR3‐K650E, and FGFR3‐V555 M, with Kd values of 17.0 ± 5.7, 39.5 ± 7.8, 11.0 ± 0.0, and 19.5 ± 4.9 nM, respectively. We also investigated the binding properties of GZD824 with FGFR1 by a CETSA. The results showed that in the presence of GZD824, the stability of FGFR1 or FGFR1‐V561F proteins, but not of the control protein GAPDH, was significantly enhanced (Figure 1C and 1D). These results show that GZD824 can inhibit the activation of FGFRs and overcome FGFR mutant resistance.
3.2. GZD824 suppresses the proliferation of FGFR1–3‐driven cancer cells in vitro
To broadly assess the antiproliferative activities of GZD824 against cancer cells with FGFRs abnormal activation, we selected more than 10 solid cancer cells, including H1581 and A204 (FGFR1 amplification), KATOIII, NCI‐H716, and SNU16 (FGFR2 amplification), RT112 (FGFR3‐TACC3), MDA‐MB‐453 (FGFR4‐Y367C mutant activation), and other FGFRs with low expression of cancer cells as negative controls. The expression and activation levels of FGFR1–4 in these cells were determined via western blot experiments (Figure S1).
The results revealed that GZD824 exerted strongly antiproliferative activity against the cancer cells harboring FGFR aberrations in vitro, especially in FGFR1–FGFR3 cell lines, with IC50 values of 4.0–91.0 nM (Table 2). However, the activity of GZD824 against A549 and MDA‐MB‐231 cancer cells was minor with IC50 values of >1.0 μM, since no FGFR expression was observed (Table 2). Under the experimental conditions, the IC50 value of GZD824 is similar to that of BGJ398, but lower than that of ponatinib.
TABLE 2.
GZD824 inhibits proliferations in different cancer cells driven by FGFRs activation
| μΜ | FGFRs status | GZD824 | BGJ398 | FGF401 | Ponatinib |
|---|---|---|---|---|---|
| H1581 | FGFR1 amplification | 0.0108 ± 0.0008 | 0.0194 ± 0.0025 | 12.885 ± 4.046 | 0.1280 ± 0.0191 |
| A204 | FGFR1 amplification | 0.0244 ± 0.0062 | / | 8.248 ± 0.708 | 0.1690 ± 0.1343 |
| KATOIII | FGFR2 amplification | 0.0098 ± 0.0012 | 0.0171 ± 0.0024 | 9.3827 ± 2.8634 | 0.0187 ± 0.0008 |
| NCI‐H716 | FGFR2 amplification | 0.0040 ± 0.0005 | 0.0042 ± 0.0008 | 11.124 ± 1.668 | 0.0264 ± 0.0029 |
| SNU16 | FGFR2 amplification | 0.0144 ± 0.0048 | 0.0186 ± 0.0063 | / | 0.0782 ± 0.0102 |
| RT112 | FGFR3‐TACC3 | 0.0910 ± 0.0057 | 0.2334 ± 0.0767 | 14.523 ± 2.363 | 0.2161 ± 0.0082 |
| MDA‐MB−453 | FGFR4‐Y367C | 0.1482 ± 0.1044 | 2.6977 ± 0.1039 | 0.0623 ± 0.0277 | 0.7067 ± 0.0264 |
| MDA‐MB−231 | / | 1.998 ± 0.7819 | >10 | >10 | 0.7011 ± 0.0699 |
| A549 | / | 3.017 ± 0.1965 | 8.7333 ± 1.9281 | >10 | 0.9896 ± 0.0541 |
| MKN−1 | / | 0.7426 ± 0.0318 | 5.107 ± 0.481 | >10 | 0.758 ± 0.072 |
The antiproliferative activities of the compounds were evaluated using CCK‐8 assay. The cells were treated with compound or 0.1% DMSO for 72 h. Each data is expressed as mean ±SD of at least three independent experiments.
3.3. GZD824 overcomes FGFR1‐V561F/M mutant resistance in vitro
On the basis of the Kd assay, we posited that GZD824 possesses the potential to overcome FGFR1 gatekeeper mutant resistance. There is, however, no commercially available FGFR1 mutant kinase to validate this. Therefore, we constructed Ba/F3 model cells stably expressing FGFR1‐WT, K656E, K656N, V561M, and V561F mutations. We evaluated the antiproliferative activities of GZD824, two approved FGFRs inhibitors (pemigatinib and erdafitinib), a selective FGFR1/2/3 inhibitor (BGJ398), an irreversible pan‐FGFR1–4 inhibitor (TAS120), and multiple kinase ponatinib inhibitors in Ba/F3 stable cells. It was shown that the two marketed FGFR inhibitors were insensitive to these four FGFR1 mutants, with a 9.3‐ to 218‐fold increase in the IC50 values. BGJ398 also displayed resistance to V561F, V561M, and K656E with IC50 values of >330 nM, and excluded FGFR1‐K656N mutation (IC50 = 0.082 ± 0.0055 μΜ). The same as the reported FGFR1‐V561M kinase inhibitory activities (18.6 vs. 1.6: V561M vs. WT), ponatinib displayed a 10‐fold increase of IC50 value (0.40 ± 0.011 μΜ vs. 0.033 ± 0.0028 μΜ) in Ba/F3‐FGFR1‐V561M compared to Ba/F3‐FGFR1‐WT cells. We also found that TAS120 was insensitive to FGFR1‐V565M and FGFR1‐V561F, with IC50 values of 1.46 ± 0.12 μΜ and 1.00 ± 0.067μM, respectively, but it retains its sensitivity to K656E (0.017 ± 0.0022 μΜ) and K656N (0.010 ± 0.0022 μΜ) (Table 3).
TABLE 3.
GZD824 overcomes FGFR1 mutant resistance in Ba/F3 models
| IC50 (μΜ) | GZD824 | Pemigatinib | Erdafitinib | BGJ398 | TAS120 | Ponatinib |
|---|---|---|---|---|---|---|
| Ba/F3‐FGFR1 | 0.0081 ± 0.0011 | 0.015 ± 0.0015 | 0.023 ± 0.003 | 0.031 ± 0.0029 | 0.0068 ± 0.0006 | 0.033 ± 0.0028 |
| Ba/F3‐FGFR1‐V561M | 0.055 ± 0.0037 | 2.31 ± 0.35 | 2.59 ± 0.26 | 4.38 ± 0.28 | 1.46 ± 0.12 | 0.40 ± 0.011 |
| Ba/F3‐FGFR1‐V561F | 0.012 ± 0.0005 | 3.28 ± 0.10 | 2.49 ± 0.56 | 4.25 ± 0.064 | 1.00 ± 0.067 | 0.056 ± 0.0090 |
| Ba/F3‐FGFR1‐K656E | 0.0206 ± 0.0008 | 0.14 ± 0.0097 | 0.24 ± 0.048 | 0.33 ± 0.0029 | 0.017 ± 0.0022 | 0.088 ± 0.028 |
| Ba/F3‐FGFR1‐K656N | 0.012 ± 0.0020 | 0.025 ± 0.0063 | 0.13 ± 0.0088 | 0.082 ± 0.0055 | 0.010 ± 0.0022 | 0.047 ± 0.0088 |
| Ba/F3(+IL‐3) | 1.71 ± 0.59 | 8.98 ± 1.40 | 3.40 ± 0.032 | 5.66 ± 0.88 | >10 | 2.38 ± 0.42 |
The antiproliferative activities of the compounds were evaluated using CCK‐8 assay. The cells were treated with compound or 0.1% DMSO for 72 h. Each data is expressed as mean ±SD of at least three independent experiments.
We found that GZD824 can strongly inhibit FGFR1‐V561M, FGFR1‐V561F, and other K656E, K656N mutants in Ba/F3 models, with IC50 values of 0.055 ± 0.0037, 0.012 ± 0.0005, and 0.0206 ± 0.0008 and 0.012 ± 0.0020 μM, respectively (Table 3). This implies that GZD824 has a strong potential to overcome FGFR1 mutant resistance.
3.4. GZD824 inhibits phosphorylation of FGFR1 and downstream proteins
To study the mechanism of action of GZD824, the effects of GZD824 on the FGFR1 signaling pathway were investigated in H1581FGFR1 overexpression, A204 FGFR1 overexpression, Ba/F3‐FGFR1, Ba/F3‐FGFR1‐V561F, Ba/F3‐FGFR1‐V561M, and A549 FGFR1 lowexpression by western blotting analysis. As shown in Figure 2, treatment with GZD824 for 4 h revealed dose‐dependent inhibition of FGFR1 phosphorylation, the downstream FRS2 and ERK1/2 proteins progressing from 3.7 nM to 300 nM in H1581, Ba/F3‐FGFR1, and Ba/F3‐FGFR1‐V561F cells.
FIGURE 2.
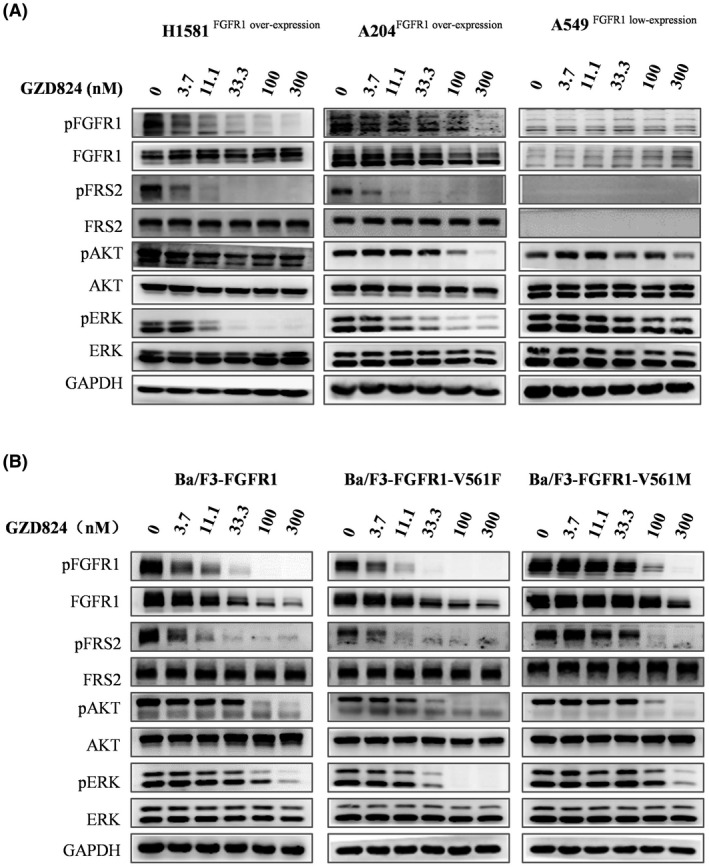
GZD824 suppresses FGFR1‐WT and V561F/M mutant signal pathway in cells (4 h). (A) Effects of GZD824 on FGFR1 signal pathway in H1581, A204, and A549 cancer cell lines. (B) Effects of GZD824 on FGFR1 signal pathway in Ba/F3 model cells. Cells were treated with or without compound GZD824 for 4 h at the indicated concentration, then harvested and lysed. Cell lysates were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and analyzed by western blot for phosphorylated FGFR1, FGFR1, phosphorylated FRS2, FRS2, phosphorylated AKT (Ser473), AKT, phosphorylated Erk, and Erk. GAPDH was used as a control
GZD824 displayed no obvious effect on the total level of FGFR1 protein expression in H1581, A204, and A549 cell lines (Figure 2A) but showed an obvious degradation of FGFR1 in all Ba/F3 stable cells at doses of 33.3 nM or higher, with an increased ubiquitination level of FGFR1 (Figure S2B). Further research validated that the proteasome inhibitor MG132 can rescue FGFR1 degradation induced by GZD824, which supports that this is a proteasome‐dependent pathway (Figure S2A). However, its effects on FGFR1 downstream signal pathway in A549 cells were very weak as a negative control.
Under the same conditions, upon treatment with 300 nM of pemigatinib, TAS120 or BGJ398 in Ba/F3‐FGFR1‐V561F and Ba/F3‐FGFR1‐V561M, no effects on activation of FGFR1 and its signal pathway were observed (Figure S3).
3.5. GZD824 induces G0/G1 phase arrest and apoptosis in Ba/F3‐FGFR1‐V561F cells
To further identify the efficacy of GZD824, the effects of GZD824 on cell cycle and apoptosis in Ba/F3‐FGFR1 and Ba/F3‐FGFR1‐V561F cell lines were detected by flow cytometry analysis. The results displayed that GZD824 dose‐dependently induces G0/G1 phase arrest and apoptosis in Ba/F3‐FGFR1 and Ba/F3‐FGFR1‐V561F. Treatment with 100 nM of GZD824 led to 70.88% and 71.13% G0/G1 phase arrest after 24 h, and 65.73% and 71.01% apoptosis after 48 h in Ba/F3‐FGFR1 and Ba/F3‐FGFR1‐V561F cells, respectively (Figure 3A and 3C). Under the same conditions, even treatment with 1000 nM of NVP‐BGJ398, pemigatinib, and TAS120 showed a weak effect on cell cycle and apoptosis in Ba/F3‐FGFR1‐V561F cells (Figure S5). WB analysis also showed that GZD824 dose‐dependently decreases the protein levels of CDK2, cyclin D2, CDK4, cyclin E, and the activating cleavage of caspase‐3 and caspase‐9 in the above FGFR1 model cells (Figure 3B and 3D).
FIGURE 3.
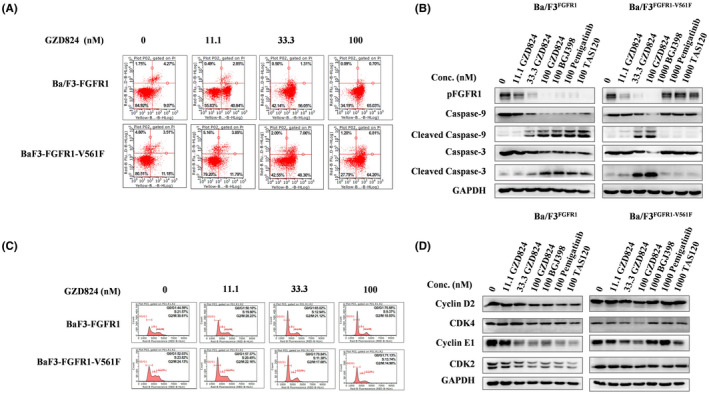
GZD824‐induced G0/G1 phase arrest and apoptosis in Ba/F3‐FGFR1‐WT and Ba/F3‐FGFR1‐V561F cells. (A) GZD824‐induced apoptosis in Ba/F3‐FGFR1 and Ba/F3‐FGFR1‐V561F cells. Cells were treated with various doses of GZD824 for 48 h before DNA labeling by Annexin V and 7‐AAD and cell apoptosis analysis by flow cytometry. Representative flow cytometry profiles were shown here. (B) Effects of GZD824 on apoptosis related proteins of capase‐3 and capase‐9 by western blotting analysis. (C) GZD824‐induced G0/G1 phase arrest in Ba/F3‐FGFR1 and Ba/F3‐FGFR1‐V561F cells. Cells were treated with various doses of GZD824 for 24 h before DNA labeling with propidium iodide and cell cycle analysis by flow cytometry. Representative flow cytometry profiles of cell cycle distribution were shown here. (D) Effects of GZD824 on cell cycle‐related proteins of Cyclin D2, CDK4, Cyclin E1, and CDK2 by western blotting analysis
3.6. GZD824 as a type II FGFR1 inhibitor overcomes FGFR1‐V561F/M mutant resistance
In order to explain the mechanism of sensitivity of GZD824 to the FGFR1‐V561M/F mutation, we performed an induced fit docking. The docking results of GZD824 with FGFR1 (PDB code: 4V04) shown in Figure 4 revealed that GZD824 as a type II inhibitor binds to the DFG‐out conformation of FGFR1‐WT, FGFR1‐V561M, and FGFR1‐V561F. The 1H‐pyrazolo[3,4‐b]pyridine core of GZD824 occupies the adenine pocket of FGFR1 kinase and forms hydrogen bond interactions with the backbone of Ala564 in the hinge region of the FGFR. The methylphenyl ring occupies the hydrophobic pocket located behind the gatekeeper residue Val561. The amide moiety is involved in two hydrogen bond interactions with the backbone nitrogen atom of Asp641 from the DFG motif and the side chain of the strictly conserved glutamate of the aC‐helix (Glu531), which is characteristic of type II inhibitors. The side chain of Phe642 is extended toward the solvent, creating an induced‐fit hydrophobic pocket into which the trifluoromethylphenyl ring binds. The terminal piperazinyl nitrogen atom of GZD824 is protonated and the resulting tertiary ammonium ion forms a hydrogen bond with the carbonyl oxygen atom of residue Ile620 in the catalytic loop. As shown in Figure 4, GZD824 binds to FGFR1‐V561M in a very similar fashion to that adopted by FGFR1‐WT. Although the 2.8 Å increase in length that occurs upon replacing a valine residue with methionine, GZD824 could participate in favorable van der Waals interactions with the gatekeeper Met without causing steric clashes. In the GZD824‐FGFR1 V561F docking complex, the side chain of Phe561 rotates to conserve the electrostatically favorable face‐to‐face π–π interaction with the methylphenyl ring of GZD824.
FIGURE 4.
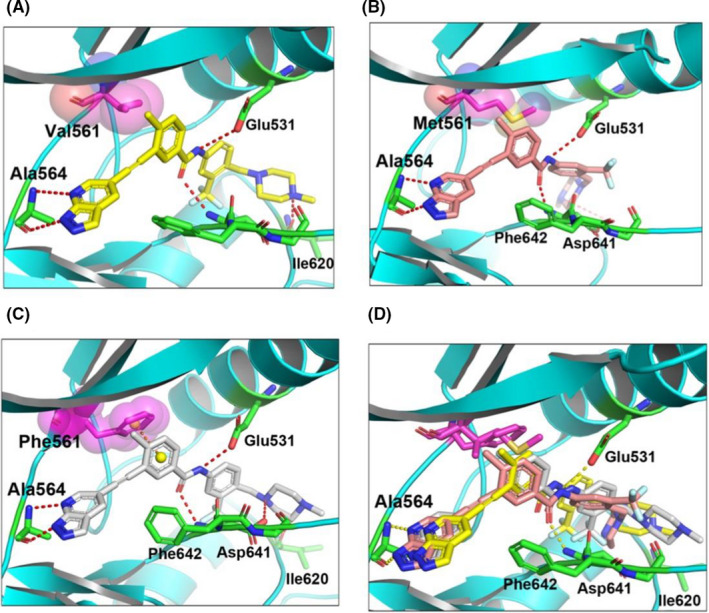
Binding mode analysis of GZD824 to FGFR1WT, FGFR1V561M, and FGFR1V561F. (A) Molecular docking of GZD824 into FGFR1WT (generated from PDB code 4V04). (B) Molecular docking of GZD824 into FGFR1V561M (generated from PDB code 4V04). (C) Molecular docking of GZD824 into FGFR1V561F (generated from PDB code 4V04). (D) Overlay of the binding conformation of GZD824 from the FGFR1WT, FGFR1V561M, and FGFR1V561F, respectively
Ponatinib binds to FGFR1 WT, FGFR1 V561M, and FGFR1 V561F in a very similar fashion to GZD824. BGJ398, pemigatinib, and TAS120 are not type II inhibitors and reasonable binding conformations are not found between these and FGFR1‐V561M or FGFR1‐V561F.
3.7. GZD824 overcomes FGFR1‐V561F and V561M mutant resistance in vivo
We further investigated the antitumor efficacy of GZD824 in Ba/F3‐FGFR1, Ba/F3‐FGFR1‐V561M, and Ba/F3‐FGFR1‐V561F mouse xenograft models. The animals were administered with vehicle, GZD824 once every 2 days at 10 mg/kg/day or 20 mg/kg/day or with BGJ398 once every day (10 mg/kg for Ba/F3‐FGFR1 and 30 mg/kg for Ba/FGFR1‐V561F) via oral gavage for 12 consecutive days. GZD824 displayed no mortality or significant loss of body weight observed during experiment (Figures S4A, S4B and S4C). GZD824 exhibited strong dose‐dependent antitumor efficacy with tumor growth inhibition (TGI) values of 76.5%, 49.8%, and 73.4% at dosages of 20 mg/kg (Bid) in Ba/F3‐FGFR1, Ba/F3‐FGFR1‐V561M, and Ba/F3‐FGFR1‐V561F, respectively (Figure 5A, 5B, and 5C). BGJ398 also displayed a TGI value of 64.2% in the Ba/F3‐FGFR1 xenograft model, but showed almost no effect on Ba/F3‐FGFR1‐V561F models even with a dose of 30 mg/kg (TGI=2.4%). IHC and TUNEL assays in the tumor tissues displayed that GZD824 inhibits the FGFR1 activation and induces apoptosis in Ba/F3‐FGFR1 and Ba/F3‐FGFR1‐V561F xenograft models (Figure 5D and 5E).
FIGURE 5.

In vivo effect of GZD824 in Ba/F3‐FGFR1, Ba/F3‐FGFR1‐V561M, and Ba/F3‐FGFR1‐V561F xenograft model. (A) Antitumor efficacy of GZD824 in Ba/F3‐FGFR1 xenograft mouse model. (B) Antitumor efficacy of GZD824 in Ba/F3‐FGFR1‐V561F xenograft mouse model. (C) Antitumor efficacy of GZD824 in Ba/F3‐FGFR1‐V561M xenograft mouse model. (D) pFGFR1, Ki67, and TUNEL staining of Ba/F3‐FGFR1 tumors harvested from mice treated with GZD824, BGJ398, or vehicle for 12 days. (E) pFGFR1, Ki67, and TUNEL staining of Ba/F3‐FGFR1‐V561F tumors harvested from mice treated with GZD824, BGJ398, or vehicle for 12 days. Tumors harvested from the mice after dosing vehicle, GZD824, or BGJ398 for 12 days. CB17‐SCID mice xenograft models were orally dosed once every 2 days (q2d) for 12 days with vehicle or GZD824, or once a day (qd) with BGJ398. Tumor volumes were measured every 2 days. Tumors were harvested from the mice at the terminal. (*p < 0.05, **p < 0.01)
4. DISCUSSION
FGFR1–FGFR4 exhibit various abnormal activations in different tumors, including overexpression, mutation and fusion, drive malignant tumor proliferation, migration, and invasion. These observations suggest that FGFR1–FGFR4 may be potential targets of broad‐spectrum targeted therapy for many cancers. The clinical treatment of FGFR2 and FGFR3 abnormal tumors has been successful, and two marketed drugs (erdafitinib and pemigatinib) have been approved to treat those two kinds of tumors, respectively. Although dozens of FGFR inhibitors have been developed, they have not been used successfully in the treatment of patients with abnormal FGFR1/4 activation and are still at the stage of clinical research.
FGFR1 can be considered a therapeutic target for many cancers from different tissues. 28 It has been reported that FGFR1 amplifications emerge in various of cancers, such as NSCLC (12%–17%), prostate cancer (16%), myxofibrosarcoma (20%), bladder cancer (9%), esophageal cancer (9%), colorectal cancer (6%), and in undifferentiated pleomorphic sarcomas (7%). 3 , 4 FGFR1 fusions such as BCR–FGFR1 29 and FGFR1OP2‐FGFR1 30 have been found in acute myeloid leukemia and other cancers. Due to resistance mediated by target mutation or bypass signal activation, treatment of FGFR1 amplified cancers with FGFR inhibitors is currently unsatisfactory. Reported data indicate that cancer cells harboring FGFR1 amplifications can acquire resistance to FGFR inhibitors mediated by overexpression of NRAS, MET amplification, mutational inactivation of PETN, and activation of AKT. 31 Gatekeeper mutations such as FGFR1‐V561M (FGFR2‐V564F/I, FGFR3‐V555M) are, however, the predominant resistant mechanism during treatment with FGFR‐targeted therapies. 19 Although many FGFR inhibitors such as erdafitinib, pemigatinib, and TAS120 have been developed, it has been reported that only LY2874455 and ponatinib can overcome FGFR1‐V561M mutant resistance.
The gatekeeper mutation could generate a steric clash, or eliminate critical hydrogen bonds required high‐affinity binding preventing inhibitors binding. Crystal structure of FGFR1‐V561M displayed that this gatekeeper mutation can stabilize the hydrophobic spine to promote catalytic activation, which was validated by FGFR1‐V561M conferring a 38‐fold increase in kcat relative to FGFR1‐WT by a kinase kinetic assay. 32 FIN‐2, FIN‐3, and TAS‐120, which have been designed to form a covalent bond with FGFR kinases to overcome FGFRs mutant resistance, were all successful in overcoming FGFR2‐K659N, C491A, and other mutations but not FGFR2‐V564F in cells or patients. In this paper, we first report the sensitives of FGFR1‐V561F to FGFR inhibitors which similar to V561M are insensitive to pemigatinib, erdafitinib, BGJ398, and TAS120. We were surprised to find that GZD824 can effectively overcome V561F mutant resistance in cells and animals with an IC50 lower than that of ponatinib. Our induced fit docking assays suggested that GZD824 possess a different binding model compared to those of type I inhibitors, which bind to FGFR1‐V561F/M in a very similar fashion to FGFR1‐WT, a type II FGFR1 inhibitor.
It has been reported that BGJ398 effectively inhibits the activation of ERK but not AKT in FGFR1‐amplified lung cancer cell lines. 1 In this research, GZD824 was found to exert properties similar to those of BGJ398 in H1581 and A204 cells, which displays obvious inhibition of pFRS2 and pERK at a dose of 3.7 nM or higher, but with no effect on pAKT, even at a dose of 300 nM. In FGFR2 overexpressed SNU‐16 and in NCI‐H716 cells, GZD824, at levels from 3.7 to 300 nM can strongly inhibit pAKT. We suggest these differences may be due to the different backgrounds of the cell lines.
In summary, we report GZD824 as a pan‐FGFR inhibitor which inhibits the signaling pathways of FGFR1 kinases and suppresses the proliferation of cancer cells harboring overexpression or mutant activation of FGFR1 in vitro and in vivo. GZD824 overcomes FGFR1‐V561F, V561M, K656E, and K656N mutant resistance, and effectively inhibits the growth of Ba/F3‐FGFR1‐V561F/M mutant xenograft tumors in vivo. GZD824 may be considered to be an effective drug to treat patients with FGFR1 abnormal activation or mutant resistance in clinical trials.
CONFLICT OF INTEREST
All authors declare that they have no conflict of interest.
ETHICAL STATEMENT
All animal studies were approved by the Institutional Animal Care and Use Committee of the Medical College of Jinan University. This article does not contain any studies with human performed by any of the authors. Informed consent was obtained from all individual participants included in the study.
Supporting information
Fig S1‐S5
ACKNOWLEDGMENTS
The authors appreciate the financial support from National Natural Science Foundation of China (21702075, 21572230, and 81973158), National Ministry of Science and Technology (SQ2019YFE010401), Guangdong Province (2019B020204002 and 2019A1515011235), Guangzhou city (202002030414), and Jinan University.
Jiang K, Tang X, Guo J, et al. GZD824 overcomes FGFR1‐V561F/M mutant resistance in vitro and in vivo. Cancer Med. 2021;10:4874–4884. 10.1002/cam4.4041
Kaili Jiang and Xia Tang contributed equally to the work.
Contributor Information
Ke Ding, Email: dingke@jnu.edu.cn.
Zhang Zhang, Email: zhang_zhang@jnu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Yuta A, Kazuyoshi W, Kenji K, et al. Resistance mediated by alternative receptor tyrosine kinases in FGFR1‐amplified lung cancer. Carcinogenesis. 2017;38(11):1063‐1072. [DOI] [PubMed] [Google Scholar]
- 2. Dai S, Zhou Z, Chen Z, Xu G, Chen Y. Fibroblast Growth Factor Receptors (FGFRs): structures and small molecule inhibitors. Cells. 2019;8(6):614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Irina SB, Nicholas CT. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318‐332. [DOI] [PubMed] [Google Scholar]
- 4. Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105‐122. [DOI] [PubMed] [Google Scholar]
- 5. Masaru K. FGFR inhibitors: effects on cancer cells, tumor microenvironment and whole‐body homeostasis (Review). Int J Mol Med. 2016;38(1):3‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barbara R, Tenzin G, Jian CZ, et al. Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol. 2016;131(6):847‐863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferone G, Song JY, Krijgsman O, et al. FGFR1 oncogenic activation reveals an alternative cell of origin of SCLC in Rb1/p53 mice. Cell Rep. 2020;30(11):3837‐3850.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Agelopoulos K, Richter GH, Schmidt E, et al. Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in Ewing sarcoma. Clin Cancer Res. 2015;21(21):4935‐4946. [DOI] [PubMed] [Google Scholar]
- 9. Dyson K, Rivera‐Zengotita M, Kresak J, et al. FGFR1 N546K and H3F3A K27M mutations in a diffuse leptomeningeal tumour with glial and neuronal markers. Histopathology. 2016;69(4):704‐707. [DOI] [PubMed] [Google Scholar]
- 10. D'Angelo A, Bagby S, Galli IC, Bortoletti C, Roviello G. Overview of the clinical use of erdafitinib as a treatment option for the metastatic urothelial carcinoma: where do we stand. Expert Rev Clin Pharmacol. 2020;13(10):1139‐1146. [DOI] [PubMed] [Google Scholar]
- 11. Weaver A, Bossaer JB. Fibroblast growth factor receptor (FGFR) inhibitors: a review of a novel therapeutic class. J Oncol Pharm Pract. 2020;29:1078155220983425. [DOI] [PubMed] [Google Scholar]
- 12. Guagnano V, Kauffmann A, Wöhrle S, et al. FGFR genetic alterations predict for sensitivity to NVP‐BGJ398, a selective pan‐FGFR inhibitor. Cancer Discov. 2012;2(12):1118‐1133. [DOI] [PubMed] [Google Scholar]
- 13. Perera TPS, Jovcheva E, Mevellec L, et al. Discovery and pharmacological characterization of JNJ‐42756493 (Erdafitinib), a functionally selective small‐molecule FGFR family inhibitor. Mol Cancer Ther. 2017;16(6):1010‐1020. [DOI] [PubMed] [Google Scholar]
- 14. Gavine PR, Mooney L, Kilgour E, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012;72(8):2045‐2056. [DOI] [PubMed] [Google Scholar]
- 15. Gozgit JM, Wong MJ, Moran L, et al. Ponatinib (AP24534), a multitargeted pan‐FGFR inhibitor with activity in multiple FGFR‐amplified or mutated cancer models. Mol Cancer Ther. 2012;1(3):690‐699. [DOI] [PubMed] [Google Scholar]
- 16. Kim KB, Chesney J, Robinson D, Gardner H, Shi MM, Kirkwood JM. Phase I/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin Cancer Res. 2011;17(23):7451‐7461. [DOI] [PubMed] [Google Scholar]
- 17. Bello E, Colella G, Scarlato V, et al. E‐3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res. 2011;71(4):1396‐1405. [DOI] [PubMed] [Google Scholar]
- 18. Hibi M, Kaneda H, Tanizaki J, et al. FGFR gene alterations in lung squamous cell carcinoma are potential targets for the multikinase inhibitor nintedanib. Cancer Sci. 2016;107(11):1667‐1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ryan MR, Sohl CD, Luo B, Anderson KS. The FGFR1 V561M gatekeeper mutation drives AZD4547 resistance through STAT3 activation and EMT. Mol Cancer Res. 2019;17(2):532‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donglou L, Qiaowan C, Yujin G, Ting Z, Wentao G. Insight into resistance mechanisms of AZD4547 and E3810 to FGFR1 gatekeeper mutation via theoretical study. Drug Des Devel Ther. 2017;11:451‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion‐positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tan L, Wang J, Tanizaki J. et al. Development of covalent inhibitors that can overcome resistance to first‐generation FGFR kinase inhibitors. Proc Natl Acad Sci USA. 2014;111(45):E4869‐E4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kalyukina M, Yosaatmadja Y, Middleditch MJ, et al. TAS‐120 cancer target binding: defining reactivity and revealing the first fibroblast growth factor receptor 1 (FGFR1) irreversible structure. ChemMedChem. 2019;14(4):494‐500. [DOI] [PubMed] [Google Scholar]
- 24. Goyal L, Shi L, Liu LY, et al. TAS‐120 overcomes resistance to ATP‐competitive FGFR inhibitors in patients with FGFR2 Fusion‐Positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019;9(8):1064‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu D, Guo M, Min X, et al. LY2874455 potently inhibits FGFR gatekeeper mutants and overcomes mutation‐based resistance. Chem Commun (Camb). 2018;54(85):12089‐12092. [DOI] [PubMed] [Google Scholar]
- 26. Ren X, Pan X, Zhang Z, et al. Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region‐Abelson (Bcr‐Abl) kinase and overcomes clinically acquired mutation‐induced resistance against imatinib. J Med Chem. 2013;56(3):879‐894. [DOI] [PubMed] [Google Scholar]
- 27. Mo C, Zhang Z, Guise CP, et al. 2‐Aminopyrimidine derivatives as new selective fibroblast growth factor receptor 4 (FGFR4) inhibitors. ACS Med Chem Lett. 2017;8(5):543‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Göke F, Franzen A, Hinz TK, et al. FGFR1 expression levels predict BGJ398 sensitivity of FGFR1‐dependent head and neck squamous cell cancers. Clin Cancer Res. 2015;21(19):4356‐4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roumiantsev S, Krause DS, Neumann CA, et al. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198‐FGFR1 and BCR‐FGFR1 fusion genes from 8p11 translocations. Cancer Cell. 2004;5(3):287‐298. [DOI] [PubMed] [Google Scholar]
- 30. Masahiro O, Kazumasa O, Makoto I, et al. The 8p11 myeloproliferative syndrome owing to rare FGFR1OP2‐FGFR1 fusion. Eur J Haematol. 2011;86(4):347‐349. [DOI] [PubMed] [Google Scholar]
- 31. Datta J, Damodaran S, Parks H, et al. Akt activation mediates acquired resistance to fibroblast growth factor receptor inhibitor BGJ398. Mol Cancer Ther. 2017;16(4):614‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sohl CD, Ryan MR, Luo BeiBei, Frey KM, Anderson KS. Illuminating the molecular mechanisms of tyrosine kinase inhibitor resistance for the FGFR1 gatekeeper mutation: the Achilles’ heel of targeted therapy. ACS Chem Biol. 2015;10(5):1319‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.